

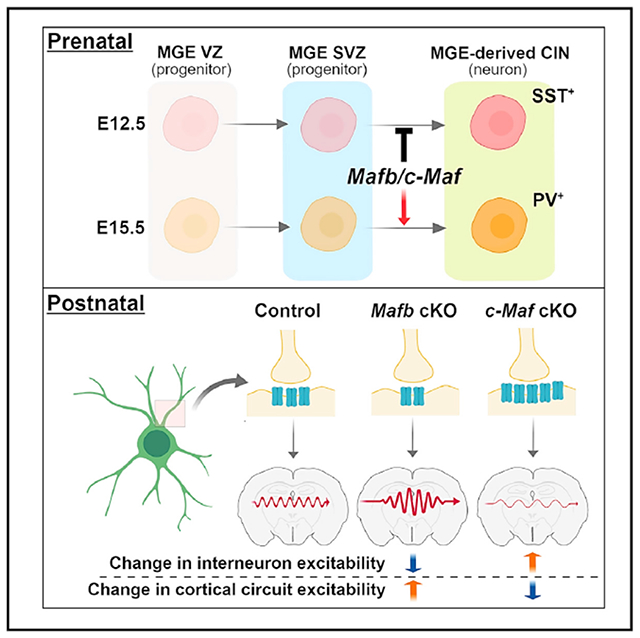
SUMMARY
Mafb and c-Maf transcription factor (TF) expression is enriched in medial ganglionic eminence (MGE) lineages, beginning in late-secondary progenitors and continuing into mature parvalbumin (PV+) and somatostatin (SST+) interneurons. However, the functions of Maf TFs in MGE development remain to be elucidated. Herein, Mafb and c-Maf were conditionally deleted, alone and together, in the MGE and its lineages. Analyses of Maf mutant mice revealed redundant functions of Mafb and c-Maf in secondary MGE progenitors, where they repress the generation of SST+ cortical and hippocampal interneurons. By contrast, Mafb and c-Maf have distinct roles in postnatal cortical interneuron (CIN) morphological maturation, synaptogenesis, and cortical circuit integration. Thus, Mafb and c-Maf have redundant and opposing functions at different steps in CIN development.
Graphical Abstract
In Brief
Using mouse genetics and ex vivo physiology studies, Pai et al. show that Mafb and c-Maf together are necessary to generate the proper numbers of parvalbumin and somatostatin GABAergic interneurons. However, in maturing interneurons, Mafb and c-Maf function divergently to control their firing properties and communication with other neurons.
INTRODUCTION
Disturbances in cortical development and maturation are thought to underlie some symptoms of neurological and neuropsychiatric disorders, such as autism spectrum disorder (ASD), epilepsy, and schizophrenia. One mechanism that is postulated to contribute to symptoms is a circuit imbalance in the excitation to inhibition (E/I) ratio (Chao et al., 2010; Han et al., 2012; Rubenstein and Merzenich, 2003; Yizhar et al., 2011). Although most cortical excitation is generated by glutamatergic projection neurons and thalamic afferents, inhibition is largely generated by locally projecting GABAergic cortical interneurons (CINs). CINs exhibit diverse morphological, connectivity, molecular, and electrophysiological properties (Huang et al., 2007; Kepecs and Fishell, 2014; Kessaris et al., 2014), which facilitate the E/I balance in distinct cortical microcircuits.
CINs are derived from progenitor zones in the subpallial telencephalon, called the medial and caudal ganglionic eminences (MGE and CGE, respectively), and the preoptic area (POA) (Gelman et al., 2011; Wonders and Anderson, 2006). The development of MGE and CGE-derived CINs are coordinated by a combination of transcription factors (TFs) expressed in these progenitor zones (Hu et al., 2017; Lim et al., 2018a). Mature CINs express molecular markers that delineate four broad subgroups: MGE-derived somatostatin (SST+) and parvalbumin (PV+), and CGE-derived vasoactive intestinal peptide (VIP+) and reelin+;SST− (Lim et al., 2018a).
Mafb and c-Maf, a part of the large Maf TF family, bind to DNA through the basic leucine zipper motif (Kataoka, 2007). Mafb and c-Maf function alone or together to control cell fate and differentiation in bone, epithelial cells, lens, macrophages, and pancreas (Lopez-Pajares et al., 2015; Nishikawa et al., 2010; Soucie et al., 2016). In the nervous system, Mafb and c-Maf have multiple functions. For example, Mafb controls embryonic hindbrain regional patterning (Cordes and Barsh, 1994) and promotes the formation of auditory ribbon synapses that are required to activate inner hair cells (Lu et al., 2011; Yu et al., 2013). c-Maf is involved in touch receptor differentiation in the peripheral nervous system (Wende et al., 2012).
Mafb and c-Maf are particularly intriguing in CIN development because their MGE expression initiates in the MGE subventricular zone (SVZ) and persists in MGE-derived interneurons but not in MGE-derived projection neurons (Cobos et al., 2006; McKinsey et al., 2013; Zhao et al., 2008). In addition, TFs that control MGE CIN development also regulate Mafb and c-Maf expression. For instance, Zfhx1b mutants have reduced c-Maf expression, Lhx6 mutants have reduced Mafb expression, and Dlx1/2 mutants have reduced Mafb and c-Maf expression (McKinsey et al., 2013; Zhao et al., 2008). Two recent reports provided evidence that (1) Mafb is preferentially expressed in a subtype of SST+ CINs (Martinotti cells), where it regulates their migration and axonal projection (Lim et al., 2018b); and (2) c-Maf promotes the generation of SST+ CINs (Mi et al., 2018), a finding that is in opposition to the combined functions of Mafb and c-Maf presented herein.
Here, we report the individual and combined functions of Mafb and c-Maf after conditional deletion in the MGE lineages with Nkx2.1-Cre and 799-CreER. Mafb;c-Maf conditional double knockout (cDKO) phenotypes provided evidence that Mafb and c-Maf compensate for each other. Notably, cDKOs generate excessive SST+ at the expense of PV+ CINs. Furthermore, cDKOs have reduced CIN numbers, probably because of a combination of mechanisms, including ectopic migration to the hippocampus and a progressive reduction in CINs during postnatal ages. However, our electrophysiological analyses of adult somatosensory cortices and in vitro assays of neonatal CINs provide evidence that Mafb and c-Maf have distinct postnatal functions in CIN maturation, synaptogenesis, and activity. Together these defects lead to alterations in neocortical circuit excitability and provide potential mechanistic insights into how these TFs operate during CIN development and maturation.
RESULTS
Mafb and c-Maf Have Overlapping and Distinct Expression Patterns in Developing CINs
We compared Mafb and c-Maf gene expression in the MGE and at later stages of CIN maturation. First, we determined Mafb and c-Maf gene expression profiles and their cellular specificity in the MGE by reanalyzing single cell RNA sequencing (scRNA-seq) data derived from wild-type (WT) E11.5, E13.5, and E15.5 MGE tissue (Chen et al., 2017). We studied the data set by Louvain clustering with Jaccard distance matrix, which revealed nine molecularly distinct clusters (Figures 1A and 1B; Table S1) (Shekhar et al., 2016). Mafb and c-Maf were expressed in some MGE progenitor cells (clusters 1, 8, and 9; Table S1). In addition, Mafb and c-Maf mRNA were co-expressed in a subset of these progenitors. Mafb and c-Maf were significantly enriched in cluster 4, which we propose corresponds to cells that will become CINs, based on their expression of multiple genes, including Cux2 and Erbb4 (Figure 1C). The expression levels of Mafb and c-Maf in cluster 4 showed that these genes were largely co-expressed in immature CINs. Thus, Mafb and c-Maf expression initiates in progenitors, and their co-expression increases in immature CINs (Figures 1C, 1D, 1F, and 1H–1S).
Figure 1. Analysis of Mafb and c-Maf Expression in Single Cells and in Tissue Sections from the MGE and in MGE-Derived Developing and Mature CINs.
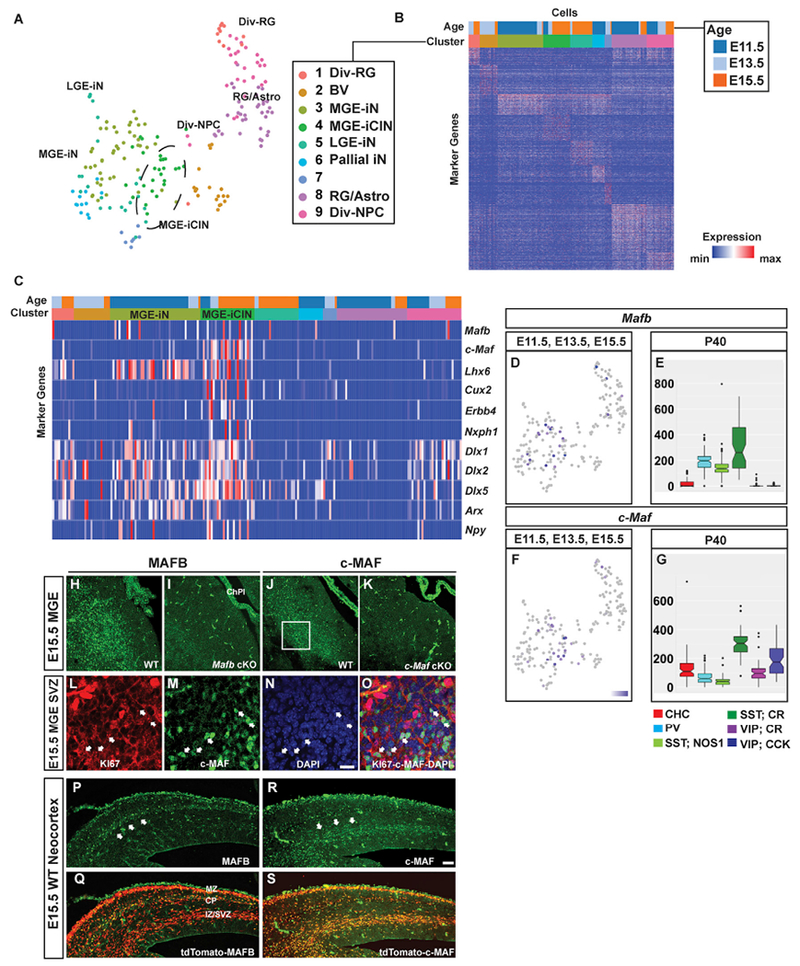
(A–G) Single-cell RNA-seq MGE and adult CIN analyses.
(A) t-stochastic neighbor embedding (t-SNE) plot representing MGE cells analyzed from age E11.5, E13.5, and E15.5 and colored by their cluster assignments. Cells with properties of MGE-derived immature CINs (iCIN) are encircled in cluster 4.
(B) Heat map representation of cluster marker genes. Please see Table S1 for the gene list.
(C) Heat map representation of the 9 clusters; it shows enrichment of Mafb and c-Maf in cluster 4, and the expression of markers for MGE-derived CINs. Notethat half of the cluster 4 CINs co-express Mafb and c-Maf.
(D–G) Expression feature plot of Mafb (D) and c-Maf (F) in MGE cells; positive cells are blue. Box plots of Mafb (E) and c-Maf (G) expression in CIN subtypes at P40.
(H and I) Immunofluorescent images of the MGE from E15.5 WT (H) and Mafb cKO (I) showing MAFB expression.
(J and K) Immunofluorescent images of the MGE from E15.5 WT (J) and c-Maf cKO (K) showing c-MAF expression.
(L–O) Confocal imaging that shows co-labeling of KI67 (L) and c-MAF (M) in the MGE SVZ (O) (boxed in J). Arrowheads point to cells that are c-MAF+ progenitors.
(P–S) Immunofluorescent images from E15.5 WT neocortex that show Nkx2.1-cre-mediated tdTomato expression merged with MAFB (P–Q) and c-MAF (R–S). Scale barin (N) and (R) represents 100 μm. Div-RG, dividing radial glia; MGE-iN/iCIN, MGE-derived immature neurons/cortical interneurons; LGE-iN, LGE-derived immature neurons; pallial iN, pallial immature neurons; RG/Astro, radial glias and astrocytes; Div-NPC, dividing neural progenitor cells; CHC, chandelier cells; CR, calretinin; VIP, vasoactive intestinal peptide; ChPl, choroid plexus; MZ, marginal zone; CP, cortical plate; IZ/SVZ, intermediate zone-subventricular zone.
We then assessed single-cell transcriptome data from postnatal day 40 (P40) CINs (Paul et al., 2017). These data showed that Mafb and c-Maf expression persisted in MGE-derived adult CINs. Mafb and c-Maf were enriched in multiple MGE-derived CIN subtypes, including chandelier cells (CHCs), PV+ basket cells (PVBCs), SST;neuronal nitric oxide synthase+ cells (SST;NOS1) and SST;CALRETININ+ (SST;CR) cells. Importantly, these two TFs are highly expressed in the SST;CR subpopulation (Figures 1E and 1G), suggesting that the broad SST+ group of CINs was determined by these TFs. Moreover, it indicated that c-Maf, but not Mafb, mRNA was detected in VIP+ CINs (CGE-derived), suggesting a divergence of Maf expression into other CIN progenitor domains.
To complement the single-cell transcriptome analysis data, we performed histochemistry to study Mafb and c-Maf RNA and protein expression (Figures 1H–1S and S1). Consistent with the scRNA-seq data, we found that MAFB and c-MAF proteins expressed in the MGE SVZ and in MGE-derived immature CINs (SVZ: Figures 1H and 1J, S1A and S1B, S1D and S1E, S1G, and S1J; CINs: Figures 1P–1S, S1C, and S1F). In the MGE SVZ, MAFB and c-MAF were expressed in KI67+ SVZ2 progenitors (Figures 1L–1O and S1M–S1AJ’). The SVZ2 is the layer of SVZ progenitors that are between the SVZ1 (adjacent to the VZ) and the layer of newborn neurons. (Petryniak et al., 2007).
CINs tangentially migrate to the cortex along multiple pathways. We explored whether MAFB+ and c-MAF+ CINs had shared or different trajectories. Immunofluorescence analyses of E15.5 WT neocortices showed that MAFB was expressed in immature CINs migrating along the marginal zone (MZ) pathway and in the cortical plate (CP) (Figures 1P and 1Q), as described in Lim et al. (2018b). On the other hand, c-MAF expression was enriched in the deep migratory pathway (Figures 1R and 1S). This suggests that MAFB and c-MAF may differentially regulate CINs migrating along the superficial and deep pathways, respectively.
Combined Loss of Mafb and c-Maf Results in Decreased MGE-Derived CINs
The functions of Mafb and c-Maf in CIN development remain largely unknown, in part because constitutive Mafb and c-Maf mutant mice die at embryonic or neonatal ages (Blanchi et al., 2003; Kawauchi et al., 1999). Thus, to examine the prenatal and postnatal functions of these TFs in MGE-derived CINs, we generated conditional mutant mice with floxed Mafb and c-Maf (Wende et al., 2012; Yu et al., 2013), combined with Nkx2.1-Cre (Xu et al., 2008) alleles, whose expression in the MGE and POA begins on embryonic day (E) ~9.5. We crossed either MafbFlox and/or c-MafFlox mice to those harboring Nkx2.1-Cre and the Ai14 allele (Madisen et al., 2010), which expresses the fluorescent protein tdTomato after CRE recombination, and subsequently generated both conditional Mafb and c-Maf single (cKOs) as well as conditional double knockouts (cDKOs). Mice were born at expected Mendelian ratios and lived into adulthood. Of note, these crosses generated both WT and conditional heterozygous states for each genotype (see details in STAR Methods). We did not detect gross phenotype differences between WT and conditional heterozygous mice. Thus, controls used in this report are either WT or mixed conditional heterozygotes, unless otherwise noted.
We began our phenotypic analysis of Maf mutant mice by assessing MGE-derived CINs and CIN subgroups from Nkx2.1-Cre-lineages (tdTomato+) at P35 in the somatosensory cortex (Figure 2). Mafb and c-Maf cKOs had modest decreases of 25% and 32%, respectively, in tdTomato+ cells (Figures 2A–2C, 2E–2G, and 2J; Mafb cKOs, p = 0.002; c-Maf cKOs, p = 0.0002). On the other hand, Maf cDKOs had a 64% reduction in the density of tdTomato+ cells (Figures 2D, 2H, and 2J; p < 0.0001). These results suggest that Mafb and c-Maf have compensatory roles in regulating the number of MGE-derived CINs.
Figure 2. CINs in Maf cDKOs Are Reduced in Numbers, Have Altered Laminar Positions, and Have a Decreased Proportion of PV+ CINs Cell Autonomously.
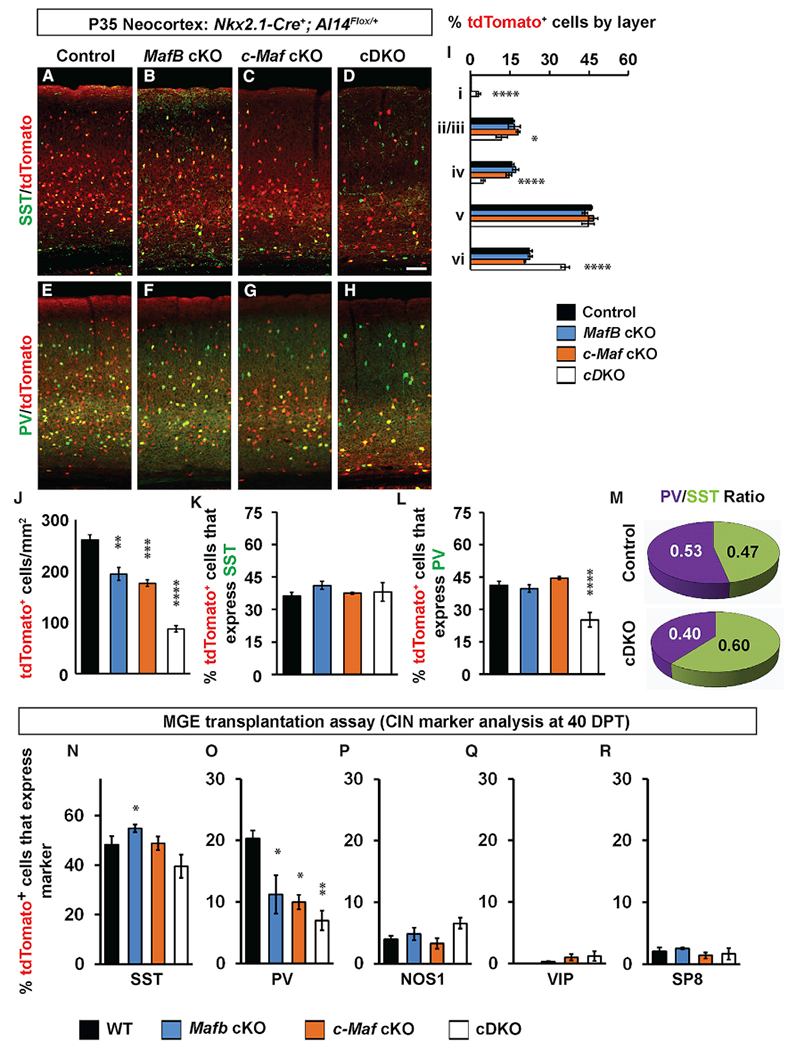
(A–D) Immunofluorescent images from P35 somatosensory cortices from 4 genotypes that show native tdTomato merged with somatostatin (SST) staining.
(E–H) Immunofluorescent images from P35 somatosensory cortices from 4 genotypes that show native tdTomato merged with parvalbumin (PV) staining.
(I) Quantification of the relative proportion of tdTomato cells that occupy cortical layers.
(J) Quantification of the number of tdTomato+ cells/mm2 in the somatosensory cortex.
(K and L) Quantification of the proportion of tdTomato+ cells that co-express either SST (K) or PV (L).
(M) Pie chart of PV+/SST+ ratio of the remaining tdTomato+ cells. n = 4 for all groups.
(N–R) MGE cell transplantation into neocortex assay to assess cell autonomy of CIN phenotypes. Quantification of the number of transplanted tdTomato+ cells/mm2 that co-express MGE or CGE CIN markers including SST (N), PV (O), NOS1 (P), VIP (Q), and SP8 (R). n = 4 for all groups.
Scale bar in (D) represents 100 μm. Data are expressed as the means ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Next, we assessed the proportion of cells that occupied each lamina of the somatosensory cortex. Mafb and c-Maf cKOs did not differ from controls, whereas the cDKOs had a greater loss in upper lamina (layers II—IV) and an increased proportion in the deep lamina (layer VI) (Figure 2I, layers II/III, p = 0.03; layer IV, p < 0.0001; layer VI, p < 0.0001), even though tdTomato+ cell densities were reduced in all layers (Figure 2J). Interestingly, in the cDKOs, MGE-derived CINs were also found in layer I, a lamina that is not populated by those cells (Figure 2I; p < 0.0001).
To determine whether loss of Mafb and c-Maf affected MGE lineage CINs equally, we determined the proportion of remaining tdTomato+ cells in the mutants that expressed either SST or PV. Although the density of tdTomato+;SST+ CINs decreased in all KOs (Figures 2A–2D; Table S2), the proportion of SST+ CINs was similar between genotypes (Figures 2J and 2K). Furthermore, the density of tdTomato+;PV+ CINs decreased in all KOs (Figures 2E–2H, Table S2); however, the proportion of tdTomato+ cells expressing PV was reduced 2-fold only in the cDKO (Figures 2J and 2L; p < 0.0001). This disproportionate decrease in PV+ CINs led to an increase in the ratio of SST to PV CINs in the cortex in cDKOs (Figure 2M). Of note, interneurons in the hippocampus and striatum also exhibited similar disproportionate decreases in the PV+ cells (Table S2).
In sum, Mafb and c-Maf together are required for controlling the appropriate number of MGE-derived CINs at P35. Furthermore, they are particularly important in promoting PV+ cortical, hippocampal, and striatal interneuron generation and/or maturation.
Cell Autonomous Role for Mafb and c-Maf in Establishing Normal Numbers of PV+ MGE-Lineage CINs
To test whether the preferential reduction of PV+ cells in the Maf cDKOs was cell autonomous, we used an MGE transplantation assay to transduce small numbers of MGE progenitor cells into a WT cortex and monitor their development in vivo (Vogt et al., 2015). E13.5 Ai14Flox/+ MGE cells, which were either WT, MafbFlox/Flox, c-MafFlox/Flox, or MafbFlox/Flox;c-MafFlox/Flox, were harvested and virally transduced with a Cre expression vector using the DlxI12b enhancer. These MGE cells were transplanted into P1 WT neocortices; 40 days later, they were analyzed for SST, PV, NOS1, VIP, and SP8 expression.
The proportion of tdTomato+ cells that were PV+ were decreased in all Maf mutant cells (Figure 2O; Mafb cKOs, 45%, p = 0.04; c-Maf cKOs, 50%, p = 0.02; cDKOs, 65%, p = 0.006). SST numbers were not changed, except for Mafb cKO (increased 14%; Figure 2N; p = 0.04). Other MGE-derived CINs, such as NOS1+ cells (Figure 2P), and CGE-derived CINs, VIP+, and SP8+, were unchanged (Figures 2Q and 2R).
These data suggest that the decrease in PV+ MGE-lineage CINs is cell autonomous and that loss of Mafs does not lead to CIN fate change from the MGE type (SST and PV) to the CGE type (SP8 and VIP).
cDKOs Have Reduced CINs and Excess Hippocampal Interneurons at P0
To identify the onset in the reduction of MGE-lineage CINs observed in cDKOs (Figure 2), we assessed tdTomato+ cells at multiple ages (Figure S2). At E13.5 and E15.5, we found no significant changes in the density of tdTomato+ neocortical cells (Figures S2A–S2H and S2W). By P0, the cDKOs had a decrease (~26%) in the density of tdTomato+ neocortical cells (Figures S2I–S2L and S2W; p = 0.046). By P7 (Figures S2M–S2P) and P16 (Figure S2Q–S2T), there were further decreases in the density of tdTomato+ neocortical cells, which reached ~64% reduction at P35 (Figure S2W).
Concomitant with the significant decrease in the density of neocortical tdTomato+ cells starting at P0, the cDKOs had a ~42% increased density of tdTomato+ cells in the hippocampus (Figures S3F–S3J; p = 0.007). However, the increase of hippocampal tdTomato+ cells was transient; it was no longer present at P16 (Table S2). By P35, the cDKOs had a comparable reduction of tdTomato+ cells in both the hippocampus and somatosensory cortex (Table S2).
We suggest that the transient increased density of tdTomato+ cells in the hippocampus could be due to an “over-migration” of CINs into that region, which may contribute to the reduction in their numbers in the neocortex.
MGE Proliferation and CIN Apoptosis Are Not Altered in cDKOs
Several mechanisms could lead to the reduction of CIN numbers (Figures 2 and S2). Although ectopic cell accumulation in the hippocampus could account for cell loss in the neocortex, it is also possible that reduced cell proliferation and/or increased cell death has a part. We focused our subsequent analyses on the cDKOs. To study MGE proliferation, we administered 5-ethynyl-2′-deoxyuridine (EdU) for 30 min to E13.5 pregnant mice to label the S-phase progenitors (schema, Figure S4A). We assessed EdU+ cell density in the VZ, SVZ1, and SVZ2 (secondary progenitors) and found no change in the cDKOs. (Figures S4B–S4D). Next, at E13.5 and E15.5, we determined the density of phospho-histone-3 (PH3+) cells to assess the numbers of M-phase progenitors and, again, found no difference (Figures S4E–S4J). These data suggested that, during peak MGE CIN generation, there were no significant differences in the number of S-phase and M-phase progenitors in the VZ and SVZ.
To determine whether increased apoptosis could contribute to the CIN reduction in cDKOs, we stained for cleaved caspase-3 (CC3), a marker of apoptosis. At E13.5 and E15.5, no differences in CC3+ cell densities were detected in the MGE or along the CIN migration route. At P0, P7, and P16, we also observed no differences in the density of tdTomato+/CC3+ cells (data not shown).
Additionally, we stained for IBA1, a marker of active microglia, to see whether increased microglia activity might contribute to the CIN reduction in cDKOs. Again, we observed no significant changes in IBA1+ microglia density in the neocortex at P7 and P16 (data not shown). Thus, we did not obtain evidence for increased apoptosis and microglia engulfment contributing to the reduction in neocortical CINs in cDKOs.
Early Born MGE Lineages Are Overrepresented in cDKO Adult Brains
In the adult (P35) cDKO somatosensory cortex, the remaining CINs maintained the normal proportion of SST+ CINs but showed a reduction of PV+ CINs. Furthermore, cDKO CINs were disproportionately reduced in superficial layers (likely late born) and increased in deep layers (likely early born) (Figure 2I).
We hypothesized that P35 cDKOs CINs were enriched for early born SST CINs. The birth date of MGE-derived CINs correlates with their cell fate and laminar position. Early born (E12.5–E13.5) MGE cells tend to occupy deep neocortical lamina and express SST, whereas later-born (E15.5) cells tend to occupy superficial lamina and express PV (Inan et al., 2012; Miyoshi et al., 2007; Pla et al., 2006). To test that hypothesis, we followed the fate of early and late-born control and cDKO MGE CINs by giving EdU at either E12.5 or E15.5 and, then, analyzing their neocortical laminar position at P35 (schema, Figure 3A). After the E12.5 EdU pulse, we detected a 2-fold increase in the proportion of double-labeled tdTomato+;EdU+ cells in cDKOs (Figure 3B; p = 0.0003). However, no differences were observed after the E15.5 pulse (Figure 3C). These data support the idea that the cDKO generates disproportionally more CINs by E12.5. The laminar distribution of tdTomato+;EdU+ CINs in these experiments showed no major differences, except for a trend for an increase in layer VI and a decrease in layers II/II/IV (Figures 3D and 3E).
Figure 3. EdU Pulse-Chase Experiments Revealed Maf cDKOs Have Increased Early Born Adult CINs and Have Precocious MGE Neurogenesis.
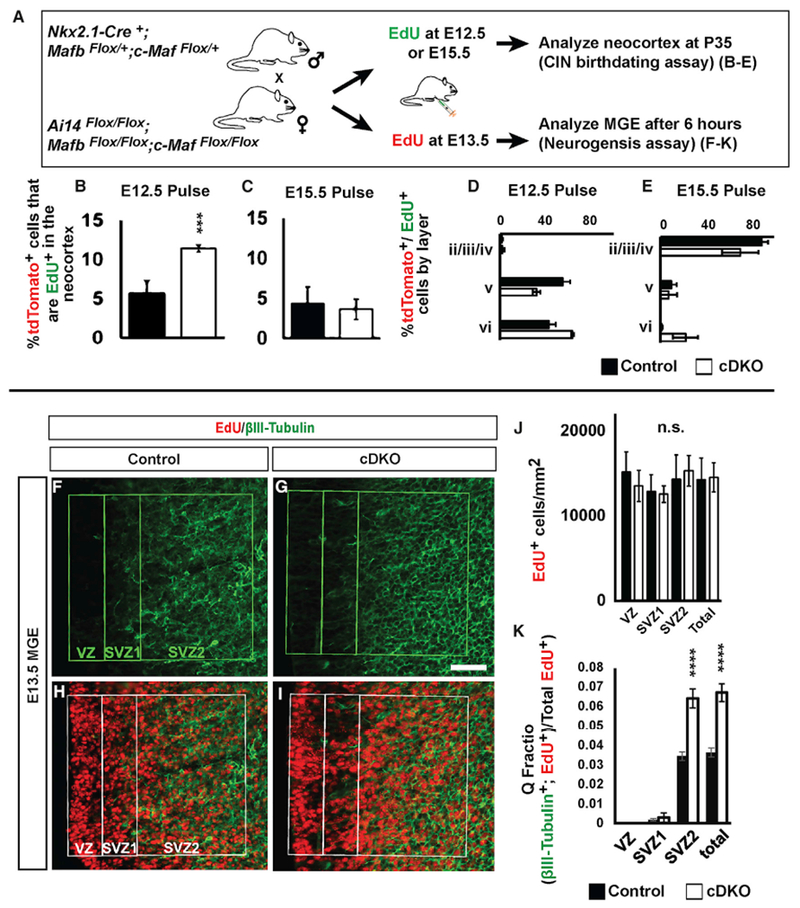
(A) Schema depicting the EdU pulse-chase assays.
(B–K) To follow CIN birthdates (B–E), EdU was injected into pregnant females when embryos were E12.5 or E15.5. The somatosensory cortex was assessed at P35. To study MGE neurogenesis (F–K), EdU was injected into pregnant females when embryos were E13.5; the embryos were analyzed 6 h later for co-expression of EdU and βIII-Tubulin, a neuronal marker.
(B and C) Quantification of EdU+;tdTomato+ double-labeled cells/mm2 from EdU pulses at either E12.5 (B) or E15.5 (C).
(D and E) Quantification, as a function of cortical lamina, of the proportion of EdU+; tdTomato+ double-labeled cells from EdU pulses at E12.5 (D) or E15.5 (E). n = 4 for all groups.
(F–I) Immunofluorescent images show the MGE co-stained with EdU (H and I) and βIII-tubulin (F and G) from control and cDKO. Boxed region indicates VZ, early SVZ (SVZ1), and late SVZ (SVZ2).
(J) Quantification of EdU+ progenitors/mm2 in the VZ, SVZ1, and SVZ2.
(K) Quantification of the Q fraction (EdU+; βIII-tubulin+/EdU+) in the VZ, SVZ1, and SVZ2.
n = 3 for all groups. Data are expressed as the means ± SEM. ***p < 0.001, ****p < 0.0001. Scale bar in (G) represents 50 μm. SVZ1, subventricular zone 1; SVZ2, subventricular zone 2; VZ, ventricular zone.
Of note, the above data could be confounded by the cell loss that occurs in the cDKO neocortices by P35 (Figure 2). To circumvent that, we used a prenatal, 6 hr, EdU pulse-chase paradigm (schema, Figure 3A) to compare neurogenesis in the control and cDKOs MGE before cell loss occurs. To that end, pregnant mice were given EdU when the embryos were E13.5; 6 hr later the embryos were sacrificed, and immunofluorescently co-labeled for EdU and the neuronal marker, βIII-tubulin (Figures 3F–3I). We quantified the percentage of EdU+ βIII-tubulin+ double-positive cells to determine the fraction of cells that left the cell cycle and became immature neurons (the quiescent (Q)-fraction) (Figure 3K) (Takahashi et al., 1996). We focused on the MGE SVZ, where newly generated neurons are present before they migrate.
As in previous experiments, we did not detect differences in the density of EdU+ cells in the MGE (Figure S4D). However, the proportion of newly post-mitotic (βIII-tubulin+;EdU+) cells (Q-fraction) generated at E13.5 increased ~2-fold in the cDKOs (Figure 3K; p = 0.0009). These data provided evidence that, although loss of Mafb;c-Maf did not change the rate of proliferation, the newly generated cells were becoming neurons at an increased rate in the cDKO. This could lead to an increase in SST+ CINs, perhaps at the expense of the PV+ CINs.
Excessive Production of SST+ CINs in cDKOs
Our earlier EdU pulse-chase experiments provided evidence that cDKOs have an increased proportion of early born CINs in the adult cortex (Figure 3B), as well as increased MGE neurogenesis at E13.5 (Figure 3K). Thus, because the early wave of CINs are mostly Sst+ (Inan et al., 2012; Miyoshi et al., 2007; Pla et al., 2006), we were interested in determining whether there were increased numbers of Sst+ cells being generated in the cDKO MGE. We characterized Sst RNA expression by in situ hybridization (ISH) at E13.5, E15.5, and P0 (Figures 4A–4C). Although there was no detectable change in Sst expression at E13.5 (Figure 4A), there was an ~2-fold increase in the density of cortical Sst+cells at E15.5 (Figures 4B and 4E; p = 0.0184). This increase was even more apparent by P0 (Figure 4C). Furthermore, at P0, Sst+ cells were clearly increased in the hippocampus (caudal) and in the cingulate (rostral) cortex (Figure 4C), consistent with the transient increase in neonatal MGE-lineage cells (tdTomato+) in those locations at P0 (Figures S3F–S3J).
Figure 4. Mafb and c-Maf Regulate the Quantity and Position (Laminar and Regional) of MGE-Derived Somatostatin+ Interneurons during Cortical and Hippocampal Development.
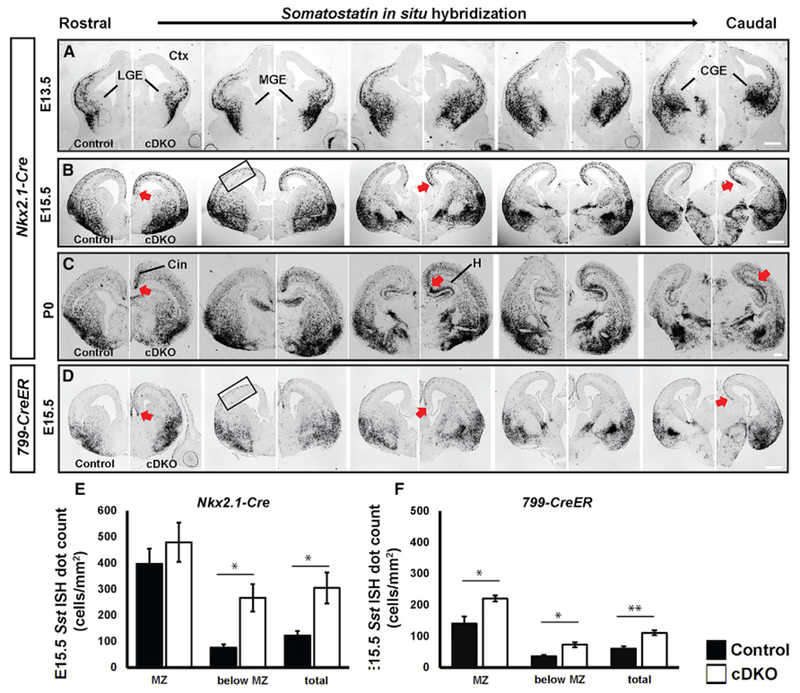
(A–C) Sst in situ hybridization at E13.5 (A), E15.5 (B), and P0 (C) in control and cDKO generated using Nkx2.1-Cre.
(D) Sst in situ hybridization at E15.5 in control and cDKO generated using 799-CreER, whose activity begins in the SVZ. Increased numbers of Sst+ CINs in the dorsomedial cortex and hippocampus are denoted by red arrows.
(E) Quantification of Sst+ CINs/mm2 by region in the neocortex in control and cDKO generated with Nkx2.1-cre. Increased Sst+ CINs were observed in laminae below the marginal zone (MZ) in cDKO at E15.5. n = 4 for control; n = 3 for cDKO
(F) Quantification of Sst+ CINs/mm2 by region in the neocortex in control and cDKO generated with 799-CreER. Increased Sst+ CINs were observed in the MZ and below the MZ in cDKO at E15.5.
n = 4 for both groups. Data are expressed as the means ± SEM. *p < 0.05, **p < 0.01. Scale bar in (A)–(D) represents 500 μm. Boxed region indicates where quantification was done. Cin, cingulate cortex; MGE, medial ganglionic eminence; LGE, lateral ganglionic eminence; H, hippocampus
To determine whether the increase in Sst+ cells in the cortex was related to the precocious neurogenesis observed at early ages in the MGE (Figure 3K), we administered EdU to pregnant mice at E12.5 and analyzed them at E15.5 in the neocortex to assess the number of SST+ and tdTomato+ cells generated at an earlier age (Schema, Figures 5A–5G). Indeed, the proportion of tdTomato+ cells that expressed SST nearly doubled in cDKOs (Figure 5H; p < 0.0001). Moreover, we found that the number of triple-labeled tdTomato+;SST+ CINs that were EdU+ also doubled in the cDKOs (Figure 5I; p = 0.0002). Overall, these data show that without Mafb and c-Maf, the MGE has a normal density of progenitors, which generate more early born SST+ CINs. Because there was no increased density in cortical tdTomato+ cells at E15.5 (Figure S2), we hypothesize that the remaining tdTomato+;SST− cells are most likely of the PV+ lineage (although that age is too young to assess PV expression).
Figure 5. Increased Numbers of SST+ Interneurons in Developing cDKO Neocortex.
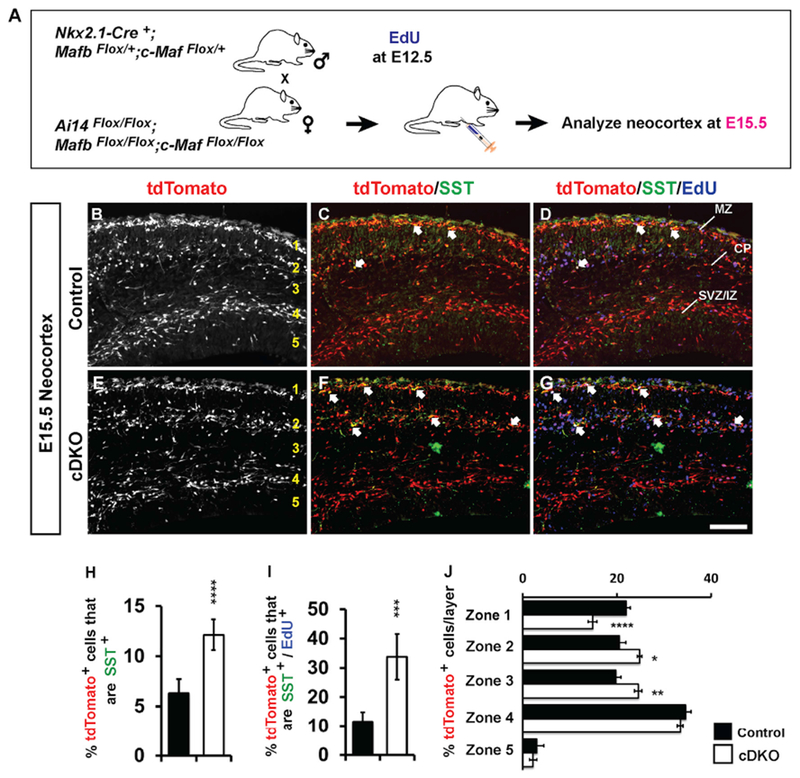
(A) Schema depicting the EdU 3-day pulse-chase experiment. EdU was injected into pregnant females when embryos were E12.5. The neocortices were assessed at E15.5.
(B–G) Immunofluorescent images of E15.5 neocortices showing tdTomato+ (B and E) cells co-stained for SST (C and F) and EdU (D and G). Arrows show double- or triple-labeled cells (C, D, F, and G).
(H) Quantification of the number of tdTomato+ cells/mm2 that are SST+.
(I) Quantification of the number of EdU-labeled tdTomato+ cells/mm2 that are SST+.
(J) Quantification of the proportion of tdTomato+ cells for the five layers labeled in (B) and (E). Marginal zone (zone 1), cortical plate and subplate (zone 2), intermediate zone (zone 3), deep migration stream (zone 4), and the ventricular zone (zone 5).
n = 3–4 for all groups. Data are expressed as the means ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Scale bars in (G) represents 100 μm.
Mafb and c-Maf Function in SVZ Progenitors to Control MGE-Derived CIN Production
We have provided evidence that the lack of Mafb and c-Maf results in Sst+ CIN overproduction in the late SVZ of MGE (SVZ2; Figure 3K). Thus, we hypothesized that Mafb and c-Maf function in the SVZ to control the production and fate of CINs. To further test that idea, we generated conditional Maf cDKOs using the 799-CreER mouse line, whose activity initiates in the SVZ of the MGE (Silberberg et al., 2016).
First, we identified, in more detail, the type(s) of MGE progenitors in which 799-CreER activity is initiated (Figure S3). We induced Cre activity with a tamoxifen injection at E11.5 and assayed CRE activity with the Ai14 reporter. We harvested the embryos at E12.5, 30 min after an EdU injection. To test whether there was 799-CreER activity in the MGE progenitors, histological sections were analyzed with antibodies to tdTomato, EdU (S-phase progenitors) and Ki67 (pan-progenitor marker). We identified tdTomato and Ki67 double-positive cells in the SVZ2 but not in the VZ or SVZ1 (Figures S3K and S3L). Thus, we provided evidence that 799-CreER activity initiates in a subset of MGE “late” progenitors. Furthermore, MAFB and tdTomato co-localize in the MGE SVZ2, suggesting MAFB expression initiates around the time that 799-CreER activity begins (Figures S3M–S3O).
Next, we used 799-CreER (tamoxifen at E11.5) to generate cDKO embryos, which were analyzed at E15.5 to test whether the mutation affected the number of SST+ CINs. Sst ISH showed an ~2-fold increase (Figures 4D and 4F; p = 0.003), which was also observed with Nkx2.1-Cre-mediated deletion (Figures 4B and 4E); similar changes were also seen in CIN laminar distribution (Figures 4E and 4F; MZ, p = 0.03; below MZ, p = 0.012).
Lastly, we used Sst-IRES-Cre (Taniguchi et al., 2011) and Ai14 (Figures S3P–S3Y) to generate cDKO mice that express tdTomato at P0 and P30 to test whether deletion of Mafb and c-Maf expression in post-mitotic SST+ lineage cells affects the numbers of SST+ CINs. In the cDKOs, we observed no change in the density of tdTomato+ CINs or tdTomato+;SST+ CINs (data not shown).
Thus, the use of 799-CreER and Sst-IRES-Cre support the conclusion that MAF proteins act in the SVZ2, and not in immature CINs, to repress the generation of SST+ CINs.
c-Maf Controls CIN Migration Alone and Together with Mafb
Mafb and c-Maf cDKOs, in addition to the increase in Sst+ CINs in immature neocortex and hippocampus, also have changes in CIN migration patterns at E15.5. We quantified CIN densities of the deep and superficial migration streams using both E15.5 Sst ISH (Figures 4E and 4F) and E15.5 tdTomato immunofluorescence (IF) (Figure 5J).
In the Nkx2.1-Cre-mediated cDKO, the density of Sst+ cells in the lamina below the MZ was increased (Figure 4E; p = 0.02). Consistent with that, the proportion of tdTomato+ CINs in the MZ (zone 1) was reduced ~30% (Figure 5J; p < 0.0001). In turn, zones 2 and 3, which included the CP and subplate, had increased proportions of cells in the cDKOs (Figure 5J; p = 0.04 and 0.003, respectively), whereas the deeper layers—zones 4 and 5—were unchanged.
We also conducted Sst ISH at E15.5 in the Mafb and c-Maf single cKOs (Figures S3A–S3D). Although there was no obvious change in total Sst+ cell density in the Mafb cKO, the c-Maf cKO showed a slight increase (Figure S3E).
Of note, tdTomato expression identified an alteration in the laminar pattern of CINs in the deep tangential migration zone at E15.5 in the c-Maf cKO and the cDKO. Although control and Mafb cKOs have tightly organized deep migration zones (arrow-heads, Figures S2E and S2F), that region is disorganized and has ~2-fold fewer CINs in the c-Maf cKOs and the cDKOs (Figures S2G and S2H; c-Maf cKO, p < 0.0001; cDKO, p = 0.0001). The observation that the c-Maf cKO has a defect in the deep migration layer correlates with the c-Maf selective expression in that location (Figures 1P and 1R). Together those alterations in CIN laminar organization, during their tangential migration, suggest that c-Maf alone, and together with Mafb, control migration of immature CINs.
To understand the mechanisms underlying the migration phenotypes, we assayed Cxcr7 RNA expression because Cxcr7 mutants have a reduction in migrating CINs in the MZ and an increase in the CP, very similar to the phenotype observed in the distribution of tdTomato+ CINs in the cDKO (Figure 5J). Indeed, in the cDKO, the cortical distribution of Cxcr7 RNA resembled that of tdTomato (Figures S5O, S5P, S5PO’, and S5P’) (Li et al., 2008; Sánchez-Alcañiz et al., 2011; Stumm et al., 2003; Wang et al., 2011). This provides evidence that Mafb and c-Maf control CIN migration independent of Cxcr7 expression, potentially through a parallel pathway and/or downstream event.
Mafb and c-Maf Loss of Function Alters CIN Action Potential Firing Properties and Synaptic Excitation
To better understand howMaf mutations affect CINs physiology, we performed whole-cell patch-clamp recordings from Nkx2.1-Cre-lineage CINs in cortical layer 5 of the somatosensory cortex in adult control and Maf mutant mice (P63–P82). CINs were visually identified for electrophysiological analyses via tdTomato expression. For assessments, where feasible, we post hoc identified fast-spiking (FS) and regular-spiking (RS) CINs, based on their firing patterns, and analyzed their cellular electrical properties separately.
Active and Passive Membrane Properties of CINs
We measured the mean firing frequency of FS and RS CINs in response to the increasing intensity of the intracellular positive current injection (F–I plots, Figure 6D). We found that, in FS cells, the maximal firing frequency was reduced in all mutant CINs compared with controls. FS CINs from all Maf mutants could not sustain firing rates greater than ~60 Hz and could not maintain firing rates for currents above 200 pA, most prominently in the Mafb cKOs. Interestingly, the F–I plots in RS CINs were similar between controls and all Maf mutants (Figure S6B). These results provide evidence that loss of Mafb and c-Maf reduces the ability of FS CINs to sustain action potential (AP) firing.
Figure 6. Synaptic Excitation and Intrinsic Excitability of Fast-Spiking CINs in Mafb cKO and c-Maf cKO and cDKO Mice.
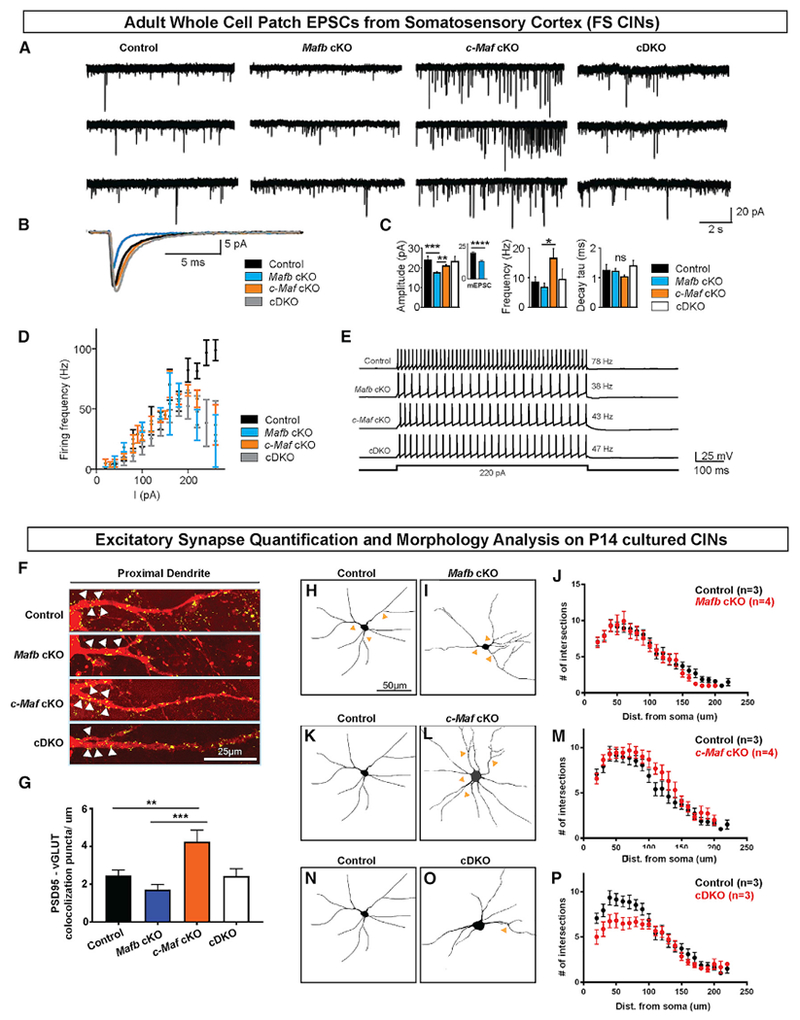
(A) Representative traces of spontaneous EPSCs (sEPSCs) in layer 5–6 fast-spiking (FS) CINs.
(B) Overlaid average sEPSCs from the representative cells depicted in (A). Note the reduced amplitude of sEPSC specifically in Mafb cKO FS CINs.
(C) Quantification (means ± SEM) of the amplitude, frequency, and decay time constant of sEPSCs (Vhold = −70 mV) in FS CINs. Note the reduced amplitude of sEPSCs in Mafb cKO compared with other genotypes and the enhanced frequency of sEPSCs in c-Maf cKO compared with other genotypes. *p < 0.05; **p < 0.01 (control, 28 cells; Mafb cKO, 36 cells; c-Maf cKO, 39 cells; cDKO, 12 cells). Inset indicates reduced amplitude of mEPSCs in Mafb cKO compared with control (control, 24 cells; Mafb cKO, 16 cells; ***p < 0.001, Mann-Whitney test).
(D) F–I curve for FS CINs—a plot of the mean action potential firing frequency as a function of current intensity injected in the FS CINs (control, 19 cells; Mafb cKO, 32 cells; c-Maf cKO, 43 cells; cDKO, 12 cells). Note the inability of all Maf mutant FS CINs to sustain firing frequencies greater than ~60 Hz, compared with control CINs that can exhibit firing rates >100 Hz. p < 0.0001 for all genotypes.
(E) Representative firing traces from FS CINs for each genotype.
(F) Confocal immunofluorescence analysis of tdTomato+ CINs grown in vitro for 14 days (14 DIV). Representative flattened Z-plane images of excitatory synapse labeling in proximal dendrites of tdTomato+ CINs from 4 genotypes. Yellow dots represent the colocalized punta of tdTomato, PSD95, and vGLUT1 staining.
(G) Quantification (means ± SEM) of excitatory synapses on proximal dendrites (within 15 μm from soma); n = 3–4 (animals) per genotype and 20–30 proximal dendrites were analyzed per group. **p < 0.01; ***p < 0.001.
(H and I) Representative images of control (H) and Mafb cKO (I) CINs at DIV14.
(J) Quantification (means ± SEM) of Sholl analysis between control and Mafb cKO CINs.
(K and L) Representative images of control (K) and c-Maf cKO (L) CINs at DIV14.
(M) Quantification (means ± SEM) of Sholl analysis between control and c-Maf cKO CINs.
(N and O) Representative images of control (N) and cDKO (O) CINs at DIV14.
(P) Quantification (means ± SEM) of Sholl analysis between control and cDKO CINs. Note the increase in neurite complexity in c-Maf cKO and decrease in neurite complexity in cDKO.
n = 3–4 (animals) for all groups; each genotype had 15–20 cells analyzed. Scale bar in (F) represents 25 μm and in (H) represents 50 μm.
In the FS CIN population, we found similar passive electrical membrane properties when comparing controls with each of the Maf mutants (Table S3, top). We also observed no strong changes in single AP properties (threshold, amplitude, and duration) in Maf mutants (Table S3, bottom). However, there were differences when Mafb and c-Maf cKOs were compared side-by-side (Table S3, bottom). Notably, Mafb cKO had lower AP threshold (p = 0.001 versus c-Maf cKO); increased AP amplitude (p = 0.025 versus c-Maf cKO); increased AP duration (p = 0.04 versus c-Maf cKO); and increased AP half duration (p = 0.02 versus c-Maf cKOs). These results suggest Mafb and c-Maf have divergent roles in regulating CIN AP properties.
In the RS CIN population, some passive electric membrane properties were different in Maf mutants (Table S3, top). For instance, Mafb cKOs had a more depolarized resting membrane potential (p = 0.01 versus control); c-Maf cKOs had decreased membrane capacitance (p = 0.04 versus control); and cDKOs had decreased input resistance (p = 0.0005 versus control). We observed similar AP properties between controls and all Maf mutants (Table S3, bottom). These results suggest that the features of RS CINs are also altered after deletion of Mafb and c-Maf, but the changes may not be as extensive as the changes seen in FS CINs.
Excitatory Synaptic Properties onto CINs
To investigate excitatory inputs onto CINs, we measured the spontaneous excitatory post-synaptic currents (sEPSCs) onto control and Maf mutant CINs (Figure 6). sEPSCs were smaller in amplitude in Mafb cKO compared with control c-Maf cKOs and cDKOs (Figures 6A and 6C; Table S4; p = 0.0004, 0.002, and 0.002, respectively), whereas the average frequency of sEPSCs was increased in c-Maf cKO compared with Mafb cKO (Figures 6A and 6C; Table S4; p = 0.015). These phenotypes were primarily observed in FS CINs. We found no significant differences in the decay time constant of the recorded sEPSCs across different genotypes (Table S4). These results suggest that the synaptic excitatory input is reduced onto Mafb cKO CINs but increased onto c-Maf cKO CINs.
To determine whether the reduced sEPSCs amplitude observed in Mafb cKO resulted from changes in pre- or post-synaptic mechanisms, we measured miniature EPSCs (mEPSCs) in controls and the Mafb cKO CINs. Similar to sEPSCs, we observed no change in the average mEPSC frequency but a significant decrease in mEPSC amplitude in Mafb cKO compared with controls (Figure 6C, inset; Table S4, bottom; p < 0.0001). These results suggest that the reduced sEPSC amplitude in the Mafb cKO CINs was likely due to a post-synaptic mechanism, rather than a reduced pre-synaptic transmitter release, and was not confounded by the increased cortical activity (Figure 7).
Figure 7. LFPs and CSDs for Neocortical Slices Illustrate Cortical Hyper-Excitability in Mafb cKOs but Not in c-Maf cKOs or DKO Mice.
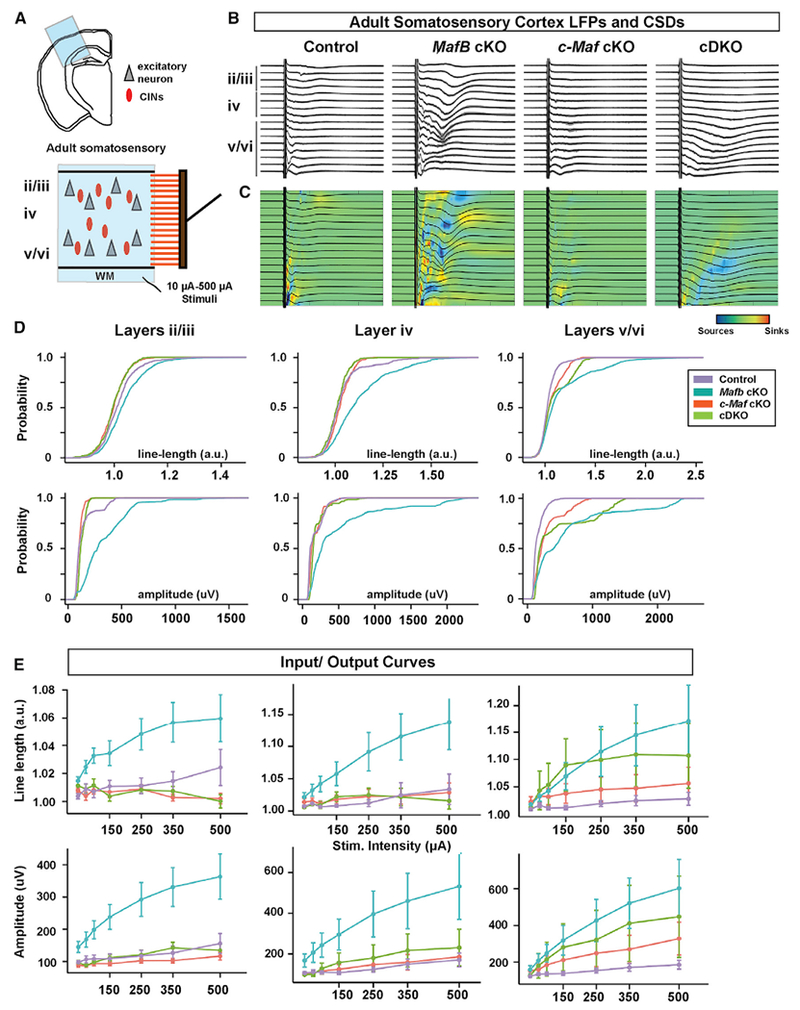
(A) Schema depicting the local field potential (LFP) multi-array experiment.
(B) Examples of average LFPs (black) overlaid on the individual LFPs recordings (gray) for representative slices.
(C) Current source densities (CSDs) for the average LFPs of the representative slices. Blue indicates a source, and red indicates a sink.
(D) Bootstrapped cumulative probability distributions of the amplitude and the line-length (see STAR Methods for details) of the LFP evoked by 500 μA stimulation of the white matter. Note that Mafb cKOs show greater excitability in all layers at 500 μA (indicated by greater line-length and amplitude in all layers compared with all genotypes), whereas c-Maf cKOs show hyporesponsiveness in superficial layers (indicated by reduced line-length and amplitude in layers 2–3 compared with control).
(E) Average LFP responses for increasing stimulation intensities of the white matter. Note that average responses for Mafb cKOs in all layers show greater intensity-dependent-responses than the other genotypes. Note that only in layers 5–6 DKOs show a trend toward enhanced excitability approaching that of Mafb cKOs. 6 controls, 7 Mafb cKOs, 6 c-Maf cKOs, and 3 cDKOs were used for the analyses.
Graphs in (E) are shown as means ± SEM; p values are in Table S5.
c-Maf cKO CINs Had an Increased Density of Excitatory Synapses and Increased Neurite Complexity In Vitro
To determine whether the changes in the Maf mutant sEPSCs were due to a change in glutamatergic synapse densities and/or CIN morphology, we grew primary cortical neurons for 14 days from control, Mafb cKO, c-Maf cKO, and cDKO P0 mice. We then analyzed soma size, neurite complexity (Sholl analysis), and the excitatory synapse density on proximal dendrites of tdTomato+ CINs (Figures 6, schema, and S6D). Soma size was similar between controls and all Maf mutants (Figure S6E). Sholl analysis revealed that the cDKOs had decreased neurite complexity (Figures 6N–6P), whereas the c-Maf cKOs had increased neurite complexity (Figures 6K–6M). Next, we studied excitatory synapses using vGLUT1 and PSD95 as pre- and post-synaptic markers (Figure 6F). CINs from c-Maf cKOs had an increased density of excitatory synapses compared with both controls and Mafb cKOs (Figure 6G; p = 0.008 and 0.0001, respectively), in agreement with an increased frequency of sEPSCs in c-Maf cKO (Figures 6A and 6C). On the other hand, the Mafb cKOs showed a trend for reduced density of excitatory synapses compared with controls and a significant decrease compared with c-Maf cKOs (Figures 6F and 6G; p = 0.0001). Of note, excitatory synapse density in the cDKOs resembled that of the control, supporting the hypothesis that Mafb and c-Maf have opposite effects on synaptogenesis, which could have important impacts on physiological phenotypes in the single and double mutants. Notably, the normal excitatory synapse density result in the cDKO was consistent with the finding that sEPSCs were similar between the control and the cDKO (Figures 6A, 6C, and 6G).
Slice Local Field Potential Analyses Showed Mafb cKOs Had Increased Neocortical Circuit Excitability, Unlike the c-Maf cKOs and cDKOs
To further assess the effect of the Maf mutations on cortical circuit excitability, we measured the local field potentials (LFPs) in acute brain slice preparations from the adult somatosensory cortex. We recorded LFPs across all cortical lamina evoked by electrical stimulation of the white matter tract (schema, Figure 7A). We performed current-source density (CSD) analysis (Aizenman et al., 1996) to determine the patterns of cortical activation between all genotypes. Notably, the CSD pattern was most significantly affected in the Mafb cKO neocortex, which showed a pattern consistent with hyper-excitability with an altered spatiotemporal pattern of synaptic sources and sinks, where darker blue and red represent more deviation from normal activation (Figure 7C). In contrast, the CSD pattern of c-Maf cKOs suggested that cortical activity was diminished (Figures 7B and 7C). Interestingly, cortical excitability of the cDKOs fell in between that of Mafb and c-Maf cKOs (Figures 7B and 7C). Quantification of the duration of the response to the stimulus, line-length, and the amplitude of the evoked LFPs across layers show the above differences between Maf mutants (Figures 7D and 7E), and detailed statistical comparisons between groups can be found in Table S5.
Thus, LFP analysis provides evidence that the Mafb cKO results in cortical circuit hyper-excitability, whereas the c-Maf cKO results in cortical circuit hypo-excitability. Notably, the finding that cDKOs have relatively normal excitability in this assay, provides further evidence that Mafb and c-Maf control excitability in divergent ways.
DISCUSSION
Mafb and c-Maf Control MGE CIN Numbers
Mafb and c-Maf are expressed in the MGE SVZ and persist in migrating immature and mature CINs, but not in pallidal projection neurons, unlike other known MGE TFs (Hu et al., 2017). Thus, we wondered whether loss of Mafb and c-Maf together might abort the specification of MGE-derived CINs; however, this was not the case, as cDKOs still generated CINs that tangentially and radially migrated to the neocortex (Figures 2 and S2). This raised the possibility that other TFs are responsible for generating CINs versus projection neurons. These TFs may coordinate with Lhx6, Nkx2-1, Dlx1/2, and Zfhx1b that have already been shown to contribute to initiating CIN specification (Anderson et al., 1997; McKinsey et al., 2013; Sussel et al., 1999; Zhao et al., 2008).
Maf cDKOs have decreased SST+ and PV+ CINs at P35 with a preferential loss of PV+ CINs (Figure 2). In addition, about 40% of the tdTomato+ cells in the cDKO do not express SST and PV (in the control group, about 20% of the tdTomato+ cells do not express SST and PV, which could be due to antibody labeling efficiency). This could be due to a change in cell fate. We explored whether the ~20% “missing” cells in the cDKO that had an alternative fate. We investigated whether the PV+ cells were converted into the Cck+ basket cells, CGE-type CINs (SP8+), other types of INs (NOS1+, NPAS1+), cholinergic striatal interneurons (ChAT+), or oligodendrocytes (OLIG2+), but we did not find an increase in these cell types in the cortex of cDKOs (data not shown). Thus, we hypothesize that ~20% of the MGE lineage CINs in the cDKO may be either some other cell type or poorly differentiated PV+ CINs. Furthermore, our MGE transplant data support that conclusion (Figure 2). Alternatively, CINs in the cDKO may have impaired maturation or abnormal responses to environmental perturbations, which, in turn, could affect SST and PV expression.
Mafb and c-Maf Provide a Brake on Neural Differentiation
Mafb and c-Maf control proliferation in hematopoietic stem cell, macrophages, and epidermal cells (Lopez-Pajares et al., 2015; Sarrazin et al., 2009; Soucie et al., 2016). Their expression in the SVZ suggests that they could regulate proliferation of secondary progenitors in the MGE. However, we did not observe such changes in the cDKO MGE (Figure S4). On the other hand, in the SVZ of cDKOs, we observed increased expression of βIII-tubulin, a marker of immature neurons at E13.5 (Figures 3I and 3K). This provides evidence that Mafb and c-Maf restrain neurogenesis. Thus, Mafb and c-Maf, by serving as a brake on neural differentiation, may regulate cell fate specification.
Mafb and c-Maf Repress SST CIN Fate
SST CINs are largely generated before PV CINs (Inan et al., 2012; Miyoshi et al., 2007; Pla et al., 2006). Here, we show that Mafb and c-Maf control that temporal sequence by restraining the production of SST CINs. The cDKOs generate excessive SST+ MGE-derived cells, many of which become CINs. As early as E15.5, there is an obvious increase in immature SST+ CINs without any increase in total numbers of MGE-derived cells (Figures 5 and S2). Moreover, we show that, at E15.5, there were increased SST+ CINs that were born around E12.5 (Figure 5). Analyses (ISH) of multiple regulators of MGE development did not yield insights into how Mafb and c-Maf repress SST fate or promote PV fate (Figure S5). Thus, we hypothesize that Mafb and c-Maf control the timing of when other factors specify SST and PV CIN fate.
Mafb and c-Maf Functions Begin in SVZ2 of the MGE to Control CIN Fate
Are SST and PV CINs produced by different MGE progenitors? There is a proposal that SST+ CINs primarily arise by direct neurogenesis from radial glial progenitors in the VZ, whereas PV+ CINs are produced by secondary progenitors in the SVZ (Petros et al., 2015). Our results are not fully consistent with that model. Given that Mafb and c-Maf repress the generation of SST CINs, it is likely that these TFs are functioning autonomously in cells that produce SST CINs. Mafb and c-Maf are expressed in the SVZ and are not detected in the VZ at E12.5 and E15.5 (Figures 1 and S1), arguing that these TFs are repressing SST CIN production in SVZ progenitors.
To further address Mafb and c-Maf functions in the SVZ, we used the recently generated 799-CreER line (Silberberg et al., 2016). Here, we demonstrated that 799-CreER activity begins in the SVZ2 of MGE (Figures S3K and S3L). SVZ2 is thought to contain the most mature progenitors of the ganglionic eminences (Petryniak et al., 2007). We used 799-CreER to generate Maf cDKOs and found that they phenocopy the increase in Sst+ CINs at E15.5 in the Nkx2.1-Cre cDKO (Figure 4). Notably, we did not observe a decrease in tdTomato+ CIN numbers using SST-IRES-Cre, unlike the Nkx2.1-Cre-generated Maf cDKOs (data not shown). This provides evidence that Mafb and c-Maf, in the SVZ2, and not in postmitotic neurons, control the decision between SST and PV CIN fate.
We hypothesize that the MGE SVZ produces both SST and PV CINs and that Mafb and c-Maf control the probability and timing of SST and PV CIN generation by repressing SST CIN fate and promoting PV CIN fate (model top, Figure S7). Moreover, contrary to our hypothesis, Mi et al. (2018) proposed that c-Maf promotes the generation of SST+ CINs based on loss-of-function and viral gain-of-function studies. We agree that adult c-Maf mutants have reduced SST+ CINs (Figure 2; Table S2). However, at E15.5, we did not detect a reduction in Sst+ CINs in the c-Maf cKO (Figure S3E), supporting the idea that the reduction is caused by postnatal CIN loss (Figure S2).
Interneurons in Maf Mutants Show Laminar and Regional Mislocalization
The combined loss of Mafb and c-Maf partially phenocopies the prenatal lamination defect of Cxcr4 and Cxcr7 mutants, which have a depletion of tangentially migrating cells in the MZ and their premature entry into the CP (Figure 5) (Li et al., 2008; Sánchez-Alcañiz et al., 2011; Wang et al., 2011). Thus, we investigated Cxcr7 expression in the cDKO but found that its expression levels appeared normal at E14.5 (Figure S5).
Lamination defects were also observed in the P35 cDKO cortices (Figure 2); they had greater numbers of CINs in deep (layer VI), and fewer CINs in superficial (layers II–IV) lamina, a phenotype also observed in the Cxcr7 cKO adult cortex (Wang et al., 2011). This further raises the possibility that the CXCR signaling pathway is regulated by Mafb and c-Maf.
The cDKOs exhibited an additional migration defect: excessive numbers of MGE-derived INs in the P0 hippocampus (Figures 4 and S3). We suggest that this may reflect a defect in the ability of migratory CINs to detect signals to stop in the neocortex, thus, resulting in continued migration into the hippocampus. However, the Maf cDKO interneuron accumulation in the hippocampus diminished over time, and by P35, the hippocampus had reduced tdTomato+ cells (Table S2). Thus, both CINs and hippocampal interneurons are reduced over time in Maf mutants.
Mafb and c-Maf Have Distinct Roles in Regulating Fast-Spiking CINs and Neocortical Circuit Function Postnatally
Our results suggest that Mafb and c-Maf have distinct functions postnatally in regulating the physiological properties of FS CINs. The finding that sEPSC and mEPSC amplitudes were smaller in the Mafb cKO suggests that Mafb promotes excitation of FS CINs (Figure 6). This result is consistent with evidence that Mafb mutant spiral ganglion neurons had reduced postsynaptic AMPA receptors (Lu et al., 2011; Yu et al., 2013). The reduced excitation of CINs in the Mafb cKO could account for the enhanced circuit excitability observed in their LFP recordings (Figure 7). On the other hand, the c-Maf cKO CINs had an increased density of excitatory synapses and their sEPSC frequency was enhanced compared with the Mafb cKO CINs, suggesting that c-Maf represses CIN excitation (Figure 6). The enhanced excitability of CINs in the c-Maf cKO could explain the lower circuit excitability in their LFP recordings (Figure 7).
Of note, the cDKO has intermediate phenotypes in the LFP, sEPSC and excitatory synapse analyses. This raises the possibility that Mafb and c-Maf have distinct transcriptional effects, potentially opposing roles, in maturing and/or mature CINs and cortical circuit excitability (model bottom, Figure S7).
The finding that Mafb and c-Maf mutations mainly affect FS CINs suggests that these Maf genes regulate network excitability mainly by regulating the FS CINs. The fact that Mafb and c-Maf cKOs as well as cDKOs all have reduced firing of FS CINs in response to current injections (Figures 6D and 6E), but Mafb and c-Maf mutations have distinct effects on synaptic excitation of these cells (Figure 6C), suggests that the opposing roles of Maf genes on cortical network excitability mainly result from their distinct effects on the synaptic, rather than intrinsic, electric properties of CINs. Future experiments, such as electrocorticographic recordings, would be useful in determining how the Mafb and c-Maf mutations affect neocortical activity and pathophysiology in vivo.
In summary, we propose that, in the MGE SVZ, Mafb and c-Maf have redundant functions in controlling the balance of SST and PV CINs generation, whereas in postnatal maturing CINs Mafb and c-Maf have opposite functions in controlling CIN physiology. Ongoing studies are aimed at elucidating the molecular mechanisms underlying these overlapping and distinct functions of Mafb and c-Maf.
STAR★METHODS
CONTACT FOR REAGENTS AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, John Rubenstein (john.rubenstein@ucsf.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
All procedures and animal care were approved and performed in accordance with the University of California San Francisco Laboratory Animal Research Center (LARC) guidelines. All mice strains have been previously published: Ai14 Cre-reporter (Madisen et al., 2010), Nkx2.1-Cre (Xu et al., 2008), Mafb flox (Yu et al., 2013), c-Maf flox (Wende et al., 2012), 799-CreER (Silberberg et al., 2016) and Sst-IRES-Cre (Taniguchi et al., 2011). Mice were backcrossed onto a CD-1 background before analyses. For timed pregnancies, noon on the day of the vaginal plug was counted as embryonic day 0.5. Mouse crosses generated both pure Mafb and c-Maf single mutants and those that were hemizygous for the other gene. We did not observe gross phenotypic differences between mice with or without the additional hemizygous allele, and these were combined together for analysis. For 799-CreER experiments, tamoxifen (5mg/40 g) was administered intraperitoneally to activate the CreER, at embryonic day 11.5. All analyses included both males and females.
For the genotype information regarding the mice used for each analysis, please see the table below. The sensitized single mutants carry a hemizygous allele for the other Maf gene.
| Figure(s) | Genotype information | |||
|---|---|---|---|---|
| Control | Mafb cKO | c-Maf cKO | cDKO | |
| Figures 1 and S3 | WTs | Pure Mafb cKOs | Pure c-Maf cKOs | n.a |
| Figure 2 Neocortical analysis | A mixture of WTs and double heterozygous controls | A mixture of pure Mafb cKOs and sensitized Mafb cKOs | c-Maf cKOs | cDKOs |
| Figures 3, 4, 5, S4, and S5 | A mixture of WTs and double heterozygous controls | n.a | n.a | cDKOs |
| Figure 6 synapse analysis; Figures S2 and S6 morphology analysis | A mixture of WTs and double heterozygous controls | A mixture of pure Mafb cKOs and sensitized Mafb cKOs | A mixture of pure c-Maf cKOs and sensitized c-Mafb cKOs | cDKOs |
| Figures 6 and 7 electrophysiology | WTs | Pure Mafb cKOs | Pure c-Maf cKOs | cDKOs |
| Figure S1 | WTs | Pure Mafb cKO | Pure c-Maf cKO | n.a |
METHODS DETAILS
EdU injections and analysis
Pregnant mice were pulsed with 5-Ethynyl-2′-Deoxyuridine (EdU), 10mg/ml (Thermo Fisher Scientific E10187), at a dose of 50mg EdU/kg body weight. For MGE S-phase progenitor quantification (EdU pulse), E12.5-E13.5 mice were sacrificed 30 minutes after EdU injection and collected in ice-cold PFA/PBS. For MAFB/c-MAF and EdU Fig.colabeling experiments, E12.5 embryos were harvested 1 hr after EdU injection. For pulse-chase experiments, mice or progeny were sacrificed at E15.5 or P35. Embryonic and postnatal brains were collected and fixed overnight in 4% PFA at 4°C, and then sunk in 30% sucrose before embedding in OCT. EdU+ cells were visualized using standard procedures in the Clik-iT EdU plus kit (Thermo Fisher Scientific C10340) that were co-stained with DAPI. For pulse-chase experiments, the same parameters were used, and the only differential factor was the time needed before analysis.
Immunofluorescence/Immunohistochemistry
All tissues were fixed with 4% PFA 1-2 hr (for > P7 tissues) or overnight (< P7 tissues), followed by 30% sucrose cryoprotection. P7, P16 and P35 fixed tissues were sectioned coronally at 40 μm and stained free-floating. All embryonic ages and P0 fixed tissue were sectioned coronally, at 20 μm, and stained on glass slides. P40 transplant tissue was sectioned coronally at 25 μm and stained on glass slides. Immunofluorescent labeling was performed with the following primary antibodies: rabbit anti-Mafb (Sigma HPA005653; 1:500), rabbit anti-c-MAF (Santa Cruz Biotechnology sc-7866; 1:500), rabbit anti-parvalbumin (Swant PV25; 1:200), rat anti-somatostatin (Millipore MAB354; 1:200), goat anti-somatostatin (Santa Cruz Biotechnology sc-7819; 1:100), rabbit anti-VIP (Immunostar 20077; 1:100), rabbit anti-nNOS (Life Technologies 61-7000; 1:200), goat anti-SP8 (Santa Cruz Biotechnology sc-104661; 1:100), mouse anti-Tuj1 (Covance MMS-435P; 1:500), goat anti-MCM2 (Santa Cruz Biotechnology sc-9839; 1:200 (Maslov, 2004), rabbit anti-KI67(Abcam ab15580; 1:500), mouse anti-KI67 (BD Biosciences 550609 ; 1:200), rabbit anti-VGLUT1 (synaptic system 135303; 1:500), rabbit anti-VGAT (synaptic system 131002; 1:200), mouse anti-GEPHRIN (synaptic system 147011; 1:500) and mouse anti-PSD95 (NeuroMab 75-028, clone ID K28/43; 1:500). The appropriate 488, 594 or 647 Alexa-conjugated secondary antibodies (1:500) were from Life Technologies. All primary and secondary antibodies were diluted in PBS containing 2.5% BSA and 0.3% Triton X-100. Sections were coverslipped with Vectashield containing DAPI (Vector labs).
In situ hybridization
In situ hyrbidization was performed as previously described (McKinsey et al., 2013). Probes included CoupTF2 (M. Tsai), Cxcr7 (ATCC MGC-18378), CyclinD2 (A. Malamacci), Lhx6 (V. Pachnis), Mafb (J. Rubenstein), c-Maf (J. Rubenstein), Nkx2.1 (J. Rubenstein), Sox6 (Open Biosystems, Clone #5269193), Sp8 (C. Belmonte), Sst (T. Lufkin) were used.
To generate the Mafb DNA vector and riboprobe, Mafb cDNA was PCR amplified from mouse genomic DNA (mixed CD-1; C57BL6J) using the following primers:
5′ GAGAGTCGACATGGCCGCGGAGCTGAGC
3′ ATATGAGCTCTCACAGAAAGAACTCGGG.
SalI and SacI restriction enzymes sites were introduced (underlined). Next, the Mafb PCR product and the vector, pB3.p11 (Addgene # 69577), were digested with SalI and SacI, and then ligated. The Mafb RNA anti-sense probe was generated by T3 RNA polymerase from a SalI linearized vector, with the size of the probe ~1kb.
MGE transplantation
A detailed protocol for this procedure is available in a methods format (Vogt et al., 2015). We bred male mice homozygous for Ai14 and were either WT (control), MafbFlox/Flox, c-MafFlox/Flox or MafbFlox/Flox; c-MafFlox/Flox to females that were Ai14 negative but WT or homozygous for each of the corresponding male alleles, respectively. These crosses yielded embryos that were either WT or homozygous for each of the Maf alleles. The embryos were collected at E13.5, dissociated and then transduced with a DlxI12b-Cre lentivirus(Vogt et al., 2015, 2018) in DMEM supplemented with 10% Fetal Bovine Serum (FBS) at 37°C and at pH ~7.2 for 30 minutes. This virus deletes the Maf genes and activates tdTomato expression from the Ai14 allele. The cells were then washed several times with DMEM/FBS pelleted. Next, the cells were loaded into the front of a beveled glass needle. P1 WT pups were anesthetized on ice and injected with ~300 nL of cells over 3-5 sites in the right hemisphere. Pups were warmed until able to move and then put back with their mom. They were aged to 40 days and then perfused. Cells were analyzed as described above.
Primary neuronal culture for analysis of dendritic arborization (Sholl) and synapses using neonatal cortex
Briefly, we bred MafbFlox/Flox, c-MafFlox/Flox, Ai14fFlox/Flox females with MafbFlox/+, c-MafFlox/+, Nkx2-1 Cre+ males to generate P0 Mafb cKO, c-Maf cKO and cDKOs. Control P0 animals were generated either through the same crossing or through Ai14fFlox/Flox females bred with Nkx2-1 Cre+ males. tdTomato+ P0 pups were pre-screened using fluorescence dissection microscope. We also collected some tdTomato− P0 pups for cell preparation. Cortical tissues were dissected in cold EBSS, followed by trypsin (Thermo Fisher Scientific 25200056) treatment for 15 minutes at 37°C. Trypsinization was inhibited using 10% FBS containing DMEM. Cells were washed once with DMEM, then resuspended in 10% FBS containing Neuralbasal-A medium (Thermo Fisher Scientific 12348017) with B27 (Thermo Fisher Scientific 17504044). Cell density was quantified using hemocytometer. tdTomato+ cell preparation was diluted using tdTomato− P0 cell preparation roughly at a ratio of 1:10. Cells were plated in poly-D-lysine and laminin coated coverslips (Corning 08-774-385) preloaded in 24-well plates and cultured in 37°C incubator for 14 days. Serum free Neuralbasal-A medium with B27 and Glutamax (Thermo Fisher Scientific 35050061) was used to maintain the cell growth. At day in vitro 14, cell culture medium was removed and replaced with freshly made 4% PFA for 15min fixation. PFA was washed off several times with 1× PBS followed by regular immunostaining protocol for synapse labeling. (1) For synapse quantification, ≥ 3 animals and 25-40 proximal dendrites were analyzed per genotype. Colocalizations were scored if pre- and post-synaptic puncta along tdTomato-labeled proximal dendrites (within 15um from soma) were fully overlapped. We performed a One way ANOVA followed by Turkey’s multiple comparisons test. Differences were regarded significant if p < 0.05. (2) For Sholl analysis, ≥ 3 animals and 15-20 neurons were analyzed per genotype. Images were processed and analyzed using FIJI software based on previously described protocol (Ferreira et al., 2014; Martin et al., 2018).
Image acquisition and processing
Immunohistochemistry images were taken using a CoolSNAP EZ Turbo 1394 digital camera (Photometrics) mounted on a Nikon Eclipse 80i microscope (Nikon Instruments) using NIS Elements acquisition software (Nikon). Images for proliferative markers were taken using Nikon Ti inverted fluorescence microscope with a CSU-W1 large field of view confocal microscope that had 20X and 60X oil objectives to visualize marker colocalization. Brightness and contrast were adjusted, and images merged using FIJI software.
Sholl analysis and excitatory synapse images were taken using the Nikon Ti inverted fluorescence microscope with CSU-W1 large field of view confocal. Images for Sholl analysis were taken using 40X oil objective while images for excitatory synapse quantification were taken using a 60X oil objective. Open source micromanager 2.0 beta was used to acquire images. Brightness/contrast adjustment, z stack image and binary image processing and Sholl analysis were all conducted using the image calculator and Sholl analysis plugins in FIJI software.
Electrophysiology
Slice preparation
Mice were euthanized with 4% isoflurane, perfused with ice-cold sucrose cutting solution containing 234 mM sucrose, 2.5 mM KCl, 1.25 mM NaH2PO4, 10 mM MgSO4, 0.5 mM CaCl2, 26 mM NaHCO3, and 11 mM glucose, equilibrated with 95% O2 and 5% CO2, pH 7.4, and decapitated. We prepared 250 μm-thick horizontal thalamic slices containing Somatosensory Barrel Cortex with a Leica VT1200 microtome (Leica Microsystems). We incubated the slices, initially at 32°C for 1 h and then at 24–26°C, in artificial cerebrospinal fluid (ACSF) containing 126 mM NaCl, 2.5 mM KCl, 1.25 mM NaH2PO4, 2 mM MgCl2, 2 mM CaCl2, 26 mM NaHCO3, and 10 mM glucose, equilibrated with 95% O2 and 5% CO2, pH 7.4 as described in (Clemente-Perez et al., 2017; Paz et al., 2011,2013).
Whole-cell patch-clamp electrophysiology
Recordings were performed as previously described (Clemente-Perez et al., 2017; Paz et al., 2011, 2013). We visually identified interneurons originating from the MGE based on their expression of tdTomato. Neurons were identified by differential contrast optics with a Zeiss (Oberkochen) Axioskop microscope and an infrared video camera. Recording electrodes made of borosilicate glass had a resistance of 2.5–4 MΩ when filled with intracellular solution. Access resistance was monitored in all the recordings, and cells were included for analysis only if the access resistance was < 25 MΩ. The access resistance was similar in all recordings (p > 0.5), suggesting that differences between genotypes were not due to the quality of the whole-cell patch-clamp recording. Spontaneous excitatory post-synaptic currents (sEPSCs) were recorded in the presence of picrotoxin (50 μM, Tocris). For sEPSCs and current-clamp recordings, the internal solution contained 120 mM potassium gluconate, 11 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 10 mM HEPES, and 1 mM EGTA, pH adjusted to 7.4 with KOH (290 mOsm). Tetrodotoxin 1 μM was added to the extracellular solution for miniature EPSC (mEPSC) recordings. The experiments were performed by blinded observers. To test for differences in the sEPSC/mEPSC amplitude, frequency, and decay tau dataset across genotypes (comparison between group means), we performed a One Way ANOVA Kruskal-Wallis followed by Dunn’s multiple comparisons test. Differences were regarded significant if p < 0.05.
F-I plots were generated using a custom MATLAB code. To test for differences in the F-I dataset across genotypes (comparison between group means), we performed a regular two-way ANOVA followed by Tukey’s post hoc test for multiple comparisons. Differences were regarded significant if p < 0.05. For statistical analysis, we included only the current pulses that were presented to all genotypes (within cell type). In addition, we only included cells which were recorded at at least two of the included current pulses. For FS cells, we analyzed responses at current pulses of 20, 60, 100, 140, 180, 220, and 260 pA. For RS cells, we analyzed responses at current pulses of 20, 60, and 100 pA.
Extracellular cortical local field potential recordings
Coronal slices (400 μm) containing Somatosensory Barrel Cortex were placed in a humidified, oxygenated interface chamber and perfused at a rate of 2 mL/min at 34°C with oxygenated ACSF prepared as described above and supplemented with 300 μM glutamine for cellular metabolic support (Clemente-Perez et al., 2017; Paz et al., 2011, 2013). Extracellular LFP recordings were obtained with a 16-channel multi-electrode array (Neuronexus) placed in the Somatosensory Barrel Cortex. The signals were sampled at 24.414 kHz. Signals were amplified at 10,000× and band-pass filtered between 100 Hz and 6 kHz using the RZ5 from Tucker-Davis Technologies (TDT). We delivered electrical stimuli to the internal capsule with a concentric bipolar tungsten electrode (50–100 kΩ, FHC) and recorded the evoked local field potential (LFP) responses. Stimulations were repeated 10–30 times in a single recording and an average LFP was calculated. To assess the cortical network excitability, electrical stimulation was delivered in the internal capsule in increasing amplitudes starting from 10 μA to 500 μA. The experiments were performed by blinded observers.
QUANTIFICATION AND STATISTICAL ANALYSIS
All bar graphs were shown as mean ± SEM. All statistical analyses were performed using Graphpad Prism (version 7) or R-Project3.1, and a p value of < 0.05 was considered significant. The specific n for each experiment as well as the post hoc test corrected p values, can be found in the Results section, in the Figure legends or in supplementary tables.
For all cell counts performed for immunohistochemistry and in situ hybridization, we used the cell counter plug-in in FIJI software. For statistical analyses, we used two methods depending on whether data were parametric or nonparametric in distribution. For parametric data, we utilized a two-tailed t test or one-way ANOVA, followed by a Tukey’s post hoc test, depending on the number of samples being compared. For any data that was normalized (i.e., cell transplants normalized to total cells transplanted or cell fate analyses where proportions were calculated) we used the non-parametric Chi-square test.
For LFP analysis: From an average of responses for each slice and intensity, we calculated line length L as the sum of the absolute differences between successive points in a 30 ms signal s just after the stimulation as L = Σi = 1N|s[i−1]−s[i]|, similar to (Esteller et al., 2001). The average amplitude was calculated as the absolute difference between the minimum and maximum of the signal as |max(s)−min(s)| from a similar window. Current source densities (CSDs) were computed as the negative of the second difference across channels divided by the square of a nominal spatial differentiation grid g as: CSD = −d(d(S))/g, where S is a matrix with each row a signal for an individual channel like,
S = (s1, 1 s1,2 s1,… s1, N s2, 1 s2,2 s2,… s2, N s…,1 s…,2 s1,… s…,N s16, 1 s16, 2 s16,… s16, N), and d(·) is the first difference operator computed by column. For more theory see (Hodgkin and Huxley, 1952).
Numerical values are given as mean ± SEM unless stated otherwise. 6 Controls, 7 Mafb cKOs, 6 c-Maf cKOs and 3 cDKOs were used. For statistical analyses, we used parametric and nonparametric tests. We assessed statistical significance, as appropriate, by performing two-way ANOVA and the Kolmogoroff-Smirnoff test using R-Project 3.1.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-PARVALBUMIN antibody | Swant | PV27; RRID:AB_2631173 |
| Rat anti-SOMATOSTATIN antibody | Millipore Sigma | MAB354; RRID:AB_2255365 |
| Rabbit anti-MAFB antibody | Millipore Sigma | HPA005653; RRID:AB_1079293 |
| Rabbit anti-c-MAF antibody | Santa Cruz Biotechnology | sc-7866; RRID:AB_638562 |
| Goat anti-SOMATOSTATIN antibody | Santa Cruz Biotechnology | sc-7819; RRID:AB_2302603 |
| Rabbit anti-VIP antibody | Immunostar | 20077; RRID:AB_1073072 |
| Rabbit anti-nNOS antibody | Life Technologies | 61-7000; RRID:AB_2313734 |
| Goat anti-SP8 antibody | Santa Cruz Biotechnology | sc-104661; RRID:AB_2194626 |
| Mouse anti-TUJ1 antibody | Covance | MMS-435P; RRID:AB_2313773 |
| Goat anti-MCM2 antibody | Santa Cruz Biotechnology | sc-9839; RRID:AB_648841 |
| Rabbit anti-KI67 antibody | Abcam | ab15580; RRID:AB_443209 |
| Mouse anti-KI67 antibody | BD Biosciences | 550609; RRID:AB_393778 |
| Rabbit anti-VGLUT1 antibody | synaptic system | 135303; RRID:AB_887875 |
| Mouse anti-PSD95 antibody | NeuroMab | 75-028, clone ID K28/43; RRID:AB_2292909 |
| Bacterial and Virus Strains | ||
| pLenti_Dlxi12b-MCS-IRES-Cre lentivirus | Vogt et al., 2018 | N/A |
| One shot DH5alpha STBL3 bacteria | Thermo Fisher | C737303 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Trypsin | Thermo Fisher Scientific | 25200056 |
| Neuralbasal-A medium | Thermo Fisher Scientific | 12348017 |
| B27 supplement | Thermo Fisher Scientific | 17504044 |
| Glutamax | Thermo Fisher Scientific | 35050061 |
| Penicillin-Streptomycin | Hyclone | SV30010 |
| poly-D-lysine and laminin coated coverslips | Corning | 08-774-385 |
| EdU | Invitrogen | E10187 |
| Click-iT Plus EdU Alexa Fluor 647 Imaging Kit | Invitrogen | C10340 |
| picrotoxin | Tocris | 1128 |
| Deposited Data | ||
| GSE94641 (MGE cells) | Chen et al., 2017 | https://doi.org/10.1038/srep45656. |
| Experimental Models: Organisms/Strains | ||
| Nkx2.1-Cre mouse line | Xu et al., 2008 | Jax labs #008661 |
| SST-IRES-Cre mouse line | Taniguchi et al., 2011 | Jax labs #013044 |
| Ai14 (Cre-dependent reporter) mouse line | Madisen et al., 2010 | Jax labs #007908 |
| MafbFlox mouse line | Yu et al., 2013 | N/A |
| c-MafFlox mouse line | Wende et al., 2012 | N/A |
| Oligonucleotides | ||
| 5′ primer for Mafb riboprobe cloning: GAGAGTCGACATGGCCGCGGAGCTGAGC | ELIM Biosciences | N/A |
| 3′ primer for Mafb riboprobe cloning: ATATGAGCTCTCACAGAAAGAACTCGGG | ELIM Biosciences | N/A |
| Recombinant DNA | ||
| pB3.p11 (for synthesize Mafb riboprobe) | Addgene | C737303 |
| Sox6 In situ probe | Open Biosystems | Clone #5269193 |
| Cxcr7 In situ probe | ATCC MGC | 18378 |
| Software and Algorithms | ||
| NIS Elements software | Nikon | N/A |
| Prism v.7 | GraphPad | N/A |
| Image-J software | NIH | N/A |
| Micromanager | https://micro-manager.org/ | N/A |
| FIJI | https://imagej.net/Fiji | N/A |
| MATLAB | https://www.mathworks.com/products/matlab.html | N/A |
| R-Project 3.1 | https://www.r-project.org/ | N/A |
| Other | ||
| CoolSNAP EZ Turbo 1394 digital camera | Photometrics | N/A |
| Nikon Eclipse 80i microscope | Nikon Instruments | N/A |
| Nikon Ti inverted fluorescence microscope | Nikon Instruments | N/A |
| Leica VT1200 microtome | Leica Microsystems | N/A |
| Zeiss (Oberkochen) Axioskop microscope | Zeiss | N/A |
| 16-channel multi-electrode | Neuronexus | N/A |
| RZ5 signal amplifier | Tucker-Davis Technologies | N/A |
| BioRender | https://biorender.com | N/A |
Highlights.
Mafb and c-Maf are expressed in MGE-derived CINs but not projection neurons
Mafb and c-Maf control the numbers of MGE-lineage CINs
Loss of Mafb and c-Maf leads to increased SST+ CIN perinatally
Mafb and c-Maf control CIN synaptic maturation antagonistically
ACKNOWLEDGMENTS
We thank Dr. Josh Huang for the mouse P40, single-cell, RNA-seq data for Mafb and c-Maf expression in interneurons. We thank Dr. Mercedes Paredes and Dr. Arturo Alvarez-Buylla for the goat-MCM2 and mouse-Ki67 antibodies. We thank UCSF Mission Bay Nikon Imaging Center for the confocal microscopy service. We thank our lab manager Carol Kim for helping with all the animal care and breeding. This work was supported by the following research grants: Nina Ireland NIMH R01 MH081880 and NIMH R37/R01 MH049428 (to J.L.R.R.); UCSF CTSI 1111111 and Alliance Funds (to D.V.); UCSF neuroscience graduate program (to E.L.-L.P.); National Science Foundation 1650113 (to A.C.-P.); and NINDS 5R00NS078118-04 and 5R01NS096369-02, Department of Defense EP150038, National Science Foundation 1608234, and the Gladstone Institutes (to J.T.P.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and five tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2019.01.031.
DECLARATION OF INTERESTS
J.L.R.R. is cofounder, stockholder, and currently on the scientific board of Neurona Therapeutics, a company studying the potential therapeutic use of interneuron transplantation.
REFERENCES
- Aizenman CD, Kirkwood A, and Bear MF (1996).A current source density analysis of evoked responses in slices of adult rat visual cortex: Implications for the regulation of long-term potentiation. Cerebral Cortex 6, 751–758. [DOI] [PubMed] [Google Scholar]
- Anderson SA, Eisenstat DD, Shi L, and Rubenstein JLR (1997). Interneuron migration from basal forebrain to neocortex. Science 278, 474–476. [DOI] [PubMed] [Google Scholar]
- Blanchi B, Kelly LM, Viemari J-C, Lafon I, Burnet H, Bévengut M, Tillmanns S, Daniel L, Graf T, Hilaire G, and Sieweke MH (2003). MafB deficiency causes defective respiratory rhythmogenesis and fatal central apnea at birth. Nat. Neurosci 6, 1091–1100. [DOI] [PubMed] [Google Scholar]
- Chao H-T, Chen H, Samaco RC, Xue M, Chahrour M, Yoo J, Neul JL, Gong S, Lu H-C, Heintz N, et al. (2010). Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature 468, 263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y-JJ, Friedman BA, Ha C, Durinck S, Liu J, Rubenstein JL, Seshagiri S, and Modrusan Z (2017). Single-cell RNA sequencing identifies distinct mouse medial ganglionic eminence cell types. Sci. Rep 7, 45656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemente-Perez A, Makinson SR, Higashikubo B, Brovarney S, Cho FS, Urry A, Holden SS, Wimer M, Dávid C, Fenno LE, et al. (2017). Distinct Thalamic Reticular Cell Types Differentially Modulate Normal and Pathological Cortical Rhythms. Cell Rep. 19, 2130–2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobos I, Long JE, Thwin MT, and Rubenstein JL (2006). Cellular patterns of transcription factor expression in developing cortical interneurons. Cereb. Cortex 16, i82–i88. [DOI] [PubMed] [Google Scholar]
- Cordes SP, and Barsh GS (1994). The mouse segmentation gene kr encodes a novel basic domain-leucine zipper transcription factor. Cell 79, 1025–1034. [DOI] [PubMed] [Google Scholar]
- Esteller R, Echauz J, Tcheng T, Litt B, and Pless B (2001). Line length: an efficient feature for seizure onset detection. In Proceedings of the 23rd Annual International Conference of the IEEE Engineering in Medicine and Biology Society Conference, Istefanopulous Y, Akay M, Kearney R, and Kahya Y, eds., pp. 1707–1710. [Google Scholar]
- Ferreira TA, Blackman AV, Oyrer J, Jayabal S, Chung AJ, Watt AJ, Sjostrom PJ, and van Meyel DJ (2014). Neuronal morphometry directly from bitmap images. Nat. Methods 11, 982–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelman D, Griveau A, Dehorter N, Teissier A, Varela C, Pla R, Pierani A, and Marin O (2011). A wide diversity of cortical GABAergic interneurons derives from the embryonic preoptic area. J. Neurosci 31, 16570–16580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S, Tai C, Westenbroek RE, Yu FH, Cheah CS, Potter GB, Rubenstein JL, Scheuer T, de la Iglesia HO, and Catterall WA (2012). Autistic-like behaviour in Scn1a+/− mice and rescue by enhanced GABA-mediated neurotransmission. Nature 489, 385–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin AL, and Huxley AF (1952).Aquantitative description of membrane current and its application to conduction and excitation in nerves. J. Physiol 117, 500–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu JS, Vogt D, Sandberg M, and Rubenstein JL (2017). Cortical interneuron development: a tale of time and space. Development 144, 3867–3878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang ZJ, Di Cristo G, and Ango F (2007). Development of GABA innervation in the cerebral and cerebellar cortices. Nat. Rev. Neurosci 8, 673–686. [DOI] [PubMed] [Google Scholar]
- Inan M, Welagen J, and Anderson SA (2012). Spatial and temporal bias in the mitotic origins of somatostatin- and parvalbumin-expressing interneuron subgroups and the chandelier subtype in the medial ganglionic eminence. Cereb. Cortex 22, 820–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kataoka K (2007). Multiple mechanisms and functions of maf transcription factors in the regulation of tissue-specific genes. J. Biochem 141, 775–781. [DOI] [PubMed] [Google Scholar]
- Kawauchi S, Takahashi S, Nakajima O, Ogino H, Morita M, Nishizawa M, Yasuda K, and Yamamoto M (1999). Regulation of lens fiber cell differentiation by transcription factor c-Maf. J. Biol. Chem 274, 19254–19260. [DOI] [PubMed] [Google Scholar]
- Kepecs A, and Fishell G (2014). Interneuron cell types are fit to function. Nature 505, 318–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessaris N, Magno L, Rubin AN, and Oliveira MG (2014). Genetic programs controlling cortical interneuron fate. Curr. Opin. Neurobiol 26, 79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Adesnik H, Li J, Long J, Nicoll RA, Rubenstein JLR, and Pleasure SJ (2008). Regional distribution of cortical interneurons and development of inhibitory tone are regulated by Cxcl12/Cxcr4 signaling. J. Neurosci 28, 1085–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim L, Mi D, Llorca A, and Marin O (2018a). Development and Functional Diversification of Cortical Interneurons. Neuron 100, 294–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim L, Pakan JMP, Selten MM, Marques-Smith A, Llorca A, Bae SE, Rochefort NL, and Marin O (2018b). Optimization of interneuron function by direct coupling of cell migration and axonal targeting. Nat. Neurosci 21, 920–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Pajares V, Qu K, Zhang J, Webster DE, Barajas BC, Siprashvili Z, Zarnegar BJ, Boxer LD, Rios EJ, Tao S, et al. (2015). A LncRNA-MAF:MAFB transcription factor network regulates epidermal differentiation. Dev. Cell 32, 693–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu CC, Appler JM, Houseman EA, and Goodrich LV (2011). Developmental profiling of spiral ganglion neurons reveals insights into auditory circuit assembly. J. Neurosci 31, 10903–10918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci 13, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin PM, Stanley RE, Ross AP, Freitas AE, Moyer CE, Brumback AC, Iafrati J, Stapornwongkul KS, Dominguez S, Kivimae S, et al. (2018). DIXDC1 contributes to psychiatric susceptibility by regulating dendritic spine and glutamatergic synapse density via GSK3 and Wnt/b-catenin signaling. Mol. Psychiatry 23, 467–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maslov AY (2004). Neural Stem Cell Detection, Characterization, and Age-Related Changes in the Subventricular Zone of Mice. J. Neurosci 24, 1726–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinsey GL, Lindtner S, Trzcinski B, Visel A, Pennacchio LA, Huylebroeck D, Higashi Y, and Rubenstein JLR (2013). Dlx1&2-dependent expression of Zfhx1b (Sip1, Zeb2) regulates the fate switch between cortical and striatal interneurons. Neuron 77, 83–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi D, Li Z, Lim L, Li M, Moissidis M, Yang Y, Gao T, Hu TX, Pratt T, Price DJ, et al. (2018). Early emergence of cortical interneuron diversity inthe mouse embryo. Science 360, 81–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi G, Butt SJB, Takebayashi H, and Fishell G (2007). Physiologically distinct temporal cohorts of cortical interneurons arise from telencephalic Olig2-expressing precursors. J. Neurosci 27, 7786–7798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa K, Nakashima T, Takeda S, Isogai M, Hamada M, Kimura A, Kodama T, Yamaguchi A, Owen MJ, Takahashi S, and Takayanagi H (2010). Maf promotes osteoblast differentiation in mice by mediating the age-related switch in mesenchymal cell differentiation. J. Clin. Invest 120, 3455–3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul A, Crow M, Raudales R, He M, Gillis J, and Huang ZJ (2017). Transcriptional Architecture of Synaptic Communication Delineates GABAergic Neuron Identity. Cell 171, 522–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz JT, Bryant AS, Peng K, Fenno L, Yizhar O, Frankel WN, Deisseroth K, and Huguenard JR (2011). A new mode of corticothalamic transmission revealed in the Gria4−/− model of absence epilepsy. Nat. Neurosci 14, 1167–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz JT, Davidson TJ, Frechette ES, Delord B, Parada I, Peng K, Deisseroth K, and Huguenard JR (2013). Closed-loop optogenetic control of thalamus as a tool for interrupting seizures after cortical injury. Nat. Neurosci 16, 64–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petros TJ, Bultje RS, Ross ME, Fishell G, and Anderson SA (2015). Apical versus Basal Neurogenesis Directs Cortical Interneuron Subclass Fate. Cell Rep. 13, 1090–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petryniak MA, Potter GB, Rowitch DH, and Rubenstein JLR (2007). Dlx1 and Dlx2 control neuronal versus oligodendroglial cell fate acquisition in the developing forebrain. Neuron 55, 417–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pla R, Borrell V, Flames N, and Marin O (2006). Layer acquisition by cortical GABAergic interneurons is independent of Reelin signaling. J. Neurosci 26, 6924–6934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstein JLR, and Merzenich MM (2003). Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2, 255–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Alcañiz JA, Haege S, Mueller W, Pla R, Mackay F, Schulz S, López-Bendito G, Stumm R, and Marin O (2011). Cxcr7 controls neuronal migration by regulating chemokine responsiveness. Neuron 69, 77–90. [DOI] [PubMed] [Google Scholar]
- Sarrazin S, Mossadegh-Keller N, Fukao T, Aziz A, Mourcin F, Vanhille L, Kelly Modis L, Kastner P, Chan S, Duprez E, et al. (2009). MafB restricts M-CSF-dependent myeloid commitment divisions of hematopoietic stem cells. Cell 138, 300–313. [DOI] [PubMed] [Google Scholar]
- Shekhar K, Lapan SW, Whitney IE, Tran NM, Macosko EZ, Kowalczyk M, Adiconis X, Levin JZ, Nemesh J, Goldman M, et al. (2016). Comprehensive Classification of Retinal Bipolar Neurons by Single-Cell Transcriptomics. Cell 166, 1308–1323. e30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silberberg SN, Taher L, Lindtner S, Sandberg M, Nord AS, Vogt D, Mckinsey GL, Hoch R, Pattabiraman K, Zhang D, et al. (2016). Subpallial Enhancer Transgenic Lines: a Data and Tool Resource to Study Transcriptional Regulation of GABAergic Cell Fate. Neuron 92, 59–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soucie EL, Weng Z, Geirsdottir L, Molawi K, Maurizio J, Fenouil R, Mossadegh-Keller N, Gimenez G, VanHille L, Beniazza M, et al. (2016). Lineage-specific enhancers activate self-renewal genes in macrophages and embryonic stem cells. Science 351, aad5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumm RK, Zhou C, Ara T, Lazarini F, Dubois-Dalcq M, Nagasawa T, Höllt V, and Schulz S (2003). CXCR4 regulates interneuron migration in the developing neocortex. J. Neurosci 23, 5123–5130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sussel L, Marin O, Kimura S, and Rubenstein JL (1999). Loss of Nkx2.1 homeobox gene function results in a ventral to dorsal molecular respecification within the basal telencephalon: evidence for a transformation of the pallidum into the striatum. Development 126, 3359–3370. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Nowakowski RS, and Caviness VS Jr. (1996). The leaving or Q fraction of the murine cerebral proliferative epithelium: a general model of neocortical neuronogenesis. J. Neurosci 16, 6183–6196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi H, He M, Wu P, Kim S, Paik R, Sugino K, Kvitsiani D, Fu Y, Lu J, Lin Y, et al. (2011). A resource of Credriver lines for genetic targeting of GABAergic neurons in cerebral cortex. Neuron 71, 995–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt D, Wu P-R, Sorrells SF, Arnold C, Alvarez-Buylla A, and Rubenstein JLR (2015). Viral-mediated labeling and transplantation of medial ganglionic eminence (mge) cells for in vivo studies. J. Vis. Exp, Published online April 23, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt D, Cho KKA, Shelton SM, Paul A, Huang ZJ, Sohal VS, and Rubenstein JLR (2018). Mouse Cntnap2 and human CNTNAP2 ASD alleles cell autonomously regulate PV+ cortical interneurons. Cereb. Cortex 28,3868–3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li G, Stanco A, Long JE, Crawford D, Potter GB, Pleasure SJ, Behrens T, and Rubenstein JLR (2011). CXCR4 and CXCR7 have distinct functions in regulating interneuron migration. Neuron 69, 61–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wende H, Lechner SG, Cheret C, Bourane S, Kolanczyk ME, Pattyn A, Reuter K, Munier FL, Carroll P, Lewin GR, et al. (2012). Thetranscription factor c-maf controls touch receptor development and function. Science 335, 1373–1376. [DOI] [PubMed] [Google Scholar]
- Wonders CP, and Anderson SA (2006). The origin and specification of cortical interneurons. Nat. Rev. Neurosci 7, 687–696. [DOI] [PubMed] [Google Scholar]
- Xu Q, Tam M, and Anderson SA (2008). Fate mapping Nkx2.1-lineage cells in the mouse telencephalon. J. Comp. Neurol 506, 16–29. [DOI] [PubMed] [Google Scholar]
- Yizhar O, Fenno LE, Prigge M, Schneider F, Davidson TJ, O’Shea DJ, Sohal VS, Goshen I, Finkelstein J, Paz JT, et al. (2011). Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature 477, 171–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu WM, Appler JM, Kim YH, Nishitani AM, Holt JR, and Goodrich LV (2013).A Gata3-Mafb transcriptional network directs post-synaptic differentiation in synapses specialized for hearing. eLife 2, e01341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Flandin P, Long JE, Cuesta MD, Westphal H, and Rubenstein JLR (2008). Distinct molecular pathways for development of telencephalic interneuron subtypes revealed through analysis of Lhx6 mutants. J. Comp. Neurol 510, 79–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.