

Abstract
More than one third of pregnant women in the USA are obese and maternal obesity (MO) negatively affects fetal development, which predisposes offspring to metabolic diseases. The placenta mediates nutrient delivery to fetuses and its function is impaired due to MO. Exercise ameliorates metabolic dysfunction due to obesity, but its effect on placental function of obese mothers has not been explored. Here C57BL/6J female mice were randomly assigned into two groups fed either a control or a high-fat diet (HFD), and then the mice of each diet were further divided into two sub-groups with/without exercise. In HFD-induced obese mice, daily treadmill exercise during pregnancy reduced body weight gain, lowered serum glucose and lipid concentration, and improved insulin sensitivity of maternal mice. Importantly, maternal exercise prevented fetal overgrowth (macrosomia) induced by MO. To further examine preventive effects of exercise on fetal overgrowth, placental vascularization and nutrient transporters were analyzed. Vascular density and the expression of vasculogenic factors were reduced due to MO but recovered by maternal exercise. On the other hand, the contents of nutrient transporters were not substantially altered by MO or exercise, suggesting that the protective effects of exercise in MO-induced fetal overgrowth were primarily due to alteration of placental vascularization and improved maternal metabolism. Furthermore, exercise enhanced down-stream insulin signaling and activated AMP-activated protein kinase in HFD placenta. Taken together, maternal exercise prevented fetal overgrowth induced by MO, which were associated with the improved maternal metabolism and placental vascularization in obese mothers with exercise.
Keywords: Maternal exercise, maternal-fetal exchange, nutrient transport, insulin resistance
Introduction
Obesity and sedentary life style are major risk factors for metabolic syndrome and complications, including cardiovascular disease, type 2 diabetes mellitus, and certain types of cancers (Hruby et al., 2016; Kerr et al., 2017; Leiva et al., 2017). Currently, over 30% of pregnant women in the United States are obese and with additional one third are overweight (Ogden et al., 2012), which predisposes their children to obesity and metabolic diseases (McCurdy et al., 2009; Vogt et al., 2014). Therefore, interventions which can prevent or reduce the development of metabolic dysfunction in offspring of obese mothers are imperative.
The placenta performs nutrient/oxygen exchanges between the mother and her fetuses, and syncytiotrophoblasts which mediate nutrient/gas exchange have high oxygen consumption rates (Carter, 2000) and energy expenditure (Jones & Rolph, 1985). The placental function is impaired due to maternal obesity (MO), which impedes vasculogenic/angiogenic development of the placenta (Stuart et al., 2018). Obesity and HFD intake further upregulate placental nutrient transporters such as GLUT1 and GLUT3, and stimulate placental mammalian target of rapamycin complex 1 (mTORC1) signaling pathway, inducing placental and fetal overgrowth (Aye et al., 2015). Moreover, offspring, overgrown during fetal development (macrosomia) due to maternal obesity, had impaired glucose tolerance, elevated fasting insulin level, and increased adiposity and metabolic diseases (Stanford et al., 2017; Barbour & Hernandez, 2018). Thus, maintaining proper vasculogenesis and placental function in obese mothers is the critical step for preventing metabolic dysfunction in the offspring (Bairagi et al., 2016).
Exercise during pregnancy is known to be beneficial for both maternal health and fetal development (Gregg & Ferguson, 2017; Stanford et al., 2017). However, the beneficial effects of exercise on placental vascularization and nutrient transport have not been examined. Exercise may facilitate placental development through activating AMP-activated protein kinase (AMPK), a key kinase increasingly implicated in cell differentiation and tissue development (Kaufman & Brown, 2016). In addition, exercise stimulates muscle to secrete myokines including apelin, and apelin mediates placental vasculogenesis and its deficiency is linked to preeclampsia (Gilbert, 2017; Ho et al., 2017). Moreover, exercise affects the down-stream signaling pathways of insulin and insulin-like growth factors, including mTORC1 signaling (Ogasawara et al., 2014), as well as suppresses low-grade inflammation associated with obesity (Ringseis et al., 2015). In the current study, we found that maternal exercise improved maternal metabolic health and prevented fetal overgrowth induced by maternal HFD; we further found that the enhanced placental vasculogenesis and improved maternal metabolic health might explain the beneficial effects of maternal exercise to fetal development.
Materials and Methods
Ethical Approval.
All animal procedures have been conducted in accordance with the guidelines of the National Institutes of Health (NIH) and according to the protocol approved by the Institute of Animal Care and Use Committees (IACUC) at Washington State University. The investigators understand the ethical principles under which the Journal of Physiology operates and that their work complies with the animal ethics checklists (Grundy, 2015).
Animals and diets.
Eight-week-old C57BL/6J female mice (The Jackson Laboratory, Bar Harbor, ME, USA) were randomly assigned into two groups (n = 12 /group) fed ad libitum either a control diet (10% energy from fat, D12450J, Research Diets, New Brunswick, NJ, USA) or an high-fat diet (60% energy from fat, D12492, Research Diets) for 8 weeks to induce obesity. When the dams in the HFD group had gained over 25% of their initial body weight (Fig 2A), showing HFD-induced obesity (Aye et al., 2015; Yang et al., 2016). At one week preconception, the diets were changed to a control diet (CON, 10% energy from fat, D12450J, Research Diets) or an obesogenic diet (HFD, 45% energy from fat, D12451, Research Diets). Female mice were mated with eight to ten-week-old male mice (The Jackson Laboratory) fed with regular chow. The mating was determined by examining vaginal smears. Additionally, both maternal groups were divided into two subgroups with/without exercise during gestation (n = 6 for each treatment). During treatments, food and water were provided ad libitum, and body weight and food intake were monitored daily. At embryonic day 18.5 (E18.5), after 5 h of fasting, pregnant mice were anesthetized by carbon dioxide inhalation and euthanized by cervical dislocation, consistent with previous reports (Zou et al., 2017). Placenta was collected after removing uterine tissue, umbilical cord, and other extraembryonic membranes. All female and male fetuses were collected and studied, following the fundamental principles for animal models (Dickinson et al., 2016) and the animals in research: reporting in vivo experiments (ARRIVE) guidelines (Kilkenny et al., 2012). Mice were housed at 22°C on 12 h-light/12 h-dark cycles (Figure 1).
Figure 2.
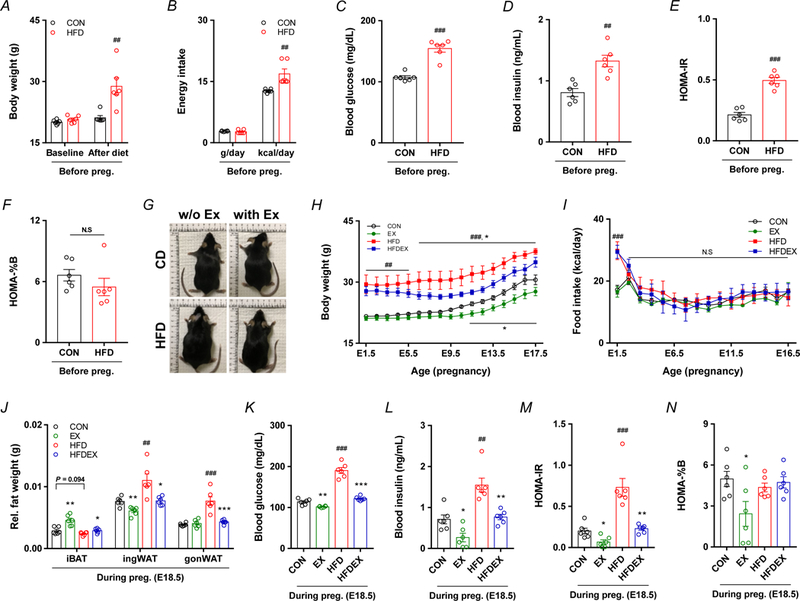
Effects of maternal obesity and exercise intervention on maternal characteristics. A-D, Data were collected following 8 weeks of diet intervention before mating. Body weight (A) and energy intake (B) of CON and HFD maternal mice before pregnancy. Blood glucose (C) and insulin (D) following 5 h of fasting in CON and HFD maternal mice before pregnancy. E-F, Calculated insulin resistance (E) and pancreatic β cell function (F) following 5 h of fasting in CON and HFD maternal mice before pregnancy. G-N, Data were collected from maternal mice at E18.5, including representative whole body images (G), body weight (H), food intake (I), and relative fat wet weight (J) in maternal mice at E18.5, as well as blood glucose (K), insulin (L), and calculated insulin resistance (M) and pancreatic β cell function (N). Data are expressed as the mean ± s.e.m. n = 6. *P < 0.05, **P < 0.01, and ***P < 0.001 in CON vs. EX or HFD vs. HFDEX; #P < 0.05, ##P <0.01, ###P < 0.001 in CON vs. HFD by two-tailed Student’s t-test (A-F, J-N) or ANOVA with post hoc Bonferroni multiple comparison analysis (H,I).
Figure 1.
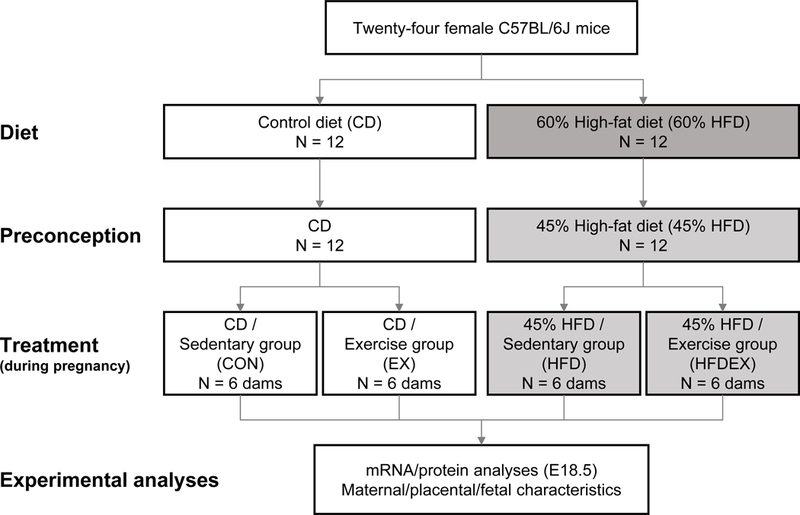
Flow chart illustrating the allocation of mice and treatments.
Endurance treadmill exercise.
To customize with exercise, all mice were subjected to flat treadmill exercise at 10 m/min for 10 min, 3 times per week before mating. Then, the maximal oxygen consumption rates (VO2max) for control and obese mice were measured using treadmill respiratory measurement system (Oxymax Fast 4 lane modular treadmill system, Columbus Instruments, Columbus, OH, USA). The exercise intensity for CON and HFD mice was set based on VO2max rates, which are commonly used in previous studies (Petrosino et al., 2016). Briefly, based on the gestation stages, maternal mice were separated into three periods, E1.5 to E7.5, E8.5 to E14.5, and E15.5 to E16.5. The VO2max rates were measured on E1.5, E8.5, and E15.5. The exercise intensity was set as follows, 40% (E1.5 to E7.5), 65% (E8.5 to E14.5), and 50% (E15.5 to E16.5) of VO2max, based on the exercise guidelines in pregnancy (Zavorsky & Longo, 2011). Each exercise regimen was composed of three steps, warming up (5 m/min for 10 min), exercise (10 to 14 m/min for 40 min), and cooling down (5 m/min for 10 min), which were performed at the same time every morning. The treadmill speeds during the exercise step were set to 11/14/12.5 m/min for E1.5-E7.5/E8.5-E14.5/E15.5-E16.5 respectively in CON+EX mice and 10/13/12 m/min for HFD+EX mice. Sedentary CON and HFD mice were placed on the treadmill for an hour daily with the speed set at 0 m/min. Mice were not subjected to exercise between E16.5 to E18.5 (two days before euthanization) in order to avoid the acute effects of exercise on samples collected.
Metabolic studies.
Two days before tissue collection (E16.5), the metabolic rates of oxygen consumption (VO2), carbon dioxide production (VCO2), respiratory exchange ratio (RER), and heat production were analyzed during 24 h (light: quiescent phase / dark: active phase) using an indirect open circuit calorimetry system (Comprehensive Lab Animal Monitoring System [CLAMS], Columbus Instruments, Columbus, OH, USA). During measurement, mice were fed ad libitum with the respective diets and water provided (Wang et al., 2017). The fat and carbohydrate oxidation was calculated as previously described (Peronnet & Massicotte, 1991).
Thermal imaging.
Surface temperatures were measured using an E6 Thermal Imaging Infrared Camera (FLIR Systems, Wilsonville, OR, USA) before euthanization at E18.5.
Hand grip strength.
A grip strength meter (Columbus Instruments, Columbus, OH, USA) was used to measure the maximal grip strength (average of 5 times measurement). In addition, the endurance grip strength was assessed by sequentially performing 10 times of measurements.
Serum analysis.
Before mating, blood samples were collected from tail tip following 5 h fasting. At E18.5, following 5 h fasting, maternal blood was further collected through cardiopuncture under deep anesthesia via carbon dioxide inhalation, and used for measuring glucose and insulin levels. Moreover, fetal blood was collected from fetuses through the retroorbital sinus into the conical tube for centrifuge. Blood glucose was measured using blood glucose monitoring system, and insulin was measured using the mouse high range insulin enzyme-linked immunosorbent assay (ELISA; ALPCO, Salem, NH, USA). The homeostatic model assessment of insulin resistance (HOMA-IR) and pancreatic β-cell function (HOMA-%B) were calculated according to the following formula: HOMA-IR [Fasting insulin in μU/mL × (fasting blood glucose in mg/dL × 0.055)/22.5] and HOMA-%B [(360 × fasting insulin in μU/mL)/(fasting blood glucose – 63)] (Matthews et al., 1985; Ghasemi et al., 2015; Lemaitre et al., 2018).
Gene expression.
The experimental approach was consistent with the minimum information for publication of quantitative real-time PCR experiments (MIQE) guidelines (Bustin et al., 2009). Briefly, at E18.5, the placenta was collected and frozen in liquid N2 and stored at −80°C until analysis. Total RNA was isolated by TRIzol reagent (Invitrogen, Grand Island, NY, USA), and cDNA was synthesized using extracted mRNA with the iScript™ cDNA Synthesis Kit (Bio-Rad, Hercules, CA, USA). Relative mRNA expression was determined by quantitative real-time PCR (IQ5, Bio-Rad) using SsoAdvanced™ Universal SYBR® Green Supermix (Bio-Rad), and 18S rRNA was used as a reference gene for normalization (Yang et al., 2016). Primer sequences are listed in Table 1.
Table 1.
qPCR primer sequences used for gene expression analyses
| Gene Name |
Forward primer sequence | Reverse primer sequence |
|---|---|---|
| Vegfa | 5’-TGGACCCTGGCTTTACTGCT-3’ | 5’-GCAGTAGCTTCGCTGGTAGA-3’ |
| Vegfr1 | 5’-TGTGCACATGACGGAAGGAA-3’ | 5’-GTATTGGTCTGCCGATGGGT-3’ |
| Fgf2 | 5’-GGCTGCTGGCTTCTAAGTGT-3’ | 5’-GTCCCGTTTTGGATCCGAGT-3’ |
| Fgfr2 | 5’-CACGACCAAGAAGCCAGACT-3’ | 5’-CTCGGCCGAAACTGTTACCT-3’ |
| Hif1a | 5’-AGGATGAGTTCTGAACGTCGAAA-3’ | 5’-CTGTCTAGACCACCGGCATC-3’ |
| Ccn1 | 5’-AGAGGCTTCCTGTCTTTGGC-3’ | 5’-CCAAGACGTGGTCTGAACGA-3’ |
| Gcm1 | 5’-GAAGGCGGACAGGCTTTGA-3’ | 5’-GTTTCACGTAGGAGTCCGGC-3’ |
| Glut1 | 5’-GCGGGAGACGCATAGTTACA-3’ | 5’-CAGCCCCGTTACTCACCTTG-3’ |
| Snat1 | 5’-GGGCATAAGGTACACCGAGG-3’ | 5’-CAACGTGCACCTGTTTACCG-3’ |
| Snat2 | 5’-ACGTGGATCCCGAAAACCAG-3’ | 5’-CCAAGGATTCCACTGCCCAC-3’ |
| Lat1 | 5’-GGGAAGGACATGGGACAAGG-3’ | 5’-GCCAACACAATGTTCCCCAC-3’ |
| Lpl | 5’-GAAAGGGCTCTGCCTGAGTT-3’ | 5’-TAGGGCATCTGAGAGCGAGT-3’ |
| Cd36 | 5’-TGAATGGTTGAGACCCCGTG-3’ | 5’-TAGAACAGCTTGCTTGCCCA-3’ |
| Fabp4 | 5’-CGACAGGAAGGTGAAGAGCATCATA-3’ | 5’-CATAAACTCTTGTGGAAGTCACGCCT-3’ |
| Igf2 | 5’-CGCCCCAGCCCTAAGATAC-3’ | 5’-GGGTATGCAAACCGAACAGC-3’ |
| Igfbp1 | 5’-TGTTTCTTGGCCGTTCCTGA-3’ | 5’-GAGAAATCTCGGGGCACGAA-3’ |
| 18S | 5’-GTAACCCGTTGAACCCCATT-3’ | 5’-CCATCCAATCGGTAGTAGCG-3’ |
Immunoblotting analysis.
Proteins were extracted from collected frozen placental tissues at −80°C using lysis buffer (100 mM Tris-HCL pH 6.8, 2.0% SDS, 20% glycerol, 0.02% Bromophenol Blue, 5% 2-mercaptoethanol, 100 mM NaF, and 1 mM Na3VO4), and the protein concentration of lysates was determined by the Bradford Assay (Bio-Rad). The following antibodies were used for the detection of phosphorylation and total protein levels: phosphorylated insulin receptor substrate 1 [p-IRS-1 (Ser789); 2389S], insulin receptor substrate 1 (IRS-1; 3407S), p-Akt (Ser473; 9271), phosphorylated AMPKα [p-AMPKα (Thr172); 2535], AMPKα (2532), phosphorylated mammalian target of rapamycin [p-mTOR (Ser2448); 2971], p-4E-BP1 (Thr37/46; 9459), 4E-BP1 (9452), p-Erk½ (Thr202/Tyr204; 4370), Erk½ (4695), GLUT1 (12939), tumor necrosis factor α (TNF-α; 3707), interleukin-1β (IL-1β; 12242), and interleukin-6 (IL-6; 12153), all of which were purchased from Cell Signaling Technology (Danvers, MA, USA). Antibodies against Akt (sc-5298), mTOR (sc-517464), vascular endothelial growth factor (VEGF; sc-7269), endogenous ligand for the G-protein coupled APJ receptor (APLN; sc-293441), and hypoxia-inducible factor 1α (HIF1α; sc-13515) were purchased from Santa Cruz Biotechnology (Dallas, TX, USA). β-Actin and β-tubulin were obtained from the Developmental Studies Hybridoma Bank (Iowa City, IA, USA). IRDye 800CW goat anti-rabbit secondary antibody (1:10,000) and IRDye 680 goat anti-mouse secondary antibody (1:10,000) were purchased from LI-COR Biosciences (Lincoln, NE, USA). Immunoblotting analysis was performed using an Odyssey infrared imaging system (Li-COR Biosciences) for detecting target proteins.
Histological analyses.
Fresh placenta tissues were fixed for 24 h at room temperature in PBS containing 4% paraformaldehyde, and then embedded in paraffin. The tissue sections at 5 μm-thickness were prepared for hematoxylin and eosin (H&E) staining or immunocytochemical (ICC) staining following deparaffinization as previously described (Wang et al., 2017). For measuring the ratio of cross-sectional area (CSA) of the junctional/labyrinth zones and CSA of the placenta, each zone was calculated using ImageJ software (National Institutes of Health, Bethesda, MD, USA). For ICC staining, sections of labyrinth zone were heated in Tris-EDTA buffer for 20 min, blocked within 5% goat serum in Tris-buffer saline (TBS) containing 0.3% Triton X-100 for 2 h. Then, samples were incubated with purified anti-mouse CD34 primary antibody (#119301; 1:100; BioLegend, San Diego, CA, USA) or a mouse monoclonal endothelial nitric oxide synthase (eNOS) primary antibody (sc-136977; 1:50; Santa Cruz Biotechnology, Dallas, TX, USA) overnight and Alexa Fluor 488 anti-rat IgG secondary antibody (#405418:1,000; BioLegend) for 1 h. Sections were mounted with a mounting medium (Vector Laboratories, Burlingame, CA, USA), and fluorescence was examined using an EVOS® FL color Imaging System (Mill Creek, WA, USA). Oil Red O staining was performed on placenta cryosections, as previously described (Louwagie et al., 2018). Placenta samples including labyrinth zone were excised, fixed with paraformaldehyde and embedded in OCT (Thermo 6502, Thermo Fisher Scientific, Waltham, MA, USA). Tissue sections at 10 μm-thickness were prepared and stained with Oil Red O. Sections were mounted with a mounting medium (Vector Laboratories, Burlingame, CA, USA), and images were obtained using an EVOS® XL Core Imaging System (Mill Creek, WA, USA).
Data presentation and statistical analyses.
Data were analyzed and visualized using GraphPad Prism 7 for Windows (GraphPad Software, San Diego, CA, USA), and expressed as the mean ± s.e.m. Statistical analyses were performed using one- or two-way ANOVA (diet×exercise), with Bonferroni post hoc analysis (SAS Institute Inc., Cary, NC, USA). P values less than 0.05 (* or #), 0.01 (** or ##) or 0.001 (*** or ###) were statistically different.
Results
Effects of maternal obesity and exercise intervention on maternal characteristics.
An overview of the experimental schematic diagram for the assignment of diets, preconception treatments, exercise, and experimental analyses is shown in Fig. 1. Before mating, HFD group had higher body weight, in accordance with higher caloric intake (Fig. 2A,B). Consistently, the fasting glucose, insulin, and HOMA-IR were significantly elevated in HFD mice (Fig. 2C–F), consistent with previous reports (Fu et al., 2016; Zou et al., 2017). After mating, HFD mice with exercise training gained less weight compared to HFD without exercise, despite of no difference in food intake (Fig. 2G–I). At E18.5, maternal mice were sacrificed following 5 h fasting. The weights of inguinal white adipose tissue (ingWAT) and gonadal WAT (gonWAT) were higher in HFD mice, which was mitigated by exercise (Fig. 2J). On the other hand, exercise intervention during gestation increased the weight of intrascapular brown adipose tissue (iBAT), but HFD did not affect iBAT weight (Fig. 2J). Blood glucose, insulin, and HOMA-IR in HFD-induced obese mothers were higher than those of control mothers; on the other hand, exercise training alleviated these changes (Fig. 2K–M). For HOMA-%B index, which indicates the insulin secretion by pancreatic β cells (Matthews et al., 1985), was decreased only in maternal mice fed with a control diet and with exercise (Fig. 2N), suggesting exercise stimulated insulin-independent utilization of glucose.
Exercise training improves muscle strength and energy expenditure in maternal mice challenged with HFD.
Maternal obesity and HFD lead to a loss of aerobic exercise capacity including reduced total exercise time and distance, but the maximal oxygen consumption during a single bout of exhaustive exercise was not altered (Fig. 3A–C). In addition, relative maximal grip strength and endurance grip strength were significantly reduced due to HFD but recovered following exercise (Fig. 3D,E). At E15.5, we further measured oxygen and CO2 consumption, and the respiratory ratio. HFD reduced oxygen and CO2 consumption, which was recovered in HFD mice with exercise. On the other hand, no significant changes in VO2 and VCO2 between CON and CON with exercise mice were detected (Fig. 3G,H). The HFD profoundly decreased the respiratory exchange ratio, showing increased utilization of fatty acids, which was not altered by exercise (Fig. 3I). Quantitatively, HFD increased the fat oxidation and decreased carbohydrate oxidation, but exercise training increased the carbohydrate oxidation of HFDEX mice in the dark cycle (Fig. 3K,L). These data were consistent with earlier reports (Romijn et al., 1993; Schrauwen et al., 2000). Moreover, the surface temperature was increased in HFD mice with exercise (Fig. 3F).
Figure 3.
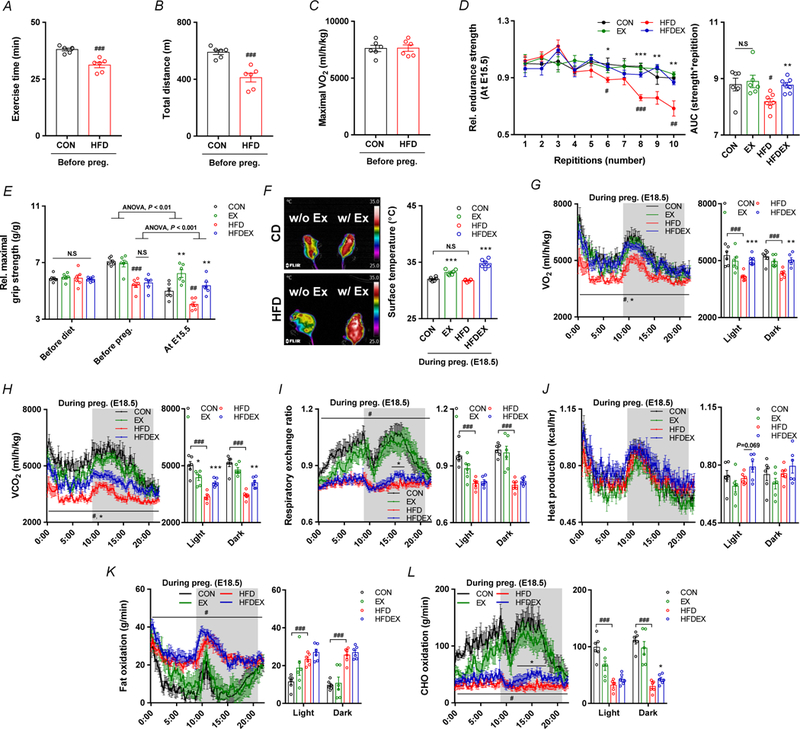
Maternal exercise training improves exercise capacity and energy expenditure in HFD-induced obese and pregnant mice. A-C, Maximal aerobic capacity test of female mice before mating, including exercise time (A), total distance (B), and maximal oxygen consumption levels (C). D-F, Muscle strength of maternal mice at E15.5: relative endurance grip strength (D) and forelimb grip strength (E), and representative thermogenic images (F). G-L, Metabolic parameters of maternal mice at E16.5: Time-resolved oxygen consumption (G), time-resolved carbon dioxide production (H), respiratory exchange ratio (RER) (I), Time-resolved heat production (J), time-resolved fat oxidation (K) and time-resolved carbohydrate oxidation (L); dark phase is marked as dark background; right inset depicts calculated means as indicated. Mean ± s.e.m. n = 6. *P < 0.05, **P < 0.01, and ***P < 0.001 in CON vs. EX or HFD vs. HFDEX; #P < 0.05, ##P <0.01, ###P < 0.001 in CON vs. HFD by two-tailed Student’s t-test (A-E, G-L), ANOVA with post hoc Bonferroni multiple comparison analysis (D, E).
Exercise intervention of obese mothers prevents fetal overgrowth.
Placental weight of obese mice did not differ from control mice, but it was higher in exercised mice (Fig. 4A). HFD increased the fetal body weight at E18.5, which was alleviated by exercise intervention (Fig. 4B). There was no difference in the litter sizes among groups (Fig. 4C). Placental efficiency, as determined by fetal weight divided by the weight of placenta, increased by HFD, consistent with a previous report (Jones et al., 2009), but decreased by exercise training (Fig. 4D). The mRNA expression of nutrient transporters was also analyzed. The mRNA expression of lipoprotein lipase (Lpl) and Cd36 (fatty acid transporter) was higher, and that of glutamine transporter Snat1 and amino acid transporter Snat2 was lower in HFD compared to CON mice at E18.5, which were reversed by exercise training (Fig. 4H). Consistently, blood glucose, insulin, and insulin resistance were elevated in fetuses from HFD mothers without exercise, which were alleviated in both female and male fetuses of HFD mothers with exercise (Fig 4E–G). However, no differences between female and male fetuses in glucose and insulin levels were observed (Fig. 4E,F). Additionally, the positive correlations in the blood glucose and insulin levels between mothers and fetuses were observed (Fig. 4J–M). To explore whether increased expression of fatty acid transporters led to lipid accumulation in the placenta, we performed Oil Red O staining. The amount of lipid droplets was higher in HFD placenta, which was prevented through maternal exercise training (Fig. 4I). Placental inflammation during pregnancy affects placental development and function, which induces fetal abnormal growth (Howell & Powell, 2017). The contents of inflammatory cytokines, TNF-α and IL-1β in the placental tissue were increased in HFD mice, while exercise down-regulated their levels. On the other hand, the protein level of IL-6 was reduced in obese mice while exercise increased its content (Fig. 4N). Unlike TNF-α and IL-1β, IL-6 has both anti-inflammatory and inflammatory effects dependent on the context, and exercise is known to elevate IL-6 levels (Petersen & Pedersen, 2005).
Figure 4.
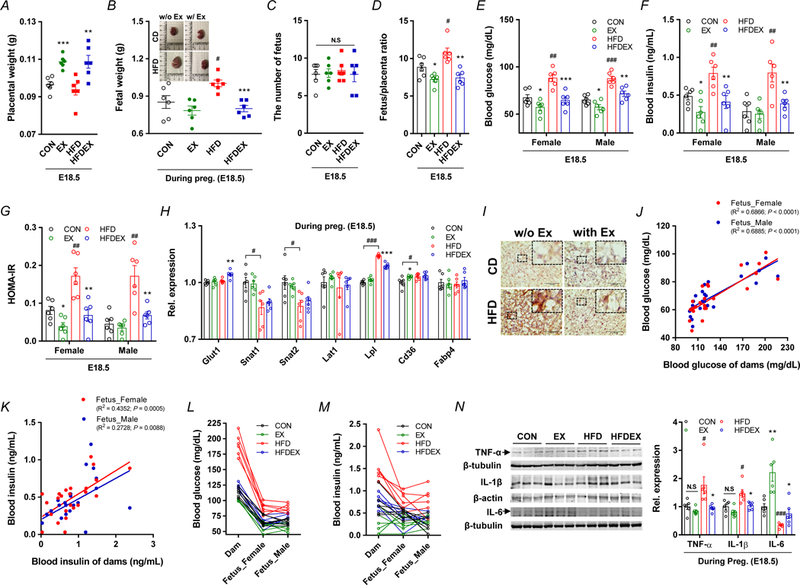
Maternal exercise training regulates placental efficiency and nutrient transport, and fetal metabolic parameters. A-D, Placental parameters of CON or HFD with/without exercise mice: placental weight (A), representative fetal images and weight (B), the number of fetuses per dam (C), and placental efficiency (D). E-G, Blood glucose (E), insulin (F), and insulin resistance (G) in E18.5 fetuses. H, mRNA levels of different nutrient transport markers in CON or HFD mice with/without exercise training. I, Representative images of Oil Red O staining in the placenta, Scale bar, 100 μm. J-M, Pearson correlations (J,K) and distributions (L,M) in the blood glucose and insulin levels between dams and fetuses. N, Cropped western blots of tumor necrosis factor α (TNF-α), interleukin-1β (IL-1β), and interleukin-6 (IL-6) in the placenta (β-tubulin or β-actin were used as the loading control, n = 6 per group). Data are expressed as the mean ± s.e.m. *P < 0.05, **P < 0.01, and ***P < 0.001 in CON vs. EX or HFD vs. HFDEX; #P < 0.05, ##P < 0.01, ###P < 0.001 in CON vs. HFD by two-tailed Student’s t-test (A-H, L) followed by multiple comparison and Pearson correlations analysis (J,K).
Exercise intervention reverses impaired placental vascularization in obese mice.
Placental vascularization is critical for its proper function (Ho et al., 2017). The area of junctional zone, which contains spongiotrophoblast cells, was decreased by HFD and the area of labyrinth zone was increased due to HFD, which were reversed by exercise training (Fig. 5A–D,I,J). The CD31, CD34, and eNOS are markers of endothelial cells (Hu et al., 2016; Lacko et al., 2017). Based on immunohistochemical CD34 and eNOS staining, and immunoblotting of CD31 of E18.5 placenta, the vascular density was reduced in HFD but recovered by exercise training (Fig. 5K–M). Recently, the APLN, also known as apelin, was identified as a potent mediator of placental vascularization (Ho et al., 2017), and the apelin content was decreased due to HFD but highly upregulated following exercise training (Fig. 5O). In agreement, factors regulating angiogenesis, including VEGF, VEGFR1, and hypoxia induced factor 1α (HIF1α) were dramatically increased by exercise training except CCN family member 1 (CCN1) which is an extracellular matrix-associated angiogenic inducer (Mo et al., 2002), and glial cells missing gene (GCM1) which is a chorion-specific transcription factor. (Fig. 5N,O).
Figure 5.
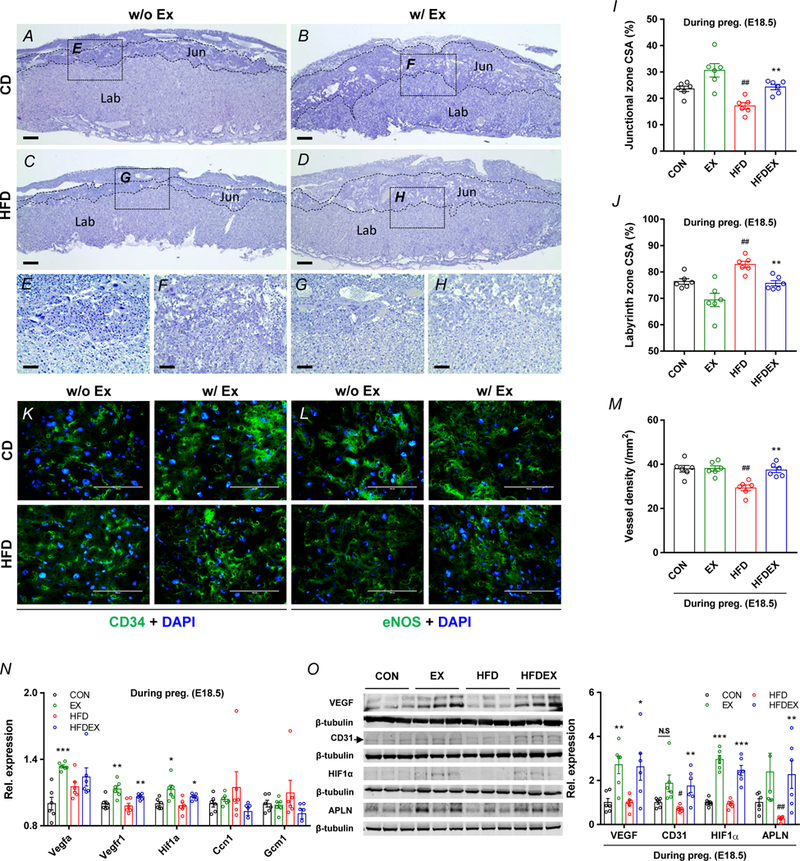
Maternal exercise training reverses impaired placental vascularization in HFD-mice. A-H,K,L, Representative images of hematoxylin and eosin (H&E), CD34 and eNOS immunocytochemistry (ICC) staining of placenta from CON or HFD mice with/without exercise, Scale bar, 500 μm (A-D), 100 μm (E-H,K,L), junctional zone (Jun) and labyrinth zone (Lab). I-J, Cross-sectional area (CSA) of junctional zone (I) and labyrinth zone (J) at E18.5 in placenta. M, Vessel density per mm2 in labyrinth zone of placenta. N, mRNA levels of different vasculogenic markers in CON or HFD mice with/without exercise. Expression was normalized by ΔCt values. O, cropped western blots of VEGF, CD31, and APLN in the placenta (β-tubulin was used as the loading control) from CON or HFD with/without exercise. Data are expressed as the mean ± s.e.m. n = 6. *P < 0.05, **P < 0.01, and ***P < 0.001 in CON vs. EX or HFD vs. HFDEX; ##P <0.01 in CON vs. HFD by two-tailed Student’s t-test followed by one-way ANOVA with post hoc Bonferroni multiple comparison analysis (I-O).
Maternal exercise training enhances AMPK, mTORC1, and insulin signaling in the placenta.
AMPK is a master regulator of cell metabolism activated by exercise (Niederberger et al., 2015), which is a critical mediator of remodeling in many tissues (Fu et al., 2016; Yang et al., 2016; Yao et al., 2017; Zhu et al., 2018). In addition, a negative correlation of placental AMPK phosphorylation in relation to body mass index (BMI) and birth weight was reported (Jansson et al., 2013). In this study, AMPK phosphorylation was downregulated in the placenta of obese mice, while exercise dramatically upregulated the phosphorylation of AMPK (Fig. 6C). On the other hand, growth factors, including insulin-like growth factor 2 (IGF-2) and fibroblast growth factor 2 (FGF2), stimulate mTORC1 signaling to enhance placental development; in addition, mTORC1 and AMPK function as nutrient and energy sensors, controlling protein synthesis (Kim et al., 2002; Hardie, 2014; Carroll & Dunlop, 2017). In the present study, the mRNA expression of growth factors, Fgf2 and Frfr2, was profoundly decreased in HFD group, but recovered by exercise (Fig. 6A). No differences in Igf2 and Igfbp1 mRNA expression were found. (Fig. 6A). As the downstream mediators of growth factor-initiated signaling, Akt phosphorylation was suppressed in the HFD placenta, but recovered by exercise (Fig. 6D). Consistently, phosphorylation of mTOR and 4E-BP1 was elevated in EX and HFD+EX mice, but no differences between CON and HFD mice were observed. (Fig. 6B). The impairment of IRS-1 and Erk phosphorylation in HFD mice was also reversed by exercise (Fig. 6D). Collectively, these data demonstrated that maternal exercise intervention activated AMPK and enhanced mTORC1 mediated growth signaling which were attenuated in response to maternal obesity.
Figure 6.
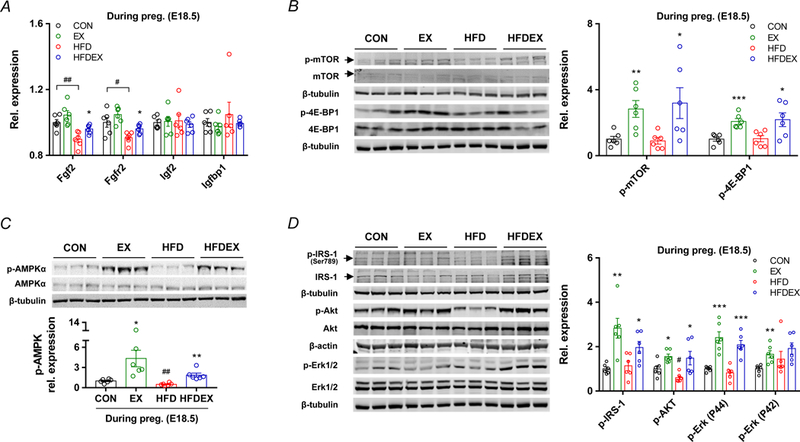
Maternal exercise training activates AMPK/mTORC1/insulin signaling in the placenta of HFD mice with exercise. A, mRNA levels of growth factors in CON or HFD mice with/without exercise mice (n = 6 per group). Expression was normalized by ΔCt values. B, cropped western blots of mTOR phosphorylation and 4E-BP1 phosphorylation in the placenta (the total mTOR or total 4E-BP1 were used as the reference). C, cropped western blots of AMPK phosphorylation in the placenta (the total AMPK was used as the reference). D, cropped western blots of IRS-1 phosphorylation, and Akt phosphorylation, and Erk½ phosphorylation in the placenta (the total IRS-1, total Akt, and total Erk½ were used as the references). Data are expressed as the mean ± s.e.m. n = 6. *P < 0.05, **P < 0.01, and ***P < 0.001 in CON vs. EX or HFD vs. HFDEX; #P < 0.05 and ##P < 0.001 in CON vs. HFD by two-tailed Student’s t-test (A-D).
Discussion
Through mediating nutrients, oxygen and waste exchange between mothers and their fetuses, adaptive remodeling in placenta has profound effects on fetal development. Maternal obesity and metabolic dysfunction is known to negatively affect fetal development, which has long-term metabolic impacts on their offspring (Zou et al., 2017). Placental function is altered by maternal obesity (Brett et al., 2014), triggering low efficient nutrient flux to the fetus and hampering fetal development independent with nutrient levels in the maternal side (Gaccioli et al., 2013). Exercise improves metabolic health of obese populations. Though the beneficial efforts of exercise in preventing the adverse effects of maternal obesity on fetal and placental development have been reported previously (Nathanielsz et al., 2013; Mangwiro et al., 2018a; Mangwiro et al., 2019), detailed mechanisms remain poorly defined. In this study, we found that maternal obesity impaired placental vasculogenesis/angiogenesis, which was reversed by maternal exercise training, preventing fetal overgrowth resulted from maternal obesity.
Placental nutrient transporters mediate cross-placental nutrient delivery. In the present study, we found that the expression of glutamine and amino acid transporters was downregulated, but fatty acid transporters were increased in HFD mice, consistent with a previous report (Nam et al., 2017). Nonetheless, the overall changes in the contents of nutrient transporters were quite small, suggesting that changes in nutrient transporter abundance might not explain observed changes in placental function. Consistently, in humans, maternal obesity elevates the umbilical vein glucose level and increases placental size, but not the abundance of nutrient transporters, suggesting placental expansion associated with obesity may explain neonatal macrosomia (Acosta et al., 2015).
In addition to the density of nutrient transporters, the placental vasculature system mediates blood circulation and thus is also critically important for nutrient transport. Poor placental vasculature due to HFD reduces oxygenation of the fetal tissues, resulting in poor neonatal survival (Hayes et al., 2012). Furthermore, chronic inflammation due to MO induces dysregulation of placental angiogenesis (Kim et al., 2018). On the other hand, exercise can suppress inflammation due to obesity (Antunes et al., 2018), which may promote placental angiogenesis (Reyes & Davenport, 2018). In addition, enlargement of junctional zone in placenta increases the spongiotrophoblast cells which is important for maintaining the endocrine function within the junctional zone (Coan et al., 2006; Burton & Fowden, 2012). Labyrinth zone, known as the fetal side in placenta and containing dense capillaries and syncytiotrophoblasts, is the site for nutrition and oxygen interaction between the mothers and her fetuses (Burton & Fowden, 2012). In the present study, the area of the labyrinth zone was increased but vessel density was reduced in HFD mice; on the other hand, exercise training reversed these adverse changes induced by MO. Consistently, exercise training increased while MO reduced eNOS content in labyrinth zone of HFD mice, which might partially explain placental dysfunction in HFD mice because eNOS deficiency blocks uteroplacental blood flow and nutrient transport (Cureton et al., 2017).
Recent studies increasingly implicate AMPK in regulating tissue development, including vascularization (Park et al., 2008). Interestingly, we observed that AMPK phosphorylation was suppressed due to maternal obesity, but there was a profound increase in AMPK phosphorylation in exercised mice, which suggests an AMPK-mediated mechanism regulating vasculogenesis and nutrient/oxygenic exchanges in placenta. Furthermore, the angiogenic markers including Vegfa and Vegfr, and a hypoxic marker, Hif1a, were elevated in exercised mice, which correlated with AMPK phosphorylation (Skeffington et al., 2016), suggesting a possible causal link between exercise-induced hypoxic/vasculogenic condition and AMPK activation (Skeffington et al., 2016). In alignment, AMPK knockdown inhibited placental nutrient transport function (Carey et al., 2014), and AMPK signaling is essential for hypoxia-induced angiogenesis in endothelial cells (Nagata et al., 2003). The effect of exercise on angiogenesis could also be mediated by apelin (Ho et al., 2017). Apelin has been considered as an exercise-induced hormone that can mediate angiogenesis in the skeletal muscle (Son et al., 2017). In addition, apelin promotes angiogenesis through activating AMPK activation in myocardial microvascular endothelial cells (Yang et al., 2014), and apelin is known to stimulate vasculogenesis in placenta (Kurowska et al., 2018). Consistently, our data showed increased AMPK phosphorylation and apelin protein level in the placenta following exercise training, demonstrating the possible role of apelin in mediating AMPK activation and placental angiogenesis stimulated by exercise.
The mTORC1-mediated signaling regulates protein synthesis, which interacts with AMPK to regulate Akt phosphorylation and protein synthesis (Inoki et al., 2012). In our study, we found that both AMPK and mTORC1 signaling was attenuated in HFD placenta, which was reversed by exercise, suggesting that maternal exercise training might prevent fetal overgrowth in obese mice via normalizing placental AMPK and mTORC1 signaling. The down-regulation of mTORC1 in HFD placenta was consistent with the previous observations that obesity and associated insulin resistance inhibit mTORC1 signaling (Ost et al., 2010; Kleinert et al., 2014). However, in a previous study, activation of placental mTORC1 signaling was observed in obese women (Jansson et al., 2013). The discrepancy in mTORC1 signaling due to obesity might be explained by the presence of insulin resistance or chronic inflammation (Jiang et al., 2014; Kleinert et al., 2014). Indeed, in insulin resistant mice, Akt and mTORC1 phosphorylation is suppressed (Li et al., 2018) and exercise can stimulate phosphorylation of Akt (Kido et al., 2017). In agreement, maternal obesity in our mouse model might be associated with insulin resistance, which was alleviated by exercise. To be conclusive, however, insulin stimulation could be applied, which might directly assess the changes in insulin sensitivity.
Finally, in order to mimic the situation in obese mothers, in this study, we induced female obese first and then mated. Therefore, maternal obesity should be the major factor inducing changes in placental function, but also includes the effect of HFD, mimicking the situation of obese mothers in humans. In this study, we only measured the vascular density based on the expression of markers and immunohistochemical staining, which showed up-regulation of vasculogenesis in placenta of obese mothers with exercise. It will be better if the placental transport function could be directly measured.
In summary, our data show that, for the first time to our knowledge, maternal exercise enhanced placental vasculogenesis and prevents fetal overgrowth, which might be mediated by activation of AMPK, mTORC1, and insulin signaling in the placenta, all of which were suppressed by maternal obesity and HFD intake. Collectively, these data support the compelling possibility that AMPK might be a key factor mediating placental function altered by obesity and exercise training.
Figure 7.
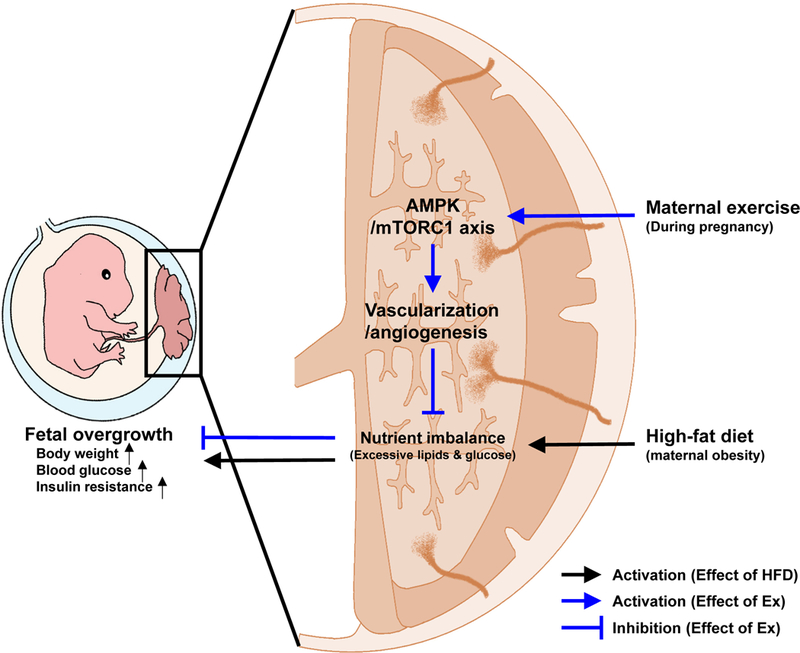
Potential schematic mechanisms of maternal exercise to prevent fetal overgrowth due to high-fat diet-induced obesity in mothers.
Key points summary.
Maternal exercise improves metabolic health of maternal mice challenged with high-fat diet.
Exercise intervention of obese mothers prevents fetal overgrowth.
Exercise intervention reverses impaired placental vascularization in obese mice.
Maternal exercise activates placental AMP-activated protein kinase, which was inhibited due to maternal obesity.
Acknowledgements
This work was funded by the National Institutes of Health (NIH R01-HD067449).
Abbreviations
- MO
maternal obesity
- HFD
high-fat diet
- mTORC1
mammalian target of rapamycin complex 1
- AMPK
AMP-activated protein kinase
- VO2max
maximal oxygen consumption rate
- VO2
oxygen consumption rate
- VCO2
carbon dioxide production
- RER
respiratory exchange ratio
- ELISA
enzyme-linked immunosorbent assay
- HOMA-IR
homeostatic model assessment of insulin resistance
- HOMA-%B
homeostatic model assessment of pancreatic β-cell function
- H&E
hematoxylin and eosin
- ICC
immunocytochemical
- ingWAT
inguinal white adipose tissue
- gonWAT
gonadal white adipose tissue
- iBAT
intrascapular brown adipose tissue
- Lpl
lipoprotein lipase; IRS-1, insulin receptor substrate 1
- TNF-α
tumor necrosis factor α; IL-1β, interleukin-1β
- IL-6
interleukin-6
- VEGF
vascular endothelial growth factor
- APLN
endogenous ligand for the G-protein coupled APJ receptor
- VEGFR1
VEGF receptor 1
- HIF1
hypoxia induced factor 1
- eNOS
endothelial nitric oxide synthase
- CCN1
CCN family member 1
- GCM1
glial cells missing gene
- IGF-2
insulin-like growth factor 2
- FGF2
fibroblast growth factor 2
Footnotes
Conflict of interest: The authors declare no competing financial interests.
References
- Acosta O, Ramirez VI, Lager S, Gaccioli F, Dudley DJ, Powell TL & Jansson T. (2015). Increased glucose and placental GLUT-1 in large infants of obese nondiabetic mothers. Am J Obstet Gynecol 212, 227.e221–227. [DOI] [PubMed] [Google Scholar]
- Antunes BM, Campos EZ, Dos Santos RVT, Rosa-Neto JC, Franchini E, Bishop NC & Lira FS. (2018). Anti-inflammatory response to acute exercise is related with intensity and physical fitness. J Cell Biochem. [DOI] [PubMed] [Google Scholar]
- Aye IL, Rosario FJ, Powell TL & Jansson T. (2015). Adiponectin supplementation in pregnant mice prevents the adverse effects of maternal obesity on placental function and fetal growth. Proc Natl Acad Sci U S A 112, 12858–12863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bairagi S, Quinn KE, Crane AR, Ashley RL, Borowicz PP, Caton JS, Redden RR, Grazul-Bilska AT & Reynolds LP. (2016). Maternal environment and placental vascularization in small ruminants. Theriogenology 86, 288–305. [DOI] [PubMed] [Google Scholar]
- Barbour LA & Hernandez TL. (2018). Maternal Lipids and Fetal Overgrowth: Making Fat from Fat. Clin Ther 40, 1638–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brett KE, Ferraro ZM, Yockell-Lelievre J, Gruslin A & Adamo KB. (2014). Maternal-fetal nutrient transport in pregnancy pathologies: the role of the placenta. Int J Mol Sci 15, 16153–16185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton GJ & Fowden AL. (2012). Review: The placenta and developmental programming: balancing fetal nutrient demands with maternal resource allocation. Placenta 33 Suppl, S23–27. [DOI] [PubMed] [Google Scholar]
- Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J & Wittwer CT. (2009). The MIQE guidelines: minimum information for publication of quantitative real-time PCR. Clin Chem 55, 611–622. [DOI] [PubMed] [Google Scholar]
- Carey EA, Albers RE, Doliboa SR, Hughes M, Wyatt CN, Natale DR & Brown TL. (2014). AMPK knockdown in placental trophoblast cells results in altered morphology and function. Stem Cells Dev 23, 2921–2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll B & Dunlop EA. (2017). The lysosome: a crucial hub for AMPK and mTORC1 signalling. Biochem J 474, 1453–1466. [DOI] [PubMed] [Google Scholar]
- Carter AM. (2000). Placental oxygen consumption. Part I: in vivo studies--a review. Placenta 21 Suppl A, S31–37. [DOI] [PubMed] [Google Scholar]
- Coan PM, Conroy N, Burton GJ & Ferguson-Smith AC. (2006). Origin and characteristics of glycogen cells in the developing murine placenta. Dev Dyn 235, 3280–3294. [DOI] [PubMed] [Google Scholar]
- Cureton N, Korotkova I, Baker B, Greenwood S, Wareing M, Kotamraju VR, Teesalu T, Cellesi F, Tirelli N, Ruoslahti E, Aplin JD & Harris LK. (2017). Selective Targeting of a Novel Vasodilator to the Uterine Vasculature to Treat Impaired Uteroplacental Perfusion in Pregnancy. Theranostics 7, 3715–3731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson H, Moss TJ, Gatford KL, Moritz KM, Akison L, Fullston T, Hryciw DH, Maloney CA, Morris MJ, Wooldridge AL, Schjenken JE, Robertson SA, Waddell BJ, Mark PJ, Wyrwoll CS, Ellery SJ, Thornburg KL, Muhlhausler BS & Morrison JL. (2016). A review of fundamental principles for animal models of DOHaD research: an Australian perspective. J Dev Orig Health Dis 7, 449–472. [DOI] [PubMed] [Google Scholar]
- Fu X, Zhu M, Zhang S, Foretz M, Viollet B & Du M. (2016). Obesity Impairs Skeletal Muscle Regeneration Through Inhibition of AMPK. Diabetes 65, 188–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaccioli F, Lager S, Powell TL & Jansson T. (2013). Placental transport in response to altered maternal nutrition. J Dev Orig Health Dis 4, 101–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghasemi A, Tohidi M, Derakhshan A, Hasheminia M, Azizi F & Hadaegh F. (2015). Cut-off points of homeostasis model assessment of insulin resistance, beta-cell function, and fasting serum insulin to identify future type 2 diabetes: Tehran Lipid and Glucose Study. Acta Diabetol 52, 905–915. [DOI] [PubMed] [Google Scholar]
- Gilbert JS. (2017). From apelin to exercise: emerging therapies for management of hypertension in pregnancy. Hypertens Res 40, 519–525. [DOI] [PubMed] [Google Scholar]
- Gregg VH & Ferguson JE 2nd. (2017). Exercise in Pregnancy. Clin Sports Med 36, 741–752. [DOI] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology. J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG. (2014). AMPK--sensing energy while talking to other signaling pathways. Cell Metab 20, 939–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes EK, Lechowicz A, Petrik JJ, Storozhuk Y, Paez-Parent S, Dai Q, Samjoo IA, Mansell M, Gruslin A, Holloway AC & Raha S. (2012). Adverse fetal and neonatal outcomes associated with a life-long high fat diet: role of altered development of the placental vasculature. PLoS One 7, e33370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho L, van Dijk M, Chye STJ, Messerschmidt DM, Chng SC, Ong S, Yi LK, Boussata S, Goh GH, Afink GB, Lim CY, Dunn NR, Solter D, Knowles BB & Reversade B. (2017). ELABELA deficiency promotes preeclampsia and cardiovascular malformations in mice. Science 357, 707–713. [DOI] [PubMed] [Google Scholar]
- Howell KR & Powell TL. (2017). Effects of maternal obesity on placental function and fetal development. Reproduction 153, R97–r108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hruby A, Manson JE, Qi L, Malik VS, Rimm EB, Sun Q, Willett WC & Hu FB. (2016). Determinants and Consequences of Obesity. Am J Public Health 106, 1656–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Li J, Zhang Q, Zheng L, Wang G, Zhang X, Zhang J, Gu Q, Ye Y, Guo SW, Yang X & Wang L. (2016). Phosphoinositide 3-Kinase (PI3K) Subunit p110delta Is Essential for Trophoblast Cell Differentiation and Placental Development in Mouse. Sci Rep 6, 28201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K, Kim J & Guan KL. (2012). AMPK and mTOR in cellular energy homeostasis and drug targets. Annu Rev Pharmacol Toxicol 52, 381–400. [DOI] [PubMed] [Google Scholar]
- Jansson N, Rosario FJ, Gaccioli F, Lager S, Jones HN, Roos S, Jansson T & Powell TL. (2013). Activation of placental mTOR signaling and amino acid transporters in obese women giving birth to large babies. J Clin Endocrinol Metab 98, 105–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Westerterp M, Wang C, Zhu Y & Ai D. (2014). Macrophage mTORC1 disruption reduces inflammation and insulin resistance in obese mice. Diabetologia 57, 2393–2404. [DOI] [PubMed] [Google Scholar]
- Jones CT & Rolph TP. (1985). Metabolism during fetal life: a functional assessment of metabolic development. Physiol Rev 65, 357–430. [DOI] [PubMed] [Google Scholar]
- Jones HN, Woollett LA, Barbour N, Prasad PD, Powell TL & Jansson T. (2009). High-fat diet before and during pregnancy causes marked up-regulation of placental nutrient transport and fetal overgrowth in C57/BL6 mice. FASEB J 23, 271–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman MR & Brown TL. (2016). AMPK and Placental Progenitor Cells. Exp Suppl 107, 73–79. [DOI] [PubMed] [Google Scholar]
- Kerr J, Anderson C & Lippman SM. (2017). Physical activity, sedentary behaviour, diet, and cancer: an update and emerging new evidence. Lancet Oncol 18, e457–e471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kido K, Yokokawa T, Ato S, Sato K & Fujita S. (2017). Effect of resistance exercise under conditions of reduced blood insulin on AMPKalpha Ser485/491 inhibitory phosphorylation and AMPK pathway activation. Am J Physiol Regul Integr Comp Physiol 313, R110–r119. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne WJ, Cuthill IC, Emerson M & Altman DG. (2012). Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. Osteoarthritis Cartilage 20, 256–260. [DOI] [PubMed] [Google Scholar]
- Kim CY, Jung E, Kim EN, Kim CJ, Lee JY, Hwang JH, Song WS, Lee BS, Kim EA & Kim KS. (2018). Chronic Placental Inflammation as a Risk Factor of Severe Retinopathy of Prematurity. J Pathol Transl Med 52, 290–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P & Sabatini DM. (2002). mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 110, 163–175. [DOI] [PubMed] [Google Scholar]
- Kleinert M, Sylow L, Fazakerley DJ, Krycer JR, Thomas KC, Oxboll AJ, Jordy AB, Jensen TE, Yang G, Schjerling P, Kiens B, James DE, Ruegg MA & Richter EA. (2014). Acute mTOR inhibition induces insulin resistance and alters substrate utilization in vivo. Mol Metab 3, 630–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurowska P, Barbe A, Rozycka M, Chmielinska J, Dupont J & Rak A. (2018). Apelin in Reproductive Physiology and Pathology of Different Species: A Critical Review. Int J Endocrinol 2018, 9170480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacko LA, Hurtado R, Hinds S, Poulos MG, Butler JM & Stuhlmann H. (2017). Altered feto-placental vascularization, feto-placental malperfusion and fetal growth restriction in mice with Egfl7 loss of function. Development 144, 2469–2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leiva AM, Martinez MA, Cristi-Montero C, Salas C, Ramirez-Campillo R, Diaz Martinez X, Aguilar-Farias N & Celis-Morales C. (2017). [Sedentary lifestyle is associated with metabolic and cardiovascular risk factors independent of physical activity]. Rev Med Chil 145, 458–467. [DOI] [PubMed] [Google Scholar]
- Lemaitre RN, Yu C, Hoofnagle A, Hari N, Jensen PN, Fretts AM, Umans JG, Howard BV, Sitlani CM, Siscovick DS, King IB, Sotoodehnia N & McKnight B. (2018). Circulating Sphingolipids, Insulin, HOMA-IR, and HOMA-B: The Strong Heart Family Study. Diabetes 67, 1663–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Yu L & Zhao C. (2018). Dioscin attenuates highfat dietinduced insulin resistance of adipose tissue through the IRS1/PI3K/Akt signaling pathway. Mol Med Rep. [DOI] [PubMed] [Google Scholar]
- Louwagie EJ, Larsen TD, Wachal AL & Baack ML. (2018). Placental lipid processing in response to a maternal high-fat diet and diabetes in rats. Pediatr Res 83, 712–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangwiro YT, Briffa JF, Gravina S, Mahizir D, Anevska K, Romano T, Moritz KM, Cuffe JS & Wlodek ME. (2018a). Maternal exercise and growth restriction in rats alters placental angiogenic factors and blood space area in a sex-specific manner. Placenta 74, 47–54. [DOI] [PubMed] [Google Scholar]
- Mangwiro YT, Cuffe JS, Mahizir D, Anevska K, Gravina S, Romano T, Moritz KM, Briffa JF & Wlodek ME. (2019). Exercise initiated during pregnancy in rats born growth restricted alters placental mTOR and nutrient transporter expression. J Physiol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangwiro YTM, Cuffe JSM, Briffa JF, Mahizir D, Anevska K, Jefferies AJ, Hosseini S, Romano T, Moritz KM & Wlodek ME. (2018b). Maternal exercise in rats upregulates the placental insulin-like growth factor system with diet- and sex-specific responses: minimal effects in mothers born growth restricted. J Physiol 596, 5947–5964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF & Turner RC. (1985). Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 28, 412–419. [DOI] [PubMed] [Google Scholar]
- McCurdy CE, Bishop JM, Williams SM, Grayson BE, Smith MS, Friedman JE & Grove KL. (2009). Maternal high-fat diet triggers lipotoxicity in the fetal livers of nonhuman primates. J Clin Invest 119, 323–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo FE, Muntean AG, Chen CC, Stolz DB, Watkins SC & Lau LF. (2002). CYR61 (CCN1) is essential for placental development and vascular integrity. Mol Cell Biol 22, 8709–8720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata D, Mogi M & Walsh K. (2003). AMP-activated protein kinase (AMPK) signaling in endothelial cells is essential for angiogenesis in response to hypoxic stress. J Biol Chem 278, 31000–31006. [DOI] [PubMed] [Google Scholar]
- Nam J, Greenwald E, Jack-Roberts C, Ajeeb TT, Malysheva OV, Caudill MA, Axen K, Saxena A, Semernina E, Nanobashvili K & Jiang X. (2017). Choline prevents fetal overgrowth and normalizes placental fatty acid and glucose metabolism in a mouse model of maternal obesity. J Nutr Biochem 49, 80–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathanielsz PW, Ford SP, Long NM, Vega CC, Reyes-Castro LA & Zambrano E. (2013). Interventions to prevent adverse fetal programming due to maternal obesity during pregnancy. Nutr Rev 71 Suppl 1, S78–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niederberger E, King TS, Russe OQ & Geisslinger G. (2015). Activation of AMPK and its Impact on Exercise Capacity. Sports Med 45, 1497–1509. [DOI] [PubMed] [Google Scholar]
- Ogasawara R, Sato K, Matsutani K, Nakazato K & Fujita S. (2014). The order of concurrent endurance and resistance exercise modifies mTOR signaling and protein synthesis in rat skeletal muscle. Am J Physiol Endocrinol Metab 306, E1155–1162. [DOI] [PubMed] [Google Scholar]
- Ogden CL, Carroll MD, Kit BK & Flegal KM. (2012). Prevalence of obesity in the United States, 2009–2010. NCHS Data Brief, 1–8. [PubMed] [Google Scholar]
- Ost A, Svensson K, Ruishalme I, Brannmark C, Franck N, Krook H, Sandstrom P, Kjolhede P & Stralfors P. (2010). Attenuated mTOR signaling and enhanced autophagy in adipocytes from obese patients with type 2 diabetes. Mol Med 16, 235–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park M, Lyons J 3rd, Oh H, Yu Y, Woltering EA, Greenway F & York DA. (2008). Enterostatin inhibition of angiogenesis: possible role of pAMPK and vascular endothelial growth factor A (VEGF-A). Int J Obes (Lond) 32, 922–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peronnet F & Masicotte D. (1991). Table of nonprotein respiratory quotient: an update. Can J Sport Sci 16, 23–29. [PubMed] [Google Scholar]
- Petersen AM & Pedersen BK. (2005). The anti-inflammatory effect of exercise. J Appl Physiol (1985) 98, 1154–1162. [DOI] [PubMed] [Google Scholar]
- Petrosino JM, Heiss VJ, Maurya SK, Kalyanasundaram A, Periasamy M, LaFountain RA, Wilson JM, Simonetti OP & Ziouzenkova O. (2016). Graded Maximal Exercise Testing to Assess Mouse Cardio-Metabolic Phenotypes. PLoS One 11, e0148010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes LM & Davenport MH. (2018). Exercise as a therapeutic intervention to optimize fetal weight. Pharmacol Res 132, 160–167. [DOI] [PubMed] [Google Scholar]
- Ringseis R, Eder K, Mooren FC & Kruger K. (2015). Metabolic signals and innate immune activation in obesity and exercise. Exerc Immunol Rev 21, 58–68. [PubMed] [Google Scholar]
- Romijn JA, Coyle EF, Sidossis LS, Gastaldelli A, Horowitz JF, Endert E & Wolfe RR. (1993). Regulation of endogenous fat and carbohydrate metabolism in relation to exercise intensity and duration. Am J Physiol 265, E380–E391. [DOI] [PubMed] [Google Scholar]
- Schrauwen P, Wagenmakers AJ, van Marken Lichtenbelt WD, Saris WH & Westerterp KR. (2000). Increase in fat oxidation on a high-fat diet is accompanied by an increase in triglyceride-derived fatty acid oxidation. Diabetes 49, 640–646. [DOI] [PubMed] [Google Scholar]
- Skeffington KL, Higgins JS, Mahmoud AD, Evans AM, Sferruzzi-Perri AN, Fowden AL, Yung HW, Burton GJ, Giussani DA & Moore LG. (2016). Hypoxia, AMPK activation and uterine artery vasoreactivity. J Physiol 594, 1357–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son JS, Kim HJ, Son Y, Lee H, Chae SA, Seong JK & Song W. (2017). Effects of exercise-induced apelin levels on skeletal muscle and their capillarization in type 2 diabetic rats. Muscle Nerve 56, 1155–1163. [DOI] [PubMed] [Google Scholar]
- Stanford KI, Takahashi H, So K, Alves-Wagner AB, Prince NB, Lehnig AC, Getchell KM, Lee MY, Hirshman MF & Goodyear LJ. (2017). Maternal Exercise Improves Glucose Tolerance in Female Offspring. Diabetes 66, 2124–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart TJ, O’Neill K, Condon D, Sasson I, Sen P, Xia Y & Simmons RA. (2018). Diet-induced obesity alters the maternal metabolome and early placenta transcriptome and decreases placenta vascularity in the mouse. Biol Reproduction 98, 795–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt MC, Paeger L, Hess S, Steculorum SM, Awazawa M, Hampel B, Neupert S, Nicholls HT, Mauer J, Hausen AC, Predel R, Kloppenburg P, Horvath TL & Bruning JC. (2014). Neonatal insulin action impairs hypothalamic neurocircuit formation in response to maternal high-fat feeding. Cell 156, 495–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Wang Z, de Avila JM, Zhu MJ, Zhang F, Gomez NA, Zhao L, Tian Q, Zhao J, Maricelli J, Zhang H, Rodgers BD & Du M. (2017). Moderate alcohol intake induces thermogenic brown/beige adipocyte formation via elevating retinoic acid signaling. FASEB J 31, 4612–4622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q, Liang X, Sun X, Zhang L, Fu X, Rogers CJ, Berim A, Zhang S, Wang S, Wang B, Foretz M, Viollet B, Gang DR, Rodgers BD, Zhu MJ & Du M. (2016). AMPK/alpha-Ketoglutarate Axis Dynamically Mediates DNA Demethylation in the Prdm16 Promoter and Brown Adipogenesis. Cell Metab 24, 542–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Zhu W, Zhang P, Chen K, Zhao L, Li J, Wei M & Liu M. (2014). Apelin-13 stimulates angiogenesis by promoting crosstalk between AMP-activated protein kinase and Akt signaling in myocardial microvascular endothelial cells. Mol Med Rep 9, 1590–1596. [DOI] [PubMed] [Google Scholar]
- Yao G, Zhang Y, Wang D, Yang R, Sang H, Han L, Zhu Y, Lu Y, Tan Y & Shang Z. (2017). GDM-Induced Macrosomia Is Reversed by Cav-1 via AMPK-Mediated Fatty Acid Transport and GLUT1- Mediated Glucose Transport in Placenta. PLoS One 12, e0170490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zavorsky GS & Longo LD. (2011). Exercise guidelines in pregnancy: new perspectives. Sports Med 41, 345–360. [DOI] [PubMed] [Google Scholar]
- Zhu MJ, Sun X & Du M. (2018). AMPK in regulation of apical junctions and barrier function of intestinal epithelium. Tissue Barriers 6, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou T, Chen D, Yang Q, Wang B, Zhu MJ, Nathanielsz PW & Du M. (2017). Resveratrol supplementation of high-fat diet-fed pregnant mice promotes brown and beige adipocyte development and prevents obesity in male offspring. J Physiol 595, 1547–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]