

Abstract
Although homozygous sickle cell disease is often clinically severe, the corresponding heterozygous state, sickle cell trait, is almost completely benign despite the fact that there is only a modest difference in sickle hemoglobin levels between the two conditions. In both conditions, hypoxia can lead polymerization of sickle hemoglobin, changes in red cell mechanical properties, and impaired blood flow. Here, we test the hypothesis that differences in the oxygen-dependent rheological properties in the two conditions might help explain the difference in clinical phenotypes. We use a microfluidic platform that permits quantification of blood rheology under defined oxygen conditions in physiologically sized microchannels and under physiologic shear rates. We find that, even with its lower sickle hemoglobin concentration, sickle trait blood apparent viscosity increases with decreasing oxygen tension and may stop flowing under completely anoxic conditions, though far less readily than the homozygous condition. Sickle cell trait blood flow becomes impaired at significantly lower oxygen tension than sickle cell disease. We also demonstrate how sickle cell trait can serve as a benchmark for sickle cell disease therapies. We characterize the rheological effects of exchange transfusion therapy by mixing sickle blood with non-sickle blood and quantifying the transfusion targets for sickle hemoglobin composition below which the rheological response resembles sickle trait. These studies quantify the differences in blood flow phenotypes of sickle cell disease and sickle cell trait, and they provide a potentially powerful new benchmark for evaluating putative therapies in vitro.
Keywords: Sickle cell disease, sickle cell trait, RBC transfusion, rheology, microfluidics
Introduction
In sickle cell disease (SCD), individuals inherit two copies of the sickle variant of the human beta globin gene (HBB), and under hypoxic conditions, the mutant sickle hemoglobin (HbS) proteins can polymerize into fibers and gels, resulting in stiffened red blood cells (RBCs).1–4 Changes in RBC mechanical properties lead to increased blood apparent viscosity and along with increased cellular adhesion and inflammation, contribute to vaso-occlusion, the primary driver of morbidity in SCD.3–4,5–6 Vaso-occlusion and its downstream pathologies contribute to a mortality rate for SCD as high as 7.3% in children under 5-years old and a significantly reduced overall life expectancy.7–11 In sickle cell trait (SCT) however, individuals inherit only a single copy of the mutant hemoglobin subunit beta gene, which results in an almost entirely clinically benign phenotype, except under rare and extreme conditions.12–23 While SCD and SCT have vastly different clinical outcomes, the differences in sickle hemoglobin composition measured in the clinical laboratory can be far more modest. The HbS fraction for SCD is typically above 80% but can be as low as ~55% in the sickling syndromes of hemoglobin SC disease or sickle-beta-plus thalassemia, and even lower in transfused SCD patients in general. By comparison, the HbS fraction in SCT typically ranges from 30–45%.24,25 It is currently unclear how such a modest reduction in hemoglobin S fraction – from levels found in many SCD patients to those typical of SCT – almost completely and permanently abrogates clinical symptoms. A more detailed understanding of the functional consequences of these modest changes in hemoglobin S fraction would likely suggest novel ways to alter or perturb the SCD clinical phenotype toward the SCT phenotype, thereby providing much-needed effective treatments.
It has been clear for decades that despite the almost uniformly benign outcomes of SCT, hemoglobin S polymer forms in RBCs from individuals with SCT. Prior studies have detected hemoglobin polymer in SCT RBCs in the physiologic range of oxygen tensions.26–29 There is also some clinical evidence that the HbS fraction in sickle trait correlates with the degree of mild impairment in kidney function, though not with risk for serious clinical outcomes.30,31 Current standard clinical screening for SCT relies on the detection of polymer in RBCs under extreme hypoxic conditions.24 Why this polymerization in SCT RBCs does not translate into adverse clinical outcomes remains unclear, but one likely factor is that under physiologic conditions oxygen tension only rarely falls below the level at which significant polymer forms in SCT RBCs. Only under extreme conditions would there be enough polymerization in SCT to cause a pathologic increase in blood apparent viscosity that would persist long enough to cause symptoms before the RBCs can be sufficiently re-oxygenated. Testing this hypothesis regarding in vivo flow properties, however, is challenging due to limitations in existing model systems. Studies in transgenic mouse models provide a means to observe blood flow under physiologically relevant conditions, but the lack of precise spatial control of oxygen tension or blood pressure in vivo makes it difficult to draw firm conclusions. Polymerization kinetics of mixtures of HbS and adult hemoglobin (HbA) have been quantified in vitro, but validated models do not currently exist to extrapolate from molecular or even single cell measurements to whole blood, which is relevant in vivo. What is required is a way to quantify blood flow under conditions that are relevant in vivo and that can be directly controlled as experimental variables. In a previous study, we quantified the flow of blood from SCD individuals using a microfluidic platform that allowed for precise control of oxygen tension in a pressure-driven system at physiologically relevant size scales, temperatures, and shear rates. Using this platform, we were able to show that blood from SCD patients shows significantly increased apparent viscosity at relatively high oxygen tensions, even in the range of oxygen tension that can be found within the arterial circulation, suggesting that hemoglobin S polymerization in SCD could lead to increased blood apparent viscosity throughout the vasculature.32 The same system can be applied to define the conditions under which hemoglobin S polymerization in SCT could lead to increased apparent viscosity.
In this study, we used our previously published microfluidic platform to quantify the rheology of SCT blood as a function of oxygen tension and to compare those results to SCD blood. Our results show that at native hematocrit and physiologic temperature and shear rate, blood from individuals with SCT exhibits oxygen-dependent rheology that is qualitatively similar to but quantitatively distinct from blood from SCD individuals. We show that the apparent viscosity of SCT blood begins to increase at significantly lower oxygen tensions than SCD blood, and we show that SCT blood from some individuals can occlude under completely anoxic conditions but not universally, unlike in SCD. We further report how simulated blood transfusion alters the rheology of SCD and show that SCT provides a useful benchmark to establish clinically effective transfusion targets.
Methods
Blood Sample Collection
Whole blood was collected from patients with either sickle cell disease or sickle cell trait in 5mL EDTA vacutainers and stored at 4°C for between a few hours and up to 4 days. Previous studies from our own lab and others have shown these storage conditions do not cause significant changes to the rheological properties of blood.32–37 Blood samples were collected from Massachusetts General Hospital, Brigham and Women’s Hospital, and the University of Minnesota Medical Center under protocols authorized by Institutional Review Boards at Partners Healthcare and the University of Minnesota. Complete blood count tests were completed on all samples using a Sysmex XE-5000 automated analyzer (Sysmex, Kobe, Japan). Hemoglobin fractions were determined using high performance liquid chromatography via a Tosoh G7 column (Tosoh Bioscience, San Francisco, California).
Microfluidic device and fabrication
For all experiments, we utilized a microfluidic platform as shown in Fig. 1A,B and described previously32,34. Briefly, we developed a multi-layered polydimethylsiloxane (PDMS) based microfluidic device that consists of three vertically separated layers: 1) a blood microchannel layer with an arteriole/venule sized cross sectional area of 15 μm x 15 μm as seen in Fig. 1C, 2) a 1.5 mm × 100 μm hydration layer to prevent blood layer evaporation, and 3) a 1.5 mm x 150 μm oxygen gas tension control layer. A 100 μm PDMS membrane separated each of the three individual layers.
Figure 1.
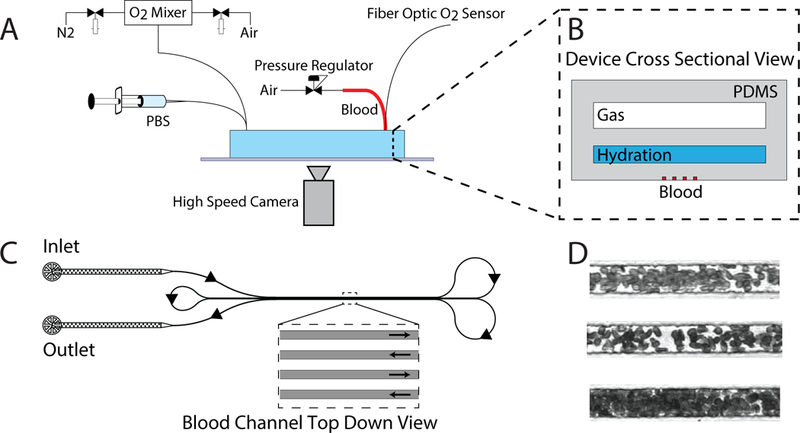
In vitro disease model to study sickle trait blood flow using a microfluidic platform. (A) Schematic of the experimental platform built around the microfluidic device including an oxygen gas mixer, PBS hydration flow regulator, pressure regulation of whole blood, oxygen sensor, and high speed camera. Insert (B) shows a cross sectional view of the microfluidic device, which comprises of the gas, hydration, and blood layers as described in methods (C). Illustration of the blood microchannels in the device. A single blood channel starts at the inlet and loops around allowing for the simultaneous imaging of 4 sections of the channel as shown by the inset. Arrows along the channels depict the directionality of the blood as it traverses through the microchannels. (D). Photographs of blood as it traverses the microfluidic channels.
Figure 2.
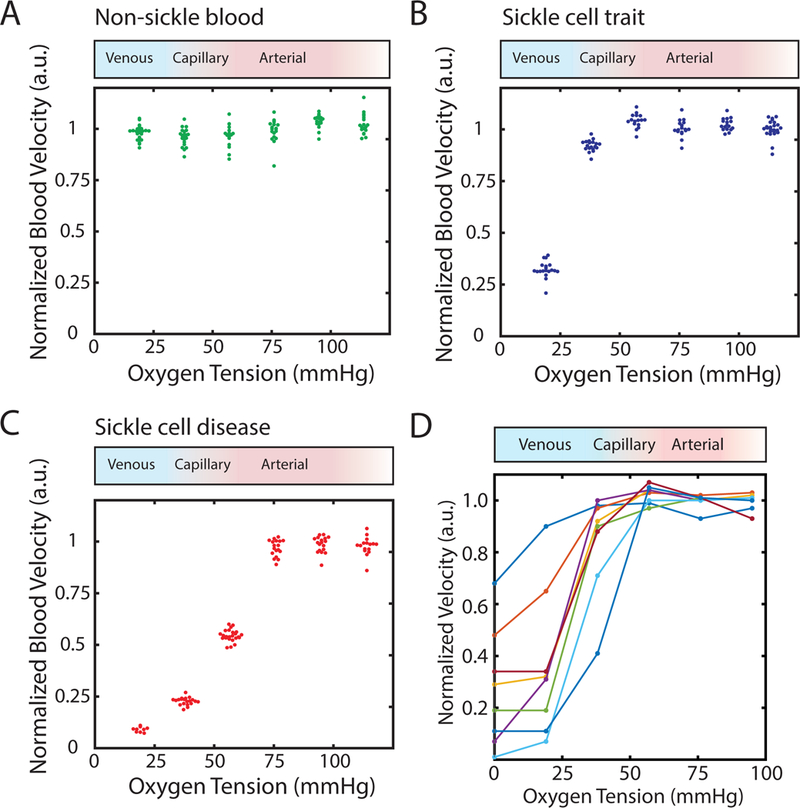
Rheological behavior of SCT blood becomes oxygen-dependent near venous oxygen tension. (A) Normalized, steady state flow velocity of a non-sickle (genotype AA) individual’s blood sample does not depend on oxygen tension. Oxygen tension was varied in a stepwise manner as described in methods. The red to blue shaded gradient above the plot corresponds to oxygen tensions typically found in arterial circulation (red) and venous circulation (blue). Supra-physiological oxygen tension (white) is also displayed towards the right side of the box. (B) Normalized steady state flow velocity of an individual with sickle cell trait is dependent on oxygen tension. At high oxygen tension (>38 mmHg), velocities are oxygen- independent and exhibit regime I flow behavior. Between 19 and 38 mmHg, flow behavior transitions to regime II where steady state velocities become impaired and are sensitive to oxygen tension. (C) Normalized steady state flow velocity for an individual with homozygous sickle cell disease is dependent on oxygen tension. High oxygen tension (>76 mmHg) results in oxygen-independent regime I flow behavior. Intermediate oxygen tension (>19 mmHg and <76 mmHg or 19<ppO2<76 mmHg) displays impaired, oxygen-dependent regime II flow behavior, and low oxygen tension (<19 mmHg) exhibit severely impaired/nearly occluded regime III behavior. (D) Normalized, median, steady state velocities for 8 different SCT blood samples. Each colored line corresponds to a unique SCT blood sample.
Fabrication of the PDMS microfluidic device consisted of three primary steps. First, soft photolithography was used to create three separate master molds for each of the different layers as described previously.32–34 To fabricate each of the individual layers of the device, we cast PDMS at an elastomer/curing agent ratio of 10:1 onto each of the master molds. For the oxygen gas layer, PDMS was cast directly on the mold to achieve a total PDMS thickness of 5 mm. For the blood microchannel and hydration layers, we used a PDMS compression molding technique as described previously to create the individual PDMS layers at their specified thicknesses.32,33 Finally, the gas, hydration, and blood microchannel layers were covalently bonded together using plasma at a power of 100 watts, an oxygen flow rate of 100 cc/minute, and an exposure time of 30 seconds. The final construct was then bonded to a clean microscope glass slide to complete device fabrication.
Experimental setup and operation
To measure blood rheology, whole blood from individuals with sickle cell disease or sickle cell trait was perfused through the blood microchannels of the microfluidic device mounted to a temperature-regulated (37°C) Zeiss Axio Vert.A1 (Carl Zeiss, Jena, Germany). Hematocrit of blood was not altered from the in vivo level to focus investigation on in vivo rheological properties. Blood was perfused at a constant 1 PSI pressure using an electronic pressure regulator (PCD-15PSIG, Alicat Scientific, Tucson, Arizona). Phosphate buffered saline was perfused through the hydration channel of the device using a syringe pump (NE-500, New Era Pump Systems, Farmingdale, New York) to prevent dehydration of the blood. Oxygen gas was perfused through the oxygen gas layer of the device at a constant 1 PSI pressure using pressure regulators (Omega, Norfolk, Connecticut). Discrete oxygen gas tensions were created by mixing 160 mmHg oxygen gas (21% O2, 5% CO2, balance N2) with 0 mmHg oxygen gas (5% CO2, balance N2) using a previously developed solenoid valve gas mixing setup that periodically duty cycles the two gases.32,33,38 Oxygen gas tension was continuously monitored on chip using a fiber optic oxygen sensor (NeoFox-GT, Ocean Optics, Dunedin, Florida). For all experiments, as blood was perfused through the blood microchannels of the device, oxygen gas tension was initially set to fully oxygenated conditions (160 mmHg). Oxygen gas tension was then decreased to 114 mmHg (15%) and held for 5 minutes. Oxygen gas tension was further decreased in 19 mmHg (2.5%) steps until 0 mmHg or until occlusive behavior was seen. Each oxygen gas tension step was held for a minimum of 5 minutes to capture stabilized, steady state behavior.
Rheological measurements
Blood rheological behavior was quantified by measuring blood flow velocity within the blood microchannels. For each experiment, videos of blood flow within the microchannels are captured in 32 frame bursts at a frame rate of 700 frames per second at a frequency of one video every 3–4 seconds. Figure 1D shows still frames from blood as it traverses the microchannels. An optical flow tracking algorithm based on a Kanade-Lucas-Tomasi feature tracker using computer vision within MATLAB (Mathworks, Natick, Massachusetts) was then implemented to calculate blood flow velocities.39 The efficacy of the tracking algorithm was validated against our previously published method.40 The velocity at each time point was defined as the average velocity in the center 50% of the channel (excludes cell velocities at the wall). Blood flow velocities were then normalized to the mean steady state velocities during fully oxygenated conditions (160 mmHg) to account for slight variations in device setup. Because collected velocity values include transients due to hemoglobin polymerization delay times or oxygen diffusion within the microfluidic device, we defined the steady state velocities for each oxygen gas tension step by considering only the middle 50–80% portion of the time interval.
For all SCT blood samples we tested, we determined a critical oxygen tension at which the blood velocity transitions from an oxygen-independent flow velocity to an oxygen-dependent (reduced) velocity (regime I/II boundary, as described in results). We first determined a confidence interval for the oxygen tension of the regime I/II boundary by determining the range of oxygen tension that we experimentally measured where mean steady state velocity decreased significantly (p<0.01). The lower bound of the confidence interval was defined as the highest oxygen tension where mean blood velocity is at least three standard deviations below the mean blood velocity of the next highest measured oxygen tension step, as well as three standard deviations below the mean blood velocity of the fully oxygenated condition (160 mmHg). The upper bound of the confidence interval was defined as the lowest oxygen tension whose mean blood velocity is not significantly different from the fully oxygenated condition. To determine the specific oxygen tension where the regime I/II boundary is located, we linearly interpolated velocities at every oxygen tension (at a ppO2=1 mmHg resolution) and found the highest oxygen tension in which the interpolated velocity is three standard deviations lower than the measured upper bound of the confidence interval. To compare between SCT and SCD groups, statistical significance was determined non-parametrically using a Mann-Whitney U test.
Results
Sickle cell trait blood rheology is oxygen dependent
We quantified the relationship between steady state blood flow velocity and oxygen gas tension for blood samples from 8 SCT patients (genotype AS) at native hematocrit and physiologic temperature. Using our microfluidic platform, we serially decreased oxygen tension in 19 mmHg steps (2.5% O2) from 114 mmHg (15% O2) down to 0 mmHg (0% O2) or until occlusive behavior was observed. At each oxygen tension step, we quantified the steady state velocity, and the results are shown in Fig. 2. As expected, blood from individuals lacking the sickle mutation (genotype AA) showed no detectable response to oxygen (Fig. 2A), whereas blood from both SCT (Fig. 2B) and SCD (Fig. 2C) individuals had flow velocities that began to decrease with decreasing oxygen tension after some oxygen tension threshold. In the representative SCT patient blood sample shown in Fig. 2B, oxygen tensions above 38 mmHg resulted in blood velocities that were independent of oxygen tension and were comparable in magnitude to genotype AA blood. Between 19 and 38 mmHg, there is a transition to an oxygen-dependent regime, where velocity decreases monotonically with oxygen tension. This behavior is qualitatively similar to that observed for the SCD sample in Fig. 2C. As expected, however, the transition to oxygen dependent flow occurs at a higher oxygen tension for the SCD sample relative to the SCT sample, and the SCD sample fully occludes at low oxygen tension, unlike this SCT sample. Additionally, we imaged individual RBCs for SCD and SCT to observe changes in RBC morphology in response to oxygen tension (Supplemental Figure S1). We found that at very low oxygen both conditions trigger morphological changes, but morphology changes first appear at higher oxygen tension in SCD than SCT.
For SCD blood samples (as seen in Supplemental Figure S2), we previously characterized three distinct functional regimes for blood flow as a function of oxygen tension: (I) at high oxygen tension, blood flow was oxygen-independent with velocities comparable to those measured in non-sickle (genotype AA) blood; (II) at intermediate oxygen tension, blood flow was oxygen-dependent, with decreasing oxygen tension leading to monotonically decreasing blood flow velocity; (III) at low oxygen tension, blood flow was oxygen-independent but with significantly reduced velocities (sometimes zero) compared to regime I.32 Figure 2D shows the normalized, median velocities for all 8 SCT blood samples as a function of oxygen tension. In all of these SCT blood samples, we see similar qualitative behavior to the homozygous SCD with blood flow also exhibiting oxygen-dependent behavior (Fig. 2B). All SCT samples we measured displayed oxygen-independent flow velocities at high oxygen tension (regime I), as well as oxygen-dependent impaired flow (regime II) at intermediate oxygen tension. However, not all of the SCT samples we quantified exhibit regime III behavior. In the 4 samples that did not exhibit regime III behavior, low oxygen tension resulted in an extension of regime II behavior where decreasing oxygen tension resulted in monotonically decreasing blood velocity. SCT blood samples that did exhibit regime III behavior, typically displayed near/full occlusion. Thus, only 50% of the SCT samples tested occluded under fully anoxic conditions (i.e., unlikely to occur other than in extreme or pathologic conditions), while 83% of the SCD samples tested occluded under anoxic conditions. Additionally, we observed that SCT samples that did occlude recovered to initial flow velocities (regime I behavior) when oxygen was reintroduced, similar to our previous observations for SCD samples.32–34,41
In addition to steady state rheology, we measured the rate of change of velocity upon rapid deoxygenation from a fully oxygenated state as described previously.42 We compared whole blood from 8 different SCT blood samples against 11 SCD blood samples and found deceleration rates to be significantly slower in SCT blood (p=0.041, Mann Whitney U non-parametric testing), as seen in Supplemental Figure S3.
Oxygen dependence of SCT blood samples correlates with HbS fraction
Previously, we quantified the in vitro oxygen tensions at which blood flow in SCD patients first became impaired and oxygen-dependent (regime I/II transition)32, and in this study, we quantified this transition for 8 unique SCT patients (Fig. 3A). For one of the samples, we observed no impaired flow until oxygen tension dropped below 19 mmHg. In four of the eight samples, flow became impaired when oxygen dropped from 38 to 19 mmHg, and for three of the eight samples flow became impaired when oxygen tension dropped from 57 to 38 mmHg. We compared this regime I/II transition oxygen tension for SCT patients to that from 12 unique SCD patient samples (Fig. 3A), and the regime I/II transition occurred at significantly (p<0.01) lower oxygen tensions in SCT patients compared to SCD patients (Fig. 3B).
Figure 3.
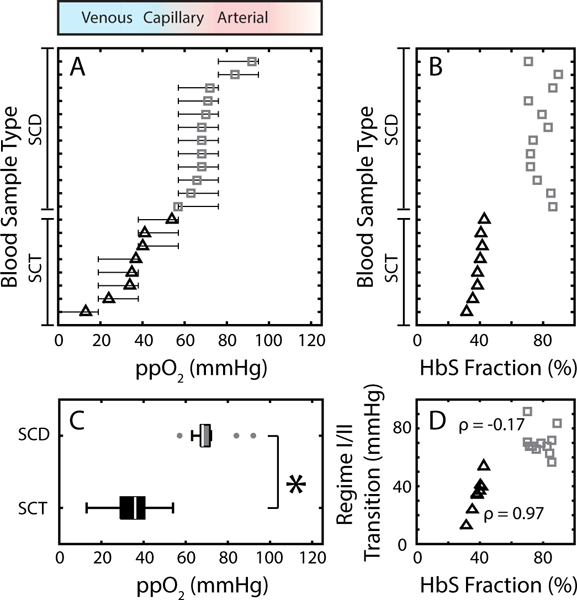
Flow behavior of SCT blood differs from SCD. (A) Regime I/II transition for SCD (gray squares) compared to SCT (black triangles). Confidence intervals are determined as outlined in the methods. The red to blue to white color gradient bar above the plot corresponds to typical oxygen tensions found in arterial circulation (red), venous circulation (blue), and supra-physiological oxygen tension (white). (B) Bulk HbS fraction for both SCD blood samples (gray squares) and SCT blood samples (black triangles) as measured by HPLC. SCD blood samples are non-transfused and all have higher HbS% compared to SCT blood samples. (C) Box plot comparing the oxygen tension at which the regime I/II transition occurs for SCD (gray box) with SCT (black box) blood samples. Non-parametric analysis (Mann Whitney-U) tests found significant differences between the two groups (p=0.0023). (D) Oxygen tension at which the regime I/II transition occurs as a function of HbS fraction for SCD (gray squares) and SCT (black triangles) blood samples. SCD blood did not exhibit a strong correlation between the regime I/II transition and HbS% (Pearson correlation coefficient, ρ=−0.17), but SCT samples displayed a strong, positive correlation (ρ=0.97).
We previously found no strong correlation between the location of the transition to impaired flow (regime I/II transition) and common hematologic parameters for SCD patients, and that finding holds for the SCD patient samples measured for this study as well (Fig. 3B,D).32 By contrast, the transition to impaired flow for SCT patient samples displayed a strong correlation (correlation coefficient, ρ=0.97) with bulk HbS fraction. The SCT sample with the lowest HbS% had the lowest regime I/II transition boundary, and the SCT samples with the highest HbS% had the highest regime I/II transition boundary. We also observed that occlusions (regime III behavior) were more likely in SCT samples with higher HbS%, and no sample with an HbS fraction less than 40% occluded at low oxygen tension. Overall, we found that 4 out of the 8 SCT patient blood samples occluded at low oxygen tension (ppO2 <= 19 mmHg).
Sufficiently transfused SS blood samples show oxygen-dependent rheology similar to that of SCT blood samples
Transfusion therapy lowers HbS in SCD patients and is one of the most common treatments for the disease.43–47 Clinical guidelines for red cell exchange procedures have a target endpoint (33% HbS48–52) that is comparable to the HbS fraction typically found in SCT. The HbS in SCT is generally assumed to be uniformly distributed across all RBCs, in contrast to the HbS in transfused SCD which is confined to the recipient’s own RBCs, and we can use our measurement system to compare the rheology of SCT blood and transfused SCD blood. For two unique SS patients, we simulated transfusion therapy by mixing their blood with ABO/Rh-matched AA blood in five volumetric ratios that include the range of clinical transfusion targets (Supplemental Table S1). We then performed the same measurements in our microfluidic platform as described above for SCT and quantified the steady state velocity response for oxygen tensions between 0 and 114 mmHg. Fig. 4A,B displays a point graph of normalized, median velocities (y-axis) as a function of oxygen tension (x-axis) for all transfusion levels for each blood sample. At transfusion levels that corresponded to simple transfusion of up to about two units of blood (30% donor blood by volume mixed with 70% recipient) and resulted in ~47% HbS fraction, the two samples exhibited nearly identical rheological behavior to untransfused blood at all oxygen tensions. For transfusions that resulted in HbS fractions of ~47%, the blood samples exhibited regimes I, II, and III flow behavior as defined previously. In contrast, decreasing the HbS fraction below the red cell exchange procedure target (<30% HbS), even without controlling HCT, yielded a large reduction in the magnitude of velocity change between oxygenated and deoxygenated conditions as seen by the differences in velocity along the x-axis of the Fig. 4A,B. Transfused samples with ~5% HbS fraction still yielded a detectable rheological response to oxygen, albeit a weaker response than other transfused specimens, as expected. Thus, we observe that transfused SCD samples show similar oxygen-dependent rheological changes that are qualitatively similar to untransfused SCD blood, but the magnitude of the rheological changes decreases with decreasing HbS fraction. Notably, we observe oxygen-dependent rheological behavior even with only 5% HbS fraction, suggesting that even a small fraction of deoxygenated HbS-containing cells is capable of increasing whole blood viscosity by a detectable amount.
Figure 4.
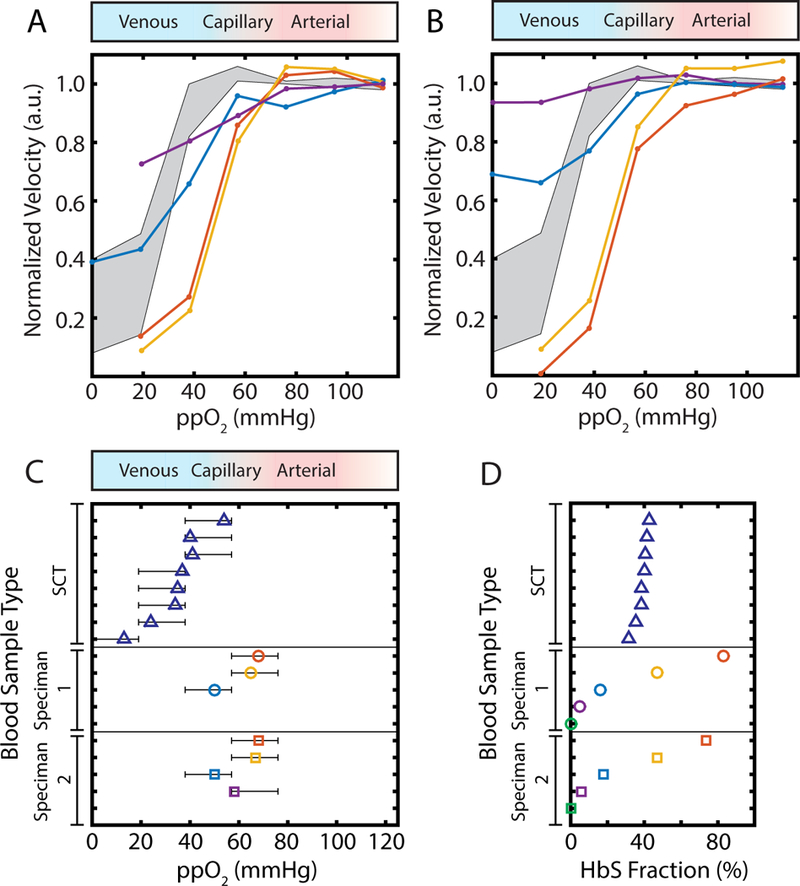
Oxygen-dependence of rheology for transfused SCD compared to SCT. (A,B) Normalized, median velocity under steady state conditions for the two transfusion blood samples we measured. Simulated transfusion specimens shown are ~5–6% (purple), ~16–18% (blue), ~45–48% (orange), and ~70–85% (red) genotype SS blood by volume. Grey, shaded region shows the interquartile range of normalized blood velocity in response to oxygen tension for all SCT blood samples. Blue to red to white color gradient bar above plot represents typical oxygen tension found in venous, arterial, and supraphysiologic circulation respectively. (C) Oxygen tension at which the regime I/II transition occurs for SCT (blue triangles) and two unique, simulated transfusion experiments (colored circles and squares, with HbS % corresponding to the colors specified in A,B). Blue to red to white color gradient bar above plot represents typical oxygen tension found in venous, arterial, and supraphysiologic circulation respectively. Confidence intervals for each regime I/II transition are determined as outlined in methods. (D) Bulk hemoglobin S fraction as quantified by HPLC is presented for SCT (blue triangles) blood samples. For simulated transfusions, HbS fraction was calculated based on the native HbS fraction of the genotype SS blood sample, hematocrit for both SS and AA samples, and the volume of blood transfused.
We also measured the regime I/II transition for all transfused specimens (Fig. 4C), and we found for some specimens that the transition to impaired flow (regime I/II transition) generally occurs at lower oxygen tension as the fraction of HbA increases (Fig. 4C). Samples transfused to ~47% HbS fraction exhibited a regime I/II transition similar to untransfused samples, but samples with 18% HbS fraction or less displayed a regime I/II transition at significantly lower oxygen tensions. The regime I/II transition was undetectable for one of the samples transfused to ~5% HbS fraction (purple dots, Fig. 4B) and for both 100% AA (0% HbS fraction) control experiments, as expected (green dots, Fig. 4C,D).
We also compared our findings for transfused samples directly with SCT samples (Fig. 4A,B), since the benefits seen in both cases are due to increased HbA and reduced HbS, but with different cellular distributions. We found that blood transfusion simulations with 18% HbS fraction or less (Fig. 4A,B blue and purple lines) yielded a biophysical response that most resembled that of SCT blood samples with the mildest oxygen-dependence (interquartile range of all SCT responses are indicated by gray, shaded regions in Fig. 4A,B). Unlike samples with SCT, samples transfused to 16–18% HbS fraction still exhibited regime III behavior at low oxygen tension. As shown by the blue lines in Fig. 4A,B, at oxygen tensions under 20 mmHg, blood flow is both impaired and oxygen-independent. Transfused blood specimens of ~47% HbS fraction (orange lines) and untransfused samples (red lines) had nearly identical rheological changes under hypoxic conditions, which were larger in magnitude than for SCT blood samples. Figure 4D shows the estimated HbS fraction for each simulated transfusion (based on HCT, native HbS %, and blood volume fractions) as well as the measured HbS fraction for all SCT samples tested. Generally, we observe that a lower HbS fraction is required in transfusions relative to SCT samples to observe similar rheological behavior.
Discussion
In this study, we quantified the oxygen-dependence of sickle trait blood rheology in physiologically-sized microchannels and under physiologically relevant shear rates using pressure-driven flow. We leveraged an experimental platform and measurement protocol previously developed for measuring homozygous sickle blood32, and directly compared oxygen-dependent rheology of SCT and SCD blood. We found that SCT blood displays similar qualitative behavior to SCD blood, with unimpaired flow at high oxygen tensions and impaired flow that is oxygen dependent at lower oxygen tensions. In general, we found that the most notable difference is that the transition to reduced flow occurs at significantly lower oxygen tension for SCT blood. Interestingly, we found that some SCT blood samples do occlude under completely anoxic conditions. From a molecular perspective, it has been shown that SCD RBCs contain more HbS polymer than SCT RBCs at the same oxygen tension.53–55 Our results are consistent with the interpretation that the apparent viscosity of a blood sample increases with increasing polymer fraction, as suggested in previous studies showing that single-RBC mechanical stiffness increased with polymer fraction.56,57 Overall, these in vitro observations are in broad agreement with prior studies detecting hemoglobin polymer in SCT cells and with clinical observation that SCT is clinically benign except under conditions of extreme hypoxia such as intense exercise or high altitude.12–23,30,31
Clinical experience suggests that under the vast majority of conditions, SCT blood remains safely within a range of oxygen tension that hemoglobin polymerization is limited to a range where any increase in blood apparent viscosity is not clinically significant. Blood flow remains sufficient to avoid symptoms, in sharp contrast to SCD, where impaired flow can occur chronically and contributes to the long term morbidity associated with the disease.7–9 Notably, we find that the median difference in the regime I/II oxygen transition between the two conditions is modest (38 mmHg), suggesting that the dramatic improvement in clinical outcome for SCT compared to SCD is achieved with a modest reduction in the oxygen threshold at which a blood sample’s apparent viscosity becomes oxygen-dependent. This modest reduction in oxygen threshold may also be cooperative with the reduced deceleration in SCT after deoxygenation, increasing the probability of SCT blood escaping the microcirculation before occlusion.
Clinical experience also suggests that the phenotype of sickle trait is virtually indistinguishable from one SCT individual to the next, while SCD is known for its clinical heterogeneity. It is therefore somewhat surprising that we find significant variation in the oxygen tension at which SCT blood flow becomes impaired. One explanation is that the regime I/II transition is sufficiently low for all SCT samples that the likelihood of occlusion in vivo is negligible, suggesting that this oxygen transition, which represents the point at which a statistically significant change in blood rheology occurs, is not the most clinically relevant characteristic of SCT blood rheology.31 More useful might be the oxygen threshold at which a clinically significant change in blood rheology occurs. Given physiologic adaptations in cardiac and vascular function, it is possible that blood apparent viscosity must double or triple persistently before clinical consequences are likely to occur. A prior study found that the degree of kidney concentration deficit in sickle trait correlated strongly with HbS fraction, suggesting a possible link between this relatively benign clinical condition and the tendency for hemoglobin to polymerize in SCT cells under the conditions of low oxygen, low pH, and high osmolality that can be found in the kidney.30 Our finding of correlation between HbS fraction in SCT and the location of the regime I/II boundary (Fig. 3D) is consistent with this interpretation and raises the possibility that differences in oxygen-dependent rheology may contribute to differences in these mild renal deficits found in individuals with sickle trait. Further investigation is warranted into the most clinically relevant characteristics of this quantified relationship between oxygen tension and in vitro blood viscosity.
Because SCT is clinically benign, the relationship between SCT blood rheology and oxygen tension provides a useful benchmark for putative therapies.29 Transfusion therapy with genotype AA blood is one of the most commonly used treatment options for SCD48–52, even though evidence for specific treatment targets, such as post-transfusion HCT or hemoglobin S fraction, is limited. Our results show that simulating transfusion of SCD can shift in vitro SCD blood rheology toward that of SCT, and that a sufficiently large transfusion of healthy RBCs (reducing the HbS fraction below ~18%) can result in an in vitro rheological response similar to or less sensitive than that of SCT blood (e.g. higher velocity under hypoxic conditions or transition to impaired flow at lower oxygen tension). We find that the HbS fraction at which these rheological improvements are observed in transfused SCD samples is lower than the HbS fraction usually found in SCT samples. We speculate that differences in cellular distribution of HbA and HbS among the RBC population are responsible. In SCD, all native cells will contain high fractions of HbS (typically >80%), and even a small fraction (~10%) of these high HbS cells is sufficient to produce measurable rheological changes. Moreover, these cells will form significant polymer at higher oxygen tensions than SCT RBCs, possibly causing polymerization and reduced flow even in the arterial circulation.32 Thus, as has been postulated for HbF58, a more uniform distribution of HbA may help explain the improved prognosis. Our simulated transfusion results suggest that a 70/30 volumetric mixture of SS and AA blood (comparable to a simple transfusion of one or two units of blood) does not have a significant effect on the oxygen dependence of SS blood rheology, but a 30/70 mixture (comparable to a red blood cell exchange) does have a significant effect. These results are qualitatively consistent with existing clinical studies which collectively provide limited evidence for the efficacy of the simple transfusion of one unit of blood compared to persuasive evidence for the efficacy of red cell exchange.47 More generally, this study demonstrates a measurement that may be useful in generating evidence for quantitative transfusion targets for SCD using SCT as a benchmark for clinical efficacy.
This in vitro quantification of oxygen-dependent rheology may also be helpful defining therapeutic targets for experimental treatments that modulate HbF fraction or hemoglobin oxygen affinity. It is challenging to define the therapeutic windows for these treatments, and an in vitro comparison of various regimens with SCT may be helpful. Future study is warranted to determine if this approach is useful for personalizing transfusion endpoints for individual SCD patients or providing a functional in vitro surrogate endpoint to assess experimental therapies involving globin gene manipulation or modulation of hemoglobin oxygen affinity.
Supplementary Material
Acknowledgements
The authors would like to thank José Valdez, Athena Geisness, Dr. Greg Vercelloti, and Dr. Bob Molokie, for the helpful discussions. For assistance with blood sample collection, identification, testing, and transport, the authors would like to thank the assistance of members of the MGH Clinical Laboratories and the MGH Clinical Research Program, including John Yablonski, Chhaya Patel, and Hasmukh Patel. The authors would also like Dr. Yvonne Data with the University of Minnesota Medical Center for assistance with blood sample collection as well as the University of Minnesota Advanced Research and Diagnostic Laboratory for assistance with blood sample identification and testing. Additionally, the authors would like to thank the Minnesota Nanofabrication Center for support with microfluidic device fabrication. Finally, the authors would like to thank all the patients and blood sample donors who helped make this work possible.
This work was supported by the National Heart, Lung, and Blood Institute (NHLBI) under grants R21HL130818, R56HL132906, and R01HL132906. X.L. was supported under American Heart Association pre-doctoral fellowship 16PRE31020025. A.C. was supported as a Good Ventures Fellow of the Life Sciences Research Foundation (LSRF). J.M.H was supported by a National Institutes of Health (NIH) Director’s New Innovator Award (DP2DK098087) and NHLBI grant HL114476. D.K.W. was supported under American Heart Association grant 13SDG6450000.
Footnotes
Conflict of interest disclosure
The authors declare no competing interests related to this work.
References
- 1.Noguchi CT, Torchi DA, Schechter AN. Determination of deoxyhemoglobin S polymer in sickle erythrocytes upon deoxygenation. Proc Natl Acad Sci USA. 1980;77(9):5487–5491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eaton WA, Hofrichter J. Hemoglobin S gelation and sickle cell disease. Blood. 1987;70(5):1245–1266. [PubMed] [Google Scholar]
- 3.Usami S, Chien S, Scholtz PM, Bertles JF. Effect of deoxygenation on blood rheology in sickle cell disease. Microvasc Res. 1975;9(3):324–334. [DOI] [PubMed] [Google Scholar]
- 4.Barabino GA, Platt MO, Kaul DK. Sickle cell biomechanics. Annu Rev Biomed Eng. 2010;12:345–367. [DOI] [PubMed] [Google Scholar]
- 5.Embury SH. The not-so-simple process of sickle cell vasoocclusion. Microcirculation. 2010;11:101–113. [DOI] [PubMed] [Google Scholar]
- 6.Tsai M, Kita A, Leach J, et al. In vitro modeling of the microvascular occlusion and thrombosis that occur in hematologic diseases using microfluidic technology. J Clin Invest. 2012:122(1):408–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Steinberg MH, Management of sickle cell disease. N Engl J Med. 1999;340(13):1021–1030. [DOI] [PubMed] [Google Scholar]
- 8.Rees DC, Williams TN, Gladwin MR. Sickle-cell disease. Lancet. 2010;316:2018–2031. [DOI] [PubMed] [Google Scholar]
- 9.Bunn HF. Pathogenesis and treatment of sickle cell disease. N Engl J Med. 1997;337(11):762–769. [DOI] [PubMed] [Google Scholar]
- 10.Makani J, Cox SE, Soka D, et al. Mortality in sickle cell anemia in Africa: a prospective cohort study in Tanzania. PLoS One. 2011;6(2):e14699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Platt OS, Brambilla DJ, Rosse WF, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med. 1994;330(23):1639–1644. [DOI] [PubMed] [Google Scholar]
- 12.Sanders WJ. A rare case of avascular necrosis in sickle cell trait: a case report. BMC Hematol. 2018;18(5):doi: 10.1186/s12878-018-0098-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jones SR, Binder RA, Donowho EM. Sudden death in sickle-cell trait. N Engl J Med. 1970;282(6):507–512. [DOI] [PubMed] [Google Scholar]
- 14.Mitchell BL. Sickle cell trait and sudden death – bringing it home. J Natl Med Assoc. 2007;99(3):300–305. [PMC free article] [PubMed] [Google Scholar]
- 15.Conn HO, Sickle-cell trait and splenic infarction associated with high-altitude flying. N Engl J Med. 1954;251(11):417–420. [DOI] [PubMed] [Google Scholar]
- 16.Chirico EN, Faës C, Connes P, Canet-Soulas E, Martin C, Pialoux V. Role of exercise-induced oxidative stress in sickle cell trait and disease. Sports Med. 2016;46(5):629–639. [DOI] [PubMed] [Google Scholar]
- 17.Kark JA, Posey DM, Schumacher HR, Ruehle CJ. Sickle-cell trait as a risk factor for sudden death in physical training. N Engl J Med. 1987;317(13):781–787. [DOI] [PubMed] [Google Scholar]
- 18.Nelson DA, Deuster PA, Carter R III, Hill OT, Wolcott VL, Kurina LM. Sickle cell trait, rhabdomyolysis, and mortality among U.S. army soldiers. N Engl J Med. 2016;375(5):435–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harris KM, Haas TS, Eichner ER, Maron BJ. Sickle cell trait associated with sudden death in competitive athletes. Am J Cardiol. 2012;110(8):1185–1188. [DOI] [PubMed] [Google Scholar]
- 20.Tsaras G, Owusu-Ansah A, Boateng FO, Amoateng-Adjepong Y Complications associated with sickle cell trait: a brief narrative review. Am J Med. 2009;122(6):507–512. [DOI] [PubMed] [Google Scholar]
- 21.Hyacinth HI, Carty CL, Seals SR, et al. Association of Sickle Cell Trait With Ischemic Stroke Among African Americans: A Meta-analysis. JAMA Neurol. 2018;doi: 10.1001/jamaneurol.2018.0571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Key NS, Connes P, Derebail VK. Negative health implications of sickle cell trait in high income countries: from the football field to the laboratory. Br J Haematol. 2015;170(1):5–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Naik RP, Derebail VK, Grams ME, et al. Association of sickle cell trait with chronic kidney disease and albuminuria in African Americans. JAMA. 2014;312(20):2115–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bain BJ. Hemoglobinopathy Diagnosis, 2nd ed Malden, MA: Blackwell Publishing; 2006. [Google Scholar]
- 25.Cameron BF, Smith DB, Cody B. Hemoglobin S levels in sickle cell trait individuals. Am J Hematol. 1984;16(2):123–127. [DOI] [PubMed] [Google Scholar]
- 26.Noguchi CT, Torchia DA, Schechter AN. Polymerization of hemoglobin in sickle trait erythrocytes and lysates. J Biol Chem. 1981;256(9):4168–4171. [PubMed] [Google Scholar]
- 27.Noguchi CT, Schechter AN. The intracellular polymerization of sickle hemoglobin and its relevance to sickle cell disease. Blood. 1981;58(6):1057–1068. [PubMed] [Google Scholar]
- 28.Hofrichter J, Eaton WA. Hemoglobin S gelation and sickle cell disease. Blood. 1987;70(5):1245–1266. [PubMed] [Google Scholar]
- 29.Li Q, Henry ER, Hofrichter J, et al. Kinetic assay shows that increasing red cell volume could be a treatment for sickle cell disease. Proc Natl Acad Sci USA. 2017;114(5):E689–E696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gupta AK, Kirchner KA, Nicholson R, et al. Effects of alpha-thalassemia and sickle polymerization tendency on the urine-concentrating defect of individuals with sickle cell trait. J Clin Invest. 1991;88(6):1963–1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kennedy AP, Walsh DA, Nicholson R, Adams JG 3rd, Steinberg MH. Influence of HbS levels upon the hematological and clinical characteristics of sickle cell trait. Am J Hematol. 1986;22(1):51–54. [DOI] [PubMed] [Google Scholar]
- 32.Lu X, Wood DK, Higgins JM. Deoxygenation reduces sickle cell blood flow at arterial oxygen tension. Biophys J. 2016;110(12):2751–2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lu X, Galarneau MM, Higgins JM, Wood DK. A microfluidic platform to study the effects of vascular architecture and oxygen gradients on sickle blood flow. Microcirculation. 2017;24(5):e12357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wood DK, Soriano A, Mahadevan L, Higgins JM, Bhatia SN. A biophysical indicator of vaso-occlusive risk in sickle cell disease. Sci Transl Med. 2012;4(123):123ra26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reinhart SA, Schulzki T, Bonetti PO, Reinhart WH. Studies on metabolically depleted erythrocytes. Clin Hemorheol Microcirc. 2014;45(2):161–173. [DOI] [PubMed] [Google Scholar]
- 36.Henkelman S, Dijkstra-Tiekstra MJ, De Wildt-Eggen J, Graaff R, Rakhorst G, van Oeveren W. Is red blood cell rheology preserved during routine blood bank storage? Transfusion. 2010;50(4):941–948. [DOI] [PubMed] [Google Scholar]
- 37.Berezina TL, Zaets SB, Morgan C, et al. Influence of storage on red blood cell rheological properties. J Surg Res. 2002;102(1):6–12. [DOI] [PubMed] [Google Scholar]
- 38.Adler M, Polinkovsky M, Gutierrez E, Groisman A. Generation of oxygen gradients with arbitrary shapes in a microfluidic device. Lab Chip. 2010;10(3):388–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shi J, Tomasi C. Good features to track. Paper presented at IEEE Conference on Computer Vision and Pattern Recognition 21 June 1994 Seattle, WA. [Google Scholar]
- 40.Higgins JM, Eddington DT, Bhatia SN, Mahadevan L. Statistical dynamics of flowing red blood cells by morphological image processing. PLoS Comput Biol. 2009;5(2):e1000288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Higgins JM, Eddington DT, Bhatia SN, Mahadevan L. Sickle cell vasoocclusion and rescue in a microfluidic device. Proc Natl Acad Sci USA. 2007;104(51):20496–20500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carden MA, Fay ME, Lu X, et al. Extracellular fluid tonicity impacts sickle red blood cell deformability and adhesion. Blood. 2017;130(24):2654–2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vichinsky EP, Earles A, Johnson RA, Hoag MS, Williams A, Lubin B. Alloimmunization in sickle cell anemia and transfusion of racially unmatched blood. N Engl J Med. 1990;322(23):1617–1621. [DOI] [PubMed] [Google Scholar]
- 44.Lanzkowsky P, Shenede A, Karayalcin G, Kim YJ, Aballi AJ. Partial exchange transfusion in sickle cell anemia. Use in children with serious complications. Am J Dis Child. 1978;132(12):1206–1208. [DOI] [PubMed] [Google Scholar]
- 45.Russell MO, Goldberg HI, Reis L, et al. Transfusion therapy for cerebrovascular abnormalities in sickle cell disease. J Pediatr. 1976;88(3):382–387. [DOI] [PubMed] [Google Scholar]
- 46.Wayne AS, Kevy SV, Nathan DG. Transfusion management of sickle cell disease. Blood. 1993;81(5):1109–1123. [PubMed] [Google Scholar]
- 47.Yawn BP, Buchanan GR, Afenyi-Annan AN, et al. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA. 2014;312(10):1033–1048. [DOI] [PubMed] [Google Scholar]
- 48.Alexy T, Pais E, Armstrong JK, Meiselman HJ, Johnson CS, Fisher TC. Rheologic behavior of sickle and normal red blood cell mixtures in sickle plasma: implications for transfusion therapy. Transfusion. 2006;46(6):912–918. [DOI] [PubMed] [Google Scholar]
- 49.Aliyu ZY Tumblin AR, Kato GJ. Current therapy of sickle cell disease. Haematologica. 2006;91(1):7–10. [PMC free article] [PubMed] [Google Scholar]
- 50.Chou ST. Transfusion therapy for sickle cell disease: a balancing act. Hematology Am Soc Hematol Educ Program. 2013:439–446. [DOI] [PubMed] [Google Scholar]
- 51.Adams RJ, McKie VC, Brambilla D, et al. Stroke prevention trial in sickle cell anemia. Control Clin Trials. 1998;19(1):110–129. [DOI] [PubMed] [Google Scholar]
- 52.Adams RJ, McKie VC, Hsu L, et al. Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial doppler ultrasonography. N Engl J Med. 1998;339(1):5–11. [DOI] [PubMed] [Google Scholar]
- 53.Sunshine HR, Hofrichter J, Eaton WA. Gelation of sickle cell hemoglobin in mixtures with normal adult and fetal hemoglobins. J Mol Biol. 1979;133(4):435–467. [DOI] [PubMed] [Google Scholar]
- 54.Sunshine HR, Hofrichter J, Ferrone FA, Eaton WA. Oxygen binding by sickle cell hemoglobin polymers. J Mol Biol. 1982;158(2):251–273. [DOI] [PubMed] [Google Scholar]
- 55.Noguchi CT, Schechter AN. Non-uniformity of intracellular polymer formation in sickle erythrocytes: possible correlation with severity of hemolytic anemia. Am J Pediatr Hematol Oncol. 1984;6(1):46–50. [PubMed] [Google Scholar]
- 56.Itoh T, Chien S, Usami S. Deformability measurements on individual sickle cells using a new system with pO2 and temperature control. Blood. 1992;79(8):2141–2147. [PubMed] [Google Scholar]
- 57.Itoh T, Chien S, Usami S. Effects of hemoglobin concentration on deformability of individual sickle cells after deoxygenation. Blood. 1995;85(8):2245–2253. [PubMed] [Google Scholar]
- 58.Steinberg MH, Chui DH, Dover GJ, Sebastiani P, Alsultan A. Fetal hemoglobin in sickle cell anemia: a glass half full? Blood. 2014;123(4):481–485. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.