

Abstract
A characteristic phenotype of congenital generalized lipodystrophy 2 (CGL2) that is caused by loss-of-function of seipin gene is mental retardation. Here, we show that seipin deficiency in hippocampal CA1 pyramidal cells caused the reduction of peroxisome proliferator-activated receptor gamma (PPARγ). Twelve-week-old systemic seipin knock-out mice and neuronal seipin knock-out (seipin-nKO) mice, but not adipose seipin knock-out mice, exhibited spatial cognitive deficits as assessed by the Morris water maze and Y-maze, which were ameliorated by the treatment with the PPARγ agonist rosiglitazone (rosi). In addition, seipin-nKO mice showed the synaptic dysfunction and the impairment of NMDA receptor-dependent LTP in hippocampal CA1 regions. The density of AMPA-induced current (IAMPA) in CA1 pyramidal cells and GluR1/GluR2 expression were significantly reduced in seipin-nKO mice, whereas the NMDA-induced current (INMDA) and NR1/NR2 expression were not altered. Rosi treatment in seipin-nKO mice could correct the decrease in expression and activity of AMPA receptor (AMPAR) and was accompanied by recovered synaptic function and LTP induction. Furthermore, hippocampal ERK2 and CREB phosphorylation in seipin-nKO mice were reduced and this could be rescued by rosi treatment. Rosi treatment in seipin-nKO mice elevated BDNF concentration. The MEK inhibitor U0126 blocked rosi-restored AMPAR expression and LTP induction in seipin-nKO mice, but the Trk family inhibitor K252a did not. These findings indicate that the neuronal seipin deficiency selectively suppresses AMPAR expression through reducing ERK-CREB activities, leading to the impairment of LTP and spatial memory, which can be rescued by PPARγ activation.
SIGNIFICANCE STATEMENT Congenital generalized lipodystrophy 2 (CGL2), caused by loss-of-function mutation of seipin gene, is characterized by mental retardation. By the generation of systemic or neuronal seipin knock-out mice, the present study provides in vivo evidence that neuronal seipin deficiency causes deficits in spatial memory and hippocampal LTP induction. Neuronal seipin deficiency selectively suppresses AMPA receptor expression, ERK-CREB phosphorylation with the decline of PPARγ. The PPARγ agonist rosiglitazone can ameliorate spatial cognitive deficits and rescue the LTP induction in seipin knock-out mice by restoring AMPA receptor expression and ERK-CREB activities.
Keywords: AMPA receptor, long-term potentiation, peroxisome proliferator-activated receptor gamma, seipin, spatial cognitive
Introduction
Congenital generalized lipodystrophy (CGL) is an autosomal-recessive disorder characterized by a near total loss of adipose tissue, insulin resistance, and hypertriglyceridemia (Szymanski et al., 2007). Genome-wide linkage analysis has identified two loci for CGL: CGL1 by mutation in the 1-acylglycerol-3-phosphate-O-acyl transferase 2 gene and CGL2 by mutation in the Berardinelli–Seip congenital lipodystrophy 2 (BSCL2) gene that encodes seipin (Magre et al., 2001; Agarwal et al., 2002). The loss of function of the seipin gene has been identified in CGL2 (Magre et al., 2001; Van Maldergem et al., 2002). Systemic seipin knock-out mice (Cui et al., 2011; Chen et al., 2012; Prieur et al., 2013) and seipin knock-out rats (Ebihara et al., 2015) display a severe lipodystrophy with a dramatic loss of fat mass, severe insulin resistance, and hypertriglyceridemia.
An important clinical feature in CGL2 patients is mental retardation with delayed cognitive development and intellectual impairment (Van Maldergem et al., 2002). Rajab et al. (2003) reported that the patients with CGL2 had sensorineural deafness and delayed cognitive development. Schuster et al. (2014) have reported that CGL2 patients have mild to moderate intellectual impairment. In contrast, subjects carrying asparagine 88 to serine (N88S) or serine 90 to leucine (S90L) develop a broad spectrum of motor neuropathy (Agarwal and Garg, 2006; Guo et al., 2013) without mental retardation (Ito et al., 2008). In addition, CGL4 patients have muscular dystrophy and delayed motor milestones that are not associated with intellectual impairment (Murakami et al., 2013). We observed anxiety- and depression-like phenotypes in neuronal seipin knock-out mice (Zhou et al., 2014). Ebihara et al. (2015) have recently reported that seipin knock-out rats show the impairment of spatial working memory. However, the underlying mechanisms remain to be elucidated.
Seipin is highly expressed in the cortex, cerebellum, hippocampus, and hypothalamus (Magre et al., 2001; Wei et al., 2013). The level of hippocampal peroxisomal proliferator-activated receptor gamma (PPARγ) is remarkably reduced in neuronal seipin knock-out mice (Zhou et al., 2014). PPARγ, a class of nuclear receptors, acts as a transcription factor involved in regulating whole-body insulin sensitivity, adipocyte differentiation, lipid storage, and glucose uptake. Knock-down of seipin in 3T3-L1 cells has been reported to reduce the expression of PPARγ to impair adipocyte homeostasis strongly, leading to lipodystrophy (Chen et al., 2009). The defective differentiation of adipocyte by the seipin knock-down is able to be rescued by the PPARγ agonist pioglitazone. PPARγ is also expressed in the CNS (Heneka et al., 2000; Moreno et al., 2004). A potential action of PPARγ in the amelioration of memory deficits has been reported in various models of cognitive deficits. For example, the cognitive impairment in Tg2576 mice, an Alzheimer's disease (AD) model, is improved by treatment with the PPARγ agonist rosiglitazone (rosi) that enhances ERK2 activity in the hippocampus (Denner et al., 2012). Kariharan et al. (2015) have reported that the intracranial delivery of rosi in type 2 diabetic mice significantly improves spatial memory and LTP induction without improvement of peripheral insulin sensitivity. Therefore, it is of great interest to investigate the involvement of reduced hippocampal PPARγ in the delayed cognitive development and intellectual impairment of CGL2 patients.
In this study, systemic seipin knock-out (seipin-sKO) mice, neuronal seipin knock-out (seipin-nKO) mice and adipose seipin knock-out (seipin-aKO) mice were used to determine whether the cognitive deficits arise from the neuronal seipin deficiency or the lipodystrophy-produced peripheral metabolic disorder. In addition, we examined the influence of neuronal seipin deficiency on basal synaptic properties and frequency-dependent LTP and the function and expression of AMPA receptor (AMPAR) and NMDA receptor (NMDAR) in hippocampal CA1 regions. Furthermore, we investigated the involvement of PPARγ in the spatial cognitive deficits and synaptic dysfunction induced by the seipin deficiency and the underlying mechanisms. The results of this study indicate that hippocampal seipin deficiency causes downregulation of AMPA receptors, leading to LTP and spatial cognitive deficits through reduced ERK-CREB activities, which can be rescued by PPARγ activation.
Materials and Methods
Experimental animals
All animal handling procedures followed the guidelines for Laboratory Animal Research of the Nanjing Medical University. The use of animals was approved by the Institutional Animal Care and Use Committee of Nanjing Medical University. The mice were maintained in a constant environmental condition (temperature 23 ± 2°C, humidity 55 ± 5%, and 12:12 h light/dark cycle). seipin-sKO, seipin-nKO, and seipin-aKO mice were generated as described previously (Cui et al., 2011; Liu et al., 2014; Zhou et al., 2014). Briefly, seipin loxP/loxP mice were generated as described previously (Cui et al., 2011). Neuron- and adipose-specific deletion of seipin exon 3 was induced by crossing mice with the loxP seipin allele to transgenic mice expressing Cre recombinase driven by a neuron-specific or an adipocyte-specific promoter (nestin-Cre or aP2-Cre; Jackson Laboratories). Progenies were screened by PCR for loss of the seipin exon 3 and presence of nestin-Cre or aP2-Cre. The genotype of seipin-nKO and seipin-aKO mice was identified by PCR using genomic DNA from their tails. When heterozygous seipin LoxP-nestin- or aP2-Cre were obtained, they were further crossed with seipin loxP/loxP homozygous mice. The progenies were screened by genomic DNA PCR for seipin loxP/loxP with nestin- or aP2-Cre, which would be used in subsequent experiments. The genotype identification of seipin-sKO, seipin-nKO mice, seipin-aKO mice was performed according to the procedures described in the above citations. All animals received a standard laboratory diet before and after all procedures.
In this study, 12-week-old male seipin-sKO mice (n = 24) and WT littermates (n = 24), male seipin-nKO mice (n = 128) and control mice (n = 128), male seipin-aKO mice (n = 24) and a-control mice (n = 24) were used at the beginning of all experiments. All mice were randomly assigned to different experimental groups: (1) behavioral tests (n = 12 mice for each group) → Western blot analyses/RT-PCR (n = 8 mice for each group, unilateral hippocampal tissue per mice was used for Western blot analyses or RT-PCR) were sequentially performed in the same cohorts; (2) electrophysiological analysis was performed in the separate cohorts (n = 8 mice for each group); (3) pharmacological experiments were examined in the separate cohorts (n = 8 mice for each group); and (4) the concentration of BDNF was measured in the difference cohorts (n = 8 mice for each group). Each experiment was performed by two experimenters blinded to the experimental groups.
Drug administration
The PPARγ agonist rosiglitazone (rosi; Enzo) was dissolved in dimethyl sulfoxide (DMSO) and then diluted in 0.9% saline to a final concentration of 0.5% DMSO. The oral administration of rosi was given daily at a dose of 5 mg/kg (Salehi-Sadaghiani et al., 2012). In addition, hippocampal slices were treated with acute perfusion of rosi (1 μm) for 30 min. The MEK inhibitor U0126 and Trk family inhibitor K252a were purchased from Sigma-Aldrich and dissolved in 0.5% DMSO. For repeated intracerebroventricular injection of U0126 (0.3 nmol/3 μl/mouse; Yang et al., 2012) or K252a (0.2 μm/3 μl/mouse; Camarena et al., 2010), a 26 G stainless-steel guide cannula (Plastics One) was implanted into the right lateral ventricle (0.3 mm posterior to bregma, 1.0 mm lateral, and 2.3 mm ventral) and anchored to the skull with 3 stainless steel screws and dental cement.
Behavioral examination
Morris water maze task.
The apparatus of the Morris water maze was a pool (diameter = 120 cm) made of white-colored plastic. Milk powder was used to render the water opaque. The apparatus was housed in a light-controlled room and maintained at a temperature of 20 ± 1°C. The pool was divided arbitrarily into four equally sized quadrants. The walls around the pool were pasted with high contrast visual cues (an X, a triangle, a circle, and a square) to allow animals to build up their spatial memory (Tsai et al., 2011). Swimming paths were analyzed by a computer system with a video camera (AXIS-90 Target/2; Beijing Sunny Instruments). On days 1–2 of training, a cylindrical dark-colored platform (7 cm in diameter) was placed 0.5 cm above the surface of the water. On days 3–7 of training, the platform was moved to the opposite quadrant of the visible platform and submerged 1 cm below the water surface. Three starting positions were used pseudorandomly. The escape latency to reach the visible platform or the hidden platform and swim distance were measured. If the mouse could not reach the platform within 90 s, the experimenter gently assisted it onto the platform and allowed it to remain there for 10 s. The mouse was then dried and returned to the cage to rest for 15 min before the next trial. After removing the platform, the probe trial was performed on day 8 of training. The mouse was released from the opposite quadrant of a hidden platform to allow swimming for 90 s to determine its search patterns. The percentage of total time spent in each quadrant was measured.
Y-maze task.
Spatial working memory performance was assessed by recording spontaneous alternation behavior in a Y-maze (Maurice et al., 1996). Y-maze task was performed at 48 h after the probe trial task. Y-maze was made of black painted wood. Each arm was 40 cm long, 13 cm high, 3 cm wide at the bottom, 10 cm wide at the top, and converged at an equal angle. Each mouse was placed at the end of one arm and allowed to move freely through the maze during an 8 min session. The series of arm entries was recorded visually and arm entry was considered to be completed when the hindpaws of the mouse were completely placed in the arm. Alternation was defined as successive entries into the three arms on overlapping triplet sets. The percentage alternation was calculated as the ratio of actual to possible alternations (defined as the total number of arm entries minus two). The scorers were blinded to the treatment groups.
Histological examination
The mice were anesthetized with chloral hydrate (400 mg/kg, i.p.) and perfused transcardially with 4% paraformaldehyde. Brains were removed and processed for paraffin embedding. Coronal sections (5 μm in thickness) were placed in gelatin-coated slides, blocked with 3% normal goat serum, and then incubated in rabbit monoclonal anti-seipin antibody (1:1000) at 4°C overnight (Jiang et al., 2014). The seipin monoclonal antibody was a kind gift from Professor Jiahao Sha (State Key Laboratory of Reproductive Medicine, Nanjing Medical University, China). After PBS rinses, the sections were incubated in biotin-labeled goat anti-rabbit IgG antibody (1:500; Bioworld Technology) for 2 h. Immunoreactivities were visualized by avidin-biotin horseradish peroxidase complex (ABC Elite; Vector Laboratories) using 3,3′-diaminobenzidine as the chromogen. The immunopositive cells/fibers were observed using a conventional light microscope (40× objective, DP70; Olympus).
Electrophysiological analysis
The mice were deeply anesthetized with isofluorane and decapitated. The brains were rapidly removed and coronal brain slices (400 μm) were cut using a vibrating microtome (Microslicer DTK 1500; Dousaka) in ice-cold cutting solution composed of the following (in mm): 94 sucrose, 30 NaCl, 4.5 KCl, 1.0 MgCl2, 26 NaHCO3, 1.2 NaH2PO4, and 10 d-glucose, pH 7.4. The hippocampal slices were continuously perfused with artificial CSF (ACSF) at 30 ± 1°C for >60 min to recover from damage. ACSF was composed of the following (in mm): 124 NaCl, 2 CaCl2, 4.5 KCl, 1 MgCl2, 26 NaHCO3, 1.2 NaH2PO4, and 10 d-glucose, with the pH was adjusted to 7.4. Both the cutting solution and ACSF were oxygenated with a gas mixture of 95% O2/5% CO2.
Field potential recording.
After a slice was submerged in a recording chamber, one of two hydraulic micromanipulators (MMW204; Narishige) mounted on the microscopic stage were used to place a bipolar tungsten stimulating electrode in Schaffer collateral. EPSP was recorded from the radium layer with a 5 MΩ resistance glass microelectrode that was filled with 2 m NaCl and connected to a preamplifier with a high-pass filter at 5 kHz. Signals were amplified by a differential AC amplifier (A-M Systems, model 1700). The EPSPs were digitized and saved using a pCLAMP system (Axon). Generally, the test stimulus was set at ∼50% of maximum stimulus intensity that evoked a saturated EPSP in each slice. First, input/output function was assessed by averaging the stimulus intensity from 0.1 to 1.5 mA. Second, paired-pulse facilitation (PPF) was measured by two stimuli with an interpulse interval (IPI) of 25–100 ms. Paired-pulse ratio (PPR) was calculated with the following formula: (EPSPS2/EPSPS1) × 100, where EPSPS1 and EPSPS2 represent EPSP slopes to the first and second stimulation, respectively. Third, LTP was induced by conditioning stimulations. After delivering conditioning stimulations, the EPSP slopes were recorded for a further period of 60 min. LTP was determined 55–60 min after delivering conditioning stimulations if the EPSP slopes were larger than 20% of baseline.
Patch-clamp recording.
Experiments were performed at 32–34°C, with the temperature maintained by an in-line heating device (Warner Instruments). AMPA-activated current (IAMPA) in hippocampal CA1 pyramidal cells was induced by the application of AMPA (Li et al., 2013b) using a rapid drug delivery system and recorded using an EPC-10 amplifier (HEKA Elektronik). The holding potential was −60 mV and the glass pipette resistance was 4–5 MΩ filled with an internal solution containing the following (in mm): CsCl 140, MgCl2 2, CaCl2 1, Tris-ATP 2, HEPES 10, and EGTA 10, pH 7.2. The hippocampal pyramidal neuron was perfused with oxygenated ACFS containing 10 μm d-APV, 1 μm strychnine, 10 μm bicuculline, and 0.1 μm TTX. In the dose–response curve, IAMPA induced by various doses (1–300 μm) of AMPA was normalized to IAMPA induced by 300 μm AMPA in the same neuron. The data were fitted to a logistic equation in which I = Imax/[1 + (EC50/C)n], with n being the Hill coefficient and EC50 being the concentration producing 50% maximal response.
NMDA-activated current (INMDA) was induced as described previously (Li et al., 2013a). The slices were perfused continually with the oxygenated calcium-free ACFS. Ten micromolar bicuculline, 10 μm NBQX, and 0.1 μm TTX were applied extracellularly. In the dose–response curve, INMDA induced by various doses (1–1000 μm) of NMDA was normalized to INMDA induced by 1000 μm NMDA in the same neuron.
Western blot analyses
Tissues obtained from the hippocampus were homogenized in 1% Nonidet P-40 lysis buffer containing 50 mm Tris, 150 mm NaCl, 0.02% NaN3, and complete protease inhibitors (Roche). The homogenates were centrifuged for 15 min at 12,000 r.p.m. (Thermo Scientific) and the supernatants were collected. Proteins (50 μg) were loaded in each lane for separation in SDS-PAGE and transferred to nitrocellulose membranes. Blotting membranes were incubated with blocking solution (5% nonfat dried milk) for 1 h at room temperature, washed three times, and then incubated with rabbit monoclonal antibodies of phosphorylation ERK1/2 or CREB (1:1000; Cell Signaling Technology), rabbit monoclonal antibodies of GluR1, GluR2, or PPARγ (1:1000; Cell Signaling Technology) at 4°C overnight. After being washed with TBST, the membranes were incubated for 1 h with HRP-labeled secondary antibodies and developed using the ECL detection kit (GE Healthcare). After visualization, the blots were stripped by incubation in a stripping buffer (Restore; Pierce) for 5 min, reblocked with 5% nonfat dried milk at room temperature for 60 min, and then incubated with rabbit monoclonal antibodies of ERK1/2 or CREB (1:1000; Cell Signaling Technology). Internal control was performed using a GAPDH antibody (1:5000; Bioworld Technology) or β-actin antibody (1:2000; Cell Signaling Technology). Western blot bands were scanned and analyzed with ImageJ.
RT-PCR
Real-time PCR was performed as described previously (Wang et al., 2009). Total RNA was isolated from the hippocampus using TRIzol reagent (Invitrogen) and reverse-transcribed into cDNA using a PrimeScript RT reagent kit (Takara) for quantitative PCR (ABI Step One Plus) in the presence of a fluorescent dye (SYBR Green I; Takara). The relative expression of genes was determined using the 2-ΔΔct method with normalization to GAPDH expression. Primer sets were used as listed in Table 1.
Table 1.
Primer sequences for RT-PCR
| Gene | Sense primer | Antisense primer |
|---|---|---|
| GluR1 | 5′-TTGCCTTAATCGAGTTCTGCTAC-3′ | 5′-GTATGGCTTCATTGATGGATTG-3′ |
| GluR2 | 5′-CCAGGGATGCAGACACCAAA-3′ | 5′-GGCTCATGAATGGCTTCGAGA-3′ |
| NR1 | 5′-TCACCTCCACCCTGGCCTCC-3′ | 5′-GCTGGCCCTCCTCCCTCTCA-3′ |
| NR2A | 5′-ATCATGGCTGACAAGGATCCGACA-3′ | 5′-TGACCAAGGAGAAGACATGCCAGT-3′ |
| NR2B | 5′-CTGCATGCGGAATACAGTC-3′ | 5′-TTGATGTAGCCTGGTTCCT-3′ |
| PSD 95 | 5′-GGTGACGACCCATCCATCTTTATC-3′ | 5′-CGGACATCCACTTCATTGACAAAC-3′ |
| TARPs (γ-8) | 5′-GGGGGCAGTGGCTCCTCAGA-3′ | 5′-CAGGCCTGCTGCCACGAACA-3′ |
| FAS | 5′-GGGTCTATGCCACGATTC-3′ | 5′-GTGTCCCATGTTGGATTTG-3′ |
| GAPDH | 5′-TGGGTGTGAACCACGAG-3′ | 5′-ACCACAGTCCATGCCATCAC-3′ |
Data analysis/statistics
The data were retrieved and processed with the software PulseFit (HEKA Elektronik) and Microcal Origin 6.1. The group data are expressed as the means ± SEM. The difference between the two groups was evaluated using Student's t test. In analyzing one-variable experiments with >2 groups, the significance of the difference was evaluated using ANOVA followed by Bonferroni's post hoc tests. Statistical analysis was performed using Stata 7 software (STATA). Differences at p < 0.05 were considered statistically significant.
Results
Neuronal seipin deficiency reduces hippocampal PPARγ level
Consistent with the previous reports (Magre et al., 2001; Ebihara et al., 2015), the seipin was highly expressed in hippocampal CA1 pyramidal cells of mice (Fig. 1A). Compared with WT mice or control mice, the levels of hippocampal PPARγ protein in seipin-sKO mice (F(1,28) = 32.179, p < 0.001; Fig. 1B) and seipin-nKO mice (F(1,28) = 20.094, p < 0.001) were significantly reduced and were not affected by the treatment with the PPARγ agonist rosi (5 mg/kg, p.o.) for 3 d (seipin-sKO: F(1,28) = 0.227, p = 0.637; seipin-nKO: F(1,28) = 0.400, p = 0.532). In contrast, the PPARγ protein level in seipin-aKO mice had no significant difference from a-control mice (F(1,28) = 0.165, p = 0.688). In addition, rosi treatment in WT mice, control mice, or a-control mice failed to alter the level of hippocampal PPARγ (p > 0.05, n = 8).
Figure 1.

Neuronal seipin deficiency reduces hippocampal PPARγ level. A, Representative images of seipin immunostaining in hippocampal CA1 region of control mice and seipin-nKO mice. str-R, Stratum radiatum; str-P, stratum pyramidale; str-O, stratum oriens. Arrows indicate seipin-immunopositive cells. Scale bars, 50 μm. B, Bars show the levels of hippocampal PPARγ in seipin-sKO mice and WT mice, seipin-nKO mice and control mice, or seipin-aKO mice and a-control mice treated with vehicle or rosi for 3 d. The densitometric values for Western blots of protein are normalized to the amounts of GAPDH and normalized again by control values. **p < 0.01 versus WT mice or control mice (two-way ANOVA).
Rosi improves seipin-deficiency-induced spatial cognitive deficits
Spatial memory was examined using place learning in the Morris water maze task. The mean latency to find visible platform on days 1–2 of training and the subsequent escape latency to reach hidden platform on days 3–7 of training were recorded in seipin-sKO mice and WT mice (Fig. 2A), seipin-nKO mice and control mice (Fig. 2C), or in seipin-aKO mice and a-control mice (Fig. 2E), respectively. To test the influence of reduced PPARγ by the seipin deficiency on spatial memory, rosi treatment (5 mg/kg, p.o.) was given starting 3 d before the behavioral examinations for 12 d. First, the latency of visible platform was not affected by the genotypes (seipin-sKO: F(1,44) = 0.016, P=0.961; seipin-nKO: F(1,44) = 0.225, p = 0.637; seipin-aKO: F(1,44) = 0.046, p = 0.831) or rosi treatment (seipin-sKO: F(1,44) = 0.010, p = 0.922; seipin-nKO: F(1,44) = 0.001, p = 0.978; seipin-aKO: F(1,44) = 0.058, p = 0.810). Repeated-measures ANOVA revealed that the escape latency of the hidden platform progressively decreased with training days in all groups (seipin-sKO: F(4,176) = 64.284, p < 0.001; seipin-nKO: F(4,176) = 84.587, p < 0.001; seipin-aKO: F(4,176) = 90.982, p < 0.001). There was a main effect of genotype for the escape latency in seipin-sKO mice (F(1,44) = 7.398, p = 0.009) and seipin-nKO mice (F(1,44) = 11.026, p = 0.002), but not in seipin-aKO mice (F(1,44) = 0.084, p = 0.774). Another main effect was rosi treatment (seipin-sKO: F(1,44) = 7.55, p = 0.009; seipin-nKO: F(1,44) = 10.021, p = 0.003). Compared with controls, seipin-sKO mice (p = 0.027) and seipin-nKO mice (p = 0.004) needed a longer time to reach the hidden platform on day 5–7 of training, which were improved by rosi treatment (seipin-sKO: p = 0.036; seipin-nKO: p = 0.009). The swimming speed during visible and hidden platform tests was not affected by the genotypes (seipin-sKO: F(1,44) = 0.080, p = 0.928; seipin-nKO: F(1,44) = 0.067, p = 0.798; seipin-aKO: F(1,44) = 0.042, p = 0.839) or rosi treatment (seipin-sKO: F(1,44) = 0.104, p = 0.749; seipin-nKO: F(1,44) = 0.029, p = 0.866; seipin-aKO: F(1,44) = 0.031, p = 0.861).
Figure 2.

Treatment with rosi improves neuronal seipin-deficiency-induced spatial cognitive deficits. In the Morris water maze test, latency (in seconds) to reach visible and hidden platform (A, C, E) and percentage of swimming time (B, D, F) in 4 quadrants (%) of platform (PQ), opposite (OQ), and right/left adjacent (R-AQ, L-AQ) in seipin-sKO mice and WT mice (A, B), seipin-nKO mice and control mice (C, D), or seipin-aKO mice and a-control mice (E, F). Tracings of typical swimming patterns on day 7 of hidden platform trainings and probe test are shown at the top. Black circles indicate the position of the platform. A, **p < 0.01 versus WT mice; #p < 0.05, ##p < 0.01 versus seipin-sKO mice (repeated-measure ANOVA). B, **p < 0.01 versus WT mice; ##p < 0.01 versus seipin-sKO mice (two-way ANOVA). C, **p < 0.01 versus control mice; #p < 0.05, ##p < 0.01 versus seipin-nKO mice (repeated-measure ANOVA). D, **p < 0.01 versus control mice; ##p < 0.01 versus seipin-nKO mice (two-way ANOVA). G, H, Alternation ratio (%) and total arm entries in Y-maze. **p < 0.01 versus WT mice or control mice; ##p < 0.01 versus seipin-sKO mice or seipin-nKO mice (two-way ANOVA).
A probe test was conducted 24 h after the hidden platform training. The swimming times spent in each quadrant (platform, opposite, and adjacent two quadrants) are shown in Figure 2, B, D, and F, respectively. There was a main effect of genotype for the swimming times of platform quadrant in seipin-sKO mice (F(1,44) = 11.926, p = 0.001) or seipin-nKO mice (F(1,44) = 8.799, p = 0.005), but not in seipin-aKO mice (F(1,44) = 1.310, p = 0.259). In addition, there was a main effect of rosi treatment (seipin-sKO: F(1,44) = 7.347, p = 0.01; seipin-nKO: F(1,44) = 9.794, p = 0.03). The seipin-sKO mice (p = 0.001) and seipin-nKO mice (p < 0.001) spent less swimming time in the platform quadrant than WT mice and control mice. rosi treatment in seipin-sKO mice (p = 0.002) and seipin-nKO mice (p < 0.001) increased swimming time in the platform quadrant.
Spatial working memory was evaluated by Y-maze test 48 h after the probe trial task. The alternation ratio and number of arm entries are shown in Figure 2, G and H. There was a main effect of genotype for alternation ratio in seipin-sKO mice (F(1,44) = 11.926, p = 0.001) or seipin-nKO mice (F(1,44) = 8.799, p = 0.005), but not in seipin-aKO mice (F(1,44) = 0.335, p = 0.566). In addition, rosi treatment was a main effect for the alternation ratio (seipin-sKO: F(1,44) = 7.565, p = 0.009; seipin-nKO: F(1,44) = 5.933, p = 0.018). The alternation ratio in seipin-sKO mice (p = 0.001) or seipin-nKO mice (p < 0.001) was reduced compared with WT mice or control mice, which was rescued by rosi treatment (seipin-sKO: p < 0.001; seipin-nKO: p < 0.001). In contrast, the number of arm entries was not altered by the genotypes (seipin-sKO: F(1,44) = 0.071, p = 0.792; seipin-nKO: F(1,44) = 0.234, p = 0.631; seipin-aKO: F(1,44) = 0.035, p = 0.853) or rosi treatment (seipin-sKO: F(1,44) = 0.196, p = 0.660; seipin-nKO: F(1,44) = 0.107, p = 0.745; seipin-aKO: F(1,44) = 0.096, p = 0.758).
Rosi rescues seipin-deficiency-impaired hippocampal synaptic function and LTP
To explore the mechanisms underlying the neuronal seipin-deficiency-impaired spatial memory, the basal property of Schaffer collateral–CA1 synaptic transmission was analyzed by plotting fractional changes in EPSP slopes against diverse stimulating intensities (0.2–1.5 mA). The repeated-measures ANOVA revealed a main effect of seipin-nKO (F(1,36) = 10.709, p = 0.002; Fig. 3A). In addition, there was a main effect of rosi treatment (F(1,36) = 4.406, p = 0.043). The input–output curve revealed that the EPSP slopes elicited by the test stimulus at 0.6–1.5 mA were less in seipin-nKO mice than those in control mice (p = 0.002), which could be corrected by rosi treatment for 3 d (p = 0.043). The PPF with an IPI ranging from 25 to 150 ms was measured to analyze the capability of presynaptic glutamate release. PPRs in seipin-nKO mice were not significantly different from those in control mice (F(1,36) = 0.113, p = 0.738; in Fig. 3B) and were not affected by rosi treatment (F(1,36) = 1.007, p = 0.322).
Figure 3.

Treatment with rosi rescues seipin-deficiency-impaired hippocampal synaptic function and LTP. A, Input–output curve at Schaffer collateral–CA1 synapses. EPSP slopes are plotted against stimulus intensity (0.2–1.5 mA). **p < 0.01 versus control mice; #p < 0.05, ##p < 0.01 versus seipin-nKO mice (repeated-measures ANOVA). Representative traces evoked by a testing stimulation (0.6 mA). B, PPR of EPSP slopes is plotted against various IPIs ranging from 25 to 150 ms in control mice and seipin-nKO mice treated with rosi or vehicle. Representative traces evoked with IPI of 50 ms. C, Induction of LTP by HFS (100 Hz, 100 pulses). Each point represents the group mean of EPSP slopes expressed as a percentage of baseline. Representative traces obtained at 1 min before HFS and 60 min after HFS. A solid arrow indicates when HFS was given. D, Induction of LTP in the presence of NMDAR antagonist MK801 in control mice and seipin-nKO mice. E, Induction of LTP in control mice and seipin-nKO mice treated with rosi for 3 d. F, Induction of LTP after the rosi application for 30 min in slices obtained from seipin-nKO mice.
The induction of Schaffer collateral–CA1 synaptic LTP was further examined by delivering a conditioning stimulation. As shown in Figure 3C, the high-frequency conditioning stimulation (100 pulses at 100 Hz) could induce ∼40% increase of EPSP slopes lasting over 60 min, indicative of “LTP”, in the slices obtained from control mice (n = 8), but not from seipin-nKO mice (n = 8). The frequency-dependent LTP induction in control mice was blocked by the NMDAR antagonist MK801 (n = 8; Fig. 3D). Interestingly, the same protocol was able to induce a stable LTP in seipin-nKO mice treated with rosi for 3 d (n = 8; Fig. 3E). In contrast, acute treatment with rosi (1 μm) for 30 min in the slices obtained from seipin-nKO mice failed to rescue the induction of LTP (n = 8; Fig. 3F).
Rosi recovers seipin deficiency-reduced AMPAR expression and activity
The next experiment was designed to examine the function and expression of AMPAR and NMDAR in hippocampal CA1 pyramidal cells using whole-cell patch-clamp recording, RT-PCR, and Western blot analysis. Typical recordings of IAMPA (100–300 μm) and INMDA (1–1000 μm) are shown in Figure 4, A and B. The densities of IAMPA in seipin-nKO mice were less than those in control mice (F(1,18) = 28.999, p < 0.001). However, there was no difference between the EC50 of the dose–response curve of IAMPA in seipin-nKO mice (30 ± 2.78 μm) and control mice (33 ± 3.52 μm; p = 0.539). In contrast, the densities of INMDA were not altered in seipin-nKO mice compared with control mice (F(1,18) = 0.807, p = 0.381). rosi treatment for 3 d could correct the reduction of IAMPA in seipin-nKO mice (vs control p = 0.370, n = 10; Fig. 4C), but failed to affect the INMDA (p = 0.727, n = 10). In addition, acute treatment with rosi (1 μm) for 30 min in the slices obtained from seipin-nKO mice had no effect on the IAMPA (p = 0.002, n = 10; Fig. 4D) or INMDA (p = 0.711, n = 10).
Figure 4.
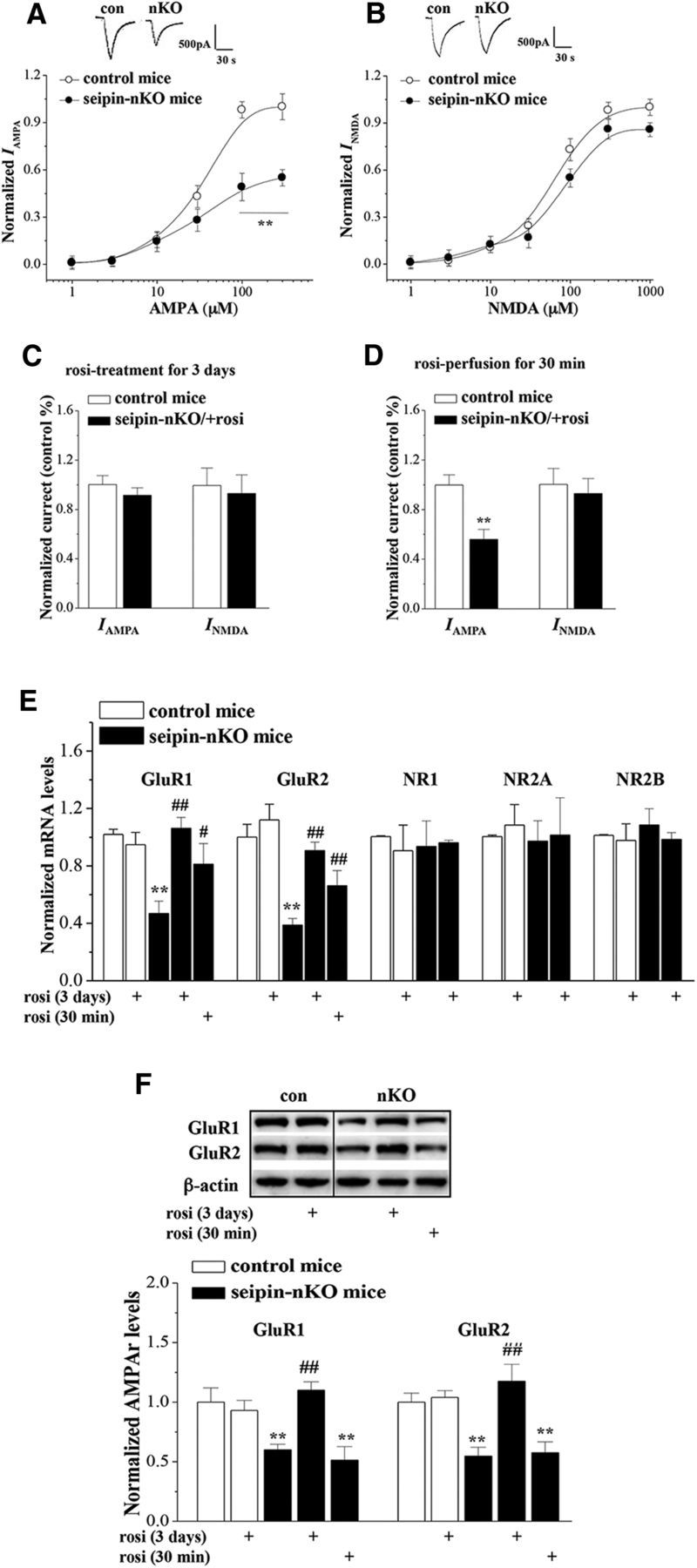
Treatment with rosi rescues seipin-deficiency-induced AMPAR downregulation. A, Dose–response curves of IAMPA in control mice and seipin-nKO mice. Each point represents the normalized current from 10 hippocampal CA1 pyramidal cells. Inset, Representative traces of IAMPA (100 μm AMPA). **p < 0.01 versus control mice (repeated-measures ANOVA). B, Dose–response curves of INMDA in control mice and seipin-nKO mice. Inset, Representative traces of INMDA (300 μm NMDA). C, D, Influence of rosi treatment for 3 d or rosi application for 30 min on IAMPA (100 μm AMPA) and INMDA (300 μm NMDA). Bars show the mean values of IAMPA and INMDA normalized by controls. E, Bars show the levels of GluR1, GluR2, NR1, NR2A, and NR2B mRNA in control mice and seipin-nKO mice or slices treated with rosi for 3 d or 30 min. The levels of mRNA are normalized by control values. **p < 0.01 versus control mice; #p < 0.05, ##p < 0.01 versus seipin-nKO mice (two-way ANOVA). F, Bars show the levels of GluR1 and GluR2 protein in control mice and seipin-nKO mice or slices treated with rosi for 3 d or 30 min. Densitometric values for Western blots of protein are normalized to the amounts of β-actin and then normalized again by control values. **p < 0.01 versus control mice; ##p < 0.01 versus seipin-nKO mice (two-way ANOVA).
Levels of hippocampal GluR1/2 mRNA and GluR1/2 protein are shown in Figure 4, E and F. There was a main effect of seipin-nKO for the GluR1 (F(1,35) = 8.014, p = 0.008) and GluR2 mRNAs (F(1,35) = 24.068, p < 0.001) and the GluR1 (F(1,35) = 4.474, p = 0.042) and GluR2 proteins (F(1,35) = 4.549, p = 0.040). Another main effect was rosi treatment for the GluR1 (F(1,35) = 10.886, p = 0.002) and GluR2 mRNAs (F(1,35) = 22.021, p < 0.001) or GluR1 (F(1,35) = 10.694, p = 0.002) and GluR2 proteins (F(1,35) = 18.011, p < 0.001). Compared with controls, the levels of GluR1/2 mRNA (p < 0.001) and GluR1/2 protein (p < 0.001) in seipin-nKO mice were significantly reduced, which could be corrected by rosi treatment for 3 d (GluR1/2 mRNA: p < 0.001; GluR1/2 protein: p < 0.001). In contrast, acute treatment with rosi (1 μm) for 30 min in the slices obtained from seipin-nKO mice could not increase the levels of GluR1 (p = 0.489, n = 8) and GluR2 proteins (p = 0.472, n = 8), although it partially corrected the reduction of GluR1 (p = 0.049, n = 8) and GluR2 mRNAs (p = 0.032, n = 8). In addition, the levels of NR1 (F(1,28) = 0.130, p = 0.721), NR2A (F(1,28) = 0.440, p = 0.512), and NR2B mRNAs (F(1,28) = 0.269, p = 0.608) in seipin-nKO mice did not differ significantly from control mice. The results indicate that the seipin deficiency suppresses AMPAR expression, leading to a reduction of IAMPA, which could be restored by activation of PPARγ.
Rosi rescues seipin-deficiency-reduced ERK-CREB phosphorylation
The levels of hippocampal phosphorylation of ERK2 (phosphor-ERK2) and cAMP response element binding protein (phosphor-CREB) are illustrated in Figure 5, A and B, respectively. There was a main effect of seipin-nKO for the phosphor-ERK2 (F(1,42) = 30.129, p < 0.001) and phosphor-CREB (F(1,56) = 15.752, p < 0.001). Another main effect was rosi treatment for the phosphor-ERK2 (F(1,42) = 22.367, p < 0.001) and phosphor-CREB (F(1,56) = 18.865, p < 0.001). The decline of phosphor-ERK2 and phosphor-CREB (p < 0.001) in seipin-nKO mice was corrected by rosi treatment for 3 d (p < 0.001). The MEK inhibitor U0126 could block the rosi-corrected phosphor-CREB in seipin-nKO mice (p < 0.001, n = 8) and attenuate the basal level of phosphor-CREB in control mice (p < 0.001, n = 8). In contrast, there was no significant main effect of seipin-nKO (F(1,28) = 0.180, p = 0.675; Fig. 5C) for the hippocampal mature BDNF. However, there was a main effect of rosi treatment (F(1,28) = 10.749, p = 0.003). The concentration of mature BDNF was increased by rosi treatment for 3 d in control mice (p = 0.025, n = 8) or seipin-nKO mice (p = 0.047, n = 8). However, inhibition of TrkB receptor by K252a in seipin-nKO mice did not affect the rosi-corrected phosphor-ERK2 (p = 0.578) and phosphor-CREB (p = 0.666).
Figure 5.

Treatment with rosi rescues seipin-deficiency-reduced ERK-CREB activities. A, B, Bars show the levels of hippocampal phosphor-ERK1/2 and phosphor-CREB in control mice and seipin-nKO mice treated with rosi, U0126, or K252a. **p < 0.01 versus control mice; ##p < 0.01 versus seipin-nKO mice; ++p < 0.01 versus rosi-treated seipin-nKO mice (two-way ANOVA). C, Bars show the level of hippocampal BDNF in control mice and seipin-nKO mice treated with rosi or vehicle. **p < 0.01 versus control mice (two-way ANOVA).
Seipin-deficiency-reduced ERK-CREB signaling impairs AMPAR expression and LTP
To determine whether the rosi-recovered ERK-CREB activity or increased BDNF is required for rosi-rescued AMPAR expression and LTP induction, seipin-nKO mice were treated with intracerebroventricular injection of U0126 or K252a at 30 min before rosi administration (Fig. 6A). The results showed that the administration of U0126 in seipin-nKO mice could block the rosi-increased GluR1 (p < 0.001, n = 8; Fig. 6B) and GluR2 mRNAs (p < 0.001, n = 8) or the rosi-restored IAMPA (p = 0.019, n = 8; Fig. 6C) and LTP (n = 8; Fig. 6D). In addition, the administration of U0126 in control mice attenuated the levels of GluR1 (p < 0.001, n = 8) and GluR2 mRNAs (p = 0.001, n = 8) and the IAMPA (p = 0.001, n = 8), which were insensitive to rosi treatment. The administration of K252a in seipin-nKO mice failed to affect the rosi-increased GluR1 (p = 0.476, n = 8) and GluR2 mRNAs (p = 0.330, n = 8) or the rosi-restored IAMPA (p = 0.932, n = 8).
Figure 6.

Seipin-deficiency-reduced ERK-CREB signaling impairs AMPAR and LTP. A, Time chart of experimental procedure. B, Effects of U0126 or K252a on rosi-restored AMPAR expression. Bars show the levels of GluR1 and GluR2 mRNA in control mice and seipin-nKO mice treated with rosi, U0126, or K252a. **p < 0.01 versus control mice; ##p < 0.01 versus seipin-nKO mice with rosi (two-way ANOVA). C, Effects of U0126 or K252a on rosi-restored IAMPA in seipin-nKO mice. Bars show IAMPA (100 μm AMPA) in hippocampal CA1 pyramidal cells in control mice and seipin-nKO mice treated with rosi, U0126, or K252a. **p < 0.01 versus control mice; ##p < 0.01 versus seipin-nKO mice with rosi (two-way ANOVA). D, Effects of U0126 on rosi-restored LTP induction in seipin-nKO mice.
Discussion
The present study provides in vivo evidence that neuronal seipin deficiency leads to spatial cognitive deficits thtat can be rescued by the activation of PPARγ. This conclusion is deduced mainly from the following observations. First, the seipin-sKO mice and seipin-nKO mice exhibited spatial cognitive deficits, but the seipin-aKO mice did not. Because seipin-sKO mice and seipin-aKO mice show insulin resistance and hypertriglyceridemia (Cui et al., 2011; Liu et al., 2014), the cognitive deterioration is unlikely to be secondary to the peripheral metabolic abnormalities. Second, the impairment of motor neuron by gain-of-function mutation in the N-glycosylation site of seipin (Ito and Suzuki, 2009) is not observed in CGL2 patients, seipin knock-out rats (Ebihara et al., 2015), or seipin-nKO mice (Zhou et al., 2014). No changes in the swim speed in Morris water maze or the number of arm entries in Y-maze were detected in seipin-sKO mice or seipin-nKO mice, indicating that seipin deficiency does not affect the motor ability. Third, seipin-sKO mice and seipin-nKO mice have been reported to display a depression-like phenotype (Zhou et al., 2014). The impairment of spatial working memory as assessed by Y-maze was observed in seipin-sKO and seipin-nKO mice and seipin knock-out rats (Ebihara et al., 2015). Fourth, the hippocampal NMDAR-dependent LTP, a cellular model for spatial learning and memory (Takeuchi et al., 2014), was impaired in seipin-nKO mice. Finally, Seipin was highly expressed in hippocampal CA1 pyramidal cells. Consistent with the report by Zhou et al. (2014), the levels of hippocampal PPARγ were remarkably reduced in seipin-sKO mice and seipin-nKO mice, but not in seipin-aKO mice. Importantly, rosi treatment could rescue the spatial memory and the LTP induction in seipin-sKO mice and seipin-nKO mice. Therefore, it is conceivable that the cognitive deterioration by the seipin deficiency might be attributed to the reduction of PPARγ.
No changes in the hippocampal size and thickness of pyramidal cell layer are found in seipin-nKO mice (Zhou et al., 2014). The seipin knock-out rats do not show a decrease in density of hippocampal neuronal cells (Ebihara et al., 2015). Neuronal PPARγ deficiency in mice is not sufficient to cause neuronal loss in hippocampus (Zhao et al., 2009). Notably, the efficiency of Schaffer collateral–CA1 synaptic transmission in seipin-nKO mice was significantly reduced. The mutation of seipin (N88S) has been found to reduce the synaptic release (Wei et al., 2014). The capability of presynaptic glutamate release in seipin-nKO mice was not affected, whereas the postsynaptic IAMPA was obviously reduced. The functional AMPAR expression is regulated by transcriptional and RNA editing mechanisms. The AMPAR is synthesized dendritically and inserted into synaptic membrane (Ju et al., 2004). The levels of hippocampal GluR1/GluR2 mRNAs and GluR1/GluR2 proteins were lower in seipin-nKO mice than those in control mice. The activation of NMDAR is known to inhibit the transcription of GluR2, leading to a decrease in dendritic and synaptic AMPAR numbers (Grooms et al., 2006). Seipin deficiency did not affect the postsynaptic INMDA. In addition, postsynaptic density-95 (PSD95) and transmembrane AMPAR regulatory proteins (TARPs) differentially interact with C-terminal end of GluR2 subunit to affect its expression (Bassani et al., 2013). In hippocampus of seipin-nKO mice, the levels of PSD95 and TARPγ-2 showed no significant difference from control mice (data not shown). In addition, the functional AMPAR number is regulated by their trafficking between endoplasmic reticulum to postsynaptic membrane via Golgi bodies. Wei et al. (2013) reported that seipin had no effects on the AMPAR trafficking. Therefore, the knock-down of the seipin gene in neuronal cells does not affect the inserting ability of AMPAR (Wei et al., 2014). Importantly, rosi treatment for 3 d in seipin-nKO mice restored AMPAR expression and recovered IAMPA and synaptic efficiency. In contrast, acute treatment with rosi for 30 min failed to alter the reduction of IAMPA andsynaptic efficiency in seipin-nKO mice but partially increased the GluR1/GluR2 mRNAs. The findings indicate that seipin deficiency suppresses the transcription of GluR1/GluR2, leading to the downregulation of AMPAR, which can be rescued by the activation of PPARγ.
The inhibition of PPARγ can significantly decrease ERK2 activation (Denner et al., 2012). rosi treatment can increase nuclear ERK2 activity, as noted by an increase in Thr202/Tyr204 phosphorylated ERK2. Hippocampal ERK2 and CREB phosphorylation in seipin-nKO mice was significantly reduced, which could be corrected by rosi treatment. The transcription factor CREB is known to regulate the expression of GluR1. ERK-CREB phosphorylation can upregulate AMPAR function (Qin et al., 2005). The decline of CREB phosphorylation is associated with a specific reduction in postsynaptic GluR1 (Borges and Dingledine, 2001). The MEK inhibitor U0126 could abolish the rosi-corrected reduction of CREB phosphorylation, AMPAR expression, and IAMPA in seipin-nKO mice. In addition, the inhibition of ERK by U0126 could attenuate the AMPAR expression and function in control mice. The results indicate that the seipin deficiency reduces the transcription of GluR1/GluR2 though reducing the ERK-CREB phosphorylation. Conversely, the activation of PPARγ is able to elevate BDNF concentration through enhancing the BDNF promoter (Wang et al., 2011; Kariharan et al., 2015). rosi treatment in seipin-nKO mice could increase the concentration of hippocampal mature BDNF, although seipin deficiency did not affect the BDNF level. BDNF has been reported to enhance the transcription activity of GluR1/GluR2 (Schratt et al., 2004; Caldeira et al., 2007), the transport of mRNAs along dendrites, and their translation at the synapse (Santos et al., 2010). However, the Trk family inhibitor K252a did not affect the rosi-corrected reduction of AMPAR expression and IAMPA in seipin-nKO mice. A recent study (Reimers et al., 2014) has reported the bidirectional effects of BDNF on AMPAR surface expression, depending on duration of exposure. The acute (30 min) treatment with BDNF can increase the cellular surface GluR1/GluR2, whereas the long-term (24 h) treatment produces an opposite effect. Therefore, the contradictory results may arise from the difference in the experimental timing. In addition, the AMPAR is assembled in the endoplasmic reticulum (ER) (Rubio and Wenthold, 1999). GluR2 colocalizes with the ER chaperone BiP (Greger et al., 2002). Ito et al. (2008) reported that the mutations (N88S/S90L) of the seipin gene could cause the formation of cytoplasmic inclusions and enhance ubiquitination, leading to ER stress. This idea is not supported because the levels of the ER stress makers BiP and CHOP in the hippocampus of seipin-nKO mice are not elevated (Zhou et al., 2014).
The AMPAR-containing GluR2 subunit is selectively permeable to Na+ and K+, accumulating evidence suggests that AMPAR plays important role during genesis of LTP in excitatory neurons (Santos et al., 2009; Schwenk et al., 2014). The LTP induction is impaired in GluR1 knock-out mice (Zamanillo et al., 1999), but not in double knock-out mice of both GluR2 and GluR3 (Meng et al., 2003). The reduction of AMPAR population on the hippocampal postsynaptic membrane may lead to poor development of LTP. Disruption of the interaction between GluR1 and some proteins (e.g., actin-binding protein 4.1N or the activator protein 2) can block the activity-dependent LTP (Shi et al., 2001). Several lines of evidence suggest that the ERK-CREB signaling pathway is critical for the NMDAR-dependent LTP induction and many forms of learning and memory (Sweatt, 2004). Chronic treatment with rosi can correct a downregulated ERK signal transduction pathway in Tg2576 mice to overcome their cognitive deficits (Denner et al., 2012). CREB, as a nuclear coactivator of PPARγ (Bugge et al., 2009; Inoue et al., 2012), integrates the convergence of the PPARγ and ERK pathways to rescue learning and memory deficits in AD mouse models (Caccamo et al., 2010). Therefore, the impairment of NMDAR-dependent LTP in seipin-nKO mice is due to the downregulation of AMPAR and ERK-CREB signaling.
In conclusion, the present study provides evidence that the activation of PPARγ by rosi treatment could rescue the hippocampal LTP and spatial cognitive deficits in seipin-sKO mice and seipin-nKO mice. rosi treatment has been reported to enhance cognition in AD mice (Denner et al., 2012). Moreover, rosi treatment in type 2 diabetic mice significantly improves spatial memory and LTP induction without improvement of peripheral insulin sensitivity (Kariharan et al., 2015). The findings indicate that this nuclear receptor is a potential therapeutic target for the cognitive deficits. This idea is strengthened by the fact that PPARγ is reduced in AD brain and certain polymorphisms in the PPARγ gene are associated with increased risk for the cognitive and intellectual impairment (Kitamura et al., 1999; Scacchi et al., 2007). Similarly, the decline of hippocampal PPARγ level was observed in seipin-sKO mice or seipin-nKO mice. rosi treatment in seipin-sKO mice or seipin-nKO mice can alleviate their anxiety- and depression-like phenotypes (Zhou et al., 2014). Furthermore, rosi treatment can protect the impairment of adult neurogenesis in hippocampal dentate gyrus of seipin-nKO mice (Li et al., 2015). Although much more work needs to be conducted, our results in this study indicate the therapeutic possibility of the PPARγ agonist on mental retardation in CGL2 patients.
Footnotes
This work was supported by National 973 Basic Research Program of China (Grant 2014CB943303 to Ling Chen) and the National Science Foundation of China (Grants 31171440, 81361120247, and 81471157 to Ling Chen).
The authors declare no competing financial interests.
References
- Agarwal AK, Garg A. Genetic basis of lipodystrophies and management of metabolic complications. Annu Rev Med. 2006;57:297–311. doi: 10.1146/annurev.med.57.022605.114424. [DOI] [PubMed] [Google Scholar]
- Agarwal AK, Arioglu E, De Almeida S, Akkoc N, Taylor SI, Bowcock AM, Barnes RI, Garg A. AGPAT2 is mutated in congenital generalized lipodystrophy linked to chromosome 9q34. Nat Genet. 2002;31:21–23. doi: 10.1038/ng880. [DOI] [PubMed] [Google Scholar]
- Bassani S, Folci A, Zapata J, Passafaro M. AMPAR trafficking in synapse maturation and plasticity. Cell Mol Life Sci. 2013;70:4411–4430. doi: 10.1007/s00018-013-1309-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borges K, Dingledine R. Functional organization of the GluR1 glutamate receptor promoter. J Biol Chem. 2001;276:25929–25938. doi: 10.1074/jbc.M009105200. [DOI] [PubMed] [Google Scholar]
- Bugge A, Grøntved L, Aagaard MM, Borup R, Mandrup S. The PPARgamma2 A/B-domain plays a gene-specific role in transactivation and cofactor recruitment. Mol Endocrinol. 2009;23:794–808. doi: 10.1210/me.2008-0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caccamo A, Maldonado MA, Bokov AF, Majumder S, Oddo S. CBP gene transfer increases BDNF levels and ameliorates learning and memory deficits in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2010;107:22687–22692. doi: 10.1073/pnas.1012851108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldeira MV, Melo CV, Pereira DB, Carvalho R, Correia SS, Backos DS, Carvalho AL, Esteban JA, Duarte CB. Brain-derived neurotrophic factor regulates the expression and synaptic delivery of alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor subunits in hippocampal neurons. J Biol Chem. 2007;282:12619–12628. doi: 10.1074/jbc.M700607200. [DOI] [PubMed] [Google Scholar]
- Camarena V, Kobayashi M, Kim JY, Roehm P, Perez R, Gardner J, Wilson AC, Mohr I, Chao MV. Nature and duration of growth factor signaling through receptor tyrosine kinases regulates HSV-1 latency in neurons. Cell Host Microbe. 2010;8:320–330. doi: 10.1016/j.chom.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Yechoor VK, Chang BH, Li MV, March KL, Chan L. The human lipodystrophy gene product Berardinelli-Seip congenital lipodystrophy 2/seipin plays a key role in adipocyte differentiation. Endocrinology. 2009;150:4552–4561. doi: 10.1210/en.2009-0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Chang B, Saha P, Hartig SM, Li L, Reddy VT, Yang Y, Yechoor V, Mancini MA, Chan L. Berardinelli-seip congenital lipodystrophy 2/seipin is a cell-autonomous regulator of lipolysis essential for adipocyte differentiation. Mol Cell Biol. 2012;32:1099–1111. doi: 10.1128/MCB.06465-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui X, Wang Y, Tang Y, Liu Y, Zhao L, Deng J, Xu G, Peng X, Ju S, Liu G, Yang H. Seipin ablation in mice results in severe generalized lipodystrophy. Hum Mol Genet. 2011;20:3022–3030. doi: 10.1093/hmg/ddr205. [DOI] [PubMed] [Google Scholar]
- Denner LA, Rodriguez-Rivera J, Haidacher SJ, Jahrling JB, Carmical JR, Hernandez CM, Zhao Y, Sadygov RG, Starkey JM, Spratt H, Luxon BA, Wood TG, Dineley KT. Cognitive enhancement with rosiglitazone links the hippocampal PPARgamma and ERK MAPK signaling pathways. J Neurosci. 2012;32:16725–16735a. doi: 10.1523/JNEUROSCI.2153-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebihara C, Ebihara K, Aizawa-Abe M, Mashimo T, Tomita T, Zhao M, Gumbilai V, Kusakabe T, Yamamoto Y, Aotani D, Yamamoto-Kataoka S, Sakai T, Hosoda K, Serikawa T, Nakao K. Seipin is necessary for normal brain development and spermatogenesis in addition to adipogenesis. Hum Mol Genet. 2015;24:4238–4249. doi: 10.1093/hmg/ddv156. [DOI] [PubMed] [Google Scholar]
- Greger IH, Khatri L, Ziff EB. RNA editing at arg607 controls AMPA receptor exit from the endoplasmic reticulum. Neuron. 2002;34:759–772. doi: 10.1016/S0896-6273(02)00693-1. [DOI] [PubMed] [Google Scholar]
- Grooms SY, Noh KM, Regis R, Bassell GJ, Bryan MK, Carroll RC, Zukin RS. Activity bidirectionally regulates AMPA receptor mRNA abundance in dendrites of hippocampal neurons. J Neurosci. 2006;26:8339–8351. doi: 10.1523/JNEUROSCI.0472-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J, Qiu W, Soh SL, Wei S, Radda GK, Ong WY, Pang ZP, Han W. Motor neuron degeneration in a mouse model of seipinopathy. Cell Death Dis. 2013;4:e535. doi: 10.1038/cddis.2013.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Klockgether T, Feinstein DL. Peroxisome proliferator-activated receptor-gamma ligands reduce neuronal inducible nitric oxide synthase expression and cell death in vivo. J Neurosci. 2000;20:6862–6867. doi: 10.1523/JNEUROSCI.20-18-06862.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue M, Tanabe H, Matsumoto A, Takagi M, Umegaki K, Amagaya S, Takahashi J. Astaxanthin functions differently as a selective peroxisome proliferator-activated receptor gamma modulator in adipocytes and macrophages. Biochem Pharmacol. 2012;84:692–700. doi: 10.1016/j.bcp.2012.05.021. [DOI] [PubMed] [Google Scholar]
- Ito D, Suzuki N. Seipinopathy: a novel endoplasmic reticulum stress-associated disease. Brain. 2009;132:8–15. doi: 10.1093/brain/awn216. [DOI] [PubMed] [Google Scholar]
- Ito D, Fujisawa T, Iida H, Suzuki N. Characterization of seipin/BSCL2, a protein associated with spastic paraplegia 17. Neurobiol Dis. 2008;31:266–277. doi: 10.1016/j.nbd.2008.05.004. [DOI] [PubMed] [Google Scholar]
- Jiang M, Gao M, Wu C, He H, Guo X, Zhou Z, Yang H, Xiao X, Liu G, Sha J. Lack of testicular seipin causes teratozoospermia syndrome in men. Proc Natl Acad Sci U S A. 2014;111:7054–7059. doi: 10.1073/pnas.1324025111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju W, Morishita W, Tsui J, Gaietta G, Deerinck TJ, Adams SR, Garner CC, Tsien RY, Ellisman MH, Malenka RC. Activity-dependent regulation of dendritic synthesis and trafficking of AMPA receptors. Nat Neurosci. 2004;7:244–253. doi: 10.1038/nn1189. [DOI] [PubMed] [Google Scholar]
- Kariharan T, Nanayakkara G, Parameshwaran K, Bagasrawala I, Ahuja M, Abdel-Rahman E, Amin AT, Dhanasekaran M, Suppiramaniam V, Amin RH. Central activation of PPAR-gamma ameliorates diabetes induced cognitive dysfunction and improves BDNF expression. Neurobiol Aging. 2015;36:1451–1461. doi: 10.1016/j.neurobiolaging.2014.09.028. [DOI] [PubMed] [Google Scholar]
- Kitamura Y, Shimohama S, Koike H, Kakimura Ji, Matsuoka Y, Nomura Y, Gebicke-Haerter PJ, Taniguchi T. Increased expression of cyclooxygenases and peroxisome proliferator-activated receptor-gamma in Alzheimer's disease brains. Biochem Biophys Res Commun. 1999;254:582–586. doi: 10.1006/bbrc.1998.9981. [DOI] [PubMed] [Google Scholar]
- Li G, Zhou L, Zhu Y, Wang C, Sha S, Xian X, Ji Y, Liu G, Chen L. Seipin knock-out in mice impairs stem cell proliferation and progenitor cell differentiation in the adult hippocampal dentate gyrus via reduction of PPARγ. Dis Model Mech. 2015;8:1615–1624. doi: 10.1242/dmm.021550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Qu W, Zhou L, Lu Z, Jie P, Chen L, Chen L. Activation of transient receptor potential Vanilloid 4 increases NMDA-activated current in hippocampal pyramidal neurons. Front Cell Neurosci. 2013a;7:17. doi: 10.3389/fncel.2013.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Yin J, Jie PH, Lu ZH, Zhou LB, Chen L, Chen L. Transient receptor potential vanilloid 4 mediates hypotonicity-induced enhancement of synaptic transmission in hippocampal slices. CNS Neurosci Ther. 2013b;19:854–862. doi: 10.1111/cns.12143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Jiang Q, Wang X, Zhang Y, Lin RC, Lam SM, Shui G, Zhou L, Li P, Wang Y, Cui X, Gao M, Zhang L, Lv Y, Xu G, Liu G, Zhao D, Yang H. Adipose-specific knockout of SEIPIN/BSCL2 results in progressive lipodystrophy. Diabetes. 2014;63:2320–2331. doi: 10.2337/db13-0729. [DOI] [PubMed] [Google Scholar]
- Magré J, Delépine M, Khallouf E, Gedde-Dahl T, Jr, Van Maldergem L, Sobel E, Papp J, Meier M, Mégarbané A, Bachy A, Verloes A, d'Abronzo FH, Seemanova E, Assan R, Baudic N, Bourut C, Czernichow P, Huet F, Grigorescu F, de Kerdanet M, et al. BSCL Working Group Identification of the gene altered in Berardinelli-Seip congenital lipodystrophy on chromosome 11q13. Nat Genet. 2001;28:365–370. doi: 10.1038/ng585. [DOI] [PubMed] [Google Scholar]
- Maurice T, Lockhart BP, Privat A. Amnesia induced in mice by centrally administered beta-amyloid peptides involves cholinergic dysfunction. Brain Res. 1996;706:181–193. doi: 10.1016/0006-8993(95)01032-7. [DOI] [PubMed] [Google Scholar]
- Meng Y, Zhang Y, Jia Z. Synaptic transmission and plasticity in the absence of AMPA glutamate receptor GluR2 and GluR3. Neuron. 2003;39:163–176. doi: 10.1016/S0896-6273(03)00368-4. [DOI] [PubMed] [Google Scholar]
- Moreno S, Farioli-Vecchioli S, Cerù MP. Immunolocalization of peroxisome proliferator-activated receptors and retinoid X receptors in the adult rat CNS. Neuroscience. 2004;123:131–145. doi: 10.1016/j.neuroscience.2003.08.064. [DOI] [PubMed] [Google Scholar]
- Murakami N, Hayashi YK, Oto Y, Shiraishi M, Itabashi H, Kudo K, Nishino I, Nonaka I, Nagai T. Congenital generalized lipodystrophy type 4 with muscular dystrophy: clinical and pathological manifestations in early childhood. Neuromuscul Disord. 2013;23:441–444. doi: 10.1016/j.nmd.2013.02.005. [DOI] [PubMed] [Google Scholar]
- Prieur X, Dollet L, Takahashi M, Nemani M, Pillot B, Le May C, Mounier C, Takigawa-Imamura H, Zelenika D, Matsuda F, Fève B, Capeau J, Lathrop M, Costet P, Cariou B, Magré J. Thiazolidinediones partially reverse the metabolic disturbances observed in Bscl2/seipin-deficient mice. Diabetologia. 2013;56:1813–1825. doi: 10.1007/s00125-013-2926-9. [DOI] [PubMed] [Google Scholar]
- Qin Y, Zhu Y, Baumgart JP, Stornetta RL, Seidenman K, Mack V, van Aelst L, Zhu JJ. State-dependent Ras signaling and AMPA receptor trafficking. Gene Dev. 2005;19:2000–2015. doi: 10.1101/gad.342205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajab A, Khaburi M, Spranger S, Kunze J, Spranger J. Congenital generalized lipodystrophy, mental retardation, deafness, short stature, and slender bones: a newly recognized syndrome? Am J Med Genet A. 2003;121A:271–276. doi: 10.1002/ajmg.a.20245. [DOI] [PubMed] [Google Scholar]
- Reimers JM, Loweth JA, Wolf ME. BDNF contributes to both rapid and homeostatic alterations in AMPA receptor surface expression in nucleus accumbens medium spiny neurons. Eur J Neurosci. 2014;39:1159–1169. doi: 10.1111/ejn.12422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio ME, Wenthold RJ. Calnexin and the immunoglobulin binding protein (BiP) coimmunoprecipitate with AMPA receptors. J Neurochem. 1999;73:942–948. doi: 10.1046/j.1471-4159.1999.0730942.x. [DOI] [PubMed] [Google Scholar]
- Salehi-Sadaghiani M, Javadi-Paydar M, Gharedaghi MH, Zandieh A, Heydarpour P, Yousefzadeh-Fard Y, Dehpour AR. NMDA receptor involvement in antidepressant-like effect of pioglitazone in the forced swimming test in mice. Psychopharmacology (Berl) 2012;223:345–355. doi: 10.1007/s00213-012-2722-0. [DOI] [PubMed] [Google Scholar]
- Santos AR, Comprido D, Duarte CB. Regulation of local translation at the synapse by BDNF. Prog Neurobiol. 2010;92:505–516. doi: 10.1016/j.pneurobio.2010.08.004. [DOI] [PubMed] [Google Scholar]
- Santos SD, Carvalho AL, Caldeira MV, Duarte CB. Regulation of AMPA receptors and synaptic plasticity. Neuroscience. 2009;158:105–125. doi: 10.1016/j.neuroscience.2008.02.037. [DOI] [PubMed] [Google Scholar]
- Scacchi R, Pinto A, Gambina G, Rosano A, Corbo RM. The peroxisome proliferator-activated receptor gamma (PPAR-gamma2) Pro12Ala polymorphism is associated with higher risk for Alzheimer's disease in octogenarians. Brain Res. 2007;1139:1–5. doi: 10.1016/j.brainres.2006.12.078. [DOI] [PubMed] [Google Scholar]
- Schratt GM, Nigh EA, Chen WG, Hu L, Greenberg ME. BDNF regulates the translation of a select group of mRNAs by a mammalian target of rapamycin-phosphatidylinositol 3-kinase-dependent pathway during neuronal development. J Neurosci. 2004;24:7366–7377. doi: 10.1523/JNEUROSCI.1739-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster J, Khan TN, Tariq M, Shaiq PA, Mäbert K, Baig SM, Klar J. Exome sequencing circumvents missing clinical data and identifies a BSCL2 mutation in congenital lipodystrophy. BMC Med Genet. 2014;15:71. doi: 10.1186/1471-2350-15-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwenk J, Baehrens D, Haupt A, Bildl W, Boudkkazi S, Roeper J, Fakler B, Schulte U. Regional diversity and developmental dynamics of the AMPA-receptor proteome in the mammalian brain. Neuron. 2014;84:41–54. doi: 10.1016/j.neuron.2014.08.044. [DOI] [PubMed] [Google Scholar]
- Shi S, Hayashi Y, Esteban JA, Malinow R. Subunit-specific rules governing AMPA receptor trafficking to synapses in hippocampal pyramidal neurons. Cell. 2001;105:331–343. doi: 10.1016/S0092-8674(01)00321-X. [DOI] [PubMed] [Google Scholar]
- Sweatt JD. Mitogen-activated protein kinases in synaptic plasticity and memory. Curr Opin Neurobiol. 2004;14:311–317. doi: 10.1016/j.conb.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Szymanski KM, Binns D, Bartz R, Grishin NV, Li WP, Agarwal AK, Garg A, Anderson RG, Goodman JM. The lipodystrophy protein seipin is found at endoplasmic reticulum lipid droplet junctions and is important for droplet morphology. Proc Natl Acad Sci U S A. 2007;104:20890–20895. doi: 10.1073/pnas.0704154104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi T, Duszkiewicz AJ, Morris RG. The synaptic plasticity and memory hypothesis: encoding, storage and persistence. Philos Trans R Soc Lond B Biol Sci. 2014;369:20130288. doi: 10.1098/rstb.2013.0288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai YW, Yang YR, Wang PS, Wang RY. Intermittent hypoxia after transient focal ischemia induces hippocampal neurogenesis and c-Fos expression and reverses spatial memory deficits in rats. PLoS One. 2011;6:e24001. doi: 10.1371/journal.pone.0024001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Maldergem L, Magré J, Khallouf TE, Gedde-Dahl T, Jr, Delépine M, Trygstad O, Seemanova E, Stephenson T, Albott CS, Bonnici F, Panz VR, Medina JL, Bogalho P, Huet F, Savasta S, Verloes A, Robert JJ, Loret H, De Kerdanet M, Tubiana-Rufi N, et al. Genotype-phenotype relationships in Berardinelli-Seip congenital lipodystrophy. J Med Genet. 2002;39:722–733. doi: 10.1136/jmg.39.10.722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Zhang L, Chen ZB, Wu JY, Zhang X, Xu Y. Icariin enhances neuronal survival after oxygen and glucose deprivation by increasing SIRT1. Eur J Pharmacol. 2009;609:40–44. doi: 10.1016/j.ejphar.2009.03.033. [DOI] [PubMed] [Google Scholar]
- Wang SH, Guo YJ, Yuan Y, Li L, Li FF, Ye KP, Huang Y. PPARgamma-mediated advanced glycation end products regulate neural stem cell proliferation but not neural differentiation through the BDNF-CREB pathway. Toxicol Lett. 2011;206:339–346. doi: 10.1016/j.toxlet.2011.07.026. [DOI] [PubMed] [Google Scholar]
- Wei S, Soh SL, Qiu W, Yang W, Seah CJ, Guo J, Ong WY, Pang ZP, Han W. Seipin regulates excitatory synaptic transmission in cortical neurons. J Neurochem. 2013;124:478–489. doi: 10.1111/jnc.12099. [DOI] [PubMed] [Google Scholar]
- Wei S, Soh SL, Xia J, Ong WY, Pang ZP, Han W. Motor neuropathy-associated mutation impairs Seipin functions in neurotransmission. J Neurochem. 2014;129:328–338. doi: 10.1111/jnc.12638. [DOI] [PubMed] [Google Scholar]
- Yang R, Chen L, Wang H, Xu B, Tomimoto H, Chen L. Anti-amnesic effect of neurosteroid PREGS in Abeta25–35-injected mice through sigma1 receptor- and alpha7nAChR-mediated neuroprotection. Neuropharmacology. 2012;63:1042–1050. doi: 10.1016/j.neuropharm.2012.07.035. [DOI] [PubMed] [Google Scholar]
- Zamanillo D, Sprengel R, Hvalby O, Jensen V, Burnashev N, Rozov A, Kaiser KM, Köster HJ, Borchardt T, Worley P, Lübke J, Frotscher M, Kelly PH, Sommer B, Andersen P, Seeburg PH, Sakmann B. Importance of AMPA receptors for hippocampal synaptic plasticity but not for spatial learning. Science. 1999;284:1805–1811. doi: 10.1126/science.284.5421.1805. [DOI] [PubMed] [Google Scholar]
- Zhao X, Strong R, Zhang J, Sun G, Tsien JZ, Cui Z, Grotta JC, Aronowski J. Neuronal PPARgamma deficiency increases susceptibility to brain damage after cerebral ischemia. J Neurosci. 2009;29:6186–6195. doi: 10.1523/JNEUROSCI.5857-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Yin J, Wang C, Liao J, Liu G, Chen L. Lack of seipin in neurons results in anxiety- and depression-like behaviors via down regulation of PPARgamma. Hum Mol Genet. 2014;23:4094–4102. doi: 10.1093/hmg/ddu126. [DOI] [PubMed] [Google Scholar]