

Abstract
The Curtius rearrangement is the thermal decomposition of an acyl azide derived from carboxylic acid to produce an isocyanate as the initial product. The isocyanate can undergo further reactions to provide amines and their derivatives. Due to its tolerance for a large variety of functional groups and complete retention of stereochemistry during rearrangement, the Curtius rearrangement has been used in the synthesis of a wide variety of medicinal agents with amines and amine-derived functional groups such as ureas and urethanes. The current review outlines various applications of the Curtius rearrangement in drug discovery and medicinal chemistry. In particular, the review highlights some widely used rearrangement methods, syntheses of some key agents for popular drug targets and FDA-approved drugs. In addition, the review highlights applications of the Curtius rearrangement in continuous-flow protocols for the scale-up of active pharmaceutical ingredients.
Keywords: amine synthesis, carbamates, carboxylic acids, Curtius rearrangement, drug discovery, medicinal chemistry
Graphical Abstract
Thanks to its tolerance for a wide range of functional groups and complete retention of stereochemistry, the Curtius rearrangement has been used in the synthesis of a variety of medicinal agents with amines and amine-derived functional groups such as ureas and urethanes. This review outlines various applications of the Curtius rearrangement in drug discovery and medicinal chemistry and highlights some widely used rearrangement methods in the syntheses of key agents for popular drug targets and FDA-approved drugs.
1. Introduction
Amines and amine derivatives such as amides, carbamates, and ureas are important structural units that are often found in bioactive molecules. The amines and amine-derived functionalities are also widely used in drug discovery and medicinal chemistry. They can be readily prepared from carboxylic acids using the Curtius rearrangement as the key reaction. The Curtius rearrangement was discovered by Julius Wilhelm Theodor Curtius in the 1890s. The rearrangement involves the decomposition of an acyl azide to produce an isocyanate.[1–3] As shown in Scheme 1, an acyl azide (2), deriving from the corresponding carboxylic acid (1) is converted by heating into the corresponding isocyanate (4), through an acyl nitrene intermediate (3). During the thermolysis, molecular nitrogen is eliminated and at the same time a [1,2]-shift of the substituent attached to the carbonyl group takes place with retention of configuration. The resulting amine (5) has a carbon less, because the last step of the reaction entails the loss of a molecule of CO2. Isocyanates can also be transformed into urethanes (6) by alcoholysis after workup or in situ by performing the rearrangement in alcoholic solvents. Alternatively, reaction of isocyanates with an amine would provide the corresponding urea derivatives (7).[4,5]
Scheme 1.

The Curtius rearrangement and its applications for the synthesis of amines and amine derivatives.
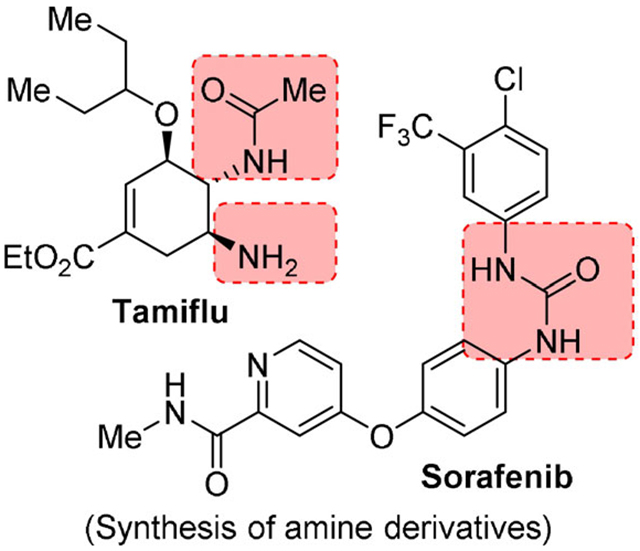
Mechanistic studies on the Curtius rearrangement have been reviewed recently.[5] Pertinent features of the Curtius rearrangement are 1) it is a concerted intramolecular rearrangement; 2) stereochemical configurations of the migrating groups are completely retained; 3) the kinetics are first order; and 4) free carbenium ions or radical intermediates are not involved.[5] The importance of the Curtius rearrangement lies in its general application to acyl azides which are readily formed from a range of aliphatic, heterocyclic, and aromatic carboxylic acids and their subsequent conversion to amines involving a one carbon loss.[6,7] These primary amines are devoid of any contamination from secondary and tertiary amines, unlike other methods for amine preparation.[8,9] Furthermore, the rearrangement proceeds with the retention of stereochemistry of the migrating carbon center. The ready availability of carboxylic acids and their easy access through functional group transformation, make the Curtius rearrangement an ideal choice for the synthesis of achiral and chiral amines. The Curtius rearrangement has been extensively employed in the synthesis of a variety of natural products and numerous biomolecules. We recently reported the applications of Curtius rearrangement in the synthesis of natural products.[5] As shown in Figure 1, Curtius rearrangement was employed in the total synthesis of (+)-zampanolide (8) to install the N-acyl hemiaminal moiety.[10] Due to the predominant role of amines and aminederived functional groups including ureas and urethanes in medicinal chemistry, the Curtius rearrangement is increasingly used in drug discovery and synthesis of drug candidates. In bioactive molecules, the amine and amine-derived functionalities often bind to a specific site of target receptors or proteins through intermolecular forces, particularly through hydrogen bonds.[6,7] Basic amines also display a wide range of physical properties including a very important role in solubilizing drugs as a free base or as water-soluble amine salts. In this context, the Curtius rearrangement has been used in a variety of applications. For example, a key Curtius rearrangement is involved for the establishment of the two neighboring nitrogen functionalities of Tamiflu (9), a potent and selective inhibitor of viral neuraminidases A and B for the treatment of influenza (H5N1 influenza virus).[11–13] The use of Curtius rearrangement is also represented in Sorafenib (10), a potent kinase inhibitor.[14,15] Here, a Curtius rearrangement was employed for the installation of the key urea moiety. In this review, we now provide an overview of the applications of the Curtius rearrangement in the development of a variety of medicinal agents and drug candidates.
Figure 1.

Examples of applications of Curtius rearrangement for the synthesis of medicinally relevant compounds. The pink frames highlight the functionalities built by using Curtius rearrangement.
2. Convenient Methods and Reagents
For the Curtius rearrangement, acyl azides are generally prepared starting from carboxylic acids or their activated derivatives such as acyl chlorides or anhydrides. A large number of mild methods and a variety of reagents have been developed over the years. These methods have been reviewed in detail.[5] In this section, we provide only a few selected convenient methods that have been used widely for medicinal chemistry applications.
One of the most employed methods is the direct conversion of carboxylic acids in a one pot procedure using diphenylphosphoryl azide (DPPA, 11).[16–18] This method displays the advantage of avoiding isolation of the generally explosive acyl azides. First, a mixed anhydride of carboxylic acid/phosphoric acid 12 is formed and azide is eliminated. Anhydride 12 then acylates the azide anion providing acyl azide 2 which undergoes rearrangement to the corresponding isocyanate 4 (Scheme 2).
Scheme 2.

Mechanism of acyl azide formation using DPPA.
A very recent application of this method was proposed by Liang and collaborators which reported a scale-up synthesis of azaindolyl-pyrimidine derivative 13, a potent inhibitor of human influenza virus replication (Scheme 3).[19] The synthesis employed the Curtius rearrangement in two key steps, both performed by using DPPA. In particular, in the first Curtius step performed on derivative 14, DPPA was used in the presence of triethylamine and benzyl alcohol as the solvent in order to provide the corresponding Cbz-protected amine which was subsequently converted to free amine 15 under catalytic hydrogenation conditions. In the second Curtius step, reaction of acid 16 with DPPA in THF followed by treatment with morpholine provided the desired urea functionality of inhibitor 13.
Scheme 3.

Use of DPPA for the preparation of inhibitor 13.
Treatment of carboxylic acid derivatives with oxalyl chloride followed by sodium azide is another widely employed method for the preparation of key acyl azide intermediates. An interesting example in this context can be found in the large-scale synthesis of oseltamivir (19) by Hayashi and co-workers.[20] The tert-butoxycarbonyl group in compound 17 was converted into the acyl azide 18 in a single pot operation (Scheme 4). Reaction with oxalyl chloride in the presence of catalytic dimethylformamide (DMF) followed by treatment with sodium azide led to acyl azide 18, a key intermediate for the oseltamivir (19) synthesis.
Scheme 4.

Use of oxalyl chloride and sodium azide protocol for the Curtius rearrangement involved in the synthesis of oseltamivir (19).
The use of thionyl chloride and DMF followed by treatment with sodium azide is also widely applied for the preparation of medicinally relevant compounds. This is exemplified in the recent development of inhibitors of the protein-tyrosine phosphatase 4A3 (PTP4A3), such as compound 22.[21] Acid 20 was converted to the key acyl azide intermediate 21 successfully employing this protocol (Scheme 5).
Scheme 5.

Use of thionyl chloride and sodium azide protocol in the synthesis of PTP4A3 inhibitor 22.
The combination of thionyl chloride and trimethylsilyl azide is another useful protocol that finds wide applications. This method is used in the synthesis of cisplatin analogues with antitumor activity (Scheme 6).[22] Derivative 23 was treated with thionyl chloride and catalytic DMF to provide acid chloride 24, which upon treatment with trimethylsilyl azide in dioxane provided the key intermediate 25. This was subsequently converted into the cisplatin analogue 26.
Scheme 6.

Use of trimethylsilyl azide in the Curtius rearrangement for the synthesis of cisplatin derivative 26.
Another effective protocol is the formation of a mixed anhydride followed by treatment with sodium azide. This protocol was successfully used for the synthesis of dihydrexidine (29), a dopamine D1 agonist using Curtius rearrangement as the key step.[23] Enantiomerically pure acid 27 was reacted with ethyl chloroformate and the resulting mixed anhydride was subsequently reacted with sodium azide. Curtius rearrangement, followed by hydrolysis of isocyanate gave amine 28 which was then converted to dihydrexidine (Scheme 7).
Scheme 7.

Use of a mixed anhydride and sodium azide for the Curtius rearrangement in the synthesis of dihydrexidine (29).
There are other recent methods and protocols that have been covered in recent reviews.[5] In this review, we now highlight applications of the Curtius rearrangement in medicinal chemistry and drug development. In particular, we will provide key examples of application of the Curtius rearrangement for the synthesis of medicinally relevant compounds or key intermediates for their preparation. Also, we will showcase its importance for the synthesis of some approved drugs. For convenience, the applications of Curtius the rearrangement will be sub-grouped based on the medicinal chemistry fields of application.
3. Application of the Curtius Rearrangement in Medicinal Chemistry
3.1. Antibacterials
Over the years a variety of antibacterial agents have been developed by targeting bacterial DNA and its associated processes, attacking bacterial metabolic pathways including protein synthesis, or interfering with bacterial cell wall synthesis and function.[24]
Quinolones are potent, broad-spectrum antibiotics widely used in medicine since the late 1980s.[25] The synthesis and biological activity of a series of quinolone antibacterials containing alkyl- and aryl-substituted pyrrolidines at C-7 was reported by Hagen and collaborators.[26] The synthesis of target quinolone derivative 33 was carried out using a Curtius rearrangement as the key step. As shown, the pyrrolidine acid 30 was converted to Boc-derivative 31 by treatment with DPPA and Et3N in tBuOH. Removal of Boc and N-benzyl groups provided 3-phenyl-3-pyrrolidinamine moiety 32. This was converted to the key quinolone derivative 33 (Scheme 8). It showed a slightly improved activity against Gram-positive bacteria and retained activity against Gram-negative strains.[26]
Scheme 8.

Synthesis of fluoroquinolone compound 33.
Later on, Domagala and co-workers employed similar chemistry to construct a variety of other quinolones to modulate activity.[27]
Kuramoto and co-workers also employed the Curtius rearrangement to explore structural modifications of the N-1 aryl moiety of the fluoroquinolones, tosufloxacin and trovafloxacin. Benzoic acid derivative 34 was converted to the nitrophenylcarbamate derivative 35 by nitration followed by Curtius rearrangement. It was converted to naphthyridones and quinolones incorporating the novel 5-amino-2,4-difluorophenyl group at the N-1 position (Scheme 9). Compound 36 showed interesting antibacterial activities against Gram-positive and Gram-negative bacteria with improved activity over clinically used compounds, including trovafloxacin.[28]
Scheme 9.

Synthesis of fluoroquinolone derivative 36.
Linezolid (41) is an antibacterial agent that has been developed to inhibit protein synthesis.[29] It is active against most Gram-positive organisms.[30] Linezolid was previously synthesized by using hazardous 3,4-difluoronitrobenzene.[31] McCarthy and co-workers envisioned an improved synthesis using benzoyl azide 37. Thus, compound 37 was refluxed in xylene with lithium bromide and tri-n-butylphosphine oxide, to provide isocyanate 38. Reaction of 38 with epoxide 39 provided the desired N-aryloxazolidone 40 in 71% yield. This was finally converted to linezolid (41) by an acid catalyzed hydrolysis of the Schiff base followed by acetylation with acetic anhydride (Scheme 10).[32]
Scheme 10.

Synthesis of the antibacterial agent linezolid (41).
3.2. Anticancer agents
With the increasing knowledge and understanding of cellular and molecular causes of cancers, anticancer drug development has become an exciting area of intense research.[33,34] Particular emphasis has been devoted to develop drugs that interfere and inhibit tumor growth by targeting specific proteins.[35] In addition, searching for new and effective ways to overcome resistance to anticancer drugs has been a subject of intense research.[36] In this section we will review application of the Curtius rearrangement in anticancer drug development.
3.2.1. Tubulin binding agents
Podophyllotoxin is known to bind to tubulin to a specific binding site different from those of vinblastine and paclitaxel.[37] Synthesis of podophyllotoxin analogues have attracted much attention to reduce toxicity and improve antitumor activity.[38,39] Hitotsuyanagi and collaborators synthesized (−)-4-aza-4-deoxypodophyllotoxin (45) from (−)-podophyllotoxin (42) through C-ring cleavage, Curtius rearrangement and intramolecular N-alkylation as the key steps. Podophyllotoxin (42) was converted to carboxylic acid 43 in eight steps. Curtius rearrangement of the carboxylic acid, followed by intramolecular N-alkylation of the resulting amine afforded tetrahydroquinoline 44 in 69% yield. This was converted to (−)-4-aza-4-deoxypodophyllotoxin 45, which showed a comparable IC50 to (−)-podophyllotoxin against P388/VCR leukemia cells (Scheme 11).[40]
Scheme 11.

Synthesis of podophyllotoxin analogue 45.
Colchicine (46) is one of the first discovered tubulin-binding agents.[41] However, its significant toxicity limited its use in cancer therapy. Fedorov and co-workers recently reported a semi-synthetic pyrrolo-allocolchicine 49 derivative starting from naturally occurring (−)-(aR,7S)-colchicine 46.[42] The synthesis involved the Curtius rearrangement as a key step. Bromination and base-catalyzed ring contraction provided 4-bromoallocolchicinic acid 47. It was subjected to the Curtius rearrangement in the presence of sodium azide providing the corresponding aniline 48 in 45% yield. This was then converted to heterocyclic derivative 49 (Scheme 12). This derivative showed cytotoxic properties at sub-nanomolar concentration, efficiently arrested the cell cycle in the G2/M phase, and showed pronounced anti-cancer activity compared to colchicine.[42]
Scheme 12.

Synthesis of novel colchicine derivative 49.
3.2.2. Epigenetic modulators
Drug discovery approaches targeting key epigenetic enzymes with small-molecule inhibitors have resulted in pre-clinical and clinical agents.[43] Histone deacetylases (HDAC) and lysine specific demethylase 1 (LSD1) are two of the major epigenetic targets in anticancer drug discovery. HDAC inhibitors can alter gene expression by inducing chromatin remodeling, and thus have the potential to reverse the epigenetic states related to cancer.[44] LSD1 can remove the methyl group from a mono- or di-methylated lysine residue of histone H3 lysine 4 (H3K4), H3K9 or a non-histone protein. Overexpression of LSD1 was found in a broad range of cancers, including leukemia, lung, prostate and breast cancers.[45]
Shah and collaborators used boswellic acids as an alternative cap group for HDAC inhibitors.[46] 11-Keto-β-boswellic acid 50 was converted to an acid chloride and subjected to a Curtius rearrangement to yield the corresponding isocyanate subsequently converted to amine 51. It was converted to hydroxamic acid derivative 52 (Scheme 13). Compound 52 showed promising HDAC inhibitory activity, induced G1 cell-cycle arrest and showed loss of mitochondrial membrane potential at significantly low concentrations.[46]
Scheme 13.

Synthesis of HDAC inhibitors.
Zhou and collaborators reported a series of cyclopropylamine-based potent LSD1 inhibitors. The synthesis involved a Curtius rearrangement to give the trans-cyclopropylamine template. Bromobenzaldehyde 53 was converted to cyclopropane carboxylic acid derivative 54. It was subjected to DPPA and Et3N in toluene in the presence of anhydrous tBuOH to provide cyclopropylamine 55 in 70% yield. Compound 55 is the key intermediate for the synthesis of all inhibitors. Alkylation of 55 with a piperazine moiety followed by Boc deprotection provided derivative 56 (Scheme 14). Compound 56 was one of the most potent LSD1 inhibitors of the series (IC50=0.064 μm) also displaying high selectivity (>280-fold) towards related enzymes monoamine oxidase (MAO) A and B.[47]
Scheme 14.

Synthesis of LSD1 inhibitors.
3.2.3. Kinase and phosphatase inhibitors
Protein phosphorylation plays key roles in many physiological processes and is often deregulated in cancer. Protein kinases and phosphatases orchestrate the phosphorylation changes that control cellular functions thus rendering these enzymes potential drug targets for the treatment of cancer.[48]
Okaniwa and co-workers developed derivatives of sorafenib (10) as dual inhibitors of BRAF(V600E) and VEGFR2 kinases for the treatment of various human cancers.[49] The synthesis involved the hydrolysis of ethyl ester 57, followed by Curtius rearrangement of the carboxylic acid in the presence of tert-butanol to give the Boc-protected amine. The Boc group was then deprotected to afford the 2-aminoimidazo[1,2-b]pyridazine derivative 58, which was then acylated to afford 59 (Scheme 15). Compound 59 served as one of the lead compounds for the structure-based design of novel RAF/VEGFR2 inhibitors bearing [5,6]-fused bicyclic scaffolds.[49]
Scheme 15.

Synthesis of sorafenib derivatives.
Routier and collaborators developed valmerins as a new family of Cyclin-Dependent Kinase (CDK) and Glycogen Synthase Kinase 3 (GSK3β) inhibitors. These compounds are characterized by a tetrahydropyrido[1,2-α]isoindolone core, which is functionalized by a terminal (het)arylurea moiety. For the synthesis of these compounds, the urea formation reactions using triphosgene or p-nitrophenylcarbamate resulted in complex mixtures. Thus, a “one-pot” Curtius rearrangement was developed to provide access to these ureas in good yields. For example, acid 60 was converted to isocyanate 61, which was immediately reacted with amine 62 to provide urea 63 in good yield. Compound 63 exhibited potent antiproliferative and antitumor activities in human tumor xenografts, inducing cell death by caspase 3 induction (Scheme 16).[50]
Scheme 16.

Synthesis of GSK3b inhibitor 63.
Helal et al. have identified novel cis-1,3-disubstituted cyclobutyl-4-aminoimidazole inhibitors that gave improved enzyme and cellular potency against CDK5/p25 with up to 30-fold selectivity over CDK2/Cyclin E.[51] Later on, Helal proposed a novel synthesis for compound 66, employing a Curtius rearrangement for the conversion of key intermediate 64 to 65 (Scheme 17).[52]
Scheme 17.

Synthesis of compound 66.
Wipf and collaborators reported the development of new small molecule inhibitors of the protein-tyrosine phosphatase 4A3 (PTP4A3).[21] One of the synthetic routes involved a key Curtius rearrangement step. Acid 20 was converted to acid chloride followed by acyl azide 21 in 44% yield over two steps. Curtius rearrangement and concomitant cyclization required high temperatures which produced thienopyridone 67. Finally, nitration followed by hydrogenation provided 22 (Scheme 18).[21] Thienopyridone 22 is the most potent known inhibitor of PTP4A3 to date and was shown to be selective for the PTP4A family over 11 other phosphatases.
Scheme 18.

Synthesis of PTP4A3 inhibitor 22.
3.2.4. Androgen receptor antagonists
Androgen deprivation employing androgen receptor (AR) antagonists is the mainstay therapy for metastatic prostate cancer.[53] A novel series of trans-N-aryl-2,5-dimethylpiperazine-1-carboxamide derivatives were developed by Kinoyama as orally potent and peripherally selective nonsteroidal AR antagonists.[54] Aryl isocyanate 70 was prepared by the Curtius rearrangement of acid 68. Reaction of arylpiperazine 71 with aryl isocyanate 70, provided urea derivative 72. The authors reported that sodium azide allowed the reaction to proceed more effectively than DPPA for isocyanate formation via acyl azide. The most potent compound 72 might be considered as a potential therapeutic for prostate cancer (Scheme 19).[54]
Scheme 19.

Synthesis of AR antagonist 72.
Balog and colleagues from Bristol-Myers Squibb discovered BMS-641988 (73) as a novel, nonsteroidal AR antagonist designed for the treatment of prostate cancer. The synthesis of 73 involved a key Curtius rearrangement step leading to an amine functionality which offered a good handle for the functionalization of the oxa-bicyclic skeleton. As described in Scheme 20, Diels–Alder cycloaddition between maleimide 74 and the MEM ester of 2,5-dimethyl-3-furoic acid 75 provided bicyclic derivative 76 in good yield. It was readily converted to acid 77. The Curtius rearrangement of 77 provided amine 78 in good yields. A series of amides, sulfamides, carbamates, ureas, and sulfonamides were prepared from amine 78, leading to the discovery of lead compound 73 (Scheme 20).[55] Compound 73 was tested in the human prostate cancer xenograft model CWR22-BMSLD1 demonstrating superior efficacy compared to bicalutamide and robust pharmacokinetic and pharmacodynamic profiles.
Scheme 20.

Synthesis of AR antagonist 73.
3.2.5. DNA binding agents
A large number of anticancer chemotherapeutic drugs are DNA-damaging agents.[56] However, the clinical potential of DNA-damaging agents is limited by adverse side effects and unfavorable drug-like properties.
Witiak and co-workers synthesized diastereomeric cyclohexanediol diamines to serve as ligands in PtII complexes for new congeners of cisplatin.[22] It was envisioned that hydroxyl substitution on the cyclohexane ring may increase aqueous solubility of the organoplatinum complex. For the synthesis of diamine derivatives a Curtius rearrangement was used as the key step.
Treatment of anhydride 79 with trimethylsilyl azide provided isocyanate 23 which was converted to acid chloride 24.
The bis-isocyanate 25 was converted to amine 80 as the HCl salt in 62% yield. The amine was converted to cyclohexanediol diamines 26, 81, 82 (Scheme 21). A series of trans-amine derivatives were prepared using similar reactions. The antitumor activity of a number of compounds was attenuated relative to cisplatin in P-388 leukemia implanted CDF1 mice.[22]
Scheme 21.

Synthesis of congeners of cisplatin.
3.3. Antiviral compounds
The advances of molecular biology and new technologies have a tremendous impact on the development of novel and effective antiviral therapies for the treatment of most viral infections.[57,58] The majority of today’s approved antiviral drugs specifically target proteins critical to viral replication.[59,60] Thus, protein structure-based drug design plays a very important role in developing many structural classes of effective antiviral drugs. Interestingly, amine and amine-derived functionalities are key features of these antiviral agents. In the context of synthesis and structure–activity relationships studies the Curtius rearrangement has been extensively employed. Here, we provide some of these applications.
3.3.1. Influenza virus
Tamiflu, a potent and selective inhibitor of viral neuraminidases A and B is widely used for the treatment of influenza (H5N1 influenza virus).[11] A concise synthesis of Tamiflu (9, oseltamivir phosphate is the orally active prodrug of Tamiflu) was reported by Shibasaki and collaborators using a Curtius rearrangement as a key step.[12] A novel barium-catalyzed asymmetric Diels–Alder reaction between compounds 83 and 84 furnished the cyclohexene frame-work (85) of Tamiflu. The Curtius rearrangement carried on with DPPA in the presence of Et3N in dry THF subsequently established the two neighboring nitrogen functionalities of Tamiflu with an excellent 95% yield. Differentiation of the amino functionalities was thus successfully achieved. The cyclic carbamate 86 was then converted to Tamiflu through eight more steps (Scheme 22).[61] Previously, when the Curtius rearrangement was attempted with TBS as the alcohol protecting group, the desired product was not obtained.
Scheme 22.

Synthesis of Tamiflu (9).
The corresponding hydroxyl diacyl azide of ester 85 was instead obtained in a 95% yield from compound 85 when the reaction was carried out at low temperatures (strictly maintained below 4°C). Products from the other isomer were not observed.[12]
Synthesis of (−)-oseltamivir (19) was also achieved by Hayashi and co-workers employing three one-pot operations with a 57% overall yield and using the Curtius rearrangement as the key step. This procedure was suitable for large-scale preparation of this important compound. The tert-butoxycarbonyl group in compound 17 was converted into the acyl azide 18 in a single pot, as shown in Scheme 23. When 18 was treated with acetic acid in acetic anhydride, a Curtius rearrangement took place, followed by acetamide formation. The Curtius rearrangement proceeded at room temperature. Reduction of the nitro group and a retro-Michael reaction were achieved in the same pot, leading to (−)-oseltamivir (19) (Scheme 23).[20]
Scheme 23.

Synthesis of oseltamivir (19).
Zutter and co-workers accomplished the synthesis of oseltamivir phosphate using a Curtius rearrangement as the key step to install the amine functionality.[62]
Liang and collaborators reported a scale-up synthesis of azaindole-pyrimidine derivative 91, which is a potent inhibitor of human influenza virus replication.[19] The synthesis employed the Curtius rearrangement in two key steps. Enzymatic desymmetrization of ester 87 using lipase AYS Amano selectively hydrolyzed the diester to its corresponding cis-cyclohexane monoacid. Initial efforts to convert this monoacid to amine 88 were not successful. Therefore, the intermediate isocyanate was trapped with benzyl alcohol, followed by hydrogenation to smoothly provide amine 88 as an HCl salt (85% yield). The SNAr displacement of sulfoxide 89 with amine 88 followed by ester hydrolysis and tosyl group deprotection using LiOH led to acid 90. A second Curtius rearrangement with DPPA/TEA, followed by the addition of morpholine furnished urea 91 in 67% yield (Scheme 24).[19]
Scheme 24.

Synthesis of influenza virus replication inhibitor 87.
3.3.2. HIV protease and integrase inhibitors
The HIV protease plays a critical role in viral replication and became a pivotal therapeutic target. Currently, there are ten HIV protease inhibitors approved by the FDA. A large number of solved HIV protease protein structures have greatly facilitated the design of new and improved inhibitors.[63,64] HIV-1 integrase enzyme catalyzes the insertion of the viral DNA into the genome of host cells. Because of the lack of its homologue in human cells and its essential role in HIV-1 replication, integrase inhibition represents an attractive therapeutic target for HIV-1 treatment.[65] Since its identification as a promising therapeutic target three inhibitors (raltegravir, elvitegravir and dolutegravir) have been approved.[66]
Wittenberger and co-workers designed C2-symmetric and pseudo-C2-symmetric inhibitors based on the inherent symmetry of the HIV-1 protease homodimer.[67] A key Curtius rearrangement step was employed in the construction of the pseudo-C2-symmetric 1,3-diamino-2-propanol core 94. As shown, hydrolysis of the imide 92 provided the N-Boc amino hydroxyl acid, which was protected with 2-methoxypropene to give the corresponding carboxylic acid. The acid was converted to the corresponding acyl azide, and subsequent Curtius rearrangement of the azide in the presence of DPPA produced the isocyanate, subsequently trapped with benzyl alcohol to give the differentially protected amine 93. Amine 93 could easily be converted to the key diamino alcohol core unit 94 by sequential deprotection steps (Scheme 25). This has been converted to a range of HIV protease inhibitors.[67]
Scheme 25.

Synthesis of diamino alcohol core 94 for HIV protease inhibitors.
Ghosh and co-workers designed and synthesized the novel dipeptide mimic core 96 for HIV protease inhibitors by using a key Curtius rearrangement of acid 95 in the presence of benzyl alcohol. As shown, carboxylic acid derivative 95 was converted to differentially protected diamine derivative 96. It has been converted to potent HIV-1 protease inhibitors such as 97 with nanomolar IC50 value (Scheme 26).[68]
Scheme 26.

Synthesis of HIV protease inhibitor core 96.
Aminoalkyl epoxides 98 and 99 have been used in the synthesis of a range of potent HIV-1 protease inhibitors. Ghosh and co-workers developed a stereocontrolled synthesis of aminoalkyl epoxide 98 using an asymmetric syn-aldol reaction and a Curtius rearrangement as the key steps. As shown, hydrolytic cleavage of oxazolidinone 100 provided β-hydroxy acid which was subjected to Curtius rearrangement to afford the oxazolidinone 101. Hydrolysis of the oxazolidinone and Boc protection resulted in intermediate 102. This was converted to the aminoalkyl epoxide 98 which is a key intermediate for the synthesis of hydroxyethylene and hydroxyethylamine isosteres for saquinavir and other potent HIV-1 protease inhibitors. Diastereomeric epoxide 99 was also obtained using the same Curtius rearrangement as a key step (Scheme 27).[69,70]
Scheme 27.

Synthesis of key epoxides 98 and 99 for HIV protease inhibitors.
Recently, Ghosh and co-workers reported a highly diastereoselective synthesis of fluorinated epoxide 106 as the key intermediate for the synthesis of novel HIV-1 protease and β-secretase inhibitors. Saponification of syn-aldol product 103 with aqueous lithium hydroperoxide followed by Curtius rearrangement of the resulting acid with DPPA in the presence of Et3N in dry benzene at 90°C afforded oxazolidinone derivative 104 in 70% yield over 2-steps. The oxazolidinone was converted to Boc-derivative 105 in a two-step sequence involving hydrolysis of 104 with aqueous KOH followed by treatment with di-tert-butyl dicarbonate and catalytic hydrogenation. The diol 105 was then converted to epoxide 106 (Scheme 28).[71]
Scheme 28.

Synthesis of key difluoroepoxide 106.
Smith and co-workers designed a novel monopyrrolinone scaffold for HIV-1 protease inhibitors to improve cellular transport properties. Stereoselective synthesis of the tetrahydroiso-quinolyl P2 side chain 109 for the inhibitor was achieved employing an intramolecular Friedel–Crafts acylation performed on a key isocyanate intermediate. As shown, Curtius rearrangement on the carboxylic acid 107 furnished the required isocyanate 108. Treatment of isocyanate with AlCl3 at 60°C afforded the required lactam 109, which was converted to the HIV-1 protease inhibitor 110 (Scheme 29).[72]
Scheme 29.

Synthesis of HIV protease inhibitor 110.
7-Benzyl-4-hydroxynaphthyridinone derivatives were prepared as HIV-integrase inhibitors by Boros and co-workers. The compounds displayed improved enzyme inhibitory activity, optimum cellular antiviral activity and reduced plasma protein binding. The best compound of the series was found to be 114, which demonstrated good oral bioavailability and in vivo clearance in rats.
A key Curtius rearrangement step was employed for the synthesis of this compound. Accordingly, intermediate 111 was treated with DPPA in tert-butanol at 80°C providing the N-Boc pyridine derivative. The Boc group was deprotected to obtain the aminopyridinecarboxylic ester 112 in 50% yield over two steps. Treatment of 112 with ethylmalonyl chloride provided the malonylamide intermediate 113 which was cyclized to the corresponding naphthyridinone by addition of sodium ethoxide. Following conversion to the N1-methyl derivative by reaction with MeI in DMF and reaction with the requisite amine led to final inhibitor 114 (Scheme 30).[73]
Scheme 30.

Synthesis of HIV integrase inhibitor 114.
3.3.3. Anti HCV agents
Hepatitis C virus (HCV) infection is a global health concern.[74] An estimated 3% of the human population worldwide is infected with HCV.[75] Since identification of this virus, the NS3 serine protease contained within the N-terminal region of the NS3 protein has been widely studied.[76] After boceprevir, detailed investigations toward a second generation protease inhibitor led to the discovery of narlaprevir (118), with improved potency, pharmacokinetic profile and physicochemical characteristics. A key Curtius rearrangement step was employed for the synthesis of this compound. In particular, acid 115 was converted in isocyanate 116, by using DPPA in triethylamine and THF. Treatment of isocyanate 116 with L-tert-leucine, in a biphasic medium gave acid 117, a key intermediate for the synthesis of inhibitor 118 (Scheme 31).[77]
Scheme 31.

Synthesis of Narlaprevir (118).
3.3.4. Nucleoside derivatives
Audran and collaborators designed methylenecyclopropane analogues of nucleosides and evaluated their potential as antiviral agents. The starting chiral methylenecyclopropane 119 was obtained by enzymatic desymmetrization of a meso-diol. The free alcohol was converted to acyl azide 120 using Jones oxidation followed by treatment with oxalyl chloride and sodium azide. The Curtius rearrangement gave an unstable isocyanate which was reacted with tert-butanol to provide carbamate 121 in 75% yield over two steps. Deprotection of carbamate and acetate functionalities and introduction of the heterocyclic base led to the required nucleoside derivative 122 (Scheme 32).[78]
Scheme 32.

Synthesis of nucleoside analogue 122.
Chu and co-workers synthesized 2′-hydroxyethylcyclopropyl carbocyclic nucleosides as potential antivirals.[79] The key alcohol intermediate 123 was oxidized to the carboxylic acid, converted to acyl azide and subjected to Curtius rearrangement to furnish urea 124. The urea was transformed to various nucleoside derivatives (e.g., 125, Scheme 33).[79]
Scheme 33.

Synthesis of nucleoside analogue 125.
3.4. Drugs for Cardiovascular Diseases: renin inhibitors, β-adrenoreceptor partial agonists and PDEIII inhibitors
The renin-angiotensin system (RAS) plays an important role in the development of cardiovascular diseases such as hypertension and heart failure. Inhibitors of renin block the RAS at its first and rate-limiting step and may therefore offer major potential benefits in blood pressure control.[80]
Plattner and collaborators prepared a series of dipeptide analogues of angiotensinogen as renin inhibitors and assessed their susceptibility to cleavage by chymotrypsin.[81] For preparation of the β, β -dimethyl Phe intermediate, Curtius rearrangement was used for the introduction of the nitrogen atom. As shown, diester 126 was hydrolyzed and subjected to Curtius rearrangement in the presence of DPPA and Et3N. The intermediate isocyanate was trapped with morpholine to provide compound 127 displaying an N-terminal urea functionality in a satisfactory 69% yield after two steps. This modified Phe derivative was coupled to the histidyl intermediate 128 using standard DCC/HOBt procedure. Compound 129 proved to be substantially stable against chymotrypsin degradation (Scheme 34).[81]
Scheme 34.

Synthesis of renin inhibitor 129.
Patel and co-workers developed a 1,1,1-trifluoromethylketone as a transition-state analogue inhibitor of the aspartyl protease renin.[82] The activated ketone functionality was important for intrinsic potency since its hydrate form could nicely serve as a transition-state analogue. For the preparation of the key trifluoroamino alcohol 132, a Curtius rearrangement of carboxylic acid 130 with DPPA in the presence of triethylamine was efficiently carried out in 75% yield. Compound 131 was then fully deprotected providing amino alcohol 132, isolated as a tosylate salt and converted to the trifluoromethylketone inhibitor 133 in a couple of further steps (Scheme 35). The title inhibitor 133 showed an IC50 value of 250 nM against human renin.[82]
Scheme 35.

Synthesis of renin inhibitor 133.
Prasad and collaborators synthesized the orally active non-peptidic renin inhibitor aliskiren (136) by using the Curtius rearrangement as the key step.[83] Carboxylic acid 134 was converted to acyl azide which rearranged to the isocyanate, then trapped with benzyl alcohol to produce the desired benzyl carbamate 135. It was converted to aliskiren (136) by lactone opening followed by cleavage of the carbamate functionality (Scheme 36).[83]
Scheme 36.

Synthesis of aliskiren (136).
More recently a novel synthesis of aliskiren was proposed by Cini and co-workers based on an unprecedented C5-C6 disconnection, which was carried out on a multigram scale in nine steps. Also in this case a key Curtius rearrangement step was involved.[84]
Acid 137, bearing the adjacent OH functionality protected as acetyl derivative, possessed all of the features required for application of the Curtius rearrangement without the risk of a concurring lactonization reaction. It was reacted with DPPA in the presence of benzyl alcohol. The reaction provided the NHCbz derivative 138 in good yield. Acidic removal of the acetate followed by Cbz/Boc exchange (benzyl hydrogenolysis in the presence of Boc2O) led to known N-Boc lactone 139. Subsequent steps led to aliskiren (136) (Scheme 37).
Scheme 37.

A recent route to aliskiren (136).
Kellam and collaborators recently reported the first β1-selective β-adrenoreceptor partial agonists for the treatment of patients with concomitant respiratory and cardiovascular diseases.[85] SAR studies suggested that an extended alkoxy side chain, alongside substituents at the meta- or para-positions of the phenylurea moiety, increases ligand affinity and β1-selectivity. For the synthesis of the desired hydroxyphenylureas, a key Curtius rearrangement was planned to provide the appropriate alkylisocyanates. Phthalimide derivative 140 was converted to the corresponding isocyanate. Addition of o-/m-aminophenol to the isocyanate afforded the corresponding ureas 141a,b. These were converted to the β1-adrenoreceptor agonists 142a,b in two additional steps (Scheme 38). It was observed that substitution in the meta- and para-positions of the phenyl ring led to higher β1-affinity (and therefore improved β1-selectivity) than substitutions at the ortho-position.[85]
Scheme 38.

Synthesis of β1 partial agonists 142a,b.
Singh and collaborators designed novel cAMP PDEIII inhibitors for the treatment of cardiovascular diseases.[86] A key Curtius rearrangement was used for the preparation of this novel class of compounds. As shown, treatment of the aminopyridinecarboxylic acid 143 with DPPA and triethylamine resulted in the formation the acyl azide 144. Curtius rearrangement of 144 provided the isocyanate 145. Intramolecular cyclization of the isocyanate with the o-amino group afforded the cyclic urea 146 (Scheme 39). Compound 146 was shown to be a very potent inhibitor of cAMP PDEIII with nanomolar range in vitro activity.[86] A series of other derivatives were prepared using the Curtius rearrangement as the key step.
Scheme 39.

Synthesis of PDEIII inhibitor 146.
3.5. Receptor ligands
3.5.1. Dopamine receptors
In order to probe the enantioselective mode of interaction of dopamine receptor(s) agonists, Kaiser and co-workers carried out the synthesis, resolution and determination of the absolute configuration of the two enantiomers of 3′,4′-dihydroxynomifensine (149), a dopamine receptor agonist.[87]
Curtius rearrangement on racemic mixed anhydride obtained from carboxylic acid 147 provided racemic amine 148, which was resolved and converted to the enantiomers of 3′,4′- dihydroxynomifensine (Scheme 40). The dopamine agonists were found to exhibit a high level of enantiospecificity in their interaction with the D1 receptor. Accordingly, it was observed that D1 dopaminergic activity was shown exclusively by the S-enantiomer.[87]
Scheme 40.

Synthesis of dopamine receptor agonist 149.
Tomioka and collaborators synthesized dihydrexidine (29, Scheme 7), a dopamine D1 agonist using Curtius rearrangement as the key step.[23] The compound showed promising properties for the treatment of Parkinson’s disease.[88]
3.5.2. NMDA receptors
Bigge and co-workers synthesized a series of octahydrophenanthrenamine derivatives as noncompetitive antagonists of the N-methyl-D-aspartate (NMDA) receptor complex, and evaluated their affinity at the phencyclidine (PCP) binding site.[89] A key Curtius rearrangement on compound 150 using DPPA afforded the angular isocyanate, which was trapped with methanol and provided the required carbamate. The isolated carbamate was hydrolyzed to provide the primary amine. It was also reduced with lithium aluminum hydride to give the methylamine derivative 151 (Scheme 41).
Scheme 41.

Synthesis of NMDA antagonist 151.
Bigge and collaborators also prepared a series of hexahydrofluorenamines as noncompetitive inhibitors of the NMDA receptor complex. As shown in Scheme 42, Curtius rearrangement of acid 152 furnished carbamate 153. The carbamate was then efficiently converted to compound 154, a rigid analogue of phencyclidine (155) behaving as a potent antagonist at the PCP site.[90]·
Scheme 42.

Synthesis of noncompetitive inhibitor of NMDA receptor complex 154.
3.5.3. Benzodiazepine receptor
A series of 3-amino-β-carboline derivatives was prepared employing a Curtius-type reaction to modulate the benzodiazepine receptor.[91] Refluxing β-CCE (156) with hydrazine hydrate in ethanol gave the corresponding hydrazide, which furnished the acyl azide on treatment with sodium nitrite. Refluxing of acyl azide in acetic acid led to rearrangement to the corresponding amine derivative in good yield. Alternatively, the azide when refluxed in anhydrous methanol furnished the methyl carbamate 157 (Scheme 43A). Compound 157 showed potent activity for the benzodiazepine receptor. 4-Amino-3-carboxy-β-carboline derivatives were also synthesized as benzodiazepine receptor antagonists. A DPPA-promoted Curtius rearrangement was used to convert carboxylic acid 158 to the required antagonist 159 (Scheme 43b).[91]
Scheme 43.

Synthesis of benzodiazepine receptor ligands.
3.5.4. Melatonin receptor
Jellimann and co-workers synthesized phenalene and acenaphthene derivatives as conformationally restricted ligands for melatonin receptors.[92] Curtius rearrangement of the acid 160 led to the corresponding isocyanate, which was readily converted to the required amine 161 in good yield. It was reacted with the propionic anhydride to provide amide 162 (Scheme 44). Many derivatives were prepared using this general route. Many of this new class of melatoninergic naphthalene derivatives displayed higher potencies than melatonin (163, picomolar affinities).[92]
Scheme 44.

Synthesis of melatonin receptor ligands.
3.5.5. GABA receptor
Gavande and collaborators reported novel γ-aminobutyric acid (GABA) analogues based on a 3-(guanido)-1-oxo-1-hydroxyphospholane structure.[93] Compound 167 exhibited high potency and selectivity, thus unveiling phosphinic acids as templates for developing more potent and selective GABAC receptor antagonists. The title compound was prepared by a modified Curtius rearrangement of the acid 164 and hydrolysis of the intermediate isocyanate 165 with aqueous HCl to afford amine 166 in 48% yield over three steps. Compound 167 was obtained by guanylation of amine 166 using formamidinesulfinic acid in the presence of a base (Scheme 45).[93]
Scheme 45.

Synthesis of GABA receptor ligand 167.
3.5.6. Glycine receptor
Johnson and co-workers synthesized a series of aminohydroxyoxazoles to investigate the potential of small, substituted heterocycles to act as potential glycine receptor agonists. Curtius rearrangement was used to synthesize one of those compounds (169) from acid 168, following the method of Poutler and Capson (Scheme 46). Although the calculated parameters were very similar when compared to glycine, considerable receptor-binding activity was not observed, highlighting the challenging nature of the glycine receptor.[94]
Scheme 46.

Synthesis of glycine agonist 169.
3.6. Carbohydrate-based compounds
Vasella and collaborators reported the synthesis of glucose-, mannose- and galactose-derived spirocyclic cyclopropylamines as potential glycosidase inhibitors. Curtius rearrangement of the O-benzylated cyclopropanecarboxylic acid 170 provided the Cbz-protected amine 172 through acyl azide 171 in 68% yield over four steps. This latter was easily converted to the desired cyclopropaneammonium chloride 173 (Scheme 47).[95]
Scheme 47.

Synthesis of spirocyclic sugar derivatives.
An efficient one-pot procedure for the synthesis of urea-linked peptidomimetics and neoglycopeptides was discovered by Sureshbabu and co-workers using Curtius rearrangement conditions (Scheme 48). The urea linkage is of special interest in peptidomimetic research and in drug discovery due to interesting hydrogen-bonding properties. The method circumvented the isolation of acyl azide and isocyanate intermediates. The rearrangement of the in situ generated acyl azide via the acyl fluoride happened under ultrasonication to produce the isocyanate, which was captured by an amine to generate a urea (general structure 175). The protocol worked well with all the common N-urethane-protected amino acids and also with galactose-6-acid. Common amino acid methyl esters worked well as the nucleophilic amine. The usefulness of the protocol was demonstrated by the synthesis of urea derivative 177, where a glycosylamine was used.[96]
Scheme 48.

Synthesis of glycopeptides.
Pseudo-sugar disaccharides were synthesized by Larsen and co-workers starting from the Diels–Alder cycloadduct 178 (Scheme 49). Acidic hydrolysis of the silyl enol ether, followed by reduction of the resulting ketone with sodium cyanoborohydride furnished the lactonic acid 179. The acid was converted to the acyl azide via the acid chloride, and Curtius rearrangement of the azide afforded the isocyanate 180 in 84% yield over three steps. Hydrolysis of the isocyanate produced the amine in low yields, due to competing hydrolysis of the lactone and the acetates. The best yields were obtained when an equimolar amount of triethylamine was used in aqueous THF. The amine was subsequently converted to the required monocarba-disaccharide 181 in two additional steps.[97]
Scheme 49.

Synthesis of pseudo-sugar disaccharides.
3.7. Miscellaneous application of Curtius rearrangement in drug discovery
Edmondson and collaborators reported a series of dipeptidyl peptidase IV (DPP-4) inhibitors for the treatment of type 2 diabetes mellitus (T2DM).[98] Inhibitor 185 exhibited excellent intrinsic inhibition of DPP-4 (IC50=6.2 nm) and oral bioavailability. Synthesis of the target inhibitor employed a Curtius rearrangement as the key step. As shown, carboxylic acid 182 upon treatment with DPPA and Et3N in the presence of tBuOH provided Boc-amine. Boc-deprotection gave amine 183 which was converted to the iodo-substituted triazolopyridine 184. Suzuki coupling of the corresponding boronate with the heterocyclic halide provided target DPP-4 inhibitor 185 (Scheme 50).[98]
Scheme 50.

Synthesis of DPP-4 inhibitor 185.
Remaining in the field of potential T2DM therapeutics, Huard and co-workers proposed a series of inhibitors of Acetyl-CoA carboxylase (ACC) useful for rebalancing the alterations in lipid metabolism associated with the pathogenesis of insulin resistance.
Compound 186 underwent Curtius rearrangement with DPPA and Et3N to provide isocyanate 187, in 91% yield. Reaction of 187 with either sec-butyllithium or tert-butyllithium at −78°C followed by Boc deprotection afforded the desired spirolactam core 188. Final amide formation reaction led to inhibitor 189 which proved to be a potent ACC inhibitor with an IC50 value of 67 nm (Scheme 51).[99]
Scheme 51.

Synthesis of ACC inhibitor 189.
Marquis and collaborators described the design, synthesis, and pharmacological characterization of potent azepanonebased inhibitors of the osteoclast-specific cysteine protease cathepsin K.[100] Having established the importance of the C4 stereocenter of azepanone ring in determining inhibitor potency, a practical asymmetric route was developed involving a Curtius rearrangement as the key step. As shown, hydroxy acid 190 was treated with DPPA and Et3N to provide oxazolidinone 191 in 61% yield. It was then converted to the cathepsin K inhibitor 192 (Scheme 52).[101]
Scheme 52.

Synthesis of cathepsin K inhibitor 192.
Pettersson and co-workers reported a novel series of conformationally constrained g-secretase modulators (GSMs).[102] Compound 196 exhibited improvement in Aβ-lowering activity, displayed very good potency and ADME profile. The synthesis used a key tandem Curtius rearrangement and Friedel–Crafts acylation. Carboxylic acid derivative 193 was converted to the acyl azide 194. Curtius rearrangement and intramolecular Friedel–Crafts cascade reactions on acyl azide 194 afforded the 5,6-disubstituted isoquinolone intermediate 195. The arylimidazole final moiety was subsequently prepared in four additional steps (Scheme 53).
Scheme 53.

Synthesis of γ-secretase modulator 196.
Grunewald and co-workers reported phenolic benzobicyclo[3.2.1]octylamines as conformationally defined analogues of tyramine as selective inhibitors for the epinephrine synthesizing enzyme phenylethanolamine N-methyltransferase (PNMT).[103] The compounds were prepared employing a Curtius rearrangement as the key step.
As shown, the bridgehead carboxylic acid 197 was converted to amine 198 by treatment with DPPA and tert-butyl alcohol, followed by hydrolysis of the mixture with hydrochloric acid. The amine was converted into the required inhibitor 199 in three additional steps (Scheme 54). It was observed that the analogues with the p-hydroxy moiety bind better to PNMT than their m-hydroxy counterparts.[103]
Scheme 54.

Synthesis of PNMT inhibitors 199.
Kulkarni and co-workers reported a microwave-based one-pot tandem synthesis of unsymmetrical ureas via a Curtius rearrangement of commercially available (hetero)aromatic acids and amines in the presence of DPPA. The process is extremely rapid (1–5 min) and allows construction of an array of unsymmetrical ureas in good to excellent yields. The authors demonstrated the scope and utility of this method by performing a gram-scale synthesis of key biologically active compounds targeting the cannabinoid 1 and α7 nicotinic acetylcholine receptors, such as compound PSNCBAM-1 (202), an allosteric modulator of the CB1 receptor, obtained in 75% yield from acid 200 and aniline 201 (Scheme 55).[104]
Scheme 55.

Synthesis of unsymmetrical ureas.
Bornmann et al. reported the synthesis of a macrocyclic compound using standard amino acids and Linked Amino Acid Mimetics (LAAMs). These macrocycles (e.g., 206) would contain a peptide targeting region and variable functional regions. They used a very effective Curtius rearrangement for the conversion of carboxylic acid LAAM-1 (203) to amine LAAM-2 (205) through PMB carbamate 204 (Scheme 56). The authors propose that varieties of LAAMs may be combined to generate macrocycle libraries for general screening or other uses such as development of protein-protein interaction inhibitors.[105]
Scheme 56.

Synthesis of macrocyclic compounds.
Podlech and co-workers developed a procedure for ring-opening of α-amino acid derived β-lactams (e.g., compound 207) with various O-, N- or S-nucleophiles, using catalytic sodium azide. When amino acid esters other than glycine (e.g., 208) were used as the nucleophile in presence of stoichiometric sodium azide, Curtius rearrangement was observed, leading to urea derivatives (e.g., 209) (Scheme 57). These compounds are of considerable interest on account of their possible use as peptidomimetics.[106,107]
Scheme 57.

Synthesis of peptidomimetics.
Reekie and co-workers recently proposed a convenient preparation of N-(indol-2-yl)amides and N-(indol-3-yl)amides which found many applications as pharmacophores, although unstable intermediates impeded previously reported syntheses. The authors employed cheap and readily available substrates in the Curtius rearrangement of indole-3-carboxazide to afford N- (indol-3-yl)amides without prior activation or modification. The reaction can be applied to alkyl and aryl carboxylic acid derivatives. Moreover, either N-substituted or 1H-indole derivatives are tolerated. This approach was extended to the preparation of N-(indol-2-yl)amides 212 from the corresponding indole-2-carboxazides 211 (Scheme 58).[108]
Scheme 58.

Synthesis of N-(indol-2-yl)amides.
3.8. Continuous-flow Curtius rearrangement
The potential safety concerns associated with accumulation of acylazide and isocyanate intermediates during scale-up of active pharmaceutical ingredients (API) represent a relevant issue to be addressed. The employment of continuous-flow processes for scale-up offers several advantages over traditional batch protocols in terms of safety, efficiency, quality and cost.
Therefore, applications of continuous-flow protocols for Curtius rearrangement in the scale-up of API have been steadily increasing over the past years. Also, the use of a continuous-flow technology enables safer, scalable, high-yielding and environmentally friendly processes.
An efficient multistep method for the continuous-flow synthesis of thieno[2,3-c]isoquinolin-5(4H)-one-A (TIQA, 217), an important building block for PARP-1 inhibitors, has been developed by Filipponi and co-workers.[109] After a Suzuki coupling reaction to generate 3-phenylthiophene-2-carboxylic acid 215, this is transformed into the corresponding acyl azide and readily cyclized by a thermal Curtius rearrangement. The authors also employed a statistical design of experiments to support the decision and enable the development of a robust and reliable protocol for large-scale preparation. The large-scale applicability of this protocol was tested by conducting the reactions on a multigram scale to produce the desired product in high yield and quality.
Accordingly, 3-bromothiophene-2-carboxylic acid (213, 10 g) was premixed with Pd(PPh3)4 (1.45 mmol) in THF-PEG-400 (pump A) and reacted with phenylboronic acid (214) in the presence of aqueous sodium hydroxide and TBAB (24.1 mmol) (pump B).The outflow was finally collected into a separatory funnel, and the water layer was acidified with HCl 37%. The resulting solid was filtered off affording 3-phenylthiophene-2-carboxylic acid (215) in 91% yield. Compound 215 was then dissolved in 1,2-dichlorobenzene in the presence of triethylamine and treated with DPPA. The formed acyl azide 216 was promptly reacted within the second coil reactor heated at 235°C. Silica gel purification provided TIQ-A (217) in 50% over-all yield (Scheme 59).
Scheme 59.

Continuous-flow synthesis of thieno[2,3-c]isoquinolin-5(4H)-one-A (TIQA, 217).
More recently, Marsini and co-workers proposed a concise and scalable synthesis of CCR1 antagonist 221 using continuous-flow technology.[110] In particular, they achieved the first example of continuous Curtius rearrangement and semi-continuous acid-isocyanate coupling for the direct synthesis of an amide from two carboxylic acid partners. This safe, robust, and green methodology produced compound 221 in high yield and quality on large scale.
In this semi-continuous-flow process, the isocyanate 219 (generated continuously at 135°C from acid 218) would flow into a mixture containing acid 220 in the presence of triethylamine and toluene, to finally provide desired amide 221 in 76% overall yield on a 100 g scale (Scheme 60).
Scheme 60.

Continuous-flow synthesis of CCR1 antagonist 221.
Ley and co-workers proposed a combination of flow and microwave chemistry methods to bromosporine (222) analogues as modulators of the histone reader BRD9. In particular, the authors implemented an automated Curtius rearrangement reaction and simplified the processing for the preparation of a large amount (40 mmol scale) of the key urethane building block 224 from acid 223 in a safer manner than the corresponding batch process (Scheme 61).[111]
Scheme 61.

Continuous-flow synthesis of key building block 224.
4. Conclusions
Modern drug discovery requires reliable and efficient methods as tools for the synthesis of complex molecules. The introduction of nitrogen in complex organic molecules is often considered one of the most challenging aspects of synthesis and medicinal chemistry. The Curtius rearrangement represents an excellent strategy for the introduction of amine-derived functionalities. In particular, the stereochemical integrity of this rearrangement can be exploited for the generation of chiral centers containing amine derivatives. The abundance of carboxylic acids and their relatively simple conversion to acyl azides also contributes to the increasing scope of this reaction. Amine functionality, amide bonds and its bioisosteres are prevalent in numerous naturally occurring and synthetic medicinal agents. The Curtius rearrangement has been strategically applied to their synthesis over the years. Furthermore, structure–activity relationship studies often entail the swapping of the carboxylic acid and amine functionalities, and the Curtius rearrangement offers a straightforward way to this strategic conversion. This review provides an overview of the applications of Curtius rearrangement in the synthesis of medicinally relevant compounds in a variety of therapeutic fields. It also provides a brief outline of the modern flow methodologies which have adapted the Curtius rearrangement to large-scale processes. Although it has been more than a century since its discovery, the Curtius rearrangement continues to grow as a widely employed protocol for the synthesis of nitrogen-containing bioactive compounds. We hope that this review will stimulate further application of the Curtius rearrangement, particularly in the areas of drug discovery and process development.
Acknowledgements
The research was supported by grants from the US National Institutes of Health (GM53386).
Biographies

Arun K. Ghosh received his BS (Honors) and MS in chemistry from the University of Calcutta and the Indian Institute of Technology, Kanpur, respectively. He obtained his PhD in chemistry in 1985 at the University of Pittsburgh. He pursued postdoctoral research with Professor E. J. Corey at Harvard University from 1985 to 1988. He was a research fellow at Merck Research Laboratories prior to joining the University of Illinois, Chicago as an assistant Professor in 1994. In 2005, he moved to Purdue University, where he is currently the Ian P. Rothwell Distinguished Professor in the Departments of Chemistry and Medicinal Chemistry. His research interests are in the areas of organic, bioorganic, and medicinal chemistry.

Margherita Brindisi graduated cum laude in 2004 from the University of Siena, Italy. In 2008 she received her PhD in pharmaceutical sciences from the same university. In 2010–2011, 2016–2017, and 2018, she worked as a research fellow in Professor Ghosh’s research group at Purdue University. There, she worked on the design and synthesis of protease inhibitors for the treatment of HIV/AIDS and Alzheimer’s disease. She is currently a researcher at the Department of Biotechnology, Chemistry, and Pharmacy at the University of Siena working on the development of novel treatments for cancer, pain, parasitic diseases, and brain disorders.

Anindya Sarkar attended Presidency College, Kolkata, India for his undergraduate studies in chemistry (Honors) and was awarded the KVPY Fellowship. He completed his Master’s degree in chemistry from the Indian Institute of Technology, Kharagpur in 2012. He began his studies at Purdue University in 2012 with a Ross Fellowship and worked in Professor Ghosh’s research group, where his work focused on the design and synthesis of potent HIV protease inhibitors and the enantioselective syntheses of alloyohimbane and yohimbane. He received his PhD in chemistry in 2017. He is currently a postdoctoral researcher at The Scripps Research Institute, San Diego in Professor Dale Boger’s research group.
Footnotes
The ORCID identification number(s) for the author(s) of this article can be found under: https://doi.org/10.1002/cmdc.201800518.
Conflict of interest
The authors declare no conflict of interest.
References
- [1].Smith PAS, Org. Reac 1946, 3, 337–449. [Google Scholar]
- [2].Curtius T, Ber. Dtsch. Chem. Ges 1890, 23, 3023–3033. [Google Scholar]
- [3].Curtius T, J. Prakt. Chem 1894, 50, 275–294. [Google Scholar]
- [4].Saunders JH, Slocombe RJ, Chem. Rev 1948, 43, 203–218. [DOI] [PubMed] [Google Scholar]
- [5].Ghosh AK, Sarkar A, Brindisi M, Org. Biomol. Chem 2018, 16, 2006–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Amino Group Chemistry: From Synthesis to the Life Sciences (Ed.: Ricci A), Wiley-VCH, Weinheim, 2008. [Google Scholar]
- [7].Amines: Synthesis Properties and Applications (Ed.: Lawrence SA), Cambridge University Press, Cambridge, 2004. [Google Scholar]
- [8].Salvatore RN, Yoon CH, Jung KW, Tetrahedron 2001, 57, 7785–7811. [Google Scholar]
- [9].Lebel H, Leogane O, Org. Lett 2006, 8, 5717–5720. [DOI] [PubMed] [Google Scholar]
- [10].Smith AB III, Safonov IG, Corbett RM, J. Am. Chem. Soc 2002, 124, 11102–11113. [DOI] [PubMed] [Google Scholar]
- [11].Lew W, Chen X, Kim CU, Curr. Med. Chem 2000, 7, 663–672. [DOI] [PubMed] [Google Scholar]
- [12].Yamatsugu K, Kamijo S, Suto Y, Kanai M, Shibasaki M, Tetrahedron Lett. 2007, 48, 1403–1406. [Google Scholar]
- [13].Davies BE, J. Antimicrob. Chemother 2010, 65, ii5–ii10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wilhelm SM, Adnane L, Newell P, Villanueva A, Llovet JM, Lynch M, Mol. Cancer. Ther 2008, 7, 3129–3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Keating GM, Santoro A, Drugs 2009, 69, 223–240. [DOI] [PubMed] [Google Scholar]
- [16].Ninomiya K, Shioiri T, Yamada S, Tetrahedron 1974, 30, 2151–2157. [Google Scholar]
- [17].Shioiri T, Yamada S, Ninomiya K, J. Am. Chem. Soc 1972, 94, 6203. [DOI] [PubMed] [Google Scholar]
- [18].Wolff O, Waldvogel SR, Synthesis 2004, 1303–1305. [Google Scholar]
- [19].Liang JL, Cochran JE, Dorsch WA, Davies I, Clark MP, Org. Process Res. Dev 2016, 20, 965–969. [Google Scholar]
- [20].Ishikawa H, Suzuki T, Hayashi Y, Angew. Chem. Int. Ed 2009, 48, 1304–1307; Angew. Chem. 2009, 121, 1330–1333. [DOI] [PubMed] [Google Scholar]
- [21].Salamoun JM, McQueeney KE, Patil K, Geib SJ, Sharlow ER, Lazo JS, Wipf P, Org. Biomol. Chem 2016, 14, 6398–6402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Witiak DT, Rotella DP, Filppi JA, Gallucci J, J. Med. Chem 1987, 30, 1327–1336. [DOI] [PubMed] [Google Scholar]
- [23].Asano Y, Yamashita M, Nagai K, Kuriyama M, Yamada K, Tomioka K, Tetrahedron Lett. 2001, 42, 8493–8495. [Google Scholar]
- [24].Anderson RJ, Groundwater PW, Todd A, Worsley AJ, Antibacterial Agents: Chemistry, Mode of Action, Mechanisms of Resistance and Clinical Applications, Wiley-VCH, Weinheim, 2012. [Google Scholar]
- [25].Redgrave LS, Sutton SB, Webber MA, Piddock LJ, Trends Microbiol. 2014, 22, 438–445. [DOI] [PubMed] [Google Scholar]
- [26].Hagen SE, Domagala JM, Heifetz CL, Sanchez JP, Solomon M, J. Med. Chem 1990, 33, 849–854. [DOI] [PubMed] [Google Scholar]
- [27].Bucsh RA, Domagala JM, Laborde E, Sesnie JC, J. Med. Chem 1993, 36, 4139–4151. [DOI] [PubMed] [Google Scholar]
- [28].Kuramoto Y, Ohshita Y, Yoshida J, Yazaki A, Shiro M, Koike T, J. Med. Chem 2003, 46, 1905–1917. [DOI] [PubMed] [Google Scholar]
- [29].Shinabarger DL, Marotti KR, Murray RW, Lin AH, Melchior EP,Swaney SM, Dunyak DS, Demyan WF, Buysse JM, Antimicrob. Agents Chemother. 1997, 41, 2132–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Brickner SJ, Barbachyn MR, Hutchinson DK, Manninen PR, J. Med. Chem 2008, 51, 1981–1990. [DOI] [PubMed] [Google Scholar]
- [31].Brickner SJ, Hutchinson DK, Barbachyn MR, Manninen PR, Ulanowicz DA, Garmon SA, Grega KC, Hendges SK, Toops DS, Ford CW, Zurenko GE, J. Med. Chem 1996, 39, 673–679. [DOI] [PubMed] [Google Scholar]
- [32].McCarthy JR, Tetrahedron Lett. 2015, 56, 6846–6847. [Google Scholar]
- [33].Hait WN, Nat. Rev. Drug Discovery 2010, 9, 253–254. [DOI] [PubMed] [Google Scholar]
- [34].Baguley BC, Kerr DJ, Anticancer Drug Development, Academic Press, Cambridge, 2002. [Google Scholar]
- [35].Avendano C, Menendez JC, Medicinal Chemistry of Anticancer Drugs, 2 Ed., Elsevier, Amsterdam, 2015. [Google Scholar]
- [36].Housman G, Byler S, Heerboth S, Lapinska K, Longacre M, Snyder N,Sarkar S, Cancers 2014, 6, 1769–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Sackett DL, Pharmacol. Ther 1993, 59, 163–228. [DOI] [PubMed] [Google Scholar]
- [38].Hitotsuyanagi Y, Naka Y, Yamagami K, Fujii A, Tahara T, J. Chem. Soc. Chem. Commun 1995, 49–50. [Google Scholar]
- [39].Hitotsuyanagi Y, Yamagami K, Fujii A, Naka Y, Ito Y, Tahara T, Bioorg. Med. Chem. Lett 1995, 5, 1039–1042. [Google Scholar]
- [40].Hitotsuyanagi Y, Kobayashi M, Morita H, Itokawa H, Takeya K, Tetrahedron Lett. 1999, 40, 9107–9110. [Google Scholar]
- [41].Lu Y, Chen J, Xiao M, Li W, Miller DD, Pharm. Res 2012, 29, 2943–2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Shchegravina ES, Maleev AA, Ignatov SK, Gracheva IA, Stein A,Schmalz HG, Gavryushin AE, Zubareva AA, Svirshchevskaya EV,Fedorov AY, Eur. J. Med. Chem 2017, 141, 51–60. [DOI] [PubMed] [Google Scholar]
- [43].Andreoli F, Barbosa AJ, Parenti MD, Del Rio A, Curr. Pharm. Des 2013, 19, 578–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Wagner JM, Hackanson B, Lubbert M, Jung M, Clin. Epigenetics 2010, 1, 117–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Chen Y, Jie W, Yan W, Zhou K, Xiao Y, Crit. Rev. Eukaryotic Gene Expression 2012, 22, 53–59. [DOI] [PubMed] [Google Scholar]
- [46].Sharma S, Ahmad M, Bhat JA, Kumar A, Kumar M, Zargar MA, Hamid A, Shah BA, Bioorg. Med. Chem. Lett 2014, 24, 4729–4734. [DOI] [PubMed] [Google Scholar]
- [47].Zhou C, Wu F, Lu L, Wei L, Pai E, Yao Y, Song Y, PLoS One 2017, 12, e0170301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Ventura JJ, Nebreda AR, Clin. Transl. Oncol 2006, 8, 153–160. [DOI] [PubMed] [Google Scholar]
- [49].Okaniwa M, Hirose M, Imada T, Ohashi T, Hayashi Y, Miyazaki T, Arita T, Yabuki M, Kakoi K, Kato J, Takagi T, Kawamoto T, Yao S, Sumita A, Tsutsumi S, Tottori T, Oki H, Sang BC, Yano J, Aertgeerts K,Yoshida S, Ishikawa T, J. Med. Chem 2012, 55, 3452–3478. [DOI] [PubMed] [Google Scholar]
- [50].Boulahjar R, Ouach A, Matteo C, Bourg S, Ravache M, le Guevel R, Marionneau S, Oullier T, Lozach O, Meijer L, Guguen-Guillouzo C, Lazar S, Akssira M, Troin Y, Guillaumet G, Routier S, J. Med. Chem 2012, 55, 9589–9606. [DOI] [PubMed] [Google Scholar]
- [51].Ahlijanian MK, Cooper CB, Helal CJ, Lau L-F, Menniti FS, Sanner MA, Seymour PA, Villalobos A (Pfizer Products Inc.), Int. PCT Pub. No. WO0210141 A1, 2002.
- [52].Helal CJ, Kang Z, Lucas JC, Bohall BR, Org Lett. 2004, 6, 1853–1856. [DOI] [PubMed] [Google Scholar]
- [53].Helsen C, Van den Broeck T, Voet A, Prekovic S, Van Poppel H, Joniau S, Claessens F, Endocr. Relat. Cancer 2014, 21, T105–118. [DOI] [PubMed] [Google Scholar]
- [54].Kinoyama I, Taniguchi N, Toyoshima A, Nozawa E, Kamikubo T, Imamura M, Matsuhisa A, Samizu K, Kawanimani E, Niimi T, Hamada N,Koutoku H, Furutani T, Kudoh M, Okada M, Ohta M, Tsukamoto S, J. Med. Chem 2006, 49, 716–726. [DOI] [PubMed] [Google Scholar]
- [55].Balog A, Rampulla R, Martin GS, Krystek SR, Attar R, Dell-John J,DiMarco JD, Fairfax D, Gougoutas J, Holst CL, Nation A, Rizzo C,Rossiter LM, Schweizer L, Shan W, Spergel S, Spires T, Cornelius G,Gottardis M, Trainor G, Vite GD, Salvati ME, ACS Med. Chem. Lett 2015, 6, 908–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Gurova K, Future Oncol. 2009, 5, 1685–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Martinez JP, Sasse F, Bronstrup M, Diez J, Meyerhans A, Nat. Prod. Rep 2015, 32, 29–48. [DOI] [PubMed] [Google Scholar]
- [58].De Clercq E, Antiviral Res. 2005, 67, 56–75. [DOI] [PubMed] [Google Scholar]
- [59].De Clercq E, Li G, Clin. Microbiol. Rev 2016, 29, 695–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].de Chassey B, Meyniel-Schicklin L, Vonderscher J, Andre P, Lotteau V, Genome Med. 2014, 6, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Yamatsugu K, Yin L, Kamijo S, Kimura Y, Kanai M, Shibasaki M, Angew. Chem. Int. Ed 2009, 48, 1070–1076; Angew. Chem. 2009, 121, 1090–1096. [DOI] [PubMed] [Google Scholar]
- [62].Zutter U, Iding H, Spurr P, Wirz B, J. Org. Chem 2008, 73, 4895–4902. [DOI] [PubMed] [Google Scholar]
- [63].Ghosh AK, Osswald HL, Prato G, J. Med. Chem 2016, 59, 5172–5208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Flexner CW, Ogden R, Protease Inhibitors in AIDS Therapy, CRC Press, Boca Raton, 2001. [Google Scholar]
- [65].Kessl JJ, McKee CJ, Eidahl JO, Shkriabai N, Katz A, Kvaratskhelia M, Viruses 2009, 1, 713–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Wong E, Trustman N, Yalong A, JAAPA 2016, 29, 36–40. [DOI] [PubMed] [Google Scholar]
- [67].Wittenberger SJ, Baker WR, Donner BG, Tetrahedron 1993, 49, 1547–1556. [Google Scholar]
- [68].Ghosh AK, Mckee SP, Thompson WJ, Darke PL, Zugay JC, J. Org. Chem 1993, 58, 1025–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Ghosh AK, Hussain KA, Fidanze S, J. Org. Chem 1997, 62, 6080–6082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Ghosh AK, Fidanze S, J. Org. Chem 1998, 63, 6146–6152. [DOI] [PubMed] [Google Scholar]
- [71].Ghosh AK, Cardenas EL, Brindisi M, Tetrahedron Lett. 2017, 58, 4062–4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Smith AB III, Cantin LD, Pasternak A, Guise-Zawacki L, Yao W, Charnley AK, Barbosa J, Sprengeler PA, Hirschmann R, Munshi S, Olsen DB, Schleif WA, Kuo LC, J. Med. Chem 2003, 46, 1831–1844. [DOI] [PubMed] [Google Scholar]
- [73].Boros EE, Edwards CE, Foster SA, Fuji M, Fujiwara T, Garvey EP,Golden PL, Hazen RJ, Jeffrey JL, Johns BA, Kawasuji T, Kiyama R,Koble CS, Kurose N, Miller WH, Mote AL, Murai H, Sato A, Thompson JB, Woodward MC, Yoshinaga T, J. Med. Chem 2009, 52, 2754–2761. [DOI] [PubMed] [Google Scholar]
- [74].Cohen J, Science 1999, 285, 26–30. [DOI] [PubMed] [Google Scholar]
- [75].Lavanchy D, Liver Int. 2009, 29, 74–81. [DOI] [PubMed] [Google Scholar]
- [76].Meanwell NA, Kadow JF, Scola PM, Annu. Rep. Med. Chem 2009, 44, 397–440. [Google Scholar]
- [77].Arasappan A, Bennett F, Bogen SL, Venkatraman S, Blackman M,Chen KX, Hendrata S, Huang Y, Huelgas RM, Nair L, Padilla AI, Pan W, Pike R, Pinto P, Ruan S, Sannigrahi M, Velazquez F, Vibulbhan B,Wu W, Yang W, Saksena AK, Girijavallabhan V, Shih NY, Kong J, Meng T, Jin Y, Wong J, McNamara P, Prongay A, Madison V, Piwinski JJ, Cheng KC, Morrison R, Malcolm B, Tong X, Ralston R, Njoroge FG, ACS Med. Chem. Lett 2010, 1, 64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Obame G, Bremond P, Pannecouque C, Audran G, Synthesis 2013, 45, 2612–2618. [Google Scholar]
- [79].Yang TF, Kim H, Kotra LP, Chu CK, Tetrahedron Lett. 1996, 37, 8849–8852. [Google Scholar]
- [80].Morishita Y, Kusano E, Cardiovasc. Hematol. Agents Med. Chem 2013, 11, 77–82. [DOI] [PubMed] [Google Scholar]
- [81].Plattner JJ, Marcotte PA, Kleinert HD, Stein HH, Greer J, Bolis G,Fung AK, Bopp BA, Luly JR, Sham HL, et al. , J. Med. Chem 1988, 31, 2277–2288. [DOI] [PubMed] [Google Scholar]
- [82].Patel DV, Rielly-Gauvin K, Ryono DE, Free CA, Smith SA, Petrillo EW Jr., J. Med. Chem 1993, 36, 2431–2447. [DOI] [PubMed] [Google Scholar]
- [83].Slade J, Liu H, Prashad M, Prasad K, Tetrahedron Lett. 2011, 52, 4349–4352. [Google Scholar]
- [84].Cini E, Banfi L, Barreca G, Carcone L, Malpezzi L, Manetti F, Marras G, Rasparini M, Riva R, Roseblade S, Russo A, Taddei M, Vitale R, Zanotti-Gerosa A, Org. Process Res. Dev 2016, 20, 270–283. [Google Scholar]
- [85].Mistry SN, Baker JG, Fischer PM, Hill SJ, Gardiner SM, Kellam B,J. Med. Chem 2013, 56, 3852–3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Singh B, Bacon ER, Robinson S, Fritz RK, Lesher GY, Kumar V, Dority JA, Reuman M, Kuo GH, Eissenstat MA, et al. , J. Med. Chem 1994, 37, 248–254. [DOI] [PubMed] [Google Scholar]
- [87].Dandridge PA, Kaiser C, Brenner M, Gaitanopoulos D, Davis LD,Webb RL, Foley JJ, Sarau HM, J. Med. Chem 1984, 27, 28–35. [DOI] [PubMed] [Google Scholar]
- [88].Stacy M, Jankovic J, Annu. Rev. Med 1993, 44, 431–440. [DOI] [PubMed] [Google Scholar]
- [89].Bigge CF, Malone TC, Hays SJ, Johnson G, Novak PM, Lescosky LJ, Retz DM, Ortwine DF, Probert AW Jr., Coughenour LL, et al. , J. Med. Chem 1993, 36, 1977–1995. [DOI] [PubMed] [Google Scholar]
- [90].Hays SJ, Novak PM, Ortwine DF, Bigge CF, Colbry NL, Johnson G, Lescosky LJ, Malone TC, Michael A, Reily MD, et al. , J. Med. Chem 1993, 36, 654–670. [DOI] [PubMed] [Google Scholar]
- [91].Dorey G, Dubois L, Prodo de Carvalho LP, Potier P, Dodd RH, J. Med. Chem 1995, 38, 189–198. [DOI] [PubMed] [Google Scholar]
- [92].Jellimann C, Mathe-Allainmat M, Andrieux J, Kloubert S, Boutin JA,Nicolas JP, Bennejean C, Delagrange P, Langlois M, J. Med. Chem 2000, 43, 4051–4062. [DOI] [PubMed] [Google Scholar]
- [93].Gavande N, Yamamoto I, Salam NK, Ai TH, Burden PM, Johnston GA, Hanrahan JR, Chebib M, ACS Med. Chem. Lett 2011, 2, 11–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Drummond J, Johnson G, Nickell DG, Ortwine DF, Bruns RF, Welbaum B, J. Med. Chem 1989, 32, 2116–2128. [DOI] [PubMed] [Google Scholar]
- [95].Bl chel C, Ramana CV, Vasella A, Helv. Chim. Acta 2003, 86, 2998–3036. [Google Scholar]
- [96].Hemantha HP, Chennakrishnareddy G, Vishwanatha TM, Sureshbabu VV, Synlett 2009, 407–410. [Google Scholar]
- [97].Larsen DS, Trotter NS, Stoodley RJ, Tetrahedron Lett. 1993, 34, 8151–8154. [Google Scholar]
- [98].Edmondson SD, Mastracchio A, Mathvink RJ, He J, Harper B, Park YJ, Beconi M, Di Salvo J, Eiermann GJ, He H, Leiting B, Leone JF,Levorse DA, Lyons K, Patel RA, Patel SB, Petrov A, Scapin G, Shang J, Roy RS, Smith A, Wu JK, Xu S, Zhu B, Thornberry NA, Weber AE, J. Med. Chem 2006, 49, 3614–3627. [DOI] [PubMed] [Google Scholar]
- [99].Huard K, Bagley SW, Menhaji-Klotz E, Preville C, Southers JA Jr.,Smith AC, Edmonds DJ, Lucas JC, Dunn MF, Allanson NM, Blaney EL, Garcia-Irizarry CN, Kohrt JT, Griffith DA, Dow RL, J. Org. Chem 2012, 77, 10050–10057. [DOI] [PubMed] [Google Scholar]
- [100].Marquis RW, Ru Y, LoCastro SM, Zeng J, Yamashita DS, Oh HJ,Erhard KF, Davis LD, Tomaszek TA, Tew D, Salyers K, Proksch J, Ward K, Smith B, Levy M, Cummings MD, Haltiwanger RC, Trescher G, Wang B, Hemling ME, Quinn CJ, Cheng HY, Lin F, Smith WW,Janson CA, Zhao B, McQueney MS, D’Alessio K, Lee CP, Marzulli A, Dodds RA, Blake S, Hwang SM, James IE, Gress CJ, Bradley BR, Lark MW, Gowen M, Veber DF, J. Med. Chem 2001, 44, 1380–1395. [DOI] [PubMed] [Google Scholar]
- [101].Lee Trout RE, Marquis RW, Tetrahedron Lett. 2005, 46, 2799–2801. [Google Scholar]
- [102].Pettersson M, Johnson DS, Subramanyam C, Bales KR, am Ende CW, Fish BA, Green ME, Kauffman GW, Mullins PB, Navaratnam T, Sakya SM, Stiff CM, Tran TP, Xie L, Zhang L, Pustilnik LR,Vetelino BC, Wood KM, Pozdnyakov N, Verhoest PR, O’Donnell CJ, J. Med. Chem 2014, 57, 1046–1062. [DOI] [PubMed] [Google Scholar]
- [103].Ye QH, Grunewald GL, J. Med. Chem 1989, 32, 478–486. [DOI] [PubMed] [Google Scholar]
- [104].Kulkarni AR, Garai S, Thakur GA, J. Org. Chem 2017, 82, 992–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Maxwell DS, Sun D, Peng Z, Martin DV, Bhanu Prasad BA, Bornmann WG, Tetrahedron Lett. 2013, 54, 5799–5801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Taubinger AA, Fenske D, Podlech J, Tetrahedron 2008, 64, 8659–8667. [Google Scholar]
- [107].Taubinger AA, Fenske D, Podlech J, Synlett 2008, 539–542. [Google Scholar]
- [108].Reekie TA, Wilkinson SM, Law V, Hibbs DE, Ong JA, Kassiou M, Org. Biomol. Chem 2017, 15, 576–580. [DOI] [PubMed] [Google Scholar]
- [109].Filipponi P, Ostacolo C, Novellino E, Pellicciari R, Gioiello A, Org. Process Res. Dev 2014, 18, 1345–1353. [Google Scholar]
- [110].Marsini MA, Buono FG, Lorenz JC, Yang BS, Reeves JT, Sidhu K,Sarvestani M, Tan ZL, Zhang YD, Li N, Lee H, Brazzillo J, Nummy LJ, Chung JC, Luvaga IK, Narayanan BA, Wei XD, Song JHJ, Roschangar F, Yee NK, Senanayake CH, Green Chem. 2017, 19, 1454–1461. [Google Scholar]
- [111].Guetzoyan L, Ingham RJ, Nikbin N, Rossignol J, Wolling M, Baumert M, Burgess-Brown NA, Strain-Damerell CM, Shrestha L, Brennan PE, Fedorov O, Knapp S, Ley SV, Med. Chem. Commun 2014, 5, 540–546. [Google Scholar]