

Abstract
Compared to traditional collision induced dissociation methods, electron capture dissociation (ECD) provides more comprehensive characterization of large peptides and proteins as well as preserves labile post-translational modifications. However, ECD experiments are generally restricted to the high magnetic fields of FTICR-MS that enable the reaction of large polycations and electrons. Here, we demonstrate the use of an electromagnetostatic ECD cell to perform ECD and hybrid ECD methods utilizing 193 nm photons (ECuvPD) or collisional activation (EChcD) in a benchtop quadrupole–Orbitrap mass spectrometer. The electromagnetostatic ECD cell was designed to replace the transfer octapole between the quadrupole and C-trap. This implementation enabled facile installation of the ECD cell, and ions could be independently subjected to ECD, UVPD, HCD, or any combination. Initial benchmarking and characterization of fragmentation propensities for ECD, ECuvPD, and EChcD were performed using ubiquitin (8.6 kDa). ECD yielded extensive sequence coverage for low charge states of ubiquitin as well as for the larger protein carbonic anhydrase II (29 kDa), indicating pseudo-activated ion conditions. Additionally, relatively high numbers of d- and w-ions enable differentiation of isobaric isoleucine and leucine residues and suggest a distribution of electron energies yield hot-ECD type fragmentation. We report the most comprehensive characterization to date for model proteins up to 29 kDa and a monoclonal antibody at the subunit level. ECD, ECuvPD, and EChcD yielded 93, 95, and 91% sequence coverage, respectively, for carbonic anhydrase II (29 kDa), and targeted online analyses of monoclonal antibody subunits yielded 86% overall antibody sequence coverage.
Graphical Abstract
The development of electron capture dissociation (ECD) by Zubarev et al.1 marked a significant advancement in tandem mass spectrometry (MS/MS) for the characterization of polypeptides and labile post-translational modifications (PTMs).2,3 ECD was performed via the reaction of low-energy electrons generated by a heated filament with multiply charged polypeptide cations within the high magnetic field of a Fourier transform ion cyclotron resonance mass spectrometer (FTICR-MS). The neutralizing reaction of electrons with protonated carbonyls of the polypeptide backbone yielded highly specific cleavage of the N–Cα bond to produce c- and z•-ions. To a lesser extent, electron capture at protonated amide nitrogen also yielded a•- and y-ions.4 ECD alleviated many of the amino acid specific cleavages that plague traditional collision induced dissociation (CID),5,6 enabling more complete characterization of peptides and localization of labile PTMs and spurring great interest in the further development of the top–down proteomics approach.
The development of activated ion ECD (AI-ECD) greatly extended the mass range of proteins amenable to characterization by ECD.7,8 Initial success for ECD was limited to relatively small proteins because of intramolecular noncovalent bonds that maintain secondary and tertiary protein structure. These noncovalent bonds prevent separation of the ECD product ions and yield charge reduced, radical precursor ions. This phenomenon is particularly troublesome for doubly charged peptides as well as large and or low charge density protein ions. Activated ion methods utilize controlled vibrational excitation via ion-neutral collisions, absorption of infrared photons (e.g., 10.6 μm, 0.1 eV per photon), and other methods that increase the internal energy of the polypeptide prior to or concomitant to ECD in order to disrupt intramolecular noncovalent bonds.4,7–14 Similarly, activated ion methods have been developed for electron transfer dissociation (ETD), the RF trapping device analog of ECD.15–20 It is important to note that the goal of supplemental activation in AI-ECD and AI-ETD is to simply disrupt noncovalent interactions and unfold higher-order polypeptide structure to enhance detection of ECD/ETD product ions (i.e., c- and z•-ions).
However, hybrid MS/MS methods have been developed, which incorporate multiple fragmentation modes that yield enhanced sequence coverage and more precise localization of PTMs. Generally, hybrid MS/MS methods combine complementary fragmentation methods with the aim to generate distinct product ions via both fragmentation modes. One recently developed method combined ETD and higher-energy collision dissociation (HCD) is termed EThcD.21 In an EThcD experiment, precursor ions are first subject to ETD, and then, the remaining unreacted precursor ions and all product ions are subject to HCD, generating a mixture of b/y-and c/z•-product-ions. EThcD substantially increased peptide sequence coverage and the confidence for peptide identifications for large-scale fractionated bottom–up proteomics experiments,21 enriched phosphopeptides22 and glycopeptides,23 and cross-linked peptides.24
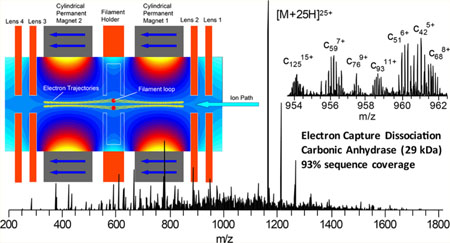
Developments to enable ECD outside the confines of high magnetic fields used in FTICR-MS over the past 15 years have yielded a number of novel implementations of ECD. Successful implementations of ECD have been demonstrated in a linear ion trap,25,26 a three-dimensional (3D) ion trap,27 a 3D digital ion trap,28 and a branched radio frequency ion trap.29 The development of an electromagnetostatic ECD cell has enabled the possibility of performing efficient ECD in a wide variety of instrument platforms using two small permanent magnets and several electrostatic lenses.30–36 The electromagnetostatic ECD cell technology has been demonstrated in triplequadrupole, Q-TOF, and recently Orbitrap mass spectrometers, in which the precursor ions were subject to a single or two passes through the ECD cell.36 Electrons were generated via a heated filament and confined to the analyte ion transmission path along the longitudinal axis of the cell through the careful matching of static electric and magnetic fields. The electrons form a cylindrical cloud that is confined radially by the magnetic field and axially by the electrostatic lenses at each end of the ECD cell (Figure 1). A small current of higher-energy electrons may leave the filament along other paths with higher energy. Precursor ions capture electrons in the amount of time required to transit the length of the ECD cell, approximately 10 μs. This experimental setup yields ECD events that are 2–3 orders of magnitude shorter than typically utilized for ECD and ETD performed in ion trapping devices because of greater overlap between the electron and analyte ion trajectories.
Figure 1.

Image of the electromagnetostatic ECD cell (circled in yellow) installed in a Q Exactive HF mass spectrometer with the C-trap removed (top panel). Schematic (bottom panel) of the electromagnetostatic ECD cell with simulated magnetic vector potential and electron trajectories in yellow along the axis of the cell. Calculations were performed with LORENTZ-EM (Integrated Engineering Software, Winnipeg, Manitoba, Canada).
Previous experiments with the electromagnetostatic ECD cell in a hybrid quadrupole–Orbitrap mass spectrometer were performed with the ECD cell between the C-trap and HCD cell.36 This experimental setup yielded two passes through the ECD cell per MS/MS acquisition and enabled efficient fragmentation of proteins and selective disulfide bond cleavage. Here, we present the development of hybrid ECD activation methods using an alternative implementation of the electromagnetostatic ECD cell in a benchtop quadrupole–Orbitrap mass spectrometer. In this work, the ECD cell replaced the small octapole between the mass selective quadrupole and C-trap. This implementation greatly simplified installation and tuning of the ECD cell and does not require modification of the instrument vacuum chamber to accommodate the cell. Refinements to the ECD cell design yielded comparable ECD efficiency for a single pass as was previously achieved for two pass ECD reactions. These developments enabled the most comprehensive characterization of model proteins up to 29 kDa and a monoclonal antibody at the subunit level reported to date.
EXPERIMENTAL SECTION
Materials and Sample Preparation.
Model proteins (ubiquitin and carbonic anhydrase II) and SILu Lite SigmaMAb (IgG1 lambda, Cat. No. MSQC4) were purchased from Sigma-Aldrich (St. Louis, MO) and used without further purification. IdeS was purchased from Promega Corporation (Madison, WI). All other chemicals were obtained from Fisher Scientific (Fairlawn, NJ). Working solutions of model proteins for direct infusion were made at 1 μM in 50:50 water/acetonitrile with 0.1% formic acid.
SigmaMAb was digested with IdeS using the manufacturer’s protocol. Briefly, SigmaMAb was reconstituted in 50 mM sodium phosphate and 150 mM NaCl (pH 6.6) at a concentration of 1 μg/μL. IdeS (50 units) was added to 50 μg of SigmaMAb and incubated for 30 min at 37 °C. F(ab′)2 and FC fragments were reduced and denatured by addition of DTT and guanidine HCl to a final concentration of 30 mM and 6 M, respectively, and incubated for 45 min at 60 °C. The reaction was quenched with 10 μL of 99.5% LC/MS grade formic acid. The reduced LC, Fd, and FC/2 fragments were diluted to 0.02 μg/μL using water with 0.2% formic acid prior to LC/MS analysis.
Mass Spectrometry.
All experiments were performed using a Q Exactive HF Orbitrap mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) modified to enable UVPD in the HCD cell in a manner similar to that previously described in Fort et al.37 Briefly, the instrument firmware was modified to enable trapping of ions in the HCD cell during HCD-MS/MS acquisitions, and a TTL signal was used to gate a pulse generator (Berkeley Nucleonics, San Rafael, CA) that produced a 30 μs pulse to trigger each 5 ns laser pulse. Photons (193 nm) were generated by an ArF excimer laser (Excistar XS 500, Coherent Inc., Santa Clara, CA). Experiments utilizing model proteins were performed by direct infusion at a flow rate of 0.5 μL/min using fused silica emitters with 20 μm tips and a spray voltage of 1.5–2 kV. An inlet capillary temperature of 300 °C, S-lens RF of 65, and protein mode were used for all experiments. Precursor ions were mass selected with an isolation width of 2 m/z. The AGC target for MS/MS acquisitions was 5E5 with a maximum injection time of 200 ms and resolving power of 120 000 or 240 000 at m/z 200. Ions were transferred to the HCD cell with a collision energy of 4 eV except for EChcD experiments, during which an appropriate collision energy was selected. Targeted LC/MS/MS for mAb characterization was performed using PRM (parallel reaction monitoring) with an inclusion list of one selected charge state for each mAb fragment, resolving power of 120 000 at m/z 200, and five microscans. MS/MS spectra for the selected charge states of the mAb fragments were acquired over the elution profiles for the respective mAb fragments.
Electromagnetostatic ECD Cell.
The ECD cell (eMSion Inc., Corvallis, OR) was designed to replace the small transfer octapole between the mass selective quadrupole and C-trap in the Q Exactive instrument platform (Figure 1 top panel). Electrical feedthroughs were added to the vacuum chamber flange behind the HCD cell. Wires that supplied the ECD cell DC voltages and filament heating current were passed alongside the HCD cell and under the C-trap. The RF leads for the octapole replaced by the ECD cell were insulated and folded back to prevent electrical shorts. The ECD cell was designed to sit in the existing octapole mounts and did not require modifications to the vacuum chamber or octapole mounts. Installation takes less than 30 min and is completely reversible. The ECD cell was powered by an eight-channel DC power supply with an integrated filament heating current source that was controlled via software on the instrument acquisition computer. The bottom panel of Figure 1 shows a schematic of the ECD cell. It is composed of two permanent magnets, four electrostatic lenses, and a loop-shaped filament. The ECD cell was first tuned for transmission with the filament initially heated to just below the start of electron emission. Once full transmission was achieved, the filament current and bias were modulated to gradually increase electron emission while simultaneously retuning the DC offsets to maintain transmission. After operating emission levels were reached, the DC potentials were tuned to yield maximum overall transmission for ECD product ions and unreacted precursor.
Placing the ECD cell in the octapole position prior to the C-trap requires transmission of analyte ions through the ECD cell for both MS and MS/MS acquisitions. MS acquisitions can be performed in two ways where the filament current or filament bias is modulated in order to eliminate or significantly reduce ECD. During MS acquisitions for off-line experiments, the filament current can be reduced to inhibit electron emission but still keep the filament hot. A preferred method for online data dependent MS/MS acquisitions would be to rapidly switch the filament bias and lens potentials to increase electron energy and significantly reduce ECD efficiency; however, this method is still under development. In the work presented here, the first method was utilized to switch between MS and MS/MS acquisitions.
Initial transmission tests of the electromagnetostatic ECD cell installed prior to the C-trap in the hybrid quadrupole–Orbitrap mass spectrometer were performed with several model peptides and proteins. Transmission efficiency through the electromagnetostatic ECD cell was characterized by comparing MS1 and HCD spectra acquired with the unmodified instrument and with the ECD cell installed. Figure S2 shows MS1 and HCD spectra of carbonic anhydrase II acquired with and without the ECD cell installed. Negligible differences in transmission were observed for carbonic anhydrase and other smaller model proteins.
Liquid Chromatography.
Antibody fragments were separated using a Waters ACQUITY UPLC system (Waters Corporation, Milford, MA) with mobile phases A and B consisting of water with 0.2% formic acid and acetonitrile with 0.2% formic acid, respectively. The analytical and trap columns were 75 μm × 50 cm and 150 μm × 5 cm, respectively, and were packed in-house with 3 μm 300 Å C2 reversed phase resin (Separation Methods Technologies, INC., Newark, DE). mAb IdeS digest (0.1 μg) was loaded onto the trap column and washed for 10 min at 5 μL/min with 1% mobile phase B. A 30 min gradient was used to elute the mAb fragments at 300 nL/min. The gradient consisted of a linear increase from 1 to 25% mobile phase B over 5 min followed by a linear increase to 35% B over 25 min.
Data Analysis.
Product ions from direct infusion MS/MS experiments and targeted LC/MS/MS experiments were matched using LcMsSpectator (https://omics.pnl.gov/software/lcmsspectator), part of the Informed Proteomics software package,38 with a product ion mass tolerance of 5 ppm for direct infusion experiments and LC/MS/MS. Annotation of product ions was restricted by isotopic fit and mass tolerance and manually validated within LcMsSpectator. Product ion maps showing protein sequence coverage were generated using LcMsSpectator. The product ion maps showed the most abundant product ions observed at each inter-residue site. MS/MS spectra for the mAb subunits were averaged over the chromatographic peak for each subunit.
RESULTS AND DISCUSSION
Ubiquitin was used for an initial characterization of ECD and hybrid ECD ion activation methods. Figure 2A shows an ECD spectrum of isolated 10+ charge state ions of ubiquitin. Considering only c/z-ions, ECD of the 10+ charge state of ubiquitin yielded 96% sequence coverage (67 c- and 65 z•-ions) with 3 of the 4 missed cleavages corresponding to sites N-terminal to proline residues. A relatively large number of other product ions types were observed, although at low abundance relative to c/z•-ions. Combined with the c/z-ions, 49 a•-, 22 b-, 17 x•-, and 47 y-ions yielded 99% sequence coverage. We can attribute these ions to the ECD process, as transfer of isolated 10+ charge state ubiquitin ions to the HCD cell with a collision energy of 4 eV does not yield product ions to any significant extent without electron emission; however, even this low level of collisional activation may have contributed to the disruption of intramolecular noncovalent interactions or mobilize radicals in the a•, z•, and charge reduced precursor ions.39
Figure 2.

ECD (A), ECuvPD with one 0.5 mJ laser pulse (B), and EChcD with CE = 22 eV (C) spectra for the 10+ charge state of ubiquitin. Spectra were acquired with 50 microscans and a resolving power of 120 000 at m/z 200.
The utility of hybrid activation methods combining ECD with HCD (EChcD) and ECD with 193 nm UVPD (ECuvPD) was also investigated using ubiquitin to benchmark performance. Figure 2B shows an ECuvPD spectrum of the 10+ charge state of ubiquitin. ECD was performed as before but with the addition of one laser pulse at 0.5 mJ, while the ions were trapped in the HCD cell prior to returning to the C-trap. A low pulse energy was chosen to avoid extensive secondary dissociation of ECD product ions. ECuvPD yielded 99% sequence coverage for the 10+ charge state of ubiquitin through the production of a combination of characteristic product ions for both ECD and UVPD, including 60 a•-, 23 b-, 64 c-, 30 x•-, 50 y-, and 58 z•-ions. Fragmentation characteristics of ubiquitin by 193 nm UVPD have been extensively studied,40 and these results are consistent with previous hybrid ETD with 193 nm UVPD (ETuvPD) results for ubiquitin.41 Figure 2C shows an EChcD spectrum for the 10+ charge state of ubiquitin, which used a collision energy of 22 eV to transfer ions to the HCD cell. EChcD yielded 29 a•-,41 b-, 52 c-, 8 x•-, 63 y-, and 26 z• ions. The combination of a•-, b-, c-, y-, and z•-ions yielded 99% sequence coverage.
Conventional ECD experiments utilized low energy electrons (i.e., < 1 eV), which yielded optimal c/z•-ion abundances; however, higher-energy electrons, termed hot ECD (HECD), were found to yield a variety of additional fragmentation pathways.42–44 Electron energies in the 3–13 eV range produced fragmentation due to electron capture, vibrational excitation via intramolecular vibrational energy redistribution (IVR), secondary fragmentation of a•- and z•- ions, and Cα–C bond cleavage. One potentially advantageous characteristic of HECD compared to ECD was the observation of secondary fragmentation of radical a•- and z•- ions to from d-and w-ions through neutral losses from amino acid side chains.42 Isoleucine and leucine yield unique side chain neutral losses, •C2H5 and •C3H7, respectively, and the presence of characteristic d- and w-ions enable differentiation of these isobaric residues. ECD of the 10+ charge state of ubiquitin yielded 12 d- and 31 w-ions that enabled differentiation of 9 of the 15 I/L residues. Figure 3 shows two examples of I/L differentiation for the 10+ charge state of ubiquitin. The observation of z•4- and w4-ions confirm L73 (Figure 3A), and the presence of z163+•- and w163+-ions confirm I61 (Figure 3B).
Figure 3.

Observation of w-ions in ECD enable differentiation of Ile and Leu residues. Zoomed in portions of an ECD spectrum of the 10+ charge state of ubiquitin showing product ions that confirm L73 (A) and I61 (B). The spectrum was acquired with 50 microscans and a resolving power of 120 000 at m/z 200. S/N for the w4-and w163+-ions are 34 and 12, respectively.
Characterization of ubiquitin by ECD, ECuvPD, and EChcD was extended to all detectable charge states of ubiquitin to determine the charge state dependencies of each method. Charge states 6+ through 13+ were analyzed using ubiquitin electrosprayed from a denaturing solution (50:50 water/ACN with 0.1% formic acid). Figure 4 shows the sequence coverage obtained for each activation methods for each charge state of ubiquitin. Remarkably, ECD alone yielded 91–96% sequence coverage (c- and z-ions only) for charge states 7+ to 13+ and 80% coverage for the 6+ charge state. Considering a•/y- and c/z•-ions, ECD yielded 95–100% sequence coverage for the 7+ to 13+ charge states and 83% for the 6+ charge state. ECuvPD yielded 99% or 100% sequence coverage for all analyzed charge states of ubiquitin. The high level of performance for ECD of lower charge state ubiquitin ions is counterintuitive, but it can be attributed to pseudo-AI-ECD conditions because of transfer of ions to the HCD cell yielding collisional unfolding. Additionally, collisional unfolding and infrared activation from the heated filament as ions transmit through the ECD cell may create pseudo-AI-ECD conditions as well. The number of unique product ions observed as a function of the precursor ion charge state for ECD and ECuvPD is shown in Figure S3. ECD yielded predominantly a•/y- and c/z•-productions with approximately equal numbers of ions observed for each charge state. ECuvPD yielded an increased number of a•-, x•-, and y-product-ions relative to ECD, and both ECD and ECuvPD yielded relatively few b-ions.
Figure 4.

Charge state dependence of sequence coverage for ECD, ECuvPD, and EChcD of ubiquitin.
The number and type of product ions observed for hybrid ion activation methods is highly dependent on activation parameters utilized for each ion activation method. We explored the dependence of sequence coverage and the number of each major product ion type by subjecting the 11+ charge of ubiquitin to EChcD with varying collision energies. A collision energy of 4 eV was used for transfer of ions to the HCD cell for ECD spectra, and 4 eV was used for the purposes of this study, and any collision energy greater than 4 eV was considered EChcD. Figure 5A,B shows the number of unique N-terminal and C-terminal product ions observed, respectively, as a function of HCD collision energy.
Figure 5.

Number of unique N-terminal product ions (A), unique C-terminal product ions (B), and sequence coverage (C) for EChcD of the 11+ charge state of ubiquitin as a function of HCD collision energy.
It is evident that there is an optimal collision energy range to obtain hybrid MS/MS spectra with maximum contributions from each MS/MS method. The greatest numbers of c/z•- and a•-ions were observed at collision energies less than 14 eV and steadily decreased with increasing collision energy. Whereas, the maximum number of b-ions was observed at 16 eV, and the greatest number of y-ions was observed at 20 eV.
Secondary dissociation was very pronounced for a•-, b-, c-, and z•-ions, while the number of y-ions decreased only slightly at collision energies above 20 eV. This trend is reflected in the observed sequence coverage as a function of collision energy (Figure 5C) for N-terminal sequence ions (a•-, b-, and c-ions) and C-terminal sequence ions (x-, y-, and z•-ions). Sequence coverage from N-terminal ions declines dramatically at collision energies greater than 20 eV, whereas sequence coverage from C-terminal ions dropped marginally up to a collision energy of 30 eV because of the persistence of y-ions or generation of y-ions from secondary dissociation of other product ions. Complementary N- and C-terminal ions pairs were observed throughout the sequence at low collision energies. At high collision energies, very few complementary N- and C-terminal ion pairs were observed, though high overall sequence coverage was achieved through short product ions from the respective termini of the protein sequence. Figure S4 shows representative mass spectra and product ion maps illustrating the two distinct ways of achieving high sequence coverage.
ECD and hybrid ECD ion activation methods were then applied to carbonic anhydrase II (29 kDa). Carbonic anhydrase II has been studied extensively with various ion activation methods including ECD, AI-ECD, plasma ECD, ETD, AI-ETD, UVPD, and others. The highest reported sequence coverage for carbonic anhydrase II was 76% by plasma ECD,12 87% by ETD,45 and 87% by 193 nm UVPD.40 In the present study, ECD of the 25+ charge state of carbonic anhydrase II yielded 64 a•-, 42 b-, 150 c-, 13 x•-, 66 y-, and 137 z•-ions. Sequence coverage produced by c/z•-ions alone was 88%, and the combination of a•/y- and c/z•-ions yielded 93% coverage with 5 ppm product ion mass tolerance. Sequence coverage as a function of product ion mass tolerance is shown in Figure S5. Figure 6A is the ECD spectrum of the 25+ charge state of carbonic anhydrase, which contains abundant ECD product ions. The corresponding product ion map for a•/y- and c/z•-product-ions is shown in Figure 6D. A total of 19 missed inter-residue cleavages were observed; however, none of the gaps in sequence coverage were greater than 2 amino acid residues in length. Of the 19 missed cleavages, 7 corresponded to missed cleavages N-terminal to proline residues. The production of a•- and y-ions enabled cleavage N-terminal to proline. However, cleavage N-terminal to proline was not preferential (i.e., not mobile proton mediated) as all of the most abundant product ions in the mass spectrum correspond to c/z•-ions distributed throughout the protein sequence.
Figure 6.

ECD (A), ECuvPD (B), and EChcD (C) spectra of the 25+ charge state of bovine carbonic anhydrase II and the corresponding product ion maps for ECD (D), ECuvPD (E), and EChcD (F). Spectra were acquired with a resolving power of 240 000 at m/z 200 and are the average of 500 microscans. ECuvPD was performed with one 0.5 mJ laser pulse per acquisition, and EChcD was performed with a collision energy of 12 eV.
ECuvPD of the 25+ charge state of carbonic anhydrase was performed using a single 0.5 mJ laser pulse per acquisition and yielded 95 a•-, 46 b-, 145 c-, 24 x•-, 68 y-, and 143 z•-ions. As expected, ECuvPD yielded additional a•- and x•-ions and roughly equivalent numbers of c-, y-, and z•-ions compared to ECD. The ECuvPD spectrum and product ion map are shown in Figure 6B and E, respectively. The combination of a•/y- and c/z•-ions yielded 95% sequence coverage with 5 ppm product ion tolerance. The additional b- and x•-ions were redundant to other sequence ions and did not yield additional sequence coverage. The gains in sequence coverage for ECuvPD compared to ECD were primarily a result of a few additional cleavages N-terminal to proline residues. EChcD with a collision energy of 12 eV was also performed on the 25+ charge state of carbonic anhydrase, and the corresponding spectrum and product ion map are shown in Figure 6C and F, respectively. EChcD yielded 53 a•-, 60 b-, 132 c-, 16 x•-, 70 y-, and 122 z•-ions with a vast majority of the new or significantly more abundant b/y-ions corresponding to cleavage N-terminal to proline or C-terminal to acidic residues. The 10 most abundant product ions in the EChcD spectrum are y-ions corresponding to cleavage N-terminal to proline residues in the C-terminal half of the protein sequence. Despite the preferential cleavages, EChcD yielded 91% sequence coverage. The 12 eV collision energy was found to be optimal in terms of overall sequence coverage, and sequence coverage declined dramatically with relatively small additional increases in collision energy due to further preferential cleavage and secondary dissociation of ECD product ions.
Finally, we compared the utility of ECD, ECuvPD, and EChcD for the online characterization of mAb subunits (LC, Fc/2, and Fd) produced by IdeS digestion and disulfide bond reduction. An LC/MS analysis was first performed to determine the elution time windows and m/z values for charge states of each subunit. Subsequent targeted LC/MS/MS analyses were then performed by acquiring MS/MS spectra for a selected charge state of each subunit over the respective subunit elution time windows. The MS/MS spectra were acquired using a resolving power of 120 000 at m/z 200 and a maximum ion injection time of 200 ms. These parameters yielded approximately 50–100 spectra averaged for each subunit with fwhm chromatographic peak widths in the range of 24–42 s. Similar analyses have been recently reported for ETD46 and 193 nm UVPD.47
Figure 7 summarizes the sequence coverage achieved for each mAb subunit using a single LC/MS/MS experiment (0.1 μg injection) for each ion activation method. ECD and ECuvPD yielded very similar results for each subunit and outperformed EChcD for all subunits. ECuvPD yielded the highest performance with 75, 79, and 59%, respectively, for the LC, Fc/2, and Fd subunits. All three activation methods precisely localized the site of G0F glycosylation to N61, and all yielded relatively low abundance of HexNAc oxonium ions (m/z 204) and other glycan fragments. This indicates a vast majority of the product ions retained the intact glycan. Overall, each activation method performed very well, and to the best of our knowledge, ECD and ECuvPD yielded greater sequence coverage for each subunit than has been previously reported for a single experiment and single activation condition (although different mAbs). Product ion maps for each activation method and mAb subunit are shown in Figures S6–S8. Combining the results for each activation method (total of three LC/MS/MS analyses) yielded 89, 92, and 76% sequence coverage for the LC, Fc/2, and Fd, respectively, and 86% sequence coverage for the mAb as a whole. Coverage of the complementarity determining regions (CDRs) in the light chain was nearly complete. ECD yielded five missed cleavages, while both ECuvPD and EChcD yielded four missed cleavages. Combined, ECD, ECuvPD, and EChcD yielded complete coverage of all three CDRs in the light chain. ECD and ECuvPD yielded comparable results for the more challenging Fd subunit with 56 and 59% sequence coverage, respectively, and EChcD yielded 46% coverage. The Fd subunit contains only five basic residues between residues 100 and 200 in the C-terminal half of the sequence. Consequently, this portion of the Fd subunit is quite difficult to sequence. However, each activation method yielded complementary fragmentation, and this is responsible for the considerable increase in sequence coverage for the combined results for the Fd subunit. ECD, ECuvPD, and EChcD yielded 11, 10, and 19 missed cleavages, respectively, in the Fd CDRs. ECD and ECuvPD each yielded complete coverage of the CDR2. Combined results for all three methods yielded three missed cleavages in the CDR1 and two missed cleavages in the CDR3.
Figure 7.

Single LC/MS/MS experiment sequence coverage produced by ECD, ECuvPD (1 pulse at 0.5 mJ), EChcD (CE = 12 eV), and combined coverage for the LC, Fc/2, and Fd subunits of SigmaMAB (MSQC4). The reported sequence coverage for the Fc/2 subunit is for the G0F glycoform. Analyses were performed for the 20+, 25+, and 24+ charge states of the LC, Fc/2, and Fd subunits, respectively.
CONCLUSIONS
We have demonstrated the unique capabilities of ECD and hybrid ECD ion activation methods (ECuvPD and EChcD) for top–down sequencing of proteins using an electromagnetostatic ECD cell implemented in a benchtop quadrupole–Orbitrap mass spectrometer. The ECD cell replaced a small transfer octapole between the mass selective quadrupole and the C-trap, greatly simplifying installation. This implementation did not require any modifications to the instrument aside from the addition of a few electrical feedthroughs and is fully reversible. ECD was performed in a single-pass reaction scheme, in which multiply charged protein ions interact with an electron cloud during transmission through the ECD cell. In this mode of operation, ECD reactions occur on the order of 10 μs, thus eliminating losses in instrument duty cycle typically associated with ECD or ETD experiments because of extended reaction times. The extensive sequence coverage achieved for low charge states and medium sized model proteins indicates that pseudo-activated ion ECD conditions were created during transmission through the ECD cell and transfer to the HCD cell. The observation of d- and w- ions enabled differentiation of leucine and isoleucine residues in proteins, which was previously reported for hot ECD (i.e., 3–13 eV electron energy) in peptides. As a result of the geometry of the electromagnetostatic ECD cell and strong charge interactions that occur within the cell, the majority of the electron cloud is thermal, but a portion of electrons emitted from the filament may assume trajectories that are accelerated off axis to create hot ECD conditions. This unique combination of pseudo-activated ion conditions and potentially elevated electron energies yielded the most extensive characterization of the model proteins up to 30 kDa and mAb subunits reported to date.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by the “High Resolution and Mass Accuracy Capability” development project at Environmental Molecular Science Laboratory (EMSL), a DOE Office of Science User Facility sponsored by the Office of Biological and Environmental Research and located at the Pacific Northwest National Laboratory and two NIH SBIR Awards to VV (R43GM122131–01 and R44GM122131–02).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at10.1021/acs.analchem.8b01901.
Instrument schematic showing the incorporation of the electromagnetostatic ECD cell in the Q Exactive instrument platform; transmission through the electromagnetostatic ECD cell; number of unique product ions observed for ECD and ECuvPD as a function of precursor ion charge state for ubiquitin; EChcD spectra and product ion maps generated with HCD collision energies of 14 and 30 eV for the 11+ charge state of ubiquitin; sequence coverage as function of product ion mass tolerance for ECD of the 25+ charge state of bovine carbonic anhydrase II; product ion maps for SigmaMAB subunits generated from a single targeted LC/MS/MS experiment using ECD, ECuvPD, and EChcD (PDF)
The authors declare the following competing financial interest(s): JBS and NM declare no competing financial interests. YV, NL, VV, and JSB are cofounders of e-MSion, Inc, which produced the ECD cell used in this study. AM is an employee of ThermoFisher Scientific, producer of Orbitrap mass spectrometers.
REFERENCES
- (1).Zubarev RA; Kelleher NL; McLafferty FW J. Am. Chem. Soc 1998, 120 (13), 3265–3266. [Google Scholar]
- (2).Kelleher NL; Zubarev RA; Bush K; Furie B; Furie BC; McLafferty FW; Walsh CT Anal. Chem 1999, 71 (19), 4250–4253. [DOI] [PubMed] [Google Scholar]
- (3).Shi SD-H; Hemling ME; Carr SA; Horn DM; Lindh I; McLafferty FW Anal. Chem 2001, 73 (1), 19–22. [DOI] [PubMed] [Google Scholar]
- (4).Breuker K; Oh H; Horn DM; Cerda BA; McLafferty FW J. Am. Chem. Soc 2002, 124 (22), 6407–6420. [DOI] [PubMed] [Google Scholar]
- (5).Gu C; Tsaprailis G; Breci L; Wysocki VH Anal. Chem 2000, 72 (23), 5804–5813. [DOI] [PubMed] [Google Scholar]
- (6).Wysocki VH; Tsaprailis G; Smith LL; Breci LA J. Mass Spectrom 2000, 35 (12), 1399–1406. [DOI] [PubMed] [Google Scholar]
- (7).Horn DM; Ge Y; McLafferty FW Anal. Chem 2000, 72 (20), 4778–4784. [DOI] [PubMed] [Google Scholar]
- (8).Ge Y; Lawhorn BG; ElNaggar M; Strauss E; Park J-H; Begley TP; McLafferty FW J. Am. Chem. Soc 2002, 124 (4), 672–678. [DOI] [PubMed] [Google Scholar]
- (9).Håkansson K; Cooper HJ; Emmett MR; Costello CE; Marshall AG; Nilsson CL Anal. Chem 2001, 73 (18), 4530–4536. [DOI] [PubMed] [Google Scholar]
- (10).Horn DM; Breuker K; Frank AJ; McLafferty FW J. Am. Chem. Soc 2001, 123 (40), 9792–9799. [DOI] [PubMed] [Google Scholar]
- (11).Håkansson K; Chalmers MJ; Quinn JP; McFarland MA; Hendrickson CL; Marshall AG Anal. Chem 2003, 75 (13), 3256–3262. [DOI] [PubMed] [Google Scholar]
- (12).Sze SK; Ge Y; Oh H; McLafferty FW Anal. Chem 2003, 75 (7), 1599–1603. [DOI] [PubMed] [Google Scholar]
- (13).Tsybin YO; Witt M; Baykut G; Kjeldsen F; Håkansson P Rapid Commun. Mass Spectrom 2003, 17 (15), 1759–1768. [DOI] [PubMed] [Google Scholar]
- (14).Mikhailov V; Cooper HJ Am. Soc. Mass Spectrom 2009, 20 (5), 763–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Syka JEP; Coon JJ; Schroeder MJ; Shabanowitz J; Hunt DF Proc. Natl. Acad. Sci. U. S. A 2004, 101 (26), 9528–9533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Pitteri SJ; Chrisman PA; Hogan JM; McLuckey SA Anal. Chem 2005, 77 (6), 1831–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Pitteri SJ; Chrisman PA; McLuckey SA Anal. Chem 2005, 77 (17), 5662–5669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Swaney DL; McAlister GC; Wirtala M; Schwartz JC; Syka JEP; Coon JJ Anal. Chem 2007, 79 (2), 477–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Ledvina AR; McAlister GC; Gardner MW; Smith SI; Madsen JA; Schwartz JC; Stafford GC; Syka JEP; Brodbelt JS; Coon JJ Angew. Chem., Int. Ed 2009, 48 (45), 8526–8528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Riley NM; Westphall MS; Coon JJ J. Am. Soc. Mass Spectrom 2018, 29, 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Frese CK; Altelaar AFM; van den Toorn H; Nolting D; Griep-Raming J; Heck AJR; Mohammed S Anal. Chem 2012, 84 (22), 9668–9673. [DOI] [PubMed] [Google Scholar]
- (22).Frese CK; Zhou H; Taus T; Altelaar AFM; Mechtler K; Heck AJR; Mohammed SJ Proteome Res. 2013, 12 (3), 1520–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Yu Q; Wang B; Chen Z; Urabe G; Glover MS; Shi X; Guo L-W; Kent KC; Li LJ Am. Soc. Mass Spectrom 2017, 28 (9), 1751–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Liu F; van Breukelen B; Heck AJ R. Mol. Cell. Proteomics 2014, 13 (10), 2776–2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Baba T; Hashimoto Y; Hasegawa H; Hirabayashi A; Waki I Anal. Chem 2004, 76 (15), 4263–4266. [DOI] [PubMed] [Google Scholar]
- (26).Satake H; Hasegawa H; Hirabayashi A; Hashimoto Y; Baba T; Masuda K Anal. Chem 2007, 79 (22), 8755–8761. [DOI] [PubMed] [Google Scholar]
- (27).Silivra OA; Kjeldsen F; Ivonin IA; Zubarev RA J. Am. Soc. Mass Spectrom 2005, 16 (1), 22–27. [DOI] [PubMed] [Google Scholar]
- (28).Ding L; Brancia FL Anal. Chem 2006, 78 (6), 1995–2000. [DOI] [PubMed] [Google Scholar]
- (29).Baba T; Campbell JL; Le Blanc JCY; Hager JW; Thomson BA Anal. Chem 2015, 87 (1), 785–792. [DOI] [PubMed] [Google Scholar]
- (30).Voinov VG; Deinzer ML; Barofsky DF Rapid Commun. Mass Spectrom 2008, 22 (19), 3087–3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Voinov VG; Beckman JS; Deinzer ML; Barofsky DF Rapid Commun. Mass Spectrom 2009, 23 (18), 3028–3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Voinov VG; Deinzer ML; Barofsky DF Anal. Chem 2009, 81 (3), 1238–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Voinov VG; Deinzer ML; Beckman JS; Barofsky DF J. Am. Soc. Mass Spectrom 2011, 22 (4), 607–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Voinov VG; Bennett SE; Beckman JS; Barofsky DF J. Am. Soc. Mass Spectrom 2014, 25 (10), 1730–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Voinov VG; Bennett SE; Barofsky DF J. Am. Soc. Mass Spectrom 2015, 26 (5), 752–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Fort KL; Cramer CN; Voinov VG; Vasil’ev YV; Lopez NI; Beckman JS; Heck AJ R. J. Proteome Res 2018, 17, 926–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Fort KL; Dyachenko A; Potel CM; Corradini E; Marino F; Barendregt A; Makarov AA; Scheltema RA; Heck AJ R. Anal. Chem 2016, 88 (4), 2303–2310. [DOI] [PubMed] [Google Scholar]
- (38).Park J; Piehowski PD; Wilkins C; Zhou M; Mendoza J; Fujimoto GM; Gibbons BC; Shaw JB; Shen Y; Shukla AK; et al. Nat. Methods 2017, 14 (9), 909–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Breuker K; Oh H; Lin C; Carpenter BK; McLafferty FW Proc. Natl. Acad. Sci. U. S. A 2004, 101 (39), 14011–14016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Shaw JB; Li W; Holden DD; Zhang Y; Griep-Raming J; Fellers RT; Early BP; Thomas PM; Kelleher NL; Brodbelt JS J. Am. Chem. Soc 2013, 135 (34), 12646–12651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Cannon JR; Holden DD; Brodbelt JS Anal. Chem 2014, 86 (21), 10970–10977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Kjeldsen F; Haselmann KF; Budnik BA; Jensen F; Zubarev RA Chem. Phys. Lett 2002, 356 (3), 201–206. [Google Scholar]
- (43).Tsybin YO; Witt M; Baykut G; Håkansson P Rapid Commun. Mass Spectrom 2004, 18 (14), 1607–1613. [DOI] [PubMed] [Google Scholar]
- (44).Zubarev RA Mass Spectrom. Rev 2003, 22 (1), 57–77. [DOI] [PubMed] [Google Scholar]
- (45).Weisbrod CR; Kaiser NK; Syka JEP; Early L; Mullen C; Dunyach J-J; English AM; Anderson LC; Blakney GT; Shabanowitz J; et al. J. Am. Soc. Mass Spectrom 2017, 28 (9), 1787–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Fornelli L; Ayoub D; Aizikov K; Beck A; Tsybin YO Anal. Chem 2014, 86 (6), 3005–3012. [DOI] [PubMed] [Google Scholar]
- (47).Cotham VC; Brodbelt JS Anal. Chem 2016, 88 (7), 4004–4013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.