

ABSTRACT
Voltage-gated Kv1.1 potassium channel α-subunits are broadly expressed in the nervous system where they act as critical regulators of neuronal excitability. Mutations in the KCNA1 gene, which encodes Kv1.1, are associated with the neurological diseases episodic ataxia and epilepsy. Studies in mouse models have shown that Kv1.1 is important for neural control of the heart and that Kcna1 deletion leads to cardiac dysfunction that appears to be brain-driven. Traditionally, KCNA1 was not believed to be expressed in the heart. However, recent studies have revealed that Kv1.1 subunits are not only present in cardiomyocytes, but that they also make an important heart-intrinsic functional contribution to outward K+ currents and action potential repolarization. This review recounts the winding history of discovery of KCNA1 gene expression and neurocardiac function from fruit flies to mammals and from brain to heart and looks at some of the salient questions that remain to be answered regarding emerging cardiac roles of Kv1.1.
KEYWORDS: Kv1.1, Kcna1, heart, action potential, epilepsy, SUDEP
Introduction
Voltage-gated potassium (Kv) channels are important regulators of membrane excitability in the brain and heart, controlling action potential initiation, propagation, shape, and repetitive firing properties in neurons and cardiomyocytes [1,2]. Many Kv channel proteins are expressed in both brain and heart tissues where they can exert a dual influence over neuronal and cardiac function and where channelopathies can potentially lead to a mixture of neurological and cardiac diseases [3]. One such neurocardiac Kv channel subunit is Kv1.1. Despite being the first mammalian Kv channel subunit to be cloned and linked to human disease, the expression and function of Kv1.1 in the heart has only recently come to light. In this review, the circuitous discovery of Kv1.1 in the heart is recounted while also discussing the neural control of cardiac function by Kv1.1. Research on Kv1.1 provides a model example of fruitful bench-to-bedside translational research, whereby basic science findings have repeatedly led to subsequent confirmatory findings in patients.
Overview of Kv channel structure
Kv channels are composed of four α-subunit proteins that assemble as homo- or hetero-tetramers to form a membrane pore that allows the selective flux of K+ ions [4]. Each α-subunit protein is composed of six transmembrane segments (S1-S6), four of which (S1-S4) comprise a voltage sensor domain and two of which (S5-S6) make up the central pore region [5]. Linking S4 and S5 is a P-loop domain that confers ion selectivity [6]. In humans, the pore-forming Kv α-subunits are encoded by 40 different genes, which belong to 12 gene subfamilies (Kv1-Kv12), some of which exhibit alternative splicing and RNA editing [7]. The α-subunit tetramers form multiprotein complexes by co-assembling with up to four cytoplasmic auxiliary β-subunits, which influence channel gating and surface expression [8]. Thus, Kv channels represent a molecularly diverse group with an array of possible channel combinations.
Discovery of Kv1.1 transcripts in brain
The first mammalian voltage-gated potassium channel α-subunit gene to be cloned, sequenced, and characterized was Kcna1, which originally was isolated from mouse brain and named MBK1 for mouse brain potassium channel 1 gene [9]. The successful initial cloning of Kcna1 (MBK1) from mouse brain was soon followed several months later by the cloning and sequencing of highly homologous isoforms from rat cortical (RCK1) and hippocampal (RBK1) cDNA libraries, and then finally from human genomic DNA (HuKi or HK1) [10–12]. Because of the disparate and potentially confusing names given to these homologous channels identified in different species, a simplified nomenclature based on sequence relatedness was adopted that assigned a gene name of Kcna1 (KCNA1 in humans) and a channel protein name of Kv1.1 (K for potassium; v for voltage-dependent; and 1.1 for the first identified member of the channel subfamily 1) [13,14].
The breakthrough discovery of Kcna1 was made possible by the prior positional cloning of the Drosophila ortholog of Kv1.1, the Shaker (Sh) gene [15–18], which provided the conserved nucleotide sequences needed to successfully screen mammalian cDNA libraries for similar transcripts [9]. The Sh locus was first identified and so named because Sh mutants exhibit leg-shaking behavior when exposed to ether anesthesia such as during fly sorting [19]. Prior to its cloning, Sh had been predicted to encode a potassium channel based on functional studies. In electrophysiology experiments, Sh mutants exhibited prolongation of action potentials in adult fly giant fiber axons (Figure 1(a)) and of neurotransmitter release at larval neuromuscular junctions, which could be mimicked in normal flies by addition of the potassium channel blocker 4-aminopyridine [20,21].
Figure 1.
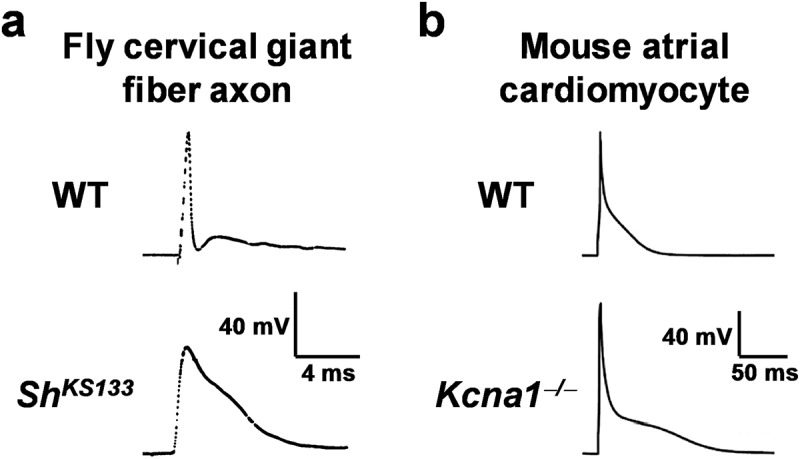
Action potential prolongation in Shaker flies and Kv1.1-deficient mice. (a) In mutant Shaker flies with the KS133 allele (ShKS133), the cervical giant fiber axon exhibits prolonged action potentials compared to normal wildtype (WT) flies due to delayed repolarization (images reproduced with permission of Mark Tanouye [20]). (b) In Kcna1–/ – mice lacking Kv1.1 channels, atrial cardiomyocytes exhibit significantly prolonged action potentials reminiscent of findings in the Shaker flies (images reproduced with permission [58]).
Despite their sequence similarities, the fly and mammalian orthologs of Kcna1 exhibit some important differences at the levels of gene regulation and function. The Drosophila Shaker locus contains 21 exons that form a single primary transcript, which is alternatively spliced to generate several distinct K+ channel proteins with differing physiological properties [22,23]. In contrast, the mammalian Kcna1 ortholog is intronless; therefore, mammals achieve Kv1 channel diversity by the presence of multiple gene loci [24]. Kv1 channel subunits can be broadly categorized into 2 functional groups: those that mediate non-inactivating delayed rectifier currents and those that mediate rapidly inactivating transient A-type currents. Shaker channels typically exhibit A-type currents due to the presence of a N-terminal protein domain that causes fast inactivation by a ball and chain mechanism [25,26]. Mammalian Kv1.1 channels lack this protein domain so they inactivate slowly unless complexed with another subunit that confers fast inactivation, such as a Kv1.4 α-subunit or a Kvβ1 subunit [27,28]. Therefore, the subunit stoichiometry of the channel complex determines the unique kinetics and gating properties of the channel. In the brain, Kv1.1 α-subunits form heterotetramers with Kv1.2, Kv1.4, and Kv1.6 [29]. The Kv1.6 α-subunits possess an N-type inactivation-prevention domain that opposes channel inactivation (i.e., the opposite of Kv1.4) to promote delayed rectifier currents [30]. Although they can form in vitro, Kv1.1 homotetramers have not been identified in vivo.
Discovery of Kv1.1 mutations causing neurological disorders
The first K+ channel gene directly linked to a human disease was KCNA1. Linkage studies and mutation analysis identified 4 different KCNA1 dominant missense mutations across 4 families that caused the neurological disease episodic ataxia type 1 (EA1), which is characterized by periodic stress-induced incoordination and myokymia (i.e., muscle rippling) [31]. These findings were later verified and studied in a mouse model of episodic ataxia that was generated by engineering animals with the same V408A mutation identified in one of the patients [32]. Following the discovery of an association between KCNA1 and EA1, Kcna1 mutation was found to cause epilepsy, first in mice and then in humans. In mice, an engineered deletion of the entire Kcna1 open reading frame to generate a null mutation (Kcna1–/–) resulted in a severe epilepsy phenotype, characterized by spontaneous tonic-clonic seizures that occurred several times daily and culminated in sudden death in about half of animals (Figure 2) [33]. The lethal seizures associated with the absence of Kv1.1 has led to Kcna1–/– mice being widely used as a model for elucidating the mechanisms underlying sudden unexpected death in epilepsy (SUDEP) [34–36]. SUDEP is generally defined as the sudden unexpected death of someone with epilepsy, who is otherwise healthy, for which no obvious cause of death can be found [37]. SUDEP is the leading cause of epilepsy-related mortality, accounting for the second most years of potential life lost among all neurological disorders [38]. Kcna1–/– mice recapitulate many of the risk factors and terminal neurocardiac patterns observed in human SUDEP victims including: (i) frequent seizures; (ii) generalized tonic–clonic seizures; (iii) early onset epilepsy; (iv) long duration of epilepsy; (v) young age; and (vi) seizure-associated cardiorespiratory arrest [33–36,39,40]. However, it should be noted that SUDEP is genetically complex and no single gene reaches genome-wide significance in exome sequencing studies [41,42].
Figure 2.
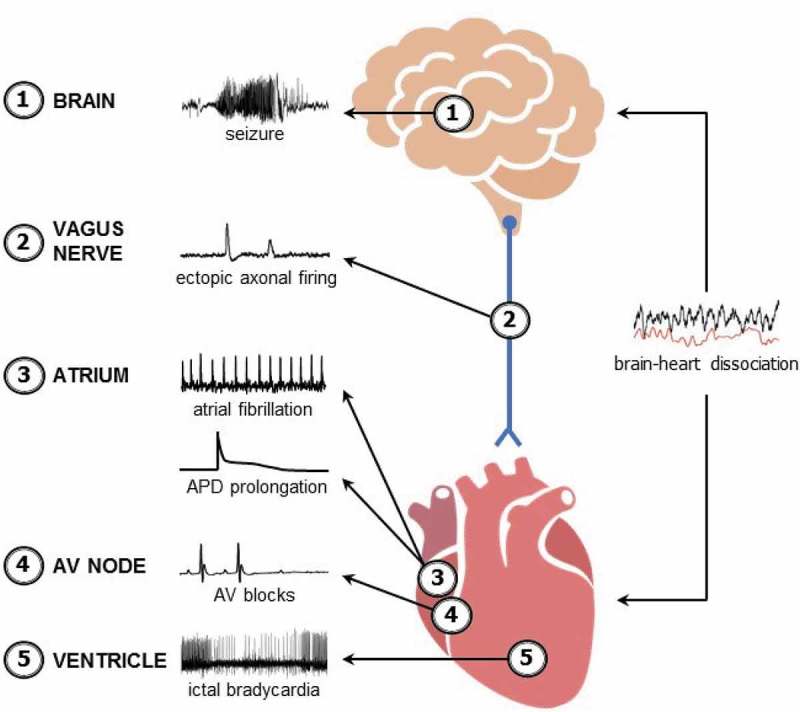
Summary of dysfunction along the brain-heart axis in Kcna1–/– mice. Kcna1–/– mice exhibit: (1) spontaneous tonic-clonic seizures which manifest as epileptiform electrographic activity in the brain [33]; (2) increased susceptibility to spontaneous ectopic action potential firing in the presence of 4-aminopyridine in myelinated vagus nerve axons [46]; (3) increased susceptibility to intracardiac-pacing induced atrial fibrillation and prolongation of action potential duration in atrial cardiomyocytes [57,58]; (4) increased frequency of atrioventricular (AV) nodal conduction blocks [35]; and (5) bradycardia during seizures [35,39]. In addition, analyses of brain-heart interaction dynamics reveals that Kcna1–/– mice have significantly decreased brain-heart association, which could be indicative of abnormal uncoupling of brain and heart activity that increases risk of sudden death [47].
The epilepsy phenotype associated with Kcna1 mutations represents an illustrative example of successful bench-to-bedside scientific discovery. About a year after the initial discovery that Kcna1 deletion causes epilepsy in mice, the first patients were identified with epilepsy due to KCNA1 loss-of-function missense mutations. In addition to EA1, these patients exhibited complex partial seizures with secondary generalization [43]. More recently, patients without ataxia have also been identified that have tonic-clonic seizures due to KCNA1 mutations [44]. Finally, a de novo copy number variant in KCNA1 was identified in a SUDEP victim, suggesting that KCNA1 mutations can increase susceptibility to SUDEP in humans [45].
Neural control of cardiac function by Kv1.1
Studies into the mechanisms of SUDEP in Kcna1–/– mice were the first to reveal cardiac abnormalities due to the absence of Kv1.1. In recordings of cardiac activity using electroencephalography (EEG) – electrocardiography (ECG) together or ECG telemetry alone, Kcna1–/– mice exhibited seizure-evoked atrioventricular (AV) conduction blocks, bradycardia, and asystole (Figure 2) [35]. SUDEP events in Kcna1–/– mice are associated with postictal bradycardia that progressively worsens leading to eventual cardiac arrest preceded by electrocortical silence [35,39]. EEG-ECG recordings also revealed that Kcna1–/– mice have interictal cardiac abnormalities including an increase in the frequency of AV blocks (Figure 2) [35]. These AV blocks can be eliminated by either autonomic blockade with co-administration of atropine and propranolol or selective parasympathetic blockade with atropine alone, but not by administration of the sympathetic blocker propranolol by itself [35]. Thus, the cardiac conduction blocks in Kcna1–/– mice appear to be brain-driven, specifically by the parasympathetic branch of the autonomic nervous system.
Measurements of heart rate variability (HRV) and vagus nerve function have revealed additional evidence of altered parasympathetic signaling underlying the cardiac abnormalities in Kcna1–/– mice. Kv1.1 channels are expressed in the myelinated axons of the vagus nerve [35,46]. When Kv1.1 is absent, myelinated vagal axons exhibit increased susceptibility to spontaneous ectopic firing in the presence of the Kv channel blocker 4-aminopyridine, indicative of hyperexcitability (Figure 2) [46]. Although in vivo heart rates (HR) appear mostly unchanged in Kcna1–/– mice, they do exhibit increases in the root mean square of successive beat-to-beat differences, a heart rate variability (HRV) measure of parasympathetic tone, suggesting increased vagal influence [35,47,48]. Unilateral transection of the vagus nerve in Kcna1–/– mice moderately increases lifespan by about 1–2 weeks, further implicating vagal mechanisms in SUDEP in this model [39].
In addition to vagal-specific aspects of cardiac control, Kcna1–/– mice also exhibit altered brain-heart interaction dynamics in EEG-ECG recordings. Typically, EEG and ECG data are analyzed separately when performing concurrent EEG-ECG recordings. However, a novel type of mathematical analysis, termed interaction dynamics, can be applied to the data to evaluate the degree of association between the brain (EEG) and heart (ECG) biosignals to reveal signs of neurocardiac dysfunction [47]. Normally, wild-type (WT) mice exhibit a negative correlation between the duration of the cardiac RR intervals of the ECG and the signal complexity (i.e., entropy) of the EEG, implying that HR and the entropy of brain signals tend to increase and decrease together [47]. In Kcna1–/– mice, this pattern is mostly abolished and the degree of association between EEG and ECG signals is significantly reduced, even during periods without seizures (Figure 2) [47]. Although the specific pathophysiology underlying this brain-heart dissociation needs to be further elucidated, the data points to a potential uncoupling of brain and heart activity in Kcna1–/– mice, which could be indicative of deleterious dysregulation of neural control of the heart and could increase risk for sudden death.
Discovery and re-discovery of Kv1.1 in the heart
Numerous older studies described Kcna1 expression in the heart, but these reports were largely dismissed at the time leading to the widespread assertion that Kv1.1 is absent in heart. The first report of Kv1.1 in the heart was in the year 1991 when Roberds and Tamkun used Northern blotting to detect Kcna1 mRNA in rat atrial tissue [49]. However, the authors hypothesized that the transcripts might arise from neuronal cells since the abundance was so low [49]. About 3 years later, Dixon and McKinnon attempted unsuccessfully to detect Kcna1 mRNA in rat atria and ventricles using an RNase protection assay method, which led them to conclude that the previous positive detection of Kcna1 mRNA in the atria by Roberds and Tamkun was likely due to contamination by neural tissues [50]. Following up on this work in rat heart, Brahmajothi et al used in situ hybridization to detect Kcna1 mRNA in cardiomyocytes isolated from ferret hearts, including sinoatrial nodal, atrial, and ventricular cells [51]. They rationalized that their fluorescent in situ hybridization technique permitted detection of low abundance Kcna1 transcripts due to the increased sensitivity of the method compared to RNase protection in the Dixon and McKinnon study. However, they also allowed for the possibility of species differences in channel expression that could explain the difference in results [51]. Finally, in 2001, London et al used Northern blots to detect Kcna1 mRNA in whole mouse heart, but they attributed this to non-myocytes since the expression bands were faint [52]. Because of the inconsistent results in these early studies and the assumption that positive detection was due to neuronal contamination, over time the accepted dogma became that Kcna1 was not expressed or functional in cardiomyocytes.
With the advent of highly sensitive PCR-based mRNA detection methods, such as real-time reverse transcriptase PCR (RT-PCR), multiple studies began to report cardiac Kcna1 transcripts using these new techniques, which began to cast doubt on the earlier conclusions that Kv1.1 is absent in the heart. Using real-time RT-PCR, Marionneau et al reported Kcna1 expression in the atria, ventricles, sinoatrial node (SAN), and atrioventricular node of mouse heart corroborating the previous work done in ferrets [51,53]. In a study of channel transcripts that are remodeled during chronic heart rate reduction, real-time RT-PCR revealed that Kcna1 mRNA levels are not only detectable in the SAN, but that they increase more than any other voltage-gated K+ channel in response to bradycardia [54]. In a comprehensive analysis of ion channel expression using a large-scale real-time quantitative RT-PCR assay, Harrell et al found Kcna1 to be the most abundantly expressed member of the Kv channel subfamily in mouse ventricles throughout perinatal development [55]. However, despite these successes using RT-PCR based methods in mouse heart, a study utilizing high-throughput real-time RT-PCR in human heart samples still found that Kv1.1 expression frequently fell below the threshold for detection [56]. Although newer and more sensitive PCR-based methods facilitated the reliable detection of Kcna1 mRNA in mouse hearts, Kv1.1 protein expression and function in the heart remained to be demonstrated.
In a series of papers by Glasscock et al, Kv1.1 protein expression was demonstrated in the heart for the first time [35,57]. Using western blotting, Kv1.1 protein was detected in whole mouse heart; however, the expression band was relatively weak and only visible in lysates with >100 μg of total protein [35]. In a follow up study, immunocytochemistry was used to reveal Kv1.1 localization in isolated atrial and ventricular myocytes from mouse hearts [57]. In atrial cells, Kv1.1 immunoreactivity showed a predominantly punctate intracellular staining pattern, whereas in ventricular cells, the staining pattern was of weaker intensity with clustering consistent with transverse (T) tubules [57]. The greater intensity of Kv1.1 immunoreactivity in atrial cells correlates with quantitative RT-PCR experiments which show that Kcna1 transcripts are about 10-fold more abundant in atrial myocytes compared to ventricular myocytes [57].
The demonstration of Kv1.1 expression at the protein level in mouse heart spurred additional investigation into the presence of Kv1.1 in human heart. First using RT-PCR and then immunoblotting, Kv1.1 mRNA and protein were detected in human atrial appendages [57]. Immunocytochemistry experiments using isolated human atrial myocytes revealed Kv1.1-positive immunoreactivity in a mostly intracellular staining pattern similar to results in mice, but also with some striations consistent with Kv1.1 localization at Z-lines where the T tubules reside [57]. Just as Kv1.1-associated epilepsy was discovered first in mice and then in humans, cardiac Kv1.1 expression was first demonstrated in mice, which led to its subsequent discovery in human heart, providing another example of successful bench-to-bedside discovery.
Intrinsic control of cardiac function by Kv1.1
The discovery of Kv1.1 in the heart opened the door to the possibility that Kv1.1 subunits make a direct intrinsic contribution to cardiac function, not only indirectly via the autonomic nervous system. The first indication that Kv1.1 could be important for intrinsic cardiac function came from experiments in Kcna1–/– mice using programmed electrical stimulation of the heart [57]. When the hearts of Kcna1–/– mice were stimulated using intracardiac burst pacing, they exhibited increased susceptibility to atrial fibrillation (AF; Figure 2). However, it should be noted that these experiments were performed in vivo with an intact nervous system, so neural influences on arrhythmogenesis cannot be entirely ruled out. To test whether Kv1.1 channels contribute to cardiac currents, patch-clamp electrophysiological recordings were performed in isolated human atrial myocytes to measure outward K+ currents [57]. In the presence of dendrotoxin-K (DTX-K), a specific blocker of Kv1.1 subunits, the peak and late outward K+ currents were significantly reduced, providing the first demonstration of functional expression of Kv1.1 in the heart and suggesting the subunits contribute to cardiac repolarization. When Kv1.1 protein was originally detected in human heart, the levels were found to significantly increase by about 75% in patients with chronic AF (cAF), suggesting disease-associated alterations in expression [57]. When outward K+ currents were measured in human atrial myocytes isolated from cAF patients, the DTX-K-sensitive portion of the current was increased two- to three-fold compared to patients in sinus rhythm [57]. This augmentation in current correlated with the increase in protein levels in cAF patients, suggesting that Kv1.1 undergoes electrical remodeling that may contribute to cAF pathomechanisms.
In a recent study, Si et al performed patch-clamp recordings of atrial cardiomyocytes isolated from Kcna1–/– mice revealing that Kv1.1-mediated currents regulate atrial repolarization and action potential morphology [58]. As observed in human atrial cells, WT mouse atrial myocytes also exhibited a significant DTX-K-sensitive outward K+ current component indicative of a contribution by Kv1.1-containing channels [58]. Importantly, DTX-K-sensitive outward K+ currents were absent in Kcna1–/– cells which lack Kv1.1, demonstrating the specificity of DTX-K for Kv1.1 and suggesting that the DTX-K-sensitive currents recorded in human cells were indeed indicative of functional Kv1.1 subunits and not some off target effect [58]. In addition to measuring outward K+ currents, Si et al also recorded the effects of Kv1.1-deficiency on cardiac action potential morphology for the first time. Kcna1–/– atrial myocytes exhibited significantly prolonged action potential durations (APD) at 90% repolarization (APD90) compared to WT cells (Figures 1(b) and 2). Interestingly, the already prolonged APD durations in Kcna1–/– mice were significantly longer in cells from the right atrium versus the left atrium, suggesting Kv1.1 may contribute to interatrial differences in repolarization that are present in mammals to maintain normal right-to-left atrial electrical conduction [58–60]. APD prolongation could also be induced in WT cells by administration of DTX-K, but not in Kcna1–/– cells, which further demonstrates the specificity of the toxin for Kv1.1 [58]. These electrophysiology studies showed that not only is the Kcna1 gene expressed in cardiomyocytes, but that channels containing Kv1.1 are functional in the heart and critical for cardiac repolarization and action potential waveform properties which could influence arrhythmogenesis. Thus, Kv1.1 represents a novel cardiac K+ channel.
Unanswered questions about Kv1.1 in the heart
Elucidation of the role of Kv1.1 in the heart is still in its early stages and several important questions remain to be explored to fully understand the effects of Kv1.1 dysfunction on cardiac disease and arrhythmia susceptibility. Mammalian hearts express several prominent repolarizing K+ currents mediated by voltage-gated K+ channels, including transient outward (Ito) and delayed rectifier currents (IK) [61]. The primary K+ channel subunits that encode these various cardiac repolarizing currents are generally thought to be known and have been extensively characterized. Maybe Kv1.1 contributes to one of these already identified currents or perhaps it encodes a subtle current component not yet described. Another important aspect of the function of Kv1.1-containing channels is subunit stoichiometry. Do Kv1.1 channels form hetero-tetramers with one of the previously identified cardiac channels to mediate one of these well known cardiac currents? If Kv1.1 forms functional homo-tetrameric channels in the heart, it would be the first in vivo demonstration of Kv1.1 homo-tetramers in any cell or tissue type. The identity of interacting auxiliary β-subunits will also need to be determined, especially since these can impact channel gating and trafficking.
Although Kcna1 deletion in mice has been associated with increased AF susceptibility, human KCNA1 mutations causing AF have not yet been reported. The lack of human patients with AF due to KCNA1 variants could be due to sampling bias or it could be indicative of a species difference in the arrhythmogenic roles of Kv1.1 in the heart. Given the bench-to-bedside trajectory that Kv1.1 studies have taken, it would be appropriate if AF-causing KCNA1 mutations were identified following the original discovery of AF susceptibility in the Kcna1–/– mouse model. In addition, the actual mechanism whereby Kv1.1 deficiency or dysfunction can increase atrial arrhythmia susceptibility requires elucidation. One possibility is that atrial APD prolongation related to Kv1.1 mutation promotes early afterdepolarizations that increase the likelihood of AF. To date, studies of Kv1.1 function and expression in the heart have focused on the atria. However, Kv1.1 mRNA and protein have been detected in the ventricles using RT-PCR and immunocytochemistry, respectively [35,51,53,55,57]. Kv1.1 channels would be expected to contribute to repolarization in ventricular cells as observed in atria, but possibly to a lesser extent since mouse ventricular expression levels are ~10% of the levels in atria. Since the ventricles can be a source of lethal arrhythmias and Kcna1–/– mice exhibit seizure-related sudden death, determining the effects of Kv1.1 mutations on ventricular arrhythmia susceptibility is especially important. Future studies will also need to address the expression of Kv1.1 in human ventricles, which may be substantially less than atria as suggested by expression studies which report low abundance mRNA levels [56].
Finally, the relationship between cardiac Kv1.1 channels and SUDEP deserves additional investigation. Although the discovery of functional Kv1.1 channels in the heart raises the possibility of an intrinsic cardiac contribution to SUDEP in the Kcna1–/– mouse model, it does not necessarily rule out brain-driven mechanisms as the primary cause. One possibility is that the absence of Kv1.1 in the heart may not be required for SUDEP, but that it renders the heart a vulnerable substrate for lethal cardiac arrhythmias triggered by seizures. Thus, the relationship between Kv1.1-deficient brain and heart needs to be examined further to determine the relative contributions of the two tissues to SUDEP and their potentially deleterious synergistic interactions.
Conclusions
In the span of the last 30 years, Kv1.1 channels have been discovered in the heart, dismissed as absent in the heart, and then re-discovered in the heart. Although the neural roles of Kv1.1 are well known, potential cardiac roles of Kv1.1 are just now being elucidated. Studies of the Kcna1 gene have been a model example of fruitful bench-to-bedside discovery. Kcna1 mutations were first found to cause epilepsy in mice which led to their discovery in human epilepsy patients. Kcna1 mutations were first found to increase susceptibility to SUDEP in mice, which then led to the successful identification of a SUDEP patient with a KCNA1 variant that was a principal risk factor in their death. Kcna1 expression was first found in rodent hearts which led to its subsequent discovery in human heart. Given this bench-to-bedside pattern of discovery, future studies in the lab on the neurocardiac roles of Kv1.1 are almost certain to uncover additional unexpected insights that will continue to get translated into potentially life-saving tools in the clinic.
Funding Statement
This study was supported by grants from the National Institutes of Health (R01 NS-100954 and R01 NS-099188).
Disclosure statement
No potential conflict of interest was reported by the author.
References
- [1].Priest BT, McDermott JS.. Cardiac ion channels. Channels. 2015;9:352–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Lai HC, Jan LY. The distribution and targeting of neuronal voltage-gated ion channels. Nat Rev Neurosci. 2006;7:548. [DOI] [PubMed] [Google Scholar]
- [3].Glasscock E. Genomic biomarkers of SUDEP in brain and heart. Epilepsy Behav. 2014;38:172–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kreusch A, Pfaffinger PJ, Stevens CF, et al. Crystal structure of the tetramerization domain of the Shaker potassium channel. Nature. 1998;392:945–948. [DOI] [PubMed] [Google Scholar]
- [5].Chen X, Wang Q, Ni F, et al. Structure of the full-length Shaker potassium channel Kv1.2 by normal-mode-based X-ray crystallographic refinement. Proc Natl Acad Sci. 2010;107:11352–11357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Heginbotham L, Abramson T, MacKinnon R. A functional connection between the pores of distantly related ion channels as revealed by mutant K+ channels. Science. 1992;258:1152–1155. [DOI] [PubMed] [Google Scholar]
- [7].Gutman GA, Chandy KG, Grissmer S, et al. International union of pharmacology. LIII. Nomenclature and molecular relationships of voltage-gated potassium channels. Pharmacol Rev. 2005;57:473–508. [DOI] [PubMed] [Google Scholar]
- [8].Pongs O, Schwarz JR. Ancillary subunits associated with voltage-dependent K+ channels. Physiol Rev. 2010;90:755–796. [DOI] [PubMed] [Google Scholar]
- [9].Tempel BL, Jan YN, Jan LY. Cloning of a probable potassium channel gene from mouse brain. Nature. 1988;332:837–839. [DOI] [PubMed] [Google Scholar]
- [10].Baumann A, Grupe A, Ackermann A, et al. Structure of the voltage-dependent potassium channel is highly conserved from Drosophila to vertebrate central nervous systems. Embo J. 1988;7:2457–2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Christie MJ, Adelman JP, Douglass J, et al. Expression of a cloned rat brain potassium channel in Xenopus oocytes. Science. 1989;244:221–224. [DOI] [PubMed] [Google Scholar]
- [12].Ramaswami M, Gautam M, Kamb A, et al. Human potassium channel genes: molecular cloning and functional expression. Mol Cell Neurosci. 1990;1:214–223. [DOI] [PubMed] [Google Scholar]
- [13].Gutman GA, Chandy KG. Nomenclature of mammalian voltage-dependent potassium channel genes. Semin Neurosci. 1993;5:101–106. [Google Scholar]
- [14].Chandy KG. Simplified gene nomenclature. Nature. 1991;352:26. [DOI] [PubMed] [Google Scholar]
- [15].Papazian DM, Schwarz TL, Tempel BL, et al. Cloning of genomic and complementary DNA from Shaker, a putative potassium channel gene from Drosophila. Science. 1987;237:749–753. [DOI] [PubMed] [Google Scholar]
- [16].Kamb A, Iverson LE, Tanouye MA. Molecular characterization of Shaker, a Drosophila gene that encodes a potassium channel. Cell. 1987;50:405–413. [DOI] [PubMed] [Google Scholar]
- [17].Pongs O, Kecskemethy N, Müller R, et al. Shaker encodes a family of putative potassium channel proteins in the nervous system of Drosophila. Embo J. 1988;7:1087–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Tempel BL, Papazian DM, Schwarz TL, et al. Sequence of a probable potassium channel component encoded at Shaker locus of Drosophila. Science. 1987;237:770–775. [DOI] [PubMed] [Google Scholar]
- [19].Kaplan WD, Trout WE. The behavior of four neurological mutants of Drosophila. Genetics. 1969;61:399–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Tanouye MA, Ferrus A, Fujita SC. Abnormal action potentials associated with the Shaker complex locus of Drosophila. Proc Natl Acad Sci U S A. 1981;78:6548–6552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Jan YN, Jan LY, Dennis MJ. Two mutations of synaptic transmission in Drosophila. Proc R Soc Lond B Biol Sci. 1977;198:87–108. [DOI] [PubMed] [Google Scholar]
- [22].Schwarz TL, Tempel BL, Papazian DM, et al. Multiple potassium-channel components are produced by alternative splicing at the Shaker locus in Drosophila. Nature. 1988;331:137–142. [DOI] [PubMed] [Google Scholar]
- [23].Timpe LC, Schwarz TL, Tempel BL, et al. Expression of functional potassium channels from Shaker cDNA in Xenopus oocytes. Nature. 1988;331:143–145. [DOI] [PubMed] [Google Scholar]
- [24].Chandy KG, Williams CB, Spencer RH, et al. A family of three mouse potassium channel genes with intronless coding regions. Science. 1990;247:973–975. [DOI] [PubMed] [Google Scholar]
- [25].Hoshi T, Zagotta WN, Aldrich RW. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science. 1990;250:533–538. [DOI] [PubMed] [Google Scholar]
- [26].Iverson LE, Tanouye MA, Lester HA, et al. A-type potassium channels expressed from Shaker locus cDNA. Proc Natl Acad Sci. 1988;85:5723–5727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Stühmer W, Ruppersberg JP, Schröter KH, et al. Molecular basis of functional diversity of voltage-gated potassium channels in mammalian brain. Embo J. 1989;8:3235–3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Rettig J, Heinemann SH, Wunder F, et al. Inactivation properties of voltage-gated K+ channels altered by presence of beta-subunit. Nature. 1994;369:289–294. [DOI] [PubMed] [Google Scholar]
- [29].Wang FC, Parcej DN, Dolly JO. Alpha subunit compositions of Kv1.1-containing K+ channel subtypes fractionated from rat brain using dendrotoxins. Eur J Biochem FEBS. 1999;263:230–237. [DOI] [PubMed] [Google Scholar]
- [30].Roeper J, Sewing S, Zhang Y, et al. NIP domain prevents N-type inactivation in voltage-gated potassium channels. Nature. 1998;391:390–393. [DOI] [PubMed] [Google Scholar]
- [31].Browne DL, Gancher ST, Nutt JG, et al. Episodic ataxia/myokymia syndrome is associated with point mutations in the human potassium channel gene, KCNA1. Nat Genet. 1994;8:136–140. [DOI] [PubMed] [Google Scholar]
- [32].Herson PS, Virk M, Rustay NR, et al. A mouse model of episodic ataxia type-1. Nat Neurosci. 2003;6:378–383. [DOI] [PubMed] [Google Scholar]
- [33].Smart SL, Lopantsev V, Zhang CL, et al. Deletion of the K(V)1.1 potassium channel causes epilepsy in mice. Neuron. 1998;20:809–819. [DOI] [PubMed] [Google Scholar]
- [34].Simeone KA, Matthews SA, Rho JM, et al. Ketogenic diet treatment increases longevity in Kcna1-null mice, a model of sudden unexpected death in epilepsy. Epilepsia. 2016;57:e178–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Glasscock E, Yoo JW, Chen TT, et al. Kv1.1 potassium channel deficiency reveals brain-driven cardiac dysfunction as a candidate mechanism for sudden unexplained death in epilepsy. J Neurosci. 2010;30:5167–5175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Dhaibar H, Gautier NM, Chernyshev OY, et al. Cardiorespiratory profiling reveals primary breathing dysfunction in Kcna1-null mice: implications for sudden unexpected death in epilepsy. Neurobiol Dis. 2019;127:502–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Nashef L, So EL, Ryvlin P, et al. Unifying the definitions of sudden unexpected death in epilepsy. Epilepsia. 2012;53:227–233. [DOI] [PubMed] [Google Scholar]
- [38].Thurman DJ, Hesdorffer DC, French JA. Sudden unexpected death in epilepsy: assessing the public health burden. Epilepsia. 2014;55:1479–1485. [DOI] [PubMed] [Google Scholar]
- [39].Moore BM, Jerry Jou C, Tatalovic M, et al. The Kv1.1 null mouse, a model of sudden unexpected death in epilepsy (SUDEP). Epilepsia. 2014;55:1808–1816. [DOI] [PubMed] [Google Scholar]
- [40].Fenoglio-Simeone KA, Wilke JC, Milligan HL, et al. Ketogenic diet treatment abolishes seizure periodicity and improves diurnal rhythmicity in epileptic Kcna1-null mice. Epilepsia. 2009;50:2027–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Leu C, Balestrini S, Maher B, et al. Genome-wide polygenic burden of rare deleterious variants in sudden unexpected death in epilepsy. EBioMedicine. 2015;2:1063–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Bagnall RD, Crompton DE, Petrovski S, et al. Exome-based analysis of cardiac arrhythmia, respiratory control, and epilepsy genes in sudden unexpected death in epilepsy. Ann Neurol. 2016;79:522–534. [DOI] [PubMed] [Google Scholar]
- [43].Zuberi SM, Eunson LH, Spauschus A, et al. A novel mutation in the human voltage-gated potassium channel gene (Kv1.1) associates with episodic ataxia type 1 and sometimes with partial epilepsy. Brain. 1999;122(Pt 5):817–825. [DOI] [PubMed] [Google Scholar]
- [44].Liguori R, Avoni P, Baruzzi A, et al. Familial continuous motor unit activity and epilepsy. Muscle Nerve. 2001;24:630–633. [DOI] [PubMed] [Google Scholar]
- [45].Klassen TL, Bomben VC, Patel A, et al. High-resolution molecular genomic autopsy reveals complex sudden unexpected death in epilepsy risk profile. Epilepsia. 2014;55:e6–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Glasscock E, Qian J, Kole MJ, et al. Transcompartmental reversal of single fibre hyperexcitability in juxtaparanodal Kv1.1-deficient vagus nerve axons by activation of nodal KCNQ channels. J Physiol. 2012;590:3913–3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Mishra V, Karumuri BK, Gautier NM, et al. Scn2a deletion improves survival and brain-heart dynamics in the Kcna1-null mouse model of sudden unexpected death in epilepsy (SUDEP). Hum Mol Genet. 2017;26:2091–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Vanhoof-Villalba SL, Gautier NM, Mishra V, et al. Pharmacogenetics of KCNQ channel activation in 2 potassium channelopathy mouse models of epilepsy. Epilepsia. 2018;59:358–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Roberds SL, Tamkun MM. Cloning and tissue-specific expression of five voltage-gated potassium channel cDNAs expressed in rat heart. Proc Natl Acad Sci U S A. 1991;88:1798–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Dixon JE, McKinnon D. Quantitative analysis of potassium channel mRNA expression in atrial and ventricular muscle of rats. Circ Res. 1994;75:252–260. [DOI] [PubMed] [Google Scholar]
- [51].Brahmajothi MV, Morales MJ, Rasmusson RL, et al. Heterogeneity in K+ channel transcript expression detected in isolated ferret cardiac myocytes. Pacing Clin Electrophysiol PACE. 1997;20:388–396. [DOI] [PubMed] [Google Scholar]
- [52].London B, Guo W, Pan X, et al. Targeted replacement of KV1.5 in the mouse leads to loss of the 4-aminopyridine-sensitive component of I(K,slow) and resistance to drug-induced qt prolongation. Circ Res. 2001;88:940–946. [DOI] [PubMed] [Google Scholar]
- [53].Marionneau C, Couette B, Liu J, et al. Specific pattern of ionic channel gene expression associated with pacemaker activity in the mouse heart. J Physiol. 2005;562:223–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Leoni A-L, Marionneau C, Demolombe S, et al. Chronic heart rate reduction remodels ion channel transcripts in the mouse sinoatrial node but not in the ventricle. Physiol Genomics. 2005;24:4–12. [DOI] [PubMed] [Google Scholar]
- [55].Harrell MD, Harbi S, Hoffman JF, et al. Large-scale analysis of ion channel gene expression in the mouse heart during perinatal development. Physiol Genomics. 2007;28:273–283. [DOI] [PubMed] [Google Scholar]
- [56].Gaborit N, Le Bouter S, Szuts V, et al. Regional and tissue specific transcript signatures of ion channel genes in the non-diseased human heart. J Physiol. 2007;582:675–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Glasscock E, Voigt N, McCauley MD, et al. Expression and function of Kv1.1 potassium channels in human atria from patients with atrial fibrillation. Basic Res Cardiol. 2015;110:505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Si M, Trosclair K, Hamilton KA, et al. Genetic ablation or pharmacological inhibition of Kv1.1 potassium channel subunits impairs atrial repolarization in mice. Am J Physiol Cell Physiol. 2018;316:C154–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Lomax AE, Kondo CS, Giles WR. Comparison of time- and voltage-dependent K+ currents in myocytes from left and right atria of adult mice. Am J Physiol Heart Circ Physiol. 2003;285:H1837–1848. [DOI] [PubMed] [Google Scholar]
- [60].Li D, Zhang L, Kneller J, et al. Potential ionic mechanism for repolarization differences between canine right and left atrium. Circ Res. 2001;88:1168–1175. [DOI] [PubMed] [Google Scholar]
- [61].Nerbonne JM. Molecular basis of functional myocardial potassium channel diversity. Card Electrophysiol Clin. 2016;8:257–273. [DOI] [PMC free article] [PubMed] [Google Scholar]