

Abstract
Background:
A mouse with hepatocyte-specific deiodinase type II (Dio2) inactivation (Alb-D2KO) is resistant to diet-induced obesity, hepatic steatosis and hypertriglyceridemia due to perinatal epigenetic modifications in the liver. This phenotype is linked to low levels of Zfp125, a hepatic transcriptional repressor that promotes liver steatosis by inhibiting genes involved in packaging and secretion of VLDL.
Methods:
Here we used chronic and binge ethanol (ETOH) in mice to cause liver steatosis.
Results:
The ETOH treatment causes a 2.3-fold increase in hepatic triglyceride content; Zfp125 levels were approximately 50% higher in these animals. In contrast, Alb-D2KO mice did not develop ETOH-induced liver steatosis. They also failed to elevate Zfp125 to the same levels, despite being on the ETOH-containing diet for the same period of time. Their phenotype was associated with 1.3–2.9-fold upregulation of hepatic genes involved in lipid transport and export that are normally repressed by Zfp125, i.e. Mttp, Abca1, Ldlr, Apoc1, Apoc3, Apoe, Apoh and Azgp1. Furthermore, genes involved in the ETOH metabolic pathway, i.e. Aldh2 and Acss2, were also 1.6–3.1-fold up regulated in Alb-D2KO-ETOH mice compared with control animals kept on ETOH.
Conclusions:
ETOH consumption elevates expression of Zfp125. Alb-D2KO animals, which have lower levels of Zfp125, are much less susceptible to ETOH-induced liver steatosis.
Keywords: deiodinase, thyroid hormone, lipogenesis, liver, steatosis, ethanol
Introduction
Alcoholism is a leading morbidity risk factor for individuals between 15–50 years of age (Lim et al., 2012). Liver steatosis, or accumulation of fat in the liver, is the earliest response to excessive ethanol (ETOH) consumption (Kondili et al., 2005), constituting an important component of alcoholic liver disease (Gao and Bataller, 2011). It can occur as macrovesicular or more rarely microvesicular fat deposition; it initiates and is most intense in the central perivenular area but it may spread further and encompass the total surface of a lobule (Crawford, 2012).
It is thought that ETOH abuse causes liver steatosis by disrupting the balance between lipogenesis and lipid droplet catabolism (lipophagy), and subsequent fatty acid oxidation. The metabolism of ETOH via alcohol dehydrogenase (ADH) to form acetaldehyde, which is subsequently oxidized by via aldehyde dehydrogenase (ALDH) (Seitz and Stickel, 2010), results in a relative excess of NADH, which may impair gluconeogenesis and slow down fatty-acid oxidation (Lieber, 1995). ETOH metabolism also blocks PPARα-mediated transcriptional activation of pathways leading to fatty acid oxidation (Galli et al., 2001). At the same time, ETOH oxidation excess activates the nuclear transcription factor SREBP-1, inducing a series of lipogenic enzymes (Lieber et al., 1966), e.g. α-glycerophosphate acyltransferase (GPAT), fatty acid synthase (FAS) and malic enzyme (ME) (Carrasco et al., 2001, Joly et al., 1973). ETOH excess also reduces glycerol 3-phosphate dehydrogenase activity (GPDH), impairing secretion of very low-density lipoprotein (VLDL) particles, all contributing factors to hepatic steatosis (Siler et al., 1998). Besides activating fatty acid synthesis and inhibiting oxidation, ETOH oxidation also slows down very low-density lipoprotein (VLDL) secretion from the liver (Rasineni and Casey, 2012).
Recently, a new mouse model that is resistant to liver steatosis caused by consumption of a high-fat diet (HFD) was described, which could also shed light into ETOH-induced liver steatosis. This mouse model is based on the inactivation of a liver enzyme that converts the prohormone thyroxine (T4) to T3 (Gereben et al., 2008). This enzyme, the type 2 deiodinase (D2), is only briefly expressed in the liver in the perinatal period but it defines future susceptibility to diet-induced hepatic steatosis through modification of the hepatic DNA methylation profile (Fonseca et al., 2015). In adult mice with liver-specific Dio2 inactivation (Alb-D2KO) there are about 3,500 differentially methylated regions (DMRs) that modify the liver transcriptome and its response to feeding on HFD. Three positive DMRs are located in the FoxO1 gene, reducing its expression by half (Fonseca et al., 2015). As a result, the expression level of the FoxoO1-inducible gene Zfp125 is also reduced by about 60%. Zfp125 is a transcriptional repressor that reduces the expression of 18 genes involved in intracellular lipid transport, structure and assembly of VLDL particles. In mice, Zfp125 expression reduces secretion of triglycerides and cholesterol efflux, causing lipid accumulation and liver steatosis (Fernandes et al., 2018).
In the present investigation we utilized the chronic–plus–binge model (Bertola et al., 2013) to test whether the Alb-D2KO mouse model is protected against ETOH-induced liver steatosis. The results show that Alb-D2KO mice do not develop liver steatosis when subjected to chronic ETOH consumption. This is associated with activation of hepatic genes that are involved in lipid export and are normally repressed by Zfp125.
Experimental Procedures
Animals:
Studies were approved by the local Institutional Animal Care and Use Committee. A mouse with hepatocyte-specific Dio2 inactivation (Alb-D2KO) was obtained by crossing the floxed D2 mouse (dio2Flx) (Fonseca et al., 2015) with a mouse expressing Cre-recombinase under the control of the albumin promoter (Cre-Alb) [B6.Cg-Tg(Alb-cre)21Mgn/J; Jackson Laboratories, Bar Harbor ME] (Postic et al., 1999). Two to three male mice with 8–9 weeks old were housed in plastic cages kept at room temperature (22°C), with a 12-h dark/light cycle. As indicated, some animals underwent a protocol of chronic/binge ethanol feeding described by Gao and collaborators (Bertola et al., 2013). Mice were initially fed control Lieber-DeCarli diet (Bio-Serv, Frenchtown, NJ) ad libitum for 5 days (from day 1 through 5) to become acclimated to a liquid diet. Subsequently, mice were allowed free access to the ethanol Lieber-DeCarli diet (Bio-Serv) containing 5% (vol/vol) ethanol for 10 days (from day 6 through 15); control groups were pair-fed an isocaloric control diet. On the early morning of day 16, ETOH-fed and control mice received a single dose of ETOH (5 g/kg body weight) or isocaloric dextrin-maltose by gavage and killed 9 h later by asphyxiation in a CO2 chamber. Immediately after killing, liver fragments were snap frozen in liquid nitrogen and stored at −80C for protein and mRNA analysis. The right medium lobes of the liver were processed for hematoxylin-eosin (H&E) staining or preserved in O.C.T. solution for oil red-O lipid staining. Two liver sections were analyzed for each animal.
Gene expression analysis:
Total RNA was extracted using the RNeasy kit (Qiagen, Valencia, CA) and quantified with a Nano-Drop spectrophotometer. 100 ng of total RNA were used for cDNA synthesis with the First Strand cDNA Synthesis Kit for RT-PCR (Roche). Genes of interest were assessed by RT-qPCR (StepOnePlus real time PCR system, Applied Bioscience) using SYBR Green Supermix (Quanta Biosciences). Standard curves consisting of 4–5 points of serially diluted mixed experimental and control group cDNA were included and the coefficient of correlation was consistently >0.98, with an amplification efficiency of 80–110%. The primers used are listed in Table S2, with 18S as internal control. Amplicon specificity was assessed through the melting curve.
Western Blot Analysis:
Liver sonicates were obtained as described previously (Fernandes et al., 2018), mixed with 4X sample loading buffer (Invitrogen), and 20–50 ug protein fractionated in 4–12% NuPAGE BisTris gels (Life Technologies). Samples were transferred to Immobilon-FL PVDF transfer membrane (Millipore) and probed overnight with antibodies, as indicated, at a 1:2,500 dilution. Fluorescent-labeled secondary antibodies (LI-COR Biosciences) were used at 1:2,500 for 1 hr. Blots were imaged and quantified using the LI-COR Odyssey instrument.
Biochemical analyses:
Individual plasma samples were processed for alanine transaminase activity (ALT) and glutamic oxaloacetic transaminase activity (GOT1) using a colorimetric assay (Abcam, Ref. ab105134 and Ref. ab 105135, Cambridge, UK). And also Individual plasma samples were processed for aspartate transaminase 1 (GOT1), liver-type arginase 1 (ARG) and sorbitol dehydrogenase (SDH) using a MILLIPLEX™ Rat Liver Injury Panel (Millipore Corporation, Billerica MA) and read on a Magpix (Millipore Sigma, MA). Liver cholesterol and plasma high-density lipoprotein (HDL) and low-density lipoprotein (LDL)/very-LDL (VLDL) cholesterol were measured using a colorimetric assay (Abcam, Ref. ab65390, Cambridge, UK). Liver triglyceride content was measured in ~200 mg liver fragments after homogenization and extraction with chloroform/methanol (2:1) and 0.05% sulfuric acid (Castillo et al., 2011). ETOH levels were measured using gas chromatography (Eriksson et al., 1977) with the following modifications: 50 uL plasma were diluted with ice-cold distilled water to a final volume of 300 uL. Next, one ml of 6.7% perchloric acid, containing 27.8 mM thiourea and 1.1 mM 2- propyl alcohol (propanol) was added to the mixture to precipitate proteins. Samples were mixed and centrifuged at 8,000 g for 5 min to sediment insoluble proteins. One ml of each supernatant was removed and placed into an airtight gas chromatography vial. The vapor phase from each sample was then processed by gas chromatography to detect and quantify acetaldehyde and ETOH. Appropriate ETOH and acetaldehyde standards identically prepared were used to quantify these compounds.
Statistical analysis:
All data were analyzed by PRISM software (GraphPad). Unless otherwise indicated, data are presented as values ± SEM (Table) or as box and whiskers plots (Figures); the Mann-Whitney U test was used when the experiment contained two independent groups; one-way ANOVA was used to compare more than two groups, followed by the Tukey test to detect differences between groups. p< 0.05 was used to reject the null hypothesis.
Results
Alb-D2KO mice are resistant to ETOH-induced liver steatosis
To test the susceptibility of Alb-D2KO mice to ETOH, all animals underwent a 10-day period of feeding on a liquid diet containing 5% (vol/vol) ETOH, preceded by a 5-day acclimation period during which the animals were fed with a liquid diet free of ETOH. In all experiments, controls included cre- and Alb-D2KO animals kept on ETOH-free pair-feeding regimen.
Selective Dio2 inactivation in the liver does not affect growth rate or the weight of brain, fat pads and liver (Fonseca et al., 2015). During the acclimation period the consumption of liquid diet was similar among all groups (Fig. 1A), and animals continued to gain weight (Table S1). Switching to ETOH-containing liquid diet did not affect dietary intake (Fig. 1A), and resulted in moderate (less than legally intoxicating) ETOH plasma levels, which were not affected by liver Dio2 inactivation (Fig. 1B). The dietary switch to ETOH also caused both cre- and Alb-D2KO mice to halt body weight gain, while pair-fed controls continued to gain weight (Fig. 1C). The fact that brain weight gain was preserved in all animals, regardless of genotype and diet, indicate that growth was not affected by ETOH feeding (Fig. 1D). In contrast, ETOH feeding reduced the size of the epididymal fat pad by approximately 40% in all animals (Fig. 1E). Liver weight was preserved in cre-mice switched to ETOH whereas in the Alb-D2KO mice it dropped slightly (Fig. 1D).
Fig. 1 – Morphometric parameters of Alb-D2KO and Cre mice treated with ETOH.

(A) daily food intake; (B) plasma ethanol levels at the end of experimental period; (C) delta body weight between day 16 and day 6; (D) liver, (E) brain and (F) epididymal fat pad ratio correct by the initial BW in Cre-ETOH and Alb-D2KO-ETOH mice; the gray area represents mean ± SD for the control group; values are shown as mean ± SEM or in a box and whiskers plot indicating median and quartiles; n=3–10/group; * p≤0.05 and **p< 0.01 vs Cre-ETOH; # p≤0.05 vs Cre-Control (Student’s t-test or one-way ANOVA).
Analyses of liver sections revealed minimal lipid deposit and inflammation, and no ballooning in all control animals (Fig. 2A-B,E-F; Table 1). However, the cre-animals that were switched to ETOH-containing diet developed intense steatosis as seen through enhanced severity, extent and lipid vesicular size, with no degeneration, inflammation or ballooning (Fig. 2C,G; Table 1). Remarkably, Alb-D2KO mice switched to ETOH diet exhibited much reduced hepatic accumulation of lipids (Fig. 2D,H; Table 1).
Fig. 2 – Liver histology of Alb-D2KO and Cre mice treated with ETOH.
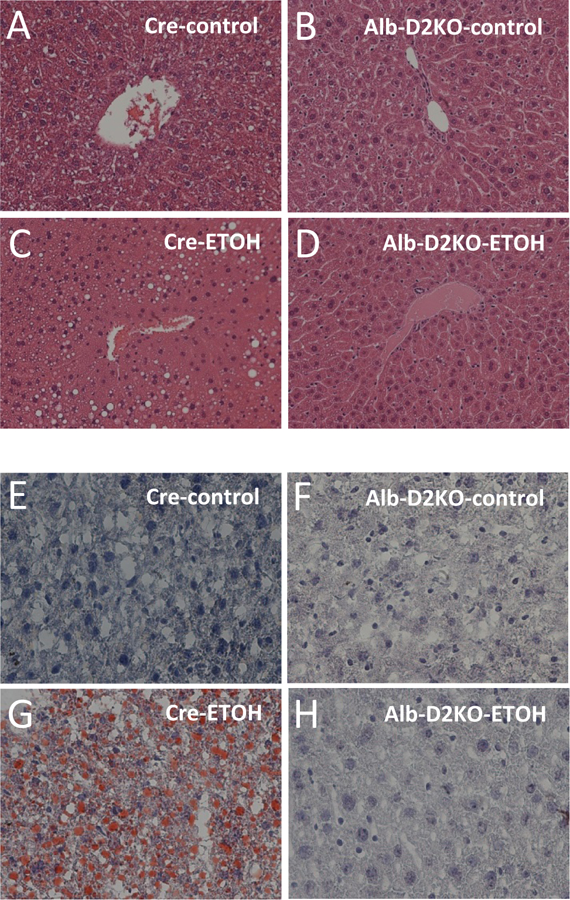
Representative microphotographs (X200 magnification) of (A-D) H&E and (E-H) oil red-O stained liver sections; n = 5 for each condition.
Table 1-.
Histological analysis of liver sections obtained from cre- and Alb-D2KO mice switched to ETOH containing diet.
| Scoring | Cre-ETOH | Alb-D2KO-ETOH | |
|---|---|---|---|
| Steatosis | Severity | 2.7±0.3b | 0.7±0.7a |
| Extent | 2.7±0.3b | 0.7±0.3a | |
| Macrovesicular | 2.7±0.3b | 0.7±0.3a | |
| Microvesicular | 2.3±0.3b | 0.7±0.3a | |
| Inflammation | Severity | No foci | No foci |
| Extent | No foci | No foci | |
| Location | No foci | No foci | |
| Ballooning | Severity | 0 | 0 |
| Extent | 0 | 0 | |
The data table uses the scoring of liver disease created by Kleiner et al. (2005): Severity = the severity of the steatosis/inflammation per hepatocyte, was score 1 to 3, (0=none, 1=mild, 2=moderate, 3=severe) and Extent = the amount of hepatocytes that have steatosis/inflammation, was score 1 to 3, (0=none, 1=1–25%, 2=26–75%, 3=76–100%); in the cre-group steatosis indices were all <0.3 and in the Alb-D2KO-group <1.0; values are the mean ± SEM of 3 independent samples.
p<0.05 vs Cre-ETOH and
p<0.01 vs Cre-control (Student’s t-test).
Quantitative measurement of hepatic lipids confirmed a ~2.3-fold increase in triglyceride content in the cre-mice fed with ETOH-containing diet (Fig. 3A). However, in the Alb-D2KO animals the hepatic triglyceride content was not elevated by ETOH consumption (Fig. 3A). Liver cholesterol content was not affected by switching cre-mice to ETOH containing diet, but in the Alb-D2KO mice there was a slight decrease in liver cholesterol (Fig. 3B). No differences in plasma triglycerides or HDL-cholesterol levels were observed in cre- or Alb-D2KO animals switched to ETOH-containing diet (Fig. 3C and D). On the other hand, serum VLDL/LDL cholesterol levels dropped in cre-mice fed with ETOH-containing diet, but in Alb-D2KO animals switched to ETOH they remained unaffected (Fig. 3E).
Fig. 3 -. Lipid profile and markers of liver function in Alb-D2KO and Cre mice treated with ETOH.

(A) triglyceride content in liver; (B) total cholesterol in liver; (C) plasma triglycerides; (D) plasma HDL cholesterol; (E) plasma LDL/VLDL cholesterol; (F) plasma ALT activity; (G) plasma GOT1 activity; (H) plasma GOT1; (I) plasma ARG1; (J) plasma SDH; the gray area represents mean ± SD for the control group; values are shown in a box and whiskers plot indicating median and quartiles or median and quartiles; n=5/group; * p≤0.05 vs Cre-ETOH; # p≤0.05 vs Cre-Control (Student’s t-test).
To further explore the phenotype of these animals, we processed plasma of all animals for alanine transaminase (ALT) and aspartate transaminase (GOT1) activity, as well as GOT1, ARD1 and SDH serum levels as indices of liver injury. No differences were observed in the animals switched to an ETOH excess, regardless of their genotype (Fig. 3F-J).
Genes involved in hepatic lipid export are up-regulated in Alb-D2KO mice by ETOH-containing diet
Next, we studied hepatic expression of lipid-related genes. Cre-mice switched to ETOH-containing diet exhibited a reduction in mRNA levels for Pparα and Srebpf1, while increased mRNA for total Lipin1 as well as Lipin1 isoforms α and β; Pgc1α mRNA levels were not affected by ETOH (Fig. 4A). In contrast, these transcriptions factors were not affected by ETOH-containing diet in the Alb-D2KO (Fig. 4A). Furthermore, the analyses of downstream targets in cre-mice revealed that feeding on ETOH-containing diet reduced hepatic mRNA levels encoding (i) the lipid transport proteins Fabp1 and Fabp5 (Fig. 4A), the triglyceride synthesis enzyme Gpat1 and LDL receptor (Fig. 4A); genes involved in other aspects of lipid metabolism were not affected (Fig. 4A). In the Alb-D2KO mice, feeding on ETOH reversed some of these changes in gene expression, i.e. elevated mRNA levels for Gpat1 and LDL receptor; furthermore, mRNA levels for Mttp and Abcg1 were also elevated (Fig. 4A).
Fig. 4 – Liver expression of selected genes in Alb-D2KO and Cre mice treated with ETOH.

(A) relative mRNA levels of selected transcriptional factors; (B) relative mRNA levels of selected genes involved in lipid metabolism; (B) relative mRNA levels of selected genes involved in lipoprotein structure and lipid mobilization; (C) relative mRNA levels of genes involved in ETOH metabolism; all results are relative to 18S or CycloB mRNA levels and normalized for the respective controls; values are shown in a box and whiskers plot indicating median and quartiles; n=4–5/group; gene abbreviations are as in the Table S2; *p< 0.05 and **p< 0.01 vs Cre-ETOH; # p≤0.05 and ## p≤0.01 vs Cre-Control (Student’s t-test).
Feeding on ETOH-containing diet did not affect mRNA levels of genes involved in lipoprotein structure or lipid mobilization such as Apoc1, Apoc3, Apoa4, Apoa5, Apoe, Apoh and Azgp1 in cre-mice (Fig. 4B). However, ETOH excess did increase expression of these genes in the Alb-D2KO animals, except for Apoa4 and Apoa5 (Fig. 4B).
mRNA levels of enzymes involved in ETOH metabolism are induced by ETOH only in Alb-D2KO mice
Only in the Alb-D2KO animals did ETOH treatment induce the hepatic expression of the Aldh2 mRNA involved in ETOH metabolism, and the Acss2 RNA that encodes the enzyme involved in the synthesis of acetyl-CoA by ~1.6-fold and ~2.6-fold, respectively (Fig. 4C). Acss1 hepatic mRNA levels were not affected by ETOH feeding in either mouse (Fig. 4C).
Zfp125 expression is induced by EOTH feeding and correlates with liver triglycerides
Hepatic Zfp125 protein levels are reduced in adult Alb-D2KO mice, explaining most of the metabolic phenotype observed in these animals (Fernandes et al., 2018). Here we saw that Zfp125 protein levels were induced by approximately 50% in the cre-animals switched to ETOH-containing diet (Fig. 5A-B). However, in the Alb-D2KO mice switched to ETOH there was induction of Zfp125 expression but the levels reached were similar to what was observed in control-cre mice (Fig. 5A-B). Next we plotted Zfp125 values against liver triglycerides for each animal, with a resulting line that indicates a high degree of correlation (Fig. 5C).
Fig. 5 – Liver expression of Zfp125 in Alb-D2KO and Cre mice treated with ETOH.

(A) quantification of relative Zfp125 protein levels in liver sonicates probed as detected by western analysis with α-actin and α-Zfp125; values are shown in a box and whiskers plot indicating median and quartiles; n=4–5/group; **p< 0.01 and ***p< 0.01 (one-way ANOVA); (B) typical blot images for hepatic actin and Zfp125 protein; (C) linear correlation between liver triglycerides levels (Fig. 3A) and Zfp125 levels (Fig. 5A-B) for each individual mouse.
Discussion
The present studies show that feeding an ETOH-containing diet elevates hepatic content of Zfp125 by approximately 50% (Fig. 5). In turn, Alb-D2KO mice, which have a distinct liver transcriptome with lower baseline hepatic levels of Zfp125 and in which the elevation of Zfp125 was limited (Fig. 5), were protected against ETOH-induced liver steatosis (Fig. 2; Table 1). Furthermore, only in Alb-D2KO was there an induction of (i) six genes that are normally repressed by Zfp125 (i.e. Mttp, Ldlr, Apoc1, Apoe, Apoh and Azgp1) (Fernandes et al., 2018) and (ii) two genes that are repressed by neonatal Dio2 expression in liver (i.e. Gapt1 and Abcg1) (Fig. 4A-B). Collectively, these genes are involved in intracellular lipid transport, as well as synthesis and secretion of lipoproteins. Overall, these studies indicate a role for perinatal Dio2 surge and Zfp125 in the pathogenesis of ETOH-induced liver steatosis in mice by slowing down hepatic lipid export. We found no evidence that ETOH-feeding accelerated hepatic lipogenesis (Fig. 5A), which could reflect the short duration of the binge model used.
Feeding ETOH promotes the reverse transport of fat stored in adipose tissue to the liver, as shown by labeling adipose tissue triglyceride molecules with deuterium (Zhong et al., 2012). This is supported by our findings that fat pads were reduced in mass after feeding an ETOH diet (Fig. 1F). The fat pad reduction caused by ETOH feeding was similar in cre- and Alb-D2KO mice, suggesting that the influx of fatty acids to the liver was similar regardless of the genotype. On the other hand, the nature of the hepatic genes differentially affected by ETOH between cre- and AlbD2KO mice (Fig. 4–5) suggests that the differences in susceptibility of cre- and Alb-D2KO mice to liver steatosis lie on the rate of lipid secretion.
ETOH consumption is known to slow down hepatic lipid secretion (McVicker et al., 2012, Venkatesan et al., 1988, Kharbanda et al., 2009). For example, in a hybrid of human fibroblasts (WI 38) and Fao rat hepatoma cells (Wif B cells), ETOH treatment reduced lipid efflux, and clearance contributing to intracellular lipid accumulation (McVicker et al., 2012). In addition, using Triton WR-1339 to inhibit clearance of circulating VLDL, hepatic secretion of VLDL was reduced in mice chronically fed with ETOH (Venkatesan et al., 1988, Kharbanda et al., 2009). How would hepatic VLDL secretion be different between cre- and Alb-D2KO mice?
Zfp125 slows down VLDL secretion and mediates most of the metabolic phenotype exhibited by Alb-D2KO mice (Fernandes et al., 2018). Zfp125 is induced by FoxO1, a transcription factor that mediates the transition between fed and fasting states in the liver (Fernandes et al., 2018). Notably, feeding an ETOH-containing diet reportedly increases hepatic FoxO1 mRNA levels and reduces the levels of inactive phospho-FoxO1 (Lieber et al., 2008), In addition, chronic ETOH feeding to mice also increases FoxO1 expression in different intestinal segments, particularly in the ileum (Wang et al., 2013). It is thus conceivable that in the present studies ETOH feeding induced Zfp125 secondary to FoxO1 activation.
One of the key targets of Zfp125 is MTP, a gene encoding the enzyme Mttp that has a central role in the assembly and secretion of apoB-containing lipoproteins, including VLDL. Studies using cell models and rats show that ETOH ingestion decreases secretion of apoB-containing lipoproteins by reducing Mttp expression, primarily by inhibiting of MTP transcription (Lin et al., 1997). The present studies provide evidence that ETOH or one of its metabolites may cause such inhibition by inducing Zfp125. First, the MTP promoter has a putative binding site for Zfp125, at approximately −600 bp (Fernandes et al., 2018). Second, MTP expression is strongly inhibited in AML12 cells that stably express Zfp125 or its human homologue ZNF670 (Fernandes et al., 2018). Third, MTP expression is reduced in livers of mice transiently expressing liposome-delivered Zfp125 or ZNF670 (Fernandes et al., 2018). Thus, it is conceivable that hepatic MTP induction by ETOH in the Alb-D2KO mice occurred because Zfp125 did not rise above control levels in these mice (Fig. 5A-B). That allowed VLDL assembly and secretion to proceed normally, thus preventing liver steatosis.
Another notable observation was a potential for enhanced capacity of Alb-D2KO mice to detoxify ETOH. Aldh2 detoxifies acetaldehyde during ethanol metabolism (Chen et al., 2014, Yoval-Sanchez and Rodriguez-Zavala, 2012). Studies in mice with global Aldh2 overexpression indicate that Aldh2 plays a beneficial role in ameliorating chronic ETOH-induced hepatic steatosis and inflammation (Guo et al., 2015). Thus, it is noteworthy that Aldh2 mRNA levels were higher in Alb-D2KO mice (Fig. 5C), along with higher levels of Acss2 that is involved in the synthesis of Acetyl-CoA (Fig. 5C). In fact, an analysis of the 5’-region upstream to the transcription start site of Aldh2 revealed the presence of two putative Zfp125 binding sites at approximately −120 bp and −950 bp (Table 2). Furthermore, four putative Zfp125 binding sites are present at approximately −580 bp, −1450 bp, −1500 bp and −1600 bp in Acss2 (Table 2). While further studies should clarify the functionality of these sites, it is remarkable that Acss1 lacks any putative Zfp125 binding sites and was not up-regulated by ETOH in Alb-D2KO mice (Fig. 4C). Taken together, these data suggest that the transcriptional changes in AlbD2KO liver include an improved ability to respond and neutralize toxicity from acetaldehyde caused by ETOH consumption.
Table 2-.
Predicted ZFP binding sites on promoter of down-regulated genes
| Description | Promoter region | |
|---|---|---|
| Aldh2 | GGCTTTGGGAGCCAGGGGTCGCGCCCCTTAGGCC | (−123/−89) |
| ATTCTGAAAACAAAGGGGTCCTGTGTGTCTCTCTG | (−950/−915) | |
| Acss2 | ATGGGAACGGAATAGGGGTGCTGTTCGGTGGGAG | (−581/−547) |
| GTGTATTTGTTCAGAGGGGTTGATGTCTCCTTGAG | (−1449/−1414) | |
| GCAGGGCTTTTCCGAGGGGTCTGAGACCTCCAGAA | (−1528/−1493) | |
| ACTGAACCCTCTCATCGGGGTTCAGATATCTTTAGT | (−1628/−1592) | |
| Consensus | ----------------------------GGGGT--------------------------- | |
Eukaryotic Promoter Database (EPD) – Mus musculus (Mouse). (http://epd.vital-it.ch/)
In conclusion, ETOH consumption in mice is associated with an elevation in hepatic Zfp125 levels, which slows down assembly and secretion of VLDL, a recognized contributing to liver steatosis. Alb-D2KO animals exhibit a unique hepatic transcriptome that includes lower levels of Zfp125 and markedly reduces susceptibility to liver steatosis. When switched to ETOH containing diet, Alb-D2KO failed to elevate Zfp125 beyond control levels and exhibited upregulation of genes involved in VLDL synthesis and secretion. These data illustrate how a neonatal peak of Dio2 in the liver affects Zfp125 expression and defines future susceptibility to ETOH-induced liver steatosis.
Supplementary Material
Acknowledgement
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases grants DK58538 and DK65055; EU H2020 THYRAGE no. 666869 and NKFIH 125247; and a Wellcome Trust Joint Investigator Award (110141/Z/15/Z).
References
- BERTOLA A, MATHEWS S, KI SH, WANG H & GAO B 2013. Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat Protoc, 8, 627–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CARRASCO MP, MARCO C & SEGOVIA JL 2001. Chronic ingestion of ethanol stimulates lipogenic response in rat hepatocytes. Life Sci, 68, 1295–304. [DOI] [PubMed] [Google Scholar]
- CASTILLO M, HALL JA, CORREA-MEDINA M, UETA C, WON KANG H, COHEN DE & BIANCO AC 2011. Disruption of thyroid hormone activation in type 2 deiodinase knockout mice causes obesity with glucose intolerance and liver steatosis only at thermoneutrality. Diabetes, 60, 1082–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN CH, FERREIRA JC, GROSS ER & MOCHLY-ROSEN D 2014. Targeting aldehyde dehydrogenase 2: new therapeutic opportunities. Physiol Rev, 94, 1–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CRAWFORD JM 2012. Histologic findings in alcoholic liver disease. Clin Liver Dis, 16, 699–716. [DOI] [PubMed] [Google Scholar]
- ERIKSSON CJ, SIPPEL HW & FORSANDER OA 1977. The occurrence of acetaldehyde binding in rat blood but not in human blood. FEBS Lett, 75, 205–8. [DOI] [PubMed] [Google Scholar]
- FERNANDES GW, BOCCO B, FONSECA TL, MCANINCH EA, JO S, LARTEY LJ, I OS, UNTERMAN TG, PREITE NZ, VOIGT RM, FORSYTH CB, KESHAVARZIAN A, SINKO R, GOLDFINE AB, PATTI ME, RIBEIRO MO, GEREBEN B & BIANCO AC 2018. The Foxo1-Inducible Transcriptional Repressor Zfp125 Causes Hepatic Steatosis and Hypercholesterolemia. Cell Rep, 22, 523–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FONSECA TL, FERNANDES GW, MCANINCH EA, BOCCO BM, ABDALLA SM, RIBEIRO MO, MOHACSIK P, FEKETE C, LI D, XING X, WANG T, GEREBEN B & BIANCO AC 2015. Perinatal deiodinase 2 expression in hepatocytes defines epigenetic susceptibility to liver steatosis and obesity. Proc Natl Acad Sci U S A, 112, 14018–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GALLI A, PINAIRE J, FISCHER M, DORRIS R & CRABB DW 2001. The transcriptional and DNA binding activity of peroxisome proliferator-activated receptor alpha is inhibited by ethanol metabolism. A novel mechanism for the development of ethanol-induced fatty liver. J Biol Chem, 276, 68–75. [DOI] [PubMed] [Google Scholar]
- GAO B & BATALLER R 2011. Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology, 141, 1572–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GEREBEN B, ZEOLD A, DENTICE M, SALVATORE D & BIANCO AC 2008. Activation and inactivation of thyroid hormone by deiodinases: local action with general consequences. Cell Mol Life Sci, 65, 570–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GUO R, XU X, BABCOCK SA, ZHANG Y & REN J 2015. Aldehyde dedydrogenase-2 plays a beneficial role in ameliorating chronic alcohol-induced hepatic steatosis and inflammation through regulation of autophagy. J Hepatol, 62, 647–56. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- JOLY JG, FEINMAN L, ISHII H & LIEBER CS 1973. Effect of chronic ethanol feeding on hepatic microsomal glycerophosphate acyltransferase activity. J Lipid Res, 14, 337–43. [PubMed] [Google Scholar]
- KHARBANDA KK, TODERO SL, WARD BW, CANNELLA JJ 3RD & TUMA DJ 2009. Betaine administration corrects ethanol-induced defective VLDL secretion. Mol Cell Biochem, 327, 75–8. [DOI] [PubMed] [Google Scholar]
- KLEINER DE, BRUNT EM, VAN NATTA M, BEHLING C, CONTOS MJ, CUMMINGS OW, FERRELL LD, LIU YC, TORBENSON MS, UNALP-ARIDA A, YEH M, MCCULLOUGH AJ, SANYAL AJ & NONALCOHOLIC STEATOHEPATITIS CLINICAL RESEARCH, N. 2005. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology, 41, 1313–21. [DOI] [PubMed] [Google Scholar]
- KONDILI LA, TALIANI G, CERGA G, TOSTI ME, BABAMETO A & RESULI B 2005. Correlation of alcohol consumption with liver histological features in non-cirrhotic patients. Eur J Gastroenterol Hepatol, 17, 155–9. [DOI] [PubMed] [Google Scholar]
- LIEBER CS 1995. Medical disorders of alcoholism. N Engl J Med, 333, 1058–65. [DOI] [PubMed] [Google Scholar]
- LIEBER CS, LEO MA, WANG X & DECARLI LM 2008. Alcohol alters hepatic FoxO1, p53, and mitochondrial SIRT5 deacetylation function. Biochem Biophys Res Commun, 373, 246–52. [DOI] [PubMed] [Google Scholar]
- LIEBER CS, SPRITZ N & DECARLI LM 1966. Role of dietary, adipose, and endogenously synthesized fatty acids in the pathogenesis of the alcoholic fatty liver. J Clin Invest, 45, 51–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIM SS, VOS T, FLAXMAN AD, DANAEI G, SHIBUYA K, ADAIR-ROHANI H, AMANN M, ANDERSON HR, ANDREWS KG, ARYEE M, ATKINSON C, BACCHUS LJ, BAHALIM AN, BALAKRISHNAN K, BALMES J, BARKER-COLLO S, BAXTER A, BELL ML, BLORE JD, BLYTH F, BONNER C, BORGES G, BOURNE R, BOUSSINESQ M, BRAUER M, BROOKS P, BRUCE NG, BRUNEKREEF B, BRYAN-HANCOCK C, BUCELLO C, BUCHBINDER R, BULL F, BURNETT RT, BYERS TE, CALABRIA B, CARAPETIS J, CARNAHAN E, CHAFE Z, CHARLSON F, CHEN H, CHEN JS, CHENG AT, CHILD JC, COHEN A, COLSON KE, COWIE BC, DARBY S, DARLING S, DAVIS A, DEGENHARDT L, DENTENER F, DES JARLAIS DC, DEVRIES K, DHERANI M, DING EL, DORSEY ER, DRISCOLL T, EDMOND K, ALI SE, ENGELL RE, ERWIN PJ, FAHIMI S, FALDER G, FARZADFAR F, FERRARI A, FINUCANE MM, FLAXMAN S, FOWKES FG, FREEDMAN G, FREEMAN MK, GAKIDOU E, GHOSH S, GIOVANNUCCI E, GMEL G, GRAHAM K, GRAINGER R, GRANT B, GUNNELL D, GUTIERREZ HR, HALL W, HOEK HW, HOGAN A, HOSGOOD HD 3RD, HOY D, HU H, HUBBELL BJ, HUTCHINGS SJ, IBEANUSI SE, JACKLYN GL, JASRASARIA R, JONAS JB, KAN H, KANIS JA, KASSEBAUM N, KAWAKAMI N, KHANG YH, KHATIBZADEH S, KHOO JP, KOK C, LADEN F, et al. 2012. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet, 380, 2224–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIN MC, LI JJ, WANG EJ, PRINCLER GL, KAUFFMAN FC & KUNG HF 1997. Ethanol down-regulates the transcription of microsomal triglyceride transfer protein gene. FASEB J, 11, 1145–52. [DOI] [PubMed] [Google Scholar]
- MCVICKER BL, RASINENI K, TUMA DJ, MCNIVEN MA & CASEY CA 2012. Lipid droplet accumulation and impaired fat efflux in polarized hepatic cells: consequences of ethanol metabolism. Int J Hepatol, 2012, 978136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- POSTIC C, SHIOTA M, NISWENDER KD, JETTON TL, CHEN Y, MOATES JM, SHELTON KD, LINDNER J, CHERRINGTON AD & MAGNUSON MA 1999. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. J Biol Chem, 274, 305–15. [DOI] [PubMed] [Google Scholar]
- RASINENI K & CASEY CA 2012. Molecular mechanism of alcoholic fatty liver. Indian J Pharmacol, 44, 299–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SEITZ HK & STICKEL F 2010. Acetaldehyde as an underestimated risk factor for cancer development: role of genetics in ethanol metabolism. Genes Nutr, 5, 121–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SILER SQ, NEESE RA, PARKS EJ & HELLERSTEIN MK 1998. VLDL-triglyceride production after alcohol ingestion, studied using [2–13C1] glycerol. J Lipid Res, 39, 2319–28. [PubMed] [Google Scholar]
- VENKATESAN S, WARD RJ & PETERS TJ 1988. Effect of chronic ethanol feeding on the hepatic secretion of very-low-density lipoproteins. Biochim Biophys Acta, 960, 61–6. [DOI] [PubMed] [Google Scholar]
- WANG Y, TONG J, ZOU D, CHANG B, WANG B & WANG B 2013. Elevated expression of forkhead box protein O1 (FoxO1) in alcohol-induced intestinal barrier dysfunction. Acta Histochem, 115, 557–63. [DOI] [PubMed] [Google Scholar]
- YOVAL-SANCHEZ B & RODRIGUEZ-ZAVALA JS 2012. Differences in susceptibility to inactivation of human aldehyde dehydrogenases by lipid peroxidation byproducts. Chem Res Toxicol, 25, 722–9. [DOI] [PubMed] [Google Scholar]
- ZHONG W, ZHAO Y, TANG Y, WEI X, SHI X, SUN W, SUN X, YIN X, SUN X, KIM S, MCCLAIN CJ, ZHANG X & ZHOU Z 2012. Chronic alcohol exposure stimulates adipose tissue lipolysis in mice: role of reverse triglyceride transport in the pathogenesis of alcoholic steatosis. Am J Pathol, 180, 998–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.