

Abstract
Munc18-1 is essential for vesicle fusion and participates in the docking of large dense-core vesicles to the plasma membrane. Recent structural data suggest that conformational changes in the 12th helix of the Munc18-1 domain 3a within the Munc18-1:syntaxin complex result in an additional interaction with synaptobrevin-2/VAMP2 (vesicle-associated membrane protein 2), leading to SNARE complex formation. To test this hypothesis in living cells, we examined secretion from Munc18-1-null mouse adrenal chromaffin cells expressing Munc18-1 mutants designed to either perturb the extension of helix 12 (Δ324–339), block its interaction with synaptobrevin-2 (L348R), or extend the helix to promote coil–coil interactions with other proteins (P335A). The mutants rescued vesicle docking and syntaxin-1 targeting to the plasma membrane, with the exception of P335A that only supported partial syntaxin-1 targeting. Disruptive mutations (L348R or Δ324–339) lowered the secretory amplitude by decreasing vesicle priming, whereas P335A markedly increased priming and secretory amplitude. The mutants displayed unchanged kinetics and Ca2+ dependence of fusion, indicating that the mutations specifically affect the vesicle priming step. Mutation of a nearby tyrosine (Y337A), which interacts with closed syntaxin-1, mildly increased secretory amplitude. This correlated with results from an in vitro fusion assay probing the functions of Munc18-1, indicating an easier transition to the extended state in the mutant. Our findings support the notion that a conformational transition within the Munc18-1 domain 3a helix 12 leads to opening of a closed Munc18-1:syntaxin complex, followed by productive SNARE complex assembly and vesicle priming.
SIGNIFICANCE STATEMENT The essential postdocking role of Munc18-1 in vesicular exocytosis has remained elusive, but recent data led to the hypothesis that the extension of helix 12 in Munc18 within domain 3a leads to synaptobrevin-2/VAMP2 interaction and SNARE complex formation. Using both lack-of-function and gain-of-function mutants, we here report that the conformation of helix 12 predicts vesicle priming and secretory amplitude in living chromaffin cells. The effects of mutants on secretion could not be explained by differences in syntaxin-1 chaperoning/localization or vesicle docking, and the fusion kinetics and calcium dependence were unchanged, indicating that the effect of helix 12 extension is specific for the vesicle-priming step. We conclude that a conformational change within helix 12 is responsible for the essential postdocking role of Munc18-1 in neurosecretion.
Keywords: adrenal chromaffin cells, amperometry, calcium uncaging, capacitance measurements, Munc18-1, SNARE proteins
Introduction
The soluble N-ethylmaleimide-sensitive factor attachment protein (SNAP) receptor (SNARE) complex, which is pivotal to the secretion of neurotransmitters, consists of the two target SNAREs syntaxin-1 and SNAP-25, and the vesicular SNARE synaptobrevin-2/VAMP2 (syb-2). Munc18-1, another protein obligatory for neurotransmitter release (Verhage et al., 2000; Voets et al., 2001), interacts with SNAREs in at least two distinct conformations. In the first, it binds via its central hydrophobic cavity to the closed conformation of syntaxin-1, where the Habc domain of syntaxin-1 occludes the SNARE motif (Dulubova et al., 1999; Misura et al., 2000; Burkhardt et al., 2008). In the second conformation, Munc18-1 binds to the assembled—or assembling—SNARE complex (Dulubova et al., 2007; Shen et al., 2007; Ma et al., 2013; Shen et al., 2015). Both binding modes include a separate interaction of the N-terminal peptide of syntaxin-1 with Munc18-1 (Shen et al., 2007; Burkhardt et al., 2008), although the exact role of this interaction is still being discussed (Burkhardt et al., 2008; Deák et al., 2009; Shen et al., 2010; Hu et al., 2011; Meijer et al., 2012; Colbert et al., 2013; Dawidowski and Cafiso, 2016).
In neuroendocrine cells and neurons, Munc18-1 plays multiple roles. Munc18-1 acts as a chaperone for syntaxin-1 (Medine et al., 2007), and vice versa (Arancillo et al., 2013), during trafficking through the ER and Golgi apparatus. Munc18-1 is also necessary for vesicle docking to the plasma membrane (PM; Voets et al., 2001). The docking phenotype can be overcome by stabilizing SNAP-25:syntaxin dimers, to which synaptotagmin-1 (Syt1) docks vesicles (de Wit et al., 2009). However, while the docking phenotype is rescued, secretion remains abrogated (de Wit et al., 2009), pointing to yet another essential function of Munc18-1 in vesicle priming or fusion (Gulyás-Kovács et al., 2007).
Data from several laboratories have converged on the conclusion that the essential stimulating function of Munc18-1 in secretion involves domain 3a (Boyd et al., 2008; Hu et al., 2011; Han et al., 2013, 2014; Parisotto et al., 2014). This is further supported by the effect of PKC phosphorylation of residues within the domain 3a, which promotes vesicle priming (Nili et al., 2006). A specific hypothesis for the role of the domain 3a has been suggested (Hu et al., 2011; Parisotto et al., 2014), as follows: the N-terminal region (amino acids 324–339) of helix 12 of Munc18-1 (within the domain 3a) can exist in the following two conformations (Fig. 1): first, a folded-back “closed” helical hairpin (Fig. 1A,B), where P335 acts as a hinge, which was identified in the Munc18-1:syntaxin crystal (Misura et al., 2000; Burkhardt et al., 2008); and, second, an extended “open” helical hairpin, where the amino acid stretch from 324 to 339 extends helix 12 (Fig. 1C). The latter conformation was identified in a crystal of Munc18-1 bound only to the N-terminal peptide of syntaxin-4 (Hu et al., 2011). The extended open helix 12 would sterically clash with closed syntaxin-1. Thus, the conformational change in domain 3a from the closed to the open helical hairpin might coincide with the transition from the closed to the open syntaxin-1 and promote SNARE complex formation. Moreover, helix 12 contains residues that interact with part of the syb-2 SNARE domain (Xu et al., 2010; Baker et al., 2015), raising the possibility that the extended helix forms a template to structure syb-2. This was recently tested in vitro using a vesicle fusion assay (Parisotto et al., 2014). In support of the model, a mutation (L348R) found to eliminate syb-2 binding abolished the fusion-promoting action of Munc18-1, whereas mutation of the “hinge” proline (P335A) increased the helicity of helix 12 and displayed a gain-of-function phenotype in vitro (Parisotto et al., 2014).
Figure 1.

Extension of helix 12 (opening of the helical hairpin) in Munc18-1. A, Crystal structure of the Munc18-1 (medium purple) bound to closed syntaxin 1 (cyan; Burkhardt et al., 2008). B, Zoom image of A showing the 11th (to the left) and 12th helix (middle right), the helical hairpin, and, with color codes, the residues mutated in this study, as follows: 324–339 in red; P335 in green; Y337 in blue; and L348 in orange. Y337 forms an H-bond (green) to the side chain of N135 in syntaxin-1. C, Crystal structure of Munc18-1 bound to the syntaxin-4 N-peptide (Martin et al., 2013) showing the extended state of helix 12. SNAREs are not included in that structure.
Here, we tested this hypothesis in live adrenal chromaffin cells. We found that the deletion of most of the helical hairpin (Δ324–339) or the disruption of syb-2 binding (L348R) impairs neurosecretion. Conversely, the P335A mutation promotes vesicular fusion by enhancing vesicle priming. Our results indicate that a conformational change in helix 12 of Munc18-1 is crucial for vesicular priming.
Materials and Methods
DNA constructs and protein purification for in vitro studies.
The following constructs were used: SNAP-25, the DNA construct (pFP247) encoding His6-tagged SNAP-25 and the protein purification were described previously (Schollmeier et al., 2011); syntaxin-1A, a full-length His6-tagged rat syntaxin-1 (encoded by pYS2) was expressed and purified as described previously (Schollmeier et al., 2011) with the following modifications: (1) recombinant protein expression was induced at 25°C for 4 h instead of 16°C overnight; (2) His6-tagged syntaxin-1 was incubated overnight with 3 ml of Ni-NTA Agarose beads (Qiagen) with a continuous flowrate of 0.5 ml/min; (3) 1.5% (w/v) sodium cholate hydrate (Sigma-Aldrich) was used instead of 1% (w/v) Triton X-100 in the washing steps; and (4) the His6-tagged protein was eluted with an imidazole gradient in the presence of 400 mm KCl; Syb-2/VAMP2, the His6-tagged VAMP2 (encoded by pTW2) was expressed as previously described (Weber et al., 1998; Malsam et al., 2012); synaptotagmin, Rat Syt1 lacking the luminal domain (encoded by pLM6) was expressed and purified as described previously (Mahal et al., 2002; Malsam et al., 2012); and Munc18-1, DNA constructs encoding GST-Munc18-1 wt and P335A (pDP2) fusion proteins were described previously (Parisotto et al., 2014). The Munc18-1 Y337A mutant was generated using QuikChange DNA mutagenesis (Qiagen) with the following primers: (1) forward 5′-CTGAAGAAAATGCCCCAGGCCCAGAAGGAGCTCAGCAA-3′; and (2) reverse 5′-TTGCTGAGCTCCTTCTGGGCCTGGGGCATTTTCTTCAG-3′. The resulting DNA construct (pTB5) was sequenced at GATC Biotech [sequencing primer forward (5′-CTGCTGCCTATTGAAAATG-3′) and reverse (5′-CATTTTCAATAGGCAGCAG-3′)] to ensure sequence integrity. Munc18-1 proteins were purified as described previously (Parisotto et al., 2014) with the following modifications: (1) 200 mm KCl instead of 135 mm KCl was used during all purification steps; and (2) the buffers did not contain 10 mm methionine.
Preparation of proteoliposomes.
Proteoliposomes were prepared as described previously (Malsam et al., 2012) with the following modification: the fluorophores rhodamine-DPPE (N-(lissamine rhodamine B sulfonyl)1,2-dipalmitoyl phosphatidylethanolamine) and NBD-DPPE (N-(7-nitro-2,1,3-benzoxadiaziole-4-yl)-1,2-dipalmitoyl phosphatidylethanolamine) were replaced by 0.5 mol% Atto488-DPPE (ATTO-TEC) and 0.5 mol% Atto550-DPPE (ATTO-TEC).
Preparation of syntaxin-giant unilamellar vesicles.
Giant unilamellar vesicles (GUVs) containing syntaxin-1 were prepared as described previously (Malsam et al., 2012) with the following three modifications: (1) Pt-coated glass slides (GeSIM mbH) were used instead of indium tin oxide-coated glass slides; (2) a buffer containing 0.5 mm 4-(2-hydroxyethyl)-1-piperazinepropanesulfonic acid, pH 7.4, and 0.5% glycerol was used for the drying of the liposomes; and (3) for the swelling reaction 2.5 V were applied for 1 h.
In vitro fusion assay.
The small unilamellar vesicle (SUV)/GUV fusion assay was performed and analyzed as described previously (Malsam et al., 2012; Parisotto et al., 2012, 2014). Briefly, syntaxin-1 GUVs (14 nmol lipid with a syntaxin 1/lipid ratio of 1:700) were preincubated in the absence or presence of Munc18-1 wild type (WT), Munc18-1 Y337A, or Munc18-1 P335A for 30 min at room temperature (RT). Subsequently, SNAP-25 (1.7 μm) and syb-2/synaptotagmin SUVs (2.5 nmol lipid with a syb-2/lipid ratio of 1:330 and a synaptotagmin-1/lipid ratio of 1:1180) were added on ice in a total volume of 100 μl, and docking was performed by with a 10 min preincubation period on ice. Atto 488 fluorescence was monitored at 37°C in a Synergy 4 plate reader (BioTek; filter wheel settings: excitation, 485/20 nm; emission, 525/20 nm) at intervals of 10 s for a total of 30 min. The fusion signal was normalized to the signal after the addition of 20 μl 5% (w/v) SDS and 5% (w/v) n-dodecyl-β-d-maltoside and by subtraction of a negative control experiment performed in the presence of the syb-2 cytoplasmic domain.
Mouse line, cell culture, constructs and, virus.
Munc18-1-null mice of either sex (Verhage et al., 2000) were obtained by crossing Munc18-1 heterozygotes and were recovered by cesarean section at embryonic day 18. Chromaffin cells were isolated from adrenal glands and were cultured as previously described (Sørensen et al., 2003b). We introduced WT or mutated Munc18-1 with Semliki Forest Virus (SFV). WT Munc18-1 and mutants were expressed from a bicistronic message containing an internal ribosomal entry site and EGFP as expression control. Mutations were introduced by overlapping primers, and all constructs were sequenced before use. The cells were exposed to the activated virus after 48–96 h of incubation and were allowed to express the protein for 6–8 h before measurements were made.
Electrophysiological recordings.
Transfected cells were placed on the stage of an Axiovert 10 microscope (Carl Zeiss), and expressing cells were identified by the green fluorescent response to 475 nm illumination by a monochromator (Polychrome IV, Till Photonics). Amperometry measurements were performed with 5-μm-diameter polyethylene-insulated carbon fibers (Thornel P-650/42, Cytec; Bruns, 2004). Fibers were pressed gently against the cell while a constant voltage of 700 mV was applied. Currents were amplified by an EPC-7 amplifier (HEKA Elektronik), filtered at 2.9 kHz, and sampled at 11.5 kHz through an auxiliary input channel of the EPC-9 patch-clamp amplifier. Cell membrane capacitance was measured simultaneously by the whole-cell patch-clamp technique described by Lindau and Neher (1988). Pulse Software (version 8.53) and an EPC-9 amplifier (HEKA Elektronik) were used in “sine+dc” mode to record and inject currents. Currents were filtered at 3 kHz and sampled at 11.5 kHz. Secretion was stimulated by UV photolysis of the Ca2+ cage nitrophenyl-EGTA by using UV light from a UV flash lamp (JML-C2, Rapp OptoElectronics) or during the “Ramp” protocol the monochromator was used for slow gradual Ca2+ uncaging; both were controlled by the Pulse Software and triggered by the EPC-9 amplifier. Ca2+ uncaging and measurements were conducted using a Fluar 40× objective (Zeiss). Fura dyes were excited alternatingly at 350 and 380 nm. Emitted light was detected by a photo diode (Till Photonics) in an area around the cell defined by a View Finder (Till Photonics). The output of the photo diode was connected to an auxiliary input channel on the EPC-9 amplifier. The signal was filtered at 3 kHz and sampled at 11.5 kHz. The Ca2+ concentration was measured with a mixture of high- and low-affinity Fura dyes (fura-4 and furaptra), as described by Voets (2000). The free Ca2+ concentrations of calibration solutions were calculated using a custom-written macro for IGOR Pro macro for IGOR Pro (WavemMetrics), assuming a Kd of 0.222 μm for BAPTA and of 80 μm for DPTA (diethylene triamine penta-acetic acid), while taking into account the additional buffering of Ca2+ by the dyes nitrophenyl-EGTA and ATP. The fluorescence signal was calibrated by performing a patch clamp on chromaffin cells with seven or eight different calibration solutions. For measurements, the patch pipette solution contained the following (in mm): 100 Cs-glutamate, 8 NaCl, 4 CaCl2, 32 HEPES, 2 Mg-ATP, 0.3 NaGTP, 5 nitrophenyl-EGTA, 1 ascorbic acid, 0.4 fura-4f, and 0.4 furaptra (all from Invitrogen), adjusted to pH 7.2 with CsOH.
For kinetic analysis of capacitance traces, a triple exponential function was fitted to individual traces using a custom-written macro in IGOR Pro (WaveMetrics), as explained previously (Sørensen et al., 2003a; Mohrmann et al., 2013). The amplitude associated with the fastest time constant (τ < 80 ms) is denoted the “fast burst,” whereas the amplitude associated with the intermediate time constant (τ > 80 ms) is denoted the “slow burst.” The third (i.e., the slowest) exponential is required to correct for the sustained component. Since the sustained component in some cells is more or less linear (i.e., the time constant is as long as or longer than the measurement time), the time constant and amplitude of this exponential are not informative and were not used directly. Instead, the linear rate of the secretion at later times (>1 s) was calculated; this is the “sustained component.” In some cells, a single exponential component was sufficient to fit the burst; this component was then assigned as a fast or a slow burst based on the fitted time constant, and the amplitude of the other component was set to zero.
Immunocytochemistry and confocal imaging.
Chromaffin cells from WT or Munc18-1-null mice expressing the different constructs for 6 h were fixed with 4% paraformaldehyde at day 3 in vitro (DIV3). The cells were permeabilized by 5 min incubation in PBS containing 0.5% Triton X-100. To block nonspecific binding, the cells were incubated for 30 min in PBS containing 0.1% Triton X-100 and 2% normal goat serum. All antibodies were diluted in this solution. The cells were incubated in the primary antibody solution [anti-Munc18 (Verhage laboratory) or anti-syntaxin (I379, a gift from the Südhof laboratory)] for 1 h at RT. After washing three times with PBS for 10 min, the cells were incubated in the secondary antibody solution (goat anti-rabbit Alexa Fluor 546, Invitrogen) for 1 h at RT. The cells were washed three times in PBS and mounted on microscopy slides with Mowiol in preparation for the confocal microscopy. Chromaffin cells were imaged with a 63× plan-neofluor lens (numerical aperture, 1.4; Carl Zeiss) on a Zeiss 510 Meta Confocal Microscope. An additional zoom factor of 5 was applied, and the images were acquired with a frame size of 1024 × 1024 pixels. A single image of the equatorial plane of the chromaffin cells was acquired. Fluorescence levels were quantified in ImageJ (National Institutes of Health), syntaxin redistribution was quantified in ImageJ using the PlasMACC plugin (Kurps et al., 2014).
Electron microscopy.
Chromaffin cells were fixed on DIV3 using aldehyde fixation protocol with 2.5% glutaraldehyde in 0.1 m cacodylate buffer, pH 7.4, for 1 h and subsequently washed with 0.1 m cacodylate buffer, pH 7.4. Due to cell detachment in high-pressure freezing/freeze substitution (HPF/FS) protocols, HPF/FS could not be applied. Since a side-by-side comparison between aldehyde fixation and HPF/FS on large dense-core vesicle docking revealed no differences in conclusions (de Wit et al., 2009), we relied on chemical fixation in this study. Postfixation was performed for 1.5 h using a 1% osmium tetroxide and 1% ruthenium tetroxide mixture in 0.1 m cacodylate buffer. Dehydration using a series of ethanol concentrations (30–100%) was performed before embedding in Epon and polymerized at least for 15 h at 65°C. The coverslip was removed from Epon resin by alternatively dipping into liquid nitrogen and hot water. On flat Epon surface region with a high-cell density monolayer was selected, cut, and mounted on prepolymerized Epon blocks for thin sectioning. Ultrathin sections (∼90 nm) were collected on single-slot, Formvar-coated copper grids, and stained with uranyl acetate and lead citrate. A JEOL 1010 transmission electron microscope was used for high-resolution imaging. A vesicle membrane attached to the plasma membrane was considered to be a docked vesicle. An infected cell was identified based on the presence of SFV (droplet shape with dense core) on the plasma membrane. Distance measurements were performed using 10,000× or 30,000× magnification images on iTEM software (EMSIS).
Statistics.
For the analysis of electron microscopy data, a multilevel comparison was used (Aarts et al., 2014). In other experiments, when more than two groups were compared we used one-way ANOVA, followed by appropriate post hoc tests corrected for multiple comparisons to locate a significant difference between any two groups. When comparing two groups, we used a two-tailed t test. The level of significance is symbolized with an asterisk (*p < 0.05, **p < 0.01, and ***p < 0.001).
Results
To investigate the role of helix 12 of Munc18-1 in live cells, we performed experiments in mouse embryonic adrenal chromaffin cells from the Munc18-1-null rescued by overexpression (OE) of different Munc18-1 mutants or the WT protein using the Semliki Forest Virus system (see Materials and Methods). Previous data showed that Munc18-1 WT OE readily (within 6 h) rescues secretion in Munc18-1-null cells (Toonen et al., 2006). The mutated amino acids are shown in Figure 1, B and C: one mutation (Δ324–339) was a deletion of 16 aa, which form most of the extended part of the α-helix 12. Another mutation was L348R, which has been shown to compromise binding between Munc18-1 and syb-2 (Parisotto et al., 2014). The P335A mutation targeted the hinge proline (P335), which does not considerably change syb-2 binding but leads to increased α-helicity of the region, thereby favoring an extended conformation (Parisotto et al., 2014). Finally, we tested an alanine mutation of Y337 (Y337A). In the closed helical hairpin conformation of Munc18-1, Y337 forms a hydrogen bond with N135 in closed syntaxin-1 and therefore might stabilize this conformation. Immunostaining for Munc18-1 after expression in Munc18-1-null chromaffin cells showed that all constructs were highly expressed, resulting in mainly diffuse cytoplasmic staining (Fig. 2A). Quantification revealed a similar expression of Munc18-1 WT protein, as well as the Δ324–339, L348R, and P335A mutants, whereas the Y337A mutant showed weaker expression, although still clearly above endogenous levels (Fig. 2B).
Figure 2.

All Munc18-1 mutants are expressed and show normal localization in Munc18-1-null cells. A, Typical example confocal images (EGFP infection marker, anti-Munc18 labeling) of wild-type chromaffin cells and Munc18-1-null cells expressing wild-type Munc18-1 (M18-WT), Munc18-1 Δ324–339 (M18-Δ324–339), Munc18-1 P335A (M18–P335A), Munc18-1 L348R (M18–L348R), Munc18-1 Y337A (M18–Y337A), or EGFP negative control, as indicated. Scale bar, 2 μm. B, Bar graph shows the average Munc18 labeling intensity [arbitrary units (a.u.), 15 cells per condition] of the chromaffin cell equatorial plane. Viral infection of Munc18-1-null cells shows a 5- to 20-fold overexpression compared with endogenous Munc18-1 labeling, ***p < 0.001 difference (ANOVA and post hoc t test with Bonferroni correction) to all conditions.
Most mutations in the helix 12 rescue syntaxin-1 localization and vesicle docking
It was suggested that the extended helix 12 in Munc18-1 is incompatible with binding to closed syntaxin-1 (Hu et al., 2011). Indeed, the P335A mutation was found to slightly destabilize the binding between Munc18-1 and monomeric syntaxin-1 (Han et al., 2014), but we did not observe this in a previous study (Parisotto et al., 2014). Impairment of the stability of the Munc18-1:syntaxin complex could potentially compromise the chaperone function of Munc18-1, which regulates syntaxin-1 transport to the plasma membrane in normal amounts. Because the local syntaxin-1 concentration is critical for neurotransmitter release (Arancillo et al., 2013), we investigated this point by immunostaining Munc18-1-null chromaffin cells expressing the different Munc18-1 mutants for syntaxin-1 (Fig. 3).
Figure 3.

Most Munc18-1 domain 3a mutants are able to restore syntaxin-1 localization at the plasma membrane. A, Typical example confocal images (EGFP infection marker and anti-syntaxin-1 labeling) of Munc18-1-null chromaffin cells expressing wild-type Munc18-1 (M18-WT), Munc18-1 Δ324–339 (M18-Δ324–339), Munc18-1 P335A (M18–P335A), Munc18-1 L348R (M18–L348R), Munc18-1 E446K (M18–E466K), Munc18-1 Y337A (M18–Y337A), or EGFP negative control, as indicated. Scale bar, 2 μm. B, Bar graph shows the distribution of the syntaxin-1 label as ratio of PM area over cytosol area in these conditions (two independent cultures, 17–24 cells). Significant differences are indicated (ANOVA and post hoc t test with Bonferroni correction, ***p < 0.001, *p < 0.05).
Expression of WT Munc18-1 in-null cells increased the targeting of syntaxin-1 to the PM, and thus the PM/cytosol ratio (Fig. 3A,B), which is in line with previous data (Gulyás-Kovács et al., 2007; Gerber et al., 2008). Increased targeting was also found for the Δ324–339, the L348R and—despite the lower expression level of this mutant (Fig. 2)—the Y337A (Fig. 3B). However, the P335A resulted in an intermediate syntaxin-1 phenotype, which was significantly different from neither the Munc18-1-null (expressing EGFP as a control) nor the Munc18-1 WT OE condition. Thus, the P335A appears to be partly compromised in chaperoning monomeric syntaxin-1 to the membrane, probably indicating a weaker binding.
Elimination of either Munc18-1 or syntaxin-1 decreases the pool of vesicles that is directly attached to the plasma membrane in aldehyde-fixed chromaffin cells, the so-called docked vesicles (Voets et al., 2001; de Wit et al., 2006). The D34N/M38V double mutation in Munc18-1 rescues both syntaxin-1 and Munc18-1 levels, whereas docking is still suppressed (Gulyás-Kovács et al., 2007); on the other hand, docking was rescued in Munc18-1-null cells by manipulations, which create ectopic syntaxin-1:SNAP-25 dimers (de Wit et al., 2009). Thus, the relationship between syntaxin-1 targeting and vesicle docking is not straightforward. We next investigated whether the mutations affected vesicle docking.
Morphometric analysis on the ultrastructural level using electron micrographs showed that Munc18-1-null cells were impaired in docking, and this was rescued by the expression of Munc18-1 WT (Fig. 4A,B). The Δ324–339, the L348R, and the P335A mutants all rescued docking to levels indistinguishable from the WT protein (Fig. 4A,B). All conditions had a similar number of total vesicles per section (Fig. 4B), showing that docking to the plasma membrane is selectively impaired in the absence of Munc18-1, not vesicle abundance (Voets et al., 2001; de Wit et al., 2009).
Figure 4.

Munc18-1 domain-3a mutants restore vesicle docking. A, Typical example micrographs of the plasma membrane area of wild-type chromaffin cells and munc18-null cells expressing wild-type Munc18-1 (M18-WT), Munc18-1 Δ324–339 (M18-Δ324–339), Munc18-1 P335A (M18–P335A), Munc18-1 L348R (M18–L348R), or EGFP negative control, as indicated. Scale bar, 200 nm. The arrowheads indicate docked vesicles (in direct contact with the plasma membrane). B, The total (left) and docked (right) number of vesicles per cross section, normalized to wild-type levels. Number of independent cultures and number of cells quantified are indicated in the bar graph. **p < 0.01 significant difference (multilevel analysis) compared with all other conditions.
Overall, these findings indicate that the mutations studied here do not lead to dramatic differences in syntaxin-1 targeting or vesicle docking—and that the reduced syntaxin-1 targeting by the P335A mutant does not translate into a detectable defect in vesicle docking.
Mutations in helix 12 impair or enhance neurosecretion
Next, we evaluated secretion from Munc18-1-null chromaffin cells overexpressing each of the mutations. Overexpression of WT Munc18-1 was used as a control. Exocytosis was triggered by uncaging Ca2+ from nitrophenyl-EGTA, while [Ca2+]i was measured using a mixture of two fura dyes (see Materials and Methods). We monitored exocytosis by parallel measurements of membrane capacitance and amperometry, which reports on released catecholamines. Uncaging Ca2+ in Munc18-1-null cells overexpressing WT Munc18-1 resulted in robust secretion (Fig. 5A, black trace). In contrast, Munc18-1-null cells have hardly any secretion at all (see Fig. 8A, gray trace; Voets et al., 2001; Toonen et al., 2006; Gulyás-Kovács et al., 2007; de Wit et al., 2009). Expression of the L438R or the Δ324–339 mutations both resulted in markedly reduced secretion (Fig. 5A). In contrast, the P335A mutation augmented secretion above WT levels (Fig. 5A). The secretion within the first half second of the Ca2+ increase is often referred to as the “exocytotic burst,” and approximately represents the fusion of those vesicles that were primed before the uncaging event. This phase was statistically different between all three mutations and the WT (mean ± SEM: WT, 140 ± 15 fF; n = 25; Δ324–339, 49 ± 6 fF; n = 30; L348R, 74 ± 8 fF; n = 23; P335A, 259 ± 25 fF; n = 28; ANOVA, p < 0.001; all groups but Δ324–339 and L348R were significantly different from each other; Fig. 5B, results of Tukey's HSD test). Total release was affected similarly (WT, 336 ± 34 fF; n = 25; Δ324–339, 98 ± 10 fF; n = 30; L348R, 154 ± 17 fF; n = 23; P335A, 547 ± 52 fF; n = 27; ANOVA, p < 0.001; Fig. 5B, results of Tukey's HSD test). Finally, the sustained release (release between 0.5 and 5.5 s after uncaging) was affected, once again P335A increased release and 324–339Δ and L348R decreased release (WT, 147 ± 20 fF; n = 25; Δ324–339, 37 ± 4 fF; n = 30; L348R, 57 ± 11 fF; n = 23; P335A, 219 ± 22 fF; n = 27; ANOVA, p < 0.001; Fig. 5B, results of Tukey's HSD test). Sustained release is assumed to originate from ongoing vesicle priming, followed immediately by fusion as long as the [Ca2+]i remains high. The preflash [Ca2+]i was not significantly different between the groups (WT, 647 ± 37 nm, n = 25; Δ324–339, 769 ± 45 nm, n = 30; L348R, 879 ± 90 nm, n = 23; P335A, 849 ± 127 nm, n = 28; ANOVA, p = 0.23). This is important, because vesicle priming—and therefore the size of the secretory burst—is Ca2+ dependent (Voets, 2000).
Figure 5.

Exocytotic release is increased by the P335A mutant and decreased by the Δ324–339 and L348R mutants. A, Ca2+-uncaging experiment (uncaging at arrow marked “Flash”) showing means for all measured cells (for n, see B–D). Top, Mean [Ca2+]i following the flash. Middle, Mean capacitance measurements. Bottom, Mean amperometric current (left ordinate axis) or amperometry charge (right ordinate axis). Measurements are of overexpressed Munc18-1 WT (black), Δ324–339 mutant (red), L348R mutant (orange), and P335A mutant (green) in Munc18-1-null chromaffin cells. B, Total release (increase in cell membrane capacitance, Δ Cm, at 5 s after the flash), burst size (ΔCm at 0.5 s after the flash), and sustained release (ΔCm at 5 s − ΔCm at 0.5 s after the flash) were all significantly different between each mutant and Munc18-1 WT overexpression. Δ324–339 and L348R decreased, whereas P335A increased secretion. C, D, The amplitudes of the fast burst (corresponding to the size of the RRP; C) and the slow burst (corresponding to the size of the SRP; D) were significantly increased in the P335A and decreased with the Δ324–339 mutant. E, F, The release kinetics (time constants) of the fast (E) and slow (F) burst were not significantly different between groups. *p < 0.05, **p < 0.01, ***p < 0.001, Tukey's HSD test following ANOVA.
Figure 8.

Release is increased in the Munc18-1 Y337A mutant when expressed in Munc18-1-null cells. A, Mean traces of all cells (for n, see B–E) for [Ca2+]i (top), ΔCm (middle), and amperometric current and charge (bottom). Measurements are from Munc18-1-null (gray), Munc18-1-null cells with Munc18-1 WT OE (black), or Munc18-1 Y337A (blue). B, No difference between WT OE and Y337A OE preflash [Ca2+]I; ANOVA, p = 0.95. C–E, Burst sizes (ΔCm at 0.5 s after flash; C), total release (ΔCm at 5 s after flash; D), and sustained release (ΔCm at 5 s − ΔCm at 0.5 s; E). Total and sustained release was significantly higher in the Y337A than in the Munc18-1 WT. #p = 0.07, *p < 0.05, **p < 0.01, ***p < 0.001, Tukey's HSD test following ANOVA.
We compared the release kinetics of the burst phase between groups by fitting a sum of two exponentials (plus a slower exponential to correct for the sustained component) to each trace, as documented previously (Sørensen et al., 2003a; Mohrmann et al., 2013). This revealed that the amplitudes of both the fast and the slow burst components were significantly reduced in the Δ324–339 mutant (Fig. 5C,D), and were significantly increased by the P335A, whereas the L348R did not reach statistical significance. The release kinetics (time constants) of the fast and slow bursts were not significantly different between groups (τfast: M18-WT, 15.7 ± 2.2 ms, n = 21; M18-Δ324–339, 19.7 ± 4.2 ms, n = 20; M18-L348R mean = 13.5 ± 2.2 ms, n = 21; M18–1-P335A, 16.2 ± 1.6 ms, n = 23; ANOVA, p = 0.44; τslow: M18-WT, 195 ± 32 ms, n = 16; M18-Δ324–339, 330 ± 71 ms, n = 22; M18-L348R, 235 ± 43 ms, n = 19; M18–1-P335A, 175 ± 22 ms, n = 20; ANOVA, p = 0.11; Fig. 5E,F). The different n values for fast and slow bursts appear because not all cells have both a fast and a slow burst. The fast-burst phase originates from release from the readily releasable pool (RRP) of vesicles, whereas the slow-burst phase originates from a vesicle pool just upstream of the RRP (Walter et al., 2013), often referred to as the slowly releasable pool (SRP). The near-proportional change in sustained release and the size of the burst, including SRP and RRP, indicate that the mutations affect a recruitment/priming step leading into the SRP from an upstream pool.
To investigate whether the Ca2+ sensitivity of release is changed by the mutations, we performed Ca2+ ramp experiments, where the [Ca2+]i was increased slowly by photo releasing Ca2+ from the cage using constant illumination oscillating between 350 and 380 nm, allowing simultaneous measurement of [Ca2+]i using the fura dyes (Sørensen et al., 2002). The secretion threshold was identified as the maximum of the second derivative of the capacitance trace. This threshold is increased by mutating synaptotagmin-1 to reduce the Ca2+ affinity (Sørensen et al., 2003a) or by mutating the synaptotagmin-1 binding sites of SNAP-25 (Mohrmann et al., 2013). Testing the Δ324–339 and the P335A mutations, we did not find differences in the release threshold (Fig. 6A,B), which was approximately the same as in Munc18-1-null cells or after the overexpression of Munc18-1 WT (null, 1.30 ± 0.42 μm, n = 20; WT, 1.41 ± 0.26 μm, n = 21; P335A, 1.17 ± 0.13 μm, n = 27; Δ324–339, 1.48 ± 0.25 μm, n = 24; ANOVA, p = 0.85). Thus, the helix 12 of Munc18-1 does not play a dominant role in setting the Ca2+ dependence of release, but rather acts to prime vesicles for release.
Figure 6.
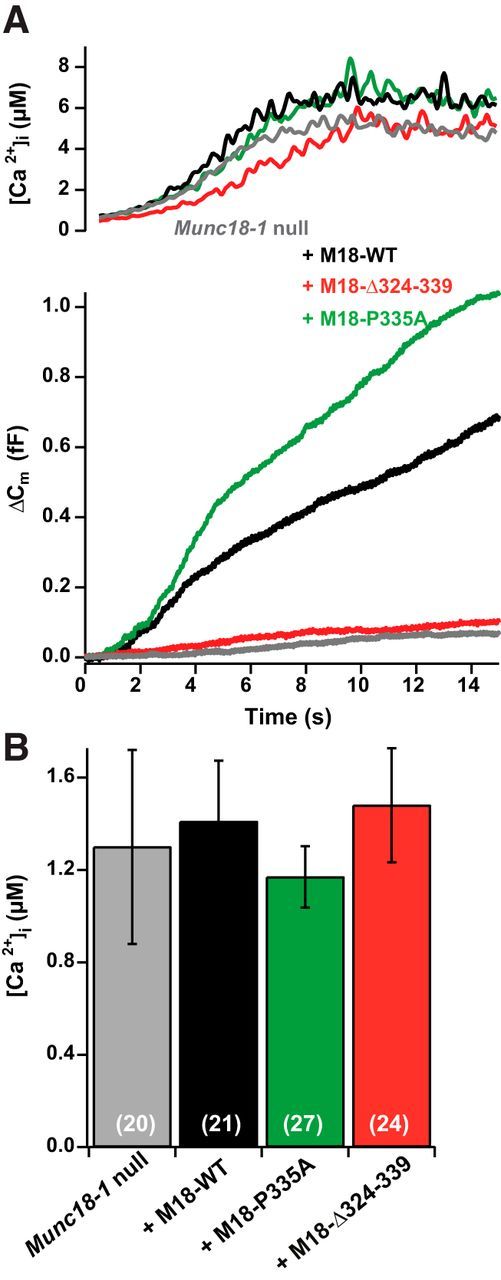
Mutations in helix 12 do not change the Ca2+ threshold of release. A, Representative [Ca2+]i and increase in cell membrane capacitance, Δ Cm, single traces for Munc18-1-null cells, and Munc18-1-null cells overexpressing Munc18-1 WT, the Δ324–339 and the P335A mutation. B, Mean Ca2+ thresholds were unchanged between conditions.
Munc18-1 Y337A mutation enhances neurosecretion
In the crystal structure of Munc18-1 bound to closed syntaxin-1 (Misura et al., 2000; Burkhardt et al., 2008), Y337 in the domain 3a of Munc18-1 is close to and expected to form a hydrogen bond with N135 of syntaxin-1 (Fig. 1B,C). A Munc18-1 Y337L mutant has previously been reported to modulate the release of single vesicles in bovine chromaffin cells (Boyd et al., 2008). We here created a Munc18-1 Y337A mutant.
To determine the functional effect of the Munc18-1 Y337A point mutation in a reconstituted membrane fusion assay, we compared this mutant with WT Munc18-1 and the gain-of-function mutant Munc18-1 P335A in an in vitro assay (Fig. 7A). All three constructs showed similar thermal unfolding temperatures, demonstrating correct protein folding (Fig. 7B). GUVs containing reconstituted syntaxin-1 and PI(4,5)P2 were preincubated with the Munc18-1 constructs, soluble SNAP-25, and SUVs containing Syt1 and syb-2, and lipid mixing was measured by a fluorescence dequenching assay (Malsam et al., 2012). In this assay, vesicle docking is mediated by Syt1–PI(4,5)P2/SNARE interactions, and SNARE complex assembly becomes a rate-limiting step (de Wit et al., 2009; Parisotto et al., 2012; Kedar et al., 2015). Munc18-1 mutants that further stabilize a closed conformation of syntaxin-1 would result in fusion inhibition. In contrast, Munc18-1 mutants that favor an open conformation of syntaxin-1 and SNARE complex assembly would increase lipid mixing. In the case of Munc18-1 WT and under the conditions chosen in this assay, the inhibitory and stimulatory functions cancel each other, and fusion kinetics are similar with and without Munc18-1 (Fig. 7C). As expected, Munc18-1 P335A, which contains an open helical hairpin conformation of domain 3a, profoundly stimulated SNARE complex assembly and lipid mixing (Fig. 7C). Munc18-1 Y337A consistently showed a weak stimulatory effect, which, however, was less pronounced than that of the P335A. Similar results were obtained at higher Munc18 concentrations (data not shown).
Figure 7.

Munc18-1 P335A and Y337A are gain-of-function mutants stimulating lipid mixing in a reconstituted SUV-GUV fusion assay. A, Incubation scheme: syntaxin-1 GUVs (20 pmol syntaxin-1, 14 nmol lipid) containing PI(4,5)P2 (2 mol%) were preincubated in the absence or presence of the indicated Munc18-1 constructs (42 pmol) for 30 min at room temperature. Soluble SNAP-25 (177 pmol) and Syt1/syb-2 SUVs (2.1 pmol Syt1, 7.5 pmol syb-2, 2.5 nmol lipid) were added and incubated for 10 min on ice to allow vesicle docking. Membrane fusion was measured in a final volume of 104 μl at 37°C. B, Melting temperatures show that both mutants are correctly folded. C, Lipid mixing was monitored by the increase in Atto 488 fluorescence for 30 min. Data were normalized to the maximum fluorescence after liposome lysis by detergent. Error bars indicate SEM. N = 3.
When overexpressed in Munc18-1-null chromaffin cells, we found a significant increase in total release and sustained release for the Y337A mutant compared with Munc18-1 WT overexpression, while the burst size displayed a trend toward larger amplitude (Fig. 8A,C–E; total release: null, 28.6 ± 6.3 fF, n = 25; WT, 330 ± 29 fF, n = 24; Y337A, 472 ± 49 fF, n = 26, ANOVA, p < 0.001; Tukey's HSD test, M18OE vs Y337A, p < 0.05; burst secretion; null, 16.6 ± 3.3 fF, n = 25; WT, 196 ± 23 fF, n = 24; Y337A, 259 ± 27 fF, n = 26; ANOVA, p < 0.001; Tukey's HSD test, M18OE vs Y337A, p = 0.07; sustained secretion: null, 12.1 ± 6.6 fF, n = 25; WT, 101 ± 10 fF, n = 24; Y337A, 160 ± 22 fF, n = 26; ANOVA, p < 0.001; Tukey's HSD test, M18OE vs Y337A, p < 0.05). We did not detect any change in the release kinetics in cells expressing the Y337A mutant (τfast: WT, 18.5 ± 2.4 ms, n = 22; Y337A, 17.3 ± 2.4 ms, n = 25; two-tailed t test, p = 0.73; τslow: WT, 331 ± 95 ms, n = 19; Y337A, 196 ± 52 ms, n = 20; two-tailed t test, p = 0.197), but we found that the RRP was significantly larger in the Y337A (WT, 91 ± 10 fF, n = 22; Y337A, 157 ± 19, n = 22; two-tailed t test, p = 0.006), whereas the SRP was unchanged (WT, 114 ± 12 fF, n = 19; Y337A, 93 ± 11 fF, n = 23; two-tailed t test, p = 0.213).
These data show that a mutation (Y337A) designed to destabilize the Munc18-1:closed-syntaxin complex increases membrane fusion both in vitro and in vivo, indicating that destabilization of the binary complex and extension of the helix 12 are both linked to vesicle priming.
Discussion
For years, a mechanistic understanding of the stimulating role of Munc18-1 in neurotransmitter release has been lacking. With the identification of domain 3a as central for the postdocking role of Munc18-1, and with the reconstitution of Munc18-1 functions in vitro (Shen et al., 2007; Parisotto et al., 2012; Ma et al., 2013), this is finally starting to change. Here, we have tested a specific hypothesis regarding the function of the helix 12 of Munc18-1 in secretion. Using rescue experiments in Munc18-1-null adrenal chromaffin cells, we found that the Munc18-1 mutant P335A, which promotes the extended state of helix 12, increased neurosecretion, whereas the mutations Δ324–339 and L348R—designed to remove the helical hairpin of helix 12 or to disrupt Munc18-1:syb-2 binding—reduced fusion dramatically. The effects of the mutations were comparable with regard to the size of the standing primed vesicle pools and the sustained component of secretion (Fig. 5B), which indicates that the forward priming rate was mainly affected. The mutations that were deleterious to secretion (L348R, Δ324–339) nevertheless rescued syntaxin-1 to levels comparable to those of Munc18-1 WT, whereas the gain-of-function P335A mutant resulted in suboptimal syntaxin-1 targeting. Thus, the effects on exocytosis upon domain 3a mutations are not secondary to syntaxin-1 targeting, which is in agreement with previous data (Martin et al., 2013); on the contrary, the true effect of P335A on vesicle fusion might be underestimated because of the partial syntaxin-1 mistargeting. This might explain why the P335A mutant supports much more fusion in vitro than Y337A (Fig. 7), whereas the augmentation of cellular secretion is more similar (Figs. 5, 8). None of the mutations affected the kinetics of fusion from the primed vesicle pools, or the calcium dependence of fusion, arguing against direct effects of helix 12 downstream of priming (Zhang et al., 2015). Furthermore, vesicle docking, which can be upstream of or parallel to priming (Verhage and Sørensen, 2008), was rescued by the mutations. Overall, our data pinpoint the effect of the mutations on the forward priming rate, rather than on steps further upstream, or the unpriming rate. This adds in vivo support to the idea that structuring and extension of the helix 12 is important for vesicle fusion and further localizes this reaction to the vesicle priming step upstream of the slowly releasable pool. This same step is supported by ubMunc13–2 in chromaffin cells (Man et al., 2015), and it is inhibited by mutations designed to interfere with N-terminal assembly of the SNARE complex (Sørensen et al., 2006; Walter et al., 2010). Overall, these data are consistent with the idea that Munc13-assisted opening of closed syntaxin-1 still bound within Munc18-1 (Ma et al., 2013) leads to SNARE complex assembly at this particular point in the exocytotic cascade.
Previous data from PC12 cells showed that deletions/insertions/mutations in helix 12 inhibited Ca2+-triggered release (Han et al., 2013, 2014; Martin et al., 2013), which is in agreement with our data. In contrast to PC12 cells (Martin et al., 2013; Han et al., 2014), the P335A mutation produced a substantial enhancement of secretion in adrenal chromaffin cells. The clearer effect of this mutation in our work might originate from the fact that we induced maximal priming by increasing preflash [Ca2+] (Voets, 2000) to obtain a larger primed pool. Furthermore, assessment of the primed vesicle pool requires stimulation and measurements to be much faster than replenishment, which is most easily achieved using Ca2+ uncaging as a stimulus and capacitance measurements as a readout, whereas biochemical release assays are too slow to clearly distinguish the primed pool.
Previous studies indicated that an N-terminal assembly step drives vesicle priming, whereas vesicle fusion is driven by assembly of the C-terminal end of the SNARE bundle (Sørensen et al., 2006; Walter et al., 2010; Gao et al., 2012), followed by zippering up of the juxtamembrane and transmembrane domains, eventually forming the postfusion SNARE complex (Stein et al., 2009). Using cross-linking and nuclear magnetic resonance (NMR), an interaction site between Munc18-1 and syb-2 was mapped to the C-terminal end of the SNARE motif and the juxtamembrane linker region of syb-2 (amino acids 87–91 or 75–95, respectively; Xu et al., 2010). In the same area, NMR studies using a lipid environment identified the second of two transiently forming helixes (amino acids 77–88; Ellena et al., 2009). If helix 12 of Munc18-1 participated in a rate-limiting step during final zippering, for instance by structuring the C-terminal syb2 helix during fusion itself, or by transducing mechanical force from the SNAREs to the lipid bilayer (Xu et al., 2010), we would have expected to identify the kinetic changes of our mutations. However, this was not observed. Instead, we propose that Munc18-1 dislodges synaptobrevin-2 from the membrane, thereby making it available for initial N-terminal contacts with its partner SNAREs (Parisotto et al., 2014). Previous mutagenesis studies in synaptobrevin-2 have revealed a large effect on vesicle priming by the insertion of additional linkers between the SNARE motif and the transmembrane domain (Kesavan et al., 2007), identifying this area as important for priming. The suggested model is further supported by the observation that the SNARE–SNARE interactions and Munc18-1 binding synergize to stimulate vesicle docking in the SUV:GUV assay (Parisotto et al., 2012). In addition, Munc18-1 might help structuring the N-terminal transient helix in syb-2, which serves as a trigger site for SNARE complex assembly (Ellena et al., 2009; Walter et al., 2010), as well as the t-SNARE complex (Ma et al., 2015). This idea is supported by two recent structures of the SM protein Vps33 bound to the native Qa- and R-SNAREs Vam3p and Nyv1, respectively (Baker et al., 2015). When combined, the two structures show the domain 3a helical hairpin extending along the N-terminal half of both SNARE domains, possibly forming a template for N-terminal SNARE complex formation.
Another mutation in the helix 12 of the 3a domain, Y337A, also led to a gain-of-function phenotype in chromaffin cells (Fig. 8). The rationale for the mutation was to probe the assumed H-bond to N137 in syntaxin-1, which might counteract the ability of the helix 12 to undergo extension and stabilize the Munc18-1:syntaxin dimer. The gain-of-function phenotype seen is in agreement with this notion. Even though the increase in secretion was numerically small, it correlated with a slight, but significant, increase in the stimulatory function of Munc18-1 in the in vitro assay. Thus, helix extension and the opening up of syntaxin-1 might be inseparable events. This is also indicated by the reduced rescue of syntaxin-1 targeting by the P335A mutation, where the helix is permanently extended and the closed syntaxin-1:Munc18-1 complex is therefore destabilized. When comparing the P335A and Y337A mutants, the former stimulated the burst phase more than the sustained phase (ratio between the factor increase of the burst and sustained phases, 1.77), whereas the opposite was the case for Y337A (ratio, 0.55). Although we should proceed with caution when mutants were not compared experimentally side by side, this is consistent with an increase in the unpriming rate in the Y337A mutant relative to the P335A. In both cases, the priming rate is increased even more, leading to more net priming at steady state. This difference between the mutations makes sense if we assume that the extension of the helical hairpin coincides with priming and the retraction of the helix represents unpriming. In the P335A, the helix might no longer retract as easily, leading to lower unpriming rates, whereas in the Y337A, due to the impaired interaction with closed syntaxin-1, both retraction and extension are still possible, and might be facilitated.
Overall, our data are consistent with the hypothesis outlined in the Introduction implying that Munc18-1 can extend its helix 12 as a template for SNARE complex assembly. For successful vesicle priming, we therefore need the 3a domain of Munc18-1, N-terminal SNARE assembly (Walter et al., 2010), the very N-terminal end of syntaxin-1 (Shen et al., 2010; Zhou et al., 2013), a Munc13 protein (Ma et al., 2015), which in the case of chromaffin cells is ubMunc13–2 (Man et al., 2015). Finally, synaptotagmin-1 (Nagy et al., 2006) and calcium activator protein for secretion (CAPS) are required for the transition between the SRP and RRP state (Liu et al., 2010), although the relative importance of CAPS binding to PI(4,5)P2 and SNAREs is currently under discussion (Khodthong et al., 2011; Nguyen Truong et al., 2014). Overall, the opening of syntaxin-1 within Munc18-1 by the extension of helix 12 appears to be the central event, which is regulated by other factors, including Munc13 proteins, in a fashion, which has yet to be unraveled.
Footnotes
This work was supported by grants from the Lundbeck Center for Biomembranes in Nanomedicine (to J.B.S.), the Danish Medical Research Council (to J.B.S.), the Novo Nordisk Foundation (to J.B.S.), the German Research Foundation (to T.H.S.), FP7-ITN CognitionNet (to S.V.S.), the European Research Council (Grant ERC-ADG-322966-DCVfusion to M.V.), the European Neuroscience Campus Network, and the European Neuroscience Campus Network (Cycle 2-project 7 to G.H.K.). We thank Anne Marie Nordvig Petersen, Robbert Zalm, Rien Dekker, and Joke Wortel for excellent technical assistance. We also thank Alessandro Moro for image analysis software.
The authors declare no competing financial interests.
References
- Aarts E, Verhage M, Veenvliet JV, Dolan CV, van der Sluis S. A solution to dependency: using multilevel analysis to accommodate nested data. Nat Neurosci. 2014;17:491–496. doi: 10.1038/nn.3648. [DOI] [PubMed] [Google Scholar]
- Arancillo M, Min SW, Gerber S, Münster-Wandowski A, Wu YJ, Herman M, Trimbuch T, Rah JC, Ahnert-Hilger G, Riedel D, Südhof TC, Rosenmund C. Titration of syntaxin1 in mammalian synapses reveals multiple roles in vesicle docking, priming, and release probability. J Neurosci. 2013;33:16698–16714. doi: 10.1523/JNEUROSCI.0187-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker RW, Jeffrey PD, Zick M, Phillips BP, Wickner WT, Hughson FM. A direct role for the Sec1/Munc18-family protein Vps33 as a template for SNARE assembly. Science. 2015;349:1111–1114. doi: 10.1126/science.aac7906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd A, Ciufo LF, Barclay JW, Graham ME, Haynes LP, Doherty MK, Riesen M, Burgoyne RD, Morgan A. A random mutagenesis approach to isolate dominant-negative yeast sec1 mutants reveals a functional role for domain 3a in yeast and mammalian Sec1/Munc18 proteins. Genetics. 2008;180:165–178. doi: 10.1534/genetics.108.090423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruns D. Detection of transmitter release with carbon fiber electrodes. Methods. 2004;33:312–321. doi: 10.1016/j.ymeth.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Burkhardt P, Hattendorf DA, Weis WI, Fasshauer D. Munc18a controls SNARE assembly through its interaction with the syntaxin N-peptide. EMBO J. 2008;27:923–933. doi: 10.1038/emboj.2008.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colbert KN, Hattendorf DA, Weiss TM, Burkhardt P, Fasshauer D, Weis WI. Syntaxin1a variants lacking an N-peptide or bearing the LE mutation bind to Munc18a in a closed conformation. Proc Natl Acad Sci U S A. 2013;110:12637–12642. doi: 10.1073/pnas.1303753110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawidowski D, Cafiso DS. Munc18-1 and the syntaxin-1 N terminus regulate open-closed states in a t-SNARE complex. Structure. 2016;24:392–400. doi: 10.1016/j.str.2016.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deák F, Xu Y, Chang WP, Dulubova I, Khvotchev M, Liu X, Südhof TC, Rizo J. Munc18-1 binding to the neuronal SNARE complex controls synaptic vesicle priming. J Cell Biol. 2009;184:751–764. doi: 10.1083/jcb.200812026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit H, Cornelisse LN, Toonen RF, Verhage M. Docking of secretory vesicles is syntaxin dependent. PLoS One. 2006;1:e126. doi: 10.1371/journal.pone.0000126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit H, Walter AM, Milosevic I, Gulyás-Kovács A, Riedel D, Sørensen JB, Verhage M. Synaptotagmin-1 docks secretory vesicles to syntaxin-1/SNAP-25 acceptor complexes. Cell. 2009;138:935–946. doi: 10.1016/j.cell.2009.07.027. [DOI] [PubMed] [Google Scholar]
- Dulubova I, Sugita S, Hill S, Hosaka M, Fernandez I, Südhof TC, Rizo J. A conformational switch in syntaxin during exocytosis: role of Munc18. EMBO J. 1999;18:4372–4382. doi: 10.1093/emboj/18.16.4372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulubova I, Khvotchev M, Liu S, Huryeva I, Südhof TC, Rizo J. Munc18-1 binds directly to the neuronal SNARE complex. Proc Natl Acad Sci U S A. 2007;104:2697–2702. doi: 10.1073/pnas.0611318104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellena JF, Liang B, Wiktor M, Stein A, Cafiso DS, Jahn R, Tamm LK. Dynamic structure of lipid-bound synaptobrevin suggests a nucleation-propagation mechanism for trans-SNARE complex formation. Proc Natl Acad Sci U S A. 2009;106:20306–20311. doi: 10.1073/pnas.0908317106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Zorman S, Gundersen G, Xi Z, Ma L, Sirinakis G, Rothman JE, Zhang Y. Single reconstituted neuronal SNARE complexes zipper in three distinct stages. Science. 2012;337:1340–1343. doi: 10.1126/science.1224492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber SH, Rah JC, Min SW, Liu X, de Wit H, Dulubova I, Meyer AC, Rizo J, Arancillo M, Hammer RE, Verhage M, Rosenmund C, Südhof TC. Conformational switch of syntaxin-1 controls synaptic vesicle fusion. Science. 2008;321:1507–1510. doi: 10.1126/science.1163174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulyás-Kovács A, de Wit H, Milosevic I, Kochubey O, Toonen R, Klingauf J, Verhage M, Sørensen JB. Munc18-1: sequential interactions with the fusion machinery stimulate vesicle docking and priming. J Neurosci. 2007;27:8676–8686. doi: 10.1523/JNEUROSCI.0658-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han GA, Bin NR, Kang SY, Han L, Sugita S. Domain 3a of Munc18-1 plays a crucial role at the priming stage of exocytosis. J Cell Sci. 2013;126:2361–2371. doi: 10.1242/jcs.126862. [DOI] [PubMed] [Google Scholar]
- Han GA, Park S, Bin NR, Jung CH, Kim B, Chandrasegaram P, Matsuda M, Riadi I, Han L, Sugita S. A pivotal role for Pro335 in balancing the dual functions of Munc18-1 domain-3a in regulated exocytosis. J Biol Chem. 2014;289:33617–33628. doi: 10.1074/jbc.M114.584805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu SH, Christie MP, Saez NJ, Latham CF, Jarrott R, Lua LH, Collins BM, Martin JL. Possible roles for Munc18-1 domain 3a and Syntaxin1 N-peptide and C-terminal anchor in SNARE complex formation. Proc Natl Acad Sci U S A. 2011;108:1040–1045. doi: 10.1073/pnas.0914906108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedar GH, Munch AS, van Weering JR, Malsam J, Scheutzow A, de Wit H, Houy S, Tawfik B, Söllner TH, Sørensen JB, Verhage M. A post-docking role of synaptotagmin 1-C2B domain bottom residues R398/399 in mouse chromaffin cells. J Neurosci. 2015;35:14172–14182. doi: 10.1523/JNEUROSCI.1911-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kesavan J, Borisovska M, Bruns D. v-SNARE actions during Ca(2+)-triggered exocytosis. Cell. 2007;131:351–363. doi: 10.1016/j.cell.2007.09.025. [DOI] [PubMed] [Google Scholar]
- Khodthong C, Kabachinski G, James DJ, Martin TF. Munc13 homology domain-1 in CAPS/UNC31 mediates SNARE binding required for priming vesicle exocytosis. Cell Metab. 2011;14:254–263. doi: 10.1016/j.cmet.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurps J, Broeke JH, Cijsouw T, Kompatscher A, van Weering JR, de Wit H. Quantitative image analysis tool to study the plasma membrane localization of proteins and cortical actin in neuroendocrine cells. J Neurosci Methods. 2014;236:1–10. doi: 10.1016/j.jneumeth.2014.07.022. [DOI] [PubMed] [Google Scholar]
- Lindau M, Neher E. Patch-clamp techniques for time-resolved capacitance measurements in single cells. Pflugers Archiv. 1988;411:137–146. doi: 10.1007/BF00582306. [DOI] [PubMed] [Google Scholar]
- Liu Y, Schirra C, Edelmann L, Matti U, Rhee J, Hof D, Bruns D, Brose N, Rieger H, Stevens DR, Rettig J. Two distinct secretory vesicle-priming steps in adrenal chromaffin cells. J Cell Biol. 2010;190:1067–1077. doi: 10.1083/jcb.201001164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C, Su L, Seven AB, Xu Y, Rizo J. Reconstitution of the vital functions of Munc18 and Munc13 in neurotransmitter release. Science. 2013;339:421–425. doi: 10.1126/science.1230473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Rebane AA, Yang G, Xi Z, Kang Y, Gao Y, Zhang Y. Munc18-1-regulated stage-wise SNARE assembly underlying synaptic exocytosis. Elife. 2015;4:e09580. doi: 10.7554/eLife.09580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahal LK, Sequeira SM, Gureasko JM, Söllner TH. Calcium-independent stimulation of membrane fusion and SNAREpin formation by synaptotagmin I. J Cell Biol. 2002;158:273–282. doi: 10.1083/jcb.200203135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malsam J, Parisotto D, Bharat TA, Scheutzow A, Krause JM, Briggs JA, Söllner TH. Complexin arrests a pool of docked vesicles for fast Ca2+-dependent release. EMBO J. 2012;31:3270–3281. doi: 10.1038/emboj.2012.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man KN, Imig C, Walter AM, Pinheiro PS, Stevens DR, Rettig J, Sørensen JB, Cooper BH, Brose N, Wojcik SM. Identification of a Munc13-sensitive step in chromaffin cell large dense-core vesicle exocytosis. Elife. 2015;4:e10635. doi: 10.7554/eLife.10635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin S, Tomatis VM, Papadopulos A, Christie MP, Malintan NT, Gormal RS, Sugita S, Martin JL, Collins BM, Meunier FA. The Munc18-1 domain 3a loop is essential for neuroexocytosis but not for syntaxin-1A transport to the plasma membrane. J Cell Sci. 2013;126:2353–2360. doi: 10.1242/jcs.126813. [DOI] [PubMed] [Google Scholar]
- Medine CN, Rickman C, Chamberlain LH, Duncan RR. Munc18-1 prevents the formation of ectopic SNARE complexes in living cells. J Cell Sci. 2007;120:4407–4415. doi: 10.1242/jcs.020230. [DOI] [PubMed] [Google Scholar]
- Meijer M, Burkhardt P, de Wit H, Toonen RF, Fasshauer D, Verhage M. Munc18-1 mutations that strongly impair SNARE-complex binding support normal synaptic transmission. EMBO J. 2012;31:2156–2168. doi: 10.1038/emboj.2012.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misura KM, Scheller RH, Weis WI. Three-dimensional structure of the neuronal-Sec1-syntaxin 1a complex. Nature. 2000;404:355–362. doi: 10.1038/35006120. [DOI] [PubMed] [Google Scholar]
- Mohrmann R, de Wit H, Connell E, Pinheiro PS, Leese C, Bruns D, Davletov B, Verhage M, Sørensen JB. Synaptotagmin interaction with SNAP-25 governs vesicle docking, priming, and fusion triggering. J Neurosci. 2013;33:14417–14430. doi: 10.1523/JNEUROSCI.1236-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy G, Kim JH, Pang ZP, Matti U, Rettig J, Südhof TC, Sørensen JB. Different effects on fast exocytosis induced by synaptotagmin 1 and 2 isoforms and abundance but not by phosphorylation. J Neurosci. 2006;26:632–643. doi: 10.1523/JNEUROSCI.2589-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen Truong CQ, Nestvogel D, Ratai O, Schirra C, Stevens DR, Brose N, Rhee J, Rettig J. Secretory vesicle priming by CAPS is independent of its SNARE-binding MUN domain. Cell Rep. 2014;9:902–909. doi: 10.1016/j.celrep.2014.09.050. [DOI] [PubMed] [Google Scholar]
- Nili U, de Wit H, Gulyas-Kovacs A, Toonen RF, Sørensen JB, Verhage M, Ashery U. Munc18-1 phosphorylation by protein kinase C potentiates vesicle pool replenishment in bovine chromaffin cells. Neuroscience. 2006;143:487–500. doi: 10.1016/j.neuroscience.2006.08.014. [DOI] [PubMed] [Google Scholar]
- Parisotto D, Malsam J, Scheutzow A, Krause JM, Söllner TH. SNAREpin assembly by Munc18-1 requires previous vesicle docking by synaptotagmin 1. J Biol Chem. 2012;287:31041–31049. doi: 10.1074/jbc.M112.386805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisotto D, Pfau M, Scheutzow A, Wild K, Mayer MP, Malsam J, Sinning I, Söllner TH. An extended helical conformation in domain 3a of the SM protein Munc18-1 provides a template for SNARE (soluble N-ethylmaleimide sensitive factor attachment protein receptor) complex assembly. J Biol Chem. 2014;289:9639–9650. doi: 10.1074/jbc.M113.514273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schollmeier Y, Krause JM, Kreye S, Malsam J, Söllner TH. Resolving the function of distinct Munc18-1/SNARE protein interaction modes in a reconstituted membrane fusion assay. J Biol Chem. 2011;286:30582–30590. doi: 10.1074/jbc.M111.269886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen C, Rathore SS, Yu H, Gulbranson DR, Hua R, Zhang C, Schoppa NE, Shen J. The trans-SNARE-regulating function of Munc18-1 is essential to synaptic exocytosis. Nat Commun. 2015;6:8852. doi: 10.1038/ncomms9852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Tareste DC, Paumet F, Rothman JE, Melia TJ. Selective activation of cognate SNAREpins by Sec1/Munc18 proteins. Cell. 2007;128:183–195. doi: 10.1016/j.cell.2006.12.016. [DOI] [PubMed] [Google Scholar]
- Shen J, Rathore SS, Khandan L, Rothman JE. SNARE bundle and syntaxin N-peptide constitute a minimal complement for Munc18-1 activation of membrane fusion. J Cell Biol. 2010;190:55–63. doi: 10.1083/jcb.201003148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sørensen JB, Matti U, Wei SH, Nehring RB, Voets T, Ashery U, Binz T, Neher E, Rettig J. The SNARE protein SNAP-25 is linked to fast calcium triggering of exocytosis. Proc Natl Acad Sci U S A. 2002;99:1627–1632. doi: 10.1073/pnas.251673298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sørensen JB, Fernández-Chacón R, Südhof TC, Neher E. Examining synaptotagmin 1 function in dense core vesicle exocytosis under direct control of Ca2+ J Gen Physiol. 2003a;122:265–276. doi: 10.1085/jgp.200308855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sørensen JB, Nagy G, Varoqueaux F, Nehring RB, Brose N, Wilson MC, Neher E. Differential control of the releasable vesicle pools by SNAP-25 splice variants and SNAP-23. Cell. 2003b;114:75–86. doi: 10.1016/S0092-8674(03)00477-X. [DOI] [PubMed] [Google Scholar]
- Sørensen JB, Wiederhold K, Müller EM, Milosevic I, Nagy G, de Groot BL, Grubmüller H, Fasshauer D. Sequential N- to C-terminal SNARE complex assembly drives priming and fusion of secretory vesicles. EMBO J. 2006;25:955–966. doi: 10.1038/sj.emboj.7601003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein A, Weber G, Wahl MC, Jahn R. Helical extension of the neuronal SNARE complex into the membrane. Nature. 2009;460:525–528. doi: 10.1038/nature08156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toonen RF, Kochubey O, de Wit H, Gulyas-Kovacs A, Konijnenburg B, Sørensen JB, Klingauf J, Verhage M. Dissecting docking and tethering of secretory vesicles at the target membrane. EMBO J. 2006;25:3725–3737. doi: 10.1038/sj.emboj.7601256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhage M, Sørensen JB. Vesicle docking in regulated exocytosis. Traffic. 2008;9:1414–1424. doi: 10.1111/j.1600-0854.2008.00759.x. [DOI] [PubMed] [Google Scholar]
- Verhage M, Maia AS, Plomp JJ, Brussaard AB, Heeroma JH, Vermeer H, Toonen RF, Hammer RE, van den Berg TK, Missler M, Geuze HJ, Südhof TC. Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science. 2000;287:864–869. doi: 10.1126/science.287.5454.864. [DOI] [PubMed] [Google Scholar]
- Voets T. Dissection of three Ca2+-dependent steps leading to secretion in chromaffin cells from mouse adrenal slices. Neuron. 2000;28:537–545. doi: 10.1016/S0896-6273(00)00131-8. [DOI] [PubMed] [Google Scholar]
- Voets T, Toonen RF, Brian EC, de Wit H, Moser T, Rettig J, Südhof TC, Neher E, Verhage M. Munc18-1 promotes large dense-core vesicle docking. Neuron. 2001;31:581–591. doi: 10.1016/S0896-6273(01)00391-9. [DOI] [PubMed] [Google Scholar]
- Walter AM, Wiederhold K, Bruns D, Fasshauer D, Sørensen JB. Synaptobrevin N-terminally bound to syntaxin-SNAP-25 defines the primed vesicle state in regulated exocytosis. J Cell Biol. 2010;188:401–413. doi: 10.1083/jcb.200907018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter AM, Pinheiro PS, Verhage M, Sørensen JB. A sequential vesicle pool model with a single release sensor and a Ca(2+)-dependent priming catalyst effectively explains Ca(2+)-dependent properties of neurosecretion. PLoS Comput Biol. 2013;9:e1003362. doi: 10.1371/journal.pcbi.1003362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber T, Zemelman BV, McNew JA, Westermann B, Gmachl M, Parlati F, Söllner TH, Rothman JE. SNAREpins: minimal machinery for membrane fusion. Cell. 1998;92:759–772. doi: 10.1016/S0092-8674(00)81404-X. [DOI] [PubMed] [Google Scholar]
- Xu Y, Su L, Rizo J. Binding of Munc18-1 to synaptobrevin and to the SNARE four-helix bundle. Biochemistry. 2010;49:1568–1576. doi: 10.1021/bi9021878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Diao J, Colbert KN, Lai Y, Pfuetzner RA, Padolina MS, Vivona S, Ressl S, Cipriano DJ, Choi UB, Shah N, Weis WI, Brunger AT. Munc18a does not alter fusion rates mediated by neuronal SNAREs, synaptotagmin, and complexin. J Biol Chem. 2015;290:10518–10534. doi: 10.1074/jbc.M114.630772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P, Pang ZP, Yang X, Zhang Y, Rosenmund C, Bacaj T, Südhof TC. Syntaxin-1 N-peptide and Habc-domain perform distinct essential functions in synaptic vesicle fusion. EMBO J. 2013;32:159–171. doi: 10.1038/emboj.2012.307. [DOI] [PMC free article] [PubMed] [Google Scholar]