

INTRODUCTION:
Hamartomatous polyposis syndromes (HPS) are rare autosomal-dominant inherited disorders associated with gastrointestinal (GI) tract and other cancers. HPS include Peutz-Jeghers syndrome (PJS), juvenile polyposis syndrome (JPS), and phosphatase and tensin homolog hamartomatous tumor syndromes (PHTS). Diagnosis, management, and outcome prediction of HPS pose a clinical challenge. To characterize genotype, phenotype, histology and outcomes of individuals with HPS.
METHODS:
A retrospective cohort study (2004–2017) of consecutive patients that were clinically diagnosed with HPS that visited a specialized GI oncology clinic. Demographic, clinicopathological, and genetic data were obtained from medical records.
RESULTS:
Fifty-two individuals from 34 families were included. Common clinical manifestations were GI bleeding (40% JPS, 23% PJS, and 25% PHTS) and bowel obstruction (46.15% PJS and 11.4% JPS). Twenty patients (38.4%) underwent surgery, 5 of whom required multiple procedures. Higher polyp burden was associated with the need for surgery (P = 0.007). Polyp histology varied widely with 69.2% of patients exhibiting histology different from the syndrome hallmark. GI cancer history was positive in 65%, 40%, and 50% of JPS, PJS, and PHTS families, respectively. Five (9.6%) patients developed cancers (one patient each had small bowel-1, colon-1, and thyroid-1, one patient had both small bowel adenocarcinoma and breast cancer, and one had both breast cancer and liposarcoma). Twenty (38.4%) patients tested positive for STK11, PTEN, SMAD4, BMPR1A, or AKT1 mutations: Sanger sequencing and multi-gene next generation sequencing panels detected mutations in 40.9% and 100% of tested cases, respectively.
DISCUSSION:
HPS patients present versatile phenotypes with overlapping clinical and histological characteristics. Polyp burden is associated with the need for surgery. Next-generation sequencing increases mutation detection.
INTRODUCTION
Hamartomatous polyps are characterized by mostly benign disorganized growth of intestinal tissue (1). They appear sporadically or as part of hereditary hamartomatous polyposis syndromes (HPS), characterized by multiple hamartomatous polyps in the gastrointestinal (GI) tract and exhibit distinct histological, clinical, genetic, and cancer predisposition features. These genetic syndromes are rare, with an incidence of 1 per 30,000–200,000 births (1). HPS syndromes include:
Peutz-Jeghers syndrome (PJS)—polyps occur throughout the GI tract but primarily in the small intestine (2). Mucocutaneous pigmentation is characteristic. Polyps may bleed or cause intussusception manifesting as bowel obstruction (2,3). There is a predisposition for GI tract, breast, pancreatic, testicular, lung, and gynecological cancers (1,4,5). The syndrome is caused by mutations in STK11 that encodes a tumor suppressor serine-threonine kinase (6).
Juvenile polyposis syndrome (JPS)—polyps occur throughout the GI tract. The most common symptom is rectal bleeding, but melena, iron deficiency anemia, and abdominal pain also occur (1,4,7). Patients are at risk for colorectal, gastric, duodenal, and pancreatic cancers (5,8). Mutated genes include SMAD4 and BMPR1A, and both are involved in tumor growth factor-β signaling pathway (9,10). SMAD4 associated JPS is associated with hereditary hemorrhagic telangiectasia as well as to massive gastric polyposis (11,12).
Cowden disease—polyps appear throughout the GI tract. Mucocutaneous hamartomas and macrocephaly occur. Patients are predisposed to breast, thyroid, renal, and endometrial cancer (4,13). Cowden disease is caused by mutations in phosphatase and tensin homolog (PTEN), a negative regulator of the PI3K signaling pathway (14). Cowden syndrome is the best described condition within the PTEN hamartomatous tumor syndromes (PHTS), alongside other syndromes such as Bannayan-Riley-Ruvalcaba syndrome and adult Lhermitte-Duclos disease (5).
The diagnosis of HPS was based until recently on clinicopathological criteria including family history of hamartomatous polyps, personal and family history of both hamartomatous polyps and associated cancers, and distinct clinical phenotypes (1). Currently, genetic diagnosis is the gold standard but achieved success only partially: 50%–80% in PJS (13,14), 60% in JPS (5), and 30%–80% in PHTS (15,16). Current next-generation sequencing (NGS) technology makes identification of rare genetic disorders readily available, which may result in higher diagnostic yield and potential discovery of new disease-causing genetic abnormalities (17,18).
Surveillance strategies have been established for each syndrome to prevent complications and cancer (5). However, due to syndrome rarity, recommendations rely on low-quality evidence and expert opinion, considering mainly the phenotype rather than the individual genetic profile (5). Additionally, there is interobserver variation among pathologists for hamartomatous polyps, which may lead to incorrect diagnoses and mistargeted gene sequencing. This may hamper surveillance, management, and optimal cancer prevention (19).
We describe herein our clinical experience with HPS as a nationwide referral center in a series of 52 individuals from 34 families. We highlight the redundancy between syndromes, potential outcome predictors, and the need for comprehensive genetic diagnoses.
METHODS
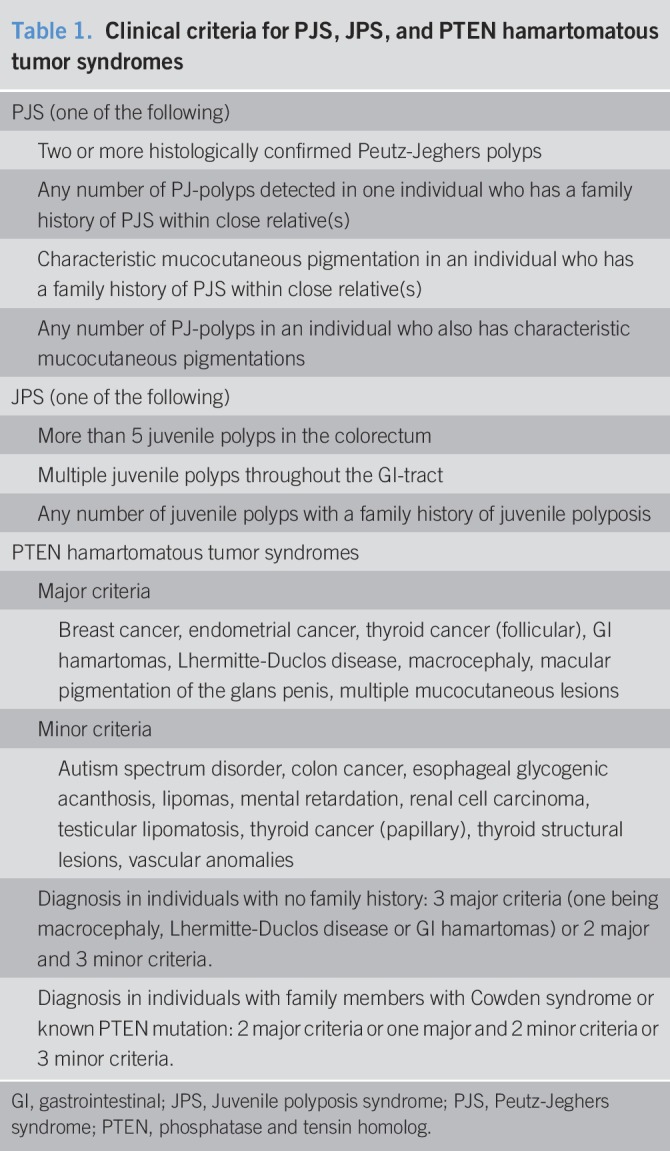
This is a retrospective cohort study. Patients with a clinical diagnosis of JPS, PJS, or PHTS were identified from our polyposis database between 2004 and 2017. Patients were diagnosed only if they fulfilled the accepted National Comprehensive Cancer Network criteria (Table 1) (1). Patients were advised endoscopic and extraintestinal surveillance tailored to the specific syndrome in accordance with the American College of Gastroenterology guidelines (5). Demographics, clinical symptoms, morbidity complications (including indication and type of GI surgery), personal and familial cancer history, genetic testing results, and mortality were documented. Data extraction was performed by a single physician into a structured uniform database.
Table 1.
Clinical criteria for PJS, JPS, and PTEN hamartomatous tumor syndromes
Composite severe clinical outcome was defined as occurrence of cancer, need for surgery, and mortality. In addition, a meticulous review of endoscopy reports over the years was performed to assess polyp number at presentation, cumulative polyp number and polyp location, size, and histology. Polyp size and numbers were based on the performing physician estimation. Main polyp sites were defined according to the location in the GI tract (stomach, small intestine, or colorectal) in which more than a third of the polyps appeared for a given patient.
We created a definition for polyp burden based on cumulative polyp numbers and maximal polyp size in order to classify low and high polyp burdens: Dozens of polyps smaller than 1 cm or up to 10 polyps smaller than 4 cm were considered low burden, other combinations were considered as high polyp burden. Patients were considered lost to follow-up if they had not attended clinic in more than 2 years. Adherence to surveillance was calculated as number of clinic visits divided by number of follow-up years.
All histologic specimens were reviewed by a specialized GI pathologist.
Genetic testing, including Sanger sequencing and multi-gene NGS panels, was performed by medically certified laboratories. All multi-gene panels included at least SMAD4, BMPR1A, STK11, PTEN, APC, MUTYH, MSH2, MSH6, PMS2, MLH1, BRCA1, and BRCA2.
Statistical analysis
Continuous variables are presented as mean ± SD and dichotomous variables as proportions. Univariate analysis was used to compare distribution of variables between surgical and nonsurgical patients, and between different groups of polyp size, number, and burden. Association between categorical variables was tested by Pearson χ2. Mann-Whitney test was used to compare distribution of continuous variables between study groups. P < 0.05 was considered statistically significant for all analyses. SPSS software was used for all analyses (IBM version 25, 2017).
RESULTS
Patients
Seventy-four patients were initially diagnosed with HPS. After examining clinical and pathological data, 52 patients from 34 families fulfilled National Comprehensive Cancer Network criteria and were included in the study (Figure 1).
Figure 1.

Study design. JPS, Juvenile polyposis syndrome; PJS, Peutz-Jeghers syndrome; Tel-Aviv Medical Center.
Thirty-five patients from 20 families had JPS, 13 patients from 10 families had PJS, and 4 patients from 4 families had PHTS. Mean follow-up time was 74.29 months (range 3–184 months), with an adherence index of 3.2 patient visits per year. Demographic, endoscopic, and histologic data of these patients are summarized in Table 2.
Table 2.
Demographic, endoscopic, and histologic data of the study population and study groups

Clinical manifestations
Table 3 summarizes the complications, surgical, and neoplastic outcomes. GI, specifically rectal bleeding, was the most common symptom, with a prevalence of 23% (3/13), 40% (14/35), and 25% (1/4) in PJS, JPS, and PHTS patients, respectively. Four JPS patients suffered from recurrent melena due to multiple bleeding stomach polyps, all underwent gastrectomy. Two PJS patients had upper GI bleeding: One had post-polypectomy hematemesis. The other had multiple ulcerated small intestine polyps, alongside with bleeding angioectasia. Of note, this patient had cirrhosis due to congenital lipodystrophy with bleeding varices that contributed to upper GI bleeding. Unfortunately, this patient was not tested genetically for PJS or for congenital lipodystrophy.
Table 3.
Complications, surgical, and neoplastic outcome

Bowel obstruction was also common and occurred in 6/13 (46.15%) PJS and 4/35 (11.4%) JPS patients. All events in the PJS group were small bowel obstructions (SBO) due to intussusception. Two patients suffered multiple episodes, one of whom underwent 4 resections and a Whipple procedure for adenocarcinoma in a duodenal polyp. In the JPS group, one patient had SBO due to intussusception caused by a cecal polyp, another had SBO with nondocumented cause which was treated conservatively, a third had colonic obstruction which necessitated hemicolectomy, and a fourth had recurrent SBO during pregnancy (possibly due to adhesions secondary to previous abdominal surgery). One PJS patient who was also on hemodialysis died due to Staphyloccocal sepsis–not related to the syndrome.
Endoscopic findings
Table 2 summarizes the endoscopic findings. The main site of polyps was the colon in JPS and PHTS (82% and 75%) and the small intestine in PJS (69%).
The majority of patients with PJS and JPS developed polyps >1 cm during follow-up (92.3% and 77.2%). Moreover, polyps >4 cm were found in 30.7% and 22.8% of PJS and JPS patients, respectively. All PHTS patients with available endoscopic data had polyps <1 cm. Associations between maximal polyp size, cumulative polyp number, and polyp burden with occurrence of malignancy, need for GI surgery, and composite severe outcome are presented in Figure 2.
Figure 2.

Association between polyp number (a), maximal polyp size (b), and polyp burden (c) with occurrence of cancer, surgery, and severe outcome.
Polyp number.
Patients with >10 polyps were more prone to the adverse composite severe outcome than those with <10 polyps (51.7% vs 15.3%; P = 0.027). However, the association with each outcome separately was not statistically significant (P = 0.159 for surgery, P = 0.317 for cancer occurrence).
Polyp size.
Polyp size did not affect the need for surgery (P = 0.267) or occurrence of cancer as 9.1% of patients with polyps <1 cm, no patients with polyps 1–4 cm, and 16.7% of patients with polyps >4 cm developed cancer (P = 0.367). There was no association of polyp size with the composite severe outcome (P = 0.505).
Polyp burden.
Fourteen patients had low polyp burden while 26 had high burden (data unavailable for 12 patients). Patients with high polyp burden required more surgical interventions compared to low polyp burden (50% vs 7.1%; P = 0.013). Polyp burden was not associated with cancer occurrence (11.5% vs 7.1%; P = 1.0). Patients with high polyp burden had more composite severe outcomes than low burden (57.7% vs 14.3%; P = 0.008).
Surgical data
Twenty patients (38.4%) underwent surgery due to complications or as a preventive measure (Table 3). Five patients had multiple surgical procedures—2 PJS patients due to recurrent intussusceptions (and one Whipple procedure as noted previously); 3 JPS patients underwent total gastrectomy due to GI bleeding and either small bowel resection or subtotal colectomy.
When comparing patients who underwent surgery to those who did not, we found that high polyp burden was associated with surgery (92.3% vs 50% of high or low polyp burden patients, respectively; P = 0.013). Other associated factors which approached statistical significance were female sex (65% vs 40.6%; P = 0.087), more than 10 polyps overall (85.7% vs 58.6%; P = 0.095), and GI bleeding (55% vs 31.3%; P = 0.089).
Cancer
Three patients (5.7%) were diagnosed with GI cancer: one PJS patient with duodenal adenocarcinoma at the age of 32 years, one JPS patient with small bowel adenocarcinoma at the age of 65 years, and one PHTS patient with malignant colonic polyps at the age of 66 years. Three patients were diagnosed with extraintestinal malignancies: one PJS patient had thyroid carcinoma at the age of 38 years, one JPS patient had breast cancer at the age of 60 years (and small bowel adenocarcinoma), and one PHTS patient had breast cancer at the age of 25 years and chest liposarcoma at the age of 30 years.
A history of GI malignancy was documented in 65% of JPS families, with up to 5 family members with known GI cancer. PJS and PHTS families had history of GI cancer in 40% and 50%, respectively. Family history of non-GI malignancies appeared in 40%, 15%, and 50% of PJS, JPS, and PHTS families, respectively (malignancies of lung, kidney, endometrium, liver, brain, and breast) (Table 3). Table 4 describes detailed comparisons between our findings and the data found in the literature review.
Table 4.
Comparison of polyp distribution, age of diagnosis, GI, and extraintestinal cancer risks to other studies

Histology
Different polyp types were identified histologically (Figure 3). Up to about 61% of PJS, 75% of JPS patients, and 50% of PHTS patients had polyp diagnoses different than the syndrome hallmark, with some patients having up to 5 or 6 different polyp types diagnosed throughout the years. Of note is the high prevalence of patients 23/52 (44.2%) that developed adenomatous polyps at any time during follow-up (mean age at diagnosis 33.5 ± 10.1), including 5/13 (38.4%) of PJS, 17/35 (48.5%) of JPS, and 1/4 (25%) of PHTS. Three of these patients (one PJS and 2 JPS) had high grade dysplasia. Occurrence of adenomas was not associated with that of cancer (P = 0.798).
Figure 3.

Variation in histologic findings of different polyps according to syndrome. PHTS, phosphatase and tensin homolog hamartomatous tumor syndromes.
Genetic tests
Thirty (57.7%) patients underwent genetic testing. Overall, a genetic mutation was identified in 20/52 (38.4% of total, 66.7% of tested) patients. Six PJS patients (46%) from 4 families had a mutation in STK11, 11 JPS patients (31.4%) from 7 families had a mutation in either BMPR1A or SMAD4, and 2 PHTS patients (50%) had a mutation in PTEN. Sanger sequencing was performed in 22/30 (73.3%) patients, and mutations were identified in 9 cases (40.9%). Six patients, of whom 3 had negative Sanger sequencing, underwent multi-gene panel testing using NGS techniques. Mutations were identified in all cases (100%). Novel mutations were an inversion in BMPR1A in a JPS patient (under validation), and a mutation in AKT1, a serine/threonine-protein kinase associated with PHTS as well as with breast, ovarian, and colorectal cancers, in a PHTS patient (22). Data regarding the type and results of the genetic test were not available for 5 patients.
HPS misclassification
Four patients had an initially incorrect clinical diagnosis according to the genetic diagnosis. Two were diagnosed as familial adenomatous polyposis but tested negative for APC mutations. Patient 1 underwent total proctocolectomy and ileoanal pouch anastomosis at the age of 18 years. Family history included a mother and a sister who died from colon cancer at a young age. Pathological revision of the surgical specimen identified juvenile polyps and genetic workup revealed BMPR1 mutation. Patient 2 had a clinical picture resembling familial adenomatous polyposis. Genetic testing showed SMAD4 mutation. Patients 3 and 4 had polyps of multiple histological subtypes and were negative for Sanger sequencing for APC, MUTYHBMPR1, SMAD4, and POLE/POLD1. Pathological review in patient 3 identified Cowden type polyps. Subsequent NGS studies in both these patients confirmed mutations in PTEN and PHTS diagnosis.
DISCUSSION
Our study summarizes the phenotypic and genotypic profiles of patients with HPS. Our main findings show significant overlap and redundancy in the clinical, endoscopic, and histologic findings, which may lead to improper diagnosis and management if genetic diagnosis is withheld. In this regard, we show that the yield of NGS was higher than standard Sanger sequencing in a small sample of cases.
GI bleeding and bowel obstruction were the most common manifestations. GI bleeding was more prevalent in JPS, consistent with previous reports (7). Bowel obstruction, particularly intussusception, was more prevalent in PJS, requiring small bowel resection in 46% of our PJS patients. These findings are consistent with a previous series of 110 PJS patients showing 69% intussusception rate during follow-up with a 50% risk at the age of 20 years (23).
Our endoscopic findings are consistent with previous reports that localized the main polyp burden to the small intestine in PJS and to the colon in JPS (1,4). Comparison of our data to that found in other studies can be seen in Table 4.
Almost a quarter of our JPS patients underwent colorectal surgery mainly due to multiple colonic polyps which were uncontrolled by endoscopy. Latchford et al. (7) described the long-term outcome of 44 JPS patients and found that 7 (15%) required GI surgery; however, only 3 were colorectal procedures, for which the indications were not specified.
Higher polyp burden was significantly associated with the need for surgery and with more cases of severe outcomes. Patients with numerous polyps were more prone to the composite outcome of surgery and malignancy. Interestingly, larger polyps did not seem to be associated with cancer development, as opposed to colonic adenoma studies (24). This might be due to the relatively low incidence of cancer in our study.
The majority of patients in all groups had different (up to 6) histological polyp types during surveillance. A previous series of 49 patients with HPS or hyperplastic polyposis also showed various polyp types. Indeed, genetic testing in combination with a review by an expert pathologist enabled the classification of 4 patients with an undefined syndrome and the reclassification of 2 patients to a different syndrome (25). This redundancy in histologic findings may be a cause for a delayed or erroneous diagnosis, which may affect surveillance and screening of patients and their families.
A significant number of patients (44.2%, mean age at diagnosis of 33.5) developed adenomatous polyps during follow-up, with 3 patients (5.76%) having high-grade dysplasia. A hamartoma-adenoma-carcinoma sequence has previously been suggested in HPS. Specifically, in JPS, genetically altered stromal cells cause changes in the microenvironment surrounding epithelial cells, which eventually lead to disturbed epithelial architecture, differentiation and proliferation (26). We did not find a correlation between occurrence of adenomas and cancer, though this could reflect the low incidence of malignancy in our study. Whether the appearance of adenomatous changes warrants a more aggressive approach has yet to be shown.
Five out of 52 (9.6%) patients developed malignant tumors during follow-up. Previous studies have shown a cumulative risk of colorectal cancer in JPS of 17–22% by the age of 35 years and 68% by the age of 60 years (8), and a mean age of 58 years for diagnosis of gastric cancer (5). In PJS, cumulative cancer risks at the age of 40 and 70 years were 20% and 76%, respectively (27). See Table 4 for a comparison of our data of cancer risk to that found in other studies. The relatively low incidence in our study group may be explained by the young age of patients (overall mean age 25.2 ± 14.2). These numbers could also be explained by the meticulous follow-up at a tertiary referral center, with a high mean adherence index of 3.2 patient visits per year. However, the relatively low mean follow-up time of 6.2 years and high number of patients lost to follow-up may be due to referrals for initial evaluation and diagnosis, after which some patients return to their community. Risk may be higher in population-based settings.
Our data demonstrate a significant family history of both GI malignancies, ranging 50%–65%, and extraintestinal malignancies, ranging 15%–50% of families. This may represent family members with an unknown diagnosis and suboptimal surveillance.
The yield of genetic tests in our study was 77% for PJS, 61% for JPS, and 100% for PHTS patients who underwent genetic testing. Other studies show similar results. Mutations in SMAD4 and BMPR1A were found in 60% of 80 JPS patients using Sanger sequencing and multiplex ligation-dependent probe amplification (28). Mutations in STK11 were identified in 52%–80% of 33–76 PJS patients, respectively (29,30). Our data in PHTS patients are scarce as we had only 4 patients and are similar to earlier reports that identified PTEN mutations in 81% of PHTS patients (16), as opposed to more recent studies that found the mutation frequency to be 34% (17).
Sanger sequencing revealed mutations in only 40.9% of cases and NGS multi-gene panels tested positive in 6/6 cases, suggesting that NGS may better diagnose rare hereditary cancer syndromes.
The unique case of BMPR1A inversion was detected using the Translational Genomics expert platform (31) on a whole genome sequencing and is currently under validation.
Aiming for an accurate genetic diagnosis in HPS patients is important, as it dictates the tailored surveillance regimen for the patient and affected family members.
CONCLUSIONS
This study supports previous data showing the severity of HPS and the need for care at highly specialized centers with use of cutting-edge genetic workup and clinical and endoscopic surveillance. Different HPS types may share characteristics such as polyp histology and burden, as well as clinical manifestations and complications, but phenotypes still show some distinction between the various syndromes. Polyp burden may be a predictor for an outcome of cancer. The multiplicity of histological diagnoses of polyps in the same patient may pose yet another challenge to distinguish between the syndromes and opt for appropriate genetic testing, thus further supporting the use of high throughput NGS testing by multi-gene panels in these cases.
CONFLICTS OF INTEREST
Guarantor of the article: Ophir Gilad, MD.
Specific author contributions: Ophir Gilad and Guy Rosner contributed equally to this work. O.G.: data collection and interpretation, and drafting of manuscript. G.R.: planning of the study, data interpretation, and drafting of manuscript. N.F.-I.: data interpretation and drafting of manuscript. S.A.-K.: data collection. H.S.: drafting of manuscript. N.G.: data interpretation and drafting of manuscript. R.K.: planning of the study, data interpretation, and drafting of manuscript.
Financial support: None.
Potential competing interests: None.
Study Highlights.
WHAT IS KNOWN
✓ HPS are rare hereditary cancer syndromes. Clinical endoscopic and histological data are scarce.
✓ Genetic diagnosis is often complex or unavailable; therefore, appropriate diagnosis may rely on clinical and histological manifestations. Correct diagnosis dictates cancer prevention by appropriate surveillance for the proband and family members.
✓ Severe outcomes of HPS include surgery and GI as well as extraintestinal malignancies and dealth.
WHAT IS NEW HERE
✓ HPS may significantly overlap in their clinical and histological characteristics, which may lead to erroneous diagnosis.
✓ NGS studies have better diagnostic yield than standard Sanger sequencing for HPS.
✓ High polyp burden is associated with severe outcomes of surgery, cancer, and mortality.
TRANSLATIONAL IMPACT
✓ Genetic diagnosis should be better performed with upfront NGS multigene panels.
✓ Patients with multiple GI polyp that are given the histological diagnosis of hamartomatous or inflammatory polyps should be revised by an expert GI pathologist to better characterize lesions due to overlap.
✓ Higher polyp burden over time predicts more severe outcome, and so, patients with high hamartomatous polyp burden should be followed more closely for complications.
REFERENCES
- 1.Stojcev Z, Borun P, Hermann J, et al. Hamartomatous polyposis syndromes. Hered Cancer Clin Pract 2013;11(1):4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Utsunomiya J, Gocho H, Miyanaga T, et al. , Peutz-Jeghers syndrome: Its natural course and management. Johns Hopkins Med J 1975;136:71. [PubMed] [Google Scholar]
- 3.Hinds R, Philp C, Hyer W, et al. Complications of childhood Peutz-Jeghers syndrome: Implications for pediatric screening. J Pediatr Gastroenterol Nutr 2004;39:219. [DOI] [PubMed] [Google Scholar]
- 4.Jelsig AM, Qvist N, Brusgaard K, et al. Hmartomatous polyposis syndromes: A review. Orphanet J Rare Dis 2014;9:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Syngal S, Brand RE, Church JM, et al. ACG clinical guideline: Genetic testing and management of hereditary gastrointestinal cancer syndromes. Am J Gastroenterol 2015;110:223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hemminki A, Markie D, Tomlinson I, et al. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature 1998;391:184. [DOI] [PubMed] [Google Scholar]
- 7.Latchford AR, Neale K, Phillips RK, et al. Juvenile polyposis syndrome: A study of genotype, phenotype, and long-term outcome. Dis Colon Rectum 2012;55:1038. [DOI] [PubMed] [Google Scholar]
- 8.Brosens LA, van Hattem A, Hylind LM, et al. Risk of colorectal cancer in juvenile polyposis. Gut 2007;56:965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Howe JR, Roth S, Ringold JC, et al. Mutations in the SMAD4/DPC4 gene in juvenile polyposis. Science 1998; 280:1086. [DOI] [PubMed] [Google Scholar]
- 10.Sayed MG, Ahmed AF, Ringold JR, et al. Germline SMAD4 or BMPR1A mutations and phenotype of juvenile polyposis. Ann Surg Oncol 2002;9:901. [DOI] [PubMed] [Google Scholar]
- 11.Gallione CJ, Repetto GM, Legius E, et al. A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4). Lancet 2004;363(9412):852. [DOI] [PubMed] [Google Scholar]
- 12.Friedl W, Uhlhaas S, Schulmann K, et al. Juvenile polyposis: Massive gastric polyposis is more common in MADH4 mutation carriers than in BMPR1A mutation carriers. Hum Genet 2002;111(1):108–11. [DOI] [PubMed] [Google Scholar]
- 13.Pilarski R. Cowden syndrome: A critical review of the clinical literature. J Genet Couns 2009;18:13. [DOI] [PubMed] [Google Scholar]
- 14.Laury AR, Bongiovanni M, Tille JC, et al. Thyroid pathology in PTEN-hamartoma tumor syndrome: Characteristic findings of a distinct entity. Thyroid 2011;21:135. [DOI] [PubMed] [Google Scholar]
- 15.Pilarski R, Stephens JA, Noss R, et al. Predicting PTEN mutations: An evaluation of Cowden syndrome and Bannayan-Riley-Ruvalcaba syndrome clinical features. J Med Genet 2011;48:505. [DOI] [PubMed] [Google Scholar]
- 16.Marsh DJ, Coulon V, Lunetta KL, et al. Mutation spectrum and genotype-phenotype analyses in Cowden disease and Bannayan-Zonana syndrome, two hamartoma syndromes with germline PTEN mutation. Hum Mol Genet 1998;7:507. [DOI] [PubMed] [Google Scholar]
- 17.Gilissen C, Hoischen A, Brunner HG, et al. Unlocking Mendelian disease using exome sequencing. Genome Biol 2011;12:228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang HH, Xie NN, Li QY, et al. Exome sequencing revealed novel germline mutations in Chinese Peutz-Jeghers syndrome patients. Dig Dis Sci 2014;59:64. [DOI] [PubMed] [Google Scholar]
- 19.Rosty C. The Role of the surgical pathologist in the diagnosis of gastrointestinal polyposis syndromes. Adv Anat Pathol 2018;25:1. [DOI] [PubMed] [Google Scholar]
- 20.National Comprehensive Cancer Network. Colorectal cancer (Version 1.2018) (https://www.nccn.org/store/login/login.aspx?ReturnURL=https://www.nccn.org/professionals/physician_gls/pdf/genetics_colon.pdf). Accessed on November 1, 2018.
- 21.Heald B, Mester J, Rybicki L, et al. Frequent gastrointestinal polyps and colorectal adenocarcinomas in prospective series of PTEN mutation carriers. Gastroenterology 2010;139(6):1927–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Orloff MS, He X, Peterson C, et al. Germline PIK3CA and AKT1 mutations in Cowden and Cowden-like syndromes. Am J Hum Genet 2013;92:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Lier MG, Mathus-Vliegen EM, Wagner A, et al. High cumulative risk of intussusception in patients with Peutz-Jeghers syndrome: Time to update surveillance guidelines? Am J Gastroenterol 2011;106:940. [DOI] [PubMed] [Google Scholar]
- 24.Martinez ME, Baron JA, Lieberman DA, et al. A Pooled analysis of advanced colorectal neoplasia diagnoses following colonoscopic polypectomy. Gastroenterology 2009;136:832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sweet K, Willis J, Zhou XP, et al. Molecular classification of patients with unexplained hamartomatous and hyperplastic polyposis. JAMA 2005;294:2465. [DOI] [PubMed] [Google Scholar]
- 26.Bosman FT. The hamartoma-adenoma-carcinoma sequence. J Pathol 1999;188:1. [DOI] [PubMed] [Google Scholar]
- 27.van Lier MG, Westerman AM, Wagner A, et al. High cancer risk and increased mortality in patients with Peutz-Jaghers syndrome. Gut 2011;60:141. [DOI] [PubMed] [Google Scholar]
- 28.Aretz S, Stienen D, Uhlhaas S, et al. High proportion of large genomic deletions and a genotype phenotype update in 80 unrelated families with juvenile polyposis syndrome. J Med Genet 2007;44:702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lim W, Hearle N, Shah B, et al. Further observations on LKB1/STK11 status and cancer risk in Peutz-Jeghers syndrome. Br J Cancer 2003;89:308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Volikos E, Robinson J, Aittomäki K, et al. LKB1 exonic and whole gene deletions are a common cause of Peutz-Jeghers syndrome. J Med Genet 2006;43:e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rappaport N, Fishilevich S, Nudel R, et al. Rational confederation of genes and diseases: NGS interpretation via GeneCards, MalaCards and VarElect. Biomed Eng Online 2017;16(Suppl 1):72. [DOI] [PMC free article] [PubMed] [Google Scholar]