

Abstract
The kidney is one of the most energy-demanding organs in the human body, and the maintenance of mitochondrial homeostasis is central to kidney function. Recent advances have led to a greater appreciation of how mitochondrial dysfunction contributes to the pathogenesis of AKI, from decreased ATP production, to enhanced mitochondrial oxidative stress, cell necrosis and apoptosis. Accumulating evidence suggests sexual dimorphism in the response to AKI with males demonstrating greater risk for developing ischemia-reperfusion and sepsis-induced kidney injury. In contrast, females may be more susceptible to nephrotoxic-AKI. There are important sex-related differences in mitochondrial respiration, biogenesis and dynamics that likely contribute to the observed sexual dimorphism in AKI. Sex hormones mediate many of these differences with multiple preclinical studies demonstrating the renoprotective actions of estrogen in many rodent models of AKI. Estrogenic control of mitochondrial biogenesis, function and reactive oxygen species (ROS) generation is discussed. Furthermore, the potential role for sex chromosomes in mediating sex differences in AKI is examined. Novel animal models such as the “four core genotypes” (FCG) mouse model provide us with important tools to study sex chromosome effects in kidney health and disease. By understanding the influences of sexual dimorphism or sex hormones on mitochondrial homeostasis and disease manifestations, we may be able to identify novel therapeutic targets and improve existing treatment options for AKI.
1. Introduction:
Acute kidney injury (AKI) remains a serious global public health problem. AKI has been reported to affect 5–17% of hospital admissions1,2 and 1–25% of ICU patients.3 Despite advances in medical care, available therapies for the prevention and treatment of AKI remain limited, and it continues to be associated with significant mortality, increased hospital length of stay and economic costs. Furthermore, recent studies indicate that AKI results in permanent kidney damage and patients who survive AKI have a greater risk of chronic kidney disease (CKD), end-stage renal disease and death after hospital discharge.4
The kidney is tasked with waste removal from the blood, regulation of fluid and electrolyte balance, reabsorption of nutrients, and maintenance of acid-base homeostasis. The kidney has the second highest mitochondrial content and oxygen consumption rates after the heart as an abundance of mitochondria is required to provide energy to drive these important processes.5 The ability of mitochondria to sense and respond to changes in nutrient availability and energy demand is critical for the maintenance of cellular homeostasis and proper functioning of the kidney. Recent advances have led to a greater appreciation of how mitochondria contribute to the pathogenesis of AKI, from decreased ATP production, to increased mitochondrial oxidative stress, cell necrosis and apoptosis. Hence, there is increased interest in exploring therapeutic strategies that ameliorate mitochondrial dysfunction to prevent and treat AKI.
There is accumulating evidence that biologic sex influences many variables that are important to kidney health, and contributes to differential injury response in patients with kidney disease. It is increasingly recognized that there are important sex-related differences in mitochondrial morphology, function, and homeostasis, and that sex differences exist in the response to AKI,6–8 progression of CKD,9 hypertension and kidney transplantation outcomes.10 This focused review highlights recent advances in our understanding of the role of mitochondrial dysfunction in the context of AKI, with special emphasis on new insights into the effects of biologic sex on intrinsic mitochondrial respiration, mitochondrial biogenesis and dynamics, and ROS homeostasis. A more complete understanding of sexual dimorphism in mitochondria function and homeostasis in the kidney could offer insights and possible therapeutic options that significantly impact our current management of AKI.
2. Mitochondrial Dysfunction in AKI:
Mitochondrial dysfunction is increasingly recognized as an initiator of and contributor to AKI. Histologically, mitochondrial matrix swelling and fragmentation have been observed in renal tubular epithelial cells in ischemia, sepsis, and drug-induced AKI.11 Other hallmark features of mitochondrial dysfunction that are observed in AKI include enhanced mitochondrial oxidative stress, a significant decrease in mitochondrial biogenesis and ATP production, and impaired mitochondrial dynamics.
Mitochondria are key sites of reactive oxygen species (ROS) generation. ROS are molecules derived from oxygen that can readily oxidize other molecules. During ATP production when electrons are passed through the mitochondrial respiratory chain, a low concentration of superoxide anions is generated. A low level of ROS is important for cell signaling and function, including eliciting proliferation and survival in response to stress conditions, but high concentrations are toxic to mitochondria and the cell.5 In ischemia-reperfusion AKI, increased ROS production occurs during reperfusion when oxygen is reintroduced into mitochondria that has sustained ischemic injury with dysregulation of the electron transport chain (ETC) and metabolic pathways, and increased electron leak. Excessive ROS can cause breaks in mitochondrial DNA (mtDNA) leading to respiratory enzymes containing mutant mtDNA-encoded defective protein subunits, and further impairment in ATP and ROS production. ROS can also cause tissue damage through the release of cytochrome C from the mitochondria triggering apoptosis or activation of the immune response through other DAMPS (damage-associated molecular patterns).12 Mitochondria have endogenous antioxidant mechanisms to counteract the excessive formation of ROS; oxidative stress activates nuclear factor erythryoid 2-related factor (NRF-2), a nuclear transcription factor that enhances the expression of mitochondrial antioxidant enzymes such as superoxide dismutase (SOD) and glutathione peroxidase. Mitochondria also contain their own pool of glutathione. Another important mitochondrial antioxidant defense mechanism involves uncoupling proteins (UCP2 in the kidneys), which catalyze protein leak to reduce membrane potential and attenuate ROS production.
Mitochondria are highly dynamic organelles that constantly undergo biogenesis, fission, fusion, and mitophagy in response to metabolic changes and signaling cues in the cell environment. Alterations in these processes have been implicated in the pathogenesis of AKI. Peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) is a master regulator of mitochondrial biogenesis. PGC-1α does not bind directly to DNA, but docks on transcription factors [e.g., nuclear respiratory factor-1 (NRF-1) and NRF-2] bound at their response elements and co-activates them to regulate the expression of target genes and mitochondrial function.13 In mouse models of AKI, PGC-1α is suppressed following injury and returns to basal levels with recovery.14,15 Furthermore, knockout of PGC-1α in mice prolongs impairment in kidney function following endotoxemia, suggesting that PGC-1α plays a critical role in renal recovery.
A precise balance between fission (when a mitochondrion divides into two daughter organelles) and fusion (when two mitochondria merge) is required for mitochondrial homeostasis. Dynamin related protein 1 (DRP1) is the main mediator of mitochondrial fission. Activation and translocation of DRP1 into the mitochondrial outer membrane has been observed in AKI, leading to mitochondrial fragmentation and apoptosis. Administration of mdivi-1, a pharmacological inhibitor of DRP1, inhibits mitochondrial fragmentation and ameliorates ischemia-reperfusion and cisplatin-induced kidney injury.16 Given the evidence for mitochondrial dysfunction in AKI, exploring therapies to maintain/restore mitochondrial homeostasis may potentially provide a tool to ameliorate AKI. Sex-specific differences in mitochondrial biology and their implications in AKI will be discussed below.
3. Evidence for Sex Differences in AKI:
There is growing evidence that sex differences influence the susceptibility, progression, and response to AKI and therapeutics. Several large studies of mixed surgical and medical populations17,18 and multiple small studies of intensive care unit patients6,19,20 found that males are more likely to have in-hospital AKI and increased mortality. Animal models have consistently shown a protective effect of female sex on the development of AKI, including models of ischemia-reperfusion injury, cardiac arrest/cardiopulmonary resuscitation, and endoplasmic reticulum stress-induced AKI.21–25 Because of this well-known resistance of females to AKI, preclinical studies have largely been performed in males. Recently however, the importance of defining pathophysiology and disease mechanisms in the context of different sexes is increasingly being integrated into biomedical research. In rat and mice models of ischemia-reperfusion kidney injury induced by clamping of the renal vein and artery, females demonstrate improved survival, less decline in renal function, and less histologic damage than males when exposed to identical ischemia times. In certain strains, the magnitude of protection observed in females is large, with females tolerating nearly twice the period of ischemia compared with males for an equivalent injury.22,26 Improved tolerance to renal ischemia in females has also been demonstrated in murine models of kidney transplantation.27 Aufhauser Jr., Wang and colleagues analyzed data from the United Network for Organ Sharing (UNOS) database, and observed the effects of biological sex on kidney transplant outcomes in humans. Ischemia-reperfusion injury occurs in all deceased donor kidney transplants and manifest as delayed graft function (DGF), defined as the need for dialysis in the first week after transplantation. The authors found that male recipients had a significantly greater risk of developing DGF compared to female recipients (odds ratio of 1.39). The authors also demonstrated that both donor and recipient sex contributed to renal ischemia tolerance in humans; grafts from male donors had higher DGF rate compared to grafts from female donors (odds ratio of 1.10).
Among septic shock patients, 60–70% develop AKI, which is associated with high mortality rates that approach 50%.28 Compared to ischemic AKI, there are few studies that examine sex effects in septic AKI. Previous studies have suggested that female sex may be associated with decreased susceptibility to sepsis, possibly due modulatory effects of the immune responses by estrogen.29–31 A recent analysis of patients with sepsis included in the Randomized Evaluation of Normal versus Augmented Level renal replacement therapy (RENAL) trial found that in patients with sepsis and severe AKI, female sex was associated with improved survival (HR 0.74 for 90-day mortality),32 suggesting that sexual dimorphism also exists in septic AKI. In contrast, the effect of biological sex on other causes of AKI, such as perioperative- and nephrotoxic-AKI, is not as clear and may be opposite to the observed effects in ischemic and septic AKI. Although large epidemiologic studies have demonstrated lower incidence of AKI after non-cardiac surgery in females compared with males,33,34 female sex has been identified repeatedly as an independent risk factor for the development of vascular or cardiothoracic surgery-associated AKI.35–37 These studies include patients undergoing aortic aneurysm repair, coronary revascularization, cardiac valve repair or replacement, and other non-transplant, non-congenital cardiac surgery involving cardiopulmonary bypass. In fact, several cardiac surgery-associated AKI risk stratification systems include female sex as a risk factor.36,38 However, a recent large meta-analysis of cardiothoracic-associated AKI studies published between 1978 and 2015 challenged the generally held consensus that female sex increases the risk for AKI after cardiac surgery.39 The authors analyzed sixty-four studies that provided sex-specific data regarding the incidence of cardiothoracic surgery-associated AKI among 1,057,412 subjects and found that women were more likely than men to develop AKI postoperatively (odds ratio 1.21). But when the analysis was restricted to studies that used the RIFLE, AKIN, or KDIGO criteria to define AKI, there was no significant sex-related difference in AKI risk; focusing on studies that used univariate versus multivariate analysis also yielded disparate results. The authors note in their discussion that these disparate conclusions may merely reflect the differing definitions of AKI. Interestingly, many studies have shown that women undergoing cardiovascular surgery show a higher prevalence of risk factors associated with poor outcomes than men, including older age, higher burden of comorbidities, worse cardiovascular status, and needing non-elective emergency procedures.40,41 As sex hormones have been postulated to contribute to sex differences in ischemic tolerance, older age and menopause may factor into the increased risk experienced by women in cardiac surgery. Sex-based biases in the delivery of healthcare may also play a role.
Many pharmacologic agents (cisplatin, aminoglycosides, and iodinated contrast) can cause AKI, and the presence of drug transporters on renal epithelia that allow for accumulation of these drugs, particularly in the proximal tubule, may facilitate tubular injury. Although several studies suggest that sexual dimorphism exists in the susceptibility to drug-induced nephrotoxicity, the results have been variable. Most studies have found that females are more susceptible to cisplatin-, aminoglycoside-, and contrast-induced kidney injury.42–44 This was attributed to sex differences in the expression of Organic Cation Transporter 2 (OCT2, a renal uptake transporter) and Multidrug and Toxin Extrusion Protein-1 and 2 (MATE1 and MATE2, renal efflux transporters that excrete these nephrotoxins into urine) on proximal renal tubules.45,46 However, a recent meta-analysis of 24 studies published between 1978 and 2015 found no effect of gender on the risk of aminoglycoside-associated nephrotoxicity.47 Moreover, two recent studies highlighted the importance of aging and changes in sex hormones on the susceptibility of females to cisplatin-induced AKI. Using a mouse-model of cisplatin-induced nephrotoxicity, Boddu and colleagues demonstrated that while young (16–17 week old) female mice were protected from AKI compared to young and aged males, aged (16–17 month old) female mice had the highest mortality.46 The other cohort study stratified patients by age and found only postmenopausal women had a significantly higher risk of kidney injury compared to men (hazard ratio 1.28).48 In fact, Joseph and colleagues examined gene expression of 30 drug transporters in normal human kidneys found no statistically significant sex-only or age-only related differences.49 However, when the participants were grouped based on both sex and age, differential expression of several drug transporter genes was observed including higher expression of OCT2 in females < 50 years compared to females ≥ 50 years. These studies suggest that sex and age impact kidney expression of drug transporters, and both factors need to be considered when examining susceptibility to drug-induced AKI.
4. Sex Hormones in AKI:
Certain mechanisms for sex differences in AKI have been proposed but the exact mechanism remains to be determined. The primary factor may be sex hormones as some epidemiologic studies demonstrate reduced renal protection in aging females post-menopause. Multiple preclinical studies have described protective actions of exogenous estrogen in different rodent models of AKI (ischemia-reperfusion injury, kidney transplantation, cardiac arrest/cardiopulmonary resuscitation, and ER stress-induced injury).21,23,24,27,50 Furthermore, ovariectomy eliminates the protective effects of female sex in models of renal ischemia, while estrogen administration to aged female mice restores tolerance to ischemic injury. Renoprotection by estrogen is also described in other forms of kidney injury, including rodent models of chronic allograft nephropathy, age-related glomerular damage, and hypertensive nephrosclerosis. The role of estrogen receptors in this process remains controversial. Some studies report significantly increased susceptibility to ischemia-reperfusion kidney injury and mortality in estrogen receptor α (ER-α) knockout female mice compared with wild-type controls.27 While others report that estrogen-mediated protection in cardiac arrest models of AKI is not affected by ER-α or ER-β deletion or blockade.51 Studies that modulate testosterone levels provide additional support for the role of sex hormones in the observed sexual dimorphism in AKI. Hodeify and colleagues demonstrated that testosterone administration to female mice increased susceptibility to ER stress-induced AKI resulting in an injury phenotype comparable to that observed in male mice.25 In ischemia-reperfusion injury, Park and colleagues showed similar results with testosterone administration; in addition, they showed that orchiectomy attenuated ischemia-reperfusion kidney injury in male mice.22,50 These studies suggest that estrogen, testosterone, and the ratio of testosterone/estrogen may all be important determinants of sex differences in AKI.
5. Potential Role for Sex Chromosomes in AKI:
More recently, studies have focused on distinguishing sex differences caused by gonadal hormones versus sex chromosome complement (XX versus XY). All sex differences arise from the inherent sexual inequality in these two chromosomes. Sex chromosome-mediated differences in phenotype may have varying mechanisms,52 including 1) Gonadal effects of the Y chromosome: The Y-linked gene Sry3 determines sex differences in the development of gonads. Genes present in both sexes cause differentiation of ovaries unless Sry is present. In males, the sex-determining region Y (SRY) protein acts as a transcription factor and initiates differentiation of testes. 2) Non-gonadal effects of the Y chromosome: Sry is expressed in adult males at times and in tissues not involved with testis determination. For example, Sry is expressed in the brain, kidney and adrenal gland, and has been reported to modulate blood pressure. The promoter sequences of angiotensinogen, renin and ACE genes all contain Sry-binding sites, suggesting that Sry may affect their expression contributing to sex-related differences in hypertension.53,54 3) Gene dosage effects of the X chromosome: Because one X chromosome is transcriptionally silenced in XX adult somatic cells, most X chromosome genes do not show large sex differences in their expression. However, some genes escape inactivation and are expressed at higher level in XX versus XY cells. These include genes that perform an array of regulatory functions and their expression is gene copy-sensitive, including histone lysine demethylases (Kdm5c and Kdm6a) and RNA helicase Ddx3x. Therefore, X gene dosage effects may mediate sex differences in kidney disease.
Until recently, separating sex chromosome effects from sex hormone effects was difficult as it requires manipulating the number of X and Y chromosomes while holding gonadal hormone levels constant. The “four core genotypes” (FCG) mouse model was created using two critical genetic manipulations: deletion of the testis-determining gene Sry from the Y chromosome,55 and insertion of a Sry transgene onto an autosome in the same mouse.56,57 In this model, the Y− chromosome is no longer testis-determining and gonadal determination rests on whether an autosome contains a functional Sry transgene or not. Four core genotypes are generated, including XX mice with ovaries, XY− mice with ovaries, XXSry mice with testes, and XY−Sry mice with testes (Figure 1). This model can discriminate between sex differences determined by gonadal type (XXSry and XY−Sry “males” differ from XX and XY− “females) versus those determined by the effects of sex chromosomes (XX and XXSry differ from XY− and XY−Sry mice).
Figure 1. Four Core Genotypes Model:

FCG mice are produced by breeding XX wild-type females with XY−Sry gonadal males that have deletion of the testis-determining gene Sry from the Y chromosome and insertion of a Sry transgene into an autosome. FCG mice allow comparison to detect the phenotypic effects of sex hormones (Sry present or absent) or sex chromosomes (XX vs. XY). Adapted from.57
There is a paucity of data regarding the role of sex chromosomes in the observed sexual dimorphism in kidney disease. However, studies utilizing the FCG model in cardiac and brain ischemic injury may provide some insights. In cardiac ischemia-reperfusion injury, Li and colleagues found that gonadectomized adult mice with two X chromosomes (XX or XXSry) have ~50% larger infarct size compared to mice with one X chromosome (XY− or XY−Sry mice), regardless of their gonadal phenotype.58 McCullough and colleagues reported similar findings in a mouse model of stroke; furthermore, XX/XXSry mice had increased microglial activation and higher serum levels of pro-inflammatory cytokines than XY−/XY−Sry mice.59 These findings suggest that the second X chromosome increases susceptibility to ischemic injury, which is paradoxical from the observed protective effect of female sex in preclinical and epidemiologic studies of ischemia-reperfusion kidney injury. Perhaps this explains the poor outcomes reported in females undergoing cardiothoracic or vascular surgery, which includes a greater proportion of older/postmenopausal women, as females may be protected by estrogens early in life but are more susceptible to ischemic injury after menopause especially because of the deleterious effects of the second X chromosome. Further studies will be required to determine the contribution to-and mechanisms by which sex chromosomes influence AKI risk and outcomes.
6. Sex Effects on Mitochondria:
Mitochondria dysfunction plays a pivotal role in the pathogenesis of AKI. There is emerging evidence suggesting that some of the sex differences in disease outcomes may be partially related to differences in mitochondrial biology. Reported sexual dimorphism in mitochondrial morphology, biogenesis, respiratory function and ROS homeostasis, and the potential role of estrogen in mediating these sex differences are discussed below and summarized in Figure 2.
Figure 2. Summary of Sex Differences in Mitochondrial Homeostasis:
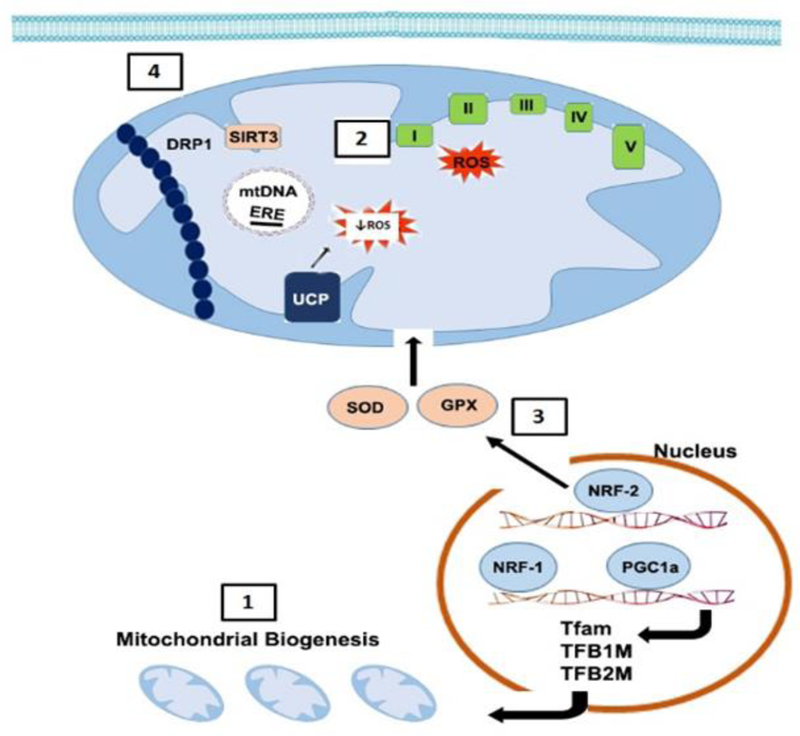
[1] Mitochondria number is greater in females likely due to enhanced biogenesis.60 Estradiol activates PGC-1α, a key regulator of mitochondrial biogenesis. Activation of PGC-1α causes its translocation to the nucleus where it coactivates NRF-1 leading to transcription of genes (including Tfam, TFB1M, TFB2M) involved in mitochondrial biogenesis.63 [2] ROS generation is greater in males likely due to lower intrinsic mitochondrial respiratory capacity and poor antioxidant defense system vs. females. Estradiol also increases the expression of mitochondrial respiratory chain complex proteins and [3] NRF-2-mediated transcription of mitochondrial antioxidants.66 [4] Increased activation and translocation of DRP1 into the mitochondrial outer membrane in AKI leads to mitochondrial fragmentation and apoptosis. SIRT3 prevents translocation of DRP1.71 Thus, lower expression of SIRT3 in males may lead to fragmented, smaller mitochondria vs. females. mtDNA, mitochondrial DNA; ERE, estrogen response element; I/II/III/IV/V, mitochondria respiratory chain complexes 1–5; UCP, uncoupling protein; SOD, superoxide dismutase; GPX, glutathione peroxidase.
Mitochondria number and morphology contribute greatly to their function. It has been reported that females have a greater number of mitochondria than males in the heart and brain, two organs that exhibit sexual dimorphism in susceptibility to ischemic injury.60 Moreover, males exhibited fragmented and smaller mitochondria relative to females. In isolated cardiac mitochondria from young mice and muscle mitochondria from young humans, higher intrinsic mitochondrial respiratory capacity is observed in females compared to males.60,61 Furthermore, Khalifa and colleagues found that these differences were associated with lower ROS (H2O2) production in female cardiac and brain tissues. In contrast, other studies report that cardiac mitochondrial activity in young mice did not vary significantly between sexes; in aged mice, mitochondrial oxygen consumption, ATP content H2O2 production and oxidative damage did not differ between males and females in the heart, skeletal muscle or liver.62 While the data suggest that sex differences in mitochondrial biology exist, there are tissue- and age-dependent variabilities. Studies to examine sex differences in kidney mitochondrial bioenergetics and ROS homeostasis are needed to elucidate its role in AKI.
As discussed above, estrogen has been shown to have renoprotective effects in many models of AKI. Classical estrogen signaling is mediated by ER-α and ER-β, members of the steroid/nuclear receptor superfamily of transcription factors. Once activated by estradiol, or an estrogen-like ligand, ERs form dimers and bind with high affinity to estrogen response elements (EREs, a 15 bp palindromic sequence) in the promoters, introns, or 3’ untranslated regions of target genes.63 ER-α and ER-β have been identified in mitochondria of various tissues. In addition, the mitochondrial genome contains DNA sequences that resemble half the palindromic nuclear ERE sequence.64 Previous studies reported that ER-α and ER-β directly bind mtDNA through these mitochondrial EREs, and 17β-estradiol (E2) treatment increased their binding. E2 treatment also increased levels of several mtDNA-encoded and nuclear DNA-encoded mitochondrial respiratory chain proteins leading to increased mitochondrial respiratory chain activity, suggesting that this is a mechanism by which female hormonal milieu affects mitochondrial function and ROS.65 Another mechanism may be mediated through upregulation of mitochondrial antioxidant proteins. Indeed, Strehlow and colleagues reported that E2 increased the expression and activity of manganese superoxide dismutase (MnSOD), an antioxidant enzyme that neutralizes the highly reactive superoxide to the less reactive hydrogen peroxide in the mitochondria, and protects cells from oxidative stress.66 Additionally, E2 induces nuclear translocation of NRF-2, providing a mechanism for E2-induced expression of antioxidant enzymes such as MnSOD.67,68 On the other hand, Kim and colleagues reported that dihydrotestosterone treatment decreased MnSOD activity in the kidney, leading to greater injury and ROS production following ischemia-reperfusion injury.69 Consistent with these observations, unpublished data from our lab show higher kidney expression of mitochondrial-targeted antioxidant proteins stanniocalcin-1 (STC1) and sirtuin-3 (SIRT3) in female mice compared with males, and that sex hormones may mediate the difference in their expression. We previously reported that STC1 inhibits ROS and protects from ischemia-reperfusion kidney injury via activation of AMPK and induction of mitochondrial SIRT3 and uncoupling protein-2.70 SIRT3 is a protein deacetylase that has been shown to regulate the function of mitochondrial respiratory chain complexes and MnSOD activity. SIRT3 may also have a functional role in mitochondrial dynamics as treatment with the AMPK agonist 5-aminoimidazole-4-carboxmide-1-β-D-ribofuranoside (AICAR) or the antioxidant agent acetyl-L-carnitine (ALCAR) restored SIRT3 expression and activity, and preserved mitochondrial integrity by preventing translocation of dynamin related protein 1 (DRP1, primary mediator of mitochondrial fission).71
Estrogen is also involved in the regulation of mitochondrial biogenesis. E2 treatment has been shown to increase transcription and protein expression of NRF-1 in different cell lines and tissues.63 NRF-1 is a nuclear-encoded transcription factor that regulates the expression of several nuclear-encoded genes that in turn regulate mitochondrial function. These include mtDNA-specific transcription factors Tfam, TFB1M, and TFB2M.72 Additional NRF-1 target genes include subunits of the mitochondrial respiratory chain complexes, and components of mtDNA transcription and replication machinery. E2 treatment has also been shown to increase expression of PGC-1α, a key regulator of mitochondrial biogenesis and coactivator of NRF-1/NRF-2 in cardiac tissue.73
7. Summary:
AKI is a common complication experienced by patients due to many different causes. It has significant implications including increased mortality and risk for the development/progression of CKD, but available therapies for the prevention and treatment of AKI remain limited. There is significant evidence that sexual dimorphism exists in the susceptibility, progression, and response to AKI and therapeutics. Furthermore, there are significant sex differences in mitochondrial biology, which play key roles in kidney health and diseases. These sex differences suggest that biomedical principles learned from the study of males may not apply equally to females. Study and direct comparison of both males and females with the purpose of finding factors that cause sex differences and prevent AKI in one sex compared to the other may help identify novel therapeutic targets and provide guidance for development of sex-directed therapies. Future investigation should further define the role of sex hormones and sex chromosomes in mediating the observed sexual dimorphism in AKI, as well as, elucidate their mechanisms of action.
8. Acknowledgements:
This work was supported by Career Development Award #BX002912 and Merit Award #BX002006 from the United States (U.S.) Department of Veterans Affairs Biomedical Laboratory Research and Development Program, and a generous gift from Dr. and Mrs. Harold Selzman. The contents do not represent the views of the U.S. Department of Veterans Affairs or the United States Government.
References:
- 1.Chertow GM, Burdick E, Honour M, Bonventre JV, Bates DW. Acute kidney injury, mortality, length of stay, and costs in hospitalized patients. J Am Soc Nephrol 2005; 16(11): 3365–70. [DOI] [PubMed] [Google Scholar]
- 2.Sawhney S, Marks A, Fluck N, Levin A, Prescott G, Black C. Intermediate and Long-term Outcomes of Survivors of Acute Kidney Injury Episodes: A Large Population-Based Cohort Study. Am J Kidney Dis 2017; 69(1): 18–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Uchino S, Kellum JA, Bellomo R, et al. Acute renal failure in critically ill patients: a multinational, multicenter study. JAMA 2005; 294(7): 813–8. [DOI] [PubMed] [Google Scholar]
- 4.Coca SG, Singanamala S, Parikh CR. Chronic kidney disease after acute kidney injury: a systematic review and meta-analysis. Kidney Int 2012; 81(5): 442–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bhargava P, Schnellmann RG. Mitochondrial energetics in the kidney. Nat Rev Nephrol 2017; 13(10): 629–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chertow GM, Lazarus JM, Paganini EP, Allgren RL, Lafayette RA, Sayegh MH. Predictors of mortality and the provision of dialysis in patients with acute tubular necrosis. The Auriculin Anaritide Acute Renal Failure Study Group. J Am Soc Nephrol 1998; 9(4): 692–8. [DOI] [PubMed] [Google Scholar]
- 7.Obialo CI, Crowell AK, Okonofua EC. Acute renal failure mortality in hospitalized African Americans: age and gender considerations. J Natl Med Assoc 2002; 94(3): 127–34. [PMC free article] [PubMed] [Google Scholar]
- 8.Wei Q, Wang MH, Dong Z. Differential gender differences in ischemic and nephrotoxic acute renal failure. Am J Nephrol 2005; 25(5): 491–9. [DOI] [PubMed] [Google Scholar]
- 9.Carrero JJ. Gender differences in chronic kidney disease: underpinnings and therapeutic implications. Kidney Blood Press Res 2010; 33(5): 383–92. [DOI] [PubMed] [Google Scholar]
- 10.Reyes D, Lew SQ, Kimmel PL. Gender differences in hypertension and kidney disease. Med Clin North Am 2005; 89(3): 613–30. [DOI] [PubMed] [Google Scholar]
- 11.Szeto HH. Pharmacologic Approaches to Improve Mitochondrial Function in AKI and CKD. J Am Soc Nephrol 2017; 28(10): 2856–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martin JL, Gruszczyk AV, Beach TE, Murphy MP, Saeb-Parsy K. Mitochondrial mechanisms and therapeutics in ischaemia reperfusion injury. Pediatr Nephrol 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Galvan DL, Green NH, Danesh FR. The hallmarks of mitochondrial dysfunction in chronic kidney disease. Kidney Int 2017; 92(5): 1051–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Portilla D, Dai G, McClure T, et al. Alterations of PPARalpha and its coactivator PGC-1 in cisplatin-induced acute renal failure. Kidney Int 2002; 62(4): 1208–18. [DOI] [PubMed] [Google Scholar]
- 15.Tran M, Tam D, Bardia A, et al. PGC-1alpha promotes recovery after acute kidney injury during systemic inflammation in mice. J Clin Invest 2011; 121(10): 4003–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brooks C, Wei Q, Cho SG, Dong Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J Clin Invest 2009; 119(5): 1275–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xue JL, Daniels F, Star RA, et al. Incidence and mortality of acute renal failure in Medicare beneficiaries, 1992 to 2001. J Am Soc Nephrol 2006; 17(4): 1135–42. [DOI] [PubMed] [Google Scholar]
- 18.Neugarten J, Golestaneh L. Female sex reduces the risk of hospital-associated acute kidney injury: a meta-analysis. BMC Nephrol 2018; 19(1): 314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Paganini EP, Halstenberg WK, Goormastic M. Risk modeling in acute renal failure requiring dialysis: the introduction of a new model. Clin Nephrol 1996; 46(3): 206–11. [PubMed] [Google Scholar]
- 20.Mehta RL, Pascual MT, Gruta CG, Zhuang S, Chertow GM. Refining predictive models in critically ill patients with acute renal failure. J Am Soc Nephrol 2002; 13(5): 1350–7. [DOI] [PubMed] [Google Scholar]
- 21.Muller V, Losonczy G, Heemann U, et al. Sexual dimorphism in renal ischemia-reperfusion injury in rats: possible role of endothelin. Kidney Int 2002; 62(4): 1364–71. [DOI] [PubMed] [Google Scholar]
- 22.Park KM, Cho HJ, Bonventre JV. Orchiectomy reduces susceptibility to renal ischemic injury: a role for heat shock proteins. Biochem Biophys Res Commun 2005; 328(1): 312–7. [DOI] [PubMed] [Google Scholar]
- 23.Tanaka R, Tsutsui H, Ohkita M, Takaoka M, Yukimura T, Matsumura Y. Sex differences in ischemia/reperfusion-induced acute kidney injury are dependent on the renal sympathetic nervous system. Eur J Pharmacol 2013; 714(1–3): 397–404. [DOI] [PubMed] [Google Scholar]
- 24.Ikeda M, Swide T, Vayl A, Lahm T, Anderson S, Hutchens MP. Estrogen administered after cardiac arrest and cardiopulmonary resuscitation ameliorates acute kidney injury in a sex-and age-specific manner. Crit Care 2015; 19: 332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hodeify R, Megyesi J, Tarcsafalvi A, Mustafa HI, Hti Lar Seng NS, Price PM. Gender differences control the susceptibility to ER stress-induced acute kidney injury. Am J Physiol Renal Physiol 2013; 304(7): F875–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hu H, Wang G, Batteux F, Nicco C. Gender differences in the susceptibility to renal ischemia-reperfusion injury in BALB/c mice. Tohoku J Exp Med 2009; 218(4): 325–9. [DOI] [PubMed] [Google Scholar]
- 27.Aufhauser DD Jr, Wang Z, Murken DR, et al. Improved renal ischemia tolerance in females influences kidney transplantation outcomes. J Clin Invest 2016; 126(5): 1968–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Feng JY, Liu KT, Abraham E, et al. Serum estradiol levels predict survival and acute kidney injury in patients with septic shock--a prospective study. PLoS One 2014; 9(6): e97967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wichmann MW, Inthorn D, Andress HJ, Schildberg FW. Incidence and mortality of severe sepsis in surgical intensive care patients: the influence of patient gender on disease process and outcome. Intensive Care Med 2000; 26(2): 167–72. [DOI] [PubMed] [Google Scholar]
- 30.Schroder J, Kahlke V, Staubach KH, Zabel P, Stuber F. Gender differences in human sepsis. Arch Surg 1998; 133(11): 1200–5. [DOI] [PubMed] [Google Scholar]
- 31.Offner PJ, Moore EE, Biffl WL. Male gender is a risk factor for major infections after surgery. Arch Surg 1999; 134(9): 935–8; discussion 8–40. [DOI] [PubMed] [Google Scholar]
- 32.O’Brien Z, Cass A, Cole L, et al. Sex and mortality in septic severe acute kidney injury. J Crit Care 2019; 49: 70–6. [DOI] [PubMed] [Google Scholar]
- 33.Grams ME, Sang Y, Coresh J, et al. Acute Kidney Injury After Major Surgery: A Retrospective Analysis of Veterans Health Administration Data. Am J Kidney Dis 2016; 67(6): 872–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kheterpal S, Tremper KK, Heung M, et al. Development and validation of an acute kidney injury risk index for patients undergoing general surgery: results from a national data set. Anesthesiology 2009; 110(3): 505–15. [DOI] [PubMed] [Google Scholar]
- 35.Katz DJ, Stanley JC, Zelenock GB. Gender differences in abdominal aortic aneurysm prevalence, treatment, and outcome. J Vasc Surg 1997; 25(3): 561–8. [DOI] [PubMed] [Google Scholar]
- 36.Thakar CV, Liangos O, Yared JP, Nelson DA, Hariachar S, Paganini EP. Predicting acute renal failure after cardiac surgery: validation and re-definition of a risk-stratification algorithm. Hemodial Int 2003; 7(2): 143–7. [DOI] [PubMed] [Google Scholar]
- 37.Bove T, Calabro MG, Landoni G, et al. The incidence and risk of acute renal failure after cardiac surgery. J Cardiothorac Vasc Anesth 2004; 18(4): 442–5. [DOI] [PubMed] [Google Scholar]
- 38.Ng SY, Sanagou M, Wolfe R, Cochrane A, Smith JA, Reid CM. Prediction of acute kidney injury within 30 days of cardiac surgery. J Thorac Cardiovasc Surg 2014; 147(6): 1875–83, 83 e1. [DOI] [PubMed] [Google Scholar]
- 39.Neugarten J, Sandilya S, Singh B, Golestaneh L. Sex and the Risk of AKI Following Cardio-thoracic Surgery: A Meta-Analysis. Clin J Am Soc Nephrol 2016; 11(12): 2113–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thakar CV, Liangos O, Yared JP, et al. ARF after open-heart surgery: Influence of gender and race. Am J Kidney Dis 2003; 41(4): 742–51. [DOI] [PubMed] [Google Scholar]
- 41.Mehta RH, Castelvecchio S, Ballotta A, Frigiola A, Bossone E, Ranucci M. Association of gender and lowest hematocrit on cardiopulmonary bypass with acute kidney injury and operative mortality in patients undergoing cardiac surgery. Ann Thorac Surg 2013; 96(1): 133–40. [DOI] [PubMed] [Google Scholar]
- 42.Sweileh WM. Gender differences in aminoglycoside induced nephrotoxicity: a prospective, hospital-based study. Curr Clin Pharmacol 2009; 4(3): 229–32. [DOI] [PubMed] [Google Scholar]
- 43.Latcha S, Jaimes EA, Patil S, Glezerman IG, Mehta S, Flombaum CD. Long-Term Renal Outcomes after Cisplatin Treatment. Clin J Am Soc Nephrol 2016; 11(7): 1173–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mehran R, Aymong ED, Nikolsky E, et al. A simple risk score for prediction of contrast-induced nephropathy after percutaneous coronary intervention: development and initial validation. J Am Coll Cardiol 2004; 44(7): 1393–9. [DOI] [PubMed] [Google Scholar]
- 45.Urakami Y, Nakamura N, Takahashi K, et al. Gender differences in expression of organic cation transporter OCT2 in rat kidney. FEBS Lett 1999; 461(3): 339–42. [DOI] [PubMed] [Google Scholar]
- 46.Boddu R, Fan C, Rangarajan S, Sunil B, Bolisetty S, Curtis LM. Unique sex- and age-dependent effects in protective pathways in acute kidney injury. Am J Physiol Renal Physiol 2017; 313(3): F740–F55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Neugarten J, Golestaneh L. The effect of gender on aminoglycoside-associated nephrotoxicity. Clin Nephrol 2016; 86(10): 183–9. [DOI] [PubMed] [Google Scholar]
- 48.Chen WY, Hsiao CH, Chen YC, et al. Cisplatin Nephrotoxicity Might Have a Sex Difference. An analysis Based on Women’s Sex Hormone Changes. J Cancer 2017; 8(19): 3939–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Joseph S, Nicolson TJ, Hammons G, Word B, Green-Knox B, Lyn-Cook B. Expression of drug transporters in human kidney: impact of sex, age, and ethnicity. Biol Sex Differ 2015; 6: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Park KM, Kim JI, Ahn Y, Bonventre AJ, Bonventre JV. Testosterone is responsible for enhanced susceptibility of males to ischemic renal injury. J Biol Chem 2004; 279(50): 52282–92. [DOI] [PubMed] [Google Scholar]
- 51.Hutchens MP, Nakano T, Kosaka Y, et al. Estrogen is renoprotective via a nonreceptor-dependent mechanism after cardiac arrest in vivo. Anesthesiology 2010; 112(2): 395–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Arnold AP, Cassis LA, Eghbali M, Reue K, Sandberg K. Sex Hormones and Sex Chromosomes Cause Sex Differences in the Development of Cardiovascular Diseases. Arterioscler Thromb Vasc Biol 2017; 37(5): 746–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ely D, Underwood A, Dunphy G, Boehme S, Turner M, Milsted A. Review of the Y chromosome, Sry and hypertension. Steroids 2010; 75(11): 747–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Turner ME, Ely D, Prokop J, Milsted A. Sry, more than testis determination? Am J Physiol Regul Integr Comp Physiol 2011; 301(3): R561–71. [DOI] [PubMed] [Google Scholar]
- 55.Lovell-Badge R, Robertson E. XY female mice resulting from a heritable mutation in the primary testis-determining gene, Tdy. Development 1990; 109(3): 635–46. [DOI] [PubMed] [Google Scholar]
- 56.Mahadevaiah SK, Odorisio T, Elliott DJ, et al. Mouse homologues of the human AZF candidate gene RBM are expressed in spermatogonia and spermatids, and map to a Y chromosome deletion interval associated with a high incidence of sperm abnormalities. Hum Mol Genet 1998; 7(4): 715–27. [DOI] [PubMed] [Google Scholar]
- 57.Arnold AP, Chen X. What does the “four core genotypes” mouse model tell us about sex differences in the brain and other tissues? Front Neuroendocrinol 2009; 30(1): 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li J, Chen X, McClusky R, et al. The number of X chromosomes influences protection from cardiac ischaemia/reperfusion injury in mice: one X is better than two. Cardiovasc Res 2014; 102(3): 375–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McCullough LD, Mirza MA, Xu Y, et al. Stroke sensitivity in the aged: sex chromosome complement vs. gonadal hormones. Aging (Albany NY) 2016; 8(7): 1432–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Khalifa AR, Abdel-Rahman EA, Mahmoud AM, et al. Sex-specific differences in mitochondria biogenesis, morphology, respiratory function, and ROS homeostasis in young mouse heart and brain. Physiol Rep 2017; 5(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cardinale DA, Larsen FJ, Schiffer TA, et al. Superior Intrinsic Mitochondrial Respiration in Women Than in Men. Front Physiol 2018; 9: 1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sanz A, Hiona A, Kujoth GC, et al. Evaluation of sex differences on mitochondrial bioenergetics and apoptosis in mice. Exp Gerontol 2007; 42(3): 173–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Klinge CM. Estrogenic control of mitochondrial function and biogenesis. J Cell Biochem 2008; 105(6): 1342–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jia G, Aroor AR, Sowers JR. Estrogen and mitochondria function in cardiorenal metabolic syndrome. Prog Mol Biol Transl Sci 2014; 127: 229–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen JQ, Eshete M, Alworth WL, Yager JD. Binding of MCF-7 cell mitochondrial proteins and recombinant human estrogen receptors alpha and beta to human mitochondrial DNA estrogen response elements. J Cell Biochem 2004; 93(2): 358–73. [DOI] [PubMed] [Google Scholar]
- 66.Strehlow K, Rotter S, Wassmann S, et al. Modulation of antioxidant enzyme expression and function by estrogen. Circ Res 2003; 93(2): 170–7. [DOI] [PubMed] [Google Scholar]
- 67.Zhu C, Wang S, Wang B, et al. 17beta-Estradiol up-regulates Nrf2 via PI3K/AKT and estrogen receptor signaling pathways to suppress light-induced degeneration in rat retina. Neuroscience 2015; 304: 328–39. [DOI] [PubMed] [Google Scholar]
- 68.Oh JY, Choi GE, Lee HJ, et al. 17beta-Estradiol protects mesenchymal stem cells against high glucose-induced mitochondrial oxidants production via Nrf2/Sirt3/MnSOD signaling. Free Radic Biol Med 2018; 130: 328–42. [DOI] [PubMed] [Google Scholar]
- 69.Kim J, Kil IS, Seok YM, et al. Orchiectomy attenuates post-ischemic oxidative stress and ischemia/reperfusion injury in mice. A role for manganese superoxide dismutase. J Biol Chem 2006; 281(29): 20349–56. [DOI] [PubMed] [Google Scholar]
- 70.Pan JS, Huang L, Belousova T, et al. Stanniocalcin-1 inhibits renal ischemia/reperfusion injury via an AMP-activated protein kinase-dependent pathway. J Am Soc Nephrol 2015; 26(2): 364–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Morigi M, Perico L, Rota C, et al. Sirtuin 3-dependent mitochondrial dynamic improvements protect against acute kidney injury. J Clin Invest 2015; 125(2): 715–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Scarpulla RC. Nuclear control of respiratory gene expression in mammalian cells. J Cell Biochem 2006; 97(4): 673–83. [DOI] [PubMed] [Google Scholar]
- 73.Murphy E, Steenbergen C. Gender-based differences in mechanisms of protection in myocardial ischemia-reperfusion injury. Cardiovasc Res 2007; 75(3): 478–86. [DOI] [PubMed] [Google Scholar]