

Abstract
Adenosine-to-inosine (A-to-I) RNA editing is a widespread and conserved post-transcriptional modification, producing significant changes in cellular function and behavior. Accurately identifying, detecting, and quantifying these sites in the transcriptome is necessary to improve our understanding of editing dynamics, its broader biological roles, and connections with diseases. Chemical labeling of edited bases coupled with affinity enrichment has enabled improved characterization of several forms of RNA editing. However, there are no approaches currently available for pull-down of inosines. To address this need, we explore acrylamide as a labeling motif and report here an acrylamidofluorescein reagent that reacts with inosine and enables enrichment of ino-sine-containing RNA transcripts. This method provides improved sensitivity in the detection and identification of inosines towards a more comprehensive transcriptome-wide analysis of A-to-I editing. Acrylamide derivatization is also highly generalizable, providing potential for the labeling of inosine with a wide variety of probes and affinity handles.
Graphical Abstract
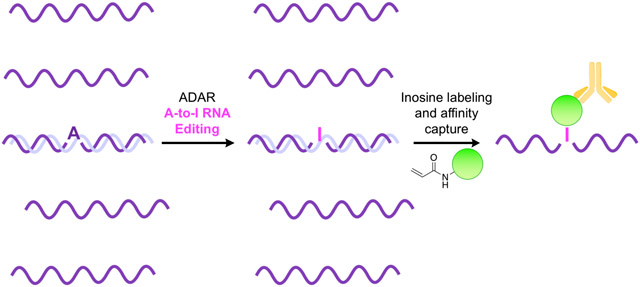
RNA is extensively edited after transcription. Adeno-sine-to-inosine (A-to-I) conversion is of one of the most common and impactful forms of editing and is catalyzed by adenosine deaminases acting on RNA (ADARs).1 Resulting inosines base pair with cytidine and are effectively decoded as guanosine by cellular machinery. A-to-I editing occurs in both coding and non-coding RNA transcripts, eliciting dramatic changes in overall cellular function and behavior. Editing of mRNA can alter protein sequence through direct modification of codons or by altering splice sites and regulatory elements in untranslated regions. A-to-I editing events are also extensive in non-coding RNAs, including microRNA and small-interfering RNA precursors, significantly altering their biosynthesis, trafficking, specificity, and gene regulation properties.2–4 Accurately identifying A-to-I RNA editing sites in the transcriptome is necessary to improve our understanding of these modifications and their biological functions. A recently developed method to map A-to-I editing locations employs chemical modification of inosines with acrylonitrile to form N1-cyanoethylinosine (Figure 1a).5, 6 Termed inosine chemical erasing sequencing (ICE-seq), this technique leverages the observation that inosine cyanoethylation inhibits Watson-Crick base pairing and effectively arrests reverse transcription at A-to-I editing sites. Resulting truncated cDNAs fail to undergo PCR amplification and are “erased” from RNA sequencing chromatograms, allowing bioinformatic detection of editing sites. Although ICE-seq has improved the accuracy and scalability of mapping and discovering A-to-I RNA editing sites, this method is also limited in sensitivity, as labeled inosine-containing transcripts cannot be enriched. Additionally, while millions of A-to-I sites have been identified across the human transcriptome, actual editing rates at these sites are highly variable and dependent on cellular and environmental cues, rendering them difficult to detect, characterize, and measure with these techniques. This is particularly true in coding RNAs, where I/A ratios can range anywhere from <0.001–5% depending on tissue type or external stimuli.7–9 Together, these challenges mask the overall prevalence and true landscape of A-to-I RNA editing across the transcriptome.
Figure 1.

Chemical labeling of inosine. (a) Acrylonitrile and (b) acrylamidofluorescein produce N1 addition products. (c) Acrylamidofluorescein labeling enables affinity capture of transcripts containing inosine.
The ability to enrich A-to-I edited transcripts from more complex total RNA samples would largely address this limitation and allow for deeper interrogation and characterization of the epitranscriptome. Approaches using chemical labeling and/or antibody immunoprecipitation to capture edited transcripts have enabled significant advances in identifying and cataloging a number of other RNA modifications, including N1- and N6-methyladenosine, 5-methylcytidine, 5-hydroxymethylcytidine, and pseudouridine (Ψ).10–18 While a previous study reported the production of antibodies targeting inosine for the enrichment of tRNAs, this method also displayed adsorptivity to other nucleobases and has not been further demonstrated in any other contexts.19 Thus, no generally applicable methods currently exist for the derivatization and/or enrichment of inosines in RNA, significantly limiting both depth and sensitivity in identifying and studying A-to-I RNA editing dynamics across the transcriptome.
In the design of a reagent for affinity capture of ino-sine-containing RNAs, we hypothesized that an acrylamide electrophile would provide similar reactivity towards inosine as acrylonitrile, while offering the structural flexibility to install an affinity handle for enrichment. To test this hypothesis, we carried out a facile synthesis to generate acrylamidofluorescein (Figure S1), as this reagent would provide both fluorescent labeling of inosines and the ability to perform affinity capture of A-to-I edited RNA transcripts using a commercially available anti-fluorescein antibody (Figure 1b,c).
After designing and synthesizing the acrylamidofluorescein reagent, we assessed initial labeling performance by reacting acrylamidofluorescein and acrylonitrile with each of the major ribonucleosides: inosine (I), pseudouridine (Ψ), uridine (U), guanosine (G), adeno-sine (A), and cytidine (C). Closely mimicking the ICE reaction conditions, a mixture comprising 50 mM ribonucleoside and 250 mM of either acrylonitrile or acrylamidofluorescein was prepared in 50:50 triethylammonium acetate:ethanol at pH 8.6. The solutions were incubated at 70 °C and the reaction was monitored by HPLC over 24 hours. As illustrated in Figure 2a, disappearance of inosine peaks is clearly shown along with the formation of a new product peak in both 254 nm and 494 nm chromatograms. This product peak was isolated and analyzed using ESI-MS and MS/MS analysis, confirming the identity of the predicted N1-fluoresceinamidoethylinosine (FAE1I) product (Figure 2b, S9). Using ribonucleoside peak areas in the chromatograms, we determined the ratio of reacted vs unreacted ribonucleoside to calculate average conversion percentages for each base at various time points over 24 hours (Figures 2c, S7).
Figure 2.

(a) Representative HPLC traces depicting the reaction between inosine and acrylamidofluorescein over 24 hours. Disappearance of inosine (I) correlates with the appearance of a new putative N1-fluoresceinacrylamidoethylinosine (FAE1I) product peak. (b) ESI-MS analysis confirming mass identity of FAE1I product. (c) Reactivity panel of acrylonitrile and acrylamidofluorescein with ribonucleosides after 24 hours. (d) Dependence of reaction rate constants on pH for the major reacting nucleosides inosine (I) and pseudouridine (Ψ).
While acrylamidofluorescein and acrylonitrile exhibit similar reactivity trends, it is clear from the data that acrylonitrile has higher reaction efficiency (Figures 2d, S7). This is likely due to the difference in electron withdrawing properties between the two reagents, which contributes significantly to the kinetics of addition reactions.20, 21 Given that the amide group is less withdrawing than the nitrile moiety, these results are then unsurprising. Regardless, acrylamidofluorescein and acrylonitrile display similar overall labeling selectivity, exhibiting major product formation with I and Ψ, minimal reactivity with U and G, and virtually no reactivity with A and C throughout extended reaction times. While both reagents display reactivity with Ψ, these observations are consistent with previous studies using acrylonitrile and serve to demonstrate the similar reactivity profiles of both acrylonitrile and acrylamidofluorescein. Indeed, the first reports of acrylonitrile-nucleoside labeling demonstrated its robust reactivity with N1 on both ino-sine and Ψ.20, 21
To further validate addition of acrylamidofluorescein at N1 of inosine, we assessed the effect of pH on reaction rates. Early characterizations of acrylonitrile reactivity with inosine showed that cyanoethylation is strongly pH dependent, suggesting N1 deprotonation is required for reactivity. Similarly, the data in Figure 2d illustrate the direct correlation between reaction rate and pH and highlight the preferred reactivity with inosine at ~pH 8.5–8.6, consistent with the known pKa values of N1 for inosine (8.7)22 and pseudouridine (9.5).23 Taken together with the MS spectra, these results strongly support the predicted N1 addition to inosine and further suggest a similar labeling mechanism of acrylamidofluorescein compared with the well characterized chemistry of acrylonitrile.
Given the promising results of our reagent with ribonucleosides, we next sought to demonstrate acrylamidofluorescein labeling of inosine in RNA oligoribonucleotides. As a test system for these studies, we chemically synthesized two short RNAs containing a 5’ Cy5 fluorescent label and an adenosine (RNA-A-Cy5) or inosine (RNA-I-Cy5) at a defined position. We subjected each of these RNAs to acrylamidofluorescein labeling and denaturing PAGE analysis. As shown in Figure 3a, fluorescein labeling is clearly observed in RNA-I-Cy5 with increasing reaction times, and the labeled product exhibits a slight decrease in migration rate. In comparison, only a faint signal is observed for RNA-A-Cy5, even after a 48 hour reaction time. Given that the presence of inosine is the only molecular difference between these two RNA strands, these data are indicative of selective fluorescein addition at this nucleotide position. Densitometric analysis was performed on the labeled RNA bands in the gels and normalized to standard amounts of fluorescein and Cy5-labeled control oligo nucleotides. These data were then used to calculate labeling yield as a function of reaction time (Figure 3b), which illustrates good selectivity for labeling of RNAI-Cy5 compared to RNA-A-Cy5. This experiment also highlights the importance of reaction time in maximizing inosine labeling efficiency while maintaining selectivity, as we observe optimal RNA-I:RNA-A labeling ratios at approximately 24 hours. While longer RNA transcripts can undergo hydrolysis in mild alkaline conditions at elevated temperatures, these data demonstrate the stability of shorter RNA segments under our reaction conditions. We envision the use of this labeling method with high-throughput RNA-seq workflows, which require fragmentation of longer RNAs prior to library preparation and amplification. This fragmentation step is employed upstream of chemical labeling and pull-down in the analogous strategies described above for mapping other RNA modifications,10, 11, 15–17, 24 and thus our results indicate compatibility with these platforms.
Figure 3.

(a) Denaturing PAGE analysis of synthetic oligoribonucleotides labeled with acrylamidofluorescein. (b) Densitometric quantification of oligoribonucleotide labeling.
Encouraged by these results, we sought to establish feasibility for our ultimate goal of enriching inosine-containing transcripts via immunoprecipitation (IP) of labeled oligonucleotides. To test this approach, we utilized the same RNA sequences from the previous experiment but labeled the inosine and adenosine variants with Cy5 and Cy3, respectively, to allow for simultaneous fluorescence-based quantification of each species. RNA-I-Cy5 and RNA-A-Cy3 were combined in varying ratios, subjected to acrylamidofluorescein labeling, and then affinity captured using an anti-fluorescein monoclonal antibody and protein A/G magnetic beads. After extensive washing, bound oligoribonucleotides were eluted and quantified using a fluorescence plate reader (Figure 4a). Final concentrations of RNA-A-Cy3 and RNA-I-Cy5 after pull-down were compared to input ratios to calculate fold-enrichment. As shown in Figure 4b, acrylamidofluorescein labeling coupled with IP enables upwards of 7-fold enrichment of inosine-containing oligoribonucleotides, with the highest enrichment factors achieved for samples containing the lowest ratios of the inosine-containing RNA.
Figure 4.

(a) Workflow for quantifying pulldown efficiency with acrylamidofluorescein labeling and immuno-precipitation. (b) Fold enrichment of inosine-containing oligoribonucleotides from varying mixtures.
Chemical modification strategies coupled with affinity capture have significantly improved the sensitivity and accuracy in sequencing, mapping, and characterizing several modified RNA bases.10–18 However, there are no extant methods for enriching A-to-I edited transcripts, greatly limiting our ability to understand the true scale and impact of A-to-I modifications on cell and tissue function. Here we address this challenge through the synthesis of a novel acrylamidofluorescein reagent that chemically labels inosine and enables the enrichment of A-to-I edited transcripts.
While the observed reactivity between acrylamidofluorescein and Ψ may seem problematic for the effective isolation and enrichment of inosine-containing transcripts from biological samples, Ψ is found predominantly in ribosomal RNAs and tRNAs, and thus effective fractionation of total RNA samples can remove significant quantities of this modified base. In coding RNAs, I also vastly outnumbers Ψ, with current estimates of ~500:1 I:Ψ.7, 17, 25 Additionally, methods have now been developed to selectively label and/or deplete Ψ from total RNA pools using biotinylated carbodiimide reagents.17 We envision that acrylamidofluorescein could be coupled with carbodiimide labeling to achieve simultaneous selective modification and separate enrichment of transcripts containing I and Ψ, respectively. We also recognize the potential to improve enrichment by reducing reactivity with the natural ribonucleosides U and G, and efforts are underway to explore alternative acrylamide structures toward this goal. Regardless, given the present lack of methods for isolating inosine-containing RNAs, the research presented here represents a critical first step toward integrating chemical labeling and enrichment methods for this important application.
A-to-I RNA editing is among the most widespread epitranscriptomic modifications and is integral to a variety of cellular processes. Additionally, direct links to malfunctions in A-to-I RNA editing are being rapidly discovered for a growing number of diseases. Robust identification and characterization of these RNA modifications is vital to understanding their biological function and dynamics. The research reported here is anticipated to advance the study of A-to-I RNA editing by enabling a more comprehensive and deeper detection of inosines in the transcriptome through pre-enrichment of edited transcripts from complex RNA mixtures. While our initial investigation utilized acrylamidofluorescein, the acrylamide scaffold offers considerable flexibility for the attachment of other affinity handles and functional probes. Thus, we envision that our labeling and affinity capture approach can be expanded into a rich toolbox for elucidating the true scale and dynamics of A-to-I editing.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the National Institutes of Health (R01GM116991 to J.M.H.). The authors thank Fred Strobel, Anna Kellner, and Brendan Deal for helpful materials, discussions and advice.
Footnotes
Supporting Information. General experimental protocols, synthetic protocols and characterization data for acrylamidofluorescein, HPLC traces and mass spectra for nucleoside labeling, tabular data for oligonucleotide pull-down. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Bass BL (2002) RNA editing by adenosine deaminases that act on RNA. Annu. Rev. Biochem 71, 817–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Valente L, and Nishikura K (2005) ADAR gene family and A-to-I RNA editing: diverse roles in posttranscriptional gene regulation. Prog. Nucleic Acid Res. Mol. Biol 79, 299–338. [DOI] [PubMed] [Google Scholar]
- (3).Kawahara Y, Zinshteyn B, Sethupathy P, Iizasa H, Hatzigeorgiou AG, and Nishikura K Redirection of silencing targets by adenosineto-inosine editing of miRNAs. Science Vol 315, 1137–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Nishikura K (2016) A-to-I editing of coding and non-coding RNAs by ADARs. Nat. Rev. Mol. Cell Biol 17, 83–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Sakurai M, Yano T, Kawabata H, Ueda H, and Suzuki T (2010) Inosine cyanoethylation identifies A-to-I RNA editing sites in the human transcriptome. Nat. Chem. Biol 6, 733–40. [DOI] [PubMed] [Google Scholar]
- (6).Sakurai M, and Suzuki T (2011) Biochemical identification of A-to-I RNA editing sites by the inosine chemical erasing (ICE) method Methods in Molecular Biology. Volume 718: RNA and DNA Editing pp 89–99, Humana Press, New York. [DOI] [PubMed] [Google Scholar]
- (7).Tan MH, Li Q, Shanmugam R, Piskol R, Kohler J, Young AN, Liu KI, Zhang R, Ramaswami G, and Ariyoshi K (2017) Dynamic landscape and regulation of RNA editing in mammals. Nature 550, 249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Paul MS, and Bass BL (1998) Inosine exists in mRNA at tissue-specific levels and is most abundant in brain mRNA. J. EMBO 17, 1120–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Yang JH, Luo X, Nie Y, Su Y, Zhao Q, Kabir K, Zhang D, and Rabinovici R (2003) Widespread inosine-containing mRNA in lymphocytes regulated by ADAR1 in response to inflammation. Immunology 109, 15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Li X, Xiong X, Wang K, Wang L, Shu X, Ma S, and Yi C (2016) Transcriptome-wide mapping reveals reversible and dynamic N 1-methyladenosine methylome. Nat. Chem. Biol 12, 311. [DOI] [PubMed] [Google Scholar]
- (11).Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob-Hirsch J, Amariglio N, and Kupiec M (2012) Topology of the human and mouse m 6 A RNA methylomes revealed by m 6 A-seq. Nature 485, 201. [DOI] [PubMed] [Google Scholar]
- (12).Edelheit S, Schwartz S, Mumbach MR, Wurtzel O, and Sorek R (2013) Transcriptome-wide mapping of 5-methylcytidine RNA modifications in bacteria, archaea, and yeast reveals m5C within archaeal mRNAs. PLoS Genet 9, e1003602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Delatte B, Wang F, Ngoc LV, Collignon E, Bonvin E, Deplus R, Calonne E, Hassabi B, Putmans P, and Awe S (2016) Transcriptome-wide distribution and function of RNA hydroxymethylcytosine. Science 351, 282–285. [DOI] [PubMed] [Google Scholar]
- (14).Schwartz S, Bernstein DA, Mumbach MR, Jovanovic M, Herbst RH, León-Ricardo BX, Engreitz JM, Guttman M, Satija R, and Lander ES (2014) Transcriptome-wide mapping reveals widespread dynamic-regulated pseudouridylation of ncRNA and mRNA. Cell 159, 148–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Carlile TM, Rojas-Duran MF, Zinshteyn B, Shin H, Bartoli KM, and Gilbert WV (2014) Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature 515, 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Lovejoy AF, Riordan DP, and Brown PO (2014) Transcriptome-wide mapping of pseudouridines: pseudouridine synthases modify specific mRNAs in S. cerevisiae. PLoS One 9, e110799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Li X, Zhu P, Ma S, Song J, Bai J, Sun F, and Yi C (2015) Chemical pulldown reveals dynamic pseudouridylation of the mammalian transcriptome. Nat. Chem. Biol 11, 592. [DOI] [PubMed] [Google Scholar]
- (18).Wu Q, Amrutkar SM, and Shao F (2018) Sulfinate based selective labeling of 5-hydroxymethylcytosine: application to biotin pull down assay. Bioconjugate Chem 29, 245–249. [DOI] [PubMed] [Google Scholar]
- (19).Inouye H, Fuchs S, Sela M, and Littauer UZ (1973) Detection of inosine-containing transfer ribonucleic acid species by affinity chromatography on columns of anti-inosine antibodies. J. Biol. Chem 248, 8125–8129. [PubMed] [Google Scholar]
- (20).Yoshida M, and Ukita T (1965) Selective modifications of inosine and ø-uridine with acrylonitrile out of the other ribonucleosides. J. Biochem 57, 818–821. [DOI] [PubMed] [Google Scholar]
- (21).Ofengand J (1967) The function of pseudouridylic acid in transfer ribonucleic acid I. the specific cyanoethylation of pseudouridine, inosine, and 4-thiouridine by acrylonitrile. J. Biol. Chem 242, 5034–5045. [PubMed] [Google Scholar]
- (22).Fox JJ, Wempen I, Hampton A, and Doerr IL (1958) Thiation of Nucleosides. I. Synthesis of 2-amino-6-mercapto-9-β-D-ribofuranosylpurine (“thioguanosine”) and Related Purine Nucleosides1. J. Am. Chem. Soc 80, 1669–1675. [Google Scholar]
- (23).Cohn WE (1960) Pseudouridine, a carbon-carbon linked ribonucleoside in ribonucleic acids: isolation, structure, and chemical characteristics. J. Biol. Chem 235, 1488–1498. [PubMed] [Google Scholar]
- (24).Suzuki T, Ueda H, Okada S, and Sakurai M (2015) Transcriptome-wide identification of adenosine-to-inosine editing using the ICE-seq method. Nat. Protoc 10, 715. [DOI] [PubMed] [Google Scholar]
- (25).Addepalli B, and Limbach PA (2011) Mass spectrometry-based quantification of pseudouridine in RNA. J. Am. Soc. Mass Spectrom 22, 1363–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.