

Abstract
Spinal cord injury (SCI) causes the release of danger signals by stressed and dying cells, a process that leads to neuroinflammation. Evidence suggests that inflammation plays a role in both the damage and repair of injured neural tissue. We show that microglia at sites of SCI rapidly express the alarmin interleukin (IL)-1α, and that infiltrating neutrophils and macrophages subsequently produce IL-1β. Infiltration of these cells is dramatically reduced in both IL-1α−/− and IL-1β−/− mice, but only IL-1α−/− mice showed rapid (at day 1) and persistent improvements in locomotion associated with reduced lesion volume. Similarly, intrathecal administration of the IL-1 receptor antagonist anakinra restored locomotor function post-SCI. Transcriptome analysis of SCI tissue at day 1 identified the survival factor Tox3 as being differentially regulated exclusively in IL-1α−/− mice compared with IL-1β−/− and wild-type mice. Accordingly, IL-1α−/− mice have markedly increased Tox3 levels in their oligodendrocytes, beginning at postnatal day 10 (P10) and persisting through adulthood. At P10, the spinal cord of IL-1α−/− mice showed a transient increase in mature oligodendrocyte numbers, coinciding with increased IL-1α expression in wild-type animals. In adult mice, IL-1α deletion is accompanied by increased oligodendrocyte survival after SCI. TOX3 overexpression in human oligodendrocytes reduced cellular death under conditions mimicking SCI. These results suggest that IL-1α-mediated Tox3 suppression during the early phase of CNS insult plays a crucial role in secondary degeneration.
SIGNIFICANCE STATEMENT The mechanisms underlying bystander degeneration of neurons and oligodendrocytes after CNS injury are ill defined. We show that microglia at sites of spinal cord injury (SCI) rapidly produce the danger signal interleukin (IL)-1α, which triggers neuroinflammation and locomotor defects. We uncovered that IL-1α−/− mice have markedly increased levels of the survival factor Tox3 in their oligodendrocytes, which correlates with the protection of this cell population, and reduced lesion volume, resulting in unprecedented speed, level, and persistence of functional recovery after SCI. Our data suggest that central inhibition of IL-1α or Tox3 overexpression during the acute phase of a CNS insult may be an effective means for preventing the loss of neurological function in SCI, or other acute injuries such as ischemia and traumatic brain injuries.
Keywords: cytokine, microglia, neurodegeneration, neuroimmunology, neuroinflammation, neutrophil
Introduction
Spinal cord injury (SCI) can lead to loss of motor and sensory function in the lower and/or upper limbs. At the site of injury, the pathology is divided into two major chronological events: the primary and secondary lesions. The second wave of tissue destruction that follows the primary mechanical insult is believed to be caused by a complex series of events leading to apoptosis, including ischemia, vascular damage, glutamate excitotoxicity, ionic dysregulation, inflammation, free radical generation, and cytokine and protease production (David and Lacroix, 2005). Damage to the CNS is associated with an almost immediate response of microglia (Davalos et al., 2005). Astrocytes, the main cellular component of the glial scar, also react quickly by exhibiting changes in gene expression, hypertrophy, and process extension (Sofroniew, 2009). The glial scar has been shown to have both protective and detrimental functions in the context of SCI (Faulkner et al., 2004; Brambilla et al., 2005, 2009).
Innate immune cells such as neutrophils and monocytes are rapidly recruited at sites of SCI (Fleming et al., 2006; Stirling and Yong, 2008; Pineau et al., 2010; Lee et al., 2011; Thawer et al., 2013). Evidence suggests that these cells could play a key role in both the damage and repair of neural tissue after an injury or ischemia (Barrette et al., 2008; Kigerl et al., 2009; Shechter et al., 2009; Stirling et al., 2009; Allen et al., 2012). The apparent contradictions among these studies may be due to the fact that different subsets of immune cells have divergent effects causing either neurotoxicity or regeneration in the injured spinal cord (David and Kroner, 2011; Bastien and Lacroix, 2014; Plemel et al., 2014). A better understanding of the roles of the various subsets of immune cells and the identification of the endogenous factors stimulating their recruitment is key for the development of efficient immunotherapies.
One important family of cytokines playing a key role in neurodegeneration is the interleukin (IL)-1 family (Allan and Rothwell, 2001; Allan et al., 2005). Two of the members of this large family, IL-1α and IL-1β, exert similar proinflammatory actions by binding to the IL-1 type 1 receptor (IL-1r1). Both IL-1α and IL-1β produced by tissue resident macrophages are required for neutrophil recruitment during cell death-induced sterile inflammation (Chen et al., 2007; Kono et al., 2010). However, the function of IL-1β has been studied more thoroughly because of its importance in the development and progression of a number of autoinflammatory and CNS diseases (Allan et al., 2005; Dinarello, 2011). Apart from its role in the initiation of gliosis and inflammation (Basu et al., 2002), studies have linked IL-1β to the production of growth factors by CNS resident cells in various disease and injury models (Herx et al., 2000; Albrecht et al., 2002, 2003; Amankulor et al., 2009). Other evidence indicates that IL-1β likely plays an important role in CNS remyelination by regulating multiple stages of oligodendrocyte development (Mason et al., 2001).
Here, we sought to determine whether cytokines of the IL-1 family are an endogenous signal initiating neuroinflammation after traumatic SCI. We uncovered a dual role for IL-1α not only in triggering inflammation in the injured spinal cord, but also in suppressing the expression of the survival factor TOX high-mobility group box family member 3 (Tox3) in oligodendrocytes. Overexpression of this transcription factor gave remarkable protection against cell death. Thus, blocking this cytokine may be a promising therapeutic avenue to prevent loss of CNS cells following SCI and other neurodegenerative conditions.
Materials and Methods
Animals.
A total of 308 mice of either sex were used in this study. C57BL/6 mice, used as controls, were purchased from Charles River Laboratories. IL-1α-, IL-1β-, and IL-1α/β-knock-out (KO) mice were obtained from Dr. Yoichiro Iwakura (Institute of Medical Science, University of Tokyo, Tokyo, Japan) and have been previously described (Horai et al., 1998). Breeders for the pIL-1b-DsRed transgenic mouse line were obtained from Dr. Akira Takashima (College of Medicine and Life Sciences, University of Toledo, Toledo, OH) and were bred in-house at the Animal Research Facility of the Centre Hospitalier de l'Université Laval Research Center and genotyped according to the method published by Matsushima et al. (2010). Finally, Cx3cr1-eGFP transgenic mice were purchased from The Jackson Laboratory. All mice had free access to food and water.
Spinal cord injury and animal treatment.
C57BL/6 (n = 75), IL-1α-KO (n = 64), IL-1β-KO (n = 60), IL-1α/β-KO (n = 13), pIL-1b-DsRed (n = 6), and Cx3cr1-eGFP (n = 18) adult mice were anesthetized with isoflurane and underwent a laminectomy at vertebral level T9–T10, which corresponds to spinal segment T10–T11. Briefly, the vertebral column was stabilized and a contusion of 50 kdyn was performed using the Infinite Horizon SCI device (Precision Systems and Instrumentation). Animals used for quantification of immune cells, either by immunofluorescence (IF) or flow cytometry, received a 70 kdyn injury. Overlying muscular layers were then sutured, and cutaneous layers were stapled. Postoperatively, animals received manual bladder evacuation twice daily to prevent urinary tract infections. Depending on the experiment performed, SCI mice were killed by transcardiac perfusion at 1, 3, 4, 6, 12, and 24 h, and 3, 4, and 38 d after contusion. C57BL/6 (n = 37), IL-1α-KO (n = 12), and IL-1β-KO (n = 12) neonatal mice were killed by either decapitation under ice anesthesia for pups of 1, 3, 7, 10, and 14 d of age, or by transcardiac perfusion under deep anesthesia induced with ketamine-xylazine for mice at postnatal day 18 (P18) and P30.
In the experiment in which we studied the effects of treatment with the IL-1r1 antagonist anakinra (100 mg/0.67 ml in prefilled syringes; Kineret, Swedish Orphan Biovitrum AB) on functional recovery after SCI, mice were injected either intravenously (anakinra diluted to a final concentration of 20 μg/μl in PBS, for a dose of 100 mg/kg per injection) or intrathecally (33 mg/kg in 5 μl of PBS). The 3 day intravenous treatment consisted of injections at 6, 24, and 48 h post-SCI, whereas the 7 day intravenous treatment consisted of injections made at the time of injury, 2 h after, and then every day until day 7. The intrathecal treatment consisted of a single injection into the cisterna magna performed 15 min after SCI using a pulled-glass micropipette connected to a 10 μl Hamilton syringe. All surgical procedures and treatments were approved by the Laval University Animal Care Committee and conducted in accordance with guidelines of the Canadian Council on Animal Care.
Flow cytometry.
Cells freshly isolated from the spinal cord of injured mice were analyzed using flow cytometry following our previously published method (de Rivero Vaccari et al., 2012). Briefly, animals were transcardially perfused with cold HBSS to remove blood from the vasculature, their spinal cords were dissected out, and a 1 cm segment centered at the site of the lesion was isolated and mechanically homogenized with a small tissue grinder. Cells were filtered through a 40 μm nylon mesh cell strainer (BD Biosciences), centrifuged at 200 × g for 10 min, washed once with HBSS, and resuspended with HBSS containing 20% fetal bovine serum (FBS; Sigma-Aldrich Canada Ltd.). For multicolor immunofluorescent labeling, cells were incubated with Mouse Fc Block (i.e., purified anti-mouse CD16/CD32; BD Biosciences) for 5 min to prevent nonspecific binding, followed by labeling for 30 min at room temperature with the following fluorescently conjugated primary antibodies: PerCP-conjugated anti-CD45 (1:33 dilution; BD Biosciences), BD HorizonTM V450-conjugated anti-CD11b (1:66; BD Biosciences), FITC-conjugated anti-Ly6C (1:100; BD Biosciences), PE-Cy7-conjugated anti-Ly6G (1:100; BD Biosciences), and APC-conjugated anti-F4/80 (1:20; AbD Serotec; for a full description of these primary antibodies, please refer to our published work; Nadeau et al., 2011). Cells were analyzed using FlowJo software (version 9.2; Tree Star Inc.) on a FACS LSRII flow cytometer (BD Biosciences). Blood-derived myeloid cells were identified based on their expression of CD45, CD11b, Ly6C, Ly6G, and F4/80, as follows: CD45hi, CD11b+, Ly6C+, Ly6G+, and F4/80− cells were considered as neutrophils; and CD45hi, CD11b+, Ly6Chi, Ly6G− and F4/80+ cells were considered as monocyte-derived M1 macrophages (de Rivero Vaccari et al., 2012).
Tissue processing and histology.
Spinal cords were collected and prepared as previously described (Pineau and Lacroix, 2007). Briefly, mice were overdosed with a mixture of ketamine-xylazine and transcardially perfused with either 1% or 4% paraformaldehyde (PFA), pH 7.4, in PBS. Spinal cords were dissected out and placed overnight in a PBS/30% sucrose solution. For each animal, a spinal cord segment of 12 mm centered over the lesion site was cut into seven series of 14-μm-thick coronal sections using a cryostat. For immunohistochemistry against 7/4 (also referred to as Ly6B; see the study by Rosas et al., 2010) and Ly6G, mice were perfused with 0.9% saline solution followed by 4% PFA, pH 9.5, in borax buffer. Spinal cords were dissected out, post-fixed for 2 d, and placed overnight in a 4% PFA-borax/10% sucrose solution until processing using a cryostat set at 30 μm thickness. All sections were collected directly onto Surgipath X-tra slides having a permanent positive charged surface (Leica Microsystems Canada) and stored at −20°C until use. To identify the lesion epicenter and quantify lesion volume, one series of adjacent sections was immunostained using a rabbit anti-GFAP (mouse) antibody (1:750 dilution; Dako Canada Inc.) and then counterstained with 4′,6′-diamidino-2-phenylindole dihydrochloride (DAPI; 1 μg/ml, Life Technologies) and FluoroMyelin red fluorescent myelin stain (1:300; Life Technologies).
Immunohistochemistry and quantification.
Cells expressing IL-1α were identified by confocal IF labeling using a goat anti-mouse IL-1α polyclonal antibody (1:100 dilution; R&D Systems) on tissue sections fixed in 1% PFA and colocalized with the myeloid cell marker CD11b (1:250; AbD Serotec) or markers of microglia such as ionized calcium-binding adaptor molecule 1 (Iba1; 1:750; Wako Chemicals) and the GFP reporter expressed under the control of the Cx3cr1 promoter (Cx3cr1-eGFP transgenic mice). The astroglial scar was visualized using the anti-GFAP antibody described above. Alexa Fluor secondary antibody conjugates (1:250; Life Technologies) were used as secondary antibodies, whereas DAPI was used for nuclear counterstaining. Immunofluorescence labeling was performed according to our previously published methods (de Rivero Vaccari et al., 2012). Sections were observed and imaged on an Olympus IX81 Fluoview FV1000 confocal microscope system equipped with 488, 543, and 633 nm lasers (Olympus Canada Inc.). Quantification of double-labeled cells was performed manually at 40× using the Olympus confocal microscope. Results were presented as the total number of IL-1α-positive (IL-1α+) cells per cross section or the percentage of IL-1α+ cells that expressed each of the two microglial markers (Iba1, GFP).
Tox3 protein expression was visualized by confocal IF microscopy using a rabbit polyclonal anti-Tox3 antibody (1:100 dilution; Novus Biologicals Canada ULC). Colocalization of Tox3 in neurons, oligodendrocytes, and endothelial cells was performed using the neuron-specific markers NeuN (1:250; EMD Millipore) and HuC/HuD (1:80; Life Technologies), the oligodendrocyte marker CC1 (also referred to as anti-APC; 1:500; Abcam) and the endothelial cell marker CD31 (also referred to as PECAM-1; 1:750; BD Biosciences), respectively. For the quantification of NeuN+ neurons and CC1+ oligodendrocytes expressing or not expressing Tox3, the number of labeled cells was estimated by the optical fractionator method using the Nova Prime software (Bioquant Image Analysis Corporation) on video images transmitted by a high-resolution Retiga QICAM fast color 1394 camera (1392 × 1040 pixels; QImaging) installed on a Nikon Eclipse 80i microscope. For this, the outline of the spinal cord gray matter (for the quantification of neurons) or white matter (for the quantification of oligodendrocytes) was traced at 10× and then sampled at 40× magnification. The counting parameters were as follows: sampling grid size, 150 × 150 μm (except for P3 mice, for which a sampling grid of 100 × 100 μm was used instead); counting frame size, 50 × 50 μm, and; dissector height, 14 μm. Cells were counted only if their nuclei lay within the dissector area, did not intersect forbidden lines, and came into the focus as the optical plane moved through the height of the dissector. Quantification was performed on a total of nine equally spaced sections (294 μm apart) spanning 3 mm centered at the lesion epicenter, since we noted in our previous work that the lesion normally extends over ∼3 mm in mice that received a 50 kdyn SCI contusion. For each section, the total number of positively stained cells was calculated by multiplying the total volume of the spinal cord gray or white matter, as measured using the Bioquant Nova Prime software, by the average number of cells per cubic millimeter. The relative number of cells that survived the trauma was then estimated by adding the total number of positively stained cells counted in each of the nine sections encompassing the lesion.
Immunoperoxidase labeling using the anti-7/4 antibody (1:800 dilution; AbD Serotec) was performed to detect both neutrophils and proinflammatory M1 macrophages in the spinal cords of injured mice, as this antibody was recently shown to be specific for detecting these two cell types in SCI tissue (de Rivero Vaccari et al., 2012). Immunoperoxidase labeling was performed on spinal cord tissue sections directly mounted onto slides using CoverWell incubation chambers (Life Technologies Inc.) and our previously published protocol (Pineau and Lacroix, 2007), with the only difference being that sections were pretreated for 15 min with proteinase K to improve immunolabeling efficiency. Following immunoperoxidase labeling, tissue sections were counterstained with Luxol fast blue to visualize damaged areas. For the quantification of neutrophils and M1 macrophages, the outline of the coronal section was traced at 10× magnification, as described above, and a grid of 50 × 50 μm positioned over the spinal cord using the Bioquant Nova Prime software. All 7/4+ cells were then counted at 20× magnification. Results were expressed as an average number of positive cells per coronal section.
All quantifications were performed blind with respect to the identity of the animals and the epineurial layer excluded from analyses.
Lesion volume analysis.
The calculation of areas of tissue damage and lesion volume after SCI was performed on one series of adjacent sections within a predetermined spinal cord segment, including the lesion epicenter and sections located up to ∼1.5 mm distal to the center of the lesion in both directions (i.e., rostral and caudal). Fourteen-micrometer-thick coronal sections were first immunostained for GFAP, and then counterstained with DAPI and FluoroMyelin to visualize the glial scar and to identify gray and white matter sparing, respectively. The analysis was performed using the BioQuant Nova Prime computerized image analysis system (with Topographer XP plug-in, Bioquant). In each section, the total area of the coronal section was first determined by manually tracing the contour at low magnification. Next, the outline of necrotic and damaged tissue was traced at higher magnifications. Areas where normal spinal cord architecture was absent, and areas containing cellular and myelin debris surrounded by the astroglial scar were considered as areas of tissue damage. The Bioquant Topographer program was then used to reconstruct the injured spinal cord and measure lesion volume.
Toluidine blue staining and g-ratio analysis.
The method used was previously described by Tremblay et al. (2010). Mice were perfused with a solution of 3.5% acrolein followed by 4% PFA in 0.1 m phosphate buffer, pH 7.4. Two-millimeter-thick cross sections of the thoracic spinal cord were left to post-fix for 2 h at 4°C in 4% PFA and then cut at a thickness of 50 μm using a vibratome (VT1000S, Leica Microsystems). Fifty-micrometer-thick sections were next treated with 1% osmium tetroxide and dehydrated into ascending concentrations of ethanol followed by propylene oxide. The tissue was embedded into Durcupan resin, and sections were cut at a thickness of 1 μm using an ultramicrotome (UC7, Leica Microsystems) and histology diamond knife. Semithin sections were collected directly onto slides, stained with 0.1% toluidine blue in sodium borate and coverslipped. G-ratios were determined by dividing the axon diameter by the axon plus myelin diameter. Quantification was performed on a total of 200 myelinated axons per animal. Half of the axons were imaged at 100× magnification in the ascending sensory tract (AST) of the dorsal column and the other half in the lateral descending rubrospinal tract (RST).
Microarray analysis.
Spinal cord segments (6 mm length centered at the impact site) were rapidly dissected, frozen in liquid nitrogen, and stored at −80°C until RNA extraction. Total RNA was isolated by TRI Reagent and purified with the GenElute mammalian total RNA miniprep kit (Sigma-Aldrich Canada Ltd.). Two hundred nanograms of total RNA was labeled using the Ambion WT Expression Kit protocol, as described by the manufacturer (Affymetrix). The quality of total RNA, cDNA synthesis, cRNA amplification, and cDNA fragmentation was monitored by microcapillary electrophoresis (Bioanalyzer 2100, Agilent Technologies). Five micrograms of fragmented single-stranded cDNA was hybridized for 16 h at 45°C with constant rotation on a GeneChip Mouse Gene 1.0 ST Array (Affymetrix). After hybridization, microarrays (n = 12) were processed using the Affymetrix GeneChip Fluidic Station 450 (protocol FS450_0007). In brief, staining was made with streptavidin-conjugated phycoerythrin (SAPE; Affymetrix), followed by amplification with a biotinylated anti-streptavidin antibody (Life Technologies), and by a second round of SAPE staining. Microarrays were scanned using a GeneChip Scanner 3000 7G (Affymetrix) that was enabled for high-resolution scanning. Images were extracted with the GeneChip Operating Software (GCOS version 1.4, Affymetrix). Quality control of microarrays was performed using the AffyQCReport software (Gautier et al., 2004).
Background subtraction and normalization of probe set intensities was performed using the method of robust multiarray analysis (RMA) described by Irizarry et al. (2003). To identify differentially expressed genes, gene expression intensity was compared using an ANOVA test with a significance threshold of <0.001 and a ≥1.5-fold change, and a Bayes smoothing approach developed for a low number of replicates (Smyth, 2004). A false discovery rate of <0.2 was used to adjust for multiple testing. This analysis was performed using the AffylmGUI Graphical User Interface for the LIMMA microarray package (Wettenhall et al., 2006), and the Partek Genomics Suite. All microarray data compliant with the MIAME guidelines (http://www.mged.org/) were deposited in the Gene Expression Omnibus database (http://www.ncbi.nlm.nih.gov/geo/) Accession Number GSE70302.
Quantitative real-time PCR.
Total RNA was isolated from spinal cords (exsanguinated) using the TRIzol method following the manufacturer protocol (Life Technologies). RNA quantity and quality were assessed using the RNA 6000 Nano LabChip and Agilent Bioanalyzer 2100. First-strand cDNA synthesis was accomplished using 5 μg of isolated RNA in a reaction containing 200 U of SuperScript III RNase H-Reverse Transcriptase (Life Technologies), 300 ng of oligo-dT18, 50 ng of random hexamers, 50 mm Tris-HCl, pH 8.3, 75 mm KCl, 3 mm MgCl2, 500 μm deoxynucleotides triphosphate, 5 mm dithiothreitol, and 40 U of Protector RNase inhibitor (Roche Diagnostics) in a final volume of 50 μl. The reaction was incubated at 25°C for 10 min and then at 50°C for 1 h, and a PCR purification kit (Qiagen) was used to purify cDNA (Life Technologies). Equal amounts of cDNA were amplified in duplicate in a final volume of 10 μl containing 5 μl of 2× LightCycler 480 SYBRGreen I Master (Roche Diagnostics), 0.5 μm forward and reverse primers and 2 μl of cDNA (10 ng/μl). The primer pairs were as follows: IL-1a, 5′-acctgcaacaggaagtaaaatttga-3′ and 5′-actgaacctgaccgtacactcctcccgacgagtaggc-3′; IL-1b, 5′-actgaacctgaccgtacaaaatgccaccttttgacagtgat-3′ and 5′-cgtcaacttcaaagaacaggtcat-3′; Tox3, 5′-cctttcagactctcagcgatcc-3′ and 5′-ctggcggtactgtgacacttgt-3′; 18S, 5′-actgaacctgaccgtacacggtacagtgaaactgcgaatg-3′ and 5′-ccaaaggaaccataactgatttaatga-3′; and Gapdh, 5′-acgggaagctcactggcatgg-3′ and 5′-atgcctgcttcaccaccttcttg-3′. The sequences chosen were selected to match only the intended gene using the GeneTools software (BioTools), and verified by BLAST analysis in GenBank. Amplification was performed using the LightCycler 480 (Roche Diagnostics) and the following conditions: 2 min at 50°C, 4 min at 95°C, followed by 45 cycles of 10 s at 95°C (denaturation), 10 s at 60°C (annealing), 12 s at 72°C (elongation), and 5 s at 74°C (reading). Amplification efficiencies were validated and normalized to 18S or Gapdh and relative amounts of mRNA levels were calculated using the standard curve method.
Behavioral analysis.
Recovery of locomotor function after SCI was quantified in an open field using the Basso Mouse Scale (BMS), according to the method developed by Basso et al. (2006). All behavioral analyses were performed blind with respect to the identity of the animals.
Cell culture and transfection.
The human oligodendrocyte cell line MO3.13 was obtained from Dr. Nathalie Arbour (Centre hospitalier de l'Université de Montréal, Montréal, QC, Canada). Cells were maintained in DMEM without sodium pyruvate (Life Technologies) supplemented with 10% FBS, 1× penicillin-streptomycin (Pen-Strep) and l-glutamine (2 mm). The pCMV6 vector into which was cloned the open reading frame of the human Tox3 transcript variant 1 fused to C-terminal c-Myc and DDK tags was purchased from OriGene (catalog #RC218824, OriGene Technologies). Cells were plated 1 d before transfection at 30,000 cells/well (24-well plate) and transfected with 0.5 μg of DNA using Lipofectamine 3000 (Life Technologies), according to the manufacturer instructions. Cells were trypsinized 48 h post-transfection, and 750 μg/ml G418 (Geneticin, Life Technologies) was added to the media for selection. After 2 weeks of selection, cells were passaged and cultured in medium containing a reduced concentration of G418 (500 μg/ml).
Hypoxia and in vitro cytotoxic assay.
To study the effect of TOX3 overexpression on cell survival, MO3.13 and TOX3-transfected MO3.13 cells were cultured in hypoxic conditions, and cell viability was measured using the XTT colorimetric assay (Roche Diagnostics), according to the manufacturer instructions. In brief, cells were plated at a density of 25,000 cells/well in a 24-well plate 1 d before the experiment. For normoxic conditions, cells were maintained in DMEM with FBS, Pen-Strep, and l-glutamine, as previously described. For hypoxic conditions, cells were cultured in DMEM, without glucose, supplemented with FBS, Pen-Strep, and l-glutamine. Hypoxic culture was performed in a sealed anaerobic chamber under vacuum to remove oxygen (5% CO2). Normoxic and hypoxic conditions were maintained for 24 h, after which cells were incubated for another 4 h with the XTT reagent at culture conditions and absorbances measured at 490 and 690 nm (reference wavelength) using the Infinite 200 Pro multimode reader (Tecan Group Ltd.).
Immunoblotting.
For quantification of protein levels, cells were plated at a density of 10,000 cells/cm2 and grown for 2 d before protein extraction. Cells were then harvested, rinsed in PBS, and lysed for 30 min on ice in lysis buffer (20 mm HEPES, pH 7.9, 300 mm KCl, 10% Glycerol, 0.1% NP-40, and 1 mm DTT) containing a protease inhibitor cocktail (Sigma-Aldrich Canada Ltd.). The cell lysate was then centrifuged, and the supernatant was collected. Ten micrograms of proteins were boiled and electrophoresed on SDS-polyacrylamide gel, followed by blotting on PVDF membranes (PerkinElmer). Membranes were blocked for 1 h with 5% dry milk in TBST buffer (50 mm Tris-HCl pH 8.0, 150 mm NaCl, 0.05% Tween-20) and then incubated overnight in 5% milk/TBST solution containing the rabbit anti-TOX3 antibody (1:500 dilution; Novus Biologicals Canada ULC). A mouse monoclonal anti-GAPDH antibody (1:5000; Applied Biological Materials) was used as a protein loading control and internal standard. Membranes were incubated with secondary antibodies conjugated to horseradish peroxidase (1:2500; Vector Laboratories) and cross-reactive bands visualized by chemiluminescence (PerkinElmer).
Statistical analysis.
Statistical evaluations were performed with one- or two-way ANOVA or repeated-measures ANOVA. Post-ANOVA comparisons were made using the Bonferroni correction. A Student's t test was performed for the statistical analysis of g-ratios and cell viability assay. All statistical analyses were performed using the GraphPad Prism software. A p value of <0.05 was considered to be statistically significant.
Results
IL-1α protein expression in the injured spinal cord precedes IL-1β and is localized in microglia at the site of injury
Previous work has suggested that IL-1α released by cells undergoing necrosis could induce sterile inflammation (Chen et al., 2007; Cohen et al., 2010; Rider et al., 2011). We thus investigated whether IL-1α protein expression could be detected early in the injured mouse spinal cord by confocal IF microscopy. While no IL-1α expression was observed in the spinal cord of adult naive (uninjured) mice, IL-1α immunostaining became detectable in the cells of the spinal cord as early as 4 h post-SCI, with the presence of 38 ± 6 cells at the lesion epicenter (Fig. 1A,B). This number had decreased to 15 ± 1 cells by 24 h (Fig. 1A,B). At 4 h post-SCI, IL-1α+ cells were mainly found at the lesion epicenter and surrounding damaged areas. On very rare occasions, a few IL-1α+ cells were found proximal or distal to the lesion site, while at 24 h all IL-1α+ cells were physically contained within the contusion site. It should be noted that most of them had a ramified morphology reminiscent of microglia (Fig. 1C–E). No IL-1α+ cells were seen at 1 h or 3 d after injury (data not shown). In these experiments, immunostaining was performed using a polyclonal anti-IL-1α antibody directed against the N-terminal (amino acids 6–166) part of murine IL-1α, thus recognizing both the mature and precursor forms of the cytokine. This analysis revealed that the IL-1α protein was detected in the cell nucleus as well as in the cytoplasm (Fig. 1C–E).
Figure 1.
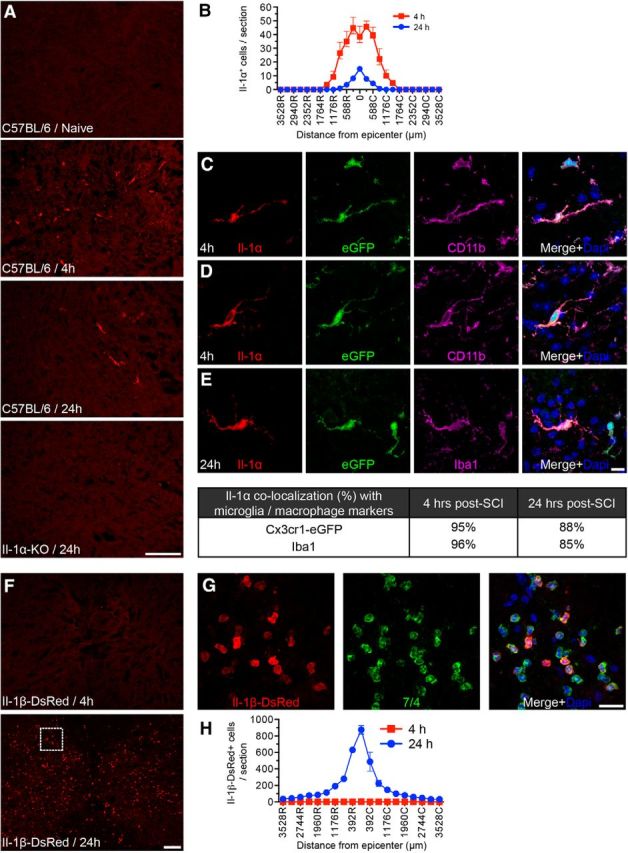
IL-1α protein expression in the injured spinal cord precedes IL-1β and is localized in microglia at the site of injury. A, Representative confocal photomicrographs showing IL-1α immunostaining (red) in the spinal cords of naive and injured C57BL/6 mice at 4 and 24 h post-SCI (n = 6–9 mice/time). Note that no IL-1α signal was detected in the injured spinal cords of IL-1α-KO mice, thus confirming antibody specificity. B, Quantification of IL-1α+ cells in spinal cord sections taken both rostral (R) and caudal (C) to the lesion epicenter at 4 and 24 h post-SCI. C–E, Confocal photomicrographs showing colocalization of the IL-1α protein (red) with the myeloid cell marker CD11b (C, D, purple) and the microglial/macrophage markers Iba1 (E, purple) and Cx3cr1-eGFP (eGFP, green cells in panels C–E). The nuclear staining with DAPI is shown in blue. Also shown is the percentage of IL-1α+ cells that express the microglia/macrophage markers Iba1 and Cx3cr1-eGFP. F, Imaging of IL-1β-producing cells (red cells) in the injured spinal cords of pIL-1b-DsRed transgenic mice at 4 and 24 h post-SCI. In these transgenic mice, the DsRed fluorescent reporter is expressed under the control of the IL-1b gene promoter. G, Confocal photomicrographs showing the colocalization of DsRed+ cells (red) with 7/4-expressing cells (green). H, Quantification of DsRed+ cells in spinal cord sections taken both rostral (R) and caudal (C) to the lesion epicenter at 4 and 24 h post-SCI (n = 3 mice/time). Scale bars: A, 100 μm; (in E) C–E, 10 μm; F, 100 μm; G, 20 μm.
Taking advantage of transgenic Cx3cr1-eGFP+/− mice in which the GFP reporter coding sequence is inserted in place of coding exons of the Cx3cr1 gene and is highly transcribed in microglia, we confirmed that the vast majority of IL-1α+ cells colocalized with GFP-expressing cells following SCI, with an average colocalization percentage of 95 ± 1% at 4 h (Fig. 1C–E). IL-1α+ cells also colocalized with the myeloid cell marker CD11b and the microglia/macrophage marker Iba1. At 4 h post-SCI, a time that precedes the entry of blood-derived macrophages, colocalization of the IL-1α protein with the Iba1 marker was estimated to be 96 ± 1%, indicating that microglia are the main source of IL-1α early after the SCI.
To examine IL-1β production, we took advantage of pIL-1b-DsRed transgenic mice in which the DsRed fluorescent protein gene is expressed under the control of the IL-1b gene promoter (pIL-1b). No DsRed expression was detected in the spinal cord of uninjured mice (data not shown). Time course experiments showed that DsRed marker expression was almost completely absent at 4 h, but increased thereafter to reach a peak at 24 h post-SCI (Fig. 1F). As for IL-1α+ cells, IL-1β-DsRed+ cells localized almost exclusively within the lesion site. However, the morphological characteristics of these cells differed drastically, as IL-1α+ cells were typically ramified and arborized, whereas IL-1β-DsRed+ cells were round. As seen in Figure 1G, DsRed+ cells colocalized with 7/4-expressing cells (i.e., neutrophils and monocyte-derived M1 macrophages), but only in rare occasions with Iba1-expressing microglia. Together, these results indicate that microglia located as sites of SCI rapidly (<4 h) respond to injury by expressing IL-1α, and that neutrophils and M1 monocytes that infiltrate the lesion site starting at 6 h further contribute to the IL-1 response by producing IL-1β.
Infiltration of neutrophils and proinflammatory M1 macrophages is equally reduced in the injured spinal cords of IL-1α- and IL-1β-KO mice
To determine to what extent these cytokines are responsible for the early recruitment of innate immune cells at sites of injury, we quantified the number of neutrophils and monocyte-derived macrophages expressing the 7/4 antigen, which is specifically expressed at the surface of both cell populations, but not in resident microglia (de Rivero Vaccari et al., 2012).
At 12 h after SCI, 1123 ± 72 cells immunopositive for 7/4 were counted at the lesion epicenter in wild-type (WT) mice (Fig. 2A). This number was reduced by 69%, 58%, and 79%, respectively, in IL-1α-KO, IL-1β-KO, and IL-1α/β-KO mice, with averages of 347 ± 79, 470 ± 45, and 237 ± 114 cells at the lesion epicenter. Similarly, the number of Ly6G+ neutrophils was reduced by ∼65%, 48% and 78%, respectively, in IL-1α-KO, IL-1β-KO, and IL-1α/β-KO mice compared with WT animals (Fig. 2B). To validate immunohistochemical data, cells were isolated from the spinal cord of contused mice at 12 h, and then characterized and quantified by flow cytometry based on their expression of the following cell-surface markers: CD45, CD11b, Ly6C, Ly6G, and F4/80. As shown in Figure 2C–E, flow cytometry data confirmed the significantly reduced presence of neutrophils (CD45hi, CD11b+, Ly6C+, Ly6G+, and F4/80−) and monocyte-derived M1 macrophages (CD45hi, CD11b+, Ly6Chi, Ly6G−, and F4/80+) in SCI mice lacking IL-1α and/or IL-1β. The number of microglia (CD45dim, CD11b+, Ly6C−, Ly6G−, and F4/80+) did not significantly differ between groups (data not shown).
Figure 2.

IL-1 deficiency reduces neutrophil and M1 monocyte infiltration in the injured spinal cord. A, B, Quantification of the number of neutrophils and proinflammatory M1 macrophages, as visualized by 7/4 (A) and Ly6G (B) immunostaining, at various rostral (R) and caudal (C) distances from the lesion epicenter at 12 h after SCI (n = 4–12 mice/group). C, D, Quantification of the proportions of M1 macrophages (C) and neutrophils (D) relative to total events in KO and WT mice (C57BL/6) at 12 h after SCI. E, Representative flow cytometry profiles showing the presence of neutrophils (black dots; CD45hi, CD11b+, Ly6C+, and Ly6G+) and monocyte-derived M1 macrophages (dark gray dots; CD45hi, CD11b+, Ly6Chi, and Ly6G−) in the spinal cords of a C57BL/6 and IL-1α/β-KO mouse at 12 h after SCI (date are representative of n = 4 mice). *,†,#p < 0.05; **,††,## p < 0.01; and ***,†††,###p < 0.001, significant difference between KO and WT mice. Two-way repeated-measures ANOVA with Bonferroni's post hoc test.
Altogether, these results suggest that cytokines of the IL-1 family are important regulators of the infiltration of innate immune cells of the granulomonocytic lineage after SCI.
Improved functional recovery and histopathological outcome in mice harboring deletion of the IL-1a gene after SCI
Neuroinflammation is considered to be one of the leading causes of secondary bystander damage in the injured spinal cord. Therefore, we next investigated whether the absence of the IL-1a or IL-1b gene affects neuropathology in SCI. Evaluation was performed in an open field using the 9-point BMS scale and the 11-point BMS subscore (Basso et al., 2006). Naive IL-1α-KO, IL-1β-KO, and WT mice all performed flawlessly, and received perfect scores on the BMS and BMS subscore scales. The situation was, however, different after a moderate (50 kdyn) traumatic SCI. As shown in Figure 3A, IL-1α-KO and IL-1β-KO mice recovered significantly better than WT mice at 1 and 3 d post-SCI. Unlike IL-1β-KO mice, IL-1α-KO mice performed significantly better than WT mice at 7 and 14 d as well. At day 14, >90% of IL-1α-KO mice were mostly coordinated and had parallel paw position at initial contact (BMS score, ≥7), compared with 0% and 10%, respectively, in WT and IL-1β-KO mice. The average BMS subscore of IL-1α-KO mice was 8.3 ± 0.4 compared with 5.4 ± 0.3 for WT mice and 4.7 ± 0.6 for IL-1β-KO mice at 14 d post-SCI (Fig. 3B). The extent and reproducibility of the early locomotor recovery seen at day 1 post-SCI in IL-1α-KO mice compared with the other two groups is best demonstrated by a dot plot showing individual BMS scores of all mice included in the experiments (n = 27 mice per group; Fig. 3C). Importantly, we found that deficiency in IL-1β attenuates the production of IL-1α, but not the opposite, in the normal and injured spinal cord (Fig. 4). This raises the possibility that some of the effects seen in IL-1β-KO mice compared with WT mice may be due to a reduction in IL-1α production.
Figure 3.

Recovery of locomotor function and spinal cord lesion volume are improved as a result of genetic or pharmacological IL-1α inhibition. A, B, Locomotor function was assessed using the BMS (A) and BMS subscore (B) over a 35 d period after SCI (n = 8–10 mice per group). C, Dot plot showing BMS scores assigned to individual mice of each mouse line at 1 d post-SCI [i.e., day postinjury (dpi)] across all experiments performed (n = 27 mice/group). Males are shown in black filled circles, and females in empty white circles. D, IL-1α-KO mice had reduced spinal cord tissue damage at 35 d after injury (n = 8–10 per group). E, F, Assessment of functional recovery using the BMS (E) and BMS subscore (F) showed that intrathecal, but not intravenous, anakinra treatment improves locomotion after SCI. Mice that received a single intrathecal infusion of the IL-1 receptor antagonist anakinra (33 mg/kg) had significantly higher BMS scores and subscores than those treated with anakinra intravenously (100 mg/kg) or PBS. Data are expressed as the mean ± SEM. *,†p < 0.05; **,††p < 0.01; and ***,†††p < 0.001. The ANOVA was performed using two-way repeated-measures ANOVA (A, B, E, F) or one-way ANOVA (C, D) followed by Bonferroni's post hoc test.
Figure 4.
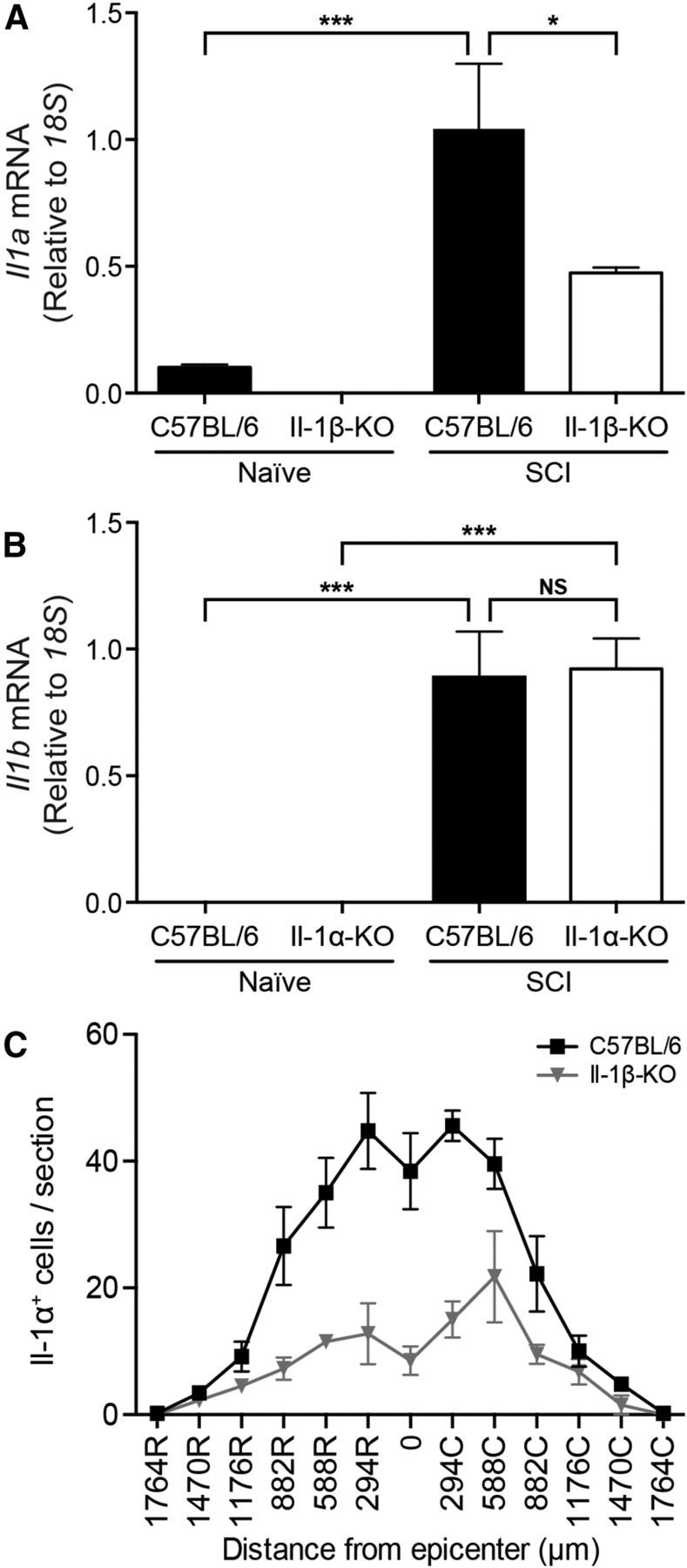
IL-1β-KO mice have reduced ll-1α levels in their spinal cords compared with WT mice, whereas IL-1α-KO mice express IL-1β normally. Analyses were performed on IL-1α and IL-1β single-knock-out animals to ascertain whether each of these genes could compensate for the loss of the other under both naive and injury conditions. A, B, qRT-PCR analysis shows decreased expression of IL-1a mRNA in IL-1β-KO mice, while no changes in IL-1b mRNA levels were detected in IL-1β-KO mice at day 1 post-SCI. The results were normalized to 18S mRNA levels. C, Quantification of IL-1α+ cells in spinal cord sections taken both rostral (R) and caudal (C) to the lesion epicenter at 4 h post-SCI in WT and IL-1β-KO mice. Data are expressed as the mean ± SEM. ***p < 0.001, *p < 0.05; one-way ANOVA with Bonferroni's post hoc test.
Strikingly, lesion volume was also reduced in IL-1α-KO mice compared with IL-1β-KO and WT mice at day 35 post-SCI, as judged by GFAP immunostaining, which delimits the lesion frontier (Fig. 3D). Together, these results suggest that deletion of the IL-1a gene contributes to better functional recovery by reducing lesion volume after SCI.
Intrathecal antagonism of IL-1 receptor promotes early locomotor recovery after SCI
We postulated that blockade of the signaling receptor for IL-1α, IL-1r1, early after SCI would improve recovery and locomotor function. We therefore compared the effectiveness of the most widely used inhibitor of IL-1 signaling, anakinra (a recombinant formulation of the natural IL-1r1 antagonist), which was administered through two different routes (intravenous vs intrathecal). Remarkably, a single intrathecal injection of anakinra given ∼15 min after the injury was effective at reducing behavioral dysfunction (Fig. 3E,F). In contrast, repeated intravenous injections of anakinra post-SCI in C57BL/6 mice failed to improve functional recovery. These data support our evidence that IL-1α is centrally produced and exerts its effects in the vicinity of the SCI lesion. Furthermore, the data strongly suggest that continuous intrathecal infusion or repeated intrathecal injections of anti-IL-1α agents could be an efficient way to prevent cell death and functional loss after CNS insult.
Increased expression of Tox3 during spinal cord development and after adult SCI in IL-1α-KO mice
To investigate the mechanism by which IL-1α mediates its detrimental effect after traumatic SCI, we analyzed the transcriptome of the injured spinal cord of IL-1α-KO, IL-1β-KO, and WT mice at 24 h after injury using GeneChip microarrays. This time point was selected because it corresponds to the earliest time at which functional recovery was detected in IL-1α-KO mice compared with the other mouse lines. As expected, transcripts from genes involved in neuroinflammation were found to be differentially regulated in the two IL-1-KO mouse lines compared with WT mice after SCI (data not shown). Surprisingly, this analysis also identified 18 genes differentially regulated in IL-1α-KO mice compared with IL-1β-KO and WT mice with no reported role in neuroinflammation (Table 1). Among those genes, the neuronal survival factor Tox3, a member of the high-mobility group box family of transcription factors that protects neurons from apoptotic cell death when overexpressed in vitro (Dittmer et al., 2011), figured prominently and was studied in greater detail.
Table 1.
Transcripts differentially regulated in the spinal cords of Il-1α-KO mice compared with Il-1β-KO and C57BL/6 mice after SCI
| Gene name | Gene symbol | C57BL/6 |
Il-Lα-KO |
Il-Lβ-KO |
|||||
|---|---|---|---|---|---|---|---|---|---|
| Mean | SD | Mean | SD | FC | Mean | SD | FC | ||
| Short coiled-coil protein | Scoc | 432.25 | 37.66 | 120.40 | 11.33 | −3.59 | 408.36 | 14.52 | −1.06 |
| WD repeat and FYVE domain containing 1 | Wdfy1 | 370.99 | 14.16 | 210.17 | 11.39 | −1.77 | 370.91 | 21.73 | −1.00 |
| Phosphatidylserine decarboxylase, pseudogene 1 | Pisd-ps1 | 883.29 | 122.76 | 556.31 | 39.28 | −1.59 | 754.47 | 79.99 | −1.17 |
| RIKEN cDNA 4933436C20 gene | 4933436C20Rik | 163.97 | 11.31 | 108.85 | 6.72 | −1.51 | 162.18 | 18.34 | −1.01 |
| ZNRD1 antisense RNA | Znrd1as | 167.69 | 5.37 | 112.35 | 26.74 | −1.49 | 190.08 | 14.97 | 1.13 |
| ELMO domain containing 2 | Elmod2 | 397.21 | 27.63 | 281.37 | 21.82 | −1.41 | 395.99 | 15.78 | −1.00 |
| tRNA methyltransferase 6 homolog (Saccharomyces cerevisiae) | Trmt6 | 342.95 | 17.67 | 244.66 | 18.48 | −1.40 | 331.38 | 33.56 | −1.03 |
| Methylthioribose-1-phosphate isomerase homolog (S. cerevisiae) | Mri1 | 343.01 | 24.14 | 258.44 | 6.92 | −1.33 | 358.50 | 21.88 | 1.05 |
| Growth arrest and DNA damage-inducible, γ interaction | Gadd45gip1 | 618.37 | 21.73 | 468.26 | 19.69 | −1.32 | 630.84 | 15.87 | 1.02 |
| G-protein-coupled receptor 34 | Gpr34 | 61.08 | 7.96 | 76.42 | 5.80 | 1.25 | 60.86 | 4.34 | −1.00 |
| Coenzyme Q9 homolog (yeast) | Coq9 | 791.47 | 54.76 | 1089.57 | 55.82 | 1.38 | 802.91 | 43.05 | 1.01 |
| Hydroxysteroid 11-β dehydrogenase 1 | Hsd11b1 | 189.43 | 7.38 | 261.01 | 21.76 | 1.38 | 211.42 | 17.90 | 1.12 |
| X-linked lymphocyte-regulated 3A | Xlr3a | 60.63 | 9.09 | 89.50 | 10.82 | 1.48 | 61.51 | 6.70 | 1.01 |
| X-linked lymphocyte-regulated 3C | Xlr3c | 51.28 | 4.89 | 76.46 | 10.82 | 1.49 | 54.50 | 4.49 | 1.06 |
| TOX high-mobility group box family member 3 | Tox3 | 277.95 | 10.26 | 419.95 | 32.27 | 1.51 | 294.00 | 13.58 | 1.06 |
| Ectonucleoside triphosphate diphosphohydrolase | Entpd4 | 1088.09 | 36.67 | 1770.74 | 37.56 | 1.63 | 1151.26 | 51.17 | 1.06 |
| X-linked lymphocyte-regulated 3B | Xlr3b | 42.24 | 3.92 | 70.96 | 12.43 | 1.68 | 46.81 | 5.98 | 1.11 |
| Predicted gene 10002 | Gm10002 | 23.45 | 0.45 | 102.69 | 8.83 | 4.38 | 23.35 | 1.24 | −1.00 |
Affymetrix GeneChip microarrays were used to generate gene expression profiles of Il-1α-KO, Il-1β-KO, and C57BL/6 mice at day 1 post-SCI. FC, Fold change compared to the C57BL/6 control group. All mice in this analysis received a spinal cord injury.
We first confirmed using quantitative real-time PCR (qRT-PCR) that Tox3 is significantly upregulated in IL-1α-KO mice compared with IL-1β-KO and WT mice at 24 h post-SCI, which is consistent with our microarray data (Fig. 5A,B). Using IF confocal microscopy, we observed that the Tox3 protein is weakly expressed in the spinal cord of WT mice under normal conditions. Nevertheless, costaining of Tox3 with various makers of CNS cells, such as the neuronal marker NeuN and the mature oligodendrocyte marker CC1, revealed that the Tox3 signal is mainly localized in the nucleus of neurons and oligodendrocytes (Fig. 5C,D). Tox3 staining was also associated with CD31+ endothelial cells, but the contribution of this cell population to the beneficial effect of IL-1α deficiency was ruled out by showing that endothelial cell survival and spinal microvascular perfusion are unaltered in IL-1α-KO mice after SCI (data not shown). Importantly, quantification of Tox3 immunostaining revealed that protein levels of the survival factor were markedly increased in the spinal cords of adult naive IL-1α-KO mice compared with those of IL-1β-KO and WT mice (Fig. 5E). The overexpression concerned mainly neurons and oligodendrocytes, with increases of ∼130% and ∼200% in signal intensity in these two cell types, respectively. This also translated into higher numbers of neurons and oligodendrocytes expressing Tox3 in the adult spinal cord of naive IL-1α-KO mice (Fig. 5F). On average, we counted 151 ± 15 NeuN+ Tox3+ cells per section in the gray matter of IL-1α-KO mice compared with 59 ± 8 and 74 ± 8 in IL-1β-KO and WT mice, respectively. The same also applied to oligodendrocytes, for which we counted 242 ± 8 CC1+ Tox3+ cells per section in the white matter of naive IL-1α-KO mice compared with 110 ± 16 and 98 ± 7 cells, respectively, in IL-1β-KO and WT mice. The total numbers of NeuN+ neurons and mature CC1+ oligodendrocytes were similar among the three mouse strains under normal, uninjured conditions (data not shown). Importantly, the deletion of IL-1 genes did not result in any differences in myelin thickness or axon caliber, as demonstrated by measurements of g-ratios in the spinal cord AST and RST (Fig. 5G–J). As expected, g-ratios were positively correlated with axon diameter in both IL-1α/β-KO and C57BL/6 mice (Fig. 5I,J). Axons in the AST of IL-1α/β-KO mice had an average g-ratio of 0.67 compared with 0.65 in C57BL/6 mice, while axons in the RST had g-ratios of 0.69 and 0.68, respectively, in IL-1α/β-KO and C57BL/6 mice. Together, these results suggest that the absence of IL-1α could mediate the protection of spinal cord neurons and/or oligodendrocytes by a mechanism distinct from its known inflammatory activities, potentially involving Tox3.
Figure 5.
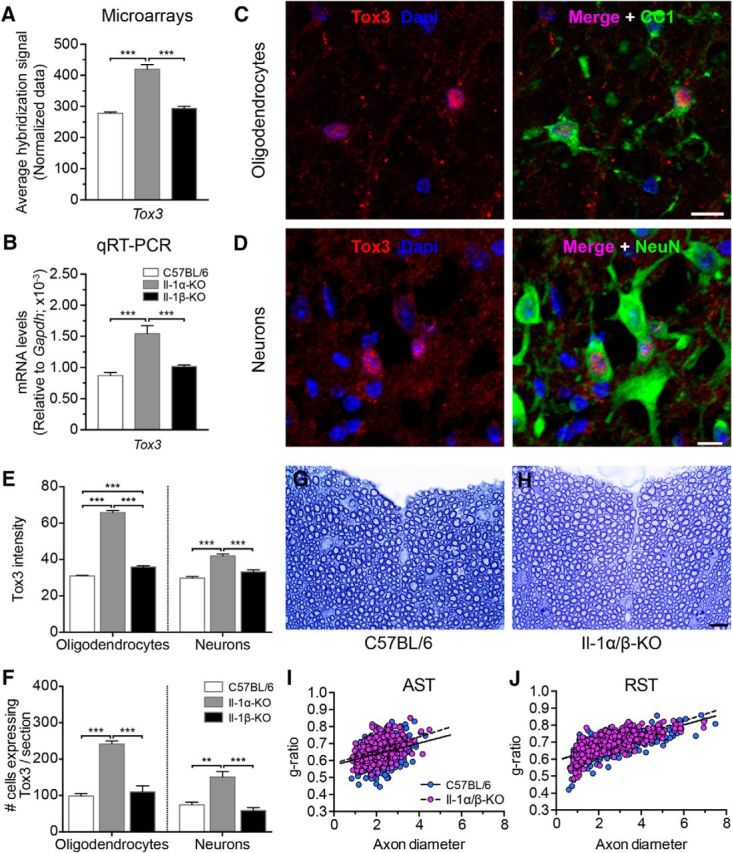
Expression of the survival factor Tox3 is increased in the injured spinal cord of IL-1α-KO mice. A, Microarray analysis of gene expression shows that Tox3 mRNA is differentially expressed (i.e., upregulated) in the spinal cords of IL-1α-KO mice compared with those of IL-1β-KO and C57BL/6 mice at day 1 post-SCI. Data are expressed as an average microarray hybridization signal (n = 4 mice per group). Normalization of probe set intensities was performed using the method of RMA. B, qRT-PCR analysis confirms the increased expression of Tox3 in the injured spinal cords of IL-1α-KO mice at day 1 post-SCI. Data are expressed as a ratio to Gapdh mRNA levels. C, D, Confocal photomicrographs showing Tox3 immunostaining (red) in the spinal cord of a naive C57BL/6 mouse. Note that the Tox3 protein is weakly expressed and localized specifically in the nucleus of oligodendrocytes (CC1+, green cells in the top right panel) and neurons (NeuN+, green cells in the bottom right panel) in adult C57BL/6 mice. The nuclear staining with DAPI is shown in blue, and the right-most panels are an overlay of the three colors. E, Quantification of Tox3 immunostaining intensity in spinal cord oligodendrocytes and neurons of adult naive mice from the three strains. F, Quantification of the total number of CC1+ oligodendrocytes and NeuN+ neurons expressing Tox3 in the spinal cord white and gray matter, respectively, of naive mice. G, H, Representative high-magnification photomicrographs showing toluidine blue-stained semithin sections prepared from the spinal cords of adult naive IL-1α/β-KO and C57BL/6 mice. The dorsal ascending sensory tract is shown in both images. I, J, Scatter plots showing g-ratios as a function of axon diameter for naive IL-1α/β-KO and C57BL/6 mice. G-ratios measurements were performed in the AST of the dorsal column (I) and lateral descending RST (J; 200 axons/mouse; n = 5 mice per group). All data are expressed as the mean ± SEM. ***p < 0.001, **p < 0.01, *p < 0.05, one-way ANOVA with Bonferroni's post hoc test. Scale bars: C, D, 10 μm; (in H) G, H, 10 μm.
To determine whether Tox3 overexpression occurs during a specific period of CNS development or later during adulthood, we performed immunostaining for Tox3, and measured protein levels in the developing spinal cords of naive IL-1α-KO, IL-1β-KO, and WT mice. As shown in Figure 6, Tox3 protein levels were increased in the spinal cords of IL-1α-KO mice compared with those of IL-1β-KO and WT mice during the postnatal period. Importantly, this overexpression translated into much higher numbers of CC1+ oligodendrocytes expressing Tox3 in the spinal cords of naive IL-1α-KO mice than in those of the other two groups of mice from P10 up to P30 (Fig. 6A,B). At P10, we counted as many as 102 ± 30 CC1+ Tox3+ oligodendrocytes per cross section in the spinal cord white matter of IL-1α-KO mice compared with only 4 ± 4 and 6 ± 4 oligodendrocytes per cross section, respectively, in IL-1β-KO and WT mice. This represents a ∼20-fold to 30-fold increase in the number of oligodendrocytes expressing Tox3 in response to deletion of the IL-1a gene. Interestingly, IL-1α-null spinal cords also contained increased the numbers of mature oligodendrocytes at P10 (Fig. 6C). However, the numbers of white matter CC1+ cells returned to control levels by P18. In contrast, the total number of NeuN+ neurons per cross section remained similar in all three mouse lines at all time points investigated postnatally, despite seeing an increased number of NeuN+ Tox3+ neurons in IL-1α-KO mice compared with IL-1β-KO and C57BL/6 mice at P10 and a trend toward significance at P18 (data not shown). Together, these results suggest that IL-1α represses Tox3 in spinal cord oligodendrocytes during early postnatal development, at ∼P10, which corresponds to the beginning of the myelination process in the mouse spinal cord.
Figure 6.

Tox3 overexpression in oligodendrocytes of IL-1α-KO mice occurs during spinal cord development. A, Confocal photomicrographs showing the presence of Tox3 immunostaining (red) in spinal cord white matter oligodendrocytes (CC1+ cells, green) of neonatal naive IL-1α-KO mice, but not C57BL/6 mice, at P10. The nuclear staining with DAPI is shown in blue. B, C, Quantification of the total numbers of CC1+ Tox3+ (B) and CC1+ (C) oligodendrocytes in the spinal cord white matter of IL-1α-KO, IL-1β-KO and C57BL/6 mice at P3, P10, P18, and P30. D, qRT-PCR analysis confirms the increased expression of IL-1a mRNA, and to a lower degree IL-1b mRNA, in the spinal cords of WT mice during the postnatal development period, at times that coincide with Tox3 overexpression in IL-1α-KO mice. qRT-PCR data are expressed as a ratio to Gapdh mRNA levels. ***p < 0.001, **p < 0.01, *p < 0.05, compared with both C57BL/6 and IL-1β-KO mice (B, C) or P1 mice (D), using two-way ANOVA with Bonferroni's post hoc test. Scale bar: A, 25 μm.
To confirm that IL-1α is normally produced during this developmental phase, we next measured mRNA levels for IL-1a and IL-1b in the spinal cords of C57BL/6 mice at various times postnatally (between P1 and P30) by qRT-PCR. mRNA levels for IL-1a, and to a much lower extent for IL-1b, were found to be robustly increased between P10 and P18 (Fig. 6D). Together, our findings indicate that deletion of the IL-1a gene promotes oligodendrocyte survival in the developing mouse spinal cord. It also raises the possibility that IL-1α inhibition may protect oligodendrocytes from secondary cell death after traumatic SCI in adult mice through the upregulation of Tox3.
Oligodendrocytes from mice lacking IL-1α upregulate the survival factor Tox3 and are protected from SCI-induced death
To investigate whether Tox3 overexpression persists in adult spinal cord neurons and oligodendrocytes following injury, we next performed dual immunostaining for Tox3 and neuron- or oligodendrocyte-specific markers (Fig. 7A). Intriguingly, Tox3 overexpression in IL-1α-KO mice was maintained in oligodendrocytes but was completely lost in neurons at 24 h after SCI, thus suggesting that the former cell type is more likely to be protected from injury. To determine whether more neurons and oligodendrocytes were protected from SCI in IL-1α-KO mice, and whether Tox3-expressing neurons/oligodendrocytes were more likely to be protected from injury-induced death, we counted these cells in spinal cord sections spanning the entire rostrocaudal extent of the lesion (i.e., ∼3 mm centered at the lesion epicenter). As expected, fewer NeuN+ neurons and CC1+ oligodendrocytes were detected in the injured compared with the uninjured spinal cord, independent of the mouse line (data not shown). On average, the neuronal and oligodendroglial cell loss in IL-1β-KO and WT (C57BL/6) mice was estimated to be ∼50% at 24 h post-SCI. Importantly, the total numbers of CC1+ oligodendrocytes, but not NeuN+ neurons, were significantly higher (by >20%) in the injured spinal cords of IL-1α-KO mice at this time (Fig. 7B). In IL-1α-KO mice, a total of 2656 ± 210 neurons and 3283 ± 209 oligodendrocytes survived the injury compared with, respectively, 2746 ± 186 and 2680 ± 135 in IL-1β-KO mice and 2759 ± 167 and 2707 ± 133 in WT mice. Another important observation is that 75% of the CC1+ oligodendrocytes that overexpress Tox3 survived the injury in IL-1α-KO mice (Fig. 7C), while the survival rate of CC1+ Tox3− oligodendrocytes was ∼40% in these same animals (Fig. 7C). CC1+ Tox3+ oligodendrocytes normally account for 35% of the total number of CC1+ oligodendrocytes in the uninjured spinal cords of IL-1α-KO mice. Importantly, the acute protection response of oligodendrocytes resulting from IL-1a gene deletion was maintained up to at least 14 d post-SCI, with a trend toward significance at day 35 (Fig. 7D). Collectively, these data suggest that Tox3 overexpression is protective in SCI, and that CC1+ Tox3+ oligodendrocytes have a greater capacity to survive the hypoxic, ischemic, and inflammatory conditions of the lesion.
Figure 7.
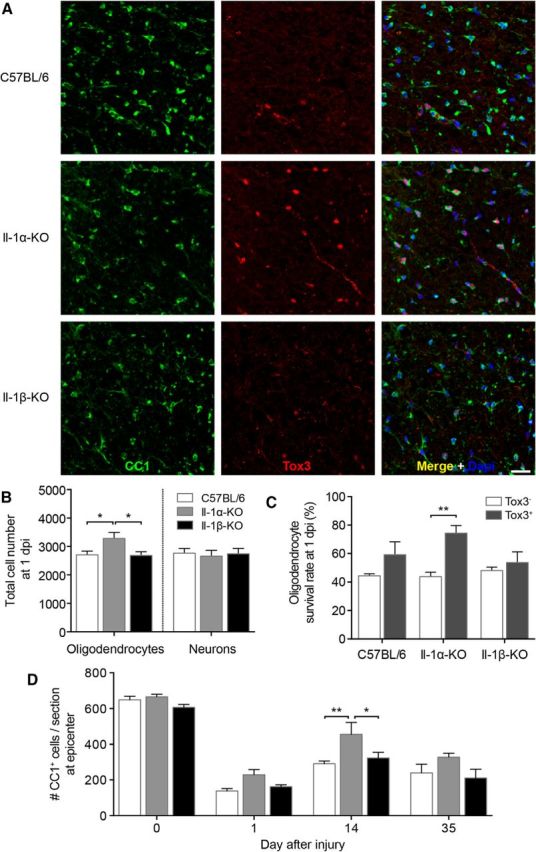
IL-1α deficiency upregulates expression of the survival factor Tox3 in oligodendrocytes and protects these cells from death after SCI. A, Representative confocal photomicrographs showing CC1 (green) and Tox3 (red) immunostaining in the injured spinal cords of C57BL/6 mice as well as IL-1α- and IL-1β-KO mice at 1 d post-injury (dpi). B, Quantification of the total number of CC1+ oligodendrocytes and NeuN+ neurons in spinal cord sections spanning the entire rostrocaudal extent of the lesion (i.e., ∼3 mm centered at the lesion epicenter) at 1 dpi. C, Percentage of spinal cord white matter CC1+ oligodendrocytes that survived the injury in relation to their expression of Tox3 for the three strains at 1 dpi. D, Quantification of the total number of CC1+ oligodendrocytes in the white matter of the spinal cord cross section located at the lesion epicenter for all three mouse lines at 1, 14, and 35 dpi. Data in B–D are expressed as the mean ± SEM. ***p < 0.001, **p < 0.01, *p < 0.05. The ANOVA was performed using a one-way ANOVA (B, C) or two-way ANOVA (D) followed by Bonferroni's post hoc test. Scale bar, 10 μm.
TOX3 overexpression protects human oligodendrocytes from hypoxia-induced apoptotic cell death
Oligodendrocytes are essential to axon myelination as well as neuronal support in the CNS (Oluich et al., 2012). After SCI, a considerable number of oligodendrocytes located close to the trauma are lost through apoptosis, in part due to the hypoxic-ischemic conditions (McTigue and Tripathi, 2008). The consequences of that cell death are the demyelination of multiple axons accompanied by the loss of axonal conduction, thus resulting in severe functional impairments in the acute phase of SCI. To further explore the role of Tox3 in oligodendrocyte survival, we next examined the effect of TOX3 overexpression in the human oligodendroglial cell line MO3.13. First, we established that human MO3.13 oligodendrocytes constitutively express the endogenous TOX3 protein (67.5 kDa; Fig. 8A), and are therefore endowed with the necessary machinery to respond to this potential protective factor. TOX3 was then overexpressed in MO3.13 cells by transfection of a vector encoding human TOX3 fused to a Myc-DDK tag. Immunoblotting using the anti-TOX3 antibody confirmed the overexpression of tagged-TOX3 (98 kDa) in the cell line and the unchanged expression of endogenous TOX3 (Fig. 8A,B). The molecular weights of the endogenous and tagged forms of the TOX3 protein are in agreement with those reported by Dittmer et al. (2011). The oligodendrocyte cultures were next subjected to 24 h of hypoxia, and cell viability was evaluated using the XTT assay. Importantly, MO3.13 oligodendrocytes overexpressing TOX3 survived in greater numbers (63 ± 4%) than either GFP-transfected cells (44 ± 5%) or untransfected cells (41 ± 2%; Fig. 8C), thus again supporting the evidence for a protective effect of TOX3 overexpression against cell death.
Figure 8.

TOX3 overexpression protects human oligodendrocytes from hypoxia-induced cell death. A, Immunoblotting using the anti-TOX3 antibody confirmed the presence of endogenous TOX3 (67.5 kDa) in untransfected and transfected human oligodendrocytes (MO3.13 cell line), as well as overexpression of TOX3-Myc-DDK (98 kDa) in stably transfected cells only. B, Quantification of the relative density of endogenous and tagged TOX3 in untransfected and transfected MO3.13 cells. C, Oligodendrocyte cultures were subjected to 24 h of hypoxia, and cell viability was quantified using the XTT assay. Note that MO3.13 oligodendrocytes overexpressing TOX3 survived in greater numbers than untransfected cells. Data are expressed as the mean ± SEM. **p < 0.01, *p < 0.05, compared with untransfected cells using a Student's t test.
Taken all together, our results indicate that IL-1α blockade protects oligodendrocytes against cell death after injury through a mechanism involving overexpression of the nuclear survival factor Tox3.
Discussion
In this study, we show that deletion of the IL-1a gene in mice results in decreased lesion volume and improved functional outcome after SCI, an effect that we ascribe to an increased survival of oligodendrocytes in damaged areas. Our data indicate that IL-1α is rapidly produced by resident microglia after SCI, which is in line with a recent report by Luheshi et al. (2011) using an ischemic brain injury model in mice. Specifically, we found that IL-1α is produced and/or released at sites of trauma, and that IL-1α production by microglia precedes both the infiltration of blood-derived innate immune cells, which we found are the main cellular source of IL-1β, and secondary damage to oligodendrocytes. Importantly, the infiltration of neutrophils and monocyte-derived macrophages is severely compromised in the injured spinal cords of IL-1α-KO mice. Our results thus support the growing body of evidence that IL-1α is a damage-associated molecular pattern molecule released by stressed or dying cells, thus enabling the recruitment of innate immune cells (Chen et al., 2007; Eigenbrod et al., 2008). However, the loss of oligodendrocytes at sites of SCI appears to be independent of the infiltration of neutrophils and macrophages, as the recruitment of these two cell populations was equally reduced in both IL-1α- and IL-1β-KO mice despite the fact that IL-1α-null mutants exhibited better locomotor recovery. Another key new finding of this study is the discovery that the survival factor Tox3 is overexpressed in oligodendrocytes as a result of deletion of the IL-1a gene, and that this overexpression occurs during the early postnatal period and lasts for the lifetime of the IL-1α-KO mouse. Furthermore, oligodendrocytes overexpressing Tox3 in the spinal cords of IL-1α-KO mice survived better than those not expressing Tox3 (in these same animals), as well as in spinal cords of IL-1β-KO and WT mice after SCI in vivo. Accordingly, in vitro data suggest that overexpression of the TOX3 gene in human oligodendrocytes enhances cell survival under hypoxic conditions.
Cohen et al. (2010) have recently reported that IL-1α associates with the chromatin and is released with the cytoplasmic content as a result of necrotic cell death, but not apoptotic cell death, following which it stimulates myeloid cell recruitment. Although our results indicate that IL-1α is released by microglia and initiates inflammation after SCI, we do not know whether this inflammatory response mediates secondary (bystander) damage. Interestingly, Allen et al. (2012) have recently shown that neutrophils acquire potentially neurotoxic properties upon transmigration across the IL-1-stimulated blood–brain barrier. This particular study suggested that neurotoxicity could be mediated by the action of neutrophil-derived proteases that are released in association with decondensed DNA, referred to as neutrophil extracellular traps. In addition, monocyte-derived M1 macrophages identified by their production of proinflammatory mediators are present in the injured spinal cord and are neurotoxic when cocultured with primary neurons (Kigerl et al., 2009). A recent cell depletion study in SCI mice has further revealed that preventing the recruitment of both neutrophils and monocytes is beneficial for functional recovery (Lee et al., 2011). The evidence provided here, however, suggests that microglia rather than neutrophils and M1 macrophages are involved in the death of oligodendrocytes. Future studies will therefore be required to determine whether microglia or other immune cells that rapidly infiltrate sites of SCI in response to IL-1α are those that contribute to secondary tissue damage.
Pro-IL-1α and pro-IL-1β are expressed as 31 kDa polypeptides. Despite long-standing evidence that the precursor form of IL-1α is biologically active, in contrast to pro-IL-1β, recent studies (Afonina et al., 2011; Zheng et al., 2013) have challenged this view by showing that pro-IL-1α has minimal activity, but its cleavage into a 17 kDa fragment by proteases such as calpain or granzyme B enhanced biological activity by up to 10-fold to 50-fold in vitro and in vivo. The processing and secretion of IL-1β is regulated by inflammasomes (Lamkanfi and Dixit, 2012), and we now know that this is also partially the case for IL-1α, at least in the context of exposition to microbial pathogen-associated molecular pattern molecules (Gross et al., 2012). Here, we show that IL-1α is rapidly (within 4 h) produced by resident microglia after SCI, and that innate immune cells that are subsequently recruited from the blood into sites of injury further amplify inflammation by producing IL-1β. It remains unknown whether the effects of IL-1α on inflammation and secondary degeneration are mediated by the proform or mature form of the cytokine. Assuming the latter, it would of great interest to identify the mechanism involved in IL-1α maturation in the context of CNS injury, especially since inhibitors of specific inflammasomes, caspase-1 and calpain, are currently being developed and tested in the clinic for other indications/disorders (Pietsch et al., 2010; Dinarello, 2011; López-Castejón and Pelegrín, 2012).
It is important to keep in mind that the IL-1β reporter mouse line has some limitations despite being a powerful tool with which to study IL-1β protein expression in vivo. One of these limitations is the slightly delayed kinetics of DsRed expression compared with IL-1β protein expression, with a lag time reported to be between 3 and 12 h, depending on the model (Matsushima et al., 2010). Another limitation is that the assessment of IL-1β protein production through the visualization of DsRed can only be performed during the induction phase of inflammation, because a certain level of fluorescence must be reached for cell detection and because the half-life and clearance of the two molecules could slightly differ. This and the fact that different species were used (rats vs mice) may explain why a previous study (de Rivero Vaccari et al., 2008) has shown that neurons are the earliest source of IL-1β, whereas we failed to detect DsRed expression (i.e., IL-1β promoter activation) in these cells after SCI.
The most important conclusion that can be drawn from the comparison of IL-1α-KO and IL-1β-KO mice is that, despite showing a similarly reduced inflammatory response after SCI, IL-1α-KO mice exhibited better recovery of locomotor abilities and had reduced lesion volume. This, combined with the early functional recovery seen at day 1 in IL-1α-KO mice, suggests that these mutant animals are receiving protection from SCI-induced neurodegeneration rather than benefiting from improved regenerative abilities. It also suggests that the improved functional recovery in injured IL-1α-KO mice is unlikely to be mediated by innate immune cells recruited from the blood. Still, we cannot completely rule out the possibility that the absence of IL-1α or IL-1β could have differentially affected the expression of specific proinflammatory and cytotoxic effectors by the remaining immune cells recruited at the lesion site. However, it must be pointed out that our microarray study failed to detect significant changes in expression levels of inflammatory genes between the two IL-1 mouse strains at day 1 post-SCI.
We found that a single intrathecal, but not intravenous, injection of anakinra administered immediately after SCI improved locomotor recovery during the first week post-treatment. This suggests that the drug either does not cross the blood–spinal cord barrier after SCI, or does so only weakly. Galea et al. (2011) have reported a slow passive penetration of anakinra into the CNS and a large variability in concentrations measured in CSF when injected intravenously at experimentally therapeutic concentrations. They estimated that only 1.6 ± 0.4% of intravenously administered anakinra crossed into the CSF of patients with subarachnoid hemorrhage. Anakinra levels were found to decrease rapidly in plasma following the cessation of intravenous treatment (half-life, 33 ± 9 min). In contrast, anakinra was more stable in the CSF, with an estimated half-life of 238 min (Galea et al., 2011). Similarly, a rat imaging study reported a low uptake of radiolabeled IL-1ra in the normal brain because of the rapid metabolism and excretion of anakinra (Cawthorne et al., 2011). As our data imply, administering anakinra directly into the CSF, which irrigates the spinal cord parenchyma, rather than systemically, may prove to be more efficient therapeutically. Intrathecal drug delivery in patients with SCI is an achievable goal, as demonstrated by the anti-Nogo clinical trial (Zorner and Schwab, 2010). Another alternative for achieving experimentally therapeutic CSF concentrations might be to systemically inject a high loading dose followed by maintenance therapy. A randomized phase II study conducted in acute stroke patients showed that an intravenous bolus dose of 100 mg of recombinant human IL-1RA followed by continuous intravenous infusion at 2 mg/kg/h over 72 h is safe and exhibited improved clinical outcomes (Emsley et al., 2005). Whether a similar intravenous regimen in SCI mice would result in superior outcomes compared with the intrathecal route remains to be investigated.
In contrast to the function of IL-1α, which remains uncertain, the role of IL-1β has been relatively well studied in the context of CNS injury (Allan and Rothwell, 2001; Allan et al., 2005). It has been proposed that IL-1 proteins have identical biological functions, given that both cytokines bind and activate the same cell surface receptor, IL-1r1, although a role for IL-1r2 in the regulation of IL-1α activity after necrosis has recently been highlighted (Zheng et al., 2013). Thus, it may come as a surprise that IL-1β-KO mice did not recover locomotor function to the same extent as IL-1α-KO mice after SCI. This could suggest that IL-1α mediates CNS cell loss independently of IL-1r1, through an as yet unidentified receptor. Interestingly, a novel isoform of the IL-1r1 accessory protein, IL-1racpb, containing a variant TIR domain whose expression is restricted to CNS neurons, has recently been implicated in neuroprotection in animal models of neuroinflammation (Smith et al., 2009). Nguyen et al. (2011) later reported that IL-1α-induced, but not IL-1β-induced, p38 phosphorylation is significantly reduced in primary neuronal cultures from IL-1racpb-KO mice. This is convincing evidence that supports the idea that IL-1α has specific effects within the CNS. Here, we have extended this work by identifying 18 genes that are regulated by IL-1α, but not by IL-1β, and through the demonstration that IL-1α regulates oligodendrocyte survival after SCI. It is important to note that the study by Smith et al. (2009) assessed IL-1racpb mRNA expression in purified neurons, microglia, and astrocytes, but not in oligodendrocytes. It is therefore possible that IL-1α may exert its effects on oligodendrocytes through a receptor other than IL-1r1, such as IL-1racpb.
Our genome-wide transcriptome analysis identified 18 genes as significantly upregulated or downregulated in IL-1α-KO mice compared with IL-1β-KO and WT mice, among which is the transcript coding for Tox3. Tox3 was recently shown to regulate Ca2+-dependent transcription in neurons through interaction with the cAMP response element-binding protein (Yuan et al., 2009). Dittmer et al. (2011) have since established that Tox3 is predominantly expressed in the CNS, where it acts as a neuronal survival factor. It was demonstrated that Tox3 overexpression in vitro protects neurons from cell death via the upregulation of antiapoptotic genes and the downregulation of proapoptotic genes. Here, our data show the presence of almost three times as many neurons and oligodendrocytes expressing Tox3 in the spinal cords of naive IL-1α-KO mice compared with IL-1β-KO and WT mice, but this difference did not translate into an increased neuronal survival in IL-1α-KO mice at day 1 post-SCI. Instead, deletion of the IL-1a gene distinctively induced the survival of mature oligodendrocytes after SCI. Recently, Miron et al. (2013) unveiled the idea that alternatively activated M2 microglia/macrophages are important for oligodendrocyte differentiation and CNS remyelination. It will be of interest in future work to determine whether the absence of IL-1α could induce a phenotypic switch in microglial polarization during the early developmental period, thus leading to gene expression changes in oligodendrocytes.
In summary, we have established that deletion of the IL-1a gene provides protection of oligodendrocytes from SCI via a mechanism that involves overexpression of the survival factor Tox3. Therefore, we propose that central inhibition of IL-1α or overexpression of Tox3 during the early acute phase of a CNS insult may be an effective means for preventing the loss of neurological function in SCI, and may be extended to a variety of other acute injuries such as ischemia and traumatic brain injuries.
Footnotes
This study was supported by grants from Wings for Life Spinal Cord Research Foundation (Project no. 48, Contract no. WFL-CA-006/11) and the Canadian Institutes of Health Research (Grant MOP-84558) to S.L. Salary support for D.B., Y.D., and S.L. was provided by the Fonds de la Recherche en Santé du Québec. We thank Nadia Fortin, Kanchan Bisht, and Samuel Boisjoly-Villeneuve for their invaluable technical assistance; and Dr. David Gosselin for his critical review of this manuscript.
References
- Afonina IS, Tynan GA, Logue SE, Cullen SP, Bots M, Lüthi AU, Reeves EP, McElvaney NG, Medema JP, Lavelle EC, Martin SJ. Granzyme B-dependent proteolysis acts as a switch to enhance the proinflammatory activity of IL-1alpha. Mol Cell. 2011;44:265–278. doi: 10.1016/j.molcel.2011.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albrecht PJ, Dahl JP, Stoltzfus OK, Levenson R, Levison SW. Ciliary neurotrophic factor activates spinal cord astrocytes, stimulating their production and release of fibroblast growth factor-2, to increase motor neuron survival. Exp Neurol. 2002;173:46–62. doi: 10.1006/exnr.2001.7834. [DOI] [PubMed] [Google Scholar]
- Albrecht PJ, Murtie JC, Ness JK, Redwine JM, Enterline JR, Armstrong RC, Levison SW. Astrocytes produce CNTF during the remyelination phase of viral-induced spinal cord demyelination to stimulate FGF-2 production. Neurobiol Dis. 2003;13:89–101. doi: 10.1016/S0969-9961(03)00019-6. [DOI] [PubMed] [Google Scholar]
- Allan SM, Rothwell NJ. Cytokines and acute neurodegeneration. Nat Rev Neurosci. 2001;2:734–744. doi: 10.1038/35094583. [DOI] [PubMed] [Google Scholar]
- Allan SM, Tyrrell PJ, Rothwell NJ. Interleukin-1 and neuronal injury. Nat Rev Immunol. 2005;5:629–640. doi: 10.1038/nri1664. [DOI] [PubMed] [Google Scholar]
- Allen C, Thornton P, Denes A, McColl BW, Pierozynski A, Monestier M, Pinteaux E, Rothwell NJ, Allan SM. Neutrophil cerebrovascular transmigration triggers rapid neurotoxicity through release of proteases associated with decondensed DNA. J Immunol. 2012;189:381–392. doi: 10.4049/jimmunol.1200409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amankulor NM, Hambardzumyan D, Pyonteck SM, Becher OJ, Joyce JA, Holland EC. Sonic hedgehog pathway activation is induced by acute brain injury and regulated by injury-related inflammation. J Neurosci. 2009;29:10299–10308. doi: 10.1523/JNEUROSCI.2500-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrette B, Hébert MA, Filali M, Lafortune K, Vallières N, Gowing G, Julien JP, Lacroix S. Requirement of myeloid cells for axon regeneration. J Neurosci. 2008;28:9363–9376. doi: 10.1523/JNEUROSCI.1447-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso DM, Fisher LC, Anderson AJ, Jakeman LB, McTigue DM, Popovich PG. Basso Mouse Scale for locomotion detects differences in recovery after spinal cord injury in five common mouse strains. J Neurotrauma. 2006;23:635–659. doi: 10.1089/neu.2006.23.635. [DOI] [PubMed] [Google Scholar]
- Bastien D, Lacroix S. Cytokine pathways regulating glial and leukocyte function after spinal cord and peripheral nerve injury. Exp Neurol. 2014;258:62–77. doi: 10.1016/j.expneurol.2014.04.006. [DOI] [PubMed] [Google Scholar]
- Basu A, Krady JK, O'Malley M, Styren SD, DeKosky ST, Levison SW. The type 1 interleukin-1 receptor is essential for the efficient activation of microglia and the induction of multiple proinflammatory mediators in response to brain injury. J Neurosci. 2002;22:6071–6082. doi: 10.1523/JNEUROSCI.22-14-06071.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brambilla R, Bracchi-Ricard V, Hu WH, Frydel B, Bramwell A, Karmally S, Green EJ, Bethea JR. Inhibition of astroglial nuclear factor kappaB reduces inflammation and improves functional recovery after spinal cord injury. J Exp Med. 2005;202:145–156. doi: 10.1084/jem.20041918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brambilla R, Hurtado A, Persaud T, Esham K, Pearse DD, Oudega M, Bethea JR. Transgenic inhibition of astroglial NF-kappaB leads to increased axonal sparing and sprouting following spinal cord injury. J Neurochem. 2009;110:765–778. doi: 10.1111/j.1471-4159.2009.06190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cawthorne C, Prenant C, Smigova A, Julyan P, Maroy R, Herholz K, Rothwell N, Boutin H. Biodistribution, pharmacokinetics and metabolism of interleukin-1 receptor antagonist (IL-1RA) using [(1)(8)F]-IL1RA and PET imaging in rats. Br J Pharmacol. 2011;162:659–672. doi: 10.1111/j.1476-5381.2010.01068.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CJ, Kono H, Golenbock D, Reed G, Akira S, Rock KL. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat Med. 2007;13:851–856. doi: 10.1038/nm1603. [DOI] [PubMed] [Google Scholar]
- Cohen I, Rider P, Carmi Y, Braiman A, Dotan S, White MR, Voronov E, Martin MU, Dinarello CA, Apte RN. Differential release of chromatin-bound IL-1alpha discriminates between necrotic and apoptotic cell death by the ability to induce sterile inflammation. Proc Natl Acad Sci U S A. 2010;107:2574–2579. doi: 10.1073/pnas.0915018107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan WB. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8:752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- David S, Kroner A. Repertoire of microglial and macrophage responses after spinal cord injury. Nat Rev Neurosci. 2011;12:388–399. doi: 10.1038/nrn3053. [DOI] [PubMed] [Google Scholar]
- David S, Lacroix S. Role of the immune response in tissue damage and repair in the injured spinal cord. In: Antel J, Birnbaum G, Hartung HP, Vincent A, editors. Clinical neuroimmunology. Ed 2. New York: Oxford UP; 2005. pp. 53–63. [Google Scholar]
- de Rivero Vaccari JP, Lotocki G, Marcillo AE, Dietrich WD, Keane RW. A molecular platform in neurons regulates inflammation after spinal cord injury. J Neurosci. 2008;28:3404–3414. doi: 10.1523/JNEUROSCI.0157-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Rivero Vaccari JP, Bastien D, Yurcisin G, Pineau I, Dietrich WD, De Koninck Y, Keane RW, Lacroix S. P2X4 receptors influence inflammasome activation following spinal cord injury. J Neurosci. 2012;32:3058–3066. doi: 10.1523/JNEUROSCI.4930-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinarello CA. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood. 2011;117:3720–3732. doi: 10.1182/blood-2010-07-273417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittmer S, Kovacs Z, Yuan SH, Siszler G, Kögl M, Summer H, Geerts A, Golz S, Shioda T, Methner A. TOX3 is a neuronal survival factor that induces transcription depending on the presence of CITED1 or phosphorylated CREB in the transcriptionally active complex. J Cell Sci. 2011;124:252–260. doi: 10.1242/jcs.068759. [DOI] [PubMed] [Google Scholar]
- Eigenbrod T, Park JH, Harder J, Iwakura Y, Núñez G. Cutting edge: critical role for mesothelial cells in necrosis-induced inflammation through the recognition of IL-1 alpha released from dying cells. J Immunol. 2008;181:8194–8198. doi: 10.4049/jimmunol.181.12.8194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley HC, Smith CJ, Georgiou RF, Vail A, Hopkins SJ, Rothwell NJ, Tyrrell PJ. A randomised phase II study of interleukin-1 receptor antagonist in acute stroke patients. J Neurol Neurosurg Psychiatry. 2005;76:1366–1372. doi: 10.1136/jnnp.2004.054882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faulkner JR, Herrmann JE, Woo MJ, Tansey KE, Doan NB, Sofroniew MV. Reactive astrocytes protect tissue and preserve function after spinal cord injury. J Neurosci. 2004;24:2143–2155. doi: 10.1523/JNEUROSCI.3547-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming JC, Norenberg MD, Ramsay DA, Dekaban GA, Marcillo AE, Saenz AD, Pasquale-Styles M, Dietrich WD, Weaver LC. The cellular inflammatory response in human spinal cords after injury. Brain. 2006;129:3249–3269. doi: 10.1093/brain/awl296. [DOI] [PubMed] [Google Scholar]
- Galea J, Ogungbenro K, Hulme S, Greenhalgh A, Aarons L, Scarth S, Hutchinson P, Grainger S, King A, Hopkins SJ, Rothwell N, Tyrrell P. Intravenous anakinra can achieve experimentally effective concentrations in the central nervous system within a therapeutic time window: results of a dose-ranging study. J Cereb Blood Flow Metab. 2011;31:439–447. doi: 10.1038/jcbfm.2010.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier L, Cope L, Bolstad BM, Irizarry RA. affy–analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004;20:307–315. doi: 10.1093/bioinformatics/btg405. [DOI] [PubMed] [Google Scholar]
- Gross O, Yazdi AS, Thomas CJ, Masin M, Heinz LX, Guarda G, Quadroni M, Drexler SK, Tschopp J. Inflammasome activators induce interleukin-1alpha secretion via distinct pathways with differential requirement for the protease function of caspase-1. Immunity. 2012;36:388–400. doi: 10.1016/j.immuni.2012.01.018. [DOI] [PubMed] [Google Scholar]
- Herx LM, Rivest S, Yong VW. Central nervous system-initiated inflammation and neurotrophism in trauma: IL-1 beta is required for the production of ciliary neurotrophic factor. J Immunol. 2000;165:2232–2239. doi: 10.4049/jimmunol.165.4.2232. [DOI] [PubMed] [Google Scholar]
- Horai R, Asano M, Sudo K, Kanuka H, Suzuki M, Nishihara M, Takahashi M, Iwakura Y. Production of mice deficient in genes for interleukin (IL)-1alpha, IL-1beta, IL-1alpha/beta, and IL-1 receptor antagonist shows that IL-1beta is crucial in turpentine-induced fever development and glucocorticoid secretion. J Exp Med. 1998;187:1463–1475. doi: 10.1084/jem.187.9.1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci. 2009;29:13435–13444. doi: 10.1523/JNEUROSCI.3257-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kono H, Karmarkar D, Iwakura Y, Rock KL. Identification of the cellular sensor that stimulates the inflammatory response to sterile cell death. J Immunol. 2010;184:4470–4478. doi: 10.4049/jimmunol.0902485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamkanfi M, Dixit VM. Inflammasomes and their roles in health and disease. Annu Rev Cell Dev Biol. 2012;28:137–161. doi: 10.1146/annurev-cellbio-101011-155745. [DOI] [PubMed] [Google Scholar]
- Lee SM, Rosen S, Weinstein P, van Rooijen N, Noble-Haeusslein LJ. Prevention of both neutrophil and monocyte recruitment promotes recovery after spinal cord injury. J Neurotrauma. 2011;28:1893–1907. doi: 10.1089/neu.2011.1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Castejón G, Pelegrín P. Current status of inflammasome blockers as anti-inflammatory drugs. Expert Opin Investig Drugs. 2012;21:995–1007. doi: 10.1517/13543784.2012.690032. [DOI] [PubMed] [Google Scholar]
- Luheshi NM, Kovács KJ, Lopez-Castejon G, Brough D, Denes A. Interleukin-1alpha expression precedes IL-1beta after ischemic brain injury and is localised to areas of focal neuronal loss and penumbral tissues. J Neuroinflammation. 2011;8:186. doi: 10.1186/1742-2094-8-186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason JL, Suzuki K, Chaplin DD, Matsushima GK. Interleukin-1beta promotes repair of the CNS. J Neurosci. 2001;21:7046–7052. doi: 10.1523/JNEUROSCI.21-18-07046.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushima H, Ogawa Y, Miyazaki T, Tanaka H, Nishibu A, Takashima A. Intravital imaging of IL-1beta production in skin. J Invest Dermatol. 2010;130:1571–1580. doi: 10.1038/jid.2010.11. [DOI] [PubMed] [Google Scholar]
- McTigue DM, Tripathi RB. The life, death, and replacement of oligodendrocytes in the adult CNS. J Neurochem. 2008;107:1–19. doi: 10.1111/j.1471-4159.2008.05570.x. [DOI] [PubMed] [Google Scholar]
- Miron VE, Boyd A, Zhao JW, Yuen TJ, Ruckh JM, Shadrach JL, van Wijngaarden P, Wagers AJ, Williams A, Franklin RJ, ffrench-Constant C. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat Neurosci. 2013;16:1211–1218. doi: 10.1038/nn.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadeau S, Filali M, Zhang J, Kerr BJ, Rivest S, Soulet D, Iwakura Y, de Rivero Vaccari JP, Keane RW, Lacroix S. Functional recovery after peripheral nerve injury is dependent on the pro-inflammatory cytokines IL-1β and TNF: implications for neuropathic pain. J Neurosci. 2011;31:12533–12542. doi: 10.1523/JNEUROSCI.2840-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen L, Rothwell NJ, Pinteaux E, Boutin H. Contribution of interleukin-1 receptor accessory protein B to interleukin-1 actions in neuronal cells. Neurosignals. 2011;19:222–230. doi: 10.1159/000330803. [DOI] [PubMed] [Google Scholar]
- Oluich LJ, Stratton JA, Xing YL, Ng SW, Cate HS, Sah P, Windels F, Kilpatrick TJ, Merson TD. Targeted ablation of oligodendrocytes induces axonal pathology independent of overt demyelination. J Neurosci. 2012;32:8317–8330. doi: 10.1523/JNEUROSCI.1053-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietsch M, Chua KC, Abell AD. Calpains: attractive targets for the development of synthetic inhibitors. Curr Top Med Chem. 2010;10:270–293. doi: 10.2174/156802610790725489. [DOI] [PubMed] [Google Scholar]
- Pineau I, Lacroix S. Proinflammatory cytokine synthesis in the injured mouse spinal cord: multiphasic expression pattern and identification of the cell types involved. J Comp Neurol. 2007;500:267–285. doi: 10.1002/cne.21149. [DOI] [PubMed] [Google Scholar]
- Pineau I, Sun L, Bastien D, Lacroix S. Astrocytes initiate inflammation in the injured mouse spinal cord by promoting the entry of neutrophils and inflammatory monocytes in an IL-1 receptor/MyD88-dependent fashion. Brain Behav Immun. 2010;24:540–553. doi: 10.1016/j.bbi.2009.11.007. [DOI] [PubMed] [Google Scholar]
- Plemel JR, Wee Yong V, Stirling DP. Immune modulatory therapies for spinal cord injury—past, present and future. Exp Neurol. 2014;258:91–104. doi: 10.1016/j.expneurol.2014.01.025. [DOI] [PubMed] [Google Scholar]
- Rider P, Carmi Y, Guttman O, Braiman A, Cohen I, Voronov E, White MR, Dinarello CA, Apte RN. IL-1α and IL-1β recruit different myeloid cells and promote different stages of sterile inflammation. J Immunol. 2011;187:4835–4843. doi: 10.4049/jimmunol.1102048. [DOI] [PubMed] [Google Scholar]
- Rosas M, Thomas B, Stacey M, Gordon S, Taylor PR. The myeloid 7/4-antigen defines recently generated inflammatory macrophages and is synonymous with Ly-6B. J Leukoc Biol. 2010;88:169–180. doi: 10.1189/jlb.0809548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shechter R, London A, Varol C, Raposo C, Cusimano M, Yovel G, Rolls A, Mack M, Pluchino S, Martino G, Jung S, Schwartz M. Infiltrating blood-derived macrophages are vital cells playing an anti-inflammatory role in recovery from spinal cord injury in mice. PLoS Med. 2009;6:e1000113. doi: 10.1371/journal.pmed.1000113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DE, Lipsky BP, Russell C, Ketchem RR, Kirchner J, Hensley K, Huang Y, Friedman WJ, Boissonneault V, Plante MM, Rivest S, Sims JE. A central nervous system-restricted isoform of the interleukin-1 receptor accessory protein modulates neuronal responses to interleukin-1. Immunity. 2009;30:817–831. doi: 10.1016/j.immuni.2009.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3:Article3. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009;32:638–647. doi: 10.1016/j.tins.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stirling DP, Yong VW. Dynamics of the inflammatory response after murine spinal cord injury revealed by flow cytometry. J Neurosci Res. 2008;86:1944–1958. doi: 10.1002/jnr.21659. [DOI] [PubMed] [Google Scholar]
- Stirling DP, Liu S, Kubes P, Yong VW. Depletion of Ly6G/Gr-1 leukocytes after spinal cord injury in mice alters wound healing and worsens neurological outcome. J Neurosci. 2009;29:753–764. doi: 10.1523/JNEUROSCI.4918-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thawer SG, Mawhinney L, Chadwick K, de Chickera SN, Weaver LC, Brown A, Dekaban GA. Temporal changes in monocyte and macrophage subsets and microglial macrophages following spinal cord injury in the lys-egfp-ki mouse model. J Neuroimmunol. 2013;261:7–20. doi: 10.1016/j.jneuroim.2013.04.008. [DOI] [PubMed] [Google Scholar]
- Tremblay ME, Riad M, Majewska A. Preparation of mouse brain tissue for immunoelectron microscopy. J Vis Exp. 2010;(41):e2021. doi: 10.3791/2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wettenhall JM, Simpson KM, Satterley K, Smyth GK. affylmGUI: a graphical user interface for linear modeling of single channel microarray data. Bioinformatics. 2006;22:897–899. doi: 10.1093/bioinformatics/btl025. [DOI] [PubMed] [Google Scholar]
- Yuan SH, Qiu Z, Ghosh A. TOX3 regulates calcium-dependent transcription in neurons. Proc Natl Acad Sci U S A. 2009;106:2909–2914. doi: 10.1073/pnas.0805555106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Humphry M, Maguire JJ, Bennett MR, Clarke MC. Intracellular interleukin-1 receptor 2 binding prevents cleavage and activity of interleukin-1alpha, controlling necrosis-induced sterile inflammation. Immunity. 2013;38:285–295. doi: 10.1016/j.immuni.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorner B, Schwab ME. Anti-Nogo on the go: from animal models to a clinical trial. Ann N Y Acad Sci. 2010;1198(Suppl 1):E22–E34. doi: 10.1111/j.1749-6632.2010.05566.x. [DOI] [PubMed] [Google Scholar]