

Abstract
Neuronal degeneration represents a pathogenetic hallmark after different brain insults, such as ischemia and status epilepticus (SE). Excessive release of glutamate triggered by pathophysiologic synaptic activity has been put forward as key mechanism in this context. In response to pathophysiologic synaptic activity, multiple signaling cascades are activated that ultimately initiate expression of specific sets of genes, which may decide between neuronal survival versus death. Recently, a core set of genes [“activity-regulated inhibitor of death” (AID) genes] including the transcription factor (TF) NPAS4 (neuronal PAS domain protein 4) has been found to provide activity-induced protection against neuronal death caused by excitotoxic stimulation. However, the downstream targets of AID action mediating neuroprotection remained so far unknown. Here, we have identified synaptotagmin 10 (Syt10), a vesicular Ca2+ sensor, as the first neuroprotective effector protein downstream of the TF NPAS4. The expression of Syt10 is strongly upregulated by pathophysiologic synaptic activity after kainic acid (KA) exposure and its absence renders mouse hippocampal neurons highly susceptible to excitotoxic insults. We found NPAS4 as critical for the increase in Syt10 levels and in turn the ability of NPAS4 to confer neuroprotection against KA-induced excitotoxicity to be severely diminished in Syt10 knock-out neurons. In summary, our results point to an important role for signaling of the NPAS4–Syt10 pathway in the neuronal response to strong synaptic activity as a consequence of excitotoxic insults.
SIGNIFICANCE STATEMENT Aberrant synaptic activity is observed in many neurological disorders and has been suggested as an important factor contributing to the pathophysiology. Intriguingly, pathophysiologic activity can also trigger signaling cascades mediating potentially compensatory neuroprotection against excitotoxic insult. Here, we identify a new neuroprotective signaling cascade involving the activity-induced transcriptional regulator NPAS4 and the vesicular Ca2+-sensor protein synaptotagmin 10 (Syt10). Syt10 is required for NPAS4 to protect hippocampal neurons against excitotoxic cell death. NPAS4 in turn controls the activity of the Syt10 gene, which is strongly induced by pathophysiologic activity. Our results uncover an entirely unexpected, novel function of Syt10 underlying the response of neurons to pathophysiologic activity and provide new therapeutic perspectives for neurological disorders.
Keywords: cell death, neuroprotection, neurotoxicity, synaptic activity, transcriptional regulation
Introduction
Excessive release of glutamate triggered by pathophysiologic synaptic activity, a process referred to as excitotoxicity, has been suggested as a key mechanism contributing to neuronal degeneration after transient or repetitive insults to the brain including status epilepticus (SE; Lau and Tymianski, 2010; Bell and Hardingham, 2011; Mehta et al., 2013). One consequence of the multiple signaling cascades activated by pathophysiologic synaptic activity is the initiation of specific gene expression programs, which in turn critically determine the decision between neuronal death or survival (Hardingham and Bading, 2010). Recently, a core set of less than a dozen genes has been identified that protects hippocampal and cortical neurons, both in vitro and in vivo, from different forms of cell death. Excitotoxic stimulation induced a transcript signature designated as “activity-regulated inhibitor of death” (AID) genes in response to transient insults, including ischemia or kainic acid-induced SE (KA-SE; Zhang et al., 2009, 2011). The AID cluster consists mainly of transcriptional regulators with neuronal PAS domain protein 4 (NPAS4) as functional key component (Zhang et al., 2009). It has been put forward that AID genes act by inducing the transcription of target genes that then execute neuroprotection but to date the molecular identity of these downstream target genes has not been resolved.
Intriguingly, the functional significance of NPAS4 is not restricted to AID-cluster control. It also plays a role in the regulation of gene programs mediating synaptic plasticity in the brain (Maya-Vetencourt, 2013; Benito and Barco, 2015; Nonaka et al., 2014). NPAS4 is an early response transcription factor of synaptic activity whose expression is strongly induced in excitatory and inhibitory neurons by Ca2+ influx through voltage-gated channels but not by increased cAMP concentrations or neurotrophic factors (Lin et al., 2008; Spiegel et al., 2014). It has been suggested to function in a wide range of processes ranging from the control of homeostatic plasticity, e.g., the formation and maintenance of inhibitory synapses (Lin et al., 2008; Spiegel et al., 2014), to the mechanisms underlying fear memory (Ramamoorthi et al., 2011). Thereby, via activating distinct gene networks, NPAS4 seems to be involved in the region- and cell-type-specific rewiring and plasticity in response to sensory information in physiologic settings independent of AID control after insults.
An intriguing AID downstream candidate gene that exhibits a strong transient upregulation in the hippocampus early after KA-SE, is synaptotagmin 10 (Syt10; Babity et al., 1997). This dramatic increase in abundance is particularly striking, as under physiologic conditions hippocampal Syt10 mRNA levels are very low. Synaptotagmins are an evolutionary conserved family of transmembrane proteins that play a role in the regulation of membrane trafficking. Members of the synaptotagmin gene family have been shown to be involved in the regulated exocytosis of vesicles and to act as Ca2+ sensors in vesicular fusion processes (Südhof, 2002; Gustavsson and Han, 2009; Cao et al., 2011; Moghadam and Jackson, 2013). Accumulating evidence suggests that defects in membrane trafficking are a hallmark of disease-associated neurodegeneration (Glavan et al., 2009; Wang et al., 2013). Together, these findings have led to the hypothesis that Syt10 fulfills a general role in seizure-induced injury responses (Glavan et al., 2009; Cao et al., 2011). However, so far the function of Syt10 in these processes and in the hippocampus has not been examined yet.
Here, we set out to investigate the link between the activity-induced stimulation of Syt10 gene expression in the hippocampus and its functional consequences. Our results show that both, hyperexcitation and the overexpression of the AID transcription factor NPAS4, increase Syt10 mRNA levels in hippocampal neurons in vitro and in vivo. Intriguingly, NPAS4 is also required for the activity-induced upregulation of Syt10 gene expression. Syt10 deficiency selectively decreases neuronal but not glial survival in response to excitotoxic stimulation. Furthermore, the ability of NPAS4 to confer neuroprotection against excitotoxicity is severely diminished in Syt10 knock-out (KO) neurons. Thus, we identify Syt10 as an essential component in the neuroprotective signaling cascade downstream of NPAS4 in response to excitotoxic stimulation.
Materials and Methods
DNA constructs and cloning.
The Syt10 promoter fragments were amplified by PCR from rat genomic DNA as template using primers with the corresponding restriction sites for subcloning into the luciferase reporter vector pGL-3 basic (Promega; pGL3–4713 FW: 5′-GCGACGCGTAGTGATTTGTAGAAAAGACCACATGA-3′, MluI; pGL3–1036 FW: 5′-GCGACGCGTGGGGTAACTTTACCATAACCTGG-3′, MluI; pGL3–306 FW: 5′-GCGACGCGTTCAGGAAGGTGTGTGGGAA-3′, MluI; pGL3–306 RV: 5′-GCGGTCGACCTTGGCTTCTGCTCGCAG-3′, SalI; RR-I FW: 5′-GCGGGATCCTAAAGGGACAAGCTTGGGCT-3′, XhoI; RR-I RV: 5′-GCGGGTCGACAAAATTCTTGATAAATACTTGCTGAA 3′, BglII; RR-5′ FW: 5′-GCGGGTACCTCTTCAGGCTCAAAATTAGTGG-3′, KpnI; RR-5′ RV: 5′-GCGGGTACCCAAACTTCAAAAACATCATGCTA-3′, KpnI; RR-3′ FW: 5′-GTCGACGTTCTTCTCGCAGAGCCTCA-3′, SalI; RR-3′ RV: 5′-GTCGACAAGTCGGGGAAAGGGATTATT-3′, SalI; RR-1/-3 FW: 5′-GCGGGTACCAGAAGTTTACCCAGCTCACCA-3′, KpnI; RR-1/-2 RV: 5′-GCGGGTACCACTCACTGTGGTAAGCATTCATT-3′, KpnI; RR-2 FW: 5′-GCGGGTACCCTGCCCAGACTGTAGATAAACG-3′, KpnI; RR-3 RV: 5′-GCGGGTACCTTATCTACAGTCTGGGCAGAACA-3′, KpnI; exon 1 FW: 5′-GCGCTCGAGTACCTGTGCCACCTCCG-3′, XhoI; exon 1 RV: 5′-GCGAAGCTTAGTTTCCGCAAGGAGGAC-3′, HindIII). The same reverse primer was used to generate plasmids −4713, −1036, and −306. The NPAS4-shRNA sequence 5′-GGTTGACCCTGATAATTTA-3′ corresponded to the previously described NPAS4-siRNA (Lin et al., 2008) and was cloned with BglII and HindIII into pAAV-U6-shRNA-CBA-hrGFP, which was kindly provided by Martin Schwarz (University of Bonn, Medical Centre, Bonn, Germany). The overexpression construct for NPAS4 was kindly provided by Tõnis Timmusk (Tallinn University of Technology, Tallinn, Estonia), for CREB, USF1, and USF2 by Priit Pruunsild (Tallinn University of Technology, Estonia), for cJun, JunB, and JunD by Marc Piechaczyk (Université de Montpellier, France) and for MEF2 by David McFadden (University of Connecticut School of Medicine, Farmington, CT). Recombinant adeno-associated virus (rAAV) viral vectors for NPAS4 and a shRNA against NPAS4 were generous gifts of Hilmar Bading (University Heidelberg, Germany).
Husbandry of mice and rats.
Mice and rats were housed under a 12 h light/dark cycle with food and water ad libitum. All experiments were performed in accordance with the guidelines of the European Union and the University of Bonn Medical Center Animal Care Committee. Time-pregnant female Wistar rats were obtained from Charles River Laboratories. Constitutive Syt10 KO mice in which exon 2 had been deleted by crossing conditional KO mice with a ubiquitous Cre-driver line were kindly provided by T. Südhof (Stanford University, Stanford, CA). Syt10 KO mice are viable and fertile and do not exhibit an overt phenotype (Cao et al., 2011).
Primary hippocampal neuron cultures.
Primary hippocampal neurons were isolated from rat pups of both sexes as described previously (Zürner et al., 2011). In brief, hippocampus was washed three to five times with HBSS (Life Technologies) and then digested with trypsine (0.025 g/ml, Life Technologies, for 20 min at 37°C). The incubation was performed for 20 min at 37°C followed by three to five washing steps with HBSS and the remaining DNA was digested with DNase I (0.001 g/ml; Roche). Next, the tissue was dissociated using cannulas (3 times 0.9 × 40 mm; 3 times 0.45 × 23 mm). The solution was passed through a Nylon cell strainer (100 μm; BD Biosciences) and the mesh was rinsed with 4–10 ml basal medium eagle (BME; Life Technologies) supplemented with 0.5% glucose (Sigma-Aldrich), 10% fetal calf serum (FCS), 2% B-27, and 0.5 mm l-glutamine (all Life Technologies) to collect all cells. After counting, the cells were plated on a 24-well cell culture plate at a density of 70,000 cells per 24-well. Neurons were cultured in a humidified incubator at 37°C and 5% CO2. Rat hippocampal neurons were stimulated with 25 μm KA for 24 h at 13–14 DIV. To expose Syt10 KO neurons to secreted factors from wild-type (WT) neurons during KA stimulation coverslips with Syt10 KO neurons were placed in dishes containing WT neurons during KA stimulation. IGF-1 rescue experiments were performed by adding 1 or 100 ng/ml IGF-1 (human, PeproTech) to Syt10 KO neurons simultaneously with KA. After 24 h, neurons were fixed, immunolabeled, and analyzed by confocal microscopy.
DNA transfection and luciferase assay.
Rat hippocampal neurons were transfected in 24-well cell-culture plates in triplicates at 5 DIV using Lipofectamine (Life Technologies) according to the manufacturer's protocol. During transfection, BME was replaced by reduced serum medium (OPTI-MEM, Life Technologies) containing the transfection solution and cells were kept at 37°C and 5% CO2. After 2 h, OPTI-MEM was substituted for the original BME. The transfection mixture contained 0.1 μg of the pGL-promoter construct expressing firefly luciferase, 0.025 μg pRL-TK with the Renilla gene (Promega), the indicated amount of expression plasmid of the respective transcription factor, and 1 μl Lipofectamine in 50 μl OPTI-MEM. The mixture was incubated at room temperature for 20 min and then added to the wells. Cells were collected 48 h after transfection and luciferase assays were performed using the Dual Luciferase Reporter Assay System (Promega). For analysis of Renilla and Firefly activities, a Glomax Luminometer (Promega) was used and the results are presented as Firefly/Renilla relative luciferase units.
RNA isolation and RT-qPCR.
mRNA from primary hippocampal neurons or from the microdissected CA1 region was isolated using Dynabeads mRNA DIRECT Micro Kit (Life Technologies) and cDNA was synthesized using RevertAid First Strand cDNA Synthesis Kit (Life Technologies) both performed following the manufacturer's specifications. The cDNA was used for quantitative real-time PCR (RT-PCR) with the Maxima SYBR Green qPCR Master Mix/ROX (Life Technologies) according to the following protocol: the reaction volume of 6.25 μl contained 3.125 μl Master Mix, 1.5 μl DEPC-H2O, 1.25 μl cDNA, and 0.1875 μl of each primer (10 pmol/ml; NPAS4 FW: 5′-GGTGTCTCAACATTCCCCTA-3′; NPAS4 RV: 5′-GTTCCCCTCCACTTCCATCT-3′; mSyp FW: 5′-TTCAGGACTCAACACCTCGGT-3′; mSyp RV: 5′-CACGAACCATAGGTTGCCAAC-3′; mSyt10 FW: 5′-TCCCTCCAGAGAATGTGGAC-3′; mSyt10 RV: 5′-AGTCCAGTTCGACACACGCCT-3′). After preincubation at 95°C for 10 min, 40 PCR cycles (15 s at 95°C, 60 s at 59°C, and 40 s at 72°C) were performed on an ABI Prism 9700HT system (Life Technologies). Experiments were performed in triplicates and analysis was performed according to the ΔΔCt method. Expression of the housekeeping gene Synaptophysin was used to normalize expression. All quantitative RT-PCR data were normalized to the respective control condition.
Viral vector production.
Recombinant AAV1/2 genomes were generated by large-scale triple transfection of HEK293 cells as described previously (van Loo et al., 2012). In brief, the adeno-associated virus (AAV) plasmid of interest (rAAV-CMV-NPAS4-FLAG, rAAV-Syn-GFP, rAAV-U6-scrambled-shRNA-hrGFP, rAAV-U6-shNPAS4-hrGFP), helper plasmids encoding rep and cap genes (pRV1 and pH21), and adenoviral helper pFΔ6 (Stratagene) were transfected using standard CaPO4 transfection. Cells were harvested ∼72 h after transfection. For crude viral extracts, cell pellets were resuspended in 1 ml DMEM (Life Technologies) supplemented with 10% (v/v) heat inactivated FCS (Hyclone), 100 units/ml penicillin/streptomycin (pen/strep) and 2 mm glutamine. Then, the cell suspension underwent four consequent freeze/thaw cycles and after a brief centrifugation, the supernatant was transferred to a new tube and stored at 4°C. For purified viruses, cell pellets were lysed in the presence of 0.5% sodium deoxycholate (Sigma-Aldrich) and 50 units/ml Benzonase endonuclease (Sigma-Aldrich). rAAV viral particles were purified from the cell lysate by HiTrapTM heparin column purification (GE Healthcare), and then concentrated using Amicon Ultra Centrifugal Filters (Millipore) until a final stock volume of 500 μl was reached. Coomassie blue staining of SDS-polyacrylamide gels loaded with 7–15 μl virus stock validated the purity of the viruses.
Infusion of AAV vectors.
AAV viral particles were infused as described previously (van Loo et al., 2012). In brief, male adult Bl6/N mice (≥55 d old; weight ≥20 g) were anesthetized with 6 mg/kg xylazine (Rompun; Bayer) plus 90–120 mg/kg ketamine (Ketavet; Pfizer), intramuscularly. Intracerebral injection of viral particles in the left and right CA1 hippocampal region was performed stereotaxically at the coordinates −2 posterior, −2 lateral, and 1.7 ventral relative to bregma. Holes the size of the injection needle were drilled into the skull, and 1 μl of viral suspension containing ∼108 transducing units was injected using a 10 μl Hamilton syringe at a rate of 100 nl/min using a microprocessor-controlled mini-pump (World Precision Instruments). After injection, the needle was left in place for 5 min before withdrawal. Fourteen days after infection, mice were decapitated under deep isoflurane anesthesia (Forene), and the CA1 region of hippocampi was microdissected. All experiments were performed in accordance with the guidelines of the University of Bonn Medical Center Animal Care Committee.
Infection of neurons with crude viral extracts.
In the respective experiments, neurons were infected 7–9 DIV with 20 μl crude viral extracts until the neurons were prepared for further analyses.
Western blot.
Proteins were separated on a 10% SDS-PAGE and transferred to Whatman Protran nitrocellulose membranes (Sigma-Aldrich). For immunolabeling, membranes were blocked for 1 h with 2% fish gelatin in 1× PBS at room temperature. Next, membranes were incubated with the respective primary antibody at 4°C overnight in 1× PBS with 0.1% Tween and the secondary antibody was used on the subsequent day in 1× PBS with 0.1% Tween and 0.01% SDS for 1 h at room temperature. The following dilutions were used: rabbit anti-Syt10 (A.M.H.W and S.S., unpublished observations) 1:250; mouse anti-β-actin 1:10,000 (ab8226, Abcam); goat anti-rabbit IRDye 800 (LI-COR Biosciences) 1:20,000; goat anti-mouse IRDye 680 (LI-COR Biosciences) 1:20,000. Western blots were scanned with an Infrared imaging system (Odyssey, LI-COR) and quantification of Western blots was performed using the software AIDA.
Immunocytochemistry.
Primary hippocampal neurons at 14 DIV were fixed for 10 min with 4% paraformaldehyde, washed three times in 1× TBS and incubated in blocking solution (0.3% Triton X-100, 10% NGS in 1× TBS) for 1 h. Then, neurons were left overnight with primary antibodies (mouse anti-MAP2, 1:500, Merck, Millipore; rabbit anti-GFAP, 1:2000, Dako) at 4°C in blocking solution. On the following morning, cells were washed three times with 1× TBS and subsequently incubation with the secondary antibodies (goat anti-mouse AlexaFluor 488, 1:400; goat anti-rabbit AlexaFluor 647, 1:400, Life Technologies) and with 0.1 μg/ml 4′,6-diamidino-2-phenylindole (DAPI; Life Technologies) was performed in blocking solution for 45 min at room temperature in the dark. Cover glasses were mounted in Mowiol 4-88 (Roth) and imaged with a confocal laser-scanning microscope (Eclipse Ti, Nikon). Cells that were positive for MAP2 or GFAP were counted with ImageJ and normalized to the total cell number (DAPI).
Statistical analysis.
Statistical analyses were performed with GraphPad Prism 6.05 software. Results were tested for statistical significance using Student's t test or one-way ANOVA followed by Tukey's post hoc test. Values were considered significant at p < 0.05. All results were plotted as mean ± SEM. Sample size (n) per experiment was calculated using power analysis, with parameters set within the accuracy of the respective experiment. All in vitro experiments were independently repeated at least two times.
Results
Pathophysiologic synaptic activity elevates Syt10 mRNA and protein levels in rat primary hippocampal neurons
Syt10 expression is strongly increased in the hippocampus after KA-SE (Babity et al., 1997). To analyze relevant signaling cascades in a controlled fashion, we turned to cell culture models in which neuronal damage is induced by activity. First, we examined whether different stimuli inducing strong synaptic activity in vitro congruently upregulate Syt10 levels in cultured primary hippocampal neurons. Depolarization of rat primary hippocampal (RH) neurons following established protocols with potassium chloride (KCl, 25 and 55 mm; Zhou et al., 2006; Pruunsild et al., 2011) resulted in a significant increase in both Syt10 mRNA and protein levels compared with the control condition as measured by quantitative RT-PCR (Fig. 1A) and quantitative immunoblotting (Fig. 1B,C). We furthermore stimulated RH neurons with 25 μm KA for 24 h and observed a strong increase in Syt10 mRNA levels after this treatment (Fig. 1D). These findings show that in RH neurons expression of Syt10 is activated by pathophysiologic synaptic activity after different stimulation paradigms.
Figure 1.

Strong pathophysiologic stimuli elevate Syt10 expression levels. A, Relative mRNA expression levels of Syt10 in RH neurons at 7 DIV. Neurons were stimulated with 25 mm KCl for 8 h and subsequently lysed for quantitative RT-PCR (N = 2, n = 3). B, Representative immunoblot of RH neurons that were stimulated at 14 DIV with 55 mm KCl for 15 min. Cells were harvested 24 h later for Western blot analysis. Here, β-actin was used to normalize protein expression. C, Western blot quantification of stimulated RH neurons as shown in B. All values were normalized to the respective control condition (N = 2, n = 2). D, Quantitative RT-PCR on mRNA extracted from KA stimulated or nonstimulated RH neurons at 14 DIV. Neurons were stimulated with 25 mm KCl for 8 h and lysed subsequently and Syt10 mRNA expression was analyzed (N = 2, n = 3). Statistical analysis was performed using Student's t test. **p ≤ 0.01, ***p ≤ 0.001. MW, Molecular weight.
Syt10 knock-out neurons are more susceptible to KA-induced cell death
One hallmark of pathophysiologic synaptic activity is the excessive release of glutamate, which is associated with exocytotoxic processes like neuronal dysfunction and cell death. To resolve the functional relevance of pathophysiologic activity-induced Syt10 expression we next examined whether Syt10 deficiency alters survival of neurons after KA treatment. To this end, we incubated primary hippocampal neurons from Syt10 littermate KO and WT mice with 25 μm KA for 24 h. Subsequently, we immunolabeled the cultures with antibodies against a neuronal [microtubule-associated protein 2 (MAP2)] and a glial [glial fibrillary acidic protein (GFAP)] marker protein as well as with DAPI to mark the cell nuclei (Fig. 2A). To quantify the cell survival rate the ratio of MAP2- or GFAP-positive cells versus the total cell number (DAPI) of the respective image was determined (Fig. 2B,C). This analysis revealed that Syt10 deficiency strongly aggravated neuronal cell death (Fig. 2B), whereas it had no impact on the survival of GFAP-positive glial cells (Fig. 2C). No difference between WT and Syt10 KO was observed in the survival of hippocampal neurons under basal conditions (Fig. 2D). These results furthermore demonstrate that the absence of Syt10 renders neurons more susceptible to KA-induced excitotoxic insults indicating a neuroprotective function for Syt10.
Figure 2.
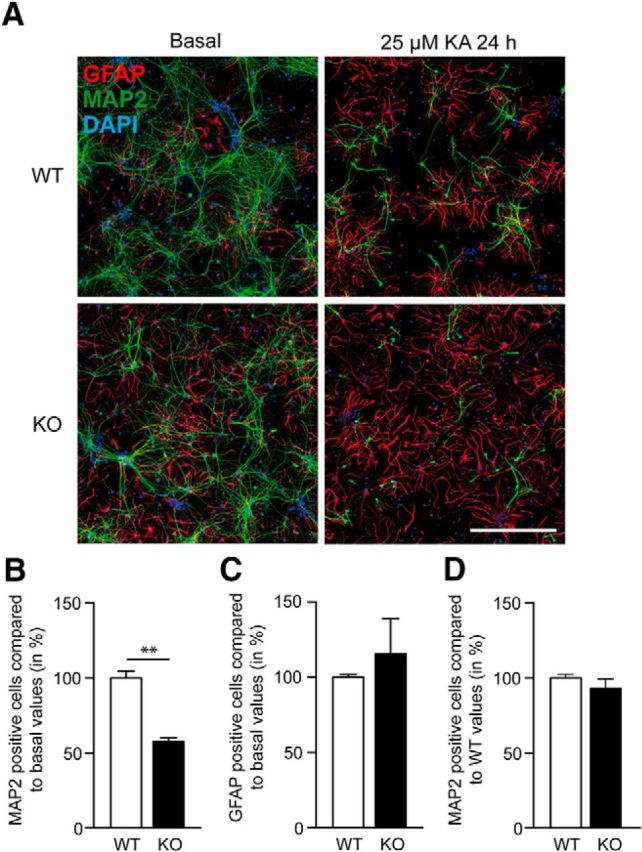
Syt10 deficiency decreases the survival rate of neurons after KA stimulation. A, Representative images of dissociated Syt10 WT and KO neurons under basal conditions (left) and following stimulation for 24 h with 25 μm KA (right). Neurons were subsequently fixed for immunostaining against the neuronal marker MAP2, the astrocytic marker GFAP and cell nuclei (DAPI). Scale bar, 500 μm. B, C, Quantification of the immunohistochemical reactions as shown in A. D, Survival of WT and Syt10 KO neurons under basal conditions. Cells that were positive for MAP2 or GFAP were counted and were normalized to the total cell number (DAPI). Then, all values were normalized to the WT basal values. Statistical analysis was performed using Student's t test. **p ≤ 0.01; N = 2, n = 3.
A secreted factor from WT neurons counteracts KA-induced cell death of Syt10 KO hippocampal neurons
One potential explanation for the increased cell death of Syt10 KO neurons upon KA treatment could be the involvement of Syt10 in the release of a neuroprotective factor. To test this hypothesis, we stimulated Syt10 KO neurons with 25 μm KA for 24 h while exposing them to medium conditioned by WT neurons under the identical stimulation paradigm. Syt10 KO neurons that had been exposed to KA in the presence of secreted factors from WT neurons had a significantly higher survival rate than neurons that were treated with KA alone (Fig. 3A). The rate of cell death under these conditions could be rescued to the level observed in WT neuronal cultures after KA treatment (Figs. 2B, 3B).
Figure 3.

Decreased survival rate of Syt10 neurons in response to KA can be rescued by a secreted factor. A, Syt10 KO neurons were incubated at 13–14 DIV for 24 h with or without 25 μm KA and in the absence or presence of medium from WT neurons. Subsequently, cells were immunolabeled for MAP2, GFAP, and DAPI and neuronal survival was quantified by normalizing the number of MAP2-positive cells to the total cell number represented by DAPI in staining. Statistical analysis was performed using one-way ANOVA followed by Tukey post hoc test. **p ≤ 0.01, ***p ≤ 0.001, N = 5, n = 3–4. B, To assess rescue efficiency the number of surviving neurons after KA incubation was normalized to the rescue condition. C, Neurons were incubated at 14 DIV for 24 h with or without 25 μm KA and IGF-1 and the level of neuronal cell death was analyzed as described in A. Statistical analysis was performed using one-way ANOVA followed by Tukey post hoc test. ***p ≤ 0.001; N = 2, n = 4.
In the olfactory bulb Syt10 has been shown to be important for the release of IGF-1 (Cao et al., 2011) and a neuroprotective function for IGF-1 has been reported for multiple types of brain injury (Fernandez and Torres-Alemán, 2012). Thus, we examined whether IGF-1 can rescue the neuronal cell death induced by KA in Syt10 KO neurons. We performed the KA stimulation of Syt10 KO neurons in the presence of 1 ng/ml or 100 ng/ml IGF-1, respectively. Neither concentration had a significant effect on the level of cell death of Syt10 KO neurons (Fig. 3C). Together, these data suggest that Syt10 is required for the secretion of one or more neuroprotective factors in response to KA-induced excitotoxicity.
The activity-induced upregulation of Syt10 gene expression is mediated by regulatory elements in its promoter region
So far, the strong induction of Syt10 gene expression by signaling cascades causing hyperexcitation and neuronal cell death, as well as the increased susceptibility to KA-induced cell death of Syt10 KO neurons suggest that Syt10 is part of an activity-induced process mediating neuroprotection. To identify this neuroprotective signaling cascade we aimed at identifying the pathway activating Syt10 gene transcription in response to activity. Therefore, we first characterized the bioinformatically identified Syt10 core promoter region (Fig. 4A) and potential upstream and downstream regulatory elements (40 kb upstream and 70 kb downstream of the Syt10 start ATG; Fig. 4A). The corresponding genomic fragments were cloned in front of a luciferase reporter plasmid (Fig. 4B) and transfected into RH neurons. Luciferase assays revealed that in accordance with the tight regulation of endogenous Syt10 expression the promoter region contained both strong enhancing and repressive regulatory elements within a region of ∼4.7 kb upstream of the ATG (Fig. 4B,D). We next examined whether also the synaptic-activity responsive regulatory element is present within this genomic region. RH neurons were transfected with luciferase reporter plasmids under control of the Syt10 promoter fragments −306, −1036, and −4713 and promoter activity was measured after depolarization with 25 mm KCl compared with control conditions (Fig. 4C). Membrane depolarization increased the relative luciferase activity of the longest promoter fragment −4713 but not of the two shorter ones suggesting that this genomic region contains transcription factor binding sites that mediate the activity-induced increase in Syt10 promoter activity. Together, these results show that the proximal promoter region around the core promoter contains positive regulatory elements that are flanked by repressive sequences. The upstream enhancer region (−3675 to −4344) is necessary and sufficient to stimulate Syt10 promoter activity in response to membrane depolarization (Fig. 4D).
Figure 4.

The Syt10 promoter contains regulatory elements mediating the activity-induced induction of Syt10 gene expression. A, Graphic representation of the rat Syt10 gene (chromosome 7). Using UCSC, 50 kb upstream and 70 kb downstream of the start ATG were analyzed. The conservation track was based on PhastCons (black bars). The lower gray bars represent pairwise alignments between the indicated species. Regions with high homology were identified and according to their position, they were named regulatory region-5′ (RR-5′), regulatory region-intron (RR-IN), regulatory region-3′ (RR-3′) and regulatory region-1, -2, and -3 (RR-1, -2, -3). Gray boxes and black box with positions (−306, −1036, −4713) indicate the fragments that were subcloned in the pGL3 basic vector carrying a luciferase reporter gene. B, Basal luciferase activity of different Syt10 promoter constructs and combined fragments consisting of −306 and a regulatory region in RH neurons. Neurons were transfected at 5 DIV, were lysed 48 h later for luciferase measurements, and Firefly/Renilla relative units were normalized to pGL3 basic. Statistical analysis was performed using a one-way ANOVA followed by Tukey post hoc test; N ≥ 2, n = 3. C, Luciferase activity of RH neurons at 7 DIV that were transfected with the indicated promoter fragments at 5 DIV. Forty hours later, cells were stimulated with 25 mm KCl for 8 h and lysis was performed directly after stimulation (N = 2, n = 3). B, C, *p ≤ 0.05, ***p ≤ 0.001, significance to pGL3 basic (B), significance to basal values (C); ###p ≤ 0.001, significance to core promoter (B). D, Graphic summary of activating (+) and repressing (−) regions of the Syt10 promoter.
The activity-regulated transcription factor NPAS4 stimulates Syt10 expression and enhances the depolarization-induced transcriptional response
To assess which TFs might be involved in the hyperexcitation-induced transcriptional upregulation of the Syt10 gene, we searched for TF binding sites, which are known to be differentially expressed after synaptic activity, including CREB, members of the activator protein 1 (AP1) transcription factor family, myocyte enhancer factor 2A, NPAS4, and upstream stimulatory factor 1 and 2 (USF1 and 2; Lyons and West, 2011). For all of these TFs, we detected predicted binding sites in the Syt10 promoter, mainly within the region −4713 bp upstream of the ATG (Fig. 5A); however, none of these sites was substantially overrepresented within the Syt10 promoter compared with its frequency of occurrence genome-wide or in all other promoter regions (data not shown). To identify functionally relevant TFs we next examined their ability to increase endogenous Syt10 expression by overexpressing the listed TFs in hippocampal neurons and measuring Syt10 mRNA levels using quantitative real-time PCR (Fig. 5B). Only NPAS4 and USF2 caused a significant increase of endogenous Syt10 expression, whereas the other TFs did not show a clear effect (Fig. 5B). As only <30% of neurons were transfected, this indicates a robust stimulatory effect of NPAS4 and USF2 on Syt10 gene expression. To test whether NPAS4 and USF2 are indeed able to activate the activity-inducible Syt10 promoter in hippocampal neurons we transfected the Syt10 −4713 luciferase construct into RH neurons together with a construct coding for NPAS4 or USF2. Whereas USF2 expression had no effect, NPAS4 increased luciferase activity of this promoter fragment by 2.5-fold (Fig. 5C) suggesting that Syt10 is a downstream target of NPAS4.
Figure 5.
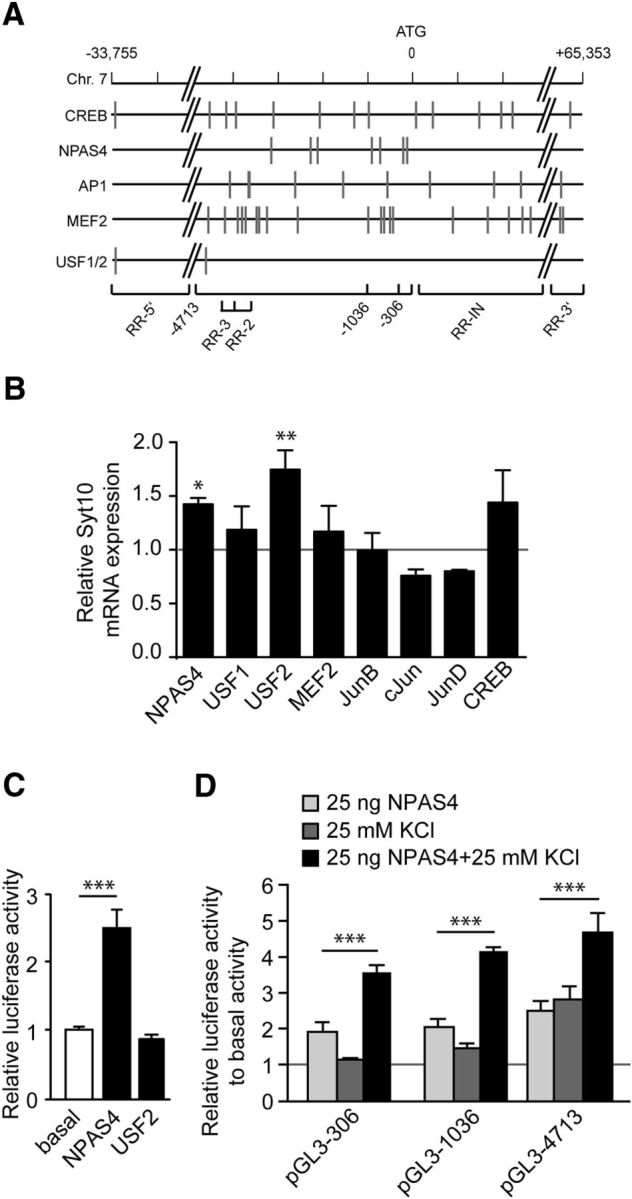
The Syt10 gene harbors functional binding sites for NPAS4. A, Graphic representation of the rat Syt10 gene with predicted transcription factor binding sites (indicated by gray vertical lines) of factors known to be involved in differential expression after synaptic activity (CREB1, NPAS4, AP1, MEF2, USF1/2). B, Quantitative RT-PCR for Syt10 mRNA expression levels from RH neurons transfected at 5 DIV with an overexpression construct for NPAS4, USF1, USF2, MEF2, JunB, cJun, JunD, or CREB. Cells were lysed 24 h (6 DIV) after transfection. All values were normalized to the respective control condition (transfection with an empty vector; gray line). Only for NPAS4 and USF2, a significant upregulation of Syt10 was observed. Statistical analysis was performed using a one-way ANOVA followed by Tukey post hoc test. *p ≤ 0.05, **p ≤ 0.01; N = 2, n = 3. C, Luciferase activity of RH neurons transfected at 5 DIV with an empty vector, 25 ng NPAS4 or 25 ng USF2 and lysed 48 h later. Firefly relative units are normalized to the basal values (mock transfected cells). NPAS4 increased the luciferase activity of the Syt10 promoter fragment −4713, whereas USF2 had no effect. Statistical analysis was performed using a one-way ANOVA followed by Tukey post hoc test. ***p ≤ 0.001; N = 3, n = 3. D, Luciferase activity of RH neurons that had been transfected with the indicated Syt10 promoter fragments and with 25 ng NPAS4 followed by KCl stimulation. Statistical analysis was performed using a one-way ANOVA followed by Tukey post hoc test. ***p ≤ 0.001; N = 2, n = 3.
NPAS4 is known to be involved in mediating transcriptional responses induced by synaptic activity (Lin et al., 2008; Zhang et al., 2009; Pruunsild et al., 2011; Ramamoorthi et al., 2011). To examine whether NPAS4 plays a role in activating the Syt10 expression triggered by hyperexcitation we analyzed the effect of NPAS4 overexpression on the depolarization-induced activity of the Syt10 promoter fragments −306, −1036, and −4713. Activity of all three promoter fragments was synergistically augmented by NPAS4 and membrane depolarization (Fig. 5D). Collectively, these findings indicate that NPAS4 is involved in mediating the activation of the Syt10 promoter in response to activity.
Endogenous Syt10 is an in vitro and in vivo downstream target of NPAS4
So far, the target genes of NPAS4 in mediating neuroprotection under activity-induced damage threat scenarios are enigmatic (Zhang et al., 2009, 2011). Our present results suggest that Syt10 may act as a downstream target gene in the NPAS4 regulated signaling cascade as potential mediator of neuroprotective action. To further scrutinize this hypothesis we tested the relevance of activity-induced NPAS4 signaling dynamics for endogenous Syt10 transcript kinetics. First, RH primary neurons were transduced with rAAV particles expressing NPAS4 and mRNA levels were measured 1 week after transduction using quantitative RT-PCR. In accordance with the promoter data a strong increase in NPAS4 mRNA levels (Fig. 6A) resulted in significantly increased endogenous Syt10 mRNA levels (Fig. 6B). Next, we analyzed whether Syt10 is also in vivo a downstream target of NPAS4. To this end rAAV-NPAS4 particles were injected into the mouse CA1 hippocampal subregion and 2 weeks after injection NPAS4 and Syt10 transcript levels in this region were determined. Intriguingly, also in vivo Syt10 gene expression levels are upregulated by NPAS4 overexpression (Fig. 6C,D). We next examined whether NPAS4 loss-of-function affects endogenous Syt10 gene expression. To this end we transduced RH neurons with rAAV particles expressing either a published shRNA against NPAS4 or a scrambled shRNA variant (Lin et al., 2008). Quantitative RT-PCR revealed a significant reduction in NPAS4, as well as Syt10 mRNA levels in the presence of the NPAS4 shRNA compared with neurons that were transduced with a scrambled shRNA (Fig. 6E,F).
Figure 6.

NPAS4 elevates Syt10 expression in vitro and in vivo. A, B, Quantitative RT-PCR on mRNA extracted from RH neurons infected with viral particles at 7 DIV encoding a rAAV-NPAS4 overexpression construct (NPAS4). Neurons were lysed at 14 DIV and mRNA expression levels of NPAS4 (A) and Syt10 (B) were measured (N = 3, n = 3). C, D, Quantitative RT-PCR on mRNA extracted from the hippocampal subregion CA1 from control (rAAV-GFP; n = 7) and rAAV-NPAS4-injected (n = 7) mice (N = 1). E, F, Quantitative RT-PCR analysis for NPAS4 (E) and Syt10 (F) on RH neurons (14 DIV) transduced at 9 DIV with either a scrambled shRNA (control shRNA) or an shRNA directed against NPAS4; N = 5, n = 3. Statistical analysis was performed using Student's t test. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001.
These results strongly argue for Syt10 as a downstream target in the activity-induced NPAS4 signaling cascade.
Syt10 is a neuroprotective downstream effector of NPAS4 in activity-induced neuronal cell death
Overexpression of NPAS4 has previously been reported to rescue activity-induced cell death of hippocampal neurons (Zhang et al., 2009). To further support our idea that Syt10 is a functional downstream effector of NPAS4 in the AID signaling cascade, we examined the ability of NPAS4 overexpression to rescue activity-induced cell death in hippocampal neurons from Syt10 KO mice. For this purpose, Syt10 WT and KO hippocampal cultures were transduced with rAAV particles expressing either GFP or NPAS4 and stimulated with KA or treated as controls. Intriguingly, the subsequent analysis of MAP2-positive neurons showed that, whereas NPAS4 almost completely rescued KA-induced cell death in Syt10 WT neurons (Fig. 7A), NPAS4 did not have the same effect in Syt10 KO neurons (Fig. 7B), where the majority of neurons died (Fig. 7B,C). In summary, our results clearly demonstrate that Syt10 acts downstream of NPAS4 in promoting neuronal survival in response to strong synaptic activity.
Figure 7.

NPAS4 rescues the KA-induced cell death only in Syt10 WT neurons. A, B, Dissociated Syt10 WT (A) and KO (B) neurons were transduced at 9 DIV with a control virus (rAAV-GFP) or with a virus encoding a rAAV-NPAS4 overexpression construct (NPAS4). At 13 DIV, neurons were stimulated, stained, and cells that were positive for MAP2 were counted with and normalized to the total cell number (DAPI). All values were normalized to the GFP basal values. C, Difference of the NPAS4-mediated rescue of kainate-treated Syt10 WT compared with Syt10 KO neurons. All values were normalized to the WT values. Statistical analysis was performed using a one-way ANOVA followed by Tukey post hoc test (n.s. > 0.05, ***p ≤ 0.001; A, B) or using Student's t test (***p ≤ 0.001; N = 1, n = 3; C).
Discussion
Excitotoxicity, the excessive stimulation of neurons by neurotransmitters such as glutamate, not only leads to neuronal damage and ultimately to cell death but also initiates neuroprotective, i.e., compensatory mechanisms. However, the molecular pathways mediating neuroprotection are still largely unknown. In this study, we found Syt10 gene expression to be strongly upregulated both by KCl-induced membrane depolarization and by exposure to KA. Neuronal activity, physiological and pathophysiological, engages a broad range of signaling pathways and transcriptional regulators to trigger complex stimulus-regulated gene expression programs (Lyons and West, 2011; Bading, 2013; Benito and Barco, 2015). In these hierarchical genetic programs NPAS4 is one of the first activity-induced TFs, which then initiate a “second wave” of gene expression including both additional immediate early transcription factors [e.g., c-fos, early growth factor 1 (Egr1)] and downstream effector molecules [e.g., activity-regulated cytoskeletal protein 3.1 (Arc/Arg3.1), brain-derived neurotrophic factor (BDNF); Maya-Vetencourt, 2013; Benito and Barco, 2015]. The dynamic architecture of the respective gene expression programs not only exerts substantial multiplicatory but also very specific effects that may critically determine the fate of cells under pathologic stress conditions. In recent years, NPAS4 has evolved as a key mediator of plasticity in the nervous system. The transcriptional programs triggered by NPAS4 are involved in diverse plasticity processes, for example in memory formation, addiction, and the response to cerebral ischemia, seizures, and brain injury (Maya-Vetencourt, 2013). In particular, NPAS4 has been shown to initiate a compensatory mechanism during phases of enhanced excitability (Lin et al., 2008) and to mediate acquired neuroprotection induced by synaptic activity (Zhang et al., 2009). Gain- and loss-of-function approaches have identified ∼300 downstream target genes of NPAS4 representing a substantial leverage and in parallel diversification potential of stress responses (Lin et al., 2008; Kim et al., 2010; Bloodgood et al., 2013; Spiegel et al., 2014). Not surprisingly, in vivo the gene program downstream of an individual transcription factor strongly depends on the molecular context and cellular state. To date the effector molecules regulating neuronal survival of hippocampal neurons in response to excitotoxic insults downstream of NPAS4 have not been resolved. Here we demonstrate that Syt10 expression is stimulated by increased NPAS4 levels in vitro and in vivo and that NPAS4 is required to maintain endogenous Syt10 expression levels (Fig. 8A). The observation that in the absence of Syt10 the ability of NPAS4 to confer neuroprotection against excitotoxic stimuli is severely diminished further supports Syt10 as the first bona fide NPAS4 downstream effector molecule in this cascade (Fig. 8B). Considering that neuronal cell death represents a complex process as well as the large number of downstream NPAS4 targets, the critical impact of a single molecule, like Syt10, for the neuroprotective action of the AID-factor NPAS4 appears highly remarkable.
Figure 8.

Model of the Syt10 functional role in neuroprotection. A, Identification of Syt10 as a new downstream target of NPAS4 during normal synaptic activity. Synaptic activity results in an increase of nuclear calcium (black arrow), leading to increased expression of AID genes (black arrow) resulting in an upregulation the NPAS4 target gene Syt10 (black arrow) eventually protecting neurons from cell death by a yet unknown secreted factor. B, Here, we claim a new function for Syt10 in neuroprotection following pathophysiologic synaptic activity. Syt10 plays an essential role in neuroprotection following pathophysiologic synaptic activity as only Syt10 WT but not Syt10 KO neurons can be rescued by NPAS4 from KA-induced cell death.
How in detail Syt10 contributes to activity-induced neuroprotection is still an open question. However, our data strongly suggests that Syt10 is part of a fusion machinery regulating the activity-dependent release of a neuroprotective factor. The identity of this or these secreted factors is so far unknown. Studies using Syt10 deficient mice have revealed a function for Syt10 as a Ca2+ sensor for activity-dependent release of the growth factor IGF-1 from nonsynaptic vesicles in the olfactory bulb (Cao et al., 2011). So far, it is unknown whether Syt10 is involved in controlling IGF-1 secretion also outside of the olfactory bulb. It was postulated that this is not universally the case as the phenotype of constitutive IGF-1 KO mice, i.e., reduced brain size, CNS hypomyelination, and loss of hippocampal granule and striatal parvalbumin-containing neurons (Baker et al., 1993; Liu et al., 1993; Beck et al., 1995), does not resemble the Syt10 KO phenotype (Cao et al., 2011). On the other side, in vivo levels of IGF-1 in the hippocampus are increased by excitotoxic stimuli (Choi et al., 2008; Wong-Goodrich and Mellott, 2008) and it is well established that IGF-1 can act as a potent neuroprotective factor in multiple types of brain injuries (Guan et al., 2003; Kaspar et al., 2003; Kazanis et al., 2004; Liu et al., 2004; Chesik et al., 2007; Torres-Aleman, 2007; Ebert et al., 2008; Miltiadous et al., 2010, 2011). Our observations indicate that IGF-1 is not the major neuroprotective secreted factor in the NPAS4–Syt10 signaling cascade as it is not able to rescue the KA-induced aggravated neuronal cell death of Syt10 hippocampal neurons by itself. However, our data do not allow us to exclude either that IGF-1 acts in concert with other secreted factors to mediate neuroprotection in response to KA or that Syt10 controls the release of IGF-1 in the hippocampus. Hence, as IGF-1 is only one of many known neuroprotective factors expressed in the hippocampus (Malva et al., 2012; Tamas et al., 2012; Zaben and Gray, 2013; Tovar-y-Romo et al., 2014), the next step will now be to determine the identity of the factor whose release is affected by Syt10 deficiency in this signaling cascade. Mechanistically, Syt10 may also be involved in the Ca2+-dependent fusion of intracellular transport vesicles as a defect in this process can as well disrupt vesicular exocytosis.
It was recently reported that in hippocampal neurons Syt10 is not present on vesicles that fuse in response to depolarization but rather on the plasma membrane (Dean et al., 2012). However, this observation was based on overexpression of a tagged Syt10 in native hippocampal neurons, in which endogenous Syt10 is only present at very low levels. Therefore, it might be hypothesized that in hippocampal neurons Syt10-dependent fusion processes only occur after strong, putatively pathophysiologic activity or even excitotoxic insults and that the respective vesicles and fusion machinery, just like Syt10, are only synthesized in response to these signals. Thereby, this machinery may represent a key cellular computing device for strong Ca2+ signaling. In the absence of such hypothesized vesicles, Syt10 would be mislocalized and therefore inactive. It will now be an important next step to identify these currently enigmatic vesicles and their cargo.
A more detailed understanding of the NPAS4–Syt10 neuroprotective signaling cascade might form the basis for the development of strategies to boost the endogenous neuroprotective machinery and thereby to novel therapeutic approaches for neurodegenerative disorders.
Footnotes
This work was supported by the Deutsche Forschungsgemeinschaft (DFG, SFB/TR3, SFB1089), the German Ministry of Research and Education (BMBF, 01GQ0806), the European Union's Seventh Framework Programme (FP7/2007-2013) under grant agreement no. 602102 (EPITARGET), and local funding (BONFOR). We thank Sabine Opitz, Lioba Dammer, Vanessa Schmitt, and Daniela Frangenberg for excellent technical assistance; Tom Südhof (Stanford University, Stanford, CA) for kindly providing Syt10 KO mice; and Hilmar Bading (University Heidelberg, Germany), Priit Pruunsild (Tallinn University of Technology, Estonia), Marc Piechaczyk (Université de Montpellier, France), David McFadden (University of Connecticut School of Medicine, Farminton, CT), and Tõnis Timmusk (Tallinn University of Technology, Estonia) for sharing plasmids.
The authors declare no competing financial interests.
References
- Babity JM, Armstrong JN, Plumier JC, Currie RW, Robertson HA. A novel seizure-induced synaptotagmin gene identified by differential display. Proc Natl Acad Sci U S A. 1997;94:2638–2641. doi: 10.1073/pnas.94.6.2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bading H. Nuclear calcium signalling in the regulation of brain function. Nat Rev Neurosci. 2013;14:593–608. doi: 10.1038/nrn3531. [DOI] [PubMed] [Google Scholar]
- Baker J, Liu JP, Robertson EJ, Efstratiadis A. Role of insulin-like growth factors in embryonic and postnatal growth. Cell. 1993;75:73–82. doi: 10.1016/S0092-8674(05)80085-6. [DOI] [PubMed] [Google Scholar]
- Beck KD, Poweli-Braxton L, Widmer HR, Valverde J, Hefti F. Igf1 gene disruption results in reduced brain size, CNS hypomyelination, and loss of hippocampal granule and striatal parvalbumin-containing neurons. Neuron. 1995;14:717–730. doi: 10.1016/0896-6273(95)90216-3. [DOI] [PubMed] [Google Scholar]
- Bell KF, Hardingham GE. The influence of synaptic activity on neuronal health. Curr Opin Neurobiol. 2011;21:299–305. doi: 10.1016/j.conb.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benito E, Barco A. The neuronal activity-driven transcriptome. Mol Neurobiol. 2015;51:1071–1088. doi: 10.1007/s12035-014-8772-z. [DOI] [PubMed] [Google Scholar]
- Bloodgood BL, Sharma N, Browne HA, Trepman AZ, Greenberg ME. The activity-dependent transcription factor NPAS4 regulates domain-specific inhibition. Nature. 2013;503:121–125. doi: 10.1038/nature12743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao P, Maximov A, Südhof TC. Activity-dependent IGF-1 exocytosis is controlled by the Ca(2+)-sensor synaptotagmin-10. Cell. 2011;145:300–311. doi: 10.1016/j.cell.2011.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesik D, Wilczak N, De Keyser J. The insulin-like growth factor system in multiple sclerosis. Int Rev Neurobiol. 2007;79:203–226. doi: 10.1016/S0074-7742(07)79009-8. [DOI] [PubMed] [Google Scholar]
- Choi YS, Cho HY, Hoyt KR, Naegele JR, Obrietan K. IGF-1 receptor-mediated ERK/MAPK signaling couples status epilepticus to progenitor cell proliferation in the subgranular layer of the dentate gyrus. Glia. 2008;56:791–800. doi: 10.1002/glia.20653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean C, Dunning FM, Liu H, Bomba-Warczak E, Martens H, Bharat V, Ahmed S, Chapman ER. Axonal and dendritic synaptotagmin isoforms revealed by a pHluorin-syt functional screen. Mol Biol Cell. 2012;23:1715–1727. doi: 10.1091/mbc.E11-08-0707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert AD, Beres AJ, Barber AE, Svendsen CN. Human neural progenitor cells over-expressing IGF-1 protect dopamine neurons and restore function in a rat model of Parkinson's disease. Exp Neurol. 2008;209:213–223. doi: 10.1016/j.expneurol.2007.09.022. [DOI] [PubMed] [Google Scholar]
- Fernandez AM, Torres-Alemán I. The many faces of insulin-like peptide signalling in the brain. Nat Rev Neurosci. 2012;13:225–239. doi: 10.1038/nrn3209. [DOI] [PubMed] [Google Scholar]
- Glavan G, Schliebs R, Zivin M. Synaptotagmins in neurodegeneration. Anat Rec (Hoboken) 2009;292:1849–1862. doi: 10.1002/ar.21026. [DOI] [PubMed] [Google Scholar]
- Guan J, Bennet L, Gluckman PD, Gunn AJ. Insulin-like growth factor-1 and post-ischemic brain injury. Prog Neurobiol. 2003;70:443–462. doi: 10.1016/j.pneurobio.2003.08.002. [DOI] [PubMed] [Google Scholar]
- Gustavsson N, Han W. Calcium-sensing beyond neurotransmitters: functions of synaptotagmins in neuroendocrine and endocrine secretion. Biosci Rep. 2009;29:245–259. doi: 10.1042/BSR20090031. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci. 2010;11:682–696. doi: 10.1038/nrn2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaspar BK, Lladó J, Sherkat N, Rothstein JD, Gage FH. Retrograde viral delivery of IGF-1 prolongs survival in a mouse ALS model. Science. 2003;301:839–842. doi: 10.1126/science.1086137. [DOI] [PubMed] [Google Scholar]
- Kazanis I, Giannakopoulou M, Philippidis H, Stylianopoulou F. Alterations in IGF-I, BDNF and NT-3 levels following experimental brain trauma and the effect of IGF-I administration. Exp Neurol. 2004;186:221–234. doi: 10.1016/j.expneurol.2003.12.004. [DOI] [PubMed] [Google Scholar]
- Kim TK, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, Harmin DA, Laptewicz M, Barbara-Haley K, Kuersten S, Markenscoff-Papadimitriou E, Kuhl D, Bito H, Worley PF, Kreiman G, Greenberg ME. Widespread transcription at neuronal activity-regulated enhancers. Nature. 2010;465:182–187. doi: 10.1038/nature09033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau A, Tymianski M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Arch. 2010;460:525–542. doi: 10.1007/s00424-010-0809-1. [DOI] [PubMed] [Google Scholar]
- Lin Y, Bloodgood BL, Hauser JL, Lapan AD, Koon AC, Kim TK, Hu LS, Malik AN, Greenberg ME. Activity-dependent regulation of inhibitory synapse development by Npas4. Nature. 2008;455:1198–1204. doi: 10.1038/nature07319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JP, Baker J, Perkins AS, Robertson EJ, Efstratiadis a. Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r) Cell. 1993;75:59–72. doi: 10.1016/S0092-8674(05)80084-4. [DOI] [PubMed] [Google Scholar]
- Liu XF, Fawcett JR, Hanson LR, Frey WH., 2nd The window of opportunity for treatment of focal cerebral ischemic damage with noninvasive intranasal insulin-like growth factor-I in rats. J Stroke Cerebrovasc Dis. 2004;13:16–23. doi: 10.1016/j.jstrokecerebrovasdis.2004.01.005. [DOI] [PubMed] [Google Scholar]
- Lyons MR, West AE. Mechanisms of specificity in neuronal activity-regulated gene transcription. Prog Neurobiol. 2011;94:259–295. doi: 10.1016/j.pneurobio.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malva JO, Xapelli S, Baptista S, Valero J, Agasse F, Ferreira R, Silva AP. Multifaces of neuropeptide Y in the brain: neuroprotection, neurogenesis and neuroinflammation. Neuropeptides. 2012;46:299–308. doi: 10.1016/j.npep.2012.09.001. [DOI] [PubMed] [Google Scholar]
- Maya-Vetencourt JF. Activity-dependent NPAS4 expression and the regulation of gene programs underlying plasticity in the central nervous system. Neural Plast. 2013;2013:683909. doi: 10.1155/2013/683909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta A, Prabhakar M, Kumar P, Deshmukh R, Sharma PL. Excitotoxicity: bridge to various triggers in neurodegenerative disorders. Eur J Pharmacol. 2013;698:6–18. doi: 10.1016/j.ejphar.2012.10.032. [DOI] [PubMed] [Google Scholar]
- Miltiadous P, Stamatakis A, Stylianopoulou F. Neuroprotective effects of IGF-I following kainic acid-induced hippocampal degeneration in the rat. Cell Mol Neurobiol. 2010;30:347–360. doi: 10.1007/s10571-009-9457-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miltiadous P, Stamatakis A, Koutsoudaki PN, Tiniakos DG, Stylianopoulou F. IGF-I ameliorates hippocampal neurodegeneration and protects against cognitive deficits in an animal model of temporal lobe epilepsy. Exp Neurol. 2011;231:223–235. doi: 10.1016/j.expneurol.2011.06.014. [DOI] [PubMed] [Google Scholar]
- Moghadam PK, Jackson MB. The functional significance of synaptotagmin diversity in neuroendocrine secretion. Front Endocrinol (Lausanne) 2013;4:124. doi: 10.3389/fendo.2013.00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonaka M, Kim R, Sharry S, Matsushima A, Takemoto-Kimura S, Bito H. Towards a better understanding of cognitive behaviors regulated by gene expression downstream of activity-dependent transcription factors. Neurobiol Learn Mem. 2014;115:21–29. doi: 10.1016/j.nlm.2014.08.010. [DOI] [PubMed] [Google Scholar]
- Pruunsild P, Sepp M, Orav E, Koppel I, Timmusk T. Identification of cis-elements and transcription factors regulating neuronal activity-dependent transcription of human BDNF gene. J Neurosci. 2011;31:3295–3308. doi: 10.1523/JNEUROSCI.4540-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramamoorthi K, Fropf R, Belfort GM, Fitzmaurice HL, McKinney RM, Neve RL, Otto T, Lin Y. Npas4 regulates a transcriptional program in CA3 required for contextual memory formation. Science. 2011;334:1669–1675. doi: 10.1126/science.1208049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegel I, Mardinly AR, Gabel HW, Bazinet JE, Couch CH, Tzeng CP, Harmin DA, Greenberg ME. Npas4 regulates excitatory–inhibitory balance within neural circuits through cell-type-specific gene programs. Cell. 2014;157:1216–1229. doi: 10.1016/j.cell.2014.03.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC. Synaptotagmins: why so many? J Biol Chem. 2002;277:7629–7632. doi: 10.1074/jbc.R100052200. [DOI] [PubMed] [Google Scholar]
- Tamas A, Reglodi D, Farkas O, Kovesdi E, Pal J, Povlishock JT, Schwarcz A, Czeiter E, Szanto Z, Doczi T, Buki A, Bukovics P. Effect of PACAP in central and peripheral nerve injuries. Int J Mol Sci. 2012;13:8430–8448. doi: 10.3390/ijms13078430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Aleman I. Targeting insulin-like growth factor-1 to treat Alzheimer's disease. Expert Opin Ther Targets. 2007;11:1535–1542. doi: 10.1517/14728222.11.12.1535. [DOI] [PubMed] [Google Scholar]
- Tovar-y-Romo LB, Ramírez-Jarquín UN, Lazo-Gómez R, Tapia R. Trophic factors as modulators of motor neuron physiology and survival: implications for ALS therapy. Front Cell Neurosci. 2014;8:61. doi: 10.3389/fncel.2014.00061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Loo KMJ, Schaub C, Pernhorst K, Yaari Y, Beck H, Schoch S, Becker AJ. Transcriptional regulation of T-type calcium channel CaV3.2: bi-directionality by early growth response 1 (Egr1) and repressor element 1 (RE-1) protein-silencing transcription factor (REST) J Biol Chem. 2012;287:15489–15501. doi: 10.1074/jbc.M111.310763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Chan CC, Cherry S, Hiesinger PR. Membrane trafficking in neuronal maintenance and degeneration. Cell Mol Life Sci. 2013;70:2919–2934. doi: 10.1007/s00018-012-1201-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong-Goodrich S, Mellott TJ, Glenn MJ, Blusztajn JK, Williams CL. Prenatal choline supplementation attenuates neuropathological response to status epilepticus in the adult rat hippocampus. Neurobiol Dis. 2008;30:255–269. doi: 10.1016/j.nbd.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaben MJ, Gray WP. Neuropeptides and hippocampal neurogenesis. Neuropeptides. 2013;47:431–438. doi: 10.1016/j.npep.2013.10.002. [DOI] [PubMed] [Google Scholar]
- Zhang SJ, Zou M, Lu L, Lau D, Ditzel DA, Delucinge-Vivier C, Aso Y, Descombes P, Bading H. Nuclear calcium signaling controls expression of a large gene pool: identification of a gene program for acquired neuroprotection induced by synaptic activity. PLoS Genet. 2009;5:e1000604. doi: 10.1371/journal.pgen.1000604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SJ, Buchthal B, Lau D, Hayer S, Dick O, Schwaninger M, Veltkamp R, Zou M, Weiss U, Bading H. A signaling cascade of nuclear calcium-CREB-ATF3 activated by synaptic NMDA receptors defines a gene repression module that protects against extrasynaptic NMDA receptor-induced neuronal cell death and ischemic brain damage. J Neurosci. 2011;31:4978–4990. doi: 10.1523/JNEUROSCI.2672-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Hong EJ, Cohen S, Zhao WN, Ho HY, Schmidt L, Chen WG, Lin Y, Savner E, Griffith EC, Hu L, Steen JA, Weitz CJ, Greenberg ME. Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron. 2006;52:255–269. doi: 10.1016/j.neuron.2006.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zürner M, Mittelstaedt T, tom Dieck S, Becker A, Schoch S. Analyses of the spatiotemporal expression and subcellular localization of liprin-α proteins. J Comp Neurol. 2011;519:3019–3039. doi: 10.1002/cne.22664. [DOI] [PubMed] [Google Scholar]