

Abstract
Clonal evolution drives tumor progression, chemoresistance and relapse in cancer. Little is known about clonal selection induced by therapeutic pressure in multiple myeloma. To address this issue, we performed large targeted sequencing of bone marrow plasma cells in 43 multiple myeloma patients at diagnosis and at relapse from exactly the same intensive treatment. The most frequently mutated genes at diagnosis were KRAS (35%), NRAS (28%), DIS3 (16%), BRAF and LRP1B (12% each). At relapse, the mutational burden was unchanged. Many of the mutations were present at the subclonal level at both time points, including driver ones. According to patients and mutations, we observed different scenarios: selection of a very rare subclone present at diagnosis, appearance or disappearance of mutations, but also stability. Our data highlight that chemoresistance and relapse could be induced by newly acquired mutations in myeloma drivers but also by (sub)clonal mutations preexisting to the treatment. Importantly, no specific mutation or rearrangement was observed at relapse, demonstrating that intensive treatment has a nonspecific effect on clonal selection in multiple myeloma. Finally, we identified 22 cases of biallelic event, including a double event deletion 17p/TP53mut.
INTRODUCTION
Multiple Myeloma (MM) is characterized by the accumulation within the bone marrow of malignant plasma cells.1 Genetically, MM is characterized by a large heterogeneity. Recent sequencing studies showed that patients present in median about 60 exonic mutations, but with a wide range, from 10 to more than 500.2–5 This inter-patient heterogeneity becomes even more complex if we look at the intra-patient level. It has been clearly shown that MM is a subclonal disease, meaning that most tumor plasma cells share a common pool of mutations, but may differ by several subclonal mutations.6–10 How will subclones evolve during MM course? It has been shown by different techniques that the major subclone at the time of diagnosis may be different from the major one observed at first relapse, which can also differ from those seen at later relapses. There is probably a competition between the clones in the bone marrow niche for survival, and several authors proposed that this competition relies on the Darwinian model.11–13 Subclones may differ by the presence of “driver” mutations (mutations that give advantages to this subclone), and/or neutral “passenger” mutations.14 As in other cancers, several kinds of clonal evolution have been described in MM: stable evolution (identical genomic profile at diagnosis and relapse), linear evolution (apparition of novel mutations at relapse, but with the same mutational architecture), and branching evolution (“disappearance” of some mutations revealing evolution from a minor undetected subclone, or appearance of novel different subclones).15, 16
The major questions are how these subclones emerge, and how (and why) some are selected. The answer is certainly not unequivocal. Local characteristics in the bone marrow niche (such as nutriment accessibility or hypoxia) may select the clone(s) with the best fitness. Some mutations may for instance generate neo-antigens that can drive immune responses. Differential proliferative capacity of subclones may also participate to the selection. Finally, chemotherapy may also play a major role, in killing the most sensitive cells but selecting the more resistant ones, and for drugs possessing mutagenic effect in directly affecting tumor cells. If the answers to the first issues are not straightforward, addressing the last issue is more feasible. To address it, we performed large targeted sequencing in a series of 43 patients with frozen samples at the time of diagnosis and first relapse from exactly the same treatment. All these 43 patients have been treated homogeneously with four cycles of Velcade®-Thalidomide-Dexamethasone (VTD) induction, followed by one high-dose melphalan with autologous stem cell support, and two cycles of VTD consolidation (VTD-MEL200-VTD). The targeted panel included 246 genes recurrently mutated in MM, 2358 single-nucleotide polymorphisms (SNPs) for copy number analyses, and the whole IGH sequence to detect all the recurrent 14q32 translocations.
METHODS
Study subjects
For this study, we selected 43 homogeneously treated myeloma patients for whom frozen CD138-enriched samples were available at the time of diagnosis and first relapse. The median time to progression was 22 months (range, 10–55). Patient characteristics are summarized in Table 1. All patients provided signed consent for these genetic analyses in accordance with the Declaration of Helsinki. This study was approved by Toulouse Ethic Committee. They all received an induction course with 4 cycles of the VTD combination (Velcade®-Thalidomide-Dexamethasone), an intensive course with melphalan 200 mg/m² followed by autologous stem cell transplant, and a consolidation phase with 2 cycles of VTD. None of them received a maintenance phase. FISH data were available for all patients at diagnosis and relapse for t(4;14) translocation and 17p deletion. The patients were treated in 20 different centers from the Intergroup Francophone du Myélome, but all the samples were sent to one central laboratory, where cell sorting, DNA extraction and sequencing were performed.
Table 1.
Patient characteristics
| Patient id. | Sex | Age at diagnosis | FISH analysis for del(17p) and t(4;14) at diagnosis | FISH analysis for del(17p) and t(4;14) at relapse | PFS (months) |
|---|---|---|---|---|---|
| 2 | M | 52 | positive for t(4;14) and del(17p) | positive for t(4;14) and del(17p) | 5 |
| 4 | M | 57 | negative | negative | 18 |
| 5 | F | 53 | negative | negative | 25 |
| 6 | M | 64 | negative | negative | 23 |
| 7 | M | 54 | positive for t(4;14) | positive for t(4;14) | 12 |
| 8 | M | 54 | negative | negative | 21 |
| 9 | F | 54 | negative | negative | 36 |
| 10 | M | 66 | positive for t(4;14) | positive for t(4;14) | 14 |
| 11 | M | 62 | negative | negative | 33 |
| 12 | M | 61 | positive for t(4;14) | positive for t(4;14) | 19 |
| 13 | F | 62 | negative | negative | 27 |
| 14 | F | 62 | positive for t(4;14) | positive for t(4;14) | 38 |
| 15 | F | 68 | negative | negative | 23 |
| 16 | M | 67 | negative | negative | 15 |
| 17 | M | 68 | negative | negative | 38 |
| 18 | M | 69 | negative | negative | 32 |
| 19 | F | 55 | negative | positive for del(17p) | 10 |
| 20 | F | 55 | negative | positive for del(17p) | 11 |
| 21 | M | 69 | negative | negative | 40 |
| 22 | M | 68 | negative | negative | 21 |
| 23 | F | 69 | negative | positive for del(17p) | 55 |
| 24 | F | 70 | negative | negative | 10 |
| 25 | F | 62 | negative | negative | 41 |
| 26 | M | 72 | negative | negative | 35 |
| 27 | M | 69 | negative | negative | 15 |
| 28 | F | 52 | negative | negative | 55 |
| 29 | M | 55 | positive for t(4;14) | positive for t(4;14) | 10 |
| 30 | M | 52 | negative | negative | 42 |
| 31 | M | 68 | negative | negative | 14 |
| 32 | F | 43 | negative | negative | 22 |
| 33 | M | 64 | negative | negative | 10 |
| 34 | M | 53 | negative | negative | 31 |
| 36 | F | 60 | positive for t(4;14) | positive for t(4;14) | 11 |
| 37 | F | 65 | negative | negative | 47 |
| 39 | M | 59 | negative | negative | 21 |
| 40 | M | 66 | negative | negative | 19 |
| 44 | M | 68 | negative | negative | 12 |
| 45 | F | 63 | negative | negative | 28 |
| 46 | M | 71 | negative | negative | 21 |
| 47 | F | 69 | negative | negative | 17 |
| 48 | M | 55 | negative | negative | 36 |
| 51 | M | 70 | negative | negative | 36 |
| 53 | M | 61 | negative | negative | 29 |
| TOTAL | Ratio M/F = 1,7 | Median= 62 | t(4;14) = 16%; del(17p) = 2% | t(4;14) = 16%; del(17p) = 9% | Median = 22 |
Targeted sequencing
Plasma cells were isolated from bone marrow using CD138+ MAC-Sorting (Miltenyi Biotec, Paris, France). Post-sorting purity was systematically checked by May-Grünwald-Giemsa staining and cytological analysis of a spin from positive fraction. Only samples with more than 80% of plasma cells after sorting were kept for DNA extraction. Median purity was 97% (range 83–100%). Constitutional control DNA was extracted from peripheral white blood cells from a pool of 12 out of 43 patients. For sequencing, libraries were prepared with the SureSelect QXT Target Enrichment for Illumina Multiplexed Sequencing kit from Agilent and we used the targeted panel recently published.17 Briefly, this panel contains all the coding sequences of 246 genes recurrently mutated in MM (supplemental Table 1), 2358 SNPs randomly dispersed on the genome for copy number analyses, and the IGH sequences to identify the IGH translocations. All the technical characteristics have been previously published.16 Pools of libraries were sequenced (paired-end 2*150) on a NextSeq500 Illumina (308x in mean for tumoral DNA samples and 239x in mean for constitutive DNA samples).
Bio-informatic and statistical analyses
The sequencing data was first aligned against the Ensembl GRCh38 reference genome using BWA (version 0.7.9a with mem option,18) with Ensembl gene annotation. Duplicated reads were removed using Picard-tools (version 1.114 with MarkDuplicates option, see https://broadinstitute.github.io/picard/). Supplemental Table 2 indicates the number of reads and the quality metrics of the sequencing data for each sample. The raw data are available on the NCBI database, with SRA accession number SRP127780 (https://www.ncbi.nlm.nih.gov/sra/SRP127780). Aligned data were prepared for mutation calling following the standard Broad Institute’s GATK pipeline (https://software.broadinstitute.org/gatk/best-practices/). Mutations were then called using MuTect (version 1.1.4,19) with additional mutation annotation from Cosmic and dbSNP databases. The 12 patients with sequenced constitutional DNA were directly compared to their matched reference. Those 12 references were then pooled to constitute a normal reference for the 31 other patients. Of note, those constitutional DNA were sequenced and pre-processed using the same pipeline as described above. Mutations flagged as “REJECT” or “UNCOVERED” as well as mutations identified in untargeted regions were removed from further analysis. Remaining mutations were further annotated using Annovar.20 Copy number variants were called using a modified version of CopyCat (https://github.com/chrisamiller/copyCat) adapted to exclude regions commonly affected by CNV in MM from normalisation benchmark. Finally, translocations were identified using BreakDancer (version 1.0,21) and manually filtered to keep only translocations involving the IGH gene region. The code used for all those computation is available in supplemental methods.
RESULTS
Mutations
From the 246 genes recurrently mutated in MM we analyzed, we found a median of 5 exonic nonsynonymous mutations per patient at diagnosis (range: 0–16) and 6 at relapse (range: 0–14). Focusing only on mutations with an allelic fraction superior to 0.1, we obtained 4 at diagnosis (range 0–12) and 5 at relapse (range 0–12). Importantly, no specific mutation that could have been selected, and thus enriched, by the treatment was observed (Figure 1 and Table 2). In agreement with previous publications on whole exome sequencing, the two most frequently mutated genes at diagnosis were KRAS (35% of the patients) and NRAS (28%), followed by DIS3 (16%), BRAF and LRP1B (12% each). All the other recurrent mutations were observed in less than 10% of the patients. If we arbitrarily consider that clonality is defined by an allelic fraction of at least 40%, these five gene mutations were clonal in 8/15, 8/12, 5/7, 1/5 and 3/5, respectively, at diagnosis (Table 2 and supplemental Figures 1 and 2). For these four genes, the mutational pattern was generally stable at relapse, with some differences. For the KRAS gene, we observed very different scenarios according to patients: stability, total disappearance of a subclonal mutation (one case), appearance of a clonal or subclonal mutation (two cases), selection of a very rare subclone with an increase to a (sub)clonal mutation at relapse (two cases) (Table 2 and supplemental figure 1). We even observed one case with a KRAS G13D mutation occurring at relapse, whereas a KRAS A146V disappeared (Figure 2 and supplemental figure 1). For NRAS, one clonal mutation at diagnosis was found to become bi-allelic at relapse, and two cases might also display the selection of a rare subclone (the mutation was identified in 1/199 reads, and ¼61 reads, respectively, reads that could either be the result of the sequencing of a rare subclone or carry sequencing errors). Of note, no disappearance of NRAS mutation was observed at relapse. Amongst DIS3 mutations present at diagnosis, one third disappeared at relapse, and no appearance was observed at relapse. Regarding BRAF mutations, all were particularly stable between diagnosis and relapse, without any disappearance or appearance.
Figure 1. Comparison of mutational profiles at diagnosis and first relapse in 43 multiple myeloma patients.
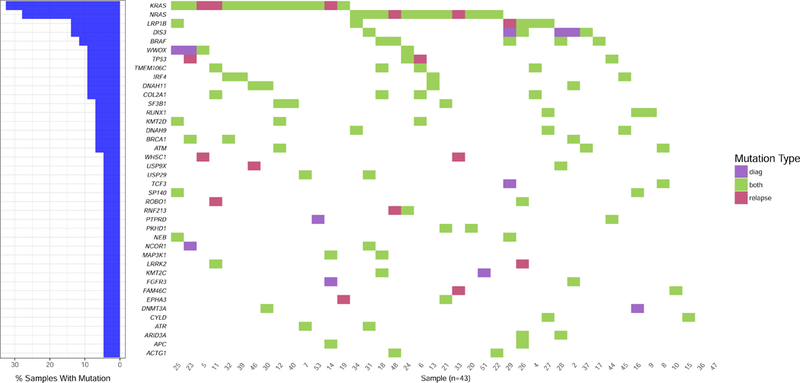
Only non-synonymous mutations found in at least two patients with an allelic fraction ≥ 0.1 are represented. Presented percentages of samples with mutations are calculated as the sum of cases with stable mutation (i.e. present at diagnosis and relapse) and cases with unstable mutation (i.e. present either at diagnosis or at relapse).
Table 2.
Mutational profile at diagnosis and relapse
| Patient id. | Mutations at diagnosis (allelic fraction ≥ 0.1) | Mutations at relapse (allelic fraction ≥ 0.1) |
|---|---|---|
| 2 | FGFR3, BRCA1, DNAH11, RNF213, PCLE1, DIS3 (sub) | FGFR3, BRCA1, DNAH11, RNF213, PCLE1, MAP4K4 (sub) |
| 4 | TMEM106C, COL2A1, LRP1B | TMEM106C, COL2A1, LRP1B |
| 5 | WWOX (bi), NOTCH1 (sub) | WWOX (bi), NOTCH1 (sub), KRAS, WHSC1, DNAH5 (sub) |
| 6 | NRAS, TMEM106C, COL2A1 | NRAS, TMEM106C, COL2A1, TP53, EP300 (sub) |
| 7 | SLAMF8, USP29, ATR, ZFHX3, FAT4, KRAS (sub) | SLAMF8, USP29, ATR, ZFHX3, FAT4, KRAS (sub) |
| 8 | TCF3, ATM (sub) | TCF3, ATM (sub) |
| 9 | RUNX1, PBRM1 | RUNX1, PBRM1 |
| 10 | EGFR, DNAH9, ATM, FAM46C (sub) | EGFR, ZFHX3, FAT4, FAM46C, BRCA2 (sub) |
| 11 | LRRK2 (bi), TMEM106C, COL2A1 | LRRK2 (bi), TMEM106C, COL2A1, KRAS (sub), ROBO1 (sub), LRP1B (sub) |
| 12 | KRAS (bi), ATM, FOXA2, SF3B1(sub) | KRAS (bi), ATM, FOXA2, SF3B1(sub) |
| 13 | NRAS, IRF4, DNAH11 | NRAS, IRF4, DNAH11 |
| 14 | NOTCH1 (bi), PTPRT, APC (sub), MAP3K1 (sub), HOX19 (sub), FGFR3 (sub) | NOTCH1 (bi), PTPRT, APC (sub), MAP3K1 (sub), HOX19 (sub), KRAS (sub) |
| 15 | DIS3 (bi), KRAS, FAT4, CYLD (sub) | DIS3 (bi), KRAS, FAT4, CYLD |
| 16 | CUL4A, RUNX1, SP140 (sub), DNMT3A (sub) | CUL4A, RUNX1, SP140 (sub) |
| 17 | MRE11A, ARIDIA, ROS1, PTPRZ1, BRAF (sub), KRAS (sub) | MRE11A, ARIDIA, ROS1, PTPRZ1, BRAF (sub) |
| 18 | COL2A1, TMEM106C (sub), MAP3K9 (sub), NRAS (sub), MAP3K1 (sub), BRAF (sub), KMT2C (sub), NOTCH1 (sub) | COL2A1, TMEM106C, MAP3K9, NRAS (sub), MAP3K1 (sub), BRAF (sub), KMT2C (sub), NOTCH1 (sub) |
| 19 | KRAS | KRAS, CREBBP (sub), ZFHX3 (sub), EPHA3 (sub), PNRC1 (sub) |
| 20 | NRAS, PRDM1, PKHD1 | NRAS, PRDM1, PKHD1 |
| 21 | TRAF3, NRAS, NF1, SF3B1, EPHA3, PKHD1 | TRAF3, NRAS, NF1, SF3B1, EPHA3, PKHD1 |
| 22 | MAGED1 (bi), NRAS, ACTG1, ERN1 | MAGED1 (bi), NRAS, ACTG1, ERN1 (sub) |
| 23 | DDX1, BRCA1, KRAS (sub), WWOX (sub), SNX7 (sub), NCOR (sub) | DDX1, BRCA1, KRAS (sub), WWOX (sub), SNX7 (sub), TP53 |
| 24 | NRAS, WWOX (bi), IKZF3, TP53, RNF213 | NRAS, WWOX (bi), IKZF3, TP53, RNF213, CYLD1 (sub) |
| 25 | LRPB1, NEB, SP140, C11orf30 (sub), WWOX (sub), KRAS A146V (sub), RAG2 (sub) | LRPB1, NEB, SP140, C11orf30 (sub), WWOX (sub), KRAS G13D |
| 26 | PI4KA (bi), BRCA2, DIS3, ARID3A, ALK, ROBO1, APC, LRPB1 (sub) | PI4KA (bi), BRCA2, DIS3, ARID3A, ALK, ROBO1, APC, LRPB1 (sub), LRKK2 (sub) |
| 27 | ARID5B, NOTCH2, CYLD, DNAH9, LRP1B, RUNX1 | ARID5B, NOTCH2, CYLD, DNAH9, LRP1B, RUNX1 |
| 28 | ARID2, EGR1, BRAF (sub), USP9X, ARID3A (sub), DIS3 (sub) | ARID2, EGR1, BRAF, USP9X, ARID3A (sub) |
| 29 | BRAF, MGA (sub), NEB (sub), DIS3, TCF3 (sub) | BRAF, MGA, NEB, TET2 (sub) |
| 30 | RNF213, NFE2L2, DNMT3A, DNAH11, KRAS (sub) | RNF213, NFE2L2, DNMT3A, DNAH11, KRAS (sub) |
| 31 | FAM46C (bi), NRAS, KMT2D, EP400, DIS3, USP29, NCOR1 (sub), ATR (R1082H) (sub), ATR (V316I) (sub), PCL0 (sub) | FAM46C (bi), NRAS (bi), KMT2D, EP400, DIS3, USP29, NCOR1 (sub), ATR (R1082H) (sub), ATR (V316I) (sub), PCL0 (sub) |
| 32 | KRAS, KMT2D, BRCA1, BRCA2, TSHZ1, PTPRT, IRF4 | KRAS, KMT2D, BRCA1, BRCA2, TSHZ1, PTPRT, IRF4 |
| 33 | TET2 (bi), USP9X, FAT1, ZFHX3, FANCD2, SETD2, NOTCH1, NOTCH4, FANCA, KRAS (sub), TRAF3 | TET2 (bi), USP9X, FAT1, ZFHX3, FANCD2, SETD2, NOTCH1, NOTCH4, FANCA, NRAS (sub), WHSC1 (sub), PRDM1 (sub), FAM46C (sub) |
| 34 | NRAS, DNAH9, ADAM29, CDKN2A, LRP1B (sub) | NRAS, DNAH9, ADAM29 (sub), CDKN2A, LRP1B, PTPRK (sub) |
| 36 | No detectable mutation | KDM5B (sub) |
| 37 | DIS3, FAT3 (sub), RPL5 (sub), ATM (sub) | DIS3, FAT3 (sub), RPL5 (sub), ATM (sub) |
| 39 | KRAS, IRF4 (sub) | KRAS, IRF4 (sub), PIK3C2G (sub) |
| 40 | KRAS (sub), BIRC3 (sub), SF3B1 (sub), IKZF1 (sub) | KRAS (sub), BIRC3 (sub), SF3B1 (sub), IKZF1 (sub) |
| 44 | TP53, PTRD, MAP3K4 (sub) | TP53, PTRD |
| 45 | CCND1 (sub), DNAH9 (sub), IRF4 (sub), NTRK2 (sub) | CCND1 (sub), DNAH9 (sub), IRF4 (sub), NTRK2 (sub) |
| 46 | KRAS, ASAP2, DNAH11 (sub) | KRAS, ASAP2, DNAH11, USP9X (sub) |
| 47 | No detectable mutation | No detectable mutation |
| 48 | ACTG1, BRAF (sub), HIST1H1C (sub) | ACTG1, BRAF, HIST1H1C (sub), NRAS (sub), RNF213 (sub) |
| 51 | NRAS (sub), KMT2C (sub) | NRAS (sub), KMT2C (sub) |
| 53 | KRAS, PTRD (sub) | KRAS |
Mutations in blue or red are unstable (present at diagnosis or relapse only, respectively). Mutations in italic are stop-gain mutations.(sub) means subclonal and (bi) means bi-allelic.
Figure 2. Comparison of genetic profiles at diagnosis and relapse in 43 multiple myeloma patients.

Circle plots from three representative patients. (A) Patient #2, (B) Patient #25, (C) Patient #51. The outer layer (black) indicates chromosomal copy-number at diagnosis, the inner circle at relapse. Mutations are represented by circles which size is proportional to the allelic fraction. Mutations found at diagnosis only are represented in blue, those found at relapse only are represented in red, and others are represented in green. The inner links represent classical translocations involving the IGH region.
Globally we observed 30 cases of mutations appearance at relapse, distributed in 42% of patients and involving 23 genes (Table 2 and supplemental Figures 1 and 2). For instance we observed the appearance of TP53 mutations at relapse in two patients. These mutations do not seem to be a clonal selection of a minor mutated clone, since they were clonal at relapse but not seen at diagnosis (0/206 and 0/593 reads). This phenomenon was also observed with WHSC1 (0/504 and 0/758 reads), MAP4K4 (0/331), DNAH5 (0/563), CREBBP (0/295), EPHA3 (0/252), PNRC1 (0/107), TET2 (0/657), PIK3C2G (0/436), PRDM1 (0/789), RNF213 (0/412), LRKK2 (0/355) or, as described below, KRAS (0/375 and 0/273). On the other hand, some appearances might have originated from the selection of a rare subclone, as in BRCA2 (9/108 reads, becoming subclonal at relapse with an allelic fraction 0,14), ROBO1 (2/378 reads, becoming subclonal at relapse with an allelic fraction = 0.25), USP9X (½02 reads, becoming subclonal at relapse with an allelic fraction = 0.18), FAM46C (2/620 reads, becoming subclonal at relapse with an allelic fraction = 0.21), and as described below KRAS (1/370 and 4/303 reads, becoming respectively subclonal with an allelic fraction = 0.17 and clonal with an allelic fraction = 0.42) and NRAS (1/199 and ¼61 reads becoming subclonal with allelic fraction = 0.35 and 0.25, respectively) (Table 2 and supplemental Figures 1 and 2).
CRBN pathway mutations, which have been shown to induce resistance to IMiDs, were observed in 7/43 patients (16%) on CUL4, IRF4, IKZF1 and IKF3 genes. All were remarkably stable between diagnosis and relapse. In contrast, none of the mutations described in having role in proteasome inhibitors resistance (XBP1, proteasome subunits) were detected in our cohort. Finally, amongst all non-synonymous mutations having allelic frequency superior or equal to 0.1 detected by the panel, we observed that the number of transversions (substitution of a purine for a pyrimidine and vice versa) was stable between diagnosis and relapse (Figure 3).
Figure 3. Comparison of transversion frequencies at diagnosis and relapse in 43 multiple myeloma patients.
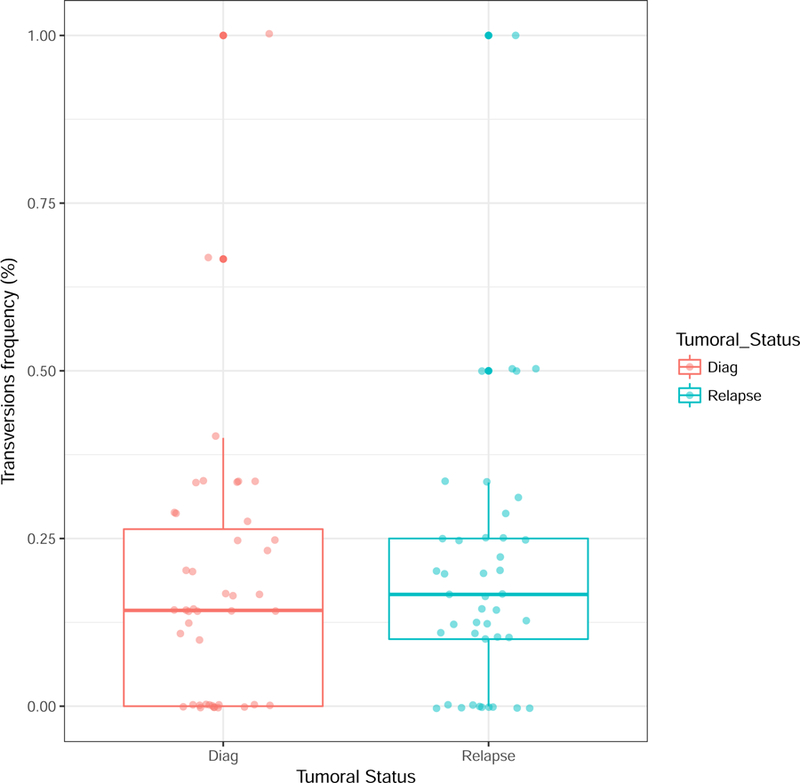
Transversions refers to the substitution of a purine for a pyrimidine and vice versa, i.e. to the substitution of an A or a G for a C or a T.
IGH translocations and copy number alteration
At diagnosis, translocations involving the IGH gene at 14q32 were found in 27 (63%) patients, including thirteen cases (30%) of t(11;14), seven cases (16%) of t(4;14), two cases (5%) of t(6;14), one case (2%) of t(14;16) and one case (2%) of t(14;20), which were the expected proportions (Table 3). The chromosomal partners were those usually described (CCND1 at 11q13, WHSC1/MMSET and FGFR3 at 4p16, CCND3 at 6p21, MAF at 16q23, and MAFB at 20q11, respectively). None of these translocations changed at relapse. We also observed 4 cases (10%) of t(8;14) involving MYC at 8q24, all of them being already present at diagnosis. The only case of concomitant translocations was the patient #51, which displays both a t(11;14) and a t(8;14) from diagnosis (Figure 2).
Table 3.
Translocations and copy number alterations
| Patient id. | Translocations 14q32 (diagnosis and relapse) | Copy number alterations at relapse |
|---|---|---|
| 2 | t(4;14) | Loss 19q |
| 4 | t(11;14) | Loss 5p, loss 8p, gain 16q |
| 5 | t(8;14) | Gain 5p,+6 partial |
| 6 | No detectable translocation | Loss 3p |
| 7 | t(4;14) | Loss 1p, gain 1q, gain 2p, −10, gain 19p, loss 19q |
| 8 | No detectable translocation | Stable |
| 9 | t(11;14) | Loss 11q |
| 10 | t(4;14) | Gain 5q |
| 11 | No detectable translocation | Gain 1q, loss 3p, loss 4q, loss 8p |
| 12 | t(4;14) | Loss 1p, gain 1q, gain 4q, gain 17q |
| 13 | t(11;14) | Loss 12q |
| 14 | t(4;14) | Loss 7q, loss 19q |
| 15 | t(14;20) | Loss 2q |
| 16 | No detectable translocation | Gain 9q, gain 11p, −13, +18, +19 |
| 17 | No detectable translocation | Loss 4q, gain 11p |
| 18 | No detectable translocation | Loss 11p |
| 19 | No detectable translocation | Loss 5q, gain 11q, loss 17p |
| 20 | No detectable translocation | Gain 1p, loss 2p, gain 6q, loss 8p, gain 9p, loss 10p, gain 11q, loss 12p, gain 13q, loss 17p, loss 19q, gain 20q |
| 21 | t(11;14) | Loss 1q, loss 8p, loss 9p, loss 9q, loss 12q, gain 14q |
| 22 | No detectable translocation | Loss 1p, gain 11p, loss 13q, loss 16q |
| 23 | No detectable translocation | Loss 6q, loss 17p |
| 24 | t(11;14) | +6, −10, loss 16q, +21 |
| 25 | No detectable translocation | +3, +8, +9, loss 13q, +18, +20, +21 |
| 26 | t(11;14) | Loss 12q |
| 27 | t(8;14) | Gain 20q, gain 21q |
| 28 | t(11;14) | Gain 1q, loss 9p, loss 12q |
| 29 | t(4;14) | Loss 1p, gain 2q, loss 6q, −10, −11, −15, gain 20q |
| 30 | No detectable translocation | Loss 1p, gain 1q, loss 5q |
| 31 | No detectable translocation | Gain 1p, gain 1q, gain 3q, loss 14q, loss 19p, loss 19q, gain 22q |
| 32 | t(11;14) | loss 2p |
| 33 | t(14;16) | -4 partial, gain 4q, loss 6q, gain 12p, loss 12q, gain 20q |
| 34 | t(11;14) | Stable |
| 36 | t(4;14) | Loss 1p, loss 3q, loss 5q, loss 6p, gain 8q, loss 11p, loss 12p, gain 12p, loss 13q, loss 20p |
| 37 | t(8;14) | Loss 8p, +9 partial |
| 39 | t(11;14) | Loss 13q, loss 15q |
| 40 | t(6;14) | Stable |
| 44 | No detectable translocation | Loss 5q, gain 6p |
| 45 | t(11;14) | Gain 1q |
| 46 | No detectable translocation | Gain 1q, loss 13q, loss 16q, gain 18q, loss 19q |
| 47 | t(11;14) | Stable |
| 48 | No detectable translocation | +6, gain 7q, −18 |
| 51 | t(11;14) + t(8;14) | Stable |
| 53 | t(6;14) | gain 7q, loss 20q |
Novel copy number alterations were observed in 38/43 patients at the time of relapse (Table 3). However, because of the relatively low SNP coverage, small changes may have been missed in the 5 patients with an apparently stable molecular karyotype. Of note the mutational pattern of these patients was also stable at relapse, and these 5 patients displayed a median progression free survival similar to the cohort (21 vs 22 months, respectively). However, most of the patients with stable mutational pattern display novel copy number alteration at relapse. The two most frequent changes were gains of 1q (8/43 cases) and losses of 1p (6/43 cases). Losses of 1p involved several regions (3 cases were del(1p32)), whereas 1q gains involved the whole long arm. Losses of 17p appeared at relapse in ¾3 cases, including one with a concomitant appearance of TP53 mutation (Patient #23). Only one deletion 17p was observed at diagnosis in our cohort (Patient #2), and this was maintained at relapse (Figure 2).
Biallelic events in relapsing samples
We then focused on putative biallelic events in relapsing samples by arbitrarily filtering on dominant mutations (allelic fraction ≥ 0.6) and we detected them in 16/43 patients, including six patients displaying each two biallelic events (Table 3). We identified 9 cases of biallelic mutations regarding WWOX (2 cases), LRRK2, KRAS, NOTCH1, DIS3, NRAS, FAM46C and TET2 genes and 12 cases of combination mutation plus copy number alteration regarding RUNX, CUL4, TRAF3, USP29, PTRPZ1, PKHD1, PRDM1, FAT1, ARID3A, PI4KA, CYLD and TP53 genes. Amongst these patients, we observed that the majority displayed the double-event from diagnosis, others displaying the “second hit” between diagnosis and relapse (patients #7, #16, #31 and 33). Finally, only one patient (#23) acquired the two events between diagnosis and relapse, a del17p/TP53mut.
DISCUSSION
Chemotherapy is considered as a driving factor of the tumor evolution process leading to relapse and chemoresistance. However, few studies have analyzed clonal evolution after treatment,22–25 and no one in myeloma after exactly the same treatment. In this study, we addressed the issue of a homogeneous therapeutic pressure on the clonal selection, by analyzing a cohort of 43 uniformly treated myeloma patients with large targeted sequencing on CD138-sorted plasma cells. In cancer in general, it has been thought that treatment may play an important role in clonal selection, by killing the most sensitive clones, and sparing the most resistant ones.26 We hypothesized that with a homogeneous treatment, relapsed samples would be enriched with specific mutated and/or rearranged clones. We found that for the mutations targeted by our panel, there was no significant change in the mutational burden of these tumors following exposure to treatment. Interestingly, no specific rearrangement or mutation was observed in the relapse samples. This observation can be interpreted in different ways. First, it could mean that chemotherapy has no specific effect on the clone selection responsible for relapse, and this selection is patient-dependent, related to internal factors such as local bone marrow and/or immune conditions. A second explanation could be that the broad therapeutic approach used in this cohort is not targeting specific pathways, whereas a more selective therapy such as a lenalidomide-dexamethasone combination could have generated different results. Finally, the more likely explanation is that the same pressure applied on a heterogeneous group induces a heterogeneous result, because it interacts differently with each acquired event within the subclones. The selection or acquisition of mutant under selection pressure of treatment has been shown to be a key driver of the development of drug resistance in other hematological malignancies (eg, BCR-ABL mutations in imatinib-treated chronic myeloid leukemia27 or BTK mutations in chronic lymphocytic leukemia patients treated with the selective inhibitor ibrutinib28). However in myeloma evolution, changes in clonal substructure may be more important than specific mutational event, as suggested by studies of smoldering myeloma progression.10
An intriguing observation was the occurrence in some patients of a clonal or subclonal population at the time of relapse, which was not at all detectable at the time of diagnosis, even when specifically curing the data for these specific mutations. For instance, two patients presented a clonal TP53 mutation at the time of relapse, which were not detectable at the time of diagnosis, at least at the level of 1/593, whereas other detected mutations were highly stable. This suggests that these TP53 mutations occurred during evolution on an ancestral tumor cell that subsequently took proliferative and/or survival advantage on the other clones to be fully clonal at relapse. However, it has been suggested that genetically resistant subclones already exist before treatment suggesting that clonal selection of preexistant populations is the main mechanism for acquired resistance to treatment.29 In our cohort, some mutations described as inducing chemoresistance increased in frequency between diagnosis and relapse, such as NRAS, KRAS and TP53 mutations, but other were absolutely stable such as BRAF and CRBN pathway lesions confirming that these mutations preexist and are not induced by treatment.25 Furthermore, we also observed some cases of apparent selection of very minor subclones such as ones displaying KRAS or NRAS mutations. Nevertheless, given the very small number of reads, a study by performing ultra-deep sequencing targeted on given mutations would be necessary to confirm the existence of these minor subclones, but also the authenticity of mutations appearance/disappearance that we observed. When mutations seem to be acquired at relapse, the next question would be the timing of this occurrence. Several hypotheses could be proposed: (i) a therapy-induced mutation, and the most plausible time would be the intensive melphalan course, since alkylating agent are known to cause DNA damages; (ii) but it could be also during the time of remission, outside any treatment impact. To address this question, we started a prospective study aiming to sort the residual tumor plasma cells before and after intensification, in order to perform sequencing on these cells. Of note, the 15 mutations we observed amongst members of the DNA-damage-repair pathway (TP53, ATM, ATR, BRCA1, BRCA2) were stable between diagnosis and relapse excepted the two cases of TP53 mutations appearance. Furthermore, the number of transversions, which have been linked to cytotoxic effect of chemotherapy in relapsed acute myeloid leukemia,22 was stable too.
Finally, we observed that many of these mutations were present at the subclonal level. Mutations are classified as subclonal on the posterior probability that the cancer cell fraction is lower than 95%.30 Because our study deals with tumor displaying either pseudodiploid karyotypes, or hyperdiploid karyotypes, and with pure samples (the median purity being 97%, the less pure of our cohort displaying 83% of purity), we considered that subclonality can be defined by an allelic fraction lower than 40%. This observation confirm previous studies revealing subclonal mutations in well-known “driver” genes, such as KRAS, NRAS or BRAF.2, 3, 25 Generally these mutations were mutually exclusive, but we observed two cases of co-occurring BRAF and NRAS mutations, as previously described.2, 3 We report here that not only these mutations are subclonal at diagnosis, but also that all of them were mainly still subclonal at relapse. These results suggest that intensive treatment fail to eradicate subclonal clones in a large majority of cases. These results may also suggest that these genes activating the RAS/MAPK pathway are not always drivers in MM development and might sometimes occur later in the disease course. Their appearance at relapse suggests that resistance to chemotherapy could be driven by acquisition of new driver mutations, inducing evolution toward fittest clones.25 From a therapeutic point of view, even if BRAF mutations were the V600E in 4/5 patients, it is important to note that in two of these four cases, the mutation was subclonal, preventing the use of the specific inhibitor vemurafenib in monotherapy in those patients.
Even if such targeted sequencing approach has already been successfully performed in untreated31 and refractory32 myeloma patients, one of the major limits of our study resides in the very principle of targeted approach: some mutations may inherently have been missed. In particular, since the 246 genes of the panel have been mainly selected in patients at diagnosis,17 mutations occurring at relapse may have gone unnoticed. However, it is worth noting that the list of genes also included many genes described at relapse, such CD40, CUL4 or PSMB5 and that the panel was validated with several MM cell lines originated from relapsed/refractory patients. Furthermore, most of the mutations inducing chemoresistance were included in the panel. Hence, we were able to notably detect mutations inducing drug resistance by triggering MAPK/ERK pathway (NRAS, KRAS, BRAF), TP53 mutations, CRBN pathway mutations inducing resistance to Imids,32, 33 XBP1 and proteasome subunits mutations described as leading to resistance to proteasome inhibitors,34 TRAF3 mutations inducing dexamethasone resistance but sensitivity to proteasome inhibitors.35 Finally, rare whole exome sequencing studies in relapsing myeloma actually found quite little in terms of new mutated genes, but rather highlighted higher prevalence of mutations in known ones, such as TP53 and RAS.25 Another limit of our study is that we only looked at one bone marrow aspirate in each patient at both time points, whereas like in solid tumors,36 a spatial heterogeneity has been demonstrated in myeloma, some focal lesions displaying several specific mutations as compared to the “random” bone marrow aspirate.37, 38
Weinhold et al. recently published a comparable study in 33 patients treated with high dose therapy strategies but not with exactly the same treatment. Paired samples were more comprehensively analyzed than we did, since gene expression profile, copy number arrays and whole exome sequencing were performed. 25 This study highlighted the critical nature of biallelic inactivation events affecting tumor suppressor genes, especially TP53 in resistance to apoptosis and increase in proliferation rate. Here we observed that amongst patients involved by biallelic events, those displaying one event at diagnosis and the second one at relapse experienced a particularly early relapse suggesting that what seems also critical is to get the second hit during or after treatment (patients #7, #16, #31 and #33: 12, 15, 14 and 10 months after diagnosis, respectively, Tables 1 and 3). However, a larger cohort and additional time points would be necessary to determinate the impact of the events kinetic. Regarding mutations of prognostic value, only those involving TP53 are widely recognized. Indeed, the two patients displaying a TP53 mutation at diagnosis had a particularly short progression free survival (10 and 12 months, Tables 1 and 2).
In conclusion, we report that no specific rearrangement or mutation detectable by the targeted panel was observed in the relapse samples of 43 patients primary treated with VTD-MEL200-VTD, suggesting that this regimen has a nonspecific effect on the clone selection. We already knew that molecular heterogeneity in multiple myeloma is huge at diagnosis, including at the patient level. This study brings another layer of complexity since each myeloma seems to evolve differently from the others at relapse, even if homogenously treated. In addition, this study highlights that chemoresistance and relapse could be induced by newly acquired mutations in myeloma drivers but also by (sub)clonal mutations preexisting to the treatment. These findings have clear potential translational implication because they suggest that personalized medicine must include and probably target the evolving nature of tumors, based on the determination of clonal evolution in each patient, including allelic fraction of targetable mutations. Future studies with other treatment strategies, in particular more selective therapy, are needed to determinate if similar results are generated.
Supplementary Material
Table 4.
Biallelic events in relapsing samples
| Patient id. | Gene | AF at diagnosis | AF at relapse | Locus | CNA at diagnosis | CNA at relapse | Conclusion |
|---|---|---|---|---|---|---|---|
| 5 | WWOX | 0.9 | 0.9 | 16q23 | No | No | biallelic mutation |
| 7 | USP29 | 0.4 | 0.8 | 19q1 | No | Deletion 60% | became homozygous mutation at relapse |
| 11 | LRRK2 | 0.9 | 0.9 | 12q12 | No | No | biallelic mutation |
| 12 | KRAS | 0.9 | 0.8 | 12p12 | No | No | biallelic mutation |
| 14 | NOTCH1 | 0.7 | 0.7 | 9q34 | Trisomy 100% | Trisomy 100% | biallelic mutation |
| 15 | DIS3 | 1.0 | 0.9 | 13q21 | No | No | biallelic mutation |
| 15 | CYLD | 0.2 | 0.7 | 16q12 | Deletion 100% | Deletion 80% | homozygous mutation |
| 16 | RUNX1 | 0.6 | 0.7 | 21q22 | Gain 50 % | Gain 50 % | amplified mutation |
| 16 | CUL4A | 0.7 | 1.0 | 13q34 | Deletion 50% | Deletion 85 % | became homozygous mutation at relapse |
| 17 | PTPRZ1 | 0.7 | 0.6 | 7q31 | Gain 50 % | Gain 50 % | amplified mutation |
| 20 | PRDM1 | 0.7 | 0.6 | 6q21 | Gain 50 % | Gain 50 % | amplified mutation |
| 20 | PKHD1 | 0.6 | 0.7 | 6p12 | Gain 50 % | Gain 50 % | amplified mutation |
| 21 | TRAF3 | 0.9 | 0.8 | 14q32 | Deletion 80% | Deletion 80% | homozygous mutation |
| 23 | TP53 | 0.0 | 0.6 | 17p13 | No | Deletion 80% | homozygous mutation |
| 24 | WWOX | 1.0 | 0.6 | 16q23 | No | No | biallelic mutation |
| 26 | ARID3A | 0.6 | 0.6 | 19p13 | Gain 50 % | Gain 50 % | amplified mutation |
| 26 | PI4KA | 1.0 | 0.8 | 22q11 | Deletion 80% | Deletion 80% | homozygous mutation |
| 27 | RUNX1 | 0.7 | 0.7 | 21q22 | Gain 50 % | Gain 50 % | amplified mutation |
| 31 | NRAS | 0.4 | 1.0 | 1p13 | No | No | became biallelic mutation at relapse |
| 31 | FAM46C | 0.7 | 1.0 | 1p12 | No | No | biallelic mutation |
| 33 | TET2 | 1.0 | 1.0 | 4q24 | No | Deletion 80% | biallelic mutation with additional deletion at relapse |
| 33 | FAT1 | 0.5 | 0.6 | 4q35 | No | Gain 20% | became amplified mutation at relapse |
ACKNOWLEDGEMENTS
This work was supported by NIH grants PO1-155258 and P50-100707 (N.M., H.A.L.); Department of Veterans Affairs Merit Review Award 1 I01BX001584-01 (N.M.) and the CAPTOR program. The CRCT Team 13 is labeled by ARC. We thank the Intergroupe Francophone du Myelome for providing patient samples and clinical data.
Footnotes
CONFLICT OF INTEREST
N.B. received honoraria and personal fees from Celgene Corporation. The remaining authors declare no competing financial interests.
REFERENCES
- 1.Palumbo A, Anderson K. Multiple myeloma. N Engl J Med 2011. March 17; 364(11): 1046–1060. [DOI] [PubMed] [Google Scholar]
- 2.Bolli N, Avet-Loiseau H, Wedge DC, Van Loo P, Alexandrov LB, Martincorena I, et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nature communications 2014; 5: 2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lohr JG, Stojanov P, Carter SL, Cruz-Gordillo P, Lawrence MS, Auclair D, et al. Widespread genetic heterogeneity in multiple myeloma: implications for targeted therapy. Cancer Cell 2014. January 13; 25(1): 91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Walker BA, Boyle EM, Wardell CP, Murison A, Begum DB, Dahir NM, et al. Mutational Spectrum, Copy Number Changes, and Outcome: Results of a Sequencing Study of Patients With Newly Diagnosed Myeloma. J Clin Oncol 2015. November 20; 33(33): 3911–3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature 2013. August 22; 500(7463): 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Egan JB, Shi C-X, Tembe W, Christoforides A, Kurdoglu A, Sinari S, et al. Whole-genome sequencing of multiple myeloma from diagnosis to plasma cell leukemia reveals genomic initiating events, evolution, and clonal tides. . Blood 2012. August 2, 2012 120 (5 ): 1060–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walker BA, Wardell CP, Melchor L, Hulkki S, Potter NE, Johnson DC, et al. Intraclonal heterogeneity and distinct molecular mechanisms characterize the development of t(4;14) and t(11;14) myeloma. Blood 2012. August 02; 120(5): 1077–1086. [DOI] [PubMed] [Google Scholar]
- 8.Keats JJ, Chesi M, Egan JB, Garbitt VM, Palmer SE, Braggio E, et al. Clonal competition with alternating dominance in multiple myeloma. Blood 2012. August 2, 2012; 120(5): 1067–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Magrangeas F, Avet-Loiseau H, Gouraud W, Lode L, Decaux O, Godmer P, et al. Minor clone provides a reservoir for relapse in multiple myeloma. Leukemia 2013. February; 27(2): 473–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Walker BA, Wardell CP, Melchor L, Brioli A, Johnson DC, Kaiser MF, et al. Intraclonal heterogeneity is a critical early event in the development of myeloma and precedes the development of clinical symptoms. Leukemia 2014. February; 28(2): 384–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morgan GJ, Walker BA, Davies FE. The genetic architecture of multiple myeloma. Nat Rev Cancer 2012. April 12; 12(5): 335–348. [DOI] [PubMed] [Google Scholar]
- 12.Nowell PC. The clonal evolution of tumor cell populations. Science 1976. October 01; 194(4260): 23–28. [DOI] [PubMed] [Google Scholar]
- 13.Burrell RA, McGranahan N, Bartek J, Swanton C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature 2013. September 19; 501(7467): 338–345. [DOI] [PubMed] [Google Scholar]
- 14.Greaves M, Maley CC. Clonal evolution in cancer. Nature 2012. January 18; 481(7381): 306–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Corre J, Munshi N, Avet-Loiseau H. Genetics of multiple myeloma: another heterogeneity level? Blood 2015. March 19; 125(12): 1870–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robiou du Pont S, Cleynen A, Fontan C, Attal M, Munshi N, Corre J, et al. Genomics of Multiple Myeloma. J Clin Oncol 2017. March 20; 35(9): 963–967. [DOI] [PubMed] [Google Scholar]
- 17.Bolli N, Li Y, Sathiaseelan V, Raine K, Jones D, Ganly P, et al. A DNA target-enrichment approach to detect mutations, copy number changes and immunoglobulin translocations in multiple myeloma. Blood Cancer Journal 2016. 09/02/online; 6: e467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010. March 01; 26(5): 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nature biotechnology 2013. March; 31(3): 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic acids research 2010. September; 38(16): e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen K, Wallis JW, McLellan MD, Larson DE, Kalicki JM, Pohl CS, et al. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nature methods 2009. September; 6(9): 677–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ding L, Ley TJ, Larson DE, Miller CA, Koboldt DC, Welch JS, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 2012. January 11; 481(7382): 506–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johnson BE, Mazor T, Hong C, Barnes M, Aihara K, McLean CY, et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 2014. January 10; 343(6167): 189–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Devarakonda S, Govindan R. Clonal evolution: multiregion sequencing of esophageal adenocarcinoma before and after chemotherapy. Cancer discovery 2015. August; 5(8): 796–798. [DOI] [PubMed] [Google Scholar]
- 25.Weinhold N, Ashby C, Rasche L, Chavan SS, Stein C, Stephens OW, et al. Clonal selection and double-hit events involving tumor suppressor genes underlie relapse in myeloma. Blood 2016. September 29; 128(13): 1735–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McGranahan N, Swanton C. Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell 2015. January 12; 27(1): 15–26. [DOI] [PubMed] [Google Scholar]
- 27.O’Hare T, Eide CA, Deininger MW. Bcr-Abl kinase domain mutations, drug resistance, and the road to a cure for chronic myeloid leukemia. Blood 2007. October 01; 110(7): 2242–2249. [DOI] [PubMed] [Google Scholar]
- 28.Woyach JA, Furman RR, Liu TM, Ozer HG, Zapatka M, Ruppert AS, et al. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N Engl J Med 2014. June 12; 370(24): 2286–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Amirouchene-Angelozzi N, Swanton C, Bardelli A. Tumor Evolution as a Therapeutic Target. Cancer discovery 2017. July 20. [DOI] [PubMed]
- 30.Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, et al. Absolute quantification of somatic DNA alterations in human cancer. Nature biotechnology 2012. May; 30(5): 413–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kortum KM, Langer C, Monge J, Bruins L, Egan JB, Zhu YX, et al. Targeted sequencing using a 47 gene multiple myeloma mutation panel (M(3) P) in −17p high risk disease. Br J Haematol 2015. February; 168(4): 507–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kortum KM, Mai EK, Hanafiah NH, Shi CX, Zhu YX, Bruins L, et al. Targeted sequencing of refractory myeloma reveals a high incidence of mutations in CRBN and Ras pathway genes. Blood 2016. September 01; 128(9): 1226–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Surget S, Lemieux-Blanchard E, Maiga S, Descamps G, Le Gouill S, Moreau P, et al. Bendamustine and melphalan kill myeloma cells similarly through reactive oxygen species production and activation of the p53 pathway and do not overcome resistance to each other. Leuk Lymphoma 2014. September; 55(9): 2165–2173. [DOI] [PubMed] [Google Scholar]
- 34.Lu S, Wang J. The resistance mechanisms of proteasome inhibitor bortezomib. Biomarker research 2013. March 01; 1(1): 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Keats J, Fonseca R, Chesi M, Schop R, Baker A, Chng W, et al. Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer Cell 2007; 12: 131–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gundem G, Van Loo P, Kremeyer B, Alexandrov LB, Tubio JM, Papaemmanuil E, et al. The evolutionary history of lethal metastatic prostate cancer. Nature 2015. April 16; 520(7547): 353–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de Haart SJ, Willems SM, Mutis T, Koudijs MJ, van Blokland MT, Lokhorst HM, et al. Comparison of intramedullary myeloma and corresponding extramedullary soft tissue plasmacytomas using genetic mutational panel analyses. Blood Cancer J 2016. May 20; 6: e426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rasche L, Chavan SS, Stephens OW, Patel PH, Tytarenko R, Ashby C, et al. Spatial genomic heterogeneity in multiple myeloma revealed by multi-region sequencing. Nature communications 2017. August 16; 8(1): 268. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.