

Abstract
Background
In contrast to multiple rare monogenetic abnormalities, a common biomarker among children with infantile autism and their parents is the discovery of serum autoantibodies directed to the folate receptor alpha (FRα) localized at blood-brain and placental barriers, impairing physiologic folate transfer to the brain and fetus. Since outcome after behavioral intervention remains poor, a trial was designed to treat folate receptor alpha (FRα) autoimmunity combined with correction of deficient nutrients due to abnormal feeding habits.
Methods
All participants with nonsyndromic infantile autism underwent a routine protocol measuring CBC, iron, vitamins, coenzyme Q10, metals, and trace elements. Serum FRα autoantibodies were assessed in patients, their parents, and healthy controls. A self-controlled therapeutic trial treated nutritional derangements with addition of high-dose folinic acid if FRα autoantibodies tested positive. The Childhood Autism Rating Scale (CARS) monitored at baseline and following 2 years of treatment was compared to the CARS of untreated autistic children serving as a reference.
Results
In this self-controlled trial (82 children; mean age ± SD: 4.4 ± 2.3 years; male:female ratio: 4.8:1), FRα autoantibodies were found in 75.6 % of the children, 34.1 % of mothers, and 29.4 % of fathers versus 3.3 % in healthy controls. Compared to untreated patients with autism (n=84) whose CARS score remained unchanged, a 2-year treatment decreased the initial CARS score from severe (mean ± SD: 41.34 ± 6.47) to moderate or mild autism (mean ± SD: 34.35 ± 6.25; paired t-test p<0.0001), achieving complete recovery in 17/82 children (20.7 %). Prognosis became less favorable with the finding of higher FRα autoantibody titers, positive maternal FRα autoantibodies, or FRα antibodies in both parents.
Conclusions
Correction of nutritional deficiencies combined with high-dose folinic acid improved outcome for autism, although the trend of a poor prognosis due to maternal FRα antibodies or FRα antibodies in both parents may warrant folinic acid intervention before conception and during pregnancy.
1. Introduction
Autism spectrum disorders represent neurodevelopmental disorders characterized by qualitative impairment of communicative and noncommunicative skills, impaired social interaction, and limited interests with stereotypies. Earlier classifications distinguished between infantile autism, Asperger syndrome, childhood disintegrative disorder, PDD-NOS, and Rett syndrome.
So far, no single etiology or common final pathway explaining the features shared by all autism spectrum disorders has been identified, although a number of diverse monogenetic, infectious, toxic or environmental causes have been associated with a minority of cases [1–5].
In the prenatal period, adequate folate delivery to the developing embryo is necessary to prevent the occurrence of neural tube defects (NTD) and possibly other congenital malformations [6]. Maternal folate deficiency increases the risk not only for NTD, but also for autism spectrum disorders (ASD) [7, 8]. Even in the presence of a normal maternal folate status, maternal serum FRα autoantibodies directed against the FRα localized at the placental barrier were shown to block adequate folate delivery across the placenta, predisposing to intrauterine folate deficiency with consequent congenital malformations or autism spectrum disorders [9–13]. Postnatal development of serum FRα-autoantibodies directed against the FRα attached to the choroid plexus epithelial cells at the blood-brain barrier causes the so-called infantile-onset cerebral folate deficiency (CFD) syndrome associated with autism in about 1/3 of cases. Subsequent studies also confirmed a high prevalence of serum FRα autoantibodies in autism spectrum disorders without neurological deficits, where these FRα antibodies were identified in both the child and its parents [9–12, 14–16].
Previous studies on autism suggested increased vulnerability to oxidative stress and decreased methylation capacity as contributory factors [17–20]. However, existing evidence was heterogeneous and inconclusive since many studies were limited by the small size [21]. Further studies postulated mitochondrial dysfunction underlying oxidative stress [14, 15]. Oxidative stress refers to an imbalance between prooxidative factors and antioxidants resulting in abundant production of reactive oxygen species (ROS), being superoxide anions, hydrogen peroxide, and hydroxyl radicals. These ROS possess highly reactive, unpaired electrons capable of initiating a cascade of biochemical reactions with damage to proteins, carbohydrates, fatty acids, lipids, and DNA molecules. Since autistic individuals often manifest feeding difficulties, it is not surprising that excess or multiple deficiencies of vitamins, metals, and trace elements will develop, part of which are essential nutrients and cofactors for intermediary brain metabolism and for antioxidant defences like cofactors of antioxidant enzymes and radical scavengers [21–25]. Thus, in addition to mitochondrial dysfunction, feeding disturbances, and malabsorption in autism may represent alternative mechanisms responsible for oxidative stress due to increased prooxidative factors and/or failing antioxidant defence mechanisms. Another aspect of feeding disturbances deranging nutrient concentrations is the negative impact of these aberrant nutrient concentrations with regard to brain development, nurturing, structure, neurometabolic processes, and regulation of gene expression.
In a previous study we found that the generation of superoxide anions in vitro catabolizes 5-methyl-tetrahydro-folate by 75% within one hour, which can be prevented through preincubation with the radical scavenger ascorbic acid [26]. This study also found that KB-cell culture exposure to superoxide anions and hydrogen peroxide reduces cellular folate incorporation mediated by FRα or RFC1 transport mechanisms. Thus transmembrane folate passage mediated by these transporters at the placenta and choroid plexus is expected to be impaired in the presence of ROS and predisposes to intrauterine folate deficiency and cerebral folate deficiency.
For these reasons, the first objective of our study design was to identify and correct aberrant nutritional derangements and in particular those markers contributing to oxidative stress mediated by elevated prooxidants and/or deficient antioxidant factors. In addition, we screened serum FRα autoantibodies in children with autism and their parents followed by high-dose folinic acid supplements upon finding positive antibody results. We have chosen a self-controlled treatment trial tailored to each individual by the findings of FRα antibodies and abnormal nutritional values. In comparison with the CARS evolution of an age- and gender-matched group of untreated patients, we have monitored the CARS at baseline and after two years of treatment where patients served as their own controls.
2. Patients and Methods
2.1. Participants
Diagnostic investigations included the ADI-R and ADOS tests, Childhood Autism Rating Scale (CARS), developmental and speech assessment, extensive psychological and psychiatric assessment, and observation at school or in the domestic situation. Each patient had a complete history, physical, and neurologic examination and underwent a brain MRI and prolonged EEG registration.
The CARS score is based on the cumulative score obtained on 15 separate items, where a score below 30 indicates absence of sufficient signs and symptoms evocative of autism, a score between 30 and 36 1/2 is compatible with mild to moderately severe autism, and a score from 37 to 60 is compatible with severe autism [27]. Routine investigations included metabolic screening measuring urinary amino acids and organic acids, and creatine and guanidinoacetate excretion. Genetic testing included Angelman syndrome, fragile-X-syndrome, the MECP2 gene defects, chromosome analysis, and array CGH to detect microdeletions or microduplications.
Only patients diagnosed with nonsyndromic infantile autism and without genetic abnormalities were recruited after exclusion of brain abnormalities, intractable epilepsy, and metabolic and recognizable genetic abnormalities or syndromes.
2.2. Design of the Self-Controlled Treatment Trial
The self-controlled intervention trial was approved by both the IRB at State University New York and Ethics committee at Liege University Hospital (Protocol: FOL040113). Partial Funding was provided by Autism Speaks (Grant #8202 to EVQ) for measuring folate receptor autoantibodies.
Recruited patients with nonsyndromic infantile autism have been divided into two groups, the untreated patients serving as a reference group (n=84) and the group of treated patients who participated and completed the self-controlled trial (n=82). Both groups have been matched according to age, gender, CARS score, and the FRα-antibody profile for child and parents. In the self-controlled treatment study conducted from 2013 to 2018, all autistic patients from both groups underwent a fasting blood drawing for complete blood count (CBC), serum and RBC folate, vitamin B12, plasma homocysteine, renal and liver function, thyroid function (TSH, T3, and T4), lactate, CPK, alkaline phosphatase, serum iron, transferrin and ferritin, calcium, magnesium, cholesterol and apolipoprotein B, copper and coeruloplasmin, zinc, manganese, selenium, coenzyme Q10, vitamin E and gamma-tocopherol, vitamin A and beta-carotene, and vitamin D. Serum samples were used to measure antigliadin antibodies and FRα autoantibodies of the binding and blocking type in the patients and their parents (see below).
Prior to the study, the age of untreated patients (n=84) varied between 1 and 16.8 years, and their CARS was plotted as a function of age, providing the evolution of CARS with progressing age among untreated patients. Likewise, the CARS of participating patients who completed the self-controlled trial (n=82) was plotted at baseline and after two years of treatment. The age of the latter patients varied between 1 and 15.9 years.
Based upon previous experience from case-control studies where antioxidant deficiencies and serum FRα antibodies were found, we outlined an adapted treatment protocol for each individual aimed at correcting nutritional derangements (deficient or excess nutrient) by oral supplements (Table 1). Blood levels were rechecked every 3-4 months during the study period of at least two years and used to adapt supplement administrations accordingly. In addition, oral administration of high-dose folinic acid was started at a dose of 0.5-1 mg/kg/day, if FRα autoantibodies came back positive [28]. Folinic acid doses could be increased up to 2 mg/kg/day with a maximum daily dose of 50 mg, if a clinical response did not occur after 6 months [14, 15, 29]. In this self-controlled trial the CARS score was repeated after therapy over two years and compared to the CARS at baseline. We also looked at the changes in CARS scores depending on the initial serum FRα autoantibody titers in the child and as a function of the eight different combinations for each family where FRα autoantibodies tested negative in the child and both parents, or FRα antibodies were only positive in the child, the mother, father, or any combination thereof.
Table 1.
The treatment protocol for the self-controlled treatment trial based upon abnormal biochemical findings and FRα autoantibodies.
| Abnormal biomarker | Daily oral supplement dosage |
| Zinc deficiency | 0.15-0.25 mg/kg zinc-sulfate |
| Selenium deficiency | 3-5 µg/kg sodium-selenite |
| Manganese deficiency | 5-10 mg/kg Vitamin C, 20 IU/kg Vitamine E, with 1 coffespoon Soya oil at night. |
| Manganese excess | idem |
| Heavy metal excess (Cu, Al, Hg, Pb) | idem |
| Raised copper/zinc ratio | idem |
| Bèta-carotene excess | idem; limit foods rich in bèta-carotene |
| Vitamin A deficiency | 600-1500 µg |
| Vitamin D (25-hydroxy-D) | 10 µg or 400 IU |
| Vitamin C deficiency | 5-10 mg/kg Vitamine C (maximal 500mg) |
| Ubiquinon-10 deficiency | 2 mg/kg co-enzyme Q10 |
| Vitamin E deficiency | 20 IU/kg |
| Gamma-Tocopherole deficiency | 1 coffeespoon soya, corn or sesame oil |
| Bèta-carotene deficiency | Consume tomato or carot juices |
| Serum folate deficiency | 0.5 mg/kg folinic acid |
| RBC folate deficiency | 0.5 mg/kg folinic acid |
| Apolipoproteine B deficiency | Supplement vitamins A D E, and vitamine K in case of secondary coagulation disorder |
| FR-alpha antibodies | Start with 0.5-1 mg/kg folinic acid daily; |
| Increase to 2 mg/kg daily without a clinical response after six months. Maximum daily dose 50 mg. |
2.3. Serum FRα Autoantibodies
The assay for both the blocking and binding FRα autoantibodies has been described previously. Blocking FRα autoantibodies were expressed as pmoles of folic acid blocked from binding to FRα per ml of serum and binding FRα autoantibodies were expressed as pmoles of IgG antibody per ml of serum [30, 31]. Serum from 30 healthy controls (age range: 1-18 years) and their parents was tested for the presence of FRα autoantibodies.
3. Results
3.1. Untreated Autism Patients
A group of 84 patients did not give consent to participate in the treatment trial but agreed to have routine laboratory testing as described above. Age (mean ± SD: 4.45 ± 2.62; range: 1-16.8 years), gender (male:female ratio: 5.46:1), and serum FR antibody profiles (71.4 % children testing positive; 30.8 % maternal and 27.4 % paternal serum samples tested positive) matched with the group of 82 who agreed to participate and completed the self-controlled treatment trial. Compared to the baseline CARS scores of the 82 treated patients, the CARS scores for the untreated 84 patients matched and showed no statistical difference in function of each age group ranging from the age group of 2 years up to the age group at or beyond 6 years (Figure 1(a)). The evolution of CARS score for the untreated patient group did not change significantly with advancing age. However, the baseline CARS for the treatment group showed a higher score for the group of children at or beyond 6 years as compared to the baseline CARS assessed for the youngest age groups at 2 and 3 years (see Figure 1(b), Table 2).
Figure 1.
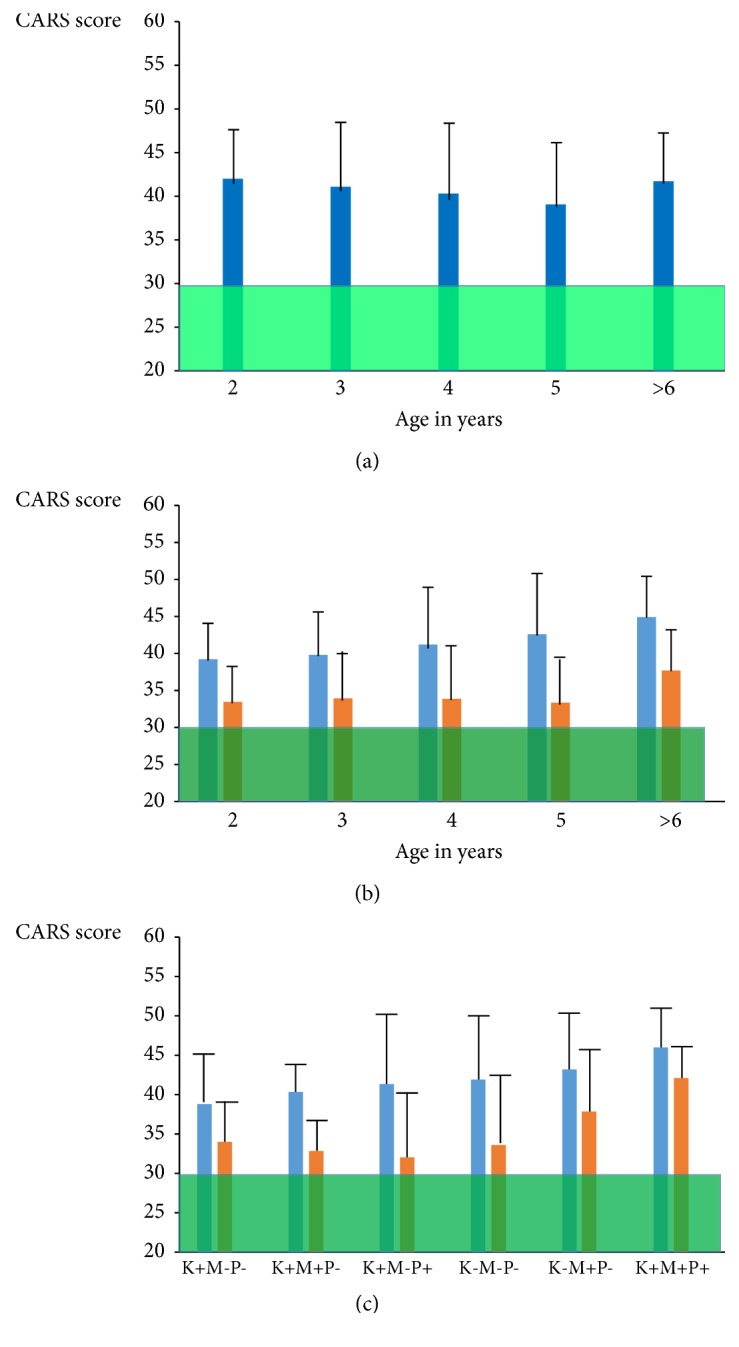
The upper figure (a) shows the plotted CARS with age for 84 untreated patients. The middle figure (b) shows the effect of treatment among 82 treated patients (blue bars represent CARS at baseline and orange bars the CARS after two years treatment). Figure (c) represents the treatment results among different groups with FR autoantibodies in the child (K), mother (M), or father (P).
Table 2.
The upper part shows the CARS score in function of age in the reference group of 84 untreated children with infantile autism. The table below shows the effect of treatment after 2 years upon the CARS score evolution in 82 children with a significant decrease for each age group. The lowest part of the table shows the CARS at baseline and after two-year treatment for 66 families in function of the profile of positive (+) or negative (-) FRα autoantibodies in the child, mother, or father. N stands for the number of subjects.
(a).
| Age | N | CARS score |
|---|---|---|
| 1 | 1 | 36.5 |
| 2 | 20 | 41.92 ± 5.4 |
| 3 | 21 | 41.12 ± 6.7 |
| 4 | 21 | 40.33 ± 7.56 |
| 5 | 9 | 39.05 ± 4.07 |
| ≥ 6 | 12 | 41.75 ± 4.25 |
(b).
| Age | N | CARS at baseline | CARS after treatment | paired t-test | p value |
|---|---|---|---|---|---|
| ( Mean ± SD) | ( Mean ± SD) | ||||
| 1 | 1 | 38 | 38 | ||
| 2 | 10 | 39.35 ± 5.2 | 33.6 ± 4.9 | 3.31 | 0.009 |
| 3 | 31 | 40.01 ± 5.8 | 33.96 ± 5.9 | 7.79 | <0.0001 |
| 4 | 19 | 41.37 ± 6.7 | 34 ± 7.9 | 4.66 | 0.0002 |
| 5 | 10 | 42.8 ± 8.2 | 33.55 ± 5.5 | 4.99 | 0.0007 |
| ≥ 6 | 11 | 45.09 ± 6.46 | 37.7 ± 5.15 | 6.02 | 0.0001 |
(c).
| FRα antibody profile | N | CARS at baseline | CARS after treatment | paired t-test | p value | ||
|---|---|---|---|---|---|---|---|
| Child | Mother | Father | ( Mean ± SD) | (Mean ± SD) | |||
| + | - | - | 23 | 39,06 ± 6,17 | 34,28 ± 4,21 | 4.86 | <0.0001 |
| + | + | - | 11 | 40,54 ± 3,71 | 33,14 ± 4,42 | 5.64 | 0.0002 |
| + | - | + | 11 | 41.5 ± 9.08 | 32.32 ± 7.6 | 4.49 | 0.0012 |
| - | - | - | 9 | 42.05 ± 6.8 | 33.88 ± 8.19 | 7.6 | <0.0001 |
| - | + | - | 5 | 43.5 ± 6.5 | 38 ± 7.06 | 3.41 | 0.027 |
| + | + | + | 7 | 46.14 ± 4.8 | 42.21 ± 3.8 | 2.61 | 0.0398 |
3.2. Self-Controlled Therapeutic Trial
After informed consent, a total of 115 children diagnosed with severe infantile autism were initially recruited for the self-controlled treatment trial during two years. Ten children were excluded from participation in the study because of underlying syndromes or genetic causes. Parents of one child were first-line cousins; one child suffered from Rubenstein-Taybi syndrome, another child and his mother had the MECP2 duplication, one child had a de novo 2p16.3 deletion containing the neurexin I (NRXN I) gene, and one patient was heterozygous for the SLC6A4 p.G56A gene and the SLC29A4 p.D326E gene, encoding the SERT and PMAT proteins, respectively. Copy number variations were found in five other children with a 12p11.21 microdeletion in one child and microduplications in four other children, located at 2q22.2-q23.3, 8p23.3, 16p11.2, and one child with two microduplications at chromosome 15q11.2 and 15q26.3. None of all investigated patients with infantile autism suffered from inborn errors of metabolism, food allergies, or celiac disease.
From 105 children with autism, 82 children (age mean ± SD: 4.4±2.3; range: 1-15.9 years; male/female ratio: 4.8-1) completed the self-controlled trial (Figure 1(b)), whereas 23 children were excluded because of incomplete test results, poor compliance, or failure to attend their follow-up. Serum contained FRα-autoantibodies in 75.6 % of all children, while these antibodies were found in 34.1% of the mothers and 29.4% of the fathers. In contrast only 3% healthy controls (age range: 1-18 years) and their parents tested positive for serum FRα autoantibodies. Considering the presence or absence of autoantibodies for each trio of child, mother, and father, eight possible combinations of a particular antibody profile existed for each trio. We only found 9/82 families (11%) where FRα antibodies tested negative in the child and both parents. In 89% of all families, FRα-autoantibodies were present in either the child and/or in one or both parents.
Based on the laboratory results, the treatment protocol for each individual consisted of supplements in combination with high-dose folinic acid (0.5-1 mg/kg/day), if FRα-autoantibodies were found. The mean for the CARS of all 82 autistic children at baseline was compatible with severe autism (mean ± SD: 41.34 ± 6.47) and after 2 years of treatment, the CARS declined significantly to a mean value at 34.35, indicating mild to moderate autism (mean ± SD: 34.35 ± 6.25; paired student t-test = 11.72, p<0.0001). In 17 out of 82 children (20.7%), the CARS after treatment dropped below a score of 30, which is consistent with the absence of autistic features. The majority (14/17) of the latter children started treatment before the age of 5 years.
The CARS score lowered significantly following treatment for each age group ranging from 2 years up to 6 years or older. However, as the baseline CARS before treatment increased significantly with advancing age from a mean ± SD of 39.35±5.2 at the age of 2 years towards 45.09±6.46 for children at 6 years or older (unpaired t-test 2.23 and p=0.038), the final CARS outcome remained higher with advancing age despite a similar therapeutic effect on autistic core signs. Therefore, the final outcome became poorer as children grew older and treatment started at a later age beyond 5 years (see Figure 1(b)). However, in the untreated group there was no significant change in the CARS score with advancing age since their mean CARS scores varied between 41.92 at 2 years and 41.75 for the children who were 6 years or older.
We evaluated the effect of treatment for all children, whose parents were negative for FRα-antibodies, as a function of FRα autoantibody titers. The drop in CARS score (Δ CARS) after treatment was significantly larger for the group of 15 autistic children whose FRα autoantibodies of the blocking type were negative or were at a low titer up to 0.44 p mol FR blocked/ml serum (Δ CARS mean ± SD: 7.4±5.22), compared to the 17 children whose FRα antibody titers were above 0.44 (Δ CARS mean ± SD: 4.2±3.36; t-test=2.08 and p=0.0455).
Each specific profile among 68 families, where positive or negative FRα antibodies were available in the child, mother, or father, was correlated with the CARS before and after treatment with supplements and folinic acid (see Figure 1(c)). Two families are not shown in Figure 1(c) and Table 2 because they represented one single family with only FRα autoantibodies in the father and one family where both parents tested positive but the child was negative. In the family where only the father tested positive for FRα autoantibodies, the initial CARS of 36 in his daughter dropped to 30 1/2 after correction of her antioxidant and vitamin B12 deficiencies. In the family where both parents tested positive, the initial CARS at 42 did not change significantly after 2 years (CARS 41.5) despite treatment.
The results for the other families demonstrated that the presence of positive maternal FRα autoantibodies tended to be associated with a higher CARS score at baseline and resulted also in a poorer final outcome after two years treatment. However, comparison of the baseline CARS scores between the group with positive maternal antibodies (N=5) and the group with absent antibodies in the child and parents (N=9) did not reach a statistically significant difference. In particular, when both parents and child tested positive, the high initial CARS score dropped after therapy but this did not represent a highly significant change (p=0.0398). Interestingly, nine families, where the child and both parents had no FRα autoantibodies but only had antioxidant or other nutrient deficiencies, had a high mean CARS score at 42.05±6.8, which declined significantly after treatment to a CARS score of 33.88±8.19.
3.3. Nutritional Derangements
The results of measured blood parameters among participating children (n=82) in the self-controlled therapeutic trial showed common deficiencies or excess of vitamins, metals, and trace elements well below or above the lower and upper boundaries of reference values established for healthy age-matched controls.
The most frequently encountered deficiencies were vitamin A (65.8%) and vitamin D (62.2%), serum iron (25.6%), ferritin (11%), serum folate (18.3%) and red blood cell folate (18.3%), and moderate apolipoprotein B deficiency (16%). The other deficiencies noted were in decreasing order, gamma-tocopherol (13.4%), manganese (8.5%), selenium (8.5%), coenzyme Q10 (7.3%), beta-carotene (6%), zinc (4.8%), vitamin C (3.6%), and vitamin E (3.6%). Elevated values were found for zinc (11%), selenium (8.5%), manganese (6%), copper (2.4%), and beta-carotene (2.4%), while a disturbed equilibrium between copper and zinc predisposed to an increased copper/zinc ratio, found in 24.3 % of all patients.
Many patients had multiple deficiencies of vitamins, metals, and trace elements simultaneously and received supplements with the advice to adapt their diet with foods rich in deficient factors. Patients with an elevated copper/zinc ratio, elevated metals (zinc, copper, selenium, manganese), or increased beta-carotene, which act as prooxidants, received treatment with high-dose vitamins C and E as radical scavengers within the lipid and aqueous compartments (Table 1).
Blood nutrient levels for elevations or deficiencies were rechecked every 3-4 months, and treatment adapted accordingly. It is interesting to note that the children with autism from 9 families where FRα antibodies were absent responded to correction of their deficient nutrient factors and showed a significant decrease of their baseline CARS from a mean ± SD at 42.05 ± 6.8 towards a mean ± SD at 33.88 ± 8.19 after treatment during two years.
4. Discussion
Our approach to improve autism outcome following correction of nutrient derangements and FRα-autoimmunity has been substantiated by the findings. It also demonstrated that a better outcome after treatment is achieved when FRα antibodies are absent or at a low titer (< 0.44 pmol FRα blocked/ ml) and if FRα autoantibodies are not present in the mother or both parents. Although in the untreated group the baseline CARS remained stable with advancing age, the baseline CARS for the treated group was significantly higher for children at or above 6 years compared to the baseline CARS at the age groups of 2 and 3 years, respectively. This difference for the baseline CARS with advancing age in the two groups is one limitation of the current study and may be attributed to the relative low number of patients in each age group. This difference in baseline CARS was the only parameter that did not match between both groups, whereas age, gender, and FRα autoantibody profiles for children and parents matched the untreated with treated groups.
Because the treatment involved correction of both nutrient derangements and FRα autoimmunity, the following question remains: which therapeutic strategy has to be indicated as the most effective with respect to clinical improvement? A limited number of autistic children from 9 families where FRα autoantibodies were absent in the child and both parents showed a significant improvement in their CARS scores following treatment of identified nutrient derangements. This underscores the important issue to consider not only treatment with high-dose folinic acid supplement for FRα autoimmunity, but also to include correction of coexisting nutrient derangements due to feeding problems, which occur frequently in patients with autism spectrum disorders. In this context, it should be stressed that coeliac disease might be responsible for nutrient malabsorption and should be considered among children with autism and specific antibody testing should be included. In our study we identified coeliac disease only in 1 child from the untreated reference group. Feeding problems with consequent nutrient derangements are a common finding in autism and have been attributed to selectivity or aversion to certain foods (particularly fruits and vegetables), combined with gustatory, olfactory, or oral sensory disturbances [21–25].
Several studies suggested that maternal folate deficiency during pregnancy increased not only the risk for neural tube defects but also the risk of autism [7, 8]. A recent study found an association between the use of maternal folic acid supplements during pregnancy and a reduced risk of autism spectrum disorders [32]. Although this and other studies support the association between folate deficiency during pregnancy and an increased risk for autism [7, 8], these studies did not consider the presence of parental FRα autoantibodies, identified by us among both mothers and fathers of children with autism [11, 16]. In the event of FRα autoantibody detection among mothers, the usually prescribed folic acid dose before and during pregnancy will probably prove largely insufficient [13].
Our self-controlled treatment trial showed that the presence of maternal FRα autoantibodies or FRα antibodies in both parents tended to be associated with a higher initial baseline CARS score among affected children with autism. Thus, this may explain that the final result and change in CARS score following 2-year treatment was less pronounced as compared to all other groups, although the small number of patients within each group did not allow a profound statistical analysis. These issues will be clarified when more patients will be included into similar treatment trials. Our findings in a minority of 7 out of 68 families (10%) identified no FRα autoantibodies in the children whereas FRα antibodies could only be detected in the mother (N=5), father (N=1), or both parents (N=1). Although feeding and nutrient problems for each child have to be taken into account, this finding suggests that parental FRα antibodies may impair folate transport into oocytes and spermatozoides and also block sufficient folate transport across the placental barrier to the embryo and fetus. Because an adequate folate pool is essential for purine and pyrimidine synthesis, and for mediating epigenetic mechanisms involving DNA methylation and histone modification, the initial embryonic development and subsequent stages of neurodevelopment will rely heavily on availability of adequate folate. Therefore, the risk of autism with its poor prognosis in the offspring associated with parental FRα antibodies warrants FRα testing among future parents followed by folinic acid intervention before conception and during pregnancy.
The common feeding disturbances associated with autism may provoke oxidative stress due to altered nutritional states where elevated metals (copper, manganese) or beta-carotene act as prooxidants through induction of Fenton chemistry. Nutritional deficiencies of radical scavenging vitamins (vitamins A, C, E, and gamma-tocopherol) as well as metals and trace elements (copper, zinc, manganese, and selenium), being cofactors of antioxidative enzymes, predispose to failing antioxidant defences. Moderate apolipoprotein B deficiency has been encountered in a significant number of autistic subjects and leads to deficient liposoluble vitamins A, D, E, and K. Deficiency of a number of vitamins and coenzyme Q10 necessary for mitochondrial metabolism, will result in mitochondrial dysfunction. Thus, oxidative stress in the brain due to mitochondrial dysfunction, elevated prooxidants, or deficient antioxidants on the one hand and FRα autoimmunity on the other hand, represent two independent variables at the basis of autism where correction of each variable showed a clinical response with a decline in the CARS score. Therefore, in addition to treatment for FRα autoimmunity [9, 10, 29], specific supplements are required to correct nutritional deficiencies in order to ameliorate intermediary metabolism and to neutralize abundant reactive oxygen species (ROS) deranging brain metabolism and function. As stated above, it appears from our findings in this study that the group of patients, where FRα antibodies tested negative in the child and its parents, benefitted only through correction of nutritional derangements as their CARS score dropped significantly.
In our study we also detected deficiencies of serum and red blood cell folate in 18.3 % of all patients. In vitro studies have supported the concept of an existing link between oxidative stress and deranged folate homeostasis. In a previous study we found that the generation of superoxide anions in vitro catabolizes 5-methyl-tetrahydrofolate by 75% within one hour, which can be prevented through preincubation with the radical scavenger ascorbic acid [26]. This study also found that KB-cells in culture exposed to superoxide anions and hydrogen peroxide reduces cellular folate incorporation mediated by FRα or RFC1 transport mechanisms. Thus transmembrane folate passage mediated by these transporters at the placenta and choroid plexus is expected to be impaired in the presence of ROS and predisposes to intrauterine folate deficiency and cerebral folate deficiency.
ROS predispose to nitrosative stress with peroxynitrite formation within neurons expressing neuronal NO-synthase leading to neuronal dysfunction and apoptosis [33–37].
Thus, in vivo superoxide anion generation promotes formation of peroxynitrite in NO-synthase positive neuronal networks (Figure 2). Neuronal folate depletion will further compromise folate-dependent de novo purine synthesis with adenosine and guanosine production. The consequent low guanosine triphosphate (GTP) synthesis, which represents the substrate for the rate-limiting enzyme GTP-cyclohydrolase I, will reduce tetrahydrobiopterin (BH4) production [38]. The low BH4 acting as cofactor for tryptophan hydroxylase, tyrosine hydroxylase, and NO-synthase will reduce these enzyme activities and diminish the production of serotonin, dopamine and NO. Moreover, in the presence of low BH4, NO-synthase will shift its enzymatic activity and start to produce the nitrosyl radical peroxynitrite instead of NO [35, 39–41]. Postmortem brain tissue studies have confirmed the presence of biomarkers for oxidative and nitrosative stress in autistic brains, because in cortical brain areas and cerebellum high concentrations of 8-hydroxy-deoxy-guanosine and 3-nitrotyrosine were found [42, 43]. Although the present study did not document disturbed metabolism of pterins, neurotransmitters, and NO, the well-known comorbidities associated with autism like ADHD, anxiety, obsessive-compulsive disorder, and sleeping problems support this hypothesis.
Figure 2.

Pathophysiology of autism based on our findings showing the impact of reactive oxygen species (ROS) at different levels of intermediary metabolism and the consequences of brain 5-methyl-tetrahydrofolate (5-methyl THF) deficiency due to FRα autoimmunity. ROS inhibits B12-methionine synthase (B12-MS) activity and stimulates cystathionine-beta-synthase (CBS) activity, shifting the homocysteine accumulation from the methionine cycle into the transsulfuration pathway with increased production of the natural antioxidant glutathione. Superoxide anions also react with NO at the level of NO-synthase (NOS1) to form peroxynitrite instead of NO, which predisposes to apoptosis and nitrosylation of tyrosine and cysteine. Nitrosative stress affects activity of tryptophan- (TPH2) and tyrosine hydroxylases (TH), the rate-limiting enzymes for serotonin and dopamine synthesis. In addition ROS catabolize 5-methyl-THF and impair folate uptake and transcellular transport across the choroid plexus and placental barriers due to interaction with FRα and RFC1 folate transporters. FRα autoantibodies also impair folate transport to the fetus and brain and predispose to brain folate deficiency with reduction of SAM production and SAM-dependent methyl-transfer reactions, reduced purine and thymidine synthesis with diminished GTP and BH4 production, which is the common cofactor of the enzymes TH, TPH2 and NOS1. Reduction of the activated methyl-group donor SAM down regulates DNA methylation and affects posttranslational modifications of histones (methylation and trimethylation of histones), thereby impeding the homeostatic balance between gene transcription and silencing. In addition folate deficiency is accompanied by overexpression of histone deacetylases, which further leads to abnormal gene silencing. The shutdown in expression of specific sets of genes will affect neuronal growth, pruning and differentiation. Abbreviations: GTPCH: GTP-cyclohydrolase I; cyst: cysteine; tyr: tyrosine; MTHFR: methylene-tetrahydrofolate reductase; RFC1: reduced folate carrier-1.
The other consequence resulting from diminished purine synthesis associated with brain folate depletion will be the low adenosine production as substrate for ATP, which can be expected to lead to mitochondrial dysfunction and also to inadequate maintenance of the mitochondrial DNA pool.
Further consequences of specific brain nutrient deficiencies predispose to oxidative stress and can affect the function of key enzymes and intermediary metabolism and predispose to alterations of DNA structure and function. Peroxynitrite inactivates the rate-limiting enzymes for serotonin and dopamine synthesis being tryptophan- and tyrosine hydroxylases via sulfhydryl oxidation at their enzyme substrate binding sites, where the protein is rich in cysteine residues [44–46]. Peroxynitrite will also nitrosylate protein tyrosine residues, but this exerts a minimal effect on enzyme activity [44]. In addition, oxidation induces neuronal tryptophan hydroxylase 2 aggregates through disulfide cross-linking [46]. Thus, generation of ROS and peroxynitrite, enhanced by folate depletion, reduces production of dopamine, NO, and in particular serotonin, which is supported by previous findings of low serotonin production known to affect about 1/3 of autistic patients [38, 47].
Exposure to oxidative stress leads to multiple adaptive mechanisms with inhibition of B12-dependent methionine synthase activity, which has a negative impact on the methionine cycle with reduced SAM production, resulting in failure of >100 methyl-transfer reactions [48, 49]. Simultaneously, oxidative stress increases the activity of cystathionine-beta-synthase which shifts homocysteine away from the methionine cycle towards the transsulfuration pathway to increase cysteine and glutathione synthesis as adaptive mechanisms to elevated oxidative stress [50]. In this context, low brain methyl-tetrahydrofolate availability due to FRα autoimmunity further inhibits methionine synthase activity and will aggravate the shift from the methionine cycle towards the transsulfuration cycle. Thus, oxidative stress will cause compensatory increases of the antioxidant glutathione and will downregulate the cellular methylation capacity.
ROS also reacts directly with DNA purine and pyrimidine components and causes DNA strand breaks, as reflected by increased oxidized DNA damage in lymphocytes. Moreover, ROS can induce oxidative DNA damage at the level of methylated CpG islands near gene promoter sites resulting in various mutations including C→T transitions, G→T transversions and CG→TT tandem mutations. In addition, ROS can convert guanosine to 8-oxo-guanosine and 5-methyl-cytosine towards 5-hydroxymethyl-cytosine [51–54]. These modified methyl-CpG sequences due to oxidative stress lead to functional loss of methylated CpG islands acting as recognition sites for methyl-CpG binding proteins (MBP), normally required to recruit histone deacetylases for chromatin condensation and gene silencing [54]. Thus, oxidative stress not only causes gene sequence alterations but also results in epigenetic changes with failure of methylated gene inactivation.
Because FRα autoantibodies block transport of methyl-tetrahydrofolate across the placenta to the fetus and the choroid plexus barrier to the brain, the consequent brain methyl-tetrahydrofolate depletion diminishes production of sufficient SAM as the activated methyl donor in over 100 methylation reactions in the brain, including the DNA-methyl-transferases which transfer methyl groups to gene CpG promoter sites which serve as recognition sites for methyl-CpG binding domain proteins (MBD) and histone deacetylases required for gene silencing. In this context, the combination of FRα autoimmunity and oxidative stress act independently or cooperate to derange epigenetic mechanisms controlling the orchestration of activation and inactivation of specific genes during neuronal development and differentiation processes.
Experiments using rat H19-7 hippocampal cell lines have shown that folate-deficient neurons have decreased proliferation rates, abnormal cell polarity, and migration, as well as failure of differentiation processes such as neurite outgrowth, and expression of glutamate receptors on dendrites, leading to abnormal synaptic function and plasticity [55]. Folate deficiency in hippocampal neuron cultures leads to decreased SAM concentrations and overexpression of histone deacetylases (HDAC 4,6,7). Histone deacetylase overexpression will consequently lead to repression of multiple developmental genes that are normally activated during specific developmental phases [56, 57]. Another consequence of folate deficiency in these hippocampal neuron cell lines is homocysteine accumulation with formation of homocysteinylated proteins (the motor proteins dynein and kinesin, among others), leading to aggregation and dysfunction of key neuronal proteins affecting normal development with possible long-lasting consequences [55, 58–61].
The consequences of folate deficiency affecting brain development may be more prominent in autistic children from mothers with folate deficiency or the presence of maternal FRα autoantibodies during pregnancy. Our finding of a higher initial baseline CARS score and less favorable outcome in these children confirms this hypothesis. In summary, the treatment response will be influenced in a negative fashion by the presence of maternal FRα autoantibodies, by late-onset treatment associated with a higher initial CARS score and in the event of elevated antibody titers. Paternal FRα antibodies may also influence the outcome and need to be further investigated, because we only identified one family.
5. Conclusion
In the pathogenesis of low-functioning autism, feeding disturbances predisposing to oxidative stress and acquisition of folate receptor autoantibodies during the pre- or postnatal period appear to play an important role by affecting intermediary metabolism and potentially deranging epigenetic control mechanisms. Early detection and appropriate therapeutic intervention is postulated to reverse core features and improve outcome.
Acknowledgments
This work has been supported by the University of Liege Autism Center and in part by grants from FNRS, Fonds National de Recherches Scientifiques # 3;4.540.09.F (to Vincent Th. Ramaekers) and Autism Speaks #8202 (to Edward V. Quadros). Study protocols were approved by the University Hospital Liège Ethics Committee and the IRB at the State University New York.
Abbreviations
- ADI-R:
Autism Diagnostic Interview-Revised
- ADOS:
Autism Diagnostic Observation Scale
- B12-MS:
B12-methionine synthase
- BH4:
Tetrahydrobiopterin
- CARS:
Childhood Autism Rating Scale
- CBS:
Cystathionine-beta-synthase
- CFD:
Cerebral folate deficiency
- cyst:
Cysteine
- FOLR1:
Folate receptor alpha gene
- FRα:
Folate receptor alpha
- GTPCH:
GTP-cyclohydrolase I
- LDL:
Low-density lipoprotein
- 5-Methyl THF:
5-Methyl-tetrahydrofolate
- MTHFR:
Methylene-tetrahydrofolate reductase
- NO:
Nitric oxide
- NOS1:
NO-synthase
- NTD:
Neural Tube Defect
- PDD-NOS:
Pervasive developmental disorder, not otherwise specified
- RFC1:
Reduces folate carrier-1
- ROS:
Reactive oxygen species
- SAM:
S-adenosyl-methionine
- TH:
Tyrosine hydroxylase
- TPH2:
Neuronal tryptophan hydroxylase
- tyr:
Tyrosine
- MTHFR:
Methylene-tetrahydrofolate reductase.
Data Availability
The data used to support the findings of this study are available from the corresponding author upon request.
Consent
All patients and volunteers received written information about the goal of the study and signed an informed consent form prior to participation.
Disclosure
The concept and design of these studies were initiated by the first author (Vincent Th. Ramaekers), in collaboration with the laboratory for immunology/biology (Edward V. Quadros, Jeffrey M. Sequeira) at the Department of Medicine, State University New York. Patients have been assessed at the Centre of Autism Liège by the child psychiatrists (Paule Philippe, Annick Jadot), child neurologist (Vincent Th. Ramaekers), psychologists (Marco DiDuca, Aurore Thomas, Céline Philippe, Marie Peters), and neuropsychologist (Géraldine Vrancken).
Conflicts of Interest
Two of the authors Jeffrey M. Sequeira and Edward V. Quadros are listed as inventors in the US patent no. 7846672B2 for the measurement of folate receptor autoantibodies, issued to the Research Foundation of the State University of New York, USA. For the other authors there were no conflicts of interest or financial interest to declare.
References
- 1.Miles J. H. Autism spectrum disorders—a genetics review. Genetics in Medicine. 2011;13(4):278–294. doi: 10.1097/GIM.0b013e3181ff67ba. [DOI] [PubMed] [Google Scholar]
- 2.Rossignol D. A. My experience learning about autism. GAHM. 2013;2(6):74–77. doi: 10.7453/gahmj.2013.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim Y. S., Leventhal B. L. Genetic epidemiology and insights into interactive genetic and environmental effects in autism spectrum disorders. Biological Psychiatry. 2015;77(1):66–74. doi: 10.1016/j.biopsych.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Frye R. E., Rossignol D. A. Identification and treatment of pathophysiological comorbidities of autism spectrum disorder to achieve optimal outcomes. Clinical Medicine Insights: Pediatrics. 2016;10:43–56. doi: 10.4137/CMPed.S38337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Park H. R., Lee J. M., Moon H. E., et al. A short review on the current understanding of autism spectrum disorders. Experimental Neurobiology. 2016;25(1):1–13. doi: 10.5607/en.2016.25.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Finnell R. H., Gould A., Spiegelstein O. Pathobiology and genetics of neural tube defects. Epilepsia. 2003;44(Supplement 3):14–23. doi: 10.1046/j.1528-1157.44.s3.5.x. [DOI] [PubMed] [Google Scholar]
- 7.Schmidt R. J., Tancredi D. J., Ozonoff S., et al. Maternal periconceptional folic acid intake and risk of autism spectrum disorders and developmental delay in the CHARGE (CHildhood Autism Risks from Genetics and Environment) case-control study. American Journal of Clinical Nutrition. 2012;96(1):80–89. doi: 10.3945/ajcn.110.004416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Surén P., Roth C., Bresnahan M., et al. Association between maternal use of folic acid supplements and risk of autism spectrum disorders in children. The Journal of the American Medical Association. 2013;309(6):570–577. doi: 10.1001/jama.2012.155925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ramaekers V. T., Blau N., Sequeira J. M., Nassogne M.-C., Quadros E. V. Folate receptor autoimmunity and cerebral folate deficiency in low-functioning autism with neurological deficits. Neuropediatrics. 2007;38(6):276–281. doi: 10.1055/s-2008-1065354. [DOI] [PubMed] [Google Scholar]
- 10.Ramaekers V. T., Wels J., Sequeira J. M., Quadros E. V., Blau N. Mitochondrial complex I encephalomyopathy and cerebral 5- methyltetrahydrofolate deficiency. Neuropediatrics. 2007;38(4):184–187. doi: 10.1055/s-2007-991150. [DOI] [PubMed] [Google Scholar]
- 11.Ramaekers V. T., Quadros E. V., Sequeira J. M. Role of folate receptor autoantibodies in infantile autism. Molecular Psychiatry. 2013;18(3):270–271. doi: 10.1038/mp.2012.22. [DOI] [PubMed] [Google Scholar]
- 12.Ramaekers V. T., Sequeira J. M., Quadros E. V. Clinical recognition and aspects of the cerebral folate deficiency syndromes. Clinical Chemistry and Laboratory Medicine. 2013;51(3):497–511. doi: 10.1515/cclm-2012-0543. [DOI] [PubMed] [Google Scholar]
- 13.Shapira I., Sequeira J. M., Quadros E. V. Folate receptor autoantibodies in pregnancy related complications. Birth Defects Research Part A - Clinical and Molecular Teratology. 2015;103(12):1028–1030. doi: 10.1002/bdra.23436. [DOI] [PubMed] [Google Scholar]
- 14.Frye R. E., Sequeira J. M., Quadros E. V., James S. J., Rossignol D. A. Cerebral folate receptor autoantibodies in autism spectrum disorder. Molecular Psychiatry. 2013;18(3):369–381. doi: 10.1038/mp.2011.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frye R. E., Delatorre R., Taylor H., et al. Redox metabolism abnormalities in autistic children associated with mitochondrial disease. Translational Psychiatry. 2013;3, article e273 doi: 10.1038/tp.2013.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Quadros E. V., Sequeira J. M., Brown W. T., et al. Folate receptor autoantibodies are prevalent in children diagnosed with autism spectrum disorder, their normal siblings and parents. Autism Research. 2018;11(5):707–712. doi: 10.1002/aur.1934. [DOI] [PubMed] [Google Scholar]
- 17.Chauhan A., Chauhan V., Brown W. T., Cohen I. Oxidative stress in autism: increased lipid peroxidation and reduced serum levels of ceruloplasmin and transferrin—the antioxidant proteins. Life Sciences. 2004;75(21):2539–2549. doi: 10.1016/j.lfs.2004.04.038. [DOI] [PubMed] [Google Scholar]
- 18.Chauhan A., Chauhan V. Oxidative stress in autism. Pathophysiology. 2006;13(3):171–181. doi: 10.1016/j.pathophys.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 19.James S. J., Melnyk S., Jernigan S., et al. Metabolic endophenotype and related genotypes are associated with oxidative stress in children with autism. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics. 2006;141(8):947–956. doi: 10.1002/ajmg.b.30366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kałużna-Czaplińska J., Jóźwik-Pruska J. Chromatographic and mass spectrometric techniques in studies on oxidative stress in autism. Journal of Chromatography B. 2016;1019:4–14. doi: 10.1016/j.jchromb.2015.12.035. [DOI] [PubMed] [Google Scholar]
- 21.Frustaci A., Neri M., Cesario A., et al. Oxidative stress-related biomarkers in autism: systematic review and meta-analyses. Free Radical Biology & Medicine. 2012;52(10):2128–2141. doi: 10.1016/j.freeradbiomed.2012.03.011. [DOI] [PubMed] [Google Scholar]
- 22.Ahearn W. H., Castine T., Nault K., Green G. An assessment of food acceptance in children with autism or pervasive developmental disorder-not otherwise specified. Journal of Autism and Developmental Disorders. 2001;31(5):505–511. doi: 10.1023/A:1012221026124. [DOI] [PubMed] [Google Scholar]
- 23.Sharp W. G., Berry R. C., McCracken C., et al. Feeding problems and nutrient intake in children with autism spectrum disorders: a meta-analysis and comprehensive review of the literature. Journal of Autism and Developmental Disorders. 2013;43(9):2159–2173. doi: 10.1007/s10803-013-1771-5. [DOI] [PubMed] [Google Scholar]
- 24.Liu X., Liu J., Xiong X., et al. Correlation between nutrition and symptoms: nutritional survey of children with autism spectrum disorder in Chongqing, China. Nutrients. 2016;8(5) doi: 10.3390/nu8050294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Malhi P., Venkatesh L., Bharti B., Singhi P. Feeding problems and nutrient intake in children with and without autism: a comparative study. The Indian Journal of Pediatrics. 2017;84(4):283–288. doi: 10.1007/s12098-016-2285-x. [DOI] [PubMed] [Google Scholar]
- 26.Opladen T., Blau N., Ramaekers V. T. Effect of antiepileptic drugs and reactive oxygen species on folate receptor 1 (FOLR1)-dependent 5-methyltetrahydrofolate transport. Molecular Genetics and Metabolism. 2010;101(1):48–54. doi: 10.1016/j.ymgme.2010.05.006. [DOI] [PubMed] [Google Scholar]
- 27.Schopler E., Reichler R. J., DeVellis R. F., Daly K. Toward objective classification of childhood autism: childhood autism rating scale (CARS) Journal of Autism and Developmental Disorders. 1980;10(1):91–103. doi: 10.1007/BF02408436. [DOI] [PubMed] [Google Scholar]
- 28.Ramaekers V. T., Rothenberg S. P., Sequeira J. M., et al. Autoantibodies to folate receptors in the cerebral folate deficiency syndrome. The New England Journal of Medicine. 2005;352(19):1985–1991. doi: 10.1056/NEJMoa043160. [DOI] [PubMed] [Google Scholar]
- 29.Frye R. E., Slattery J., Delhey L., et al. Folinic acid improves verbal communication in children with autism and language impairment: a randomized double-blind placebo-controlled trial. Molecular Psychiatry. 2016;23(2):247–256. doi: 10.1038/mp.2016.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rothenberg S. P., Da Costa M. P., Sequeira J. M., et al. Autoantibodies against folate receptors in women with a pregnancy complicated by a neural-tube defect. The New England Journal of Medicine. 2004;350(2):134–142. doi: 10.1056/NEJMoa031145. [DOI] [PubMed] [Google Scholar]
- 31.Sequeira J. M., Ramaekers V. T., Quadros E. V. The diagnostic utility of folate receptor autoantibodies in blood. Clinical Chemistry and Laboratory Medicine. 2013;51(3):545–554. doi: 10.1515/cclm-2012-0577. [DOI] [PubMed] [Google Scholar]
- 32.Wang M., Li K., Zhao D., Li L. The association between maternal use of folic acid supplements during pregnancy and risk of autism spectrum disorders in children: A meta-analysis. Molecular Autism. 2017;8(1, article 51) doi: 10.1186/s13229-017-0170-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lipton S. A., Singel D. J., Stamler J. S. Nitric oxide in the central nervous system. Progress in Brain Research. 1994;103:359–364. doi: 10.1016/S0079-6123(08)61149-8. [DOI] [PubMed] [Google Scholar]
- 34.Dawson V. L. Nitric oxide: role in neurotoxicity. Clinical and Experimental Pharmacology and Physiology. 1995;22(4):305–308. doi: 10.1111/j.1440-1681.1995.tb02005.x. [DOI] [PubMed] [Google Scholar]
- 35.Beckman J. S., Koppenol W. H. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. American Journal of Physiology-Cell Physiology. 1996;271(5):C1424–C1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- 36.Jomova K., Vondrakova D., Lawson M., Valko M. Metals, oxidative stress and neurodegenerative disorders. Molecular and Cellular Biochemistry. 2010;345(1-2):91–104. doi: 10.1007/s11010-010-0563-x. [DOI] [PubMed] [Google Scholar]
- 37.Kumar A., Singh R. L., Babu G. N. Cell death mechanisms in the early stages of acute glutamate neurotoxicity. Neuroscience Research. 2010;66(3):271–278. doi: 10.1016/j.neures.2009.11.009. [DOI] [PubMed] [Google Scholar]
- 38.Ramaekers V. T., Thöny B., Sequeira J. M., et al. Folinic acid treatment for schizophrenia associated with folate receptor autoantibodies. Molecular Genetics and Metabolism. 2014;113(4):307–314. doi: 10.1016/j.ymgme.2014.10.002. [DOI] [PubMed] [Google Scholar]
- 39.Beckman J. S., Beckman T. W., Chen J., Marshall P. A., Freeman B. A. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proceedings of the National Acadamy of Sciences of the United States of America. 1990;87(4):1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Milstien S., Katusic Z. Oxidation of tetrahydrobiopterin by peroxynitrite: Implications for vascular endothelial function. Biochemical and Biophysical Research Communications. 1999;263(3):681–684. doi: 10.1006/bbrc.1999.1422. [DOI] [PubMed] [Google Scholar]
- 41.Choi Y. K., Tarazi F. I. Alterations in dopamine and glutamate neurotransmission in tetrahydrobiopterin deficient spr-/- mice: relevance to schizophrenia. BMB Reports. 2010;43(9):593–598. doi: 10.5483/BMBRep.2010.43.9.593. [DOI] [PubMed] [Google Scholar]
- 42.Sajdel-Sulkowska E. M., Xu M., Koibuchi N. Increase in cerebellar neurotrophin-3 and oxidative stress markers in autism. The Cerebellum. 2009;8(3):366–372. doi: 10.1007/s12311-009-0105-9. [DOI] [PubMed] [Google Scholar]
- 43.Sajdel-Sulkowska E. M., Xu M., McGinnis W., Koibuchi N. Brain region-specific changes in oxidative stress and neurotrophin levels in autism spectrum disorders (ASD) The Cerebellum. 2011;10(1):43–48. doi: 10.1007/s12311-010-0223-4. [DOI] [PubMed] [Google Scholar]
- 44.Kuhn D. M., Aretha C. W., Geddes T. J. Peroxynitrite inactivation of tyrosine hydroxylase: mediation by sulfhydryl oxidation, not tyrosine nitration. The Journal of Neuroscience. 1999;19(23):10289–10294. doi: 10.1523/JNEUROSCI.19-23-10289.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kuhn D. M., Geddes T. J. Peroxynitrite inactivates tryptophan hydroxylase via sulfhydryl oxidation. Coincident nitration of enzyme tyrosyl residues has minimal impact on catalytic activity. The Journal of Biological Chemistry. 1999;274(42):29726–29732. doi: 10.1074/jbc.274.42.29726. [DOI] [PubMed] [Google Scholar]
- 46.Kuhn D. M., Sykes C. E., Geddes T. J., Jaunarajs K. L. E., Bishop C. Tryptophan hydroxylase 2 aggregates through disulfide cross-linking upon oxidation: Possible link to serotonin deficits and non-motor symptoms in Parkinson's disease. Journal of Neurochemistry. 2011;116(3):426–437. doi: 10.1111/j.1471-4159.2010.07123.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Adamsen D., Ramaekers V., Ho H. T. B., et al. Autism spectrum disorder associated with low serotonin in CSF and mutations in the SLC29A4 plasma membrane monoamine transporter (PMAT) gene. Molecular Autism. 2014;5(1, article 43) doi: 10.1186/2040-2392-5-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ludwig M. L., Matthews R. G. Structure-based perspectives on B12-dependent enzymes. Annual Review of Biochemistry. 1997;66:269–313. doi: 10.1146/annurev.biochem.66.1.269. [DOI] [PubMed] [Google Scholar]
- 49.Zhang Y., Hodgson N. W., Trivedi M. S., et al. Decreased brain levels of vitamin B12 in aging, autism and schizophrenia. PLoS ONE. 2016;11(1) doi: 10.1371/journal.pone.0146797.e0146797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Banerjee R., Evande R., Kabil Ö., Ojha S., Taoka S. Reaction mechanism and regulation of cystathionine β-synthase. Biochimica et Biophysica Acta. 2003;1647(1-2):30–35. doi: 10.1016/s1570-9639(03)00044-x. [DOI] [PubMed] [Google Scholar]
- 51.Lee D.-H., O'Connor T. R., Pfeifer G. P. Oxidative DNA damage induced by copper and hydrogen peroxide promotes CG→TT tandem mutations at methylated CpG dinucleotides in nucleotide excision repair-deficient cells. Nucleic Acids Research. 2002;30(16):3566–3573. doi: 10.1093/nar/gkf478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wilson I. M., Davies J. J., Weber M., et al. Epigenomics: mapping the methylome. Cell Cycle. 2006;5(2):155–158. doi: 10.4161/cc.5.2.2367. [DOI] [PubMed] [Google Scholar]
- 53.Toyokuni S., Akatsuka S. Pathological investigation of oxidative stress in the post-genomic era. Pathology International. 2007;57(8):461–473. doi: 10.1111/j.1440-1827.2007.02127.x. [DOI] [PubMed] [Google Scholar]
- 54.Valinluck V., Tsai H.-H., Rogstad D. K., Burdzy A., Bird A., Sowers L. C. Oxidative damage to methyl-CpG sequences inhibits the binding of the methyl-CpG binding domain (MBD) of methyl-CpG binding protein 2 (MeCP2) Nucleic Acids Research. 2004;32(14):4100–4108. doi: 10.1093/nar/gkh739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Akchiche N., Bossenmeyer-Pourié C., Kerek R., et al. Homocysteinylation of neuronal proteins contributes to folate deficiency-associated alterations of differentiation, vesicular transport, and plasticity in hippocampal neuronal cells. The FASEB Journal. 2012;26(10):3980–3992. doi: 10.1096/fj.12-205757. [DOI] [PubMed] [Google Scholar]
- 56.Haberland M., Montgomery R. L., Olson E. N. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nature Reviews Genetics. 2009;10(1):32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hirabayashi Y., Gotoh Y. Epigenetic control of neural precursor cell fate during development. Nature Reviews Neuroscience. 2010;11(6):377–388. doi: 10.1038/nrn2810. [DOI] [PubMed] [Google Scholar]
- 58.Mattson M. P. Establishment and plasticity of neuronal polarity. Journal of Neuroscience Research. 1999;57(5):577–589. doi: 10.1002/(SICI)1097-4547(19990901)57:5<577::AID-JNR1>3.0.CO;2-H. doi: 10.1002/(SICI)1097-4547(19990901)57:5<577::AID-JNR1>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 59.Conde C., Cáceres A. Microtubule assembly, organization and dynamics in axons and dendrites. Nature Reviews Neuroscience. 2009;10(5):319–332. doi: 10.1038/nrn2631. [DOI] [PubMed] [Google Scholar]
- 60.Sibrian-Vazquez M., Escobedo J. O., Lim S., Samoei G. K., Strongin R. M. Homocystamides promote free-radical and oxidative damage to proteins. Proceedings of the National Acadamy of Sciences of the United States of America. 2010;107(2):551–554. doi: 10.1073/pnas.0909737107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Guéant J.-L., Namour F., Guéant-Rodriguez R.-M., Daval J.-L. Folate and fetal programming: a play in epigenomics? Trends in Endocrinology & Metabolism. 2013;24(6):279–289. doi: 10.1016/j.tem.2013.01.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data used to support the findings of this study are available from the corresponding author upon request.