

SUMMARY
Cancer extracellular vesicles (EVs) are highly heterogeneous, which impedes our understanding of their function as intercellular communication agents and biomarkers. To deconstruct this heterogeneity, we analyzed extracellular RNAs (exRNAs) and extracellular proteins (exPTNs) from size fractionation of large, medium, and small EVs and ribonucleoprotein complexes (RNPs) from mouse glioblastoma cells by RNA sequencing and quantitative proteomics. mRNA from medium-sized EVs most closely reflects the cellular transcriptome, whereas small EV exRNA is enriched in small non-coding RNAs and RNPs contain precisely processed tRNA fragments. The exPTN composition of EVs and RNPs reveals that they are closely related by vesicle type, independent of their cellular origin, and single EV analysis reveals that small EVs are less heterogeneous in their protein content than larger ones. We provide a foundation for better understanding of segregation of macromolecules in glioma EVs through a catalog of diverse exRNAs and exPTNs.
Graphical Abstract
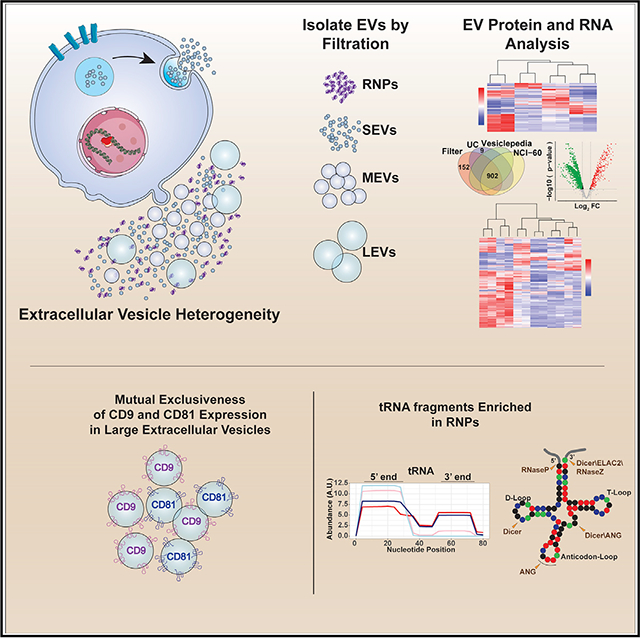
In Brief
Extracellular vesicles (EVs) are highly heterogeneous. Using genetically defined mouse glioblastoma tumor cells, Gyuris et al. employ a differential filtration approach to isolate EVs based on size and establish the differential distribution of RNA and protein between EVs and ribonucleoprotein complexes in genetically distinct contexts.
INTRODUCTION
Extracellular vesicles (EVs) are a heterogeneous array of 40- to 1000-nm diameter plasma-membrane enclosed particles that are released by all cells and have recently been described as agents of intercellular communication, capable of modifying cells within their microenvironment (Mathivanan et al., 2010; Théry, 2011). Mounting evidence suggests that this process plays a role in cancer development and maintenance (Bebelman et al., 2018; Becker et al., 2016) and therefore offers opportunities for therapeutic and diagnostic interventions. EVs are also highly heterogeneous in their cargo composition, a characteristic thought to reflect their diverse cellular provenance (Rabinowits et al., 2009; Simpson et al., 2009; Skog et al., 2008; Taylor and Gercel-Taylor, 2008). Microvesicles and exosomes (broadly referred herein as EVs), as well as non-vesicular ribonucleoprotein complexes (RNPs), contain RNA, DNA, and proteins; a comprehensive catalog of these macromolecules in relation to EV subtypes and RNPs is complicated by the differences introduced by different isolation strategies (Konoshenko et al., 2018). EVs’ intrinsic heterogeneity is problematic in terms of identifying biomarkers or testing EV-based therapeutic vehicles. Integrating the understanding of physical and molecular EV heterogeneity, relative to their cell of origin, is a crucial step toward accomplishing many of EVs promises.
Glioblastoma multiforme (GBM) is an incurable malignant brain cancer with a median survival of 14 months and a 5-year survival of less than 3% (Davis, 2016; Omuro and DeAngelis, 2013). Characterization of the genomic landscape of GBM revealed common genetic mutations and signaling abnormalities that are recognized as drivers of GBM’s characteristic uncontrollable growth, invasiveness, angiogenesis, and resistance to therapy (Brennan et al., 2013; Brat et al., 2015; Eckel-Passow et al., 2015; McLendon et al., 2008; Verhaak et al., 2010). GBMs of the classical subtype overexpress wild-type or mutated epidermal growth factor receptor (EGFR), an event often associated with loss of the INK4a/ARF (Cdkn2a locus) and PTEN tumor suppressor genes (Brennan et al., 2013; Verhaak et al., 2010). The pro-neural subclass of GBM is characterized by overexpression of the receptor tyrosine kinase PDGFRA along with loss of the P53 tumor suppressor gene (Brat et al., 2015; Eckel-Passow et al., 2015). We previously showed that mice genetically engineered to mimic these genomic events faithfully model classical and proneural GBMs (Acquaviva et al., 2011; Jun et al., 2012; Jun et al., 2018; Zhou et al., 2018; Zhu et al., 2009), providing a powerful platform for studies of glioma EV biology in a genetically defined context. The inaccessible, sensitive location of GBMs necessitates the development of a rapid and dynamic biomarker-based diagnostic modality to detect disease progression, response to treatment and guide therapeutic options. Plasma-accessible circulating EVs and RNPs from GBM patients represent an excellent modality for liquid biopsy-based biomarker development.
In this study, we applied a physical separation method to isolate EVs and RNPs and profiled their proteomic and transcriptomic contents from EGFR- and PDGFRA-driven mouse primary GBM cell cultures. We identified exPTNs and exRNAs that are enriched in EVs and RNPs relative to EV subtype. Our data contribute to the foundation to a better understanding of heterogeneity of EVs.
RESULTS
Cellular Transformation of Genetically Defined Primary Astrocytes Reduces EV Secretion
Cancer patients have been observed to present with higher circulating EV concentrations than normal donors (Logozzi et al., 2009), suggesting that cancer cells produce more EVs than their normal counterpart cells. To test whether this is a cell autonomous effect, we directly ascertained whether EV secretion is increased during the process of cellular transformation using nano-particle tracking analysis (NTA) and determined the size and numbers of EVs secreted from normal astrocytes and during their immortalization and transformation into GBMs. For this, we established normal mouse primary astrocyte cultures from conditional p53lox/lox (Marino et al., 2000) P0 mice and used them to derive isogenic immortalized p53−/− astrocytes through infection with adenovirus-Cre. Changes in EV release dynamics brought about by adenovirus infection were controlled for by infecting the p53lox/lox cultures with an adeno-GFP virus. Immortalized Cdkn2a−/− primary astrocytes were generated from the constitutive Cdkn2a null mice (Serrano et al., 1996).
Using NTA, we determined the number (Figure 1A) and size distribution (mode vesicle size) (Figure 1B) of vesicles within conditioned media collected from senescent p53lox/lox and p53lox/lox Ad-GFP astrocytes, immortalized p53−/− and Cdkn2a−/− astrocytes and several primary cultures of mouse GBMs established from our classical/mesenchymal EGFR; Cdkn2a−/−; PTEN−/− (Acquaviva et al., 2011; Jun et al., 2012; Zhu et al., 2009) and proneural PDGFRA;p53−/− (Jun et al., 2018; Zhou et al., 2018) genetically engineered mouse GBM models. We found that infection with adenovirus did not affect the secretion of EVs (control 47,554 ± 33,073 vesicles/mL/cell compared with Ad-GFP 60,922 ± 59,612 vesicles/mL/cell) but that immortalization of cells through loss of p53 or Cdkn2a function resulted in a 5-fold decrease in vesicle release (9,530 ± 3,599 vesicles/mL/cell, and 10,405 ± 3,586 vesicles/mL/cell respectively). Comparatively, fully transformed cells shed on average 17-fold fewer vesicles than normal astrocytes (EGFR 2,077 ± 2,290 vesicles/mL/cell, PDGFRA 2,251 ± 2,210 vesicles/mL/cell) (Figure 1A). Although the number of vesicles released by a given primary cell culture differs within a genotype in a culture-specific manner, the ranges between the EGFR and PDGFRA genotypes are comparable and not statistically different. We then analyzed the average size of EVs released by normal and immortalized astrocytes and the genetically engineered mouse glioma lines. We found that EV average sizes are statistically similar among the cultures with a trend toward slightly larger EVs from transformed GBM cultures when compared with normal or immortalized astrocytes (mode vesicle size: normal wild-type astrocytes 105.91 ± 18.71 nm, EGFR 142.71 ± 16.0 nm, and PDGFRA 162.70 ± 26.34 nm) (Figure 1B). Unexpectedly, PDGFRA GBM-derived vesicles are slightly larger than EGFR GBM-derived and normal and immortalized astrocyte vesicles (Figure 1B). Note that the culture conditions did not result in significant changes in growth or viability of the cells suggesting that the observed decreases in EV release are less likely due to cellular stress or excessive cell death (Figure S1). These results demonstrate that during the process of immortalization and transformation, cells secrete fewer EVs, and those EVs trend to slightly larger sizes. To explore genotype-specific RNA and protein cargos of various sized EVs, we selected a primary cell culture from each genotype that represented a mid-range of EVs released (EGFR wild-type [WT]-1, PDGFRA-1) for further studies.
Figure 1. Extracellular Vesicle Analysis and Separation.

(A and B) Nanosight nanoparticle tracking analysis (NTA) of primary mouse astrocytes and GBM cell cultures of the indicated genotypes. Multiple EGFR wild-type (WT), mutant EGFRvIII, and PDGFRA-driven primary cultures were analyzed. Each cell culture was grown in triplicate and each triplicate was individually imaged three times (30 s each). The p532lox, p532lox+AdGFP, p532lox+AdCre, and Cdkn2a−/− cultures were performed on two independent cell cultures for each and results were pooled. The individual EGFR and PDGFRA GBM lines were analyzed on at least two independent repeats on separate days. Boxplots represent an average of three NTA readings per sample and are plotted as quartiles of the dataset for each cell culture. Averages were analyzed for statistical significance using Welch’s unequal variance’s t test, two tailed, compared with control astrocytes.
(A) Number of vesicles produced per volume of conditioned media normalized to number of cells over a period of 24 hours of incubation within vesicle-depleted culture media. *p = 0.0003, **p = 7.79 × 10−5.
(B) Mode vesicle size for the indicated cell cultures as in (A). *p = 2.35 × 10−8, **p = 2.56 × 10−13.
(C) Schema of vesicle isolation procedure by sequential filtration. Cell culture supernatants from primary glioblastoma cell lines were cleared of cellular debris and dead cells by centrifugation, and vesicles were partitioned by filtering through consecutively smaller pore-sized filters (2 μm,0.8 μm, 0.22 μm, 0.02 μm) and free protein complexes were concentrated with a 3 kDa size exclusion column. Proteins (blue) were isolated from the 0.8 μm, 0.22 μm, 0.02 μm, and 3 kDa fractions and RNA (red) was isolated from the 0.22 μm, 0.02 μm, and 3 kDa fractions and processed for proteomics and RNA-seq, respectively.
(D) Representative photomicrograph of western blotting protein markers that specify different vesicle populations. Equal loading of total protein for each fraction was determined by bicinchoninic acid (BCA) and samples analyzed by silver staining and adjusted accordingly to ensure equal protein loading. Western blot was repeated at least twice and samples originated from a mouse EGFR WT primary GBM culture.
(E) Representative photomicrographs of negative stain transmission electron microscopy (TEM) of EVs from ultracentrifugation (UC) and filter isolation. Vesicles in the size range of the filter pore sizes are observed in the LEV (0.8 μm), MEV (0.22 μm) and SEV (0.02 μm) fractions. Scale bars: 500 nm; SEV inset: 25 nm. TEM was repeated at least twice on biological replicates from a mouse EGFR WT primary GBM culture.
(F) Frequency and size (vesicle diameter in nm) distribution of vesicles from negative stain TEM in (E). The number of vesicles measured: SEV n = 118; MEV n = 51; LEV n = 30; and UC n = 291. The median size in nm for each category is indicated with an asterisk (*).
See also Figure S1.
Sequential Filtration-Based Isolation of Extracellular Vesicles
The size and composition of EVs are heterogeneous in nature due to their bio-origin (Théry, 2011). Current EV and macromolecular cargo (protein, DNA, and RNA) isolation protocols include ultracentrifugation, precipitation using polymers (e.g., polyethylene glycol) or fractionation including density gradient and gel filtration (Colombo et al., 2014; Konoshenko et al., 2018). These techniques have disadvantages when examining EV contents. Ultracentrifugation generally results in a highly heterogenous output of EVs, and fractionation approaches are typically associated with low yields of material. To better define the relationships among cargo and EV size and provenance and to separate EVs and membrane-free RNP complexes, we utilized a sequential filtration protocol with decreasing pore sizes (Wei et al., 2017) to separate large extracellular vesicles (LEVs; ≥ 0.8 μm), medium extracellular vesicles (MEVs; ≥ 0.22 μm), small extracellular vesicles (SEVs; ≥ 0.02 μm, including exosomes) and a 3kDa-concentrated non-vesicular RNP fraction to study their protein and exRNA cargo content (Figure 1C). We performed this technique from condition media harvested from EGFR-positive and PDGFRA-positive mouse GBM cells and immunoblotted proteins harvested from these fractions for established EV-enriched markers to confirm the subtype enrichment of the fractions and the absence of cellular contamination in the EV and RNP fractions (Figure 1D).
Negative stain transmission electron microscopy (TEM) applied to vesicle preparations captured on the 0.8 μm,0.22 μm, and 0.02 μm filters revealed size and morphology consistent with those reported for LEVs, MEVs, and SEVs (exosomes), respectively (Figure 1E) (Hurwitz et al., 2016a; Rider et al., 2016; Wei et al., 2017). TEM also showed a more uniform distribution in size in the filtrates compared with the complex, heterogeneous mixture observed in 100,000 ×g ultracentrifugation (UC) (Figure 1E). Quantification of size distributions of vesicles from TEM for each filter fraction and UC (100,000 × g) reveals a separation of vesicles accordingly (Figure 1F). Together, these results validate the filtration protocol approach for effectively separating vesicles by subtypes from mouse primary GBM cell cultures and allows for in-depth analysis of cargo content.
Mouse GBM EVs Contain Specific Protein Cargo
We performed quantitative tandem mass spectrometry (TMT) on cellular, LEV, MEV, SEV (containing exosomes), and RNP fractions isolated from our EGFR;Cdkn2a−/−; PTEN−/− and PDGFRA;p53−/− GBM cell cultures to characterize the differential protein content of EVs and to unveil protein markers of vesicle subtypes. Although the cells are cultured in low bovine serum (0.1% v/v) prior to harvesting EVs, we noticed that there remains a substantial amount of BSA in our EV preparations (Figure S2A), a suboptimal condition for TMT. To eliminate contamination from serum albumin, we used the methionine analog azidohomoalanine (AHA) to label newly synthesized cellular proteins that can be purified using click chemistry-based methods (Ma and Yates, 2018). Detection of AHA-biotin-labeled proteins using IR800-labeled streptavidin confirms the absence of labeled BSA in all EV fractions harvested from conditioned media and demonstrates the labeling of a wide range of proteins (Figure S2A). This versatile approach also allows for affinity purification of AHA-labeled proteins for downstream MS application. AHA-labeled proteins from EGFR;Cdkn2a−/−; PTEN−/− and PDGFRA; p53−/− GBM cell cultures and their corresponding LEV, MEV, SEV, and RNP fractions were purified and analyzed by isobaric label-based quantitative mass spectrometry (MS) to measure expression of proteins (Data S1) (McAlister et al., 2014; Ting et al., 2011). We observed that the yield of peptides from the MEV fraction (≥ 0.22 μm) was consistently lower than from the other fractions, which introduced normalization biases in the depth of the quantitative analysis. Since there were no uniquely enriched proteins in this fraction when compared with LEVs, the MEVs were excluded in repeat experiments (Figures S2B and S2C; Data S1). In doing so, a total of 2,178 proteins were reproducibly identified in the cellular, LEV, SEV (exosomes), and RNP fractions of both the EGFR;Cdkn2a−/−; PTEN−/− and PDGFRA;p53−/− GBMs.
We found that most of the proteins were more abundant in the cellular fractions than in the EV fractions (Figure S2D), though it is noteworthy to point out that proteins were selected for and identified in the TMT data by their presence across all the fractions. Therefore, this dataset represents a catalog of proteins that are reproducibly found in EVs, even in the cases where the abundance is greater in the cellular fraction. Of the 2,178 proteins, there were 366 proteins that were consistently present at higher levels in EVs than in cells (Figure S2E). Proteins with a reproducible fold change (FC) log2 > 1 in EVs relative to cells at a p value < 0.05 were considered to be enriched, and proteins appearing in multiple fractions were filtered to reveal 366 unique proteins exported to vesicles and/or RNPs. Unsupervised clustering of these proteins displays vesicle-specific grouping, indicative of differential protein content in LEVs, SEVs (exosomes), and RNPs (Figure 2A).
Figure 2. Distinct Proteome Composition of Filtered Extracellular Vesicles.

(A) Unsupervised hierarchical clustering analysis of Z score mean protein expression values for EV-and RNP-enriched proteins. A combined dataset of 366 proteins significantly enriched over cellular fraction was derived from each vesicle-RNP fraction from the 2,178 dataset to create the heatmap. Location of proteins used in (C) below are indicated in right margin.
(B) Pearson correlation coefficients of a pairwise analysis of expression levels between cellular, EV, and RNP levels from the EGFR and PDGFRA GBM cell cultures. The values of the coefficient are indicated and are marked with background colors. Pearson correlation coefficients > 0.91 and < −0.91 are significant (p < 0.05) and correlation coefficients > 0.7 and < −0.7 are trend suggestive.
(C) Validation of MS data by immunoblot analysis of EV- and RNP-specific proteins derived from (A).
See also Figure S2.
Pairwise Pearson’s correlation of genotype-normalized, relative abundance values showed that the protein content of each fraction was more similar by the type of vesicle, rather than by the genotype of the GBM cultures (Figure 2B). Western blotting of representative vesicle-enriched proteins along with canonical EV (exosome) markers CD81 and CD63 confirmed the MS data (Figure 2C). These results suggest the existence of mechanisms that specifically partition proteins into vesicles and RNPs as our data show that protein cargo composition of EVs is not a simple reflection of cellular levels. Additionally, EV content is not solely dictated by the genotype of the GBM but also by the vesicle type. The EV-enriched proteins that we observed may represent vesicle type specific biomarkers.
To gain insight into potential functional roles of the vesicle type-enriched proteins, we used ontology and pathway analyses using DAVID (Database for Annotation, Visualization, and Integrated Discovery) (v6.8) and the EV subtype-enriched protein lists derived from our 366 proteins dataset. We investigated which protein classes were significantly (p value < 0.05) increased in abundance among LEVs, SEVs (exosomes), RNPs, and cells. Analyzing the Biological Process category with the LEV, SEV (exosomes), and RNP proteins, we observed that proteins associated with cell adhesion were present in all three fractions, and proteins associated with angiogenesis and extracellular matrix were observed preferentially in LEV and SEV fractions (Figure S2F). Interestingly, we observed proteins that characterized several biological processes that were observed in only one fraction subtype. For example, SEVs (exosomes) contained proteins that associated with response to hypoxia, glycosaminoglycan biosynthetic process, integrin-mediated signaling pathways, positive regulation of cell substrate adhesion, and negative regulation of TGFβ signaling pathways. On the other hand, the RNPs contained proteins that were strongly associated with semaphorin-plexin signaling pathway, negative chemotaxis, negative regulation of axon extension in axon guidance, negative regulation of endopeptidase activity, regulation of cell growth, negative regulation of peptidase activity, cell migration, wound healing, and axon guidance. We also found that protein markers of extracellularity (extracellular matrix, region, space), basement membrane as well as extracellular exosomes were enriched in all three fractions (Figure S2F). In addition, SEV proteins associated with groups of proteins that represent the following categories; membranes, cell surface, Golgi apparatus, endoplasmic reticulum, and extracellular vesicles to name a few. Proteins involved in calcium ion binding, heparin binding, integrin binding and insulin-like growth factor binding were found in all three fractions (Figure S2F). Interestingly, we noticed that proteins associated with chemoattractant activity were observed in LEVs only whereas proteins associated with chemorepellent activity were found in RNPs only. This antithetic association perhaps reflects function of RNPs and LEVs in our mouse GMB cells.
Protein Cargo from Filtered SEVs Is Similar to Ultracentrifuged EVs
By far the most commonly used approach to isolate EVs is through differential UC. Larger vesicles are first pelleted at 10,000 × g, and smaller vesicles are isolated from a subsequent 100,000 × g for 2–24 hours (Konoshenko et al., 2018). To determine the degree of similarity between UC isolated vesicles and our differential filtration approach, we subjected conditioned media from AHA-treated EGFR and PDGFRA cultures as described above to 10,000 × g and 100,000 × g UC to isolate EVs and harvested AHA-labeled proteins for TMT quantitative proteomic analysis. In doing so, a total of 1,130 proteins were reproducibly identified in the cellular, 10,000 × g, and 100,000 × g fractions of both the EGFR;Cdkn2a−/−; PTEN−/− and PDGFRA;p53−/− GBMs.
Similar to the filtered fractions above, we found that most of the proteins were more abundant in the cellular fractions than in the UC pellets (Figure 3A). Again, the identified proteins represent those that are reproducibly found in all fractions, regardless of enrichment. Like the filter isolation, pairwise Pearson’s correlation of genotype-normalized, relative abundance values showed that the protein content of each UC fraction was more similar by the UC fraction, rather than by the genotype of the GBM cultures (Figure 3B). Of the 1,130 identified, 407 unique proteins were exported to either or both 10,000 × g and 100,000 × g UC with a reproducible FC log2 > 1 in EVs relative to cells at a p value < 0.05 (Figure S3A). Unsupervised clustering of these proteins displays UC vesicle-specific grouping regardless of genotype, indicative of differential protein content in 10,000 × g and 100,000 × g UC vesicles (Figure 3C). These results reinforce the concept that biological mechanisms are involved to specifically partition proteins into different sized vesicles and that protein cargo composition of EVs is not a simple reflection of cellular levels.
Figure 3. exPTNs from Ultracentrifugation EVs Overlap with SEVs.

(A) Unsupervised hierarchical clustering analysis of the z-score proportional protein expression values of 1,130 proteins identified in cells, 10,000 × g and 100,000 × g protein fractions. MS spectral count values were Z score normalized prior to cluster analysis.
(B) Pearson correlation coefficients of a pairwise analysis of expression levels between cellular, 10,000 ×g and 100,000 × g protein levels from the EGFR and PDGFRA GBM cell cultures. The values of the coefficient are indicated and are marked with background colors. Pearson correlation coefficients > 0.91 and < −0.91 are significant (p < 0.05) and correlation coefficients > 0.7 and < −0.7 are trend suggestive.
(C) Unsupervised hierarchical clustering analysis of Z score protein expression values for UC-enriched proteins. A combined dataset of 407 proteins significantly enriched over cellular fraction was derived from each UC fraction from the 1,130 dataset to create the heatmap.
(D) Venn diagram of unique and common proteins identified in filtered and UC EVs isolated from conditioned medium from EGFR and PDGFRA GBM cells. Data represent three independent biological replicates.
(E) Unsupervised hierarchical clustering analysis of Z score mean protein expression values for vesicle enriched proteins from filter and UC-enriched proteins. The combined dataset of 993 proteins from (D) was used create the heatmap.
(F) Pearson correlation coefficients of a pairwise analysis of expression levels of the 993 proteins shared between cellular, LEV, SEV, and 10,000 × g and 100,000 × g from the EGFR and PDGFRA GBM cell cultures. The values of the coefficient are indicated and are marked with background colors. Pearson correlation coefficients > 0.91 and < −0.91 are significant (p < 0.05), and correlation coefficients > 0.7 and < −0.7 are trend suggestive.
(G) Unsupervised clustering of 30 proteins, displayed on a vesicle-type basis, extracted from the 181 EV expressed proteins not previously described in Vesiclepedia or NCI-60 vesicle proteomic datasets.
(H) Unsupervised clustering of the 9 UC-unique proteins, displayed on a UC fraction-type basis.
See also Figure S3.
To compare the filtration method with UC, we first determined the overlap of proteins identified between the two methods. We found that 87.9% (993/1,130) of the proteins detected in the UC vesicles were also present in the filtered pools (Figure 3D). Unsupervised clustering of genotype-normalized mean expression values of the cells and vesicle fractions demonstrate overlap between the isolation techniques (Figure 3E). The 100,000 × g vesicles more closely resemble the SEVs (≥ 0.02 um), with the 10,000 × g fraction diverging from these exosome-containing isolates. The LMVs constitute vesicles with a unique proteomic profile, largely excluded from the SEVs, but with constituents found in both the 10,000 × g and 100,000 × g (Figures 3E and 3F). Accordingly, representative proteins revealed to be enriched in the LEV (Dnajb9, Dnajb11, Sec24d) separate in the 0.02 μm in the 0.8 μm fraction, but not between 10,000 × g and 100,000 × g fractions. By contrast, canonical exosomal markers (CD9, CD81, and CD63) are found enriched in both the 0.02 μm and 100,000 × g, but not in the 0.8 μm and 10,000 × g fractions (Figure S3B).
Finally, a comparison of our filtration and UC vesicle MS protein content datasets with those of Vesiclepedia (Kalra et al., 2012) and the NCI-60 reference cancer panel cell lines (Hurwitz et al., 2016b) demonstrated that 91.6% (1,972/2,153) of the proteins identified in our filtrated EVs and 96.6% (1,086/1,124) of the proteins identified in our UC EVs have been reported in those studies (Figure S3C). Of the proteins we identified, 152 from filtered EVs alone, 29 from filtered and UC EVs, and 9 from UC EVs alone (for a total of 190 proteins) were not present in Vesiclepedia nor observed in the NCI-60 databases (Figure S3C). Of these 181 filtered and UC proteins, 16.6% (30/181) were enriched in LEVs, SEVs (exosomes), RNPs, 10,000 × g and 100,000 × g when compared with cellular levels, and 6 out of the 9 UC-only proteins were enriched in UC vesicles (Figures 3G and 3H). Overall, comparative analysis of our datasets to curated, public EV MS-based datasets not only validated our MS data but also added valuable information to the existing protein EV databases.
Genotype-Centric Protein Contents in Vesicles
In addition to the robust inter-vesicle differences in protein cargo composition that we observed, there also exists genotypic differences in protein enrichment within each vesicle type between the EGFR;Cdkn2a−/−; PTEN−/− and PDGFRA;p53−/− GBM cultures. Analyzing inter-genotype protein cargo differences on a vesicle specific level, we identified proteins that are enriched in both a vesicle-centric and genotype-specific manner (Figures 4A and 4B; Tables S1–S4). We compared EGFR with PDGFRA-derived fractions (cells, LEVs, SEVs [including exosomes], RNPs, 10,000 × g and 100,000 × g) for significant enrichment (cut-off set to a FC of ± 2 (− 1 ≥ log2 FC ≥ 1). The overlap of significantly enriched proteins between the fractions is limited. Most proteins displayed significant enrichment in only one fraction, and few proteins displayed enrichment in more than one fraction (Tables S1–S4). Notably, we observed proteins that are genotype- and fraction-enriched. For example, Ptx3, Fbln1, Efemp1, Igfbp2, and Igfbp6 are highly enriched in PDGFRA LEVs, SEVs (including exosomes), and RNPs but not in cells. Similarly, Tpm1 and Tpm3 are enriched in PDGFRA SEVs (including exosomes), and Tfpi and Dkk3 are enriched in PDGFRA SEVs (including exosomes) and RNPs (Table S1). Conversely, Mmp1a, Postn, Gfra2, Igfbp3, and Gdf15 are highly enriched in EGFR LEVs, SEVs (including exosomes), and RNPs. Serpine 1 and 2, Klk10, and Pcdh20 are enriched in EGFR SEVs (including exosomes) and RNPs, and Sema6a, L1cam and Spp1 are enriched in EGFR RNPs (Table S1). Similarly, we observed proteins that are enriched in 10,000 × g and 100,000 × g UC vesicles in a genotype-centric manner (Figure 4B; Tables S3 and S4). Combining the genotype-specific protein differences observed with the vesicle-specific EV markers represents a valuable approach in characterizing the genotypic makeup of the tumor cells from which they originate.
Figure 4. Genotype-centric EV Markers.

(A) Volcano plot representations of the differentially expressed proteins in a pairwise comparison of EGFR to PDGFRA cells, LEVs, SEVs (exosomes), and RNPs. The significance cut-off was set to a FDR of 0.05 (−log(adj.P.val ≥ 1.3), the biological cut-off was set to a fold change of ± 2 fold (−1 ≥ log2 FC ≥ 1), the top and bottom 10% differentially expressed proteins are labeled with their corresponding gene ID. The four different color codes used represent insignificant proteins (gray), both biologically and statistically significant proteins preferentially enriched in PDGFRA cells-EVs-RNPs (blue) and preferentially enriched in EGFR cells-EVs-RNPs (red), and statistically but not biologically significant proteins enriched in EGFR or PDGFRA (black).
(B) Volcano plot representation of the differentially expressed proteins in a pairwise comparison of EGFR to PDGFRA cells, 10,000 ×g and 100,000 ×g ultracentrifuged EVs, analyzed as in(A) above.
Single Vesicle Analysis Reveals Heterogeneity within and between Fractions
EVs are heterogeneous in nature, both physically (size, charge distribution) and molecularly (lipid composition, cargo content). Bulk proteomic-based assessment of vesicle composition offers a discovery window for potential markers that are associated with vesicle sizes and types. However, proteomics does not address distribution and quantification of a given marker expression on a per vesicle basis. To ascertain the degree of heterogeneity within given vesicle-sized fractions, we used single EV analysis (SEA) (Lee et al., 2018) on LEV and SEV (including exosomes) fractions isolated from EGFR-positive mouse GBM tumor cell cultures for expression of the canonical EV markers CD81 and CD9. In addition, we extracted two markers from our proteomics, Gja1 (GAP junction alpha 1, also known as conexin43) as a protein preferentially enriched in LEVs and SEVs (including exosomes) and Nono (non-POU domain-containing octamer-binding protein) as a protein enriched in LEV fraction (Figure S4A). We first characterized LEVs and SEVs based on paired expression of CD81 with CD9 and Nono with Gja1 (Figures 5A, 5B, and S4B). We found that in the SEV preparation,95.9% of vesicles were singly positive for CD9, 3.3% were singly positive for CD81, and 0.8% were double positive (Figure 5B). This suggests that filtration-prepared SEVs (including exosomes) are almost uniformly CD9 positive. This is less true in the LEV-filtered vesicles fraction where we observed that 48% of vesicles expressed CD9, 50.8% CD81, and very few (1.2%) of vesicles dually expressed CD9 and CD81. In both LEVs and SEVs (including exosomes), the degree of vesicles that are dually expressing CD9 and CD81 is very low, perhaps suggesting mechanistic reasons for this mutual exclusiveness.
Figure 5. Single Vesicle Analysis of EV Fractions.

(A) Representative photomicrographs of immunofluorescence labeling of EV markers from large extracellular vesicles (LEVs) and small extracellular vesicles (SEVs) filtered fractions isolated from EGFR primary cultures. Pixel shift controls for co-registration of markers in overlay.
(B) Quantitative analysis (percentage) of singly or dually positive vesicles on a per vesicle basis for expression of CD9 and CD81 or Nono and Gja1 in LEV and SEV fractions.
(C) Quantitative analysis of singly, dually or triple combinations of indicated markers on a per vesicle basis for expression of CD9, CD81, Nono, and Gja1 in LEV and SEV fractions.
A total of 643 vesicles were analyzed from the LEV fraction and 1,648 vesicles were analyzed for the SEV fraction.
See also Figure S4.
Our proteomics results demonstrated that Nono is highly enriched in LEVs and less in SEVs (exosomes), whereas Gja1 is enriched in both LEVs and SEVs (exosomes) (Figure S4A). At the single vesicle level, we observed substantially more Nono-positive vesicles in LEV than in SEV (exosomes) fractions (20.2% versus 1.1%) (Figure 5B) and relatively similar levels of Gja1-positive vesicles in both LEV and SEV (exosomes) fractions (72.3% versus 93.6%) (Figure 5B). We further refined our analysis of single vesicle marker expression by combining all four markers into one analysis. Both LEV and SEV (exosomes) fractions contained a significant number of vesicles that were single positive for Gja1 and double-positive for Gja1 and CD9 (Figure 5C). Fewer vesicles were Gja1 and CD81 double-positive than Gja1 and CD9 double-positive. Similarly, co-expression of CD9 or CD81 with Nono was restricted to CD9, with very few (1% in LEV and absent in SEV [exosomes]) CD81:Nono double-positive vesicles.
Finally, most of the Gja1:Nono double-positive vesicles also expressed CD9 but not CD81 (Figure 5C). These results suggest that CD9- and CD81-positive vesicles appear to be produced in a mutually exclusive manner, that SEVs (exosomes) are predominantly CD9 positive, and that approximately half of LEV vesicles are either CD9 or CD81 positive. Using two proteomics-derived markers (Gja1 and Nono), we further examined their exclusiveness at the single vesicle level to discover that very few are double-positive. This approach of combining filtered vesicles bulk proteomics to single vesicle analysis offers an opportunity to better understand the relationship between molecular and physical heterogeneity and highlights the need for a higher resolution, single vesicle approach in EV-based biomarker analysis.
The Extracellular RNA Landscape of EVs and RNPs Exhibit Distinct Composition
To complement the proteomic characterization of cellular, EV, and RNPs released from our EGFR and PDGFRA-derived mouse GBM primary cultures, we isolated cellular and vesicle extracellular RNA (exRNA) under the same vesicle filtration conditions described above and determined the cellular and exRNA landscapes of long and short RNAs using RNA sequencing (RNA-seq). The amount of exRNA released ranged between 220 and 8400 pg/mL accumulated in culture over 48 hours (Figure S5A). When compared with the amount of cellular RNA isolated from the producing cells (10 million cells, between 10 to 25 mg), roughly 0.004% to 0.15% of cellular RNA depending on the vesicle fraction, is accumulated in the extracellular space in 2 days, which consistent with previous observations (Wei et al., 2017). The total exRNA yield did not vary substantially among the EGFR and PDGFRA cultures (Figure S5A) and the proportion of exRNA associated with different extracellular complexes also did not vary between the cultures, suggesting a consistency in the properties of vesicular RNA release of the two genotypes. We and others have previously demonstrated that the major RNA species of EVs is ribosomal RNA (rRNA) (Wei et al., 2017). As a result, the RNA samples were processed for rRNA depletion before long and small RNA libraries were created from cellular and extracellular complexes (MEVs, SEVs [exosomes], and RNPs) and deep-sequenced to determine the repertoires of cellular and extracellular RNAs (Figure S5B). The number of reads for each RNA class were normalized to the total number of non-rRNA reads within the library, thus alleviating effects due to variability in rRNA depletion efficiencies.
EVs and RNPs Exhibit Distinct RNA Composition
Comprehensive sequencing analyses of the long RNA libraries for both cellular and extracellular RNAs derived from both genotypes revealed that between 67% to 92% of the reads from the RNA libraries mapped to the genome (Figure 6A). The unmapped reads consisted largely of sequences that contained mismatches to genomic sequences. We classified all reads generated from the long RNA libraries (generally transcripts > 100 nt) as mRNA, pseudogene, long non-coding RNA (lncRNA), mitochondrial mtRNA, RNA decay, intronic, small RNA, or other RNAs (Figure 6A; Table S5). In both EGFR and PDGFRA cultures, the most abundant RNA species were lncRNA and mRNA reads, accounting for 51%–94% and 6%–23% of long RNA libraries total reads, respectively (Figure 6A). The reads mapping to mRNAs are likely to originate from mRNA fragments as we have previously shown that full-length mRNAs of approximately < 1000 nt can be amplified from EVs, but larger full-length mRNAs are not consistently amplified from EVs, suggesting that most exRNA reads correspond to long mRNAs transcripts that are fragmented (Wei et al., 2017).
Figure 6. Relative Composition of Long RNA Classes in Cellular and EV Fractions.

(A) Long RNA sequencing analysis of cellular, MEV, SEV (exosomal), and RNP RNAs from EGFR and PDGFRA GBM primary cultures. The top panel shows total read numbers of mappable reads (dark gray) and unmapped reads (red). The numbers above each column represent the percentage of mappable reads. The bottom panel shows the relative RNA composition in long RNA libraries. The data were normalized to the total number of annotated non-rRNA reads. Details of individual RNA species making up the RNA categories are found in Table S5.
(B) Unsupervised hierarchical clustering analysis of Z score vesicle-enriched lncRNA expression values for cells, EV- and RNP-enriched lncRNAs in EGFR, and PDGFRA GBM cultures.
(C) Degree of EV/RNP enrichment for selected lncRNAs. RNA-seq read counts for annotated lncRNAs were normalized for each sample. For each lncRNA, cell values were subtracted from MEVs, SEVs (exosomal), and RNPs. Positive values (green) represent enrichment for a given lncRNA in the indicated fraction, and negative values (red) represent depletion.
The MEV Fraction Most Closely Reflects Cellular RNA Composition
Circulating cancer-derived exRNA can serve as biomarkers for diagnostics, prognostics, and monitoring of disease. Knowing which EV fraction most closely mirrors the aberrant cellular transcriptomes of cancers would be most valuable for such applications. To examine which extracellular complexes (MEVs, SEVs [exosomes], or RNPs) represent the closest proxy of GBM cellular transcriptomes, we performed pairwise Pearson correlations analyses of their RNA composition. Using mRNA read data for each class showed that for both EGFR and PDGFRA, the MEV mRNA fractions were more closely related to their corresponding cellular mRNA fractions than either the SEV (exosomal) or RNP fractions (Figure S5C).
The observation that the MEV fraction provides a more accurate peripheral readout of the source cell mRNA content than SEV (exosomes) and RNPs holds true using human glioma stem cell cultures (GSCs) mRNA sequences. In all four cultures examined, the Pearson correlation coefficient of MEV is higher than that of SEVs (exosomes) and RNPs and in line with the coefficients observed in mouse EGFR and PDGFRA GBM MEVs (Figure S5D). Overall, MEVs appear to be a suitable source for exRNA biomarker discovery, and are better representative of their cell of origin and have the potential to outperform SEVs (exosomes) as vehicles of extracellular mRNA.
Enrichment of lncRNA Species in Vesicles
By far, the most common RNA species observed in our EGFR and PDGFRA GBM libraries from cells, MEVs, SEVs (exosomes), and RNPs are lncRNAs. lncRNAs have recently garnered significant attention owing to their multifunctional faceted roles in many aspects of biology (Kopp and Mendell, 2018; Schmitt and Chang, 2016). To identify lncRNA species that potentially play a role in modulating the GBM microenvironment, we focused on lncRNAs that are preferentially enriched in MEVs, SEVs (exosomes), and RNPs when compared with their cellular expression levels. Unsupervised hierarchical clustering analysis of z-score extracellular compartment-enriched lncRNA expression values demonstrate distinct clusters segregating according to genotype (EGFR versus PDGFRA) and fraction (cells, MEVs, SEVs [exosomes], and RNPs) (Figure 6B). As a substantial number of lncRNAs remain to be characterized, we highlight species with known function (Figure 6C). Gas5 and Snhg1 both appear to be solely found in EGFR MEVs whereas Snhg12, Pvt1 and Lncpint are enriched in EGFR GBM-derived SEVs (exosomes). Snhg5 is highly enriched in EGFR SEV (exosomes) and PDGFRA MEVs. Mkln1os is observed only in PDGFRA SEVs (exosomes) and Dancr and Jpx are enriched in both EGFR and PDGFRA RNPs and to a lesser extent in SEVs (exosomes) of both genotypes (Figure 6C). These results demonstrate that the lncRNA composition of MEVs, SEVs (exosomes), and RNPs is not uniform depending on the genotype of the cell, suggesting a differential mechanism for lncRNA loading into EVs that depends on the oncogenic driver of the GBM.
Diversity in Extracellular Small RNAs
Sequencing reads of the small RNA libraries for both cellular and extracellular RNAs derived from EGFR and PDGFRA genotypes were mapped to the most accurately annotated miRNA database (miRBase), and subsequently to other databases with equal mapping priority. In total, all annotated RNA species were categorized into 12 classes (Figure 7A; Table S6). Between 82% and 96% of the reads from the RNA libraries mapped to the genome (Figure 7A). Overall, miRNAs accounted for the majority of reads (between 41% and 93%) in both EGFR and PDGFRA cultures of cellular, MEV, and SEV (exosomes) fractions (Figure 7A) but were relatively enriched in the cellular and SEV (exosomes) fractions. qRT-PCRs quantification of selected miRNAs correlated well with the distribution between the cellular and SEV (exosomes) fractions seen in the RNA-seq results (Figures S6A–S6C), supporting the accuracy of our RNA-seq analysis.
Figure 7. Relative Composition of Short RNA Classes in Cellular and EV Fractions.

(A) Short RNA sequencing analysis of cellular, MEV, SEV (exosomal), and RNP small RNAs from EGFR and PDGFRA GBM primary cultures. The top panel shows total read numbers of mappable reads (dark gray) and unmapped reads (red). The bottom panel shows the relative RNA composition of the small RNA libraries. The data were normalized to the total number of annotated non-rRNA reads. Details of individual RNA species making up the RNA categories are found in Table S6.
(B) Unsupervised hierarchical clustering analysis of z-score vesicle-enriched tRNA gene clusters expression values for cells, MEV-, SEV- (exosomal), and RNP-enriched tRNAs in EGFR and PDGFRA GBM cultures. Individual tRNA genes making up the tRNA categories are found in Table S7.
(C) Depth plot of a representative example of relative abundance (on a per nucleotide-basis) of a given tRNA sequence (Gly-GCC) gene cluster, demonstrating precisely processed fragments of tRNA in cells and RNPs.
(D) Graphical representation of expression levels by qRT-PCR of 5′ and 3′ directed custom Taqman probes for Gly-GCC tRNA gene from RNA isolated from EGFR and PDGFRA GBM primary cultures of cells and RNPs. Bar graphs represent an average of three qRT-PCR readings per sample. Averages were analyzed for statistical significance using Student’s t test, two tailed. *p < 0.0001, **p = 0.001, ***p = 0.005, and ****p = 0.012.
Non-vesicular RNPs are highly enriched in tRNA gene sequences, making up 36% and 51% of EGFR and PDGFRA RNP fractions, respectively (Figure 7A). EGFR-driven culture RNPs are also enriched in Y RNA fragments, a species of RNA of largely unknown function. This analysis of our RNA-seq data indicates that RNP fractions have a highly distinctive RNA repertoire when compared with cellular, MEV, and SEV (exosomes) fractions. Whereas tRNA and Y RNA fragments dominate the composition of RNPs, miRNAs are more prevalent in cells, MEVs, and SEVs (exosomes), an observation in agreement with our previously published results in human GBM (Wei et al., 2017).
tRNA Fragments Are Abundant and Enriched in RNPs
Close to half of the mappable reads in RNPs mapped to tRNA gene sequences. tRNA genes with common anti-codons have highly similar sequences, and because the read lengths are shorter than the entire tRNA sequences, we pooled tRNA genes into clusters based on anticodons and sequence similarity (Pantano et al., 2016) (Table S7). Unsupervised clustering analysis of vesicle-RNP-enriched tRNA gene clusters demonstrates an RNP-centric over-abundance of tRNA fragments (Figure 7B; Figure S6D).
Because tRNAs are 68–176 nt and the libraries were constructed from 15–65 nt RNAs, we reasoned that the reads mapping to tRNAs represent fragments rather than full-length transcripts. This led to the analysis of the coverage of the reads mapped to full-length tRNAs and revealed the presence of precisely processed fragments with differential levels in cells and RNP fractions (Figure 7C). Using Gly-GCC as an example, we validated the observed expression levels between cells and RNPs for both 5′ and 3′ tRNA fragments (TRFs) by qRT-PCR (Figure 7D). tRNA fragments processing sites are located within the loop domains that are known to bind several proteins (Köhn et al., 2013; Kowalski and Krude, 2015; O’Brien et al., 1993) and are the targets of known RNases (Figure S6E) (Balatti et al., 2017; Kumar et al., 2016; Lyons et al., 2018). Our data demonstrate that RNPs are enriched in TRFs, suggesting a role for these fragments in intercellular communication.
DISCUSSION
EVs are highly heterogeneous in origin and size, and in their protein, RNA, and DNA molecular cargo. Additional complexity exists within the type of exRNAs (mRNA, miRNA, lncRNA, and other RNA species) within EVs, and little is known about EV cargo heterogeneity relative to their size and origin. Our work is significant because it contributes to the understanding of the distinct RNA and protein profiles of LEVs, MEVs, SEVs (exosomes), and RNPs in the context of EGFR- and PDGFRA-driven GBM. We demonstrate that vesicles of defined sizes contain unique, vesicle-characteristic exPTN and exRNA cargos and that exPTN cargo content among vesicle classes is less influenced by cell of origin than by vesicle subtype. This observation does not hold true for exRNA content however, suggesting that protein cargo may be more influenced by or subject to mechanisms of EV creation and release than exRNA. Our results establish a landscape of exRNA and exPTN from EVs and RNPs of genetically distinct GBM tumor cells that can be leveraged for the identification of vesicle and genotype-centric biomarkers.
Another significant aspect of our work is the demonstration at the single vesicle level of mutual exclusiveness of EV biomarkers expression for both canonical (CD9 and CD81) and the identified biomarkers Nono and Gja1. We observed that LEVs are either CD9- or CD81-positive with CD9:CD81 double-positive vesicles almost nonexistent. Whereas CD9 is mostly an SEV (exosomes) marker, we found that it also marks ~50% of LEVs. This may suggest intrinsic and mechanistic differences in biogenesis of CD9-positive exosomes and CD9-positive LEVs. Single vesicle analysis also shows that CD81 is consistently found to mark LEVs. In line with our proteomics data, we observed Nono proteins mostly in LEVs and Gja1 on both LEVs and SEV (exosomes) with few double-positive vesicles. Most LEVs and SEV (exosomes) are Gja1 single-positive (55%–75%) vesicles and fewer are double Gja1-CD9-positive vesicles. Approximately half of Nono+ vesicles are also CD9 positive. At this time, the fixed and static nature of our methodology for single vesicle analysis prevents molecular characterization of these CD9-positive SEVs (exosomes) and CD9-negative and CD81-positive LEVs to determine their exPTN and exRNA content. However, recent advances in modified flow cytometry-based techniques and sorting (Higginbotham et al., 2016; Mastoridis et al., 2018; Morales-Kastresana et al., 2017) would further refine heterogeneity of vesicle populations based on surface marker combinations.
Perhaps a surprising observation is that cellular immortalization arising from loss of p53 or Cdkn2a (Ink4a/ARF) tumor suppressor function resulted in a 5-fold decrease in vesicle release, and fully transformed cells shed approximately 17-fold fewer vesicles than wild-type astrocytes (Figure 1A). This is counter to prevailing observations that suggest that cancer patients have more circulating EVs than non-cancer–bearing healthy individuals (Beach et al., 2014; Park et al., 2013; Taylor and Gercel-Taylor, 2008; Zöller, 2013). Despite this seeming inconsistency, our results are in line with the observation that p53 mediates vesicular release in response to cellular stress (Yu et al., 2006) and that loss of p53 function leads to reduction in vesicle release. The functional overlap between Cdkn2a gene product ARF and p53 likely explain why loss of Cdkn2a also contributes to lower vesicle release. Our data support the notion that the higher EVs concentrations seen in cancer patients versus healthy controls perhaps result from a systemic, non-tumor origin instead of cancer cell-produced vesicles.
An interesting observation from our work is the preferential enrichment of tRNA sequences in the RNP fractions. Alignment of tRNA read sequences and abundance reveals distinct processing of tRNA transcripts into 5′ and 3′ tRFs. The most commonly observed extracellular species of tRFs are 30- to 32-nt-long and are evolutionarily conserved molecules (Anderson and Ivanov, 2014) produced by the ribonuclease A family member angiogenin (ANG) (Lyons et al., 2017). ANG is a multi-functional RNase known to regulate angiogenesis, cell proliferation, and viability of cancer cells (Sheng and Xu, 2016). ANG is upregulated in GBM (Eberle et al., 2000; Xia et al., 2015) and has been observed to be secreted by glioma cells (Skog et al., 2008). The functionality of ANG cleavage of tRNAs is emerging. Specific 5′ tRFs have been reported to perform functions often associated with regulation of gene expression in stress response. For instance, tRFs repress protein translation (Ivanov et al., 2011; Yamasaki et al., 2009) and modulate stress response by inducing formation of stress granules (Emara et al., 2010). They may also function in a miRNA-like manner (Kumar et al., 2014) and promote cell survival through their interaction with cytochrome c (Cyt c) during apoptosis (Lyons et al., 2017; Saikia et al., 2014). Further studies on the functions of these precisely processed, highly enriched extracellular tRFs represents an exciting avenue in glioma RNA biology.
The key upshot of our work is an expansion of the repertoire of exRNAs and exPTNs released by genetically defined GBMs in distinct vesicular and RNP mediums, with functional and biomarker potential. Our work challenges the generally assumed prevalent role of miRNA as sole mediator of exRNA intercellular communication and points to the need for further in-depth investigation into other classes of exRNAs. Our studies also demonstrate the importance of better defining heterogeneity at the molecular level to leverage EVs and RNPs impact on the physiology of recipient cells and use as biomarkers.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Al Charest (acharest@bidmc.harvard.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Glioblastoma cell lines and astrocytes primary cell cultures
EGFR-driven (Acquaviva et al., 2011; Jun et al., 2012; Zhu et al., 2009) and PDGFRA-driven (Jun et al., 2018; Zhou et al., 2018) mouse GBM primary cell cultures have been described previously. Briefly, the mouse GBM tumors were dissected, disaggregated in trypsin (0.25%) for 15 minutes at 37°C, strained through a 100-μm cell strainer (Falcon) and plated on 0.2% gelatin-coated cell culture plates. Cells for both genotypes were maintained in DMEM supplemented with 10% EV-cleared Fetal Bovine Serum (FBS) (Atlanta Biologics) and penicillin-streptomycin. Constitutive Cdkn2a null primary astrocytes and conditional p532lox primary astrocytes were isolated according to published protocols (Schildge et al., 2013). Note that the resulting cell cultures for the Cdkn2a−/− are immortalized whereas cultures from the conditional p532lox mice (Marino et al., 2000) are normal and exhibit replicative senescence after several passages (~P5). To immortalize the normal p532lox cultures, an adenovirus expressing Cre recombinase was used to infect low passage p532lox cultures, resulting in deletion of p53. To control for adenovirus-mediated confounding effects, the p532lox cultures were infected with adenovirus GFP as control. The identity of the gender/sex of the animals used to derive the primary cultures is unknown due to omission of recording this information at the time of establishment of the cultures. The Cdkn2a−/− and p53 primary cultures experiments were performed on two independent isolates. For RNA isolation, 1 × 106 cells were plated in a 15 cm dish and grown in 10% serum-containing media to 80% confluency. The media was replaced with DMEM with 0.1% FBS for 8 hours then changed to fresh DMEM with 0.1% FBS for 16 hours where indicated. Cells were treated similarly for protein isolation, except for the collection duration was increased to 16 hours for the initial 0.1% FBS treatment and 24 hours for the vesicle collection. To assess growth and cell viability under these conditions, cells were plated in triplicates and at time 0, treated with 0,1% FBS for 16 hours. As controls, cells were allowed to continue growth in 10% FBS. At 16 hour, cells were trypsinized, incubated with trypan blue, and counted. Viability was established by calculating the ratio of viable cells over total cells.
METHOD DETAILS
Characterization of vesicles by NTA
For vesicle quantitation and size distribution, cells were seeded in triplicate, grown to 80% confluency, washed twice with PBS and grown overnight in phenol red free DMEM with 0.1% FBS. Vesicles were then harvested in fresh phenol red free DMEM with 0.1% FBS for 24 hours. Conditioned media was cleared of live cells and cellular debris by centrifucation at 300 × g and 2000 × g respectively. The cleared media was imaged undiluted using a Nanosight LM10 (Malvern Instruments, Malvern, UK) with camera level = 15 and screen gain = 1.0. Video recordings were analyzed using the NTA 3.1 software with screen gain = 3.0. Each conditioned media was imaged three times and averaged. Following media collection, the cells were counted to give the denominator for the vesicles/ml/cells calculation. Each well was paired for cells and vesicles and the corresponding value was averaged over the biological triplicate to yield the vesicle quantitation for each cell line. The EGFR and PDGFRA glioma lines were analyzed on at least two independent repeats.
Vesicles fractionation
Vesicles and RNPs from cell culture supernatants were isolated by sequential filtration as described (Wei et al., 2017). Briefly, the conditioned media was first depleted of whole cells (live and dead) by centrifugation at 300 × g for 10 minutes and 2000 × g for 15 minutes, respectively. The cleared conditioned media was then passed sequentially through a series of syringe filters with decreasing pore size, 2 mm (Whatman), 0.8 μm (Millipore), 0.22 μm (Millipore), and 0.02 μm (Whatman) and the flow-through media cleared of EVs by filtration was further concentrated using a size-exclusion spin column centrifuged for 1 hour at 4200 × g using a 3 kDa size-exclusion spin column (Amicon, Millipore) to capture membrane-free multiprotein and RNP complexes. This serial filtration series isolated large extracellular vesicles (LEVs), medium extracellular vesicles (MEVs), small extracellular vesicles (SEVs- exosomes) and RNP complexes respectively.
Transmission electron microscopy
Following serial filtration, the captured vesicles were eluted by reverse flow in approximately 200 μL of PBS, further pelleted by 100,000 × g ultracentrifugation for 120 min at 4°C and resuspended in 50 μL of PBS. For visualization by TEM, the eluted vesicles were adsorbed to Formvar-carbon coated electron microscopy grids, washed with PBS and imaged on Tecnai G2 Spirit BioTWIN microscope at 80 kV.
ULTRACENTRIFUGATION
Like for the filter fractionation, conditioned media was cleared of cells as above and EVs from the cleared media were prepared by ultracentrifugation, first at 10,000 ×g (8,700 rpm in SW28 rotor, Beckman Coulter), then the supernatant of that spin at 100,000 ×g (27,000 rpm in SW28 rotor, Beckman Coulter). The pellet for the 10,000 ×g spin was combined by spinning at 13,000 rpm in a bench-top micro-centrifuge and the pellet for the 100,000 ×g spin was washed once with PBS and re-pelleted at 100,000 ×g.
Azidohomoalanine (AHA) protein labeling and protein isolation
To circumvent serum albumin protein overwhelming mass spectrometry analysis, cellular proteins were labeled with AHA and enriched using the Click-iT ® system. As above, cells were grown to 80% confluency, then DMEM with 0.1% FBS for 16 hours. To deplete cells of methionine, the cells were treated for 1 hour in depletion medium (DMEM without methionine and cysteine; GIBCO) supplemented with 0.1% FBS, L-cysteine, L-glutamine and penicillin-streptomycin. Newly synthesized proteins were labeled in the same medium containing 0.1 mM L-AHA (Thermofisher) for 24 hours. Vesicles from 160 mL conditioned media were isolated by sequential filtration as described above. The captured vesicles were lysed on filters in RIPA buffer and AHA-labeled proteins were purified using the Click-iT® Protein Enrichment Kit (Invitrogen) according to the manufacturer’s protocol. Briefly, the entire volume eluted protein from each fraction was combined with of 200 μL of agarose slurry and 1 mL of the reaction solution for the Click-iT® reaction and incubated (end-over-end rotating) for 18 hours at room temperature. The resin was washed with 1.8 mL water, then resupended in 1 mL SDS buffer and 10 mM dithiothreitol (DTT) (Bio-Rad) and reduced at 70°C for 15 min. After removal of the SDS/DTT buffer, the slurry was alkylated using 1 mL SDS buffer and 7.4 mg iodoacetamide (Bio-Rad) for 30 min in the dark. The resin was transferred to a spin column and washed 5 times with 2 mL of SDS buffer, 5 times with 2 mL of 8M urea/100mM Tris, pH8 and 5 times with 2 mL of 20% acetonitrile. The washed resin was re-suspended in digestion buffer (100 mM Tris, pH8, 2 mM CaCl2 and 10% acetonitrile) and processed for mass spectrometry.
On-bead digestion and TMT labeling
All liquid reagents were HPLC grade. The washed resin was resuspended in 500 uL of digestion buffer (200 mM EPPS pH 8.5 in 10% acetonitrile) and digested at room temperature with LysC (1:100 protease-to-protein ratio) followed by Trypsin (1:100 protease-to-protein ratio) and incubated for 6h at 37°C. Then, the tubes were centrifuged at 5000 g for 5 min, the supernatants were collected and the resin was washed once using digestion buffer, centrifuged and then collected. Digests were acidified using formic acid and the peptides were desalted by C18 solid phase extraction (Sep-Pak, Waters). Peptides were dried, resuspended in 200 mM EPPS pH 8.5, 30% acetonitrile (v/v) and labeled with TMT reagents (Thermo Fischer Scientific) for 1h at room temperature. The reaction was quenched with hydroxylamine to a final concentration of 0.3% (v/v). The TMT-labeled samples were pooled at a 1:1:1:1:1:1:1:1 ratio across the 8 samples. The pooled sample was vacuum centrifuged to near dryness and subjected to C18 solid-phase extraction (Sep-Pak, Waters).
OFFLINE FRACTIONATION
The pooled sample was fractionated using the Pierce High pH Reversed-Phase Peptide Fractionation Kit. Ten fractions were collected using: 10, 12.5, 15, 17.5, 20, 22.5, 25, 27.5, 30 and 50% acetonitrile. The fractions were consolidated into 5 samples: 10 and 50, 12.5 and 30, 15 and 27.5, 17.5 and 25 and 20 and 22.5%. Samples were subsequently acidified with formic acid and dried. Each fraction was desalted via StageTip, dried again, and reconstituted in 5% acetonitrile, 5% formic acid for LC-MS/MS processing.
Liquid chromatography and tandem mass spectrometry
Mass spectrometry data were collected using an Orbitrap Fusion or Lumos mass spectrometer (ThermoFisher Scientific, San Jose, CA) coupled to a Proxeon EASY-nLC 1200 liquid chromatography (LC) pump (ThermoFisher Scientific, San Jose, CA). Peptides were separated on a 100 μm inner diameter microcapillary column packed with ≈40 cm of Accucore150 resin (2.6 μm, 150Å, ThermoFisher Scientific, San Jose, CA). For each analysis, we loaded ≈2 μg onto the column and separation was achieved using a 2.5h h gradient of 7 to 27% acetonitrile in 0.125% formic acid at a flow rate of ≈550 nL min−1. Each analysis used an SPS-MS3-based TMT method (McAlister et al., 2014; Ting et al., 2011), which has been shown to reduce on interference compared to MS2-based quantification (Paulo et al., 2016). The scan sequence began with an MS1 spectrum (Orbitrap; resolution 120000; mass range 400 – 1400 m/z; automatic gain control (AGC) target 5 × 105; maximum injection time 100 ms). Precursors for MS2/MS3 analysis were selected using a Top10 method. MS2 analysis consisted of collision-induced dissociation (quadrupole ion trap; AGC 2 × 104; normalized collision energy (NCE) 35; maximum injection time 150 ms). Following acquisition of each MS2 spectrum, we collected an MS3 spectrum using our recently described method in which multiple MS2 fragment ions were captured in the MS3 precursor population using isolation waveforms with multiple frequency notches (McAlister et al., 2014). MS3 precursors were fragmented by high-energy collision-induced dissociation (HCD) and analyzed using the Orbitrap (NCE 65; AGC 1 × 105; maximum injection time 150 ms, resolution was 50000 at 200 Th).
Mass spectrometry data analysis
Mass spectra were processed using a Sequest-based pipeline (Huttlin et al., 2010). Spectra were converted to mzXML using a modified version of ReAdW.exe. Database searching included all entries from the mouse UniProt database (2014-07-02). This database was concatenated with one composed of all protein sequences in the reversed order. Searches were performed using a 50 ppm precursor ion tolerance for total protein-level profiling. The product ion tolerance was set to 0.9 Da. These wide mass tolerance windows were chosen to maximize sensitivity in conjunction with Sequest searches and linear discriminant analysis (Beausoleil et al., 2006; Huttlin et al., 2010). TMT tags on lysine residues and peptide N termini (+229.163 Da) and carbamidomethylation of cysteine residues (+57.021 Da) were set as static modifications, while oxidation of methionine residues (+15.995 Da) was set as a variable modification. Peptide-spectrum matches (PSMs) were adjusted to a 1% false discovery rate (FDR) (Elias and Gygi, 2007). PSM filtering was performed using a linear discriminant analysis, as described previously (Huttlin et al., 2010), while considering the following parameters: XCorr, ΔCn, missed cleavages, peptide length, charge state, and precursor mass accuracy. For TMT-based reporter ion quantitation, we extracted the summed signal-to-noise (S/N) ratio for each TMT channel and found the closest matching centroid to the expected mass of the TMT reporter ion. PSMs were identified, quantified, and collapsed to a 1% peptide false discovery rate (FDR) and then collapsed further to a final protein-level FDR of 1%. Moreover, protein assembly was guided by principles of parsimony to produce the smallest set of proteins necessary to account for all observed peptides. Proteins were quantified by summing reporter ion counts across all matching PSMs, as described previously (Huttlin et al., 2010). PSMs with poor quality, MS3 spectra with more than six TMT reporter ion channels missing, or no MS3 spectra were excluded from quantification (McAlister et al., 2012). Protein quantitation values were exported for further analysis. Each reporter ion channel was summed across all quantified proteins and normalized assuming equal protein loading of all eight samples.
To integrate this study MS dataset to those of Vesiclepedia and NCI-60 cancer cell line panel, the gene name list of the identified 2178 proteins and vesicle-enriched exported proteins were matched to the gene names in those databases. 98% (2134/2178) were successfully matched and the remaining unmatched represented either mouse-specific genes or unresolved inter-species nomenclature differences.
Go term analysis
To identify proteins from EVs/RNPs enriched in Biological Process (BP), Molecular Function (MF) and Cellular Component (CC), we utilized the Database for Annotation, Visualization, and Integrated Discovery (DAVID) v7.0 (Huang et al., 2009a, 2009b) with GOTERMs BP, MF and CC. All terms with a p-value (Benjamini or Benjamini-Hochberg adjusted) less than 0.05 were considered significant and ranked by the number of proteins identified in the group.
Western Blotting
Protein from the various filters were harvested by on-filter RIPA lysis buffer incubation and quantitated by BCA. Equivalent amounts of protein from each fraction were loaded on SDS-PAGE gels and analyzed for equal loading by silver staining. Corrected equal amounts of protein were run and examined by western blotting. Antibodies used were Dnajb11 (PA5–43002), Sec24d (14687S), Nono (PA5–18514), Pdlim7 (10221–1-AP), Gja1 (3512S), CD63 (sc-5275), CD81 (sc-166029), RNaseT2 (sc-393729), PCNA (13110S), Flotillin (18634S), La (5034S), NPM (3542S), HSP90 (4874S), Ro60 (AV40534), Anti-TSG101 (ab30871) and detected using cognate species Li-Cor secondary antibodies.
Single EV Immobilization and Imaging
EVs were biotinylated by incubation with 333 μM EZ-Link Sulfo-NHS-LC-Biotin (Thermo Fisher Scientific) for 30 min at room temperature. Excess biotins were removed by Zeba Spin Desalting Column, 7K MWCO (Thermo Fisher Scientific). Biotinylated EVs were captured on a neutravidin-coated glass slide, which was prepared according to a previously described method (Lee et al., 2018). Following 30-min incubation at room temperature, the slide was washed and further incubated with fixation/permeabilization buffer (BD Biosciences; 15 min at room temperature) and then with a blocking buffer (0.2% BSA in PBS; 20 min at room temperature). For EV labeling, a cocktail of fluorescence-labeled antibodies was introduced. Samples were incubated for 90 min at room temperature and washed with 0.2% BSA in PBS. A BX-63 upright fluorescent microscope (Olympus) with a 100X oil objective was used for imaging. All the fluorescence images were taken under the same acquisition setting (i.e., objective, exposure time, camera setting, illumination).
Preparation of Antibody Cocktail for Single EV Imaging
Antibody cocktail was prepared immediately before the experiment and kept in darkness. All antibodies were diluted into 1:50 in0.2% BSA-PBS. CD9-AF647 was purchased from Biolegend. CD81-AF488 was purchased from Santa Cruz Biotechnology. Gja1-AF594 and Nono-AF680 were prepared using the Lightning-Link® Rapid Conjugation System Alexa Fluor® 594 (335–0030) and Alexa Fluor 680 Antibody Labeling (A20188) Kits, respectively.
Data Analysis
Image analysis was performed using Cellprofiler 3.0. Masks were created at every EV location and the signal of interest was isolated in 4 channels. At each EV position, mean intensities for each protein marker were obtained; histograms and heatmaps were generated using these values.
RNA and libraries preparation, sequencing and annotations
Following filtering, total RNA was isolated on-filters from all fractions as recommended by miRCURY protocol, with on-column DNase treatment (QIAGEN, Germany). Total cellular RNA was isolated from the corresponding source EGFR and PDGFRA cultures, and analyzed in parallel. The concentrations of cellular and extracellular RNA were determined by spectrophotometer (NanoDrop 2000) and Quant-iT RiboGreen RNA Assay Kit (Thermo Fisher Scientific), respectively. The RNA quality was examined using Agilent 2100 Bioanalyzer (Agilent, CA) and the RNA Integrity Number (RIN) estimated.
Total RNA, either 40–200 ng of exRNA, or 2 mg of cellular RNA, was rRNA-depleted using the Ribo-Zero rRNA Removal Kits (Illumina, CA). One quarter of the rRNA-depleted RNA was fragmented to 100–500 nt using the 5 × First-Strand Buffer (Clontech, CA), and utilized for the long RNA library construction by SMARTer Stranded RNA-Seq Kit (Clontech). The remaining RNA was used as input for the NEBNext Multiplex Small RNA Library Prep Set for Illumina (New England Biolabs), with size selection of 15–65 nt inserts for small RNA libraries. The quality of libraries was examined using the Agilent DNA 1000 kit at the Agilent 2100 Bioanalyzer instrument, and cDNA quantified by qRT-PCR. The libraries were sequenced on HiSeq 2000 (Illumina) with single read 50 cycles at the Beijing Genomics Institute (BGI, China).
Sequenced reads for both small RNA-Seq and long RNA-Seq were analyzed using bcbio-nextgen (https://github.com/bcbio/bcbio-nextgen), a python toolkit providing for fully automated high throughput sequencing analysis. Raw reads were examined for quality issues using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) to ensure library generation and sequencing were suitable for further analysis. From the FastQC files, adaptor sequences, other contaminant sequences (such as polyA tails and low quality sequences with PHRED quality scores less than five) were trimmed from reads using atropos (Didion et al., 2017). Trimmed reads were aligned to UCSC build mm10 of the Mus musculus genome, augmented with transcript information from Ensembl release GRCm38.84 using STAR (Dobin et al., 2013). Alignments were checked for evenness of coverage, rRNA content, genomic context of alignments (for example, alignments in known transcripts and introns), complexity and other quality checks using a combination of FastQC, Qualimap (García-Alcalde et al., 2012), MultiQC (Ewels et al., 2016) and custom code within the bcbio-nextgen pipeline. In parallel, Transcripts Per Million (TPM) measurements per isoform were generated by quasialignment using Salmon (Patro et al., 2017). Differential expression at the gene level was called with DESeq2 (Love et al., 2014), preferring to use counts per gene estimated from the Salmon quasialignments by tximport (Soneson et al., 2015). The DEGreport Bioconductor package was used for QC and clustering analysis (https://doi.org/10.18129/B9.bioc.DEGreport).
For small RNA-seq, we focused on miRNA analysis but the small RNA seq dataset includes also t-RNAs and pi-RNAs. Trimmed reads were aligned to miRBase v21 to the specific species with seqbuster (Pantano et al., 2010). In addition, the trimmed reads were aligned to the Mus musculus genome (version mm10) using STAR (Dobin et al., 2013). The aligned reads were analyzed with seqcluster (Pantano et al., 2011) to characterize the whole small RNA transcriptome and classify reads into rRNA, miRNA, repeats, genes, tRNAs and others from the UCSC annotation (Mangan et al., 2014). Alignments were checked for evenness of coverage, rRNA content, genomic context of alignments (for example, alignments in known transcripts and introns), complexity and other quality checks using a combination of FastQC, MultiQC (Ewels et al., 2016) and custom code within the bcbio-nextgen pipeline. Data were loaded into R using isomiRs Bioconductor package (Ramos et al., 2017) to get normalized expression values.
RNaseq Data Analysis
For the analysis of class composition in both small and long RNA libraries, the total abundance of corrected non-rRNA was used for normalization between the samples. The hierarchical clustering analyses were performed on log2-transformed, normalized read counts using the Limma R package. Venn diagrams were generated using identified differentially localized gene names input into the venn function in the gplots R package and further annotated in Adobe Illustrator.
Quantitative reverse transcription PCR
For qPCR validation of RNA-Seq, RNA was isolated as above. Validation of specific small RNAs from the RNA-Seq data was undertaken using TaqMan smallRNA Assays, which use target-specific stem-loop primers specific to the mature small RNA of interest. 10 ng of total RNA from each fraction was reverse transcribed using the TaqMan MicroRNA Reverse Transcription Kit. Individual small RNAs were PCR amplified using TaqMan Universal PCR Master Mix according to manufacturer recommended cycling conditions. miRNAs were targeted using pre-designed TaqMan miRNA Assays, while tRNAs were analyzed using Custom TaqMan Small RNA Assays designed against the most abundant tRNA fragment from that gene.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed using GraphPad Prism (GraphPad Software, San Diego). Values are given as mean ± SEM or SD as indicated. Numbers of experimental replicates are given in the figure legends. When two groups were compared, significance was determined using an unpaired two-tail t test. A p value < 0.05 is considered as statistical significance.
DATA AND CODE AVAILABILITY
Proteomics data files are available in Data S1. Datasets for RNaseq have been deposited to GEO under – GSE123414.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-Dnajb11 | ThermoFisher | Cat# PA5-43002; RRID: AB_2576759 |
| anti-Sec24d | Cell Signaling | Cat# 14687S; RRID: AB_2798574 |
| anti-Pdlim7 | Proteintech | Cat# 10221-1-AP; RRID: AB_2161789 |
| anti-Nono | ThermoFisher | Cat# PA5-18514; RRID: AB_10983564 |
| anti-Gja1 | Cell Signaling | Cat# 3512S; RRID: AB_2294590 |
| anti-CD81 | Santa Cruz Biotechnology | Cat# sc-166029; RRID: AB_2275892 |
| anti-CD63 | Santa Cruz Biotechnology | Cat# sc-5275; RRID: AB_627877 |
| anti-RNaseT2 | Santa Cruz Biotechnology | Cat# sc-393729 |
| anti-Flotillin-1 | Cell Signaling | Cat# 18634S; RRID: AB_2773040 |
| anti-La | Cell Signaling | Cat# 5034S; RRID: AB_10620954 |
| anti-PCNA | Cell Signaling | Cat# 13110S; RRID: AB_2636979 |
| anti-NPM | Cell Signaling | Cat# 3542S; RRID: AB_2155178 |
| anti-HSP90 | Cell Signaling | Cat# 4874S; RRID: AB_2121214 |
| anti-Ro60 | Sigma | Cat# AV40534; RRID: AB_1857491 |
| anti-Anti-TSG101 | Abeam | Cat# ab30871; RRID: AB_2208084 |
| anti-CD9-AF647 | BioLegend | Cat# 124809; RRID: ABJ279319 |
| anti-CD81(B11)-AF488 | Santa Cruz Biotechnology | Cat# sc-166029 AF488 |
| Bacterial and Virus Strains | ||
| Adenovirus-Cre | University of Iowa | Cat# Ad5CMVCre |
| Adenovirus-GFP | University of Iowa | Cat# Ad5CMVeGFP |
| Critical Commercial Assays | ||
| TaqMan® MicroRNA Reverse Transcription Kit | ThermoFisher | Cat# 4366596 |
| Custom TaqMan® Small RNA Assay - GlyGCC-8195 (CTMFWZ3) | ThermoFisher | Cat#4440418 (GCATTGGTGGTTCAGTG GTAGAATTCTCGCC) |
| Custom TaqMan® Small RNA Assay - GlyGCC-8193 (CTNKRKZ) | ThermoFisher | Cat# 4440418 (TGCCACGCGGGAGG CCCGGGTTCGGT) |
| mmu-miR-148a-3 TaqMan Probe | ThermoFisher | Cat# 4427975 - AssayID 000470 |
| mmu-miR-146b-5 TaqMan Probe | ThermoFisher | Cat# 4427975 - AssayID 001097 |
| mmu-miR-669c-5 TaqMan Probe | ThermoFisher | Cat# 4427975 - AssayID 002646 |
| mmu-miR-206-3 TaqMan Probe | ThermoFisher | Cat# 4440887 - AssayID 000510 |
| mmu-miR-21a-5 TaqMan Probe | ThermoFisher | Cat# 4427975 - AssayID 000397 |
| Click-IT® AHA (L-Azidohomoalanine) | ThermoFisher | Cat# C10102 |
| Click-iT® Protein Enrichment Kit | ThermoFisher | Cat# C10416 |
| Lightning-Link® Rapid Conjugation System Alexa Fluor® 594 | Novus Biologicals | Cat# 335-0030 |
| APEX™ Alexa Fluor 680 Antibody Labeling Kit | ThermoFisher | Cat# A20188 |
| miRCURY RNA Isolation Kits - Cell & Plant | Exiqon | Cat# 300110 |
| SMARTer® Stranded RNA-Seq Kit | Clontech | Cat# 634839 |
| NEBNext Multiplex Small RNA Library Prep Set for Illumina | New England BioLabs | Cat# E7300L |
| Deposited Data | ||
| RNA sequencing data | This Paper | GSE123414 |
| Proteomics data | This Paper | Data S1 |
| Experimental Models: Cell Lines | ||
| Mouse Primary Glioblastoma Cell Lines | BIDMC | N/A |
| Software and Algorithms | ||
| R | The R Foundation | https://www.r-project.org/ |
| bcbio-nextgen | Babraham Bioinformatics | https://github.com/bcbio/bcbio-nextgen |
| FastQC | Bioconductor | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| atropos | Bioconductor | https://github.com/jdidion/atropos |
| STAR Aligner | Bioconductor | https://github.com/alexdobin/STAR |
| Qualimap | Bioconductor | qualimap.bioinfo.cipf.es/ |
| MultiQC | Bioconductor | https://multiqc.info/ |
| salmon | Bioconductor | https://combine-lab.github.io/salmon/ |
| DESeq2 | Bioconductor | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| tximport | Bioconductor | https://bioconductor.org/packages/release/bioc/html/tximport.html |
| DEGreport | Bioconductor | https://bioconductor.org/packages/release/bioc/html/DEGreport.html |
| seqbuster | Bioconductor | https://github.com/lpantano/seqbuster |
| seqcluster | Bioconductor | https://github.com/lpantano/seqcluster |
| isomiRs | Bioconductor | https://bioconductor.org/packages/release/bioc/html/isomiRs.html |
| limma | Bioconductor | http://bioconductor.org/packages/release/bioc/html/limma.html |
| Cellprofiler 3.0 | Cellprofiler | https://cellprofiler.org/ |
| NanoSight NTA software, v3.1 | Malvern Panalytical | https://www.malvernpanalytical.com/ |
| Other | ||
| Puradisc 25 mm GMF Syringe Filter, GD2, 2.0 μm | GE Life Sciences | Cat# 6783-2520 |
| EMD Millipore Millex Sterile Syringe Filters - 0.80 μm | EMD Millipore | Cat# SLAA033SS |
| EMD Millipore Millex Sterile Syringe Filters - 0.22 μm | EMD Millipore | Cat# SLGV033RS |
| Anotop 25 mm Sterile Syringe Filters, 0.2 μm | GE Life Sciences | Cat# 6809-2102 |
| EMD Millipore Amicon Ultra-15 Centrifugal Filter Units | EMD Millipore | Cat# UFC900308 |
Highlights.
Extracellular vesicles (EVs) are heterogeneous in their origins and contents
Expression of CD9 and CD81 are mutually exclusive at the single EV level
Cells shed fewer EVs during the process of immortalization and transformation
tRNA fragments are enriched in ribonucleoprotein complexes
ACKNOWLEDGMENTS
This publication is part of the NIH Extracellular RNA Communication Consortium paper package and was supported by the NIH Common Fund’s exRNA Communication Program. The authors would like to thank Dr. L. Pantano for bioinformatics consultation and members of the Charest lab for helpful discussion and inputs. J.N.-P. (CVU 289937) was partially supported by the Consejo Nacional de Ciencia y Tecnología (CONACyT) of Mexico. This work was supported by NIH grants CA179563 (A.C. and A.M.K.), CA069246 (A.C., R.W., and H.L.), and CA185137 (A.C. and S.G.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j. celrep.2019.05.089.
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPORTING CITATIONS
The following references appear in the Supplemental Information: Hurwitz et al. (2016b); Kalra et al. (2012); Pathan et al. (2019); Wei et al. (2017).
REFERENCES
- Acquaviva J, Jun HJ, Lessard J, Ruiz R, Zhu H, Donovan M, Woolfenden S, Boskovitz A, Raval A, Bronson RT, et al. (2011). Chronic activation of wild-type epidermal growth factor receptor and loss of Cdkn2a cause mouse glioblastoma formation. Cancer Res. 71, 7198–7206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson P, and Ivanov P (2014). tRNA fragments in human health and disease. FEBS Lett. 588, 4297–4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balatti V, Pekarsky Y, and Croce CM (2017). Role of the tRNA-Derived Small RNAs in Cancer: New Potential Biomarkers and Target for Therapy. Adv. Cancer Res. 135, 173–187. [DOI] [PubMed] [Google Scholar]
- Beach A, Zhang HG, Ratajczak MZ, and Kakar SS (2014). Exosomes: an overview of biogenesis, composition and role in ovarian cancer. J.Ovarian Res 7, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beausoleil SA, Villén J, Gerber SA, Rush J, and Gygi SP (2006). A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol 24, 1285–1292. [DOI] [PubMed] [Google Scholar]
- Bebelman MP, Smit MJ, Pegtel DM, and Baglio SR (2018). Biogenesis and function of extracellular vesicles in cancer. Pharmacol. Ther 188, 1–11. [DOI] [PubMed] [Google Scholar]
- Becker A, Thakur BK, Weiss JM, Kim HS, Peinado H, and Lyden D (2016). Extracellular Vesicles in Cancer: Cell-to-Cell Mediators of Metastasis. Cancer Cell 30, 836–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brat DJ, Verhaak RG, Aldape KD, Yung WK, Salama SR, Cooper LA, Rheinbay E, Miller CR, Vitucci M, Morozova O, et al. ; Cancer Genome Atlas Research Network (2015). Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N. Engl. J. Med 372, 2481–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, et al. ; TCGA Research Network (2013). The somatic genomic landscape of glioblastoma. Cell 155, 462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo M, Raposo G, and Théry C (2014). Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol 30, 255–289. [DOI] [PubMed] [Google Scholar]
- Davis ME (2016). Glioblastoma: Overview of Disease and Treatment. Clin. J. Oncol. Nurs 20, S2–S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didion JP, Martin M, and Collins FS (2017). Atropos: specific, sensitive, and speedy trimming of sequencing reads. PeerJ 5, e3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberle K, Oberpichler A, Trantakis C, Krupp W, Knüpfer M, Tschesche H, and Seifert V (2000). The expression of angiogenin in tissue samples of different brain tumours and cultured glioma cells. Anticancer Res. 20 (3A), 1679–1684. [PubMed] [Google Scholar]
- Eckel-Passow JE, Lachance DH, Molinaro AM, Walsh KM, Decker PA, Sicotte H, Pekmezci M, Rice T, Kosel ML, Smirnov IV, et al. (2015). Glioma Groups Based on 1p/19q, IDH, and TERT Promoter Mutations in Tumors. N. Engl. J. Med 372, 2499–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias JE, and Gygi SP (2007). Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214. [DOI] [PubMed] [Google Scholar]
- Emara MM, Ivanov P, Hickman T, Dawra N, Tisdale S, Kedersha N, Hu GF, and Anderson P (2010). Angiogenin-induced tRNA-derived stress-induced RNAs promote stress-induced stress granule assembly. J. Biol. Chem>. 285, 10959–10968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewels P, Magnusson M, Lundin S, and Käller M (2016). MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Alcalde F, Okonechnikov K, Carbonell J, Cruz LM, Götz S, Tarazona S, Dopazo J, Meyer TF, and Conesa A (2012). Qualimap: evaluating next-generation sequencing alignment data. Bioinformatics 28, 2678–2679. [DOI] [PubMed] [Google Scholar]
- Higginbotham JN, Zhang Q, Jeppesen DK, Scott AM, Manning HC, Ochieng J, Franklin JL, and Coffey RJ (2016). Identification and characterization of EGF receptor in individual exosomes by fluorescence-activated vesicle sorting. J. Extracell. Vesicles 5, 29254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Sherman BT, and Lempicki RA (2009a). Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Sherman BT, and Lempicki RA (2009b). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Hurwitz SN, Conlon MM, Rider MA, Brownstein NC, and Meckes DG Jr. (2016a). Nanoparticle analysis sheds budding insights into genetic drivers of extracellular vesicle biogenesis. J. Extracell. Vesicles 5, 31295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurwitz SN, Rider MA, Bundy JL, Liu X, Singh RK, and Meckes DG Jr. (2016b). Proteomic profiling of NCI-60 extracellular vesicles uncovers common protein cargo and cancer type-specific biomarkers. Oncotarget 7, 86999–87015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villén J, Haas W, Sowa ME, and Gygi SP (2010). A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143, 1174–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov P, Emara MM, Villen J, Gygi SP, and Anderson P (2011). Angiogenin-induced tRNA fragments inhibit translation initiation. Mol. Cell 43, 613–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun HJ, Acquaviva J, Chi D, Lessard J, Zhu H, Woolfenden S, Bronson RT, Pfannl R, White F, Housman DE, et al. (2012). Acquired MET expression confers resistance to EGFR inhibition in a mouse model of glioblastoma multiforme. Oncogene 31, 3039–3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun HJ, Appleman VA, Wu HJ, Rose CM, Pineda JJ, Yeo AT, Delcuze B, Lee C, Gyuris A, Zhu H, et al. (2018). A PDGFRα-driven mouse model of glioblastoma reveals a stathmin1-mediated mechanism of sensitivity to vinblastine. Nat. Commun 9, 3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalra H, Simpson RJ, Ji H, Aikawa E, Altevogt P, Askenase P, Bond VC, Borràs FE, Breakefield X, Budnik V, et al. (2012). Vesiclepedia: a compendium for extracellular vesicles with continuous community annotation. PLoS Biol. 10, e1001450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhn M, Pazaitis N, and Hüttelmaier S (2013). Why YRNAs? About Versatile RNAs and Their Functions. Biomolecules 3, 143–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konoshenko MY, Lekchnov EA, Vlassov AV, and Laktionov PP (2018). Isolation of Extracellular Vesicles: General Methodologies and Latest Trends. BioMed Res. Int 2018, 8545347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp F, and Mendell JT (2018). Functional Classification and Experimental Dissection of Long Noncoding RNAs. Cell 172, 393–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalski MP, and Krude T (2015). Functional roles of non-coding Y RNAs. Int. J. Biochem. Cell Biol. 66, 20–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Anaya J, Mudunuri SB, and Dutta A (2014). Meta-analysis of tRNA derived RNA fragments reveals that they are evolutionarily conserved and associate with AGO proteins to recognize specific RNA targets. BMC Biol. 12, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Kuscu C, and Dutta A (2016). Biogenesis and Function of Transfer RNA-Related Fragments (tRFs). Trends Biochem. Sci 41, 679–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Fraser K, Ghaddar B, Yang K, Kim E, Balaj L, Chiocca EA, Breakefield XO, Lee H, and Weissleder R (2018). Multiplexed Profiling of Single Extracellular Vesicles. ACS Nano 12, 494–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logozzi M, De Milito A, Lugini L, Borghi M, Calabrò L, Spada M, Perdicchio M, Marino ML, Federici C, Iessi E, et al. (2009). High levels of exosomes expressing CD63 and caveolin-1 in plasma of melanoma patients. PLoS ONE 4, e5219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons SM, Fay MM, Akiyama Y, Anderson PJ, and Ivanov P (2017). RNA biology of angiogenin: Current state and perspectives. RNA Biol. 14, 171–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons SM, Fay MM, and Ivanov P (2018). The role of RNA modifications in the regulation of tRNA cleavage. FEBS Lett. 592, 2828–2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, and Yates JR 3rd. (2018). Proteomics and pulse azidohomoalanine labeling of newly synthesized proteins: what are the potential applications? Expert Rev. Proteomics 15, 545–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangan ME, Williams JM, Kuhn RM, and Lathe WC 3rd. (2014). The UCSC Genome Browser: What Every Molecular Biologist Should Know. Curr. Protoc. Mol. Biol 107, 19.9.1–19.9.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino S, Vooijs M, van Der Gulden H, Jonkers J, and Berns A (2000). Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev. 14, 994–1004. [PMC free article] [PubMed] [Google Scholar]
- Mastoridis S, Bertolino GM, Whitehouse G, Dazzi F, Sanchez-Fueyo A, and Martinez-Llordella M (2018). Multiparametric Analysis of Circulating Exosomes and Other Small Extracellular Vesicles by Advanced Imaging Flow Cytometry. Front. Immunol 9, 1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathivanan S, Ji H, and Simpson RJ (2010). Exosomes: extracellular organelles important in intercellular communication. J. Proteomics 73, 1907–1920. [DOI] [PubMed] [Google Scholar]
- McAlister GC, Huttlin EL, Haas W, Ting L, Jedrychowski MP, Rogers JC, Kuhn K, Pike I, Grothe RA, Blethrow JD, and Gygi SP (2012). Increasing the multiplexing capacity of TMTs using reporter ion isotopologues with isobaric masses. Anal. Chem 84, 7469–7478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlister GC, Nusinow DP, Jedrychowski MP, Wühr M, Huttlin EL, Erickson BK, Rad R, Haas W, and Gygi SP (2014). MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal. Chem 86, 7150–7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLendon R, Friedman A, Bigner D, Van Meir EG, Brat DJ, Mastrogianakis M, Olson JJ, Mikkelsen T, Lehman N, Aldape K, et al. ; Cancer Genome Atlas Research Network (2008). Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455, 1061–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales-Kastresana A, Telford B, Musich TA, McKinnon K, Clayborne C, Braig Z, Rosner A, Demberg T, Watson DC, Karpova TS, et al. (2017). Labeling Extracellular Vesicles for Nanoscale Flow Cytometry. Sci. Rep 7, 1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien CA, Margelot K, and Wolin SL (1993). Xenopus Ro ribonucleoproteins: members of an evolutionarily conserved class of cytoplasmic ribonucleoproteins. Proc. Natl. Acad. Sci. USA 90, 7250–7254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omuro A, and DeAngelis LM (2013). Glioblastoma and other malignant gliomas: a clinical review. JAMA 310, 1842–1850. [DOI] [PubMed] [Google Scholar]
- Pantano L, Estivill X, and Martí E (2010). SeqBuster, a bioinformatic tool for the processing and analysis of small RNAs datasets, reveals ubiquitous miRNA modifications in human embryonic cells. Nucleic Acids Res. 38, e34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantano L, Estivill X, and Martí E (2011). A non-biased framework for the annotation and classification of the non-miRNA small RNA transcriptome. Bioinformatics 27, 3202–3203. [DOI] [PubMed] [Google Scholar]
- Pantano L, Friedländer MR, Escaramís G, Lizano E, Pallarès-Albanell J, Ferrer I, Estivill X, and Martí E (2016). Specific small-RNA signatures in the amygdala at premotor and motor stages of Parkinson’s disease revealed by deep sequencing analysis. Bioinformatics 32, 673–681. [DOI] [PubMed] [Google Scholar]
- Park JO, Choi DY, Choi DS, Kim HJ, Kang JW, Jung JH, Lee JH, Kim J, Freeman MR, Lee KY, et al. (2013). Identification and characterization of proteins isolated from microvesicles derived from human lung cancer pleural effusions. Proteomics 13, 2125–2134. [DOI] [PubMed] [Google Scholar]
- Pathan M, Fonseka P, Chitti SV, Kang T, Sanwlani R, Van Deun J, Hendrix A, and Mathivanan S (2019). Vesiclepedia 2019: a compendium of RNA, proteins, lipids and metabolites in extracellular vesicles. Nucleic Acids Res. 47 (D1), D516–D519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patro R, Duggal G, Love MI, Irizarry RA, and Kingsford C (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulo JA, O’Connell JD, and Gygi SP (2016). A Triple Knockout (TKO) Proteomics Standard for Diagnosing Ion Interference in Isobaric Labeling Experiments. J. Am. Soc. Mass Spectrom. 27, 1620–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinowits G, Gerçel-Taylor C, Day JM, Taylor DD, and Kloecker GH (2009). Exosomal microRNA: a diagnostic marker for lung cancer. Clin. Lung Cancer 10, 42–46. [DOI] [PubMed] [Google Scholar]
- Ramos M, Schiffer L, Re A, Azhar R, Basunia A, Rodriguez C, Chan T, Chapman P, Davis SR, Gomez-Cabrero D, et al. (2017). Software for the Integration of Multiomics Experiments in Bioconductor. Cancer Res. 77, e39–e42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rider MA, Hurwitz SN, and Meckes DG Jr. (2016). ExtraPEG: A Polyethylene Glycol-Based Method for Enrichment of Extracellular Vesicles. Sci. Rep 6, 23978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saikia M, Jobava R, Parisien M, Putnam A, Krokowski D, Gao XH, Guan BJ, Yuan Y, Jankowsky E, Feng Z, et al. (2014). Angiogenin-cleaved tRNA halves interact with cytochrome c, protecting cells from apoptosis during osmotic stress. Mol. Cell. Biol 34, 2450–2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schildge S, Bohrer C, Beck K, and Schachtrup C (2013). Isolation and culture of mouse cortical astrocytes. J. Vis. Exp 71, 50079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt AM, and Chang HY (2016). Long Noncoding RNAs in Cancer Pathways. Cancer Cell 29, 452–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano M, Lee H, Chin L, Cordon-Cardo C, Beach D, and DePinho RA (1996). Role of the INK4a locus in tumor suppression and cell mortality. Cell 85, 27–37. [DOI] [PubMed] [Google Scholar]
- Sheng J, and Xu Z (2016). Three decades of research on angiogenin: a review and perspective. Acta Biochim. Biophys. Sin. (Shanghai) 48, 399–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson RJ, Lim JW, Moritz RL, and Mathivanan S (2009). Exosomes: proteomic insights and diagnostic potential. Expert Rev. Proteomics 6, 267–283. [DOI] [PubMed] [Google Scholar]
- Skog J, Würdinger T, van Rijn S, Meijer DH, Gainche L, Sena-Esteves M, Curry WT Jr., Carter BS, Krichevsky AM, and Breakefield XO (2008). Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 10, 1470–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soneson C, Love MI, and Robinson MD (2015). Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res. 4, 1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor DD, and Gercel-Taylor C (2008). MicroRNA signatures of tumor-derived exosomes as diagnostic biomarkers of ovarian cancer. Gynecol. Oncol 110, 13–21. [DOI] [PubMed] [Google Scholar]
- Théry C (2011). Exosomes: secreted vesicles and intercellular communications. F1000 Biol. Rep 3, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting L, Rad R, Gygi SP, and Haas W (2011). MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat. Methods 8, 937–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, et al. ; Cancer Genome Atlas Research Network (2010). Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17, 98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Z, Batagov AO, Schinelli S, Wang J, Wang Y, El Fatimy R, Rabinovsky R, Balaj L, Chen CC, Hochberg F, et al. (2017). Coding and non-coding landscape of extracellular RNA released by human glioma stem cells. Nat. Commun 8, 1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia W, Fu W, Cai X, Wang M, Chen H, Xing W, Wang Y, Zou M, Xu T, and Xu D (2015). Angiogenin promotes U87MG cell proliferation by activating NF-κB signaling pathway and downregulating its binding partner FHL3. PLoS ONE 10, e0116983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki S, Ivanov P, Hu GF, and Anderson P (2009). Angiogenin cleaves tRNA and promotes stress-induced translational repression. J. Cell Biol. 185, 35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Harris SL, and Levine AJ (2006). The regulation of exosome secretion: a novel function of the p53 protein. Cancer Res. 66, 4795–4801. [DOI] [PubMed] [Google Scholar]
- Zhou S, Appleman VA, Rose CM, Jun HJ, Yang J, Zhou Y, Bronson RT, Gygi S, and Charest A (2018). Chronic platelet derived growth factor receptor signaling exerts control over initiation of protein translation in glioma. Life Sci. Alliance 1, e201800029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Acquaviva J, Ramachandran P, Boskovitz A, Woolfenden S, Pfannl R, Bronson RT, Chen JW, Weissleder R, Housman DE, and Charest A (2009). Oncogenic EGFR signaling cooperates with loss of tumor suppressor gene functions in gliomagenesis. Proc. Natl. Acad. Sci. USA 106, 2712–2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zöller M (2013). Pancreatic cancer diagnosis by free and exosomal miRNA. World J. Gastrointest. Pathophysiol 4, 74–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Proteomics data files are available in Data S1. Datasets for RNaseq have been deposited to GEO under – GSE123414.