

Abstract
Introduction:
Macrophage migration inhibitory factor (MIF), a pluripotent immune regulator, is an emerging mediator in Alcohol-related Liver Disease (ALD). MIF is associated with ALD progression through its chemokine- and cytokine-like activities.
Methods:
Mechanistic studies into the role of MIF in ethanol-induced liver injury were performed in Mif-/- mice and in C57BL/6J mice treated with a small molecule MIF antagonist, MIF098, after Gao-Binge (acute-on-chronic) ethanol feeding, an ethanol feeding protocol associated with hepatic neutrophilia and induction of the Unfolded Protein Response (UPR).
Results:
The MIF axis, e.g MIF and MIF receptors CD74, CXCR2, CXCR4 and CXCR7, was enhanced in livers of alcoholic hepatitis (AH) patients as compared to healthy controls. Mif-/- mice were protected from hepatocellular injury after Gao-Binge feeding, independent of neutrophilia and inflammation, but was associated with the UPR. Interestingly, the UPR signature in AH patients and in mice following Gao-Binge feeding was biased towards cell death with increased expression of pro-cell death CHOP and decreased pro-survival GRP78. The UPR and liver injury 6h after binge was prevented in both Mif-/- mice and in MIF098-treated mice. However, both MIF interventions led to increased liver injury and exacerbated the hepatic UPR 9h after binge. Induction of upstream UPR signaling and expression of CHOP protein by thapsigargin in AML-12 hepatocytes was blunted by coexposure to MIF098, directly connecting MIF to UPR in hepatocytes.
Conclusion:
The current study revealed that, in addition to its cytokine/chemokine functions, MIF is an upstream regulator of UPR in response to ethanol feeding in mice. Importantly, both MIF and UPR can either protect or contribute to liver injury, dependent upon the stage or severity of ethanol-induced liver injury.
Keywords: Alcohol-related liver disease, MIF, neutrophil, CHOP
INTRODUCTION
Alcohol-related Liver Disease (ALD) is a progressive disorder that is responsible for nearly 20,000 deaths per year (Garcia-Saenz-de-Sicilia et al., 2017, Louvet et al., 2017, Serste et al., 2018). The current therapies for ALD are limited, ineffective in many patients, and primarily focus on decreasing inflammation (Orman et al., 2013). Inflammation underlies the onset and progression of many chronic diseases, including ALD. Pro-inflammatory cytokines, e.g. tumor necrosis factor-α (TNFα) and interleukin 1-β (IL-1β), contribute to the pathophysiology of ethanol-induced liver injury in animals and are associated with ALD progression in humans (Nagy, 2015). Chemokines are chemotactic cytokines crucial to coordinating immune cell infiltration into infected, diseased or damaged tissues (Marra and Tacke, 2014). The C-X-C subfamily of chemokines, e.g. IL-8 family, are associated with liver injury and patient mortality in human AH (Dominguez et al., 2009, Zimmermann et al., 2011) and monocyte chemoattractant protein-1 (MCP-1/CCL2) is a critical chemokine/steatokine in ethanol-induced liver injury in mice (Mandrekar et al., 2011).
Macrophage Migration Inhibitory Factor (MIF), a master immune regulator, also contributes to liver injury in ethanol-fed mice, and correlates with patient mortality in AH (Barnes et al., 2013, Marin et al., 2017). MIF is a pluripotent cytokine (Bernhagen et al., 1993, Bacher et al., 1997, Calandra et al., 2000, Leng et al., 2003) with roles in autoimmune inflammatory disease such as rheumatoid arthritis and systemic lupus erythematosus (SLE) (Sreih et al., 2011) and in infectious disease such as malaria (Awandare et al., 2009). MIF can be protective or deleterious in disease. For example, MIF’s role in SLE is dichotomous (Sreih et al., 2011). High MIF expression protects from disease development, but after disease onset, low MIF expression appears protects from SLE-associated end-organ injury. MIF’s diverse biology is due, at least in part, to its interactions with multiple receptors. Increased cytokine production and inflammatory signaling occurs through its cognate receptor CD74, while chemokine activity is mediated through the C-X-C chemokine receptors CXCR2, CXCR4, and CXCR7 (Bernhagen et al., 2007). MIF can also modulate intracellular pathways by interaction with JAB1 (Serste et al., 2018).
MIF is linked to the progression of ALD via MIF-dependent cytokine and chemokine activities (Barnes et al., 2013, Marin et al., 2017). Mif-/- mice are protected from liver injury and leukocyte recruitment after chronic Lieber-DeCarli ethanol feeding (Barnes et al., 2013). Furthermore, the concentration of MIF in suprahepatic serum, obtained during transjugular liver biopsies, positively correlates to increased liver dysfunction in AH patients, suggesting MIF plays a contributing role to AH (Marin et al., 2017). The Gao-Binge (acute-on-chronic, NIAAA model) model of ethanol feeding was used in the current study as it better recapitulates certain aspects of the phenotype of AH in humans, e.g. exacerbated liver injury, inflammation, and neutrophilia, as compared to chronic Lieber-DeCarli ethanol feeding (Bertola et al., 2013a). Gao-binge feeding also induces the Unfolded Protein Response (UPR), a cellular reaction to noxious stimuli that reestablishes tissue homeostasis acutely, but can become pathophysiological with sustained activation (Cai et al., 2017). The UPR is thought to contribute to chronic liver injury in ALD, and a few reports find UPR activation in the livers of severe AH patients (Altamirano et al., 2014, French et al., 2017). The UPR is also associated with ethanol-mediated liver injury in mice. For example, CCAAT-enhancer-binding protein homologous protein (CHOP) knockout mice are partially protected from hepatocellular injury after chronic intragastric ethanol feeding (Ji et al., 2005). The UPR is also associated with neutrophil-mediated liver injury after Gao-Binge feeding (Cai et al., 2017).
Given the emerging importance of the UPR in ALD and the association of the UPR with liver inflammation after ethanol feeding in mice, the purpose of this study was to determine if MIF was associated with the UPR in Gao-Binge ethanol feeding, revealing a hitherto unknown role for MIF in both cellular stress and in ALD. In AH patients, enhanced expression of MIF and MIF receptors was higher than in healthy controls and the UPR signature was detected with an imbalance towards cell death. In mice, MIF was not associated with liver inflammation or leukocyte recruitment after Gao-Binge, but was intrinsically connected to the ethanol-induced UPR in the liver. This study also included a novel small molecule inhibitor of MIF, MIF098 (Hare et al., 2010, Sauler et al., 2015) to antagonize MIF activity in Gao-Binge feeding and lend insight into the potential therapeutic impact of MIF neutralization in ALD. Mif-/- mice and MIF098-treated mice were protected from the peak of UPR induction, and hepatocellular injury after Gao-Binge feeding. In contrast, liver injury and UPR induction was higher in Mif-/- and MIF098-treated mice compared to respective controls at a time when binge-associated injury was waning in control mice. MIF also was critical to UPR induction after chronic Lieber-DeCarli ethanol feeding. This study strengthened the association of the MIF axis in liver dysfunction in AH patients, connected the UPR to MIF and showed the UPR differentially impacted ALD in a stage-specific manner.
Materials and Methods
Ethanol feeding models in mice
Eight to ten week old female C57BL/6J mice were purchased from Jackson Laboratories (Bar Harbor, ME). Mif-/- mice on a C57BL/6 background were obtained from Dr. R. Bucala (Yale University, New Haven, CT, USA) and a breeding colony was established and maintained at the Cleveland Clinic. All procedures using animals were approved by the Cleveland Clinic Institutional Animal Care and Use Committee. Gao-binge ethanol feeding was carried out as previously described, with minor modifications (Bertola et al., 2013a, Bertola et al., 2013b). Mice received conditioning gavages of sterile saline at random times on days -12, -8 and -4 relative to ethanol binge. On Day 11, pair-fed and ethanol-fed mice (5% ethanol v/v) were gavaged with an equivalent volume of 5g/kg maltose or 5g/kg ethanol in water, respectively. Mice were anesthetized at 1, 2, 3, 6, or 9h post gavage, blood was collected in non-heparinized syringes from the posterior vena cava, livers excised after a brief perfusion and mice were euthanized by exsanguination. In other experiments, pair- or ethanol-fed C57BL/6J mice were treated with a small-molecule MIF inhibitor MIF098 (3-(3-hydroxybenzyl)-5-methylbenzooxazol-2-one) ,at 60 mg/kg dose, administered i.p. in a PEG400/β-cyclodextran vehicle (VEH, 10 µL/gm body weight) or VEH control at -18h and -1h to the gavage on day 11 and euthanized 6h after ethanol binge. The MIF098 dosing was chosen based on previous studies (Sauler et al., 2015). Chronic Lieber-DeCarli feeding was carried out as previously described (Barnes et al., 2013). In brief, mice were fed ethanol in increasing concentrations for 25 days total as follows: 5.5% (total kcal) ethanol for 2 days, 11% for 2 days (4d), 22% for 1 week (11d), 27% for 1 week (18d), and finally 32% for 1 week (25d). Control mice were pair-fed an identical liquid diet with iso-calorically substituted with maltose dextrans in place of ethanol. Portions of each liver were fixed in formalin or frozen in optimal cutting temperature (OCT) compound (Sakura Finetek U.S.A., Inc., Torrance, CA) for histology, stored in RNAlater (Ambion) per manufacturer’s recommendations, or flash frozen in liquid nitrogen to be stored at -80°C for analysis at a later time. Blood was transferred to EDTA-containing tubes for plasma isolation to be stored at -80°C until further analysis.
Biochemical Assays
Plasma alanine aminotransferase (ALT) and aspartate aminotransferase (AST) activities were assayed with enzymatic assay kits from Sekisui Diagnostics (Framingham, MA), per manufacturer’s instructions. Liver triglycerides were determined by assay kits purchased from Pointe Scientific Inc. (Lincoln Park, MI).
Patient liver samples from Early AH Transplant Tissue Resource
Liver tissue was obtained from the NIAAA-supported Clinical Resource for Alcoholic Hepatitis Investigations at Johns Hopkins University (R24AA025017). De-identified liver tissue samples were obtained from explanted liver tissue in patients with severe AH during transplant or from healthy control, donor livers. Biopsies were snap frozen in liquid nitrogen and stored at -80° C. Written, informed consent was obtained from each patient included in the study and the study protocol conforms to the ethical guidelines of the 1975 Declaration of Helsinki as reflected in a priori approval by the Institutional Review Boards at Johns Hopkins Medical Institutions. Descriptive data for this cohort have been reported previously (Khanova et al., 2017).
RNA isolation and quantitative Real-Time Polymerase Chain Reaction (qPCR)
RNA was isolated from mouse liver tissue stored in RNAlater or from human liver tissue stored at -80°C using RNeasy Mini Kits (Qiagen, Germantown, MD). Liver RNA was reverse transcribed and analyzed on a QuantStudio5 qPCR machine. The relative messenger RNA (mRNA) was determined using gene-specific primers listed in Supplemental Table 1 by the ΔΔCt method, normalized to 18S rRNA.
Protein Isolation and Western Blotting
Frozen liver tissue from mice and human samples were homogenized in RIPA lysis buffer (Barnes et al., 2013) with protease and phosphatase inhibitors added and protein concentrations were assayed using the DC Protein Assay (BioRad, Hercules, CA). Liver lysates were then separated on 12% polyacrylamide gels and used for western blot analysis with antibodies against CHOP (#5554), phosphoSer51 eIF2α (#3597), total eIF2α (#9722) (Cell Signaling, Danvers, MA); MIF (#TP234) (Torrey Pines Biolabs, Inc, Secaucus, NJ), PDI (#610946), GRP78 (#610978) (BD Biosciences, San Jose, CA). HSC70 (sc-7298) (Santa Cruz Biotechnology, Dallas, TX), β-actin (#4967), (Cell Signaling) and GAPDH (MAB374) (Millipore Sigma, St. Louis, MO) were used as loading controls. Signal intensities were quantified using Eastman Kodak Co. Image Station 4000R.
Immunohistochemistry
Formalin-fixed liver sections were paraffin-embedded, sectioned, coded and stained with hematoxylin and eosin. Formalin-fixed paraffin-embedded sections were deparaffinized and stained with NIMPR14 (#ab2557, Abcam, Cambridge, MA) to determine neutrophil accumulation. At least 3 images were taken per tissue section and quantification of positive staining was performed using ImagePro Plus software (Media Cybernetics, Silver Springs, MD) as previously described (Barnes et al., 2013). Specificity of staining was confirmed by comparing sections incubated with PBS instead of primary antibody.
Myeloperoxidase (MPO) activity assay and neutrophil RNA isolation
MPO activity was measured in flash-frozen liver tissue as previously described (Cassini-Vieira et al., 2015). Neutrophils were isolated from bone marrow (Supplemental methods), resuspended in RPMI +10% FBS, and plated in 96-well plates at a density of 2x106 cells per mL. After 1h, allowing cells to acclimate to plating, neutrophils were challenged with or without 100 ng/mL bacterial lipopolysaccharide (#L-2654, Millipore Sigma) for 90 minutes. The plate was briefly spun to pellet cells, the cell-free supernatant was collected to perform the MPO activity assay (#600620, Cayman Chemical, Ann Arbor, MI) per manufacturer’s instructions. RPMI + 10% FBS served as a background control. Both MPO assays were terminated at 5 minutes after substrate addition. Activity was determined by change in O.D. at 450 nM over 5 minutes.
After removal of supernatant, neutrophils were washed with cold PBS, pelleted by centrifugation and resuspended in RNA lysis buffer RLT (Qiagen, Germantown, MD) for RNA isolation with RNeasy mini kits. Expression of neutrophil-associated mRNA was determined with gene-specific primers listed in Table S1.
Statistics
Values shown in all figures represent the mean ± SEM. Analysis of variance (ANOVA) was performed using the general linear models procedure (SAS, Carey, IN). Data were log-transformed if necessary to obtain a normal distribution. Post-hoc comparisons were made by least square means testing. p values of less than 0.05 were considered significant.
Results
Enhanced MIF axis and imbalanced UPR in livers of AH patients.
The concentration of MIF is increased in the sera of ALD (Marin et al., 2017, Kumagi et al., 2001). Expression of MIF protein was also increased in livers of AH patients as compared to healthy controls (Fig. 1A), whereas expression of Mif mRNA was unchanged in livers of AH patients. Expression of mRNA for MIF receptors CD74, CXCR2 and CXCR4, but not CXCR7, was robustly increased over healthy controls (Fig. 1B). In contrast, expression of mRNA for MIF receptors was modestly increased in peripheral mononuclear cells from AH patients and healthy controls (Supplemental Fig. 1). Taken together, the data are consistent with an enhanced MIF axis in the liver, and not infiltrating leukocytes in AH patients. This is consistent with previous reports using bone marrow transplants that suggested the hepatocytes were the source of pathophysiological MIF in ALD (Marin et al., 2017).
Figure 1. The MIF axis is upregulated in livers of AH patients.
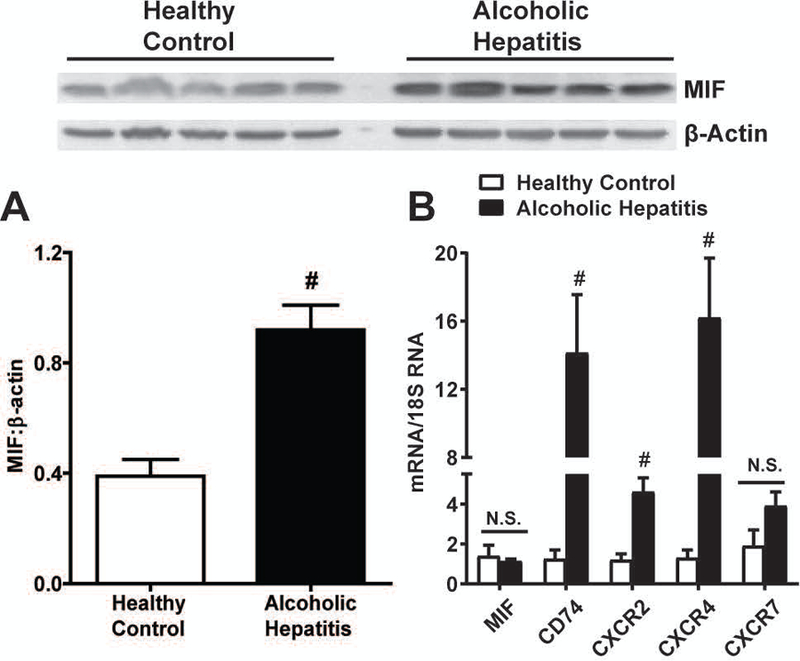
A) Expression of MIF protein was measured in livers of AH patients and healthy controls (HC) by western blot (n=5 for HC and AH) Densitomtery was carried out with β-Actin as a loading control. B) Expression of Mif, CD74, CXCR2, CXCR4 and CXCR7 mRNA was determined in livers using qRT-PCR and normalized to human 18S RNA. Data are represented as fold change of AH vs. HC. # - p<0.05
Gao-Binge Feeding Induced Dynamic Liver Injury
Gao-binge ethanol feeding in mice is associated with enhanced liver injury and neutrophil accumulation that recapitulates some features of human AH. Since MIF acts as an upstream regulator of leukocyte recruitment and hepatic inflammation in chronic Lieber-DeCarli ethanol feeding, we assessed the role of MIF in liver injury following Gao-Binge ethanol feeding. A rapidly developing state of liver injury was induced when mice were challenged with an acute ethanol binge (5g/kg) after chronic ethanol feeding (Fig. 2). The hepatocellular injury markers, plasma ALT and AST, were both increased in circulation from after binge, plateauing between 3h and 6h after binge and decreasing to background levels 9h after binge (Fig. 2A-B). Liver triglyceride (TG) accumulation was also increased in ethanol-fed mice, but was not further changed by ethanol binge (Fig. 2C). Expression of mRNA for cytokines TNFα and IL-6, as well as the chemokine MCP-1, was unchanged after Gao-Binge. In contrast, hepatic expression of IL-1β mRNA was increased at 3h and 6h after binge. The hepatic expression of neutrophil chemokines CXCL-1 and CXCL-2 mRNA, murine members of the IL-8 chemokine family, were both increased at 3h and 6h after binge, with expression of CXCL-1 mRNA remaining increased 9h after binge (Fig. 2D). Adhesion markers CD62E and ICAM-1 were also increased at 6h after binge. Expression of neutrophil-selective surface marker Ly6G mRNA and the neutrophil receptor CXCR2 mRNA was increased in Gao-Binge after ethanol binge (Fig. 2D). Immunohistochemical staining with the neutrophil detection antibody NIMPR14 was increased at 3h after Gao-binge and remained elevated through 9h (Fig. 2E). In aggregate, the specific hepatic inflammatory signature after Gao-Binge feeding in our study was characteristic of neutrophil recruitment, consistent with other studies (Bertola et al., 2013a, Bertola et al., 2013b, Cai et al., 2017, Roh et al., 2015).
Figure 2. Dynamics of liver injury and hepatic neutrophilia in Gao-Binge.

C57BL/6J mice were allowed free access to diets containing ethanol (n=5–6) or pair-fed (n=4) control diets per the Gao-Binge ethanol feeding model for 10 days. On Day 11, mice were euthanized at the indicated times after gavage with ethanol or control maltose. Hepatocellular injury was determined by activity of A) ALT and B) AST in circulation. C) Hepatic triglyceride content was measured in liver homogenate. D) Expression of TNFα, IL-1β, IL-6, Ly6G, CXCR2, MCP-1, CXCL-2, CXCL-1, CD62E, and ICAM-1 mRNA determined in mouse livers using qRT-PCR. Gene expression was normalized to 18S RNA and expressed as fold change over pair-fed controls. Paraffin-embedded liver sections were stained with an antibody against murine neutrophils, NIMPR14, E) 3h, 6h, and 9h after ethanol binge. Images were acquired with a 20X objective. Neutrophil accumulation was determined by relative quantification of NIMPR14 staining. Values are means ± SEM. Values with different superscripts are different within Gao-Binge over time, p<0.05. # p<0.05 vs. pair-fed control at each time.
MIF does not influence liver inflammation after Gao-Binge
The role of MIF in Gao-Binge was next interrogated 6h and 9h after ethanol binge in Mif-/- mice (Fig. 3) as neutrophil accumulation and maximal hepatic injury was observed 6h after binge and resolution of binge-associated injury occurred 9h after binge in C57BL/6J mice. Mif-/- mice had lower plasma ALT 6h after binge, but plasma ALT was then increased over controls 9h after binge, revealing a delayed injury response to Gao-Binge (Fig. 3A). Mif deletion did not decrease inflammatory chemokine expression in the liver (Fig. 3B), with expression of neutrophil chemokine CXCL1 and Ly6G mRNA independent of genotype, whereas CXCR2 mRNA was increased in Mif-/- mice. NIMPR14 staining indicated equivalent neutrophil recruitment at this time as well (Fig. 3C), suggesting MIF was not required for neutrophil recruitment or chemokine expression after Gao-Binge feeding. Next, mice were treated with a small-molecule MIF inhibitor that prevents MIF-mediated signaling and MIF-CD74 complex formation (MIF098) prior to ethanol binge (Hare et al., 2010, Sauler et al., 2015). MIF098-exposed mice had reduced ALT 6h after binge, but plasma ALT was equivalent to VEH-treated ethanol-binged controls at 9h (Fig. 3D). MIF098 treatment did not alter Gao-Binge induced CXCL1 or Ly6G mRNA expression, neutrophil accumulation, but expression of CXCR2 mRNA was increased (Fig. 3E-F).
Figure 3. MIF deletion or MIF inhibition delays liver injury after Gao-Binge feeding independent of hepatic neutrophilia.

A-C) C57BL/6J, Mif-/- were allowed free access to ethanol-containing (n=8) or pair-fed (n=5) control diets per Gao-Binge feeding protocol. D-F) C57BL/6J mice were acutely treated with MIF098 (n=6) prior to ethanol binge. Mice were euthanized 6h and 9h after Gao-Binge. A,D) Plasma ALT was assayed enzymatically and B,E) expression of neutrophil-associated CXCL1, CXCR2 and Ly6G mRNA was determined by qRT-PCR. C,F) NIMPR14 staining was performed to determine hepatic neutrophilia at the indicated times. Representative images were acquired with a 20X objective. Values are means ± SEM. Values with different superscripts are significantly different, p<0.05, # p<0.05
Since genetic MIF deficiency or treatment with a MIF inhibitor protected from injury 6h after binge independent of neutrophil accumulation, neutrophil activity might be impaired in Mif-/- mice. Accumulation of CD45+Ly6G+CD11b+ cells (activated, transmigrated neutrophils) (Supplemental Fig. 2) was independent of genotype (Fig. 4A). Neutrophil myeloperoxidase (MPO) activity in livers of mice after Gao-Binge feeding was not increased by Gao-Binge feeding, in contrast to livers of mice challenged with low-dose lipopolysaccharide (LPS) (Fig. 4B). Both basal and LPS-stimulated expression of MPO mRNA was equivalent in neutrophils isolated from bone marrow of C57BL/6J or Mif-/- mice (Fig. 4C). MPO release into cell culture supernatants was also similar in neutrophils from C57BL/6J or Mif-/- mice (Fig. 4D). Taken together, these data demonstrated that MIF deficiency did not impair neutrophil recruitment or activation in Gao-Binge.
Figure 4. Neutrophil activation is unaffected by MIF deficiency.
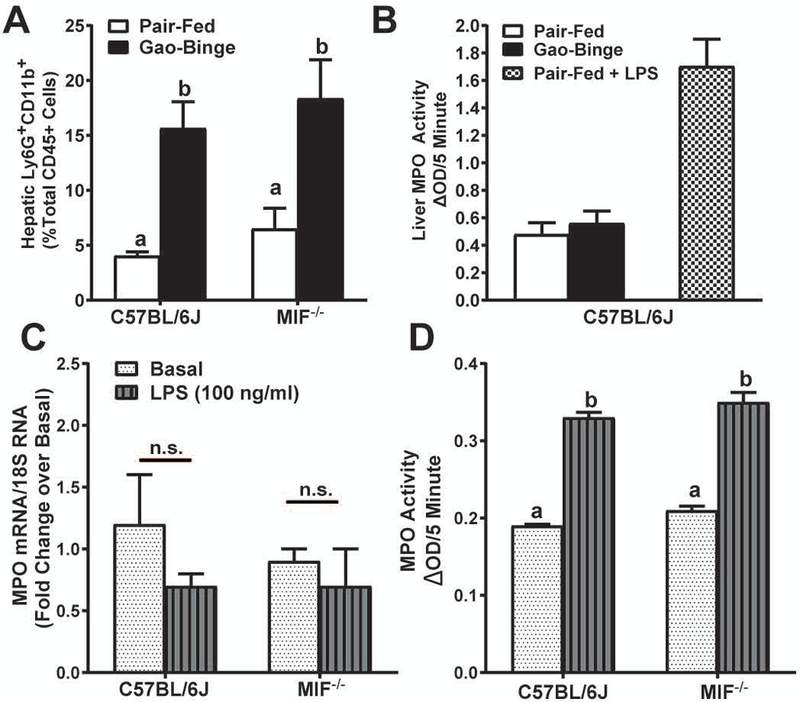
Liver nonparenchymal cells were isolated from C57BL/6J or Mif-/- 6h after ethanol binge and stained for flow cytometry analysis. A) Activated neutrophils were determined by gating Ly6G+CD11b+ cells from total CD45+ cells. B) Liver MPO activity was determined in livers of C57BL/6J 6h after binge or after low-dose LPS. Neutrophils were isolated from bone marrow of chow-fed C57BL/6J or Mif-/- mice. C) Expression of MPO mRNA in isolated neutrophils was determined using qRT-PCR and normalized to 18S RNA expression. D) MPO release from neutrophils was determined in cell culture supernatants following LPS challenge by colorimetric assay. Values are means ± SEM. Values with different superscripts are significantly different, p<0.05
Gao-Binge Induces Hepatic UPR
Since MIF did not affect neutrophil recruitment or activation after Gao-Binge feeding, we next hypothesized that MIF was required for hepatic injury via hepatocellular stress, e.g. the Unfolded Protein Response (UPR). Indeed, UPR was induced by chronic ethanol feeding and then further modulated by ethanol binge (Fig. 5). Phosphorylation of eukaryotic initiation factor-2α (p-eIF2α), expression of CCAAT-enhancer-binding protein homologous protein (CHOP) and glucose-related peptide 78 (GRP78) protein were increased in the livers of mice after 10-day ethanol feeding (0h) as compared to pair-fed controls (Fig. 5A). p-eIF2α and expression of CHOP protein peaked at 3h after binge and was returned to pre-binge (0h) levels 9h after binge (Fig. 5AB). Expression of GRP78 protein, however, was suppressed 1h-3h after ethanol binge and returned to pre-binge level by 9h (Fig. 5AB). Increased hepatic mRNA expression of UPR genes including CHOP, GRP78, FGF21 and spliced XBP-1 occurred at 6h and 9h after binge (Fig. 5C).
Figure 5. Timecourse of UPR induction in the liver after Gao-Binge feeding and UPR induction in AH patients.

C57BL/6J mice were euthanized at 0h-9h after ethanol binge per Gao-Binge ethanol feeding and UPR was determined A-B) Expression of phosphorylated eIF2α, CHOP and GRP78 were determined by western blot. Densitomtery was carried out with β-Actin as a loading control. C) Expression of UPR-associated Chop, Grp78, Fgf21 and spliced XBP-1 mRNA was determined by qRT-PCR. Gene expression was normalized to 18S RNA and expressed as fold change over pair-fed controls. D) UPR activation in livers of AH patients was determined by western blot. Expression of phosphorylated eIF2α, CHOP, PDI and GRP78 was normalized to β-actin and represented as fold over healthy controls. E) Expression of Chop and Grp78 mRNA was determined by qRT-PCR and normalized to 18S RNA. Values are means ± SEM. Values with different superscripts are significantly different, p<0.05. # p<0.05 vs. HC or PF.
The UPR signature in livers of AH patients (Fig. 5D-E) mirrored what was observed in mice. p-eIF2α and expression CHOP protein were increased in the livers of human AH patients as compared to healthy controls (Fig. 5D). Expression of pro-survival GRP78 and PDI proteins was decreased (Fig. 5D). Expression of CHOP and GRP78 mRNA was unchanged in AH patients (Fig. 5E). Taken together, this suggested the UPR in livers of AH patients and in ethanol-fed mice was biased toward the pro-cell death mechanisms of UPR.
MIF Deficiency Delayed Hepatic UPR induction after Gao-Binge
The role of MIF in the UPR was next examined in Mif-/- mice as well as in C57BL/6J mice treated with MIF098. At 6h after binge, p-eIF2α and CHOP protein were increased in C57BL/6J mice, but not in Mif-/- mice (Fig. 6A). Expression of GRP78 protein, however, was not different in Mif-/- mice. Expression of spliced XBP-1, CHOP, GRP78 and DR5 mRNA was increased 6h after binge in C57BL/6J mice, but not in Mif-/- mice, whereas FGF21 mRNA was increased in both genotypes (Fig. 6B). At 9h after binge, Mif-/- mice had an induced UPR (Fi.g 6C-D). Expression of CHOP protein was increased in both C57BL/6J and Mif-/- mice by 9h (Fig. 6C). Expression of spliced XBP-1, CHOP, GRP78 and DR5 mRNA had resolved C57BL/6J mice, but showed a delayed increased in Mif-/- mice (Fig 6D). MIF098 treatment yielded a similar phenotype at 6h and 9h after binge with respect to expression of CHOP and GRP78 protein (Fig. 6E-H). The early increase in p-eIF2α and CHOP protein 6h after binge was prevented in MIF098-treated mice as compared to VEH controls (Fig. 6E). The UPR signature was increased for spliced XBP-1, CHOP, GRP78, DR5 and FGF21, but expression of CHOP mRNA trended toward a decrease whereas DR5 mRNA was prevented by MIF098 (Fig. 6F). At 9h after binge, a delayed increase in expression of CHOP protein was detected in MIF098-treated mice (Fig. 6G) and expression of most UPR genes was also increased compared to 6h (Fig. 6H). Taken together, genetic MIF deficiency and MIF inhibition delayed the binge-associated UPR signature after Gao-Binge in mice.
Figure 6. MIF delays the binge-associated UPR in mice.

UPR was determined in livers of A-D) C57BL/6J or Mif-/- as well as E-H) C57BL/6J mice treated with MIF098 or VEH at 6h and 9h after Gao-Binge. A,C,E,G) Phosphorylation of eIF2α, expression of CHOP and GRP78 were determined by western blot and normalized to β-Actin as a loading control at the indicated times. B,D,F,H) Expression of spliced XBP-1, CHOP, Grp78, Dr5, and FGF21 mRNA was determined by qRT-PCR and normalized to 18S RNA and represented as fold change over pair-fed controls at the indicated times. Values are means ± SEM. Values with different superscripts are significantly different, p<0.05, # - p<0.05 vs pair-fed.
MIF is required for expression of CHOP protein after Chronic Ethanol Feeding
Since Mif-/- are protected from liver injury in chronic Lieber-DeCarli ethanol feeding (Barnes et al., 2013), we next determined if MIF was required for UPR induction after Lieber-DeCarli ethanol feeding in mice. Expression of p-eIF2α and CHOP protein was robustly upregulated in the livers of ethanol-fed C57BL/6J mice after 25 days of chronic ethanol feeding (6% v/v). This increase was prevented in Mif-/- mice (Fig. 7A). Expression of GRP78 protein was detected but unchanged by ethanol feeding (Fig. 7A) and GRP78 expression was independent of genotype. A modest increase in spliced XBP-1 mRNA was detected in C57BL/6J that was prevented in Mif-/- mice, another indicator that MIF played a role in ethanol-induced hepatic UPR activation. Expression of CHOP, GRP78, DR5 or FGF21 mRNA, however, was unaffected by ethanol feeding and was independent of genotype (Fig. 7B). Taken together, these data demonstrate that MIF mediates increased p-eIF2α, expression of CHOP protein and spliced XBP-1 mRNA in chronic Lieber-DeCarli ethanol feeding.
Figure 7. Upregulation of UPR required MIF after Chronic Lieber-DeCarli ethanol feeding.

UPR was determined in livers of C57BL/6J or Mif-/- mice after chronic, Lieber-DeCarli ethanol feeding. A) Expression of p-eIF2α, CHOP and GRP78 was determined 25 days (6% v/v EtOH) after Lieber-DeCarli ethanol feeding and normalized to β-actin. B) Expression of spliced XBP-1, CHOP, Grp78, Dr5 and FGF21 mRNA was determined by qRT-PCR and normalized to 18S RNA. Values are means ± SEM. # - p<0.05 vs Mif-/-
MIF Inhibition prevents induction of CHOP protein in hepatocytes
In order to test the direct role of MIF in UPR induction in hepatocytes, AML-12 murine hepatocytes were exposed to a known UPR inducer, thapsigargin (Oslowski and Urano, 2011). Of note, AML-12 cells are typically grown in DMEM/F12 medium that is supplemented with dexamethasone. Since glucocorticoids are anti-inflammatory, antagonize MIF activity, and prevent MIF release from cells (Alourfi et al., 2005, Gaber et al., 2011), AML-12 cells were treated with thapsigargin in medium without dexamethasone to determine if MIF was required for UPR induction. Indeed, with dexamethasone included in culture medium, thapsigargin-induced CHOP protein and p-eIF2α was blunted in cells (Supplemental Fig. 3). A thapsigargin dose response in AML-12 hepatocytes cultured without dexamethasone revealed a robust increase in expression of CHOP and spliced XBP-1 mRNA 6h after thapsigargin exposure (Fig. 8). Cotreatment with MIF098 (20 µM) or VEH (0.05% DMSO) prevented the thapsigargin-dependent increase in p-eIF2α and CHOP protein expression (Fig. 8C-D) but did not prevent increased expression of CHOP mRNA or XBP-1 splicing (Fig. 8A-B). These data are consistent with in vivo data suggesting that MIF is required for the maximal phosphorylation of eIF2α and expression of CHOP protein in the UPR (Figs. 6,7).
Figure 8. MIF is required for CHOP expression in thapsigargin-treated hepatocytes.

AML-12 murine hepatocytes were treated with VEH (0.05% DMSO) or MIF098 (20 µM) then exposed to thapsigargin (0.1–1.0 µM) for 6h. A) Expression Chop and B) spliced Xbp-1 mRNA was determined by qRT-PCR and normalized to 18S RNA. C) Expression of p-eIF2α and D) CHOP were determined 6h after thapsigargin exposure and normalized to GAPDH. Values are means ± SEM. Values with different superscripts are significantly different, p<0.05
DISCUSSION
MIF, a potent cytokine and chemokine, contributes to the progression of many chronic diseases like autoimmune hepatitis (Assis et al., 2014) and cancer (Bozzi et al., 2017). The current study identified a novel contribution of MIF to ALD progression, with MIF acting as a regulator of the UPR. Importantly, the increased expression of MIF protein (Fig. 1) in livers of AH patients is consistent with previous reports of increased MIF in the sera of ALD patients (Marin et al., 2017, Kumagi et al., 2001). The observation that hepatic neutrophilia and neutrophil activation were unaffected by genetic MIF deficiency or pharmacological MIF inhibition in Gao-Binge (Fig.3-4) was unexpected, suggesting a complex relationship between neutrophils, MIF and liver injury in Gao-Binge and likely in AH. Instead, we find that MIF participates in the UPR. Furthermore, depending on the context of ethanol exposure in mice or stage of ALD in humans the data indicate that MIF and/or the UPR could drive restoration of homeostasis or cell death.
Neutrophils present frequently in livers of AH patients, and it is generally accepted that neutrophils contribute to liver injury in AH (Ramaiah and Jaeschke, 2007, Xu et al., 2014). Expression of the human IL-8 chemokine family mRNA is increased in AH patients (Dominguez et al., 2009) and was markedly increased in livers of AH patients in the current study (Fig. S1), consistent with robust neutrophil accumulation in livers of AH patients. Neutrophils likely contribute to liver injury in AH and other hepatopathies through generation of reactive oxygen species via myeloperoxidase release upon neutrophil degranulation (Ramaiah and Jaeschke, 2007, Xu et al., 2014). In Gao-Binge ethanol feeding, neutrophils are commonly associated with liver injury. Hepatic neutrophilia and liver injury are prevented following: Gao-binge feeding in CD62E-/- mice (Bertola et al., 2013b), in C57BL/6J mice treated with a CXCR2 antagonist (Roh et al., 2015), or when CXCL1 is neutralized (Chang et al., 2015). However, accumulating evidence suggest neutrophils might also participate in tissue repair in AH. For example, in some populations of AH patients, higher numbers of hepatic neutrophils correlated to an improved 90-day survival rate (Bissonnette et al., 2017, Altamirano et al., 2014, Mathurin et al., 1996). Therefore, despite the strong association between neutrophils and collateral tissue damage in AH, they might also be pivotal to resolution of injury.
The current study suggests that the susceptibility of hepatocytes to ROS from ethanol metabolism and/or neutrophil-derived ROS might be intrinsically MIF-dependent, since neutrophil accumulation and activation were unaffected by genetic MIF deficiency or pharmacological MIF inhibition (Fig. 3-4). In addition, higher levels of hepatic MIF expression and circulating MIF in human AH are also correlated to liver dysfunction and higher patient mortality, suggesting the full extent of liver injury in AH is, at least in part, MIF-dependent. The upregulation of MIF following ethanol exposure is prevented in Cyp2E1-/- mice (Barnes et al., 2014), suggesting MIF is a proximal sensor and/or effector of ROS generated from ethanol metabolism in hepatocytes. It is therefore possible that MIF acts as a hepatic danger signal in ALD, since ALD pathophysiology is strongly linked to increased oxidative stress via ethanol metabolism (Barnes et al., 2014) or from the neutrophil-derived oxidative burst in the liver (Ramaiah and Jaeschke, 2007)
Rather than controlling neutrophilia, MIF augmented the UPR associated with Gao-Binge ethanol feeding via a delay in onset of UPR (Cai et al., 2017). UPR is an adaptive physiological process that is induced following exposure to noxious stimuli, e.g. alcohol, ROS, fatty acids (Cazanave et al., 2011), as well as in human nonalcoholic steatohepatitis (Lake et al., 2014). UPR is a balance between pro-cytotoxic and pro-resolution arms (Ji, 2014, Schonthal, 2012). Chronic upregulation of pro-cytotoxic factors in UPR, including CHOP and DR5, could eventually shift the balance of UPR towards cytotoxicity (Ji, 2014, Cazanave et al., 2011, Lake et al., 2014) rather than effective resolution of injury. This maladaptive UPR signature is observed in rodent models of hepatic lipotoxicity and CHOP-/- mice are protected from ethanol-induced liver injury in several studies (Ji et al., 2005, Cai et al., 2017) However, increased expression of chaperones, like GRP78, or antioxidant genes like PDI can lead to the appropriate refolding of proteins affected by intracellular redox changes to resolve UPR in cells. For example, liver injury is exacerbated in Grp78-/- after ethanol feeding, illustrating the differential roles UPR can have in ethanol-induced liver injury (Ji et al., 2011). Indeed, the combined loss of GRP78 expression with increased CHOP expression (Fig. 5) could underscore the unique binge-associated hepatocellular injury after Gao-Binge.
Phosphorylation of eIF2α, CHOP protein induction, DR5 mRNA induction, and XBP-1 splicing in livers (Fig. 6) after Gao-Binge was delayed in Mif-/- mice and MIF098-treated mice, suggesting that multiple arms of the UPR are activated by MIF. MIF signaling is a likely upstream ethanol-induced stressor of the endoplasmic reticulum. As MIF098 prevents the MIF-CD74 interaction, signals downstream of CD74, rather than CXCR2, CXCR4 or CXCR7, likely activate or enhance UPR-associated signaling. The augmented UPR in binged C57BL/6J mice was transient, returning to levels consistent chronic Lieber-DeCarli feeding 9h after binge (Fig. 6-7). However, a delay in the binge-associated UPR after Gao-binge in Mif-/- or MIF098-treated mice might have delayed the onset and resolution of injury, suggesting that a timely UPR is required to quickly resolve the injury in mice.
MIF was consistently required for CHOP protein expression, independent of expression of CHOP mRNA in vivo and in AML-12 cells (Figs. 6-8). The role of MIF in facilitating full expression of CHOP protein in response to ethanol feeding could occur at multiple levels. MIF signaling via MAPKs through CD74 might upregulate CHOP and subsequent CHOP-dependent DR5 expression. However, it is unlikely that these signals will impact the post-transcriptional regulation of the expression of CHOP protein. Instead, MIF might interact with CHOP to promote protein stability and/or maturation, as MIF acts as a chaperone in other contexts. For example, MIF interacts with superoxide dismutase-1 (SOD1) in experimental ALS (Israelson et al., 2015) and suppressed misfolded SOD1 accumulation by direct protein-protein interaction, allowing for proper folding of SOD1. MIF also directly interacts with the proapoptotic gene bcl-2-like protein 11, commonly known as BIM, to protect from apoptosis induction and cytochrome c release in cells (Liu et al., 2008).
MIF was also involved in UPR following chronic Lieber-DeCarli feeding as ethanol-induced p-eIF2α and expression of CHOP protein was largely prevented in Mif-/- mice (Fig. 7). This is consistent with stage-specific roles for both MIF and the UPR in ALD. The role of UPR in human ALD is not fully understood and a recent study in human liver samples detailed increased CHOP protein in livers of AH patients (French et al., 2017). Our data from analysis of explanted liver tissue from AH patients also found induction of CHOP expression (pro-cytotoxic) and a suppression of GRP-78 and PDI expression (pro-resolution) (Fig. 5D). It is therefore possible that an UPR signature that is biased towards increased cytotoxicity (CHOP/DR5) versus diminished resolution (GRP78/PDI) contribute to liver injury in AH. In less acutely severe cases of ALD, however, a persistent activation of UPR might underlie chronic hepatocellular injury.
Taken together this study revealed a novel role for MIF in response to multiple modes of ethanol feeding and connected MIF to the UPR. The many roles of MIF in ALD give insight into the complex interplay between inflammation, danger signals and stress responses during the progression of ALD. Taken together, our data suggest that anti-MIF therapy could lead to a worse short-term prognosis in an acute episode of AH, while chronic liver injury in ALD might be lessened with targeted anti-MIF therapies.
Supplementary Material
Acknowldegements:
This work was supported in part by NIH grants; P50 AA024333, U01AA020821 and RO1AA023722 (LEN); AR049610 (RB); F32AA024595 and K99AA026648 (KLP).
Abbreviations:
- ALD
alcohol-related liver disease
- MIF
macrophage migration inhibitory factor
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- UPR
unfolded protein response
- SLE
systemic lupus erythematosus
- TNFα
tumor necrosis factor-α
- IL-1β
interleukin-1β
- MCP-1
monocyte chemoattractant protein-1
- p-eIF2α
Phosphorylation of eukaryotic initiation factor-2α
- CHOP
CCAAT-enhancer-binding protein homologous protein
- GRP78
glucose-related peptide 78
- DR5
death receptor 5
- FGF21
fibroblast growth factor 21
- sXBP-1
spliced X-box binding protein 1
Footnotes
The authors have no conflicts of interest, financial or otherwise, related to the research to declare.
References
- ALOURFI Z, DONN RP, STEVENS A, BERRY A, MCMASTER A & RAY DW 2005. Glucocorticoids suppress macrophage migration inhibitory factor (MIF) expression in a cell-type-specific manner. J Mol Endocrinol, 34, 583–95. [DOI] [PubMed] [Google Scholar]
- ALTAMIRANO J, MIQUEL R, KATOONIZADEH A, ABRALDES JG, DUARTE-ROJO A, LOUVET A, AUGUSTIN S, MOOKERJEE RP, MICHELENA J, SMYRK TC, BUOB D, LETEURTRE E, RINCON D, RUIZ P, GARCIA-PAGAN JC, GUERRERO-MARQUEZ C, JONES PD, BARRITT AST, ARROYO V, BRUGUERA M, BANARES R, GINES P, CABALLERIA J, ROSKAMS T, NEVENS F, JALAN R, MATHURIN P, SHAH VH & BATALLER R 2014. A histologic scoring system for prognosis of patients with alcoholic hepatitis. Gastroenterology, 146, 1231–9 e1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ASSIS DN, LENG L, DU X, ZHANG CK, GRIEB G, MERK M, GARCIA AB, MCCRANN C, CHAPIRO J, MEINHARDT A, MIZUE Y, NIKOLIC-PATERSON DJ, BERNHAGEN J, KAPLAN MM, ZHAO H, BOYER JL & BUCALA R 2014. The role of macrophage migration inhibitory factor in autoimmune liver disease. Hepatology, 59, 580–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AWANDARE GA, MARTINSON JJ, WERE T, OUMA C, DAVENPORT GC, ONG’ECHA JM, WANG W, LENG L, FERRELL RE, BUCALA R & PERKINS DJ 2009. MIF (macrophage migration inhibitory factor) promoter polymorphisms and susceptibility to severe malarial anemia. J Infect Dis, 200, 629–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BACHER M, MEINHARDT A, LAN HY, MU W, METZ CN, CHESNEY JA, CALANDRA T, GEMSA D, DONNELLY T, ATKINS RC & BUCALA R 1997. Migration inhibitory factor expression in experimentally induced endotoxemia. Am J Pathol, 150, 235–46. [PMC free article] [PubMed] [Google Scholar]
- BARNES MA, MCMULLEN MR, ROYCHOWDHURY S, PISANO SG, LIU X, STAVITSKY AB, BUCALA R & NAGY LE 2013. Macrophage migration inhibitory factor contributes to ethanol-induced liver injury by mediating cell injury, steatohepatitis, and steatosis. Hepatology, 57, 1980–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BARNES MA, ROYCHOWDHURY S & NAGY LE 2014. Innate immunity and cell death in alcoholic liver disease: role of cytochrome P4502E1. Redox Biol, 2, 929–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BERNHAGEN J, CALANDRA T, MITCHELL RA, MARTIN SB, TRACEY KJ, VOELTER W, MANOGUE KR, CERAMI A & BUCALA R 1993. MIF is a pituitary-derived cytokine that potentiates lethal endotoxaemia. Nature, 365, 756–9. [DOI] [PubMed] [Google Scholar]
- BERNHAGEN J, KROHN R, LUE H, GREGORY JL, ZERNECKE A, KOENEN RR, DEWOR M, GEORGIEV I, SCHOBER A, LENG L, KOOISTRA T, FINGERLE-ROWSON G, GHEZZI P, KLEEMANN R, MCCOLL SR, BUCALA R, HICKEY MJ & WEBER C 2007. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat Med, 13, 587–96. [DOI] [PubMed] [Google Scholar]
- BERTOLA A, MATHEWS S, KI SH, WANG H & GAO B 2013a. Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat Protoc, 8, 627–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BERTOLA A, PARK O & GAO B 2013b. Chronic plus binge ethanol feeding synergistically induces neutrophil infiltration and liver injury in mice: a critical role for E-selectin. Hepatology, 58, 1814–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BISSONNETTE J, ALTAMIRANO J, DEVUE C, ROUX O, PAYANCE A, LEBREC D, BEDOSSA P, VALLA D, DURAND F, AIT-OUFELLA H, SANCHO-BRU P, CABALLERIA J, GINES P, BOULANGER CM, BATALLER R & RAUTOU PE 2017. A prospective study of the utility of plasma biomarkers to diagnose alcoholic hepatitis. Hepatology, 66, 555–563. [DOI] [PubMed] [Google Scholar]
- BOZZI F, MOGAVERO A, VARINELLI L, BELFIORE A, MANENTI G, CACCIA C, VOLPI CC, BEZNOUSSENKO GV, MILIONE M, LEONI V, GLOGHINI A, MIRONOV AA, LEO E, PILOTTI S, PIEROTTI MA, BONGARZONE I & GARIBOLDI M 2017. MIF/CD74 axis is a target for novel therapies in colon carcinomatosis. J Exp Clin Cancer Res, 36, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CAI Y, XU MJ, KORITZINSKY EH, ZHOU Z, WANG W, CAO H, YUEN PS, ROSS RA, STAR RA, LIANGPUNSAKUL S & GAO B 2017. Mitochondrial DNA-enriched microparticles promote acute-on-chronic alcoholic neutrophilia and hepatotoxicity. JCI Insight, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CALANDRA T, ECHTENACHER B, ROY DL, PUGIN J, METZ CN, HULTNER L, HEUMANN D, MANNEL D, BUCALA R & GLAUSER MP 2000. Protection from septic shock by neutralization of macrophage migration inhibitory factor. Nat Med, 6, 164–70. [DOI] [PubMed] [Google Scholar]
- CASSINI-VIEIRA P, MOREIRA CF, DA SILVA MF & BARCELOS LS 2015. Estimation of Wound Tissue Neutrophil and Macrophage Accumulation by Measuring Myeloperoxidase (MPO) and N-Acetyl-β-D-glucosaminidase (NAG) Activities. Bio-protocol, 5, e1662. [Google Scholar]
- CAZANAVE SC, MOTT JL, BRONK SF, WERNEBURG NW, FINGAS CD, MENG XW, FINNBERG N, EL-DEIRY WS, KAUFMANN SH & GORES GJ 2011. Death receptor 5 signaling promotes hepatocyte lipoapoptosis. J Biol Chem, 286, 39336–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHANG B, XU MJ, ZHOU Z, CAI Y, LI M, WANG W, FENG D, BERTOLA A, WANG H, KUNOS G & GAO B 2015. Short- or long-term high-fat diet feeding plus acute ethanol binge synergistically induce acute liver injury in mice: an important role for CXCL1. Hepatology, 62, 1070–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DOMINGUEZ M, MIQUEL R, COLMENERO J, MORENO M, GARCIA-PAGAN JC, BOSCH J, ARROYO V, GINES P, CABALLERIA J & BATALLER R 2009. Hepatic expression of CXC chemokines predicts portal hypertension and survival in patients with alcoholic hepatitis. Gastroenterology, 136, 1639–50. [DOI] [PubMed] [Google Scholar]
- FRENCH SW, MASOUMINIA M, SAMADZADEH S, TILLMAN BC, MENDOZA A & FRENCH BA 2017. Role of Protein Quality Control Failure in Alcoholic Hepatitis Pathogenesis. Biomolecules, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GABER T, SCHELLMANN S, EREKUL KB, FANGRADT M, TYKWINSKA K, HAHNE M, MASCHMEYER P, WAGEGG M, STAHN C, KOLAR P, DZIURLA R, LOHNING M, BURMESTER GR & BUTTGEREIT F 2011. Macrophage migration inhibitory factor counterregulates dexamethasone-mediated suppression of hypoxia-inducible factor-1 alpha function and differentially influences human CD4+ T cell proliferation under hypoxia. J Immunol, 186, 764–74. [DOI] [PubMed] [Google Scholar]
- GARCIA-SAENZ-DE-SICILIA M, DUVOOR C, ALTAMIRANO J, CHAVEZ-ARAUJO R, PRADO V, DE LOURDES CANDOLO-MARTINELLI A, HOLANDA-ALMEIDA P, BECERRA-MARTINS-DE-OLIVEIRA B, FERNANDEZ-DE-ALMEIDA S, BATALLER R, CABALLERIA J & DUARTE-ROJO A 2017. A Day-4 Lille Model Predicts Response to Corticosteroids and Mortality in Severe Alcoholic Hepatitis. Am J Gastroenterol, 112, 306–315. [DOI] [PubMed] [Google Scholar]
- HARE AA, LENG L, GANDAVADI S, DU X, COURNIA Z, BUCALA R & JORGENSEN WL 2010. Optimization of N-benzyl-benzoxazol-2-ones as receptor antagonists of macrophage migration inhibitory factor (MIF). Bioorg Med Chem Lett, 20, 5811–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ISRAELSON A, DITSWORTH D, SUN S, SONG S, LIANG J, HRUSKA-PLOCHAN M, MCALONIS-DOWNES M, ABU-HAMAD S, ZOLTSMAN G, SHANI T, MALDONADO M, BUI A, NAVARRO M, ZHOU H, MARSALA M, KASPAR BK, DA CRUZ S & CLEVELAND DW 2015. Macrophage migration inhibitory factor as a chaperone inhibiting accumulation of misfolded SOD1. Neuron, 86, 218–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JI C 2014. New Insights into the Pathogenesis of Alcohol-Induced ER Stress and Liver Diseases. Int J Hepatol, 2014, 513787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JI C, KAPLOWITZ N, LAU MY, KAO E, PETROVIC LM & LEE AS 2011. Liver-specific loss of glucose-regulated protein 78 perturbs the unfolded protein response and exacerbates a spectrum of liver diseases in mice. Hepatology, 54, 229–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JI C, MEHRIAN-SHAI R, CHAN C, HSU YH & KAPLOWITZ N 2005. Role of CHOP in hepatic apoptosis in the murine model of intragastric ethanol feeding. Alcohol Clin Exp Res, 29, 1496–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KHANOVA E, WU R, WANG W, YAN R, CHEN Y, FRENCH SW, LLORENTE C, PAN SQ, YANG Q, LI Y, LAZARO R, ANSONG C, SMITH RD, BATALLER R, MORGAN T, SCHNABL B & TSUKAMOTO H 2017. Pyroptosis by caspase1¼-gasdermin-D pathway in alcoholic hepatitis in mice and patients. Hepatology [DOI] [PMC free article] [PubMed]
- KUMAGI T, AKBAR F, HORIIKE N & ONJI M 2001. Increased serum levels of macrophage migration inhibitory factor in alcoholic liver diseases and their expression in liver tissues. Clin Biochem, 34, 189–93. [DOI] [PubMed] [Google Scholar]
- LAKE AD, NOVAK P, HARDWICK RN, FLORES-KEOWN B, ZHAO F, KLIMECKI WT & CHERRINGTON NJ 2014. The adaptive endoplasmic reticulum stress response to lipotoxicity in progressive human nonalcoholic fatty liver disease. Toxicol Sci, 137, 26–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LENG L, METZ CN, FANG Y, XU J, DONNELLY S, BAUGH J, DELOHERY T, CHEN Y, MITCHELL RA & BUCALA R 2003. MIF signal transduction initiated by binding to CD74. J Exp Med, 197, 1467–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIU L, CHEN J, JI C, ZHANG J, SUN J, LI Y, XIE Y, GU S & MAO Y 2008. Macrophage migration inhibitory factor (MIF) interacts with Bim and inhibits Bim-mediated apoptosis. Mol Cells, 26, 193–9. [PubMed] [Google Scholar]
- LOUVET A, LABREUCHE J, ARTRU F, BOUTHORS A, ROLLAND B, SAFFERS P, LOLLIVIER J, LEMAITRE E, DHARANCY S, LASSAILLY G, CANVA-DELCAMBRE V, DUHAMEL A & MATHURIN P 2017. Main drivers of outcome differ between short term and long term in severe alcoholic hepatitis: A prospective study. Hepatology, 66, 1464–1473. [DOI] [PubMed] [Google Scholar]
- MANDREKAR P, AMBADE A, LIM A, SZABO G & CATALANO D 2011. An essential role for monocyte chemoattractant protein-1 in alcoholic liver injury: regulation of proinflammatory cytokines and hepatic steatosis in mice. Hepatology, 54, 2185–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARIN V, POULSEN K, ODENA G, MCMULLEN MR, ALTAMIRANO J, SANCHO-BRU P, TIRIBELLI C, CABALLERIA J, ROSSO N, BATALLER R & NAGY LE 2017. Hepatocyte-derived macrophage migration inhibitory factor mediates alcohol-induced liver injury in mice and patients. J Hepatol, 67, 1018–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARRA F & TACKE F 2014. Roles for chemokines in liver disease. Gastroenterology, 147, 577–594 e1. [DOI] [PubMed] [Google Scholar]
- MATHURIN P, DUCHATELLE V, RAMOND MJ, DEGOTT C, BEDOSSA P, ERLINGER S, BENHAMOU JP, CHAPUT JC, RUEFF B & POYNARD T 1996. Survival and prognostic factors in patients with severe alcoholic hepatitis treated with prednisolone. Gastroenterology, 110, 1847–53. [DOI] [PubMed] [Google Scholar]
- NAGY LE 2015. The Role of Innate Immunity in Alcoholic Liver Disease. Alcohol Res, 37, 237–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ORMAN ES, ODENA G & BATALLER R 2013. Alcoholic liver disease: pathogenesis, management, and novel targets for therapy. J Gastroenterol Hepatol, 28 Suppl 1, 77–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OSLOWSKI CM & URANO F 2011. Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Methods Enzymol, 490, 71–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAMAIAH SK & JAESCHKE H 2007. Role of neutrophils in the pathogenesis of acute inflammatory liver injury. Toxicol Pathol, 35, 757–66. [DOI] [PubMed] [Google Scholar]
- ROH YS, ZHANG B, LOOMBA R & SEKI E 2015. TLR2 and TLR9 contribute to alcohol-mediated liver injury through induction of CXCL1 and neutrophil infiltration. Am J Physiol Gastrointest Liver Physiol, 309, G30–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAULER M, ZHANG Y, MIN JN, LENG L, SHAN P, ROBERTS S, JORGENSEN WL, BUCALA R & LEE PJ 2015. Endothelial CD74 mediates macrophage migration inhibitory factor protection in hyperoxic lung injury. FASEB J, 29, 1940–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHONTHAL AH 2012. Endoplasmic reticulum stress: its role in disease and novel prospects for therapy. Scientifica (Cairo), 2012, 857516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SERSTE T, CORNILLIE A, NJIMI H, PAVESI M, ARROYO V, PUTIGNANO A, WEICHSELBAUM L, DELTENRE P, DEGRE D, TREPO E, MORENO C & GUSTOT T 2018. The prognostic value of Acute-on-Chronic Liver Failure during the course of severe alcoholic hepatitis. J Hepatol [DOI] [PubMed]
- SREIH A, EZZEDDINE R, LENG L, LACHANCE A, YU G, MIZUE Y, SUBRAHMANYAN L, PONS-ESTEL BA, ABELSON AK, GUNNARSSON I, SVENUNGSSON E, CAVETT J, GLENN S, ZHANG L, MONTGOMERY R, PERL A, SALMON J, ALARCON-RIQUELME ME, HARLEY JB & BUCALA R 2011. Dual effect of the macrophage migration inhibitory factor gene on the development and severity of human systemic lupus erythematosus. Arthritis Rheum, 63, 3942–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- XU R, HUANG H, ZHANG Z & WANG FS 2014. The role of neutrophils in the development of liver diseases. Cell Mol Immunol, 11, 224–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZIMMERMANN HW, SEIDLER S, GASSLER N, NATTERMANN J, LUEDDE T, TRAUTWEIN C & TACKE F 2011. Interleukin-8 is activated in patients with chronic liver diseases and associated with hepatic macrophage accumulation in human liver fibrosis. PLoS One, 6, e21381. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.