

Abstract
Background
Among patients with nondialysis-dependent chronic kidney disease (NDD-CKD) and iron-deficiency anemia (IDA), ferric citrate increases hemoglobin and iron parameters and reduces serum phosphate and fibroblast growth factor 23 (FGF23), a key phosphate-regulating hormone. We conducted post hoc analyses of a phase 3 trial to explore associations between iron replacement, serum phosphate changes and FGF23 regulation.
Methods
We employed multivariable regression and longitudinal mixed-effects models to identify and confirm, respectively, whether baseline demographic and laboratory variables were associated with ferric citrate-induced changes in serum phosphate or FGF23 concentrations. We employed path analyses to determine whether changes in FGF23 concentrations were mediated via changes in serum phosphate and/or transferrin saturation (TSAT).
Results
We analyzed a total of 117 and 115 ferric citrate-treated and placebo-treated patients, respectively. At 16 weeks, ferric citrate significantly reduced serum phosphate versus placebo (P = 0.006) only among patients with elevated baseline serum phosphate (≥4.5 mg/dL) and did not reduce serum phosphate among patients with baseline serum phosphate within the population reference range. Ferric citrate reduced intact FGF23 and C-terminal FGF23 partially via changes in TSAT (for C-terminal FGF23) and serum phosphate (for intact FGF23) and partially via unknown/unmeasured mechanisms.
Conclusions
Ferric citrate reduced serum FGF23 concentrations (partially via effects on serum phosphate and iron balance) and did not reduce serum phosphate among patients with baseline serum phosphate concentrations within the population reference range.
Keywords: ferric citrate, FGF23, iron-deficiency anemia, nondialysis-dependent chronic kidney disease, serum phosphate
INTRODUCTION
With progressive loss of kidney function, patients with chronic kidney disease (CKD) frequently develop complications, including iron deficiency and anemia. Iron deficiency and associated iron-deficiency anemia (IDA) diminish self-reported health-related quality of life as a result of symptoms such as fatigue, irritability and restless leg syndrome [1] and are risk factors for cardiovascular morbidity and mortality [2–4]. Mineral metabolism disorders, including hyperphosphatemia, are also common and consequential among patients with CKD, and these disorders are associated with an increased risk of fracture, cardiovascular disease and death [5–10].
Hyperphosphatemia in CKD is typically accompanied by an increase in a key phosphate-regulating hormone, fibroblast growth factor 23 (FGF23), which is stimulated by a variety of factors, including increases in parathyroid hormone (PTH), calcium, phosphorus, 1,25-dihydroxyvitamin D and inflammation [11]. FGF23 induces phosphaturia, thereby reducing serum phosphate; however, higher serum FGF23 concentrations are independently associated with CKD progression [12], left ventricular hypertrophy, congestive heart failure [13] and death [14]. There is evidence suggesting that interventions that reduce FGF23 are associated with cardiovascular benefits in patients with CKD; in a post hoc analysis of the Evaluation of Cinacalcet Hydrochloride Therapy to Lower Cardiovascular Events trial, patients with dialysis-dependent CKD and secondary hyperparathyroidism who received cinacalcet for 20 weeks had significantly lower FGF23 concentrations compared with patients who received placebo, and these reductions were associated with lower risks of major cardiovascular events, including mortality [15].
FGF23 is regulated at the transcriptional level and by intracellular proteolytic cleavage and is measured as biologically active intact FGF23 (iFGF23) and C-terminal FGF23 (cFGF23), which includes intact and proteolytically cleaved inactive cFGF23 [16]. In CKD, FGF23 transcription is increased but cleavage is impaired, which results in elevated circulating cFGF23 and iFGF23 [16, 17]. Among patients with IDA and CKD, however, FGF23 regulation is complicated by underlying iron deficiency. In the setting of iron deficiency and normal kidney function, both FGF23 transcription and intracellular proteolytic cleavage are stimulated, therefore cFGF23 is elevated but iFGF23 is unchanged [14, 16–19]. However, among patients with IDA and CKD, cFGF23 and iFGF23 are both elevated [20, 21], but it is unclear how these clinical factors (iron deficiency and inflammation in CKD) interact to alter FGF23 regulation.
Ferric citrate, an iron-based oral phosphate binder, is approved by multiple regulatory agencies for the control of serum phosphate in adult patients with CKD and is approved in the USA for the control of serum phosphate in adult patients with CKD on dialysis and as an iron replacement product indicated for the treatment of IDA in adult patients with CKD not on dialysis [22]. In a phase 3 randomized, placebo-controlled trial among patients with nondialysis-dependent CKD (NDD-CKD) and IDA [21], patients who received ferric citrate for 16 weeks achieved significant increases in hemoglobin, transferrin saturation (TSAT) and ferritin compared with patients who received placebo. Reductions in serum phosphate with ferric citrate were modest but statistically significant compared with placebo (relative change from baseline to Week 16, −0.21 mg/dL; P = 0.02). Both cFGF23 and iFGF23 decreased significantly from baseline to Week 16 in ferric citrate–treated patients compared with placebo-treated patients.
In this post hoc analysis, we first identified baseline demographic, clinical and laboratory variables associated with changes in serum phosphate or FGF23. Second, we investigated ferric citrate effects on serum phosphate concentrations by baseline serum phosphate tertiles, by baseline CKD stage, by baseline FGF23 tertiles and by ferric citrate dose increases. Third, given the concomitant effects of ferric citrate on serum phosphate and iron deficiency, we explored whether ferric citrate reduced FGF23 via ferric citrate–mediated changes in serum phosphate and/or TSAT using path analyses.
MATERIALS AND METHODS
Study design, patient population and treatment
This previously reported phase 3 trial (ClinicalTrials.gov identifier: NCT02268994) was a randomized, double-blind, placebo-controlled trial with a 16-week randomized efficacy period and an 8-week extension period among patients with NDD-CKD and IDA [21]. Eligibility criteria included an estimated glomerular filtration rate (eGFR; Modification of Diet in Renal Disease study formula) <60 mL/min/1.73 m2, hemoglobin 9.0–11.5 g/dL inclusive, ferritin ≤200 ng/mL, TSAT ≤25%, serum phosphate ≥3.5 mg/dL and intolerance of or inadequate response to prior oral iron supplementation. Ferric citrate was started at 3 × 1 g tablets daily and titrated at Weeks 4, 8 and 12 by an additional 3 tablets daily to achieve a ≥ 1.0 g/dL hemoglobin increase from baseline, up to a maximum of 12 tablets daily. We conducted all analyses on the intention-to-treat population, defined as patients who received one or more dose of study drug with one or more postbaseline laboratory value, using data from the 16-week efficacy period.
This study was conducted in accordance with the Declaration of Helsinki and International Conference on Harmonisation Good Clinical Practice guidelines. All patients provided written informed consent before enrollment.
Study endpoints and assessments
Demographics and clinical variables were collected at baseline (Table 1). Most laboratory parameters were assessed at baseline and Weeks 1 and 2 and then every 2 weeks until Week 16. FGF23 and intact PTH (iPTH) were assessed at baseline and Weeks 8 and 16. cFGF23 was measured in plasma using the second-generation carboxyterminal enzyme-linked immunosorbent assay (ELISA; Immunotopics, San Clemente, CA, USA), which recognizes iFGF23 and C-terminal fragments. iFGF23 was measured in serum using an ELISA against the intact protein (Kainos, Japan).
Table 1.
Baseline demographics and disease characteristics (intention-to-treat population)
| Characteristics | Ferric citrate | Placebo |
|---|---|---|
| (n = 117) | (n = 115) | |
| Age (years) | 65.6 (11.2) | 65.3 (13.0) |
| Male gender, n (%) | 41 (35.0) | 44 (38.3) |
| Race, n (%) | ||
| White | 78 (66.7) | 81 (70.4) |
| Black or African American | 38 (32.5) | 31 (27.0) |
| Other | 1 (0.9) | 3 (2.6) |
| Hispanic or Latino ethnicity, n (%) | 29 (24.8) | 23 (20.0) |
| Body mass index (kg/m2) | 34.4 (8.2) | 33.5 (8.2)a |
| Patients with diabetes, n (%) | 85 (72.6) | 81 (70.4) |
| eGFR (mL/min/1.73 m2) | 27.8 (13.0) | 29.5 (12.8) |
| 30 to <60 (CKD Stage 3), n (%) | 47 (40.2) | 53 (46.1) |
| 15 to <30 (CKD Stage 4), n (%) | 55 (47.0) | 49 (42.6) |
| <15 (CKD Stage 5), n (%) | 15 (12.8) | 13 (11.3) |
| Hemoglobin (g/dL) | 10.4 (0.7) | 10.4 (0.8) |
| Transferrin saturation (%) | 20.2 (6.4) | 19.6 (6.6) |
| Ferritin (ng/mL) | 85.9 (55.7) | 81.7 (58.3) |
| Serum phosphate (mg/dL) | 4.2 (0.9) | 4.1 (0.7) |
| ≤3.8, n (%) | 43 (36.8) | 44 (38.3) |
| 3.9–4.4, n (%) | 38 (32.4) | 37 (32.1) |
| ≥4.5, n (%) | 36 (30.8) | 34 (29.6) |
| Calcium (mg/dL) | 9.3 (0.7) | 9.3 (0.7) |
| Bicarbonate (mEq/L) | 21.0 (3.9) | 21.6 (3.7) |
| Albumin (g/dL) | 4.0 (0.4) | 4.0 (0.4) |
| iPTH (pg/mL) | 103 [15–586] | 92 [21–551] |
| C-terminal FGF23 (RU/mL) | 364 [27–6480]b | 306 [34–8355]c |
| Intact FGF23 (pg/mL) | 134 [23–9562] | 134 [23–7253]a |
n = 114. bn = 116. cn = 113. Data are mean (SD) or median [min–max] unless otherwise specified.
Statistical analysis
We conducted all analyses in R version 3.4.3 (R Project for Statistical Computing, Vienna, Austria), with statistical significance determined at an α of 0.05 without adjustment for multiple comparisons.
Baseline associations with changes in serum phosphate or FGF23
We identified baseline variables associated with changes in serum phosphate, cFGF23 or iFGF23 at 16 weeks in two steps. First, we selected baseline variables and their interactions with treatment using multivariable linear regression. Second, for baseline variables with treatment interactions, we conducted confirmatory mixed-effects models for repeated measures (MMRM) analyses.
In multivariable linear regression analyses, we selected candidate baseline variables (Table 1) associated with outcomes at 16 weeks based on forward selection and backward elimination using Akaike information criterion (AIC); treatment was fixed in all models and baseline serum phosphate, cFGF23 and iFGF23 were fixed in models examining changes in serum phosphate, cFGF23 and iFGF23, respectively. Missing data for changes in FGF23 at Week 16 were imputed using the last observation carried forward (LOCF) method. Potential interactions between each baseline variable and treatment were tested sequentially by repeating the stepwise AIC multivariable models including one treatment-by-baseline covariate interaction at a time. We performed sensitivity analyses to assess the consistency of associations with changes in serum phosphate, cFGF23 or iFGF23 by comparing results using different methods of handling missing data (observed cases, LOCF and random forest multiple imputation) and alternative multivariable model selection techniques (Supplementary data). For analyses with cFGF23 or iFGF23 as dependent variables, these variables were rank-transformed before analysis because the data were heavily right-skewed [23].
For baseline variables for which treatment interaction terms were retained by stepwise AIC in multivariable models, we conducted confirmatory MMRM analyses. In contrast to multivariable regression at the single 16-week time point, MMRM uses all available data at all time points, which provides better estimates of the trajectory of change in outcome over time and how this is influenced by baseline variable level and by its interaction with treatment. For MMRMs with serum phosphate as the response, we analyzed absolute serum phosphate values over time by treatment and by baseline covariate tertiles in order to evaluate the effect of baseline covariates and their interaction with treatment on clinically relevant absolute serum phosphate concentrations. For MMRMs with cFGF23 and iFGF23 as the response, we analyzed changes from baseline in cFGF23 and iFGF23 by treatment and by continuously distributed baseline covariates. The MMRM modeling strategy for FGF23 variables differed from that for serum phosphorus for the following reasons. First, because FGF23 variables were rank-transformed, we considered the change in rank to be more meaningful than absolute rank. Second, because FGF23 variables were measured less frequently than serum phosphorus, we obtained greater statistical power using continuously distributed baseline covariates in these models. For each response variable, population marginal means [least square means (LSMs)] and 95% confidence intervals (CIs) were then estimated at levels of categorical baseline covariates (tertiles) or at the 25th and 75th percentile values of continuously distributed baseline covariates for each treatment arm by visit. Within each baseline covariate tertile or percentile, ferric citrate versus placebo LSM differences, 95% CIs and P-values at Week 16 were calculated. Further details on MMRM specifications are described in the Supplementary data.
Ferric citrate dose–response
An additional MMRM assessed ferric citrate dose–response by estimating serum phosphate ≥7 days after a stepwise dose increase (from 0 to 3, 3 to 6, 6 to 9 or 9 to 12 g/day) among ferric citrate–treated patients stratified by baseline serum phosphate tertile.
Path analyses
To test whether ferric citrate reduced FGF23 indirectly via ferric citrate–mediated changes in serum phosphate and/or TSAT, we employed path analyses using the lavaan package version 0.5-23.1097 (R Project). Path analyses using observed data from randomized controlled trials with repeated laboratory measurements afford the opportunity of testing implied temporal causal associations between treatment, changes in these laboratory measures and outcome [24]. In the current study, data on laboratory measures were collected at baseline and at 2-week intervals thereafter up to 16 weeks (for FGF23, data were available at baseline and 8 and 16 weeks). Thus changes up to 14 weeks, which represent cumulative changes from baseline over the entire 14-week period, were used to assess the influence of earlier temporal changes in serum phosphate or TSAT on later changes in FGF23 outcomes at 16 weeks. A structural equation path analysis model was specified (Figure 1). In the model, if the regression pathway coefficient (c) is significant and the products of both indirect pathway coefficients, (a × b) and (d × e), are not significant, then this could represent either a direct effect of treatment on the change in FGF23 or an indirect effect by unknown/unmeasured mechanisms. If the product of the indirect pathway coefficients (a × b) is significant and both (c) and the product of the indirect pathway coefficients (d × e) are not significant, then full mediation by the change in serum phosphate is declared. If the product of the indirect pathway coefficients (d × e) is significant and both (c) and the product of the indirect pathway coefficients (a × b) are not significant, then full mediation by the change in TSAT is declared. If both direct and indirect pathway coefficients are significant, then partial mediation is declared [25]. All variables were standardized before entry into the path analyses, which employed weighted LSMs and variance-adjusted estimation with robust standard errors. We addressed the potential influence of missing data by comparing analyses using observed values, LOCF and random forest multiple imputation.
FIGURE 1.

Structural equation model specification for path analyses.
RESULTS
Patients and baseline characteristics
We enrolled 234 patients (ferric citrate, n = 117; placebo, n = 117). Among patients randomized to receive placebo, one did not receive the study drug and one received the drug but had no postbaseline laboratory assessments, therefore the modified intention-to-treat population included 232 patients. At baseline, serum phosphate concentrations were within the population reference range of 2.5–4.5 mg/dL in 69.2% of ferric citrate– and 70.4% of placebo-treated patients and an eGFR <15 mL/min/1.73 m2 (CKD Stage 5) was present in 12.8% and 11.3%, respectively (Table 1). Baseline cFGF23 and iFGF23 varied widely (minimum–maximum, 27–8355 RU/mL and 23–9562 pg/mL, respectively). Additional baseline clinical characteristics are reported elsewhere [21].
Baseline associations with serum phosphate
We identified baseline serum phosphate and baseline eGFR as strongly associated (both P < 0.001) with serum phosphate change at 16 weeks in multivariate analysis (Table 2). We confirmed significant treatment interactions for baseline serum phosphate (P = 0.021), eGFR (P = 0.002), cFGF23 (P = 0.022) and iFGF23 (P = 0.041) on absolute serum phosphate concentrations at 16 weeks in MMRM analyses. Longitudinal plots of serum phosphate concentrations by baseline serum phosphate tertiles (Figure 2A and B) demonstrated that ferric citrate significantly reduced serum phosphate versus placebo at 16 weeks only among patients with baseline serum phosphate concentrations above the population reference range (≥4.5 mg/dL tertile); LSM difference −0.47 (95% CI −0.80 to −0.14; P = 0.006). Among patients with baseline serum phosphate concentrations within the reference range, ferric citrate did not significantly reduce serum phosphate versus placebo at 16 weeks; LSM differences 0.12 (95% CI −0.19–0.43; P = 0.438) for the 3.9–4.4 mg/dL tertile and −0.17 (95% CI −0.46–0.11; P = 0.236) for the ≤3.8 mg/dL tertile. Longitudinal plots of serum phosphate concentrations by baseline eGFR thresholds corresponding to CKD Stages 3–5 (Figures 2C and D) demonstrated that ferric citrate significantly reduced serum phosphate versus placebo at 16 weeks only among patients with baseline CKD Stages 4 and 5, LSM differences −0.73 (95% CI −1.30 to −0.16; P = 0.013) and −0.32 (95% CI −0.58 to −0.05; P = 0.020), respectively, and not among patients with baseline CKD Stage 3, LSM difference 0.08 (95% CI−0.18–0.34; P = 0.566). Results by baseline FGF23 variables are shown in Supplementary data, Figure S1.
Table 2.
Multivariable regression analysis for baseline associations with change in serum phosphate, cFGF23 and iFGF23 at 16 weeksa
| Outcome/variables | Selection method | β coefficient (95% CI) | P-value |
|---|---|---|---|
| Change in serum phosphateb | |||
| Treatment: FC versus placebo | Fixed | −0.21 (−0.38 to −0.05) | 0.012 |
| Serum phosphate, mg/dL | Fixed | −0.58 (−0.69 to −0.47) | <0.001 |
| eGFR, mL/min/1.73 m2 | Stepwise AIC | −0.01 (−0.02 to −0.01) | <0.001 |
| Albumin, g/dL | Stepwise AIC | −0.26 (−0.47 to −0.04) | 0.023 |
| Change in cFGF23c,d | |||
| Treatment: FC versus placebo | Fixed | −33.8 (−46.3 to −21.3) | <0.001 |
| cFGF23b | Fixed | −0.57 (−0.69 to −0.45) | <0.001 |
| Age, years | Stepwise AIC | −0.86 (−1.41 to −0.32) | 0.002 |
| Diabetes: yes versus no | Stepwise AIC | −16.2 (−30.3 to −2.1) | 0.025 |
| eGFR, mL/min/1.73 m2 | Stepwise AIC | −1.53 (−2.07 to −0.99) | <0.001 |
| Change in iFGF23c,e | |||
| Treatment: FC versus placebo | Fixed | −30.2 (−44.4 to −15.9) | <0.001 |
| iFGF23b | Fixed | −0.42 (−0.61 to −0.23) | <0.001 |
| Sex: male versus female | Stepwise AIC | 17.7 (3.1–32.3) | 0.018 |
| Race: non-white versus whitef | Stepwise AIC | 24.4 (9.1–39.8) | 0.002 |
| eGFR, mL/min/1.73 m2 | Stepwise AIC | −1.03 (−1.78 to −0.28) | 0.008 |
| Serum phosphate, mg/dL | Stepwise AIC | −8.5 (−19.1–2.2) | 0.122 |
| Log cFGF23 | Stepwise AIC | 7.0 (−1.5–15.6) | 0.109 |
Missing data were imputed using the last observation carried forward method.
Sensitivity analyses demonstrated stable associations, irrespective of the method for missing data handling or multivariable model selection (Supplementary data, Tables S1 and S2).
Owing to non-normal distribution of FGF23, rank transformations were employed.
Sensitivity analyses demonstrated similar associations among different methods for missing data handling or multivariable model selection (Supplementary data, Tables S3 and S4).
Sensitivity analyses demonstrated similar associations across most methods for missing data handling or multivariable model selection (Supplementary data, Tables S5 and S6).
Of the non-white category, 94.5% were Black or African American.
FC, ferric citrate.
FIGURE 2.

Mean trajectories of serum phosphate during 16 weeks by treatment (ferric citrate versus placebo) estimated at (A) baseline serum phosphate tertiles (ferric citrate curves), (B) baseline serum phosphate tertiles (placebo curves), (C) baseline eGFR level–CKD stage (ferric citrate curves) and (D) baseline eGFR level–CKD stage (placebo curves). Data are LSM (SE) serum phosphate estimates derived from a mixed-effects model for repeated measures analysis with fixed-effects terms for treatment (ferric citrate versus placebo), baseline covariate, visit, treatment × visit interaction, treatment × covariate interaction and treatment × visit × covariate interaction. aP = 0.006 versus placebo. bP = 0.438 versus placebo. cP = 0.236 versus placebo. dNormal laboratory reference range of serum phosphate. eP = 0.013 versus placebo. fP = 0.020 versus placebo. gP = 0.566 versus placebo. BL, baseline; SE, standard error; SP, serum phosphate.
Ferric citrate dose–response
Dose–response analysis confirmed that among patients with baseline serum phosphate within the population reference range, serum phosphate remained within this range, irrespective of ferric citrate dose increases. Among patients with baseline serum phosphate above the population reference range (≥4.5 mg/dL), ferric citrate dose increases progressively lowered serum phosphate to within the population reference range (Figure 3).
FIGURE 3.
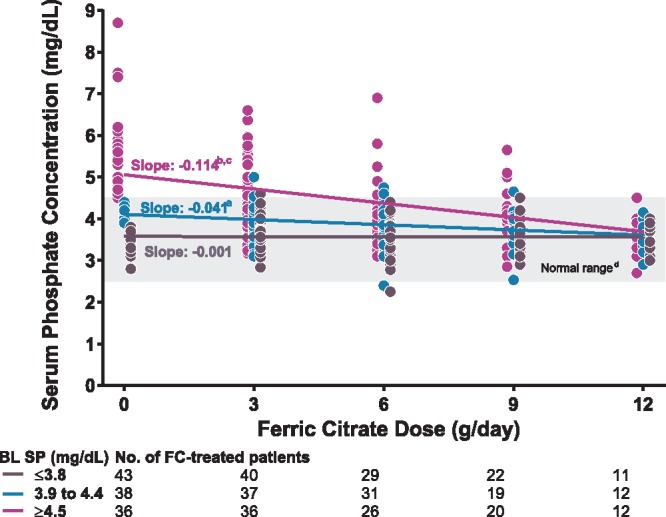
Serum phosphate (SP) concentrations after dose increases of ferric citrate (FC) by baseline phosphate tertile (FC-treated patients only). For each tertile color coded as shown in the legend, circles indicate individual patient serum phosphate concentrations and the lines indicate the mixed-model regression line of best fit. aP = 0.013 versus lowest tertile slope. bP < 0.001 versus lowest tertile slope. cP < 0.001 versus middle tertile slope. dNormal laboratory reference range of SP. BL, baseline.
Baseline associations with change in C-terminal FGF23
We identified treatment, baseline cFGF23 and baseline eGFR as strongly associated (all P < 0.001) with cFGF23 change at 16 weeks in multivariate analysis (Table 2). We confirmed that only baseline TSAT had a significant interaction with treatment (P = 0.028) on the change in cFGF23 at 16 weeks in MMRM analysis. Patients with lower baseline TSAT (25th percentile) achieved earlier cFGF23 reductions than patients with higher baseline TSAT (75th percentile); the ferric citrate versus placebo LSM difference in cFGF23 was significantly greater at 8 weeks only among patients with lower baseline TSAT, whereas this difference was significantly greater at 16 weeks, irrespective of baseline TSAT level (Table 3).
Table 3.
MMRM estimates for change in cFGF23 and iFGF23 overall and by baseline variables with significant treatment × week 16 interaction terms
| MMRM estimates | Ferric citrate | Placebo | Difference |
|---|---|---|---|
| (n = 117) | (n = 115) | (ferric citrate versus placebo) | |
| Change in cFGF23a | |||
| Week 8 | |||
| n | 98 | 91 | |
| Median change (Q1, Q3) | −27.9 (−179.3, 49.8) | 8.8 (−71.1, 59.4) | −36.7 |
| MMRM rank estimates (95% CI) | |||
| Overall | 87.8 (76.9, 98.6) | 102.0 (90.7, 113.2) | −14.2 (−29.8, 1.4) |
| By baseline TSATb | |||
| 15% | 78.8 (65.0, 92.6) | 103.0 (89.5, 116.6) | −24.3 (−43.6, −4.9)* |
| 24% | 93.5 (81.4, 105.7) | 100.9 (87.6, 114.3) | −7.4 (−25.5, 10.7) |
| Week 16 | |||
| n | 85 | 79 | |
| Median change (Q1, Q3) | −66.0 (−209.3, −5.2) | 7.5 (−38.3, 61.4) | −73.5 |
| MMRM rank estimates (95% CI) | |||
| Overall | 68.6 (59.0, 78.2) | 98.7 (88.8, 108.7) | −30.1 (−43.9, −16.3)*** |
| By baseline TSATb | |||
| 15% | 60.5 (48.6, 72.4) | 88.8 (77.2, 100.4) | −28.3 (−44.9, −11.6)** |
| 24% | 73.8 (63.3, 84.2) | 107.6 (96.3, 119.0) | −33.9 (−49.3, −18.5)*** |
| Change in iFGF23a | |||
| Week 8 | |||
| n | 98 | 92 | |
| Median change (Q1, Q3) | −16.2 (−43.2, 9.8) | 4.3 (−21.4, 34.2) | −20.5 |
| MMRM rank estimates (95% CI) | |||
| Overall | 83.8 (73.1, 94.5) | 107.9 (96.9, 119.0) | −24.1 (−39.6, −8.7)** |
| By baseline iPTHb | |||
| 64 pg/mL | 85.8 (72.5, 99.2) | 106.4 (93.6, 119.1) | −20.5 (−39.0, −2.1)* |
| 169 pg/mL | 81.9 (68.5, 95.2) | 109.8 (94.7, 125.0) | −28.0 (−48.2, −7.8)** |
| Week 16 | |||
| n | 86 | 80 | |
| Median change (Q1, Q3) | −22.3 (−48.0, 6.6) | 2.9 (−23.2, 46.5) | −25.2 |
| MMRM rank estimates (95% CI) | |||
| Overall | 71.0 (61.3, 80.7) | 97.6 (87.6, 107.7) | −26.6 (−40.6, −12.7)*** |
| By baseline iPTHb | |||
| 64 pg/mL | 77.4 (65.7, 89.1) | 88.6 (77.5, 99.8) | −11.3 (−27.4, 4.9) |
| 169 pg/mL | 65.2 (53.8, 76.6) | 113.3 (99.9, 126.6) | −48.1 (−65.6, −30.5)*** |
Owing to non-normal distribution of FGF 23, rank transformations were employed.
Estimated at the 25th and 75th percentile values of these continuously distributed baseline variables.
P < 0.5, **P < 0.01, ***P < 0.001.
Q, quartile.
Baseline associations with change in intact FGF23
We identified treatment and baseline iFGF23 as strongly associated (both P < 0.001) with iFGF23 change at 16 weeks in multivariate analysis (Table 2). Baseline serum phosphate was also associated with iFGF23 change at 16 weeks (P = 0.014; Table 2). We confirmed that only baseline iPTH had a significant interaction with treatment (P = 0.005) on the change in iFGF23 at 16 weeks in MMRM analysis. The patients with higher baseline iPTH (75th percentile) achieved larger iFGF23 reductions with ferric citrate versus placebo at 16 weeks than those with lower baseline iPTH (25th percentile) (Table 3).
Path analyses
To further understand the contribution of the change in serum phosphate and the change in iron parameters to the change in FGF23, we used path analyses to assess the influence of earlier temporal changes in serum phosphate and TSAT (up to 14 weeks) on later changes in FGF23 outcomes (at 16 weeks).
Path analyses of cFGF23 changes at 16 weeks identified significant partial mediation by the change in TSAT up to 14 weeks plus additional significant effects of ferric citrate treatment not mediated by the change in serum phosphate or TSAT (Figure 3A). Of note, although the more stringent test of partial mediation by the change in serum phosphate up to 14 weeks (a × b) was not significant, the individual regression coefficients for treatment effect on the change in serum phosphate at 14 weeks (a) and the effect of the change in serum phosphate at 14 weeks on the change in cFGF23 at 16 weeks (b) were both significant (Figure 4A).
FIGURE 4.

Path analyses exploring the change in serum phosphate and TSAT at 14 weeks as mediators of ferric citrate (FC)-induced reduction in (A) cFGF23 at 16 weeks and (B) iFGF23 at 16 weeks. •P < 0.10, *P < 0.05, **P < 0.01, ***P < 0.001. Obs, observed value analysis; RFI, random forest imputation analysis.
Path analyses of iFGF23 changes at 16 weeks identified significant partial mediation by serum phosphate change up to 14 weeks, but not by TSAT change up to 14 weeks, plus additional significant effects of ferric citrate treatment not mediated by the change in serum phosphate or TSAT (Figure 4B).
DISCUSSION
This post hoc analysis demonstrated that ferric citrate–induced reductions in serum phosphate were more pronounced among patients with higher baseline serum phosphate and lower baseline kidney function (eGFR). Among patients with baseline serum phosphate within the population reference range, serum phosphate remained within this range, irrespective of treatment with ferric citrate or placebo or dose of ferric citrate. Among patients with baseline serum phosphate above the population reference range, ferric citrate treatment reduced serum phosphate to within this reference range.
Phosphate binders, including ferric citrate, diminish dietary phosphorus absorption, leading to an increase in gastrointestinal phosphate excretion. Among phosphate binder–treated patients with mild to moderately impaired kidney function and normal to slightly elevated serum phosphate concentrations, serum phosphate reductions are modest and levels generally remain within the population reference range because of several compensatory homeostatic mechanisms, including decreased urinary phosphate excretion [26, 27]. Among phosphate binder–treated patients with severely impaired kidney function and overt hyperphosphatemia, for whom compensatory homeostatic mechanisms are impaired [5], serum phosphate is reduced yet often remains above the population reference range because of the kidneys’ inability to respond optimally to phosphaturic stimuli, including those mediated by PTH and FGF23. Our current findings are consistent with this model.
Ferric citrate treatment reduced FGF23, which is elevated in iron deficiency and CKD [16]. MMRM analyses, which examined baseline associations with longitudinal changes in FGF23, showed that ferric citrate–induced cFGF23 and iFGF23 reductions were associated with baseline TSAT and baseline iPTH, respectively. Ferric citrate reduced cFGF23 earlier in the time course of therapy among patients with lower baseline TSAT versus those with higher baseline TSAT, suggesting that faster repletion of iron stores among patients with initially low iron stores may have provided a stronger stimulus for cFGF23 reduction. Ferric citrate reduced iFGF23 to a greater extent among patients with higher baseline iPTH, likely as a result of upregulation of FGF23 when PTH is high [11].
Path analyses, which examined the dynamic interplay between earlier temporal changes in serum phosphate and TSAT with later changes in FGF23, suggested that ferric citrate reduced FGF23 indirectly via changes in TSAT and serum phosphate and via unknown/unmeasured mechanisms. Ferric citrate effects on cFGF23 were partially mediated by TSAT changes and effects on iFGF23 were partially mediated by serum phosphate changes.
These results provide insights into the regulation of FGF23 by ferric citrate treatment. FGF23 regulation in CKD and IDA is incompletely understood. Inflammation and changes in bone and mineral metabolism stimulate FGF23 transcription in CKD, but proteolytic cleavage is impaired [16, 17], resulting in high cFGF23 and iFGF23. Thus in advanced CKD with associated hyperphosphatemia, nearly all circulating FGF23 is iFGF23 [28, 29]. In contrast, iron deficiency stimulates both FGF23 transcription and cleavage [14, 16–19]. Thus cFGF23 is increased while iFGF23 is maintained at usual levels in iron-deficient patients with normal or near-normal kidney function. By reducing serum phosphate and correcting iron deficiency, ferric citrate acts on both FGF23 stimuli to reduce iFGF23 and cFGF23. Our path analysis suggested that ferric citrate acts via changes in serum phosphate to reduce iFGF23 and via changes in TSAT to reduce cFGF23, findings consistent with the expected effects of hyperphosphatemia and iron deficiency on iFGF23 and cFGF23, respectively.
We also observed significant reductions in FGF23 by ferric citrate that were not mediated by changes in serum phosphate or TSAT. It is unclear if this represents a hitherto unknown direct pharmacologic effect of ferric citrate or represents indirect mediation by unmeasured variables. Although we did not measure the effects on inflammation or vitamin D in this study, other hypothetical mechanisms might include ferric citrate attenuation of the effects of inflammation on FGF23 or modulation of the effects on FGF23 via modification of the vitamin D–FGF23 axis above and beyond effects mediated via phosphate balance. Inflammation upregulates iFGF23, which itself stimulates inflammation, both directly and indirectly, via suppressing 1,25-dihydroxyvitamin D production [11, 17]. By reducing both cFGF23 and iFGF23, ferric citrate may attenuate potential inflammation–FGF23 positive feedback loops. These speculative mechanisms require further investigation.
It is unclear whether ferric citrate alters FGF23 transcription, cleavage or both. In a study of intravenous iron supplementation in iron-deficient patients with elevated cFGF23 and normal iFGF23 at baseline, both iron dextran and ferric carboxymaltose lowered cFGF23; however, only ferric carboxymaltose increased iFGF23 [30]. An increase in iFGF23 was also observed with intravenous iron polymaltose [31], supporting the hypothesis that the carbohydrate moiety of these intravenous iron supplements may impair FGF23 cleavage [30, 32]. Hypophosphatemia and related adverse events were also commonly observed in studies of iron polymaltose and ferric carboxymaltose [31, 33], likely as a result of the increase in iFGF23 induced by these agents. Of note, ferric citrate differs from these intravenous iron formulations, having a very low incidence of hypophosphatemia (<1%) when used for IDA in patients with CKD [21, 34]. In the present study, both cFGF23 and iFGF23 were reduced, suggesting that ferric citrate reduces FGF23 transcription without impairing cleavage, which might explain the low incidence of hypophosphatemia with ferric citrate. These findings are consistent with prior studies in which ferric citrate decreased both cFGF23 and iFGF23 from baseline [35, 36], with minimal changes in serum phosphate [27, 35, 37].
The analyses presented here have several strengths. Laboratory determinations were consistently obtained at regular intervals, in accordance with the trial protocol. We employed several analytic methods to determine the associations among parameters of bone and mineral metabolism, iron status and FGF23 and we found consistent results. Results were neither dependent on the analytic method nor the method’s means of handling missing data. We believe that the complexity of the relations between these biological processes lends itself well to path analyses, which have not yet been widely used in the nephrology literature. The baseline characteristics of the trial population suggest that the findings are generalizable to patients with CKD Stages 3–5 seen in general nephrology outpatient practices. Limitations were the post hoc exploratory nature of these analyses, the relatively short 16-week randomized study period and the lack of measures of vitamin D and urinary phosphate. Longer trials, such as the 48-week COMPASS trial (ClinicalTrials.gov identifier: NCT03236246) may be useful for assessing the long-term effect of ferric citrate on FGF23 or serum phosphate and on clinical outcomes, including cardiovascular events, among patients with NDD-CKD and IDA.
Among patients with NDD-CKD and IDA, ferric citrate reduced serum FGF23 concentrations directly and indirectly (via effects on serum phosphate and iron balance). Ferric citrate reduced serum phosphate among patients with higher baseline serum phosphate but not among patients with baseline serum phosphate within the population reference range. The clinical implications of these concomitant, diverse and potentially beneficial effects on biochemical surrogates are under active investigation.
Supplementary Material
ACKNOWLEDGEMENTS
Vasupradha Vethantham, PhD, of inScience Communications, Springer Healthcare (New York, NY, USA), provided editorial support funded by Keryx Biopharmaceuticals. Additional statistical support was provided by Prometrika.
FUNDING
This study was supported by Keryx Biopharmaceuticals.
AUTHORS’ CONTRIBUTIONS
G.A.B., P.E.P., S.F., K.U., J.F.N. and G.M.C. were involved in the conceptualization and methodology of this study. G.A.B. and P.E.P. were involved in the study investigation. P.E.P., K.U. and J.F.N. conducted the formal analysis. J.G.M. conducted the statistical analyses. Data curation was performed by K.U. R.D.L., K.U. and J.F.N. were involved with visualization and supervision of the study. The original manuscript draft was prepared by G.A.B., G.M.C., J.G.M. and R.D.L. G.M.C., J.G.M., R.D.L. and K.U. prepared critical revisions. All authors contributed to the interpretation and discussion of results and reviewed and edited the manuscript.
CONFLICT OF INTEREST STATEMENT
G.A.B. has served as a consultant for Akebia, Amgen, Ardelyx, AstraZeneca, Celgene, Daiichi Sankyo, Keryx, Relypsa and Sanofi; has ownership interest in Ardelyx and Nephroceuticals; has received research support from Keryx and has received honoraria from Akebia, Amgen, AstraZeneca, Celgene, Daiichi Sankyo, Keryx and Sanofi. P.E.P. has served as a consultant for AstraZeneca, Keryx, Relypsa, Sandoz and Vifor Pharma and has received honoraria from Keryx, Relypsa, Sandoz and ZS Pharma. S.F. has served as a consultant for AstraZeneca, Keryx and Rockwell; has received research funding from Amgen, AstraZeneca and Janssen and has received honoraria from AstraZeneca, Keryx and Rockwell. J.G.M. is an employee of inScience Communications, Springer Healthcare, which was contracted by Keryx to provide statistical support for this post hoc analysis. R.D.L., K.U. and J.F.N. are employees of Keryx and have ownership interest in Keryx. G.M.C. has served as a consultant for Akebia, AMAG, Amgen, Ardelyx, AstraZeneca, Gilead and Keryx; has ownership interest in Ardelyx, Durect, Outset, Physiowave and PuraCath Medical; has received research support from Amgen, Janssen, the National Institute of Diabetes and Digestive and Kidney Diseases and the National Heart, Lung, and Blood Institute and has received honoraria from the American Society of Nephrology.
REFERENCES
- 1. Ganz T, Nemeth E.. Iron balance and the role of hepcidin in chronic kidney disease. Semin Nephrol 2016; 36: 87–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Portoles J, Gorriz JL, Rubio E. et al. The development of anemia is associated to poor prognosis in NKF/KDOQI stage 3 chronic kidney disease. BMC Nephrol 2013; 14: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. van Nooten FE, Green J, Brown R. et al. Burden of illness for patients with non-dialysis chronic kidney disease and anemia in the United States: review of the literature. J Med Econ 2010; 13: 241–256 [DOI] [PubMed] [Google Scholar]
- 4. Babitt JL, Lin HY.. Mechanisms of anemia in CKD. J Am Soc Nephrol 2012; 23: 1631–1634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Block GA, Ix JH, Ketteler M. et al. Phosphate homeostasis in CKD: report of a scientific symposium sponsored by the National Kidney Foundation. Am J Kidney Dis 2013; 62: 457–473 [DOI] [PubMed] [Google Scholar]
- 6. Vervloet MG, Sezer S, Massy ZA. et al. The role of phosphate in kidney disease. Nat Rev Nephrol 2017; 13: 27–38 [DOI] [PubMed] [Google Scholar]
- 7. Kestenbaum B, Sampson JN, Rudser KD. et al. Serum phosphate levels and mortality risk among people with chronic kidney disease. J Am Soc Nephrol 2005; 16: 520–528 [DOI] [PubMed] [Google Scholar]
- 8. Bellasi A, Mandreoli M, Baldrati L. et al. Chronic kidney disease progression and outcome according to serum phosphorus in mild-to-moderate kidney dysfunction. Clin J Am Soc Nephrol 2011; 6: 883–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zoccali C, Ruggenenti P, Perna A. et al. Phosphate may promote CKD progression and attenuate renoprotective effect of ACE inhibition. J Am Soc Nephrol 2011; 22: 1923–1930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Isakova T. Comparison of mineral metabolites as risk factors for adverse clinical outcomes in CKD. Semin Nephrol 2013; 33: 106–117 [DOI] [PubMed] [Google Scholar]
- 11. David V, Francis C, Babitt JL.. Ironing out the cross talk between FGF23 and inflammation. Am J Physiol Renal Physiol 2017; 312: F1–F8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fliser D, Kollerits B, Neyer U. et al. Fibroblast growth factor 23 (FGF23) predicts progression of chronic kidney disease: the Mild to Moderate Kidney Disease (MMKD) Study. J Am Soc Nephrol 2007; 18: 2600–2608 [DOI] [PubMed] [Google Scholar]
- 13. Scialla JJ, Xie H, Rahman M. et al. Fibroblast growth factor-23 and cardiovascular events in CKD. J Am Soc Nephrol 2014; 25: 349–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wolf M. Update on fibroblast growth factor 23 in chronic kidney disease. Kidney Int 2012; 82: 737–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Moe SM, Chertow GM, Parfrey PS. et al. Cinacalcet, fibroblast growth factor-23, and cardiovascular disease in hemodialysis: the Evaluation of Cinacalcet HCl Therapy to Lower Cardiovascular Events (EVOLVE) trial. Circulation 2015; 132: 27–39 [DOI] [PubMed] [Google Scholar]
- 16. Wolf M, White KE.. Coupling fibroblast growth factor 23 production and cleavage: iron deficiency, rickets, and kidney disease. Curr Opin Nephrol Hypertens 2014; 23: 411–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. David V, Martin A, Isakova T. et al. Inflammation and functional iron deficiency regulate fibroblast growth factor 23 production. Kidney Int 2016; 89: 135–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Farrow EG, Yu X, Summers LJ. et al. Iron deficiency drives an autosomal dominant hypophosphatemic rickets (ADHR) phenotype in fibroblast growth factor-23 (FGF23) knock-in mice. Proc Natl Acad Sci USA 2011; 108: E1146–E1155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Imel EA, Peacock M, Gray AK. et al. Iron modifies plasma FGF23 differently in autosomal dominant hypophosphatemic rickets and healthy humans. J Clin Endocrinol Metab 2011; 96: 3541–3549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Block GA, Fishbane S, Rodriguez M. et al. A 12-week, double-blind, placebo-controlled trial of ferric citrate for the treatment of iron deficiency anemia and reduction of serum phosphate in patients with CKD stages 3–5. Am J Kidney Dis 2015; 65: 728–736 [DOI] [PubMed] [Google Scholar]
- 21. Fishbane S, Block GA, Loram L. et al. Effects of ferric citrate in patients with nondialysis-dependent CKD and iron deficiency anemia. J Am Soc Nephrol 2017; 28: 1851–1858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Keryx Biopharmaceuticals. AURYXIA (ferric citrate) tablets: US prescribing information, last updated November 2017. https://www.auryxia.com/hyperphosphatemia/wp-content/uploads/Auryxia_PI_Keryx.pdf (accessed 22 March 2018)
- 23. Cuzick J. Rank Regression. Encyclopedia of Biostatistics. Hoboken, NJ: John Wiley & Sons, 2005 [Google Scholar]
- 24. Bryan A, Schmiege SJ, Broaddus MR.. Mediational analysis in HIV/AIDS research: estimating multivariate path analytic models in a structural equation modeling framework. AIDS Behav 2007; 11: 365–383 [DOI] [PubMed] [Google Scholar]
- 25. Rosseel Y. lavaan: an R package for structural equation modeling. J Stat Software 2012; 48: 1–36 [Google Scholar]
- 26. Block GA, Wheeler DC, Persky MS. et al. Effects of phosphate binders in moderate CKD. J Am Soc Nephrol 2012; 23: 1407–1415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Block GA, Chertow GM, Fishbane S. et al. Auryxia (Ferric Citrate) Effectively Reduces Serum Phosphate and Fibroblast Growth Factor 23 (FGF23) Levels in Patients With Stages 3-5 Chronic Kidney Disease (CKD). Am J Kidney Dis 2015; 65: B1–B14 [abstract 35]) [Google Scholar]
- 28. Shimada T, Urakawa I, Isakova T. et al. Circulating fibroblast growth factor 23 in patients with end-stage renal disease treated by peritoneal dialysis is intact and biologically active. J Clin Endocrinol Metab 2010; 95: 578–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Weber TJ, Liu S, Indridason OS, Quarles LD.. Serum FGF23 levels in normal and disordered phosphorus homeostasis. J Bone Miner Res 2003; 18: 1227–1234 [DOI] [PubMed] [Google Scholar]
- 30. Wolf M, Koch TA, Bregman DB.. Effects of iron deficiency anemia and its treatment on fibroblast growth factor 23 and phosphate homeostasis in women. J Bone Miner Res 2013; 28: 1793–1803 [DOI] [PubMed] [Google Scholar]
- 31. Schouten BJ, Hunt PJ, Livesey JH. et al. FGF23 elevation and hypophosphatemia after intravenous iron polymaltose: a prospective study. J Clin Endocrinol Metab 2009; 94: 2332–2337 [DOI] [PubMed] [Google Scholar]
- 32. White KE, Hum JM, Econs MJ.. Hypophosphatemic rickets: revealing novel control points for phosphate homeostasis. Curr Osteoporos Rep 2014; 12: 252–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Prats M, Font R, Garcia C. et al. Effect of ferric carboxymaltose on serum phosphate and C-terminal FGF23 levels in non-dialysis chronic kidney disease patients: post-hoc analysis of a prospective study. BMC Nephrol 2013; 14: 167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chertow GM, Block GA, Neylan JF. et al. Safety and efficacy of ferric citrate in patients with nondialysis-dependent chronic kidney disease. PLoS One 2017; 12: e0188712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Iguchi A, Kazama JJ, Yamamoto S. et al. Administration of ferric citrate hydrate decreases circulating FGF23 levels independently of serum phosphate levels in hemodialysis patients with iron deficiency. Nephron 2015; 131: 161–166 [DOI] [PubMed] [Google Scholar]
- 36. Iguchi A, Yamamoto S, Yamazaki M. et al. Effect of ferric citrate hydrate on FGF23 and PTH levels in patients with non-dialysis-dependent chronic kidney disease with normophosphatemia and iron deficiency. Clin Exp Nephrol 2017; 22: 789–796 [DOI] [PubMed] [Google Scholar]
- 37. Fishbane S, Block GA, Loram LC. et al. Auryxia (Ferric Citrate), An Oral Iron-Based Phosphate Binder Significantly Improves Serum Iron Measures in Patients With Stage 3, 4 and 5 Chronic Kidney Disease (CKD). Am J Kidney Dis 2015; 65: B1–B14 [abstract 83] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.