

OBJECTIVES:
Celiac disease (CD) is a chronic enteropathy characterized by an autoimmune reaction in the small intestine of genetically susceptible individuals. The underlying causes of autoimmune reaction and its effect on host metabolism remain largely unknown. Herein, we apply lipidomics to elucidate the early events preceding clinical CD in a cohort of Finnish children, followed up in the Type 1 Diabetes Prediction and Prevention study.
METHODS:
Mass spectrometry–based lipidomics profiling was applied to a longitudinal/prospective series of 233 plasma samples obtained from CD progressors (n = 23) and healthy controls (n = 23), matched for human leukocyte antigen (HLA) risk, sex, and age. The children were followed from birth until diagnosis of clinical CD and subsequent introduction of a gluten-free diet.
RESULTS:
Twenty-three children progressed to CD at a mean age of 4.8 years. They showed increased amounts of triacylglycerols (TGs) of low carbon number and double bond count and a decreased level of phosphatidylcholines by age 3 months as compared to controls. These differences were exacerbated with age but were not observed at birth (cord blood). No significant differences were observed in the essential TGs.
DISCUSSION:
Our preliminary findings suggest that abnormal lipid metabolism associates with the development of clinical CD and occurs already before the first introduction of gluten to the diet. Moreover, our data suggest that the specific TGs found elevated in CD progressors may be due to a host response to compromised intake of essential lipids in the small intestine, requiring de novo lipogenesis.
INTRODUCTION
Celiac disease (CD) is a chronic, systemic, autoimmune enteropathy triggered by dietary gluten and related prolamins from rye and barley in genetically susceptible individuals (1). Approximately 90%–95% of patients with CD express human leukocyte antigen (HLA)-DQ2 protein, whereas the remaining (5%–10%) express HLA-DQ8 (2). CD is characterized by a wide range of gastrointestinal and extraintestinal symptoms that include diarrhea, weight loss, abdominal distention, malabsorption, and iron deficiency anemia (1). Serologic tests such as measurement of serum immunoglobulin A (IgA) and/or immunoglobulin G (IgG) tissue transglutaminase antibodies, IgA endomysial antibodies, and deamidated gliadin peptide antibodies (IgG class) are performed for screening and diagnosis of CD. In addition, a biopsy of small intestine is still required in many countries to confirm the diagnosis (3).
The incidence of CD and other autoimmune diseases such as type 1 diabetes (T1D) has been increasing in children and adults over the past decades (4,5). The occurrence of these autoimmune diseases is higher in the Nordic countries (6) than elsewhere, with the highest prevalence of CD occurring in Sweden (29/1,000 by age 12 years) and the highest incidence rate of T1D occurring in Finland (64/100,000/year for children younger than 15 years) (7,8). Approximately 10% of patients with T1D develop overt CD (9). On the other hand, people with CD are at risk of T1D before age 20 years (10). These autoimmune diseases share common, predisposing alleles in the class II HLA-region as the DR3-DQ2 and DR4-DQ8 haplotypes (11).
Recent studies show that in addition to genetic predisposition and exposure to dietary gluten, other factors such as the composition of the intestinal microbiota, birth delivery mode, infant feeding, and the use of antibiotics may also affect the onset of CD (12). Thus, the early pathogenesis of CD is still poorly understood, and the identification of molecular signatures associated with progression to overt CD remains an unmet medical need (13). The health burden of CD in terms of quality of life, complications, mortality, and cost of treatment is considerable, meaning that its prevention has become, in the last decade, an important area of research.
Metabolomics is the study of small (<1,500 Da) molecules and their functions in cells, tissues, and body fluids (14). Metabolomic studies in adults diagnosed with active CD (15) identified marked changes in serum and urine metabolic profiles, along with altered intestinal microbiota (16–18). Interestingly, a recent, small prospective study suggests an altered early trajectory of the gut microbiome (ages 4 and 6 months) in children who later progressed to CD (19).
Lipids are a structurally and functionally highly diverse group of metabolites with many important biological functions including as components of cell membranes, intermediates in signaling pathways, and as energy sources. The emergence of lipidomics, a discipline closely related to metabolomics, has enabled researchers to study lipidomes and lipid metabolism at the systems level (20,21). Because of their diverse roles in biological systems, it is not surprising that lipid-related disturbances occur in many common diseases. In our previous lipidomics studies, for example, we found that dysregulated lipid metabolism precedes islet autoimmunity and overt disease in children who later progressed to T1D (22,23).
Here, we applied a lipidomics approach, in a longitudinal study setting, with the aim of elucidating the early events preceding onset of overt CD.
METHODS
Study design and protocol
The children included in the present study are from the Type 1 Diabetes Prediction and Prevention (DIPP) cohort, which is an ongoing prospective study initiated in 1994. In the DIPP study, parents of newborn infants at the University Hospitals of Turku, Tampere, and Oulu in Finland are asked for permission to screen the child for HLA alleles conferring the risk of T1D, using umbilical cord blood. Families of children identified as having an increased HLA-conferred risk of T1D are invited to join the study. Our analysis included children born at Tampere University Hospital between August 1999 and September 2005. During that period, 23,839 children were screened at birth for increased risk of T1D, and 2,642 eligible children were enrolled in the follow-up and had at least 2 visits to the study clinic. These children carry the high-risk HLA DQB1*02/*03:02 genotype or the moderate-risk HLA-DQB1*03:02/x genotype (x ≠ DQB1*02, 03:01, or 06:02). More than 1,200 of these children took part in the DIPP-CD study. The DIPP study in Tampere followed the children at regular intervals at the ages of 3, 6, 12, 18, and 24 months and subsequently at intervals of 12 months for children without T1D-related autoantibodies. At each visit, the families were interviewed for diet, infections, growth, and important family-related issues, and the children gave a nonfasting venous blood sample. The children were followed for 4 CD-related antibodies, anti-tissue transglutaminase (anti-tTG), anti-endomysium, anti-gliadin (AgA-IgG and AgA-IgA), and anti-reticulin antibodies, and for T1D-associated autoantibodies, islet cell antibody. If a sample was positive for islet cell antibody, insulin autoantibody, antibodies against tyrosine phosphatase-like protein (IA-2A), and glutamate decarboxylase were measured from all samples taken from that child. From the beginning of 2003, all samples were measured for the 4 T1D-associated autoantibodies.
All children participating in this study were of white origin. They were initially identified based on positivity for IgG class tissue transglutaminase antibodies. As a consequence, IgA-deficient subjects were excluded. One of the mothers had CD. Furthermore, the children in the CD follow-up cohort were annually screened for anti-tTG antibodies (tTGAs) using a commercial kit (Celikey Pharmacia Diagnostics, Freiburg, Germany). If any given child's sample was found to be positive for tTGA, all of that child's previous and following samples were analyzed for the entire set of CD-related antibodies. A duodenal biopsy was recommended for all tTGA-positive children. If the biopsy was consistent with the ESPGHAN criteria of 1990, a gluten-free diet (GFD) was recommended. Three of the CD cases were also diagnosed with T1D, one just before the diagnosis of CD and the other ones after the diagnoses of CD (62 and 76 months later).
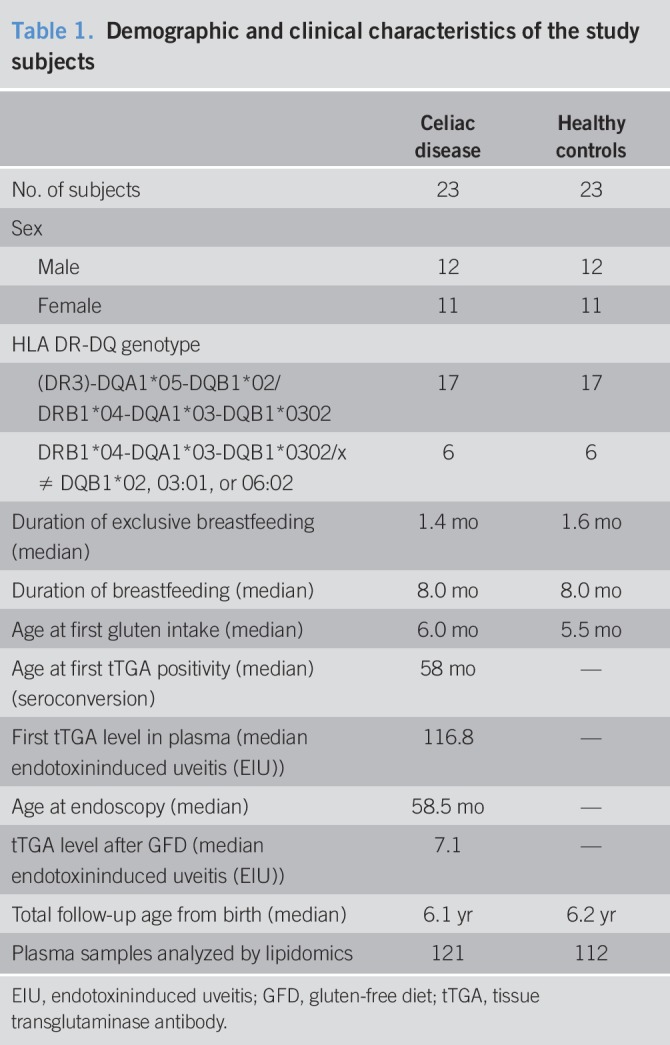
We randomly selected 23 children (12 males and 11 females) with biopsy-proven CD (progressors) and a control for each progressor matched for age, sex, and with the same risk HLA alleles, and living in the Tampere region throughout the whole follow-up period. None of the controls were diagnosed with T1D. The original HLA screening for patients with CD and suitable controls was also completed to a “full-house” DQ typing to verify the presence of DQA1*05 in selected DQB1*02-positive children (24). These clinical and genetic annotations of the participants are given in Table 1. Furthermore, the children were exclusively breastfed until 1.6 months (median), and some were exclusively breastfed up to age 8.0 months. The first exposure to gluten in this study was at a median age of 6.0 months. The children (case and control) had comparable (nominal and nonsignificant difference) energy (kJ), fat (g), carbohydrate (oat, rye, and wheat), and gluten intakes (see Figures S1 and S2, Supplementary Digital Content 1, http://links.lww.com/CTG/A44).
Table 1.
Demographic and clinical characteristics of the study subjects
Maternal total diet during pregnancy and lactation was ascertained by a validated food frequency questionnaire (25). Infant feeding was studied by structured questionnaires, which families filled in at home, and which were checked during the clinic visits. The child's diet was assessed with 3-day food records at 3, 6, and 12 months, followed by 2, 3, 4, and 6 years of age visits. The food records were posted before the study visit or given at the previous visit with detailed, written instructions. The families and daycare personnel were instructed to write down all the foods, drinks, and dietary supplements the child consumed, with the amounts and brand names during 1 weekend day and 2 weekdays. The study nurses checked the records at the study visit and helped to estimate any incomplete portion sizes with a picture booklet. The collection, processing, and calculation of food consumption data have been described previously in detail (26). The mothers of CD progressors and controls had comparable body mass indexes and energy, fat, and carbohydrate intakes during pregnancy and lactation (see Figures S3 and S4, Supplementary Digital Content 1, http://links.lww.com/CTG/A44).
Altogether, 121 plasma samples from children developing CD and 112 plasma samples from matched, healthy controls were analyzed, averaging more than 5 prospective follow-up samples for each child. Log-transformed intensities of the lipid measurements from these are given in Figures S5 and S6 (Supplementary Digital Content 1, http://links.lww.com/CTG/A44).
Analysis of molecular lipids
A total of 233 plasma samples were randomized and extracted using a modified version of the Folch procedure, similar to a protocol described recently (27). Promptly after extraction, 10 μL of 0.9% NaCl and 120 μL of CHCl3:MeOH (2:1, v/v) containing 2.5 μg·mL−1 internal standard solution (for quality control and normalization purposes) were added to 10 μL of each plasma sample. A detailed protocol of plasma lipid profiling and data preprocessing is given in Supplementary Notes (see Supplementary Digital Content 1, http://links.lww.com/CTG/A44).
Statistical methods
The samples from CD progressors and controls were divided into different age groups based on time difference between the date of the sample withdrawn and date of birth of the subject (see Figure S7, Supplementary Digital Content 1, http://links.lww.com/CTG/A44). If more than 2 samples from the same case matched a time interval, the closest was taken. The data were then log2 transformed. Homogeneity of the samples was assessed by principal component analysis (28), and no outliers were detected.
R software (http://www.r-project.org) was used for data analysis and visualization. Principal component analysis was performed using “prcomp()” function included in the “stats” package. The effect of different factors such as age, sex, and condition (healthy or CD) on the lipidomics data set was evaluated. The data were centered to zero mean and unit variance. The relative contribution of each factor to the total variance in the data set was estimated by fitting a linear regression model, where the normalized intensities of metabolites were regressed to the factor of interest, and thereby, median marginal coefficients (R2) were estimated. This analysis was performed using “scater” package (see Figure S8, Supplementary Digital Content 1, http://links.lww.com/CTG/A44). Partial least squares discriminant analysis (PLS-DA) (29) and variable importance in projection (VIP) scores (30) were estimated by an array of functions coded in “ropls” package. Moreover, the PLS-DA models were cross-validated (31) by 7-fold cross-validation as implemented in “ropls” package, and Q2 (an estimate of model's predictability) and R2 were obtained.
The longitudinal profiles of the lipids in the samples obtained from CD progressors and matched healthy controls were compared using linear mixed-effects (LMEs) models (32) as implemented in the “lme()” function of “nlme” package. The intensity of a metabolite (ŷ) in a sample (j) is a function of multiple factors such as follow-up age, sex, case/control, and subject-wise variability. The clinical and genetic factors of the children were matched and standardized in this study (Table 1); other factors such as subject-wise variability can be modeled and derived using LME models. The LME models were restricted to constant terms with the fixed effect being CD progression/nonprogression (control), age, sex, and the random effect being subject-wise variation in comparison to the group-specific mean level. Fitted LME models showed that sex difference has nominal effect on the metabolite intensities. The fully parametrized model was compared with a null model using analysis of variance (ANOVA) (33) (“aov()” as deployed in the “stats” package). The lipid profiles that changed significantly (P < 0.05) were subjected to false discovery rate (FDR) adjustment using p-adjust(). Lipid profiles with FDR <0.05 were listed.
Next, post hoc analysis using the Tukey test for honest significant difference (HSD) was performed on these selected lipids to see, if at all, they are changing between progressors and nonprogressors at a particular age. A list of differentially altered lipids that also showed HSD (P values < 0.05) between CD progressors and healthy controls at a particular age was marked. These lipids were considered for further analysis.
HSD was performed using “TukeyHSD()” function deployed in the “stats” package. Loess regression was performed using “loess()” deployed in the “stats” package. The Spearman correlation coefficient was calculated using “rcorr()” function implemented in “Hmisc” package. “Heatmap.2()” and “boxplot()” were used for data visualization.
RESULTS
Global plasma lipidome in progression to CD
The complete lipidomics data set was first explored using multivariate analysis. Among other factors affecting the lipidome, age was found to be a major confounding factor (see Figure S8, Supplementary Digital Content 1, http://links.lww.com/CTG/A44). PLS-DA (29) of lipidomics data from all 233 longitudinal samples suggested that children who later progressed to CD (progressors) may have different lipid profiles in comparison to their matched healthy controls (Figure 1a). Different classes of lipids such as cholesterol esters (CEs), phosphatidylcholines (PCs), lysophosphatidylcholines, phosphatidylethanolamines, phosphatidylinositols, sphingomyelins (SMs), and triacylglycerols (TGs) were affected (regression coefficient [±0.05] and VIP scores (30) >1) (Figure 1b).
Figure 1.

Classification of CD and healthy controls, based on their lipidome. (a) PLS-DA score plot showing difference in the lipidome between CD progressors (cyan) and healthy controls (magenta). (b) A regression plot of the lipids and their VIP scores obtained from the PLS-DA model. The lipids are grouped and color coded by their classes. (c) PLS-DA score plot showing difference in the lipidome in CD progressors along the age. (d) A regression plot of the lipids and their VIP scores at age 3 months. The lipids are grouped and color coded by their classes. CD, celiac disease; PLS-DA, partial least squares discriminant analysis; VIP, variable importance in projection.
Differences between cases and controls were observed already by age 3 months, for example, before the introduction of gluten to the diet (Figure 1c). TGs, PCs, and CEs were mostly affected (regression coefficient [±0.05] and VIP (30) scores >1) at this age (Figure 1d). The cord plasma lipidome clustered distinctly from other age groups (see Figure S9, Supplementary Digital Content 1, http://links.lww.com/CTG/A44). However, no significant differences (HSD, P values > 0.05) in the cord plasma lipids were observed between CD progressors and their matched healthy controls (Figure 2a).
Figure 2.

Number of acyl carbons in TGs. (a) Heatmap showing log2 FCs of significantly altered (FDR-adjusted P values < 0.05) longitudinal lipid profiles between CD progressors and matched healthy controls, as identified by LME models. Here, blue and red depict downregulated and upregulated lipid intensities in CD progressor as compared to their matched healthy controls, respectively, and white depicts no change. The lipids that changed specifically (HSD, P values < 0.05) at a particular age, as identified by the Tukey test for HSD, are highlighted with a star “*”. (b and c) Correlation plots of log ratio or fold changes of TGs and the number of acyl chain bonds incurred by them at early (3 months) and later ages (72 months), i.e., when gluten-free diet has been started. R denotes ranked correlation coefficients. (d and e) Correlation plots of log ratio of TGs and the number of acyl chain carbon atoms in TGs at the same age. The significantly changed (P < 0.05) TGs between CD progressors and healthy controls (at any age) are marked in dark pink. CD, celiac disease; FC, fold change; FDR, false discovery rate; HSD, honest significant difference; LME, linear mixed effect; TG, triacylglycerol.
Molecular lipids in progression to overt CD
Longitudinal analysis of lipidomic profiles at the individual lipid level identified 80 molecular lipids (from a total of 239) occurring at significantly different levels over time between CD progressors and healthy controls (FDR-adjusted P values < 0.05) (Figure 2a).
The level and direction of regulation of TGs depend on their chemical structure. There was an association between the fold change in TGs (CD progressors vs healthy controls) and the TG double bond count as well as the TG carbon number (Figure 2b–e). At age 3 months, TGs with lower double bond and carbon numbers were found to have increased in CD progressors as compared to matched, healthy controls, with coefficients (R = −0.13; P values = 0.19) and (R = −0.54; P values = 0.0001), respectively, for double bond count and carbon number (Figure 2b, d). Interestingly, at a later age, these TGs with lower double bond and carbon numbers were downregulated with (R = 0.79; P values = 0.08) and (R = 0.44; P values = 0.0002), respectively (Figure 2c, e). Notably, 4 such TGs were elevated in progressors at age 3 months (HSD, P values < 0.05). No significant changes were detected for dietary TGs, such as those containing polyunsaturated fatty acids. Representative longitudinal profiles for selected significantly altered lipids between CD progressors and healthy controls are shown in Figure 3.
Figure 3.

Longitudinal lipid profiles that are significantly different between CD progressors and healthy controls. Lipid profiles in CD progressors are marked with black color and healthy controls with orange color, along with 95% confidence intervals, as shown by the area shaded around the curves. (a–f) shows longitudinal profiles of CE(18:1), LPC(18:0), TG(47:0), SM(d32:1), PC(36:3), TG(18:2/18:2/18:2) or TG(18:3/18:2/18:1), respectively.
Impact of tissue transglutaminase antibodies on lipidomic profiles
Tissue transglutaminase (tTG) is a multifunctional, calcium-dependent enzyme (Enzyme Commission number [EC] 2.3.2.13) that plays an important role in the pathogenesis of CD. Antibodies produced against tTG, anti-tissue transglutaminase antibodies (tTGA), are used as serological markers with high sensitivity (99%) and reasonable specificity (>90%) for the diagnosis of CD (34–36).
We examined tTGA titers in 23 CD progressors at 2 different stages: (i) immediately after the seroconversion for tTGA and (ii) 6–12 months after both seroconversion and introduction of GFD. First, we identified at least 7 “essential” TGs, i.e., TGs of dietary origin, in our lipidomics data set. The identities of these TGs were validated using external sources including both the Human Metabolome Database (37) and FooDB.ca (http://foodb.ca/). Spearman correlation analysis was then performed between the levels of selected TGs (34 significantly changed nonessential and 7 nonsignificantly changed essential TGs) and PCs (18 PCs + 2 lysophosphatidylcholines) with measured tTGA levels at these stages. The aim was to determine whether the systemic levels of TGs and PCs are associated with antibody titers during CD progression. A positive correlation (Spearman rank coefficient, δavg = +0.174; minimum P value = 0.01) was found between the TG and tTGA levels after seroconversion (Figure 4a). However, a negative correlation was found at a later age, where the levels of tTGA decreased considerably (δavg = −0.163; minimum P-value = 0.05). In addition, at least 16 PCs were negatively correlated at these 2 different time points (δavg = −0.02 and −0.13; minimum P values = 0.01 and 0.02, respectively) (Figure 4b). It is known that GFD decreases tTGA levels in patients with CD (38), while it also improves the total cholesterol levels and high density lipoprotein profiles without any significant increase in low density lipoproteins (39).
Figure 4.

Correlation between plasma TGs and tTGA titers in CD progressors. (a) Heatmap showing the Spearman rank coefficient estimated between significantly changed TG levels and tTGA titer in the plasma of CD progressors immediately after the SC and 6–12 months after the SC or introduction of GFD. Red and blue depicts positive and negative correlations. (b) Heatmap showing the Spearman rank coefficient estimated between significantly changed PC and plasma tTGA titer in CD progressors after SC and GFD. CD, celiac disease; GFD, gluten-free diet; PC, phosphatidylcholine; SC, seroconversion; TG, triacylglycerol; tTGA, tissue transglutaminase antibody.
Impact of gluten on molecular lipids
We then divided the cohort into 3 subgroups: (i) before the introduction of gluten in the diet (3 months), (ii) after the introduction of gluten (12–36 months), and (iii) after the introduction of GFD (72 months, CD progressors only).
We observed a decrease in the total essential TG level in the plasma of the CD progressors after gluten intake (Figure 5a; see Figure S10A, Supplementary Digital Content 1, http://links.lww.com/CTG/A44). Introduction of GFD reversed this trend, but the changes were not significant (ANOVA, P values > 0.05) (Figure 5a, e). On the other hand, levels of nonessential endogenous TGs were decreased (ANOVA, P values = 0.004) in the plasma of the CD progressors after commencement of gluten intake and even after the diagnosis of clinical CD and introduction of GFD (P values = 0.001). The trend in the change of nonessential TGs was also observed in healthy controls (P values = 0.007 and 0.01, respectively) (Figure 5b, e; see Figure S10B, Supplementary Digital Content 1, http://links.lww.com/CTG/A44). Moreover, there was a difference (P < 0.05) in nonessential TGs levels between CD progressors and healthy controls as predicted by the Tukey test for HSD (Figure 2a). These findings suggest that dysregulation of lipid metabolism might occur at an early stage of CD progression, even before the introduction of gluten in the diet.
Figure 5.

Lipids that are significantly changed after gluten intake and by introduction of GFD. (a–d) Boxplots showing total log intensities of essential TGs, nonessential TGs, PCs, and SMs in the CD progressors and healthy controls before (3 months) and after introduction of gluten (12–36 months) in the diet and after introduction of GFD (72 months). (e) Grid map of P values obtained from ANOVA models by combining all possible conditions before/after the gluten intake in CD progressors and healthy controls is given. The 3 different conditions are “BA-Glut”, before and after gluten; “AGlut-AGFD”, after gluten and after gluten-free diet; and “BGlut-AGFD”, before gluten and after gluten-free diet. ANOVA, analysis of variance; CD, celiac disease; GFD, gluten-free diet; PC, phosphatidylcholine; TG, triacylglycerol; SM, sphingomyelin.
PCs were elevated both in the progressors (P < 0.004) and controls (P < 0.007) after the commencement of gluten intake (Figure 5c, e; see Figure S10C, Supplementary Digital Content 1, http://links.lww.com/CTG/A44). PCs are the major class of phospholipids that form the cellular constituents required for the assembly of biological membranes. In contrast to healthy controls, no significant differences in the SM level were observed in the CD progressors after commencement of gluten intake. A difference in the plasma SM level was observed only at a later age after the introduction of GFD (Figure 5d, e; see Figure S10D, Supplementary Digital Content 1, http://links.lww.com/CTG/A44).
DISCUSSION
We identified systematic differences in plasma lipidomes between children who progressed to clinical CD during the follow-up as compared to children who remained healthy. These differences were observed before the first exposure to gluten in the diet and before the first signs of CD-associated autoimmunity. The dysregulation of plasma lipidome in CD progressors was dominated by complex lipids such as PCs, TGs, and CEs. Because there is no evidence of gut damage in any studies performed so far in individuals who are autoantibody negative for tTGA, it is unlikely that these systematic differences are caused by gluten from breast milk or other sources in the infants' diet causing damage to the gut immediately after birth. An earlier lipidomics study in the PreventCD cohort did not find significant differences between CD progressors and matched controls at age 4 months (40). However, that study used a targeted analysis focusing on a subset of phospholipids and acyl-carnitines and did not measure TGs and CEs, where the major changes were found to have occurred in our study.
In this study, several nonessential TGs were upregulated in CD progressors as early as age 3 months, i.e., before the first introduction of gluten to the diet. However, no significant changes in dietary TGs were found at this age. Lipid malabsorption is believed to be a side effect of flattened villi in the small intestine (15,17). However, our data suggest that lipid-related abnormalities are not caused by CD-related villous atrophy in the gut. One plausible explanation that arises is that lipid malabsorption can lead to a reduction in cholesterol-transporting lipoproteins and secretion of apolipoprotein-A1 (41,42). A decrease in the level of CEs in CD progressors during the first 3 months after birth supports this explanation. Moreover, CEs were upregulated at a later age after the introduction of GFD (Figures 2a and 3a). Lipid malabsorption may thus occur at a very early age in CD progressors and, therefore, de novo lipogenesis (43), as reflected by increased levels of TGs with low carbon number and double bond content, may be necessarily increased to compensate for said compromised lipid uptake. Interestingly, this phenomenon was not observed at birth (i.e., in the cord blood data), thus suggesting that the observed lipid dysregulation is not an inborn phenomenon.
Furthermore, an inverse relationship between endogenous TGs and tTGA titer after seroconversion and introduction of GFD in the CD progressors also reaffirms the involvement of TGs and other phospholipids (PCs) in CD progression. Indeed, there was an increase in cholesterol levels after the introduction of GFD, and there was a decrease in tTGA titers, in agreement with previous studies (38,39). Presumably, these lipids may play a protective role against seroconversion to tTGA positivity and CD progression.
We found, in earlier studies, that T1D is preceded by dysregulation of lipid metabolism (22,44,45). The key findings from these studies were that islet autoimmunity and overt T1D are preceded by diminished phospholipid and TG levels. Although there are some similarities at an early age in phospholipid profiles in T1D and, as shown in the current study, CD progressors, no such similarities exist for TGs. The increase of TGs with low carbon number and double bond count does seem to be specific to CD progression. These TGs are associated, in adults, with elevated liver fat in nonalcoholic fatty liver disease (46,47), reflecting increased de novo lipogenesis and adipose tissue lipolysis. In fact, patients with CD have, interestingly, been found to be at an increased risk of nonalcoholic fatty liver disease (48). Our findings may thus offer an explanation for this association.
The main limitation of this study is the relatively small number of children included, and for that reason, our findings should be considered preliminary and hypothesis generating. However, the sample size is mitigated by the longitudinal study setting, with 5 prospective samples, from birth until after the introduction of GFD, on average, from each child. Moreover, the subjects of our study were well matched with their controls for genetic and environmental factors. In the current study, the participants were initially screened for HLA-conferred susceptibility to T1D. There was an overrepresentation of DR3/DR4-heterozygous (n = 17) and DR4-positive subjects (n = 6) in the study series. In a general population, most CD cases are DR3 positive. In our study, there were no significant differences in the lipidomic profiles between these 2 HLA groups. Nevertheless, because of this selection bias, our findings may not be translatable to other populations and thus need to be confirmed in a setting more representative of a general population.
In summary, our study suggests that lipid-related abnormalities in CD progressors demonstrably occur before the first introduction of gluten to the diet. These changes may be related to impaired lipid absorption and de novo lipogenesis, with both of these being part of the host response. Our preliminary findings may have important clinical implications for the detection of subjects at risk of CD and for the understanding of its early pathogenesis.
Data accessibility
The lipidomics data sets and the clinical metadata generated in this study have been submitted to MetaboLights (49) and can be located using accession number (MTBLS729). The appropriate clinical metadata were linked to the lipidomics data set using the ISA-creator package from MetaboLights.
Ethical approval and informed consent
The Ethics Committee of Tampere University Hospital approved the study. Written informed consent was obtained from the parents for HLA screening, autoantibody analysis, and intestinal biopsies.
CONFLICTS OF INTEREST
Article guarantor: Dr. Matej Orešič, PhD.
Specific author contributions: M.O. and M.K. designed and supervised the study. C.C. performed lipidomics experiments, which were supervised by T.H. P.S. analyzed the data. S.V., S.S., H.H., J.L, J.T, and R.V. contributed to the design of the clinical study. P.S. and M.O. wrote the manuscript. All authors critically reviewed and approved the final manuscript.
Financial support: This study was supported by the Academy of Finland (Centre of Excellence in Molecular Systems Immunology and Physiology Research—SyMMyS, Decision No. 250114, to M.O. and M.K.; and Personalised Health 2014 programme project, Decision No. 292568).
Potential competing interests: None declared.
Study Highlights.
WHAT IS KNOWN
✓ Previous studies have shown that individuals with clinical CD have altered lipid profiles as compared to nongenetically and non–age-matched control groups.
✓ Introduction of gluten to one's diet has always been considered to be the main trigger for this disease, yet it has been unknown what are the other contributing factors needed for disease initiation.
WHAT IS NEW HERE
✓ Our study suggests that there are alterations in lipid metabolism before first signs of autoimmunity or disease symptoms in subjects who later progress to clinical CD as compared to healthy controls with the same HLA-associated risk. The dysregulated lipid profiles were observed already at age 3 months between CD progressors and healthy controls, i.e., before the first introduction of gluten in the diet. Furthermore, these differences exacerbated with age but were not observed at birth.
TRANSLATIONAL IMPACT
✓ The plasma lipid markers identified in this study may help to predict and isolate early changes in the lipidome of the children, who later progresses to CD.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the families participating in the DIPP study for making this study possible; the expert staff of the DIPP study for their excellent work with the participating research families and sample collection; Professor Olli Simell for his important scientific contribution to the DIPP study; Dawei Geng for technical assistance in lipidomic analysis; and Aidan McGlinchey for editing the manuscript.
Footnotes
SUPPLEMENTARY MATERIAL accompanies this paper at http://links.lww.com/CTG/A44
REFERENCES
- 1.Ludvigsson JF, Leffler DA, Bai JC, et al. The Oslo definitions for coeliac disease and related terms. Gut 2013;62:43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Romanos J, Van Diemen CC, Nolte IM, et al. Analysis of HLA and non-HLA alleles can identify individuals at high risk for celiac disease. Gastroenterology 2009;137:834–40.e3. [DOI] [PubMed] [Google Scholar]
- 3.Kelly CP, Bai JC, Liu E. Advances in diagnosis and management of celiac disease. Gastroenterology 2015;148:1175–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leffler K, Catassi C, Reunanen A, et al. The prevalence of celiac disease in Europe: Results of a centralized, international mass screening project. Ann Med 2010;42:587–95. [DOI] [PubMed] [Google Scholar]
- 5.Patterson CC, Dahlquist GG, Gyürüs E, et al. Incidence trends for childhood type 1 diabetes in Europe during 1989–2003 and predicted new cases 2005–20: A multicentre prospective registration study. Lancet 2009;373:2027–33. [DOI] [PubMed] [Google Scholar]
- 6.Rewers M. Epidemiology of celiac disease: What are the prevalence, incidence, and progression of celiac disease? Gastroenterology 2005;128:S47–S51. [DOI] [PubMed] [Google Scholar]
- 7.Myléus A, Ivarsson A, Webb C, et al. Celiac disease revealed in 3% of Swedish 12-year-olds born during an epidemic. J Pediatr Gastroenterol Nutr 2009;49:170–6. [DOI] [PubMed] [Google Scholar]
- 8.Knip M. Descriptive epidemiology of type 1 diabetes—is it still in? Diabetologia 2012;55:1227–30. [DOI] [PubMed] [Google Scholar]
- 9.Rewers M, Liu E, Simmons J, et al. Celiac disease associated with type 1 diabetes mellitus. Endocrinol Metab Clin North Am 2004;33:197–214, xi. [DOI] [PubMed] [Google Scholar]
- 10.Ludvigsson JF, Ludvigsson J, Ekbom A. Celiac disease and risk of subsequent type 1 diabetes: A general population cohort study of children and adolescents. Diabetes Care 2006;29:2483–8. [DOI] [PubMed] [Google Scholar]
- 11.Montgomery DJ, Plagnol V, Walker NM, et al. Shared and distinct genetic variants in type 1 diabetes and celiac disease. N Engl J Med 2008;359:2767–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kemppainen KM, Lynch KF, Liu E, et al. Factors that increase risk of celiac disease autoimmunity after a gastrointestinal infection in early life. Clin Gastroenterol Hepatol 2017;15:694–702.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scanlon SA, Murray JA. Update on celiac disease: Etiology, differential diagnosis, drug targets, and management advances. Clin Exp Gastroenterol 2011;4:297–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hollywood K, Brison DR, Goodacre R. Metabolomics: Current technologies and future trends. Proteomics 2006;6:4716–23. [DOI] [PubMed] [Google Scholar]
- 15.Solakivi T, Kaukinen K, Kunnas T, et al. Serum fatty acid profile in celiac disease patients before and after a gluten-free diet. Scand J Gastroenterol 2009;44:826–30. [DOI] [PubMed] [Google Scholar]
- 16.di Cagno R, de Angelis M, de Pasquale I, et al. Duodenal and faecal microbiota of celiac children: Molecular, phenotype and metabolome characterization. BMC Microbiol 2011;11:219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bertini I, Calabrò A, De Carli V, et al. The metabonomic signature of celiac disease. J Proteome Res 2009;8:170–7. [DOI] [PubMed] [Google Scholar]
- 18.Sellitto M, Bai G, Serena G, et al. Proof of concept of microbiome-metabolome analysis and delayed gluten exposure on celiac disease autoimmunity in genetically at-risk infants. PLoS One 2012;7:e33387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Olivares M, Walker AW, Capilla A, et al. Gut microbiota trajectory in early life may predict development of celiac disease. Microbiome 2018;6:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hyötyläinen T, Orešič M. Systems biology strategies to study lipidomes in health and disease. Prog Lipid Res 2014;55:43–60. [DOI] [PubMed] [Google Scholar]
- 21.Hyötyläinen T, Ahonen L, Pöhö P. Lipidomics in biomedical research-practical considerations. Biochim Biophys Acta Mol Cell Biol Lipids 2017;1862:800–3. [DOI] [PubMed] [Google Scholar]
- 22.Orešič M, Simell S, Sysi-Aho M, et al. Dysregulation of lipid and amino acid metabolism precedes islet autoimmunity in children who later progress to type 1 diabetes. J Exp Med 2008;205:2975–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lamichhane S, Ahonen L, Dyrlund TS, et al. Dynamics of plasma lipidome in progression to islet autoimmunity and type 1 diabetes—type 1 diabetes prediction and prevention study (DIPP). Sci Rep 2018;8:10635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ilonen J, Kiviniemi M, Lempainen J, et al. Genetic susceptibility to type 1 diabetes in childhood–estimation of HLA class II associated disease risk and class II effect in various phases of islet autoimmunity. Pediatr Diabetes 2016;17:8–16. [DOI] [PubMed] [Google Scholar]
- 25.Erkkola M, Karppinen M, Javanainen J, et al. Validity and reproducibility of a food frequency questionnaire for pregnant Finnish women. Am J Epidemiol 2001;154:466–76. [DOI] [PubMed] [Google Scholar]
- 26.Virtanen SM, Nevalainen J, Kronberg-Kippilä C, et al. Food consumption and advanced β cell autoimmunity in young children with HLA-conferred susceptibility to type 1 diabetes: A nested case-control design. Am J Clin Nutr 2012;95:471–8. [DOI] [PubMed] [Google Scholar]
- 27.Pedersen HK, Forslund SK, Gudmundsdottir V, et al. A computational framework to integrate high-throughput “-omics” datasets for the identification of potential mechanistic links. Nat Protoc 2018;1. [DOI] [PubMed] [Google Scholar]
- 28.Carey RN, Wold S, Westgard JO. Principal component analysis: An alternative to "referee" methods in method comparison studies. Anal Chem 1975;47:1824–9. [DOI] [PubMed] [Google Scholar]
- 29.Rosipal R, Krämer N. Overview and recent advances in partial least squares. In: International Statistical and Optimization Perspectives Workshop “Subspace, Latent Structure and Feature Selection”; 2005. Springer, 2005, pp 34–51. [Google Scholar]
- 30.Farrés M, Platikanov S, Tsakovski S, et al. Comparison of the variable importance in projection (VIP) and of the selectivity ratio (SR) methods for variable selection and interpretation. J Chemom 2015;29:528–36. [Google Scholar]
- 31.Westerhuis JA, Hoefsloot HC, Smit S, et al. Assessment of PLSDA cross validation. Metabolomics 2008;4:81–9. [Google Scholar]
- 32.Pinheiro J, Bates D, DebRoy S, et al. Linear and Nonlinear mixed effects models. R package version 3. 2014. [Google Scholar]
- 33.Heiberger RM, Freeny AE, Chambers JM. Analysis of variance; designed experiments. In: Statistical Models in S: Routledge, 2017, pp 145–93. [Google Scholar]
- 34.Oxentenko AS, Murray JA. Celiac disease: Ten things that every gastroenterologist should know. Clin Gastroenterol Hepatol 2015;13:1396–9. [DOI] [PubMed] [Google Scholar]
- 35.Esposito C, Paparo F, Caputo I, et al. Anti-tissue transglutaminase antibodies from coeliac patients inhibit transglutaminase activity both in vitro and in situ. Gut 2002;51:177–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hill PG, McMillan SA. Anti-tissue transglutaminase antibodies and their role in the investigation of coeliac disease. Ann Clin Biochem 2006;43:105–17. [DOI] [PubMed] [Google Scholar]
- 37.Wishart DS, Feunang YD, Marcu A, et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res 2018;46:D608–D617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Agardh D, Lynch K, Brundin C, et al. Reduction of tissue transglutaminase autoantibody levels by gluten-free diet is associated with changes in subsets of peripheral blood lymphocytes in children with newly diagnosed coeliac disease. Clin Exp Immunol 2006;144:67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brar P, Kwon GY, Holleran S, et al. Change in lipid profile in celiac disease: Beneficial effect of gluten-free diet. Am J Med 2006;119:786–90. [DOI] [PubMed] [Google Scholar]
- 40.Kirchberg FF, Werkstetter KJ, Uhl O, et al. Investigating the early metabolic fingerprint of celiac disease–a prospective approach. J Autoimmun 2016;72:95–101. [DOI] [PubMed] [Google Scholar]
- 41.Capristo E, Addolorato G, Mingrone G, et al. Low-serum high-density lipoprotein-cholesterol concentration as a sign of celiac disease. Am J Gastroenterol 2000;95:3331–2. [DOI] [PubMed] [Google Scholar]
- 42.Farnetti S, Zocco MA, Garcovich M, et al. Functional and metabolic disorders in celiac disease: New implications for nutritional treatment. J Med Food 2014;17:1159–64. [DOI] [PubMed] [Google Scholar]
- 43.Ameer F, Scandiuzzi L, Hasnain S, et al. De novo lipogenesis in health and disease. Metab Clin Exp 2014;63:895–902. [DOI] [PubMed] [Google Scholar]
- 44.Orešič M, Gopalacharyulu P, Mykkänen J, et al. Cord serum lipidome in prediction of islet autoimmunity and type 1 diabetes. Diabetes 2013:62:3268–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.La Torre D, Seppänen-Laakso T, Larsson HE, et al. Decreased cord-blood phospholipids in young age-at-onset type 1 diabetes. Diabetes 2013, 62:3951–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Orešič M, Hyötyläinen T, Kotronen A. Prediction of non-alcoholic fatty-liver disease and liver fat content by serum molecular lipids. Diabetologia 2013;56:2266–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Luukkonen PK, Zhou Y, Sädevirta S, et al. Hepatic ceramides dissociate steatosis and insulin resistance in patients with non-alcoholic fatty liver disease. J Hepatol 2016;64:1167–75. [DOI] [PubMed] [Google Scholar]
- 48.Reilly NR, Lebwohl B, Hultcrantz R, et al. Increased risk of non-alcoholic fatty liver disease after diagnosis of celiac disease. J Hepatol 2015;62:1405–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Haug K, Salek RM, Conesa P, et al. MetaboLights: An open-access general-purpose repository for metabolomics studies and associated meta-data. Nucleic Acids Res 2013;41:D781–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The lipidomics data sets and the clinical metadata generated in this study have been submitted to MetaboLights (49) and can be located using accession number (MTBLS729). The appropriate clinical metadata were linked to the lipidomics data set using the ISA-creator package from MetaboLights.