

Abstract
Elucidation of the allosteric pathways in proteins is a computational challenge that strongly benefits from combination of atomistic molecular dynamics (MD) simulations and coarse-grained analysis of the complex dynamical network of chemical interactions based on graph theory. Here, we introduce and assess the performances of the dynamical perturbation network analysis of allosteric pathways in a prototypical V-type allosteric enzyme. Dynamical atomic contacts obtained from MD simulations are used to weight the allosteric protein graph, which involves an extended network of contacts perturbed by the effector binding in the allosteric site. The outcome showed good agreement with previously reported theoretical and experimental extended studies and it provided recognition of new potential allosteric spots that can be exploited in future mutagenesis experiments. Overall, the dynamical perturbation network analysis proved to be a powerful computational tool, complementary to other network-based approaches that can assist the full exploitation of allosteric phenomena for advances in protein engineering and rational drug design.
Graphical Abstract
INTRODUCTION
The characterization of allosteric mechanisms in proteic systems is a challenging task due to the intrinsically complex and elusive nature of protein allostery.1,2 The allosteric phenomena, ubiquitous in biology and not exclusive of proteins, have been shown to feature both structural and energetic origins.3,4 Statistical ensemble models rooted in the historical phenomenological models of allostery5,6 have suggested a unifying view of the operational allosteric mechanisms.7,8 Still, to fully exploit the potential of allosteric phenomena for protein engineering and rational drug design, where allosteric systems (and particularly enzymes) can be manipulated to inhibit/enhance their (catalytic) activity or new allosteric sites can be discovered,9–15 system-specific information is required.
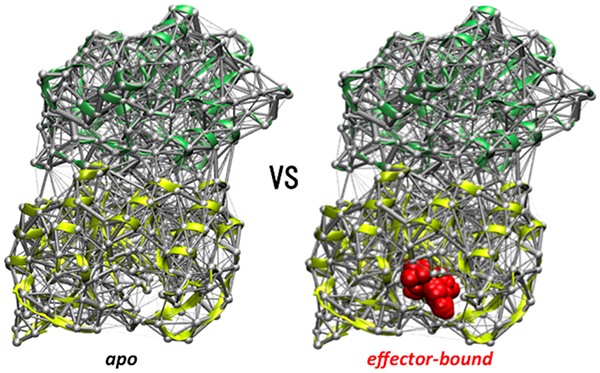
The fundamental process occurring in allosteric enzymes is the binding of an effector ligand at the allosteric site distant from the functional active site, enabling the regulation of the corresponding enzymatic function; see Figure 1. Modulation of functions in allosteric enzymes is linked to the communication from the active to the allosteric site,4,13,16 with effector-induced changes of residues dynamics and protein disorder altering either the affinity of the substrate for the active site (K-type) or the reaction rate (V-type) of the enzymes. The allosteric signal has been found to propagate through conserved amino acid residues17–19 and, in general, it is expected to involve physicochemical interactions between “allostery-related” residues that comprise various secondary structure elements, defining (multiple) “allosteric pathways” of the proteic systems.20
Figure 1.

(A) IGPS allosteric (V-type) enzyme, with substrate (in gray) binding in the active site of the HisH glutaminase domain (in green) and the effector (PRAFR, in red) binding in the allosteric site at the bottom of the HisF cyclase domain (in yellow). Previously recognized secondary structure elements belonging to the IGPS allosteric pathways are shown (in orange), linking the allosteric and active sites at sideR of the enzyme. (B) Example of a 3D representation of the IGPS protein network, with nodes of the graph located at the α carbon atoms of the enzyme and edges connecting nodes weighted by the number of contacts between residues pairs.
Classical molecular dynamics (MD) simulations provide invaluable information on protein dynamics at atomistic resolution, representing a fundamental tool for the elucidation of such allosteric pathways,21–23 whose experimental detailed characterization is certainly extremely challenging. While MD simulations enclose the dynamical information underpinning the allosteric effects,24 analyzing complex networks of interactions between (a generally large number of) fluctuating amino acid residues and finding the allosteric signal paths within the wiring of such a network call for help from graph theory techniques. Network analysis of MD trajectories that incorporate allosteric motions has delivered, in fact, characterization of allosteric pathways and identification of allostery-related amino acid residues in various biological systems,25–32 and helped rational discovery of allosteric modulators.33,34 In particular, combining nuclear magnetic resonance (NMR) relaxation dispersion experiments with community analysis of dynamical networks,35 based on mutual information on correlated protein motions obtained from MD simulations, we have revealed the allosteric pathways of the imidazole glycerol phosphate synthase (IGPS) enzyme from the thermophile Thermotoga maritima; see Figure 1.25
IGPS is a prototype allosteric enzyme absent in mammals but involved in essential biochemical pathways (histidine and purine synthesis) of pathogens, and thus, it is a potential target for antifungal, antibiotic, and herbicide development.36–38 As shown in Figure 1, two tightly associated proteins constitute the IGPS V-type allosteric enzyme: (i) the HisH glutamine amidotransferase that catalyzes the hydrolysis of the substrate (glutamine) and (ii) the HisF cyclase where the effector PRFAR, i.e., N′-[(5′,-phosphoribulosyl)formimino]-5-amino-imidazole-4-carboxamide-ribonucleotide, binds without affecting the glutamine binding affinity in HisH but accelerating its hydrolysis by ca. 5000-fold.39 Our synergistic theoretical and experimental investigations suggested secondary structure elements and key residues involved in the allosteric signal propagation induced by the PRFAR binding to the apo IGPS protein. The IGPS allosteric mechanism involves a sequence of interactions that alter the dynamics of specific regions in one side of the IGPS complex (sideR; see Figure 1), including hydrogen bonds in the flexible loop1 and hydrophobic interactions in the fβ2 strand at the HisF allosteric site, ionic interactions at the HisF/HisH interface involving the fα2, fα3, and hα1 helices, as well as hydrogen bonding between the Ω-loop and a conserved (49-PGVG) sequence adjacent to the active site, namely, the oxyanion strand. These effector-induced interactions were shown to alter the overall HisF/HisH relative fluctuations (named breathing motion), promoting rotation of the conserved oxyanion strand associated with an inactive-to-active allosteric transition. The outcome of the community network analysis stimulated experimental muta-genesis studies focused on the suggested allostery-related amino acid residues,40 as well as rational design of allosteric inhibitors able to knockout the IGPS allosteric signal propagation by interfering with the suggested allosteric pathways.34 The proposed community network analysis employed the correlations of motion between residue pairs (in close contact) to weight the protein graph, resulting in a communication network where the betweenness centrality measure can decipher the most important nodes that transfer the allosteric signal. While proving to be an extremely powerful and transferable approach that has been employed to other allosteric systems,2,27 this tool was revealed to be not very user-friendly and was particularly tedious to use when applied to large proteic systems. Very recently, we have proposed an alternative tool to the community network analysis that introduced the eigenvector centrality metric to analyze the correlated motions obtained from the MD simulations, providing a cost-effective approach that properly captures the IGPS allosteric pathways and allows the user to disentangle contributions to allostery due to short- or long-range correlations.28 Nevertheless, both the betweenness and eigenvector centrality measures have been used to analyze protein graphs weighted by the correlated motions of α carbon atoms. These correlations certainly comprise only part of the network of interactions that are altered upon effector binding. Here, we explore the use of inter-residue physical contacts to build the weighted protein network, thus moving from a physical to a geometrical measure that tracks down and approximates the chemical interactions between residues. This type of weighted contact network analysis has been successfully used to infer protein dynamics and to determine structural robustness to mutations in proteins, it being powerful to understand how a local change can produce global changes that are associated with retention or loss of protein functions.41–43 Here, we propose to use this weighted network approach to study allostery and to compute local perturbations of contacts induced by the effector binding, which are expected to propagate in the allosteric enzymes through protein dynamics. The use of unweighted networks based on a binary measure of dynamical contacts could be also envisioned to this aim, possibly providing a more coarse-grained picture of the effector-induced dynamical contacts with respect to networks weighted with the number of atomic contacts. In particular, taking advantage of the atomistic details contained in MD simulations one can account for dynamical contacts and their effector-induced modifications by averaging the number of contacts for each residue pair along a MD trajectory, using this information to weight the protein network. A similar approach, based on dynamical network of inter-residues contacts, has been used to reveal the allosteric effects of mutations in the catalytic activity of the Cyclophilin A enzyme, proving to be potentially able to identify key residues in the allosteric signal propagation.44 Here, we propose the use of the dynamical contact network approach to study allosteric perturbations induced by effector binding, instead of mutations, performing a dynamical perturbation network analysis of IGPS allostery. IGPS is, indeed, a prototypical allosteric enzyme whose allosteric pathways have been previously characterized in detail by means of MD simulations and network models and validated by NMR and biochemical and mutagenesis experiments, providing an ideal system to assess the performances of the perturbation network analysis for capturing allostery.
COMPUTATIONAL DETAILS
In this work, we used structural models of the apo and PRFAR-bound IGPS complexes and MD simulations that have been described elsewhere,25 in order to fairly compare the results of the perturbation networks with those of the previously reported community network analysis. In our previous analysis we have showed that the time-averaged weighted networks, based on MD trajectories 100 ns (ns) long, adequately describe the dynamical networks, capturing the protein conformational changes induced by effector binding during the early dynamics of the IGPS complexes.25 Therefore, previously obtained MD trajectories, including four independent simulations of 100 ns for the apo IGPS protein and four independent simulations of 100 ns for the PRFAR-bound IGPS complex, have been used.25 MD simulations of the IGPS complexes were based on the AMBER-ff99SB45 force field for the IGPS protein and on the generalized Amber force field46 for the PRFAR ligand, using the NAMD2 software package.47 Production run MD simulations succeeded a pre-equilibration procedure involving slow heating to 303 K, gradual release of atomic positions constraints, and subsequent unconstrained MD simulations of 4 ns in the canonical NVT ensemble using Langevin dynamics. Production runs were performed in the NPT ensemble at 303 K and 1 atm (using the Langevin piston) for 100 ns after reaching the equilibrium volume (i.e., after ca. 2–3 ns). Periodic boundary conditions and the particle mesh Ewald method48 were employed, with van der Waals interactions calculated using a switching distance of 10 Å and a cutoff of 12 Å. A multiple time-stepping algorithm49,50 was adopted, with bonded, short-range nonbonded, and long-range electrostatic interactions were evaluated at every one, two and four time steps, respectively, using a time step of integration set to 1 fs.
Protein Weighted Networks.
In the protein network each node represents an amino acid residue (see Figure 1), with connections between nodes (namely, the graph edges) being defined according to atomic proximity: for each pair of residues, if there exists a couple of atoms, one in each residue, whose distance is below a given distance cutoff, then the two atoms satisfy the “contact condition” and the two corresponding nodes/residues are linked by an edge. In line with previously reported perturbation network analysis,41,42 we used a 5 Å distance cutoff to define the contact condition, this choice allowing a fair comparison with previously reported community network analysis,25 where the same distance cutoff has been adopted.25 The effect of the distance cutoff parameter on the perturbation network analysis will deserve further investigation for application of the proposed network approach to other allosteric systems. The protein weighted network is then built by assigning to each edge (linking the ith and jth residues) a weight wij, which equals the number of contacts between two residues, i.e., the number of atom pairs that satisfy the contact condition between the ith and jth residues (see Figure S1 in the Supporting Information). To compute the number of contacts among the IGPS residues in the apo and PRFAR-bound complexes, and thus the corresponding contact weighted networks, we used the atomic coordinates extracted every 100 ps from the MD trajectories. The choice of the time interval to extract the atomic coordinates (and thus to compute the number of contacts) is bound to that one adopted in the community network analysis,25 in order to provide a consistent comparison between the two different approaches. In particular, after concatenating the four independent simulations per each IGPS system (apo and effector bound) the number of atomic contacts are computed by averaging over the corresponding MD frames. If an edge is not present in a given frame, i.e., if two residues do not satisfy the contact condition in that very frame, its weight is set to zero and it will be still averaged with its weights at the remaining frames. As we will illustrate in the Results section, the computation of atomic contacts could include all protein atoms or it could exclude just the hydrogen atoms.
Dynamical Perturbation Network.
The procedure described above generates two weighted contact networks, one for the apo protein and one for the PRFAR-bound complex, each one containing (in their average weights) information on the contacts dynamics of all residues pairs in the corresponding IGPS protein. As shown in Figure 2, a weighted network representing the perturbations of the contacts dynamics induced by the effector binding, i.e., the dynamical perturbation network, can be constructed by considering as edge weight for each residues pair the differences in weights (weight link) between the two IGPS proteins, i.e., using the perturbation weight (wp = wPRFAR - wAPO) to build the network. To simplify the visual inspection of such perturbation network, the edges are colored in red if PRFAR binding induces an increase in weight (wp > 0), i.e., an increase in number of atomic contacts for a given residue pair, and in blue if PRFAR binding instead reduces the contact weight (wp < 0); see Figure 2. To allow visualization of the 2D and/or 3D representations of the IGPS perturbation network, which contains around 104 edges, a weight link threshold (wt) can be applied so that only the edges whose weight is greater than the chosen weight threshold, i.e., |wp| > wt are kept for visualization. If a node loses all its edges during the subtraction process, it is also removed from the graph representation for simplicity. The impact of the weight threshold values on the graph visualization changes according to criterion used to compute the number of contacts. For instance, excluding hydrogen atoms from the count of atomic contacts reduces significantly the average weights values in each protein network and consequently also the weights in the dynamical perturbation network, allowing wt values of 5 or 6 to be large enough to make the number of edges to visualize being less than one hundred. To obtain a similar number of edges while including all atoms in the counts of atomic contacts requires much larger weight thresholds (wt > 20).
Figure 2.

Construction of the IGPS perturbation network. The average contact weights of residue pairs of the apo IGPS are subtracted from that of the effector-bound binary complex. 3D representations of the average and perturbation networks and a corresponding close-up view are depicted. Reduction and increase of the number of contacts between residue pairs upon PRFAR binding are indicated with blue and red links, respectively. The widths of the links in each average and perturbation network are normalized to facilitate their visualizations.
RESULTS AND DISCUSSION
Figure 3 shows the perturbation network associated with effector (PRFAR) binding to the IGPS protein, using a weight threshold wt = 6 and considering only contacts between heavy atoms. Notably, the PRFAR perturbations are spread over different regions of the enzyme and reach also HisH residues located quite far from the effector site in HisF.
Figure 3.

Perturbation network associated with PRFAR binding to IGPS, using a weight threshold wt = 6 for the network visualizations. (A) 2D projection and (B) 3D representation of the perturbation network, showing reduction (blue lines) and increase (red lines) of the number of contacts between heavy atoms upon PRFAR binding. Perturbations associated with previously reported allosteric pathways25 are highlighted with black circles.
At the effector binding site, perturbations can be found at both sideL and sideR of the enzyme due to the hydrogen bonds created by the PRFAR molecule at these sides. In fact, it has been shown25 that the hydroxyl groups of the PRFAR glycerol moiety create a hydrogen bonding network with the fG202 residue at the end of fβ7 (see Figure 4 in ref25). The highly conserved fG202 residue is indeed detected by our network analysis, which further shows propagation of this perturbation across sideL. The fT142 and fR133 residues appear as central nodes for PRFAR signal propagation at HisF sideL. At sideR, the perturbation network analysis indicates that upon PRFAR binding contacts in the fβ8-fα8′ turn of HisF are significantly affected, with an increase of contact between the fA224 and fF227 hydrophobic residues. Indeed, the glycerol side phosphate group of PRFAR is known to be involved in hydrogen bonds with the backbone of fA224 and the fS225 side chain located in the fβ8-fα8′ turn.18,25 Notably, near the fβ8-fα8′ turn is located the important loopl, for which the perturbation network analysis shows drastic modifications of contacts upon effector binding, in agreement with previous results (see Figure S2 in the Supporting Information for direct comparison).25 In particular, the loss of contacts in the loop, associated with residues fK19, fF23, fL26, and fR27 (blue lines in Figure 3) is compensated by an increase of contacts between residues fD11, fK19, fG20, and fH228. Thus, the invariant fKl9 plays a central role in the perturbation network being crucial for the signal transduction at sideR of HisF, as demonstrated by experimental biochemical data on the fK19A mutant.40 In fact, our network analysis allows recognition of important interactions between the highly conserved fD11 (in fβl) and fK19, occurring only upon PRFAR binding (see Figure S2 in the Supporting Information) and suggesting the participation of fβl in the allosteric pathways and fDll as another possible allosteric spot in IGPS.
As shown in Figure 3, while in HisF the increase of contacts (red lines) induced by the effector binding is almost compensated by a few contact losses (total weight gain is ca. 19), in HisH most of the perturbations are characterized by contact losses (blue lines, with total weight loss ca. 111). Among the pairs that feature contact loss in HisH, it is worth highlighting the hN12-hN15 pair connecting the Ω-loop and the hα1 helix, two secondary structure elements that have been indicated among the allosteric pathways and a crucial connection already pointed up by the community network analysis.25
The HisF/HisH interface also features perturbation of relevant contacts upon effector binding, in agreement with the change in breathing motion between apo and PRFAR-bound IGPS previously reported.25 As pointed out by previous analysis of MD simulations, the allosteric effect of PRFAR expresses at the protein-protein interface as rearrangement of the ionic interactions among the fE67, fE71, and hR18 residues, with rupture of the hR18-fE67 salt bridge (connecting the hα1 and fα2 helices) upon effector binding (see Figure S3 in the Supporting Information).25 Notably, the contact loss in the hR18-fE67 ion pair interaction appears as one of the largest perturbations in the network (see Figure 3a) and it is accompanied by other significant changes in sideR. In particular, the fE91-fR95 salt bridge within the fα3 helix is also detected by the perturbation network analysis, in agreement with the fact that both hR18-fE67 and fE91-fR95 salt bridges represent the most relevant changes in ionic interactions at sideR associated with the allosteric pathways (see Figure S3 in the Supporting Information).5 Other interactions at the HisF/HisH interface are evidenced by the perturbation network analysis: (i) contacts between the hY136 residue51 in hβ8 and residues fV248, fR249, and fL250 in the C-terminal domain of HisF and (ii) two contact pairs connecting the hα1 and hα4 helices with the fα2-fβ3 turn, i.e., hR22-fQ72 and hR187-fD74, respectively. The interactions involving the polar hY136 residue show a global increase of the number of contacts of this residue with HisF, upon effector binding. This is due to the change of H-bonding between hY136 and fN247, which brings hY136 closer to the flexible HisF C-terminus (see Figure S4 in the Supporting Information). These changes of contacts comprise fR249, a highly conserved residue involved in the π-cation hW123-fR249 molecular hinge,18 but are not associated with formation/disruption of very strong interactions that might alter significantly the IGPS structure. Still, the observed rearrangement of the HisF C-terminus involving the molecular hinge is in line with modification of the relative HisF/HisH (breathing) motion, an indirect effect associated with the disruption of the hR18-fE67 interface salt bridge. However, a contact loss is observed for the hR22-fQ72 pair upon effector binding, which involves hα1 and the fα2-fβ3 turn, respectively, and it appears to be directly related to the breaking of the adjacent hR18-fE67 salt bridge also connecting hα1 with HisF. The hR22-fQJ2 contact loss is somehow compensated by the formation of a nearby hR187-fD74 salt bridge, involving the hα4 helix. The contacts encompassing residues hR22, hR187, fE67, fQ72, and fD74 are all located at sideR of the HisF/HisH interface, which has been indicated as a crucial region for the IGPS allosteric communication and thus deserves a more detailed analysis.
Figure 4 shows the perturbation network representation using weight threshold wt = 5 that allows a detailed view of the interactions involved in the important region around the hR18-fE67 salt bridge. In addition to detecting the hR18-fE67 salt-bridge breaking, a recognized effect of PRFAR binding inducing separation of the hα1-fα2 elements,25 the perturbation network analysis also indicates that propagation of the allosteric signal through the HisF/HisH interface involves ionic interactions that were not previously detected. In particular, the formation of the hR187-fD74 salt bridge that connects hα4 helix with the fα2-fβ3 turn in the PRFAR-bound complex is concomitant with the breaking of the hR22-fD74 salt bridge between the hα1 and the fα2-fβ3 turn, which is thus involved in the modifications of ionic interactions promoted by the hR18-fE67 salt-bridge disruption (see Figure S3 in the Supporting Information).25 Notably, these results are in agreement with NMR dispersion experiments indicating that residues in the fα2-fβ3 turn (e.g., fI73 and fI75) are among those that have the largest dynamical changes upon effector binding.52 Therefore, we propose that the fα2-fβ3 turn and the hα4 helix are secondary structure elements that are involved in the allosteric communication in IGPS and that residues hR22, hR187, and fD74 are potentially good candidates for mutagenesis experiments.
Figure 4.

3D representation of the perturbation network in the region close to the hR18-fE67 salt bridge. A weight threshold wt = 5 is used for the network visualization. The perturbations associated with PRFAR binding show the relative reduction (blue lines) and increase (red lines) of the number of contacts between heavy atoms. The hR18-fE67 salt-bridge rupture upon effector binding is associated with modifications of polar and ionic interactions between the hα1 and hα4 helices and the fα2-fβ3 turn, along with contact losses and partial unfolding at the beginning of hα1 helix, where the Ω-loop is located.
In the proximity of the sideR interface region, the hN12 and hN15 residues belonging to hα1 and Ω-loop, respectively, have been suggested by the community network analysis to be important for the IGPS allostery,25 allowing communication between the hα1 helix and the HisH active site via the Ω-loop. Figure 4 shows that the hN12-hN15 contact loss captured by the perturbation network is associated with other PRFAR-induced losses, i.e., the contacts in the hR18-hM14 and hR18-fE67 pairs. Overall, these modifications induced by PRFAR binding involve a partial unfolding of hα1 helix as a response to the hR18-fE67 salt-bridge rupture (see Figure S5 in the Supporting Information) and propagate toward the HisH active site via the Ω-loop.
The perturbation network analysis, thus, is quite useful for capturing the propagation of the PRFAR allosteric signals, providing direct visualization of allosteric effects as changes in the residue contacts. The above analysis based on the contacts between heavy atoms, indeed, detected most of the secondary structure elements in the known allosteric pathways,25 including loop1, fα2, fα3, hα1, and Ω-loop, and indicated new secondary structures encompassing fβ1, fα2-fβ3 turn, and hα4 along with other key residues, like fK19, fD11, fD74, hR22, and hR187. Nevertheless, two important elements of the allosteric pathways, namely, the fβ2 strand in HisF and the 49-PGVG sequence in HisH active site, are not observed even among the nondescribed perturbations appearing in the computed network (see Figure 3). The missing secondary structure elements involve hydrophobic interactions (between fβ2 and loop1) and backbone hydrogen bonds (between 49-PGVG and Ω-loop), suggesting that the omission of hydrogen atoms (H′s) in the count of residue contacts might be the reason for such a lack of detection of these important perturbations. However, H′s are usually discarded in perturbation network analysis of mutated proteins because they are not resolved in X-ray structures and their presence significantly increases the number of contacts for each pair, adding sizable noise in the data analysis. To limit such a drawback, here we considered inclusion of the hydrogens in the perturbation networks while separating the analysis of backbone atoms (that do not contain many H′s) from that of amino acid side chains.
Figure 5 shows the perturbation network analysis restricted to the backbone atoms while including hydrogens. This analysis allows focusing on the effector perturbations induced in the IGPS backbone. The backbone network shares some features with the perturbation network analysis of heavy atom contacts but it also highlights some perturbations previously overlooked. The backbone analysis, in fact, confirms the presence of strong perturbations in the PRFAR binding site, with detection of residues fG202 and fA224 and the H-bonds redistribution in loop1, as previously described. However, new perturbations stand out when the side chain contacts are removed from the network. In particular, the invariant fS201 and the highly conserved fG202, fG203, and fG205 residues of the SGGXG sequence at the fβ7-fα7 turn all feature perturbed backbone H-bonds. These perturbations can be viewed as a consequence of the hydrogen bonding network rearrangements induced by the PRFAR glycerol hydroxyls and phosphate groups at sideL of the effector binding site.25 Moreover, the backbone analysis also catches the increase of contacts among the highly conserved residues fA224, fF227, and fH228, which is associated with a partial folding of the fβ8—fα8′ turn at sideR of the PRFAR binding site.
Figure 5.

Perturbation network associated with PRFAR binding to IGPS, computed only for backbone atoms (including hydrogens) and using a weight threshold wt = 5 for the network visualizations. 2D projection (A) and 3D representation (B) of the network, showing reduction (blue lines) and increase (red lines) of the number of contact atoms upon PRFAR binding.
It is worth noting that the fD11-fK19 ion-pair contact, strongly reinforced in the presence of PRFAR, unexpectedly appears in the backbone perturbation network. This result provides direct evidence of this interaction being not associated with the formation of a fD11-fK19 salt bridge (as could be expected for an ion pair) but to hydrogen bonding between the fD11 side chain and the fK19 backbone; see Figure 6.
Figure 6.

(A) Perturbation network around residue fK19 associated with PRFAR binding to IGPS, computed for all atoms (including hydrogens) and using a weight threshold of wt = 19 for visualization (left panel) and (B) representative configuration of the H-bonding network in the PRFAR-bound complex associated with the fD11, fK19, and fS225 residues, also showing the partial folding of the fβ8–fα8′ turn.
The backbone interactions perturbed at the HisF/HisH interface are rather limited and are restricted to the highly flexible HisF N-terminus (fM1), getting in contact with the hβ7 and the hβ9 strands, i.e., with the backbone of residues hE125 and hE157, respectively. However, important backbone perturbations are found in a localized region of HisH, remarkably close to the active site. In fact, as shown in Figure 5a, the backbone network analysis clearly catches the allosteric effect associated with the 49-PGVG (oxyanion) strand that, as previously shown,25 loses contacts with the Ω-loop due to the hydrogen bond breaking between hP10 and hV51 (see Figure 9 in ref25). Notably, three residues of the conserved 49-PGVG sequence (i.e., hP49, hG50, and hV5l) are found to lose contacts with Ω-loop residues hG9, hP10, and hG11, in line with the fact that the separation of these two secondary structure elements is associated with rotation of the oxyanion strand near the substrate binding site. Beyond the remarkable ability of the perturbation network to retrieve the allosteric effects in the active site, this analysis also suggests effector-induced alterations that were overlooked in previous studies. In fact, the loss in contacts between 49-PGVG and the Ω-loop appears to be associated with a partial unfolding of the hα2 helix (which is next in sequence to the oxyanion strand), featuring the decrease of contacts between hH53, hF54, hE56, hG57, hR59, and hR60 residues, only slightly compensated by the strengthening of the hG9-hG57 interaction. The partial unfolding of helix hα2 is contiguous and it assists the mechanistically relevant oxyanion strand flip and it should be thus considered as part of the allosteric pathways.
The perturbation network analysis using heavy atom contacts did not show a significant number of interactions among hydrophobic residues being affected by the effector binding. By including hydrogens in the count of contacts, the percentage of hydrophobic interactions that participate in the perturbation network increases (see Figure S5 in the Supporting Information). However, as mentioned above, the whole network including H′s represents a challenging graph to analyze since it contains a large number of contacts and a sizable amount of noise. As mentioned in the method section, in order to produce 2D (or 3D) representations that can be visually inspected (e.g., with number of perturbed pair <100) a large weight threshold (wt around 20) has to be applied to such a network (see Figure S6 in the Supporting Information). More than looking at the whole network including H′s, a more effective analysis can be performed by inspection of specific clusters of perturbations. For instance, Figure 6 shows the analysis of local perturbations around the key fK19 residue in loop1, indicating that rearrangements of contacts in loop1 are connected to residue fL50, previously reported as part of a hydrophobic cluster in the fβ2 strand,25 via the fD11 residue. Moreover, the modifications of the fD11-fK19 contact upon effector binding are correlated with the partial folding of the fβ8-fα8’ turn, as detected by the backbone analysis but here involving residues fS225 and fH228. Notably, it has been shown that fS225 is H-bonded to the glycerol phosphate group of PRFAR,25 and thus we performed a detailed investigation of the fS225-PRFAR H-bonds in relation to the fK19 residue along the MD simulations. We found that the fD11-fK19 contact modified upon effector binding promotes the formation of a H-bond network between fK19 (in loop1) and fS225 (in the fβ8-fα8’ turn) and the glycerol phosphate group of the PRFAR. All these observations explain the inhibition of allosteric signals in the K19A mutant40 and confirm the importance of both fD11 and fK19 residues for the allosteric communication in HisF. At the same time, the outcome claims for inclusion of the folded fβ8-fα8′ turn as a secondary structure element of the IGPS allosteric pathways.
Finally, by limiting the perturbation network analysis to the contacts among side chains (while including hydrogen atoms), some interesting features stand out at the HisF/HisH interface. In particular, the hM121 residue stands out in the side chain network (see Figure S7 in the Supporting Information) as it features several contact perturbations with the invariant fR5, fK99, and fE167 residues that belong to the ammonia tunnel gate of the HisF barrel51 and with the highly conserved fD98 of the structurally important fD98-hK181 salt-bridge anchor.25,51 It has been previously shown that the PRFAR binding, indeed, alters the dynamics of these conserved residues that are associated with important structural features of the complex IGPS enzyme.25,34 Thus, these results demonstrate that the perturbation network analysis of side chains can catch most of the structurally important conserved residues that are perturbed by the effector binding.
In summary, as shown in Figure 7, the perturbation network is a powerful tool for the characterization of the IGPS allosteric pathways based on analysis of MD trajectories, allowing recognition of previously overlooked allosteric spots. In particular, the use of the perturbation network approach showed that with just the analysis of the heavy atom contacts most of the secondary structure elements involved in the allosteric pathways are already detected. In addition to that, the involvement of the fβ1, fα2-fβ3 turn, and hα4 secondary structures (and related key residues) in the allosteric signal propagation has been recognized by perturbation of heavy atom contacts. The addition of hydrogen atoms in the contact counting and the concomitant restriction of the analysis to the backbone atoms readily provided the detection of folding/unfolding events during the MD simulations that are strictly connected to the signal propagation, including partial folding of the fβ8-fα8′ turn in the effector binding site and the partial unfolding of the hα2 helix in the proximity of the substrate binding site.
Figure 7.

Representation of the secondary structure elements involved in the allosteric pathways as predicted by previously reported studies (in light orange) and by the perturbation network analysis in this work (in light orange and red).
CONCLUSIONS
The dynamical perturbation network analysis has been proposed and assessed for the investigation of allosteric pathways in the IGPS enzyme, a prototype allosteric system that involves known allostery-relevant amino acid residues and secondary structure elements. The network analysis of dynamical inter-residue atomic contacts, obtained from averaging several independent MD simulations of the apo and effector-bound IGPS complexes, is an effective tool, as shown by the good agreement with previously reported community network analysis based on mutual information on protein-correlated motions. In fact, limiting the count of atomic contacts to heavy atoms already provided detection of strong effector-induced perturbations in the loop1, fα2, fα3, hα1, and Ω-loop secondary structure elements at the IGPS sideR, known to be involved in the allosteric signal propagation. Furthermore, the dynamical perturbation network analysis of heavy atom contacts also suggested previously overlooked residues fD11, fD74, hR22, and hR187 (located in the fβ1, fα2—fβ3 turn, and hα4 elements at sideR) as potential targets for future mutagenesis studies. Addition of hydrogen atoms in the computation of atomic contacts increases the complexity of the perturbation network, whose analysis has been separated in contributions from the backbone and the side chains atoms. The backbone network analysis, while sharing some features with the perturbation network analysis of heavy atoms contacts, highlighted some unknown allosteric perturbations, including the partial folding of the fβ8—fα8′ turn in the effector binding site and the partial unfolding of the hα2 helix in the proximity of the active site. Remarkably, restriction to the backbone atoms (including hydrogens) demonstrated how such network analysis provides rapid detection of folding/unfolding events induced by the effector binding that only time-consuming and tedious comparative analysis of MD trajectories can accomplish. However, the perturbation network analysis restricted to side chains contacts retrieved the structurally most important and highly conserved residues whose interactions are perturbed by the effector binding. Overall, by providing good agreement with previous theoretical and experimental studies and by recognition of new potential allosteric spots in the IGPS enzyme, the dynamical perturbation network analysis proved to be a powerful computational tool, complementary to other effective network-based methodologies for the characterization of allosteric pathways.
Supplementary Material
ACKNOWLEDGMENTS
The authors acknowledge the support of the Institut Rhônalpin des systemes complexes, IXXI-ENS-Lyon, Lyon, France. V.S.B. acknowledges support from the NIH grant GM106121 and supercomputer resources from NERSC. I.R. acknowledges the use of HPC resources of the “Pôle Scientifique de Modélisation Numérique” (PSMN) at the École Normale Supérieure de Lyon, France.
Biography

Ivan Rivalta, born in Catanzaro, Italy, received his Laurea and Ph.D. in Chemistry at the Università della Calabria, Italy. After a visiting postdoctoral fellowship at the Department of Chemistry and Applied Biosciences of ETH Zurich (Lugano, Switzerland), he was Associate Research Scientist first at the Chemistry Department of Yale University (USA) and subsequently at the Dipartimento di Chimica “G. Ciamician” of the Alma Mater Studiorum, Università di Bologna, Italy. In 2014, he was appointed as CNRS permanent Researcher in the Laboratoire de Chimie UMR-5182 at the École Normale Supérieure de Lyon, France. Since 2018, he is Associate Professor of Physical Chemistry at the Dipartimento di Chimica Industriale “Toso Montanari” of the Alma Mater Studiorum, Università di Bologna, Italy. His research concerns the development and application of theoretical methods and computational techniques for the study of chemical and photochemical phenomena, with a focus on biological and biomimetic systems.
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jpcb.9b01294.
Protein contact network modeling, Additional results including hydrogen bonds and ionic interactions modifications, analysis of pair contacts types and significant contact perturbations in the ammonia gate (PDF)
Published as part of The Journal of Physical Chemistry virtual special issue “Young Scientists”.
REFERENCES
- (1).Changeux JP 50 Years of Allosteric Interactions: The Twists and Turns of the Models. Nat. Rev. Mol. Cell Biol 2013, 14, 819–829. [DOI] [PubMed] [Google Scholar]
- (2).Fenton AW Allostery: An Illustrated Definition for the ‘Second Secret of Life’. Trends Biochem. Sci 2008, 33, 420–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Laskowski RA; Gerick F; Thornton JM The Structural Basis of Allosteric Regulation in Proteins. FEBS Lett 2009, 583, 1692–1698. [DOI] [PubMed] [Google Scholar]
- (4).Motlagh HN; Wrabl JO; Li J; Hilser VJ The Ensemble Nature of Allostery. Nature 2014, 508, 331–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Monod J; Wyman J; Changeux JP On the Nature of Allosteric Transitions: A Plausible Model. J. Mol. Biol 1965, 12, 88–118. [DOI] [PubMed] [Google Scholar]
- (6).Koshland DE; Nemethy G; Filmer D Comparison of Experimental Binding Data and Theoretical Models in Proteins Containing Subunits. Biochemistry 1966, 5, 365–368. [DOI] [PubMed] [Google Scholar]
- (7).Hilser VJ; Wrabl JO; Motlagh HN Structural and Energetic Basis of Allostery. Annu. Rev. Biophys 2012, 41, 585–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Tsai CJ; Nussinov R A Unified View of “How Allostery Works”. PLoS Comput. Biol 2014, 10, e1003394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Christopoulos A Allosteric Binding Sites on Cell-Surface Receptors: Novel Targets for Drug Discovery. Nat. Rev. Drug Discovery 2002, 1, 198–210. [DOI] [PubMed] [Google Scholar]
- (10).Wootten D; Christopoulos A; Sexton PM Emerging Paradigms in Gpcr Allostery: Implications for Drug Discovery. Nat. Rev. Drug Discovery 2013, 12, 630–644. [DOI] [PubMed] [Google Scholar]
- (11).Taly A; Corringer PJ; Guedin D; Lestage P; Changeux JP Nicotinic Receptors: Allosteric Transitions and Therapeutic Targets in the Nervous System. Nat. Rev. Drug Discovery 2009, 8, 733–750. [DOI] [PubMed] [Google Scholar]
- (12).Gohara DW; Di Cera E Allostery in Trypsin-Like Proteases Suggests New Therapeutic Strategies. Trends Biotechnol 2011, 29, 577–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Nussinov R; Tsai CJ Allostery in Disease and in Drug Discovery. Cell 2013, 153, 293–305. [DOI] [PubMed] [Google Scholar]
- (14).Makhlynets OV; Raymond EA; Korendovych IV Design of Allosterically Regulated Protein Catalysts. Biochemistry 2015, 54, 1444–1456. [DOI] [PubMed] [Google Scholar]
- (15).Lisi GP; Manley GA; Hendrickson H; Rivalta I; Batista VS; Loria JP Dissecting Dynamic Allosteric Pathways Using Chemically Related Small-Molecule Activators. Structure 2016, 24, 1155–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Kar G; Keskin O; Gursoy A; Nussinov R Allostery and Population Shift in Drug Discovery. Curr. Opin. Pharmacol 2010, 10, 715–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Süel GM; Lockless SW; Wall MA; Ranganathan R Evolutionarily Conserved Networks of Residues Mediate Allosteric Communication in Proteins. Nat. Struct. Biol 2003, 10, 59–69. [DOI] [PubMed] [Google Scholar]
- (18).Amaro RE; Sethi A; Myers RS; Davisson VJ; Luthey-Schulten ZA A Network of Conserved Interactions Regulates the Allosteric Signal in a Glutamine Amidotransferase. Biochemistry 2007, 46, 2156–2173. [DOI] [PubMed] [Google Scholar]
- (19).Bruschweiler S; Schanda P; Kloiber K; Brutscher B; Kontaxis G; Konrat R; Tollinger M Direct Observation of the Dynamic Process Underlying Allosteric Signal Transmission. J. Am. Chem. Soc 2009, 131, 3063–3068. [DOI] [PubMed] [Google Scholar]
- (20).del Sol A; Tsai CJ; Ma B; Nussinov R The Origin of Allosteric Functional Modulation: Multiple Pre-Existing Pathways. Structure 2009, 17, 1042–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Feher VA; Durrant JD; Van Wart AT; Amaro RE Computational Approaches to Mapping Allosteric Pathways. Curr. Opin. Struct. Biol 2014, 25, 98–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Martin NE; Malik S; Calimet N; Changeux JP; Cecchini M Un-Gating and Allosteric Modulation of a Pentameric Ligand-Gated Ion Channel Captured by Molecular Dynamics. PLoS Comput. Biol 2017, 13, e1005784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Markwick PR; McCammon JA Studying Functional Dynamics in Bio-Molecules Using Accelerated Molecular Dynamics. Phys. Chem. Chem. Phys 2011, 13, 20053–20065. [DOI] [PubMed] [Google Scholar]
- (24).De Vivo M; Masetti M; Bottegoni G; Cavalli A Role of Molecular Dynamics and Related Methods in Drug Discovery. J. Med. Chem 2016, 59, 4035–4061. [DOI] [PubMed] [Google Scholar]
- (25).Rivalta I; Sultan MM; Lee NS; Manley GA; Loria JP; Batista VS Allosteric Pathways in Imidazole Glycerol Phosphate Synthase. Proc. Natl. Acad. Sci U. S. A 2012, 109, E1428–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Ricci CG; Silveira RL; Rivalta I; Batista VS; Skaf MS Allosteric Pathways in the Pparγ-Rxrα Nuclear Receptor Complex. Sci. Rep 2016, 6, 19940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Palermo G; Ricci CG; Fernando A; Basak R; Jinek M; Rivalta I; Batista VS; McCammon JA Protospacer Adjacent Motif-Induced Allostery Activates Crispr-Cas9. J. Am. Chem. Soc 2017, 139, 16028–16031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Negre CFA; Morzan UN; Hendrickson HP; Pal R; Lisi GP; Loria JP; Rivalta I; Ho J; Batista VS Eigenvector Centrality for Characterization of Protein Allosteric Pathways. Proc. Natl. Acad. Sci. U. S. A 2018, 115, E12201–E12208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Sethi A; Eargle J; Black AA; Luthey-Schulten Z Dynamical Networks in Trna: Protein Complexes. Proc. Natl. Acad. Sci. U. S. A 2009, 106, 6620–6625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Gasper PM; Fuglestad B; Komives EA; Markwick PR; McCammon JA Allosteric Networks in Thrombin Distinguish Procoagulant Vs. Anticoagulant Activities. Proc. Natl. Acad. Sci. U. S. A 2012, 109, 21216–21222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Blacklock K; Verkhivker GM Computational Modeling of Allosteric Regulation in the Hsp90 Chaperones: A Statistical Ensemble Analysis of Protein Structure Networks and Allosteric Communications. PLoS Comput. Biol 2014, 10, e1003679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Stolzenberg S; Michino M; LeVine MV; Weinstein H; Shi L Computational Approaches to Detect Allosteric Pathways in Transmembrane Molecular Machines. Biochim. Biophys. Ada, Biomembr 2016, 1858, 1652–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Wagner JR; Lee CT; Durrant JD; Malmstrom RD; Feher VA; Amaro RE Emerging Computational Methods for the Rational Discovery of Allosteric Drugs. Chem. Rev 2016, 116, 6370–6390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Rivalta I; Lisi GP; Snoeberger NS; Manley G; Loria JP; Batista VS Allosteric Communication Disrupted by a Small Molecule Binding to the Imidazole Glycerol Phosphate Synthase Protein-Protein Interface. Biochemistry 2016, 55, 6484–6494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Manley G; Rivalta I; Loria JP Solution Nmr and Computational Methods for Understanding Protein Allostery. J.Phys. Chem. B 2013, 117, 3063–3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Chaudhuri BN; Lange SC; Myers RS; Chittur SV; Davisson VJ; Smith JL Crystal Structure of Imidazole Glycerol Phosphate Synthase: A Tunnel through a (Beta/Alpha)(8) Barrel Joins Two Active Sites. Structure 2001, 9, 987–997. [PubMed] [Google Scholar]
- (37).Breitbach K; Kohler J; Steinmetz I Induction of Protective Immunity against Burkholderia Pseudomallei Using Attenuated Mutants with Defects in the Intracellular Life Cycle. Trans. R. Soc. Trop. Med. Hyg 2008, 102 (Suppl 1), S89–94. [DOI] [PubMed] [Google Scholar]
- (38).Gomez MJ; Neyfakh AA Genes Involved in Intrinsic Antibiotic Resistance of Acinetobacter Baylyi. Antimicrob. Agents Chemother 2006, 50, 3562–3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Myers RS; Jensen JR; Deras IL; Smith JL; Davisson VJ Substrate-Induced Changes in the Ammonia Channel for Imidazole Glycerol Phosphate Synthase. Biochemistry 2003, 42, 7013–7022. [DOI] [PubMed] [Google Scholar]
- (40).Lisi GP; East KW; Batista VS; Loria JP Altering the Allosteric Pathway in Igps Suppresses Millisecond Motions and Catalytic Activity. Proc. Natl. Acad. Sci. U. S. A 2017, 114, E3414–E3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Vuillon L; Lesieur C From Local to Global Changes in Proteins: A Network View. Curr. Opin. Struct. Biol 2015, 31, 1–8. [DOI] [PubMed] [Google Scholar]
- (42).Dorantes-Gilardi R; Bourgeat L; Pacini L; Vuillon L; Lesieur C In Proteins, the Structural Responses of a Position to Mutation Rely on the Goldilocks Principle: Not Too Many Links, Not Too Few. Phys. Chem. Chem. Phys 2018, 20, 25399–25410. [DOI] [PubMed] [Google Scholar]
- (43).Achoch M; Dorantes-Gilardi R; Wymant C; Feverati G; Salamatian K; Vuillon L; Lesieur C Protein Structural Robustness to Mutations: An in Silico Investigation. Phys. Chem. Chem. Phys 2016, 18, 13770–13780. [DOI] [PubMed] [Google Scholar]
- (44).Doshi U; Holliday MJ; Eisenmesser EZ; Hamelberg D Dynamical Network of Residue-Residue Contacts Reveals Coupled Allosteric Effects in Recognition, Catalysis, and Mutation. Proc. Natl. Acad. Sci. U. S. A 2016, 113, 4735–4740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Case DA; Cheatham TE 3rd; Darden T; Gohlke H; Luo R; Merz KM Jr.; Onufriev A; Simmerling C; Wang B; Woods RJ The Amber Biomolecular Simulation Programs. J. Comput. Chem 2005, 26, 1668–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Wang J; Wolf RM; Caldwell JW; Kollman PA; Case DA Development and Testing of a General Amber Force Field. J. Comput. Chem 2004, 25, 1157–1174. [DOI] [PubMed] [Google Scholar]
- (47).Phillips JC; Braun R; Wang W; Gumbart J; Tajkhorshid E; Villa E; Chipot C; Skeel RD; Kale L; Schulten K Scalable Molecular Dynamics with NAMD. J. Comput. Chem 2005, 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Darden T; York D; Pedersen L Particle Mesh Ewald: An NLog(N) Method for Ewald Sums in Large Systems. J. Chem. Phys 1993, 98, 10089–10092. [Google Scholar]
- (49).Grubmuller H; Heller H; Windemuth H; Schulten K Generalized Verlet Algorithm for Efficient Molecular Dynamics Simulations with Long-Range Interactions. Mol. Simul 1991, 6, 121–142. [Google Scholar]
- (50).Schlick T; Skeel RD; Brunger AT; Kale LV; Board JA; Hermans J; Schulten K Algorithmic Challenges in Computational Molecular Biophysics. J. Comput. Phys 1999, 151, 9–48. [Google Scholar]
- (51).Douangamath A; Walker M; Beismann-Driemeyer S; Vega-Fernandez MC; Sterner R; Wilmanns M Structural Evidence for Ammonia Tunneling across the (Beta/Alpha)(8) Barrel of the Imidazole Glycerol Phosphate Synthase Bienzyme Complex. Structure 2002, 10, 185–193. [DOI] [PubMed] [Google Scholar]
- (52).Lipchock JM; Loria JP Nanometer Propagation of Millisecond Motions in V-Type Allostery. Structure 2010, 18, 1596–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.