

Abstract
Acquisition of neuronal polarity is a complex process involving cellular and molecular events. The second messenger cAMP is involved in axonal specification through activation of protein kinase A. However, an alternative cAMP-dependent mechanism involves the exchange protein directly activated by cAMP (EPAC), which also responds to physiological changes in cAMP concentration, promoting activation of the small Rap GTPases. Here, we present evidence that EPAC signaling contributes to axon specification and elongation. In primary rat hippocampal neurons, EPAC isoforms were expressed differentially during axon specification. Furthermore, 8-pCPT, an EPAC pharmacological activator, and genetic manipulations of EPAC in neurons induced supernumerary axons indicative of Rap1b activation. Moreover, 8-pCPT-treated neurons expressed ankyrin G and other markers of mature axons such as synaptophysin and axonal accumulation of vGLUT1. In contrast, pharmacological inhibition of EPAC delayed neuronal polarity. Genetic manipulations to inactivate EPAC1 using either shRNA or neurons derived from EPAC1 knock-out (KO) mice led to axon elongation and polarization defects. Interestingly, multiaxonic neurons generated by 8-pCPT treatments in wild-type neurons were not found in EPAC1 KO mice neurons. Altogether, these results propose that EPAC signaling is an alternative and complementary mechanism for cAMP-dependent axon determination.
SIGNIFICANCE STATEMENT This study identifies the guanine exchange factor responsible for Rap1b activation during neuronal polarization and provides an alternate explanation for cAMP-dependent acquisition of neuronal polarity.
Keywords: axon, axon initial segment, cytoskeleton, EPAC signaling, neuronal polarity, Rap1b signaling
Introduction
The establishment of a polarized morphology and the functional specialization of different cellular compartments are essential to the functions of neurons. Acquisition of polarity occurs when a single neurite projecting from the cell body undergoes rapid growth to become the axon (Conde and Cáceres, 2009; González-Billault et al., 2012). Axonal development is dependent on multiple protein complexes, which include cell surface receptors such as the TrkB receptor (Nakagawara, 2001; Huang and Reichardt, 2003; Cheng et al., 2011b), signaling proteins [e.g., the phosphoinositide 3-kinase (PI3K) signaling pathway; Shi et al., 2003], and cytoskeleton modulators [e.g., mPar3/mPar6/PKC atypical (aPKC) complex, Cdc42/Rac; Lin et al., 2000; Shi et al., 2003; Nishimura et al., 2005; Montenegro-Venegas et al., 2010]. The role for a defined protein can be classified as a determinant, mediator, or modulator of axonal elongation (Cheng and Poo, 2012). In addition, axon specification and elongation depend on the concerted function of positive and negative molecules involved in axon extension (e.g., BDNF-PI3K/PIP3/Rho GTPases, LKB1; Huang and Reichardt, 2003; Shelly et al., 2007) and retraction (e.g., GSK3β, Sema3A; Gärtner et al., 2006; Shelly et al., 2011), respectively. Interestingly, Shelly et al. (2010) investigated the role of the second messengers cAMP and cGMP in neuronal polarity and how subtle coordinated changes in their cellular levels contribute to the differentiation of an axon and multiple dendrites. The investigators found that localized increases in cAMP and cGMP in undifferentiated neurites in cultured hippocampal neurons promote and suppress axon formation, respectively. A local elevation of cAMP in one of the neurites induces a decrease in cAMP in the remaining neurites of the same neuron (Shelly et al., 2010). The generation of cAMP leads to activation of protein kinase A (PKA), which mediates axon elongation (Shelly et al., 2010). Interestingly, it has been found that PKA plays a role in the repulsion of growth cones in embryonic dorsal root ganglion (DRG) neurons, suggesting that cAMP-dependent signaling mediated by PKA and EPAC may induce opposing effects in some neurons (Murray et al., 2009).
Intracellular changes in cAMP can also activate other proteins. Recently, a family of novel cAMP effector proteins named EPACs (exchange proteins directly activated by cAMP) has been identified (de Rooij et al., 1998). The EPAC protein family is composed of EPAC1 (cAMP-GEF-I) and EPAC2 (cAMP-GEF-II), which are guanine exchange factors (GEFs) for the monomeric G-proteins Rap1 and Rap2, respectively (Kawasaki et al., 1998a; Kawasaki et al., 1998b; de Rooij et al., 1998). Interestingly, along with PKA, EPAC/Rap1 mediates cAMP signaling in mammalian cells (Kiermayer et al., 2005).
The discovery of EPAC1 and EPAC2 has altered profoundly the prevailing beliefs about cAMP signaling, which historically had been associated only with PKA. EPAC signaling is involved in many physiological processes in neurons, such as apoptosis in mouse cortical neurons (Suzuki et al., 2010), neurotransmitter release in rat hippocampal neurons (Gekel and Neher, 2008), axonal guidance (Murray et al., 2009), and growth of neurites in rat DRG neurons (Murray and Shewan, 2008). Whether EPAC proteins participate in cAMP-dependent axon specification and elongation will depend on changes in the activity of their molecular targets, particularly Rap1B (Rehmann et al., 2008). The localization of the GTPase Rap1B at the distal tip of a single neurite is a crucial step in determining which neurite will become the axon. The sequential activation of Rap1B and Cdc42 GTPases is necessary for the establishment of polarity in hippocampal neurons. Rap1B and Cdc42 GTPases are activated downstream of PI3K signaling in a mechanism involving the polarity complex Par3/Par6/aPKC (Schwamborn and Püschel, 2004). However, the identity of the GEF protein involved in Rap1B activation during polarization of hippocampal neurons has remained elusive. Here, we describe the role of EPAC proteins as axon-promoting factors and analyze their ability to connect Rap1B activation with changes in intracellular levels of cAMP.
Materials and Methods
Animals.
Sprague Dawley pregnant rats were euthanized at embryonic day 18 (E18) with a lethal injection of ketamine/xylazine mixture. EPAC1 knock-out (KO) mice (C57BL/6-SV129 background) were generated as described in detail previously (Oldenburger et al., 2014; Laurent et al., 2015). Pregnant C57BL/6 wild-type (WT) and female EPAC1 KO mice were euthanized by isoflurane anesthesia (5%). All experiments were approved by the Bioethical Committee of the Faculty of Sciences, University of Chile, according to the ethical rules of the Biosafety Policy Manual of the National Council for Scientific and Technological Development (FONDECYT) and the University of Groningen Committee for Animal Experimentation.
Antibodies and chemicals.
The following antibodies were used in this work: EPAC1 (immunofluorescence, 1:150, rabbit, Santa Cruz Biotechnology: H-70 sc-25632; Western blot, 1:300, mouse, Cell Signaling Technology: 5B1 catalog #4155), EPAC2 (immunofluorescence, 1:150, rabbit, Santa Cruz Biotechnology: H-220 catalog #sc-25633; Western blot, 1:300, mouse, Cell Signaling Technology: 5D3 catalog #4156), Rap1B (1:300, mouse, BD Biosciences: catalog #610195), α-tubulin (1:10000, mouse, Sigma-Aldrich: catalog #T6199), β-actin C4, Santa Cruz Biotechnology: catalog #sc-47778), β-III-tubulin (Tuj1; 1:1000, mouse, Promega: catalog catalog #G1712A), MAP2 (1:500, rabbit, Millipore: catalog #AB5622), Tau1 (1:500, mouse, Millipore: catalog #MAB3420), SMI-31 (1:500, mouse, Covance: catalog #14835101), phospho-PKA Substrate (1:1000, rabbit, Cell Signaling Technology: catalog #9624S), RhoA (1:1000, rabbit, Cytoskeleton: catalog #ARH03-A), AnkG (1:150, mouse, Santa Cruz Biotechnology: catalog #sc-12719), Synaptophysin I (1:1000, mouse, Synaptic Systems: catalog #101011), horseradish peroxidase (HRP)-conjugated anti-mouse IgG (1:5000, donkey, Jackson ImmunoResearch: catalog #15-035-150), HRP-conjugated anti-rabbit (IgG, 1:5000, donkey, Jackson ImmunoResearch: catalog #711-035-152), and Alexa Fluor 488 Phalloidin, 1:500, Invitrogen, catalog #A12379).
For immunofluorescence experiments, we used the following secondary antibodies: Alexa Fluor 488-conjugated anti-mouse IgG (1:600, donkey, Invitrogen: catalog #A21202), Alexa Fluor 546-conjugated anti-rabbit IgG (1:600, donkey, Invitrogen: catalog #A10040), Alexa Fluor 646-conjugated anti-mouse IgG (1:600, donkey, Invitrogen: catalog #31573), and Alexa Fluor 633-conjugated anti-rabbit IgG (1:500, goat, Invitrogen, catalog #A-21070).
Other reagents used include the following: DMSO (Sigma-Aldrich, catalog #472301), 8-pCPT-2′-O-Me-cAMP (8-pCPT; Biolog, catalog #C041-05), protein kinase A inhibitor (14–22) amide, myristoylated (PKI; Tocris Bioscience, catalog #2546), ESI-09 (Biolog, catalog #B133), ESI-05 (Biolog, catalog #M092), forskolin (Tocris Bioscience, catalog #1099). Lipofectamine 2000 (Invitrogen, catalog #11668019), protease inhibitor cocktail tablets (Roche, catalog #04693159001), glutathione Sepharose 4B medium (GE Healthcare, catalog #17-0756-01), and Pierce ECL Western blotting substrate (Thermo Fisher Scientific, catalog #32106).
DNA constructs.
pCAG-RFP and Rap1GAP-RFP were kindly provided by John Cooper (Fred Hutchinson Cancer Research Center, Seattle, Washington). RalGDS-GFP was kindly provided by Johannes Bos (University of Utrecht, Utrecht, The Netherlands). vGLUT1-Venus was kindly provided by Nils Brose (Max Planck Institute, Gottingen, Germany). EPAC1 constructs [WT, dominant-negative (DN), and constitutively active (CA)] EPAC1 mutants) were subcloned into pMT2-HA (Lopez de Jesus et al., 2006). EPAC1 shRNA was obtained from OpenBiosystems (gene set: pGIPZ-tGFP Rapgef3 shRNA, set RMM4532-EG56508).
Cell cultures and transfection.
Primary cultures of rat and mouse hippocampal neurons and rat cortical neurons were prepared as described previously (Kaech and Banker, 2006). Briefly, hippocampal and cortical neurons were isolated from Sprague Dawley rat embryos and C57BL/6J WT and EPAC1 KO mice embryos at E18 and dissociated using 0.25% (w/v) trypsin (Invitrogen) for 30 min at 37°C, followed by trituration and mixing with a fire-polished pipette in plating medium (MEM; Invitrogen) with 0.6% d-glucose (Merck, catalog #108342), 10% horse serum, and 1% penicillin–streptomycin (Invitrogen). Hippocampal cells were plated at 1 × 105 cells/cm2 in 60 mm tissue culture dishes coated with poly-l-lysine (1 mg/ml; Sigma-Aldrich) and on coverslips previously coated with poly-l-lysine (1 mg/ml) at 1.5 × 104 cells/cm2 in plating medium. After 1 h, the medium was replaced with neurobasal maintenance medium (Invitrogen) supplemented with 2% B27 (Invitrogen), GlutaMAX (Invitrogen), and 1% penicillin–streptomycin in the absence of serum. Cortical neurons were plated at 1 × 105 cells/cm2 in 100 mm tissue culture dishes coated with poly-l-lysine (1 mg/ml); after 1 h, the plating medium was replaced with neurobasal maintenance medium supplemented as described above. After 18, 36, 48, and 72 h in culture, hippocampal neurons were fixed for immunofluorescence and processed for protein extraction. Cortical neurons were maintained for 3 d and then used for pull-down assays. N2A and COS-7 cells were cultured in DMEM containing 10% fetal bovine serum and 1% penicillin–streptomycin.
Transfection of rat hippocampal neurons (1.5 × 106 cells) with vGLUT1-Venus and red fluorescent protein (RFP, 5 μg of DNA) was performed according to instructions provided with the Amaxa Nucleofector Kit for rat neurons (catalog #VPG 1003) using program O-003, as suggested by the manufacturers. The RalGDS-GFP WT, DN, and CA EPAC1, RFP, Rap1GAP, and GFP plasmids (0.5 μg of DNA) were transfected using neurobasal serum-free medium and Lipofectamine 2000 (0.75 μl) as per the manufacturer. The medium was changed after 2 h to neurobasal maintenance medium and cultures were incubated for 48 or 72 h.
Rap1GAP transfection of N2A cells using 4 μg of DNA/60 mm dish and RFP, WT, DN and CA EPAC1 and Rap1Gap transfection of COS-7 cells using 8 μg of DNA/100 mm dish were performed with OptiMEM (Invitrogen) and Lipofectamine 2000 as per the manufacturer. The medium was changed after 4 h to fresh, serum-containing DMEM and the cultures were incubated for 48 h. All cultures were grown in a humidified culture incubator at 37°C, 5% CO2.
Treatments.
PKA inhibition with PKI (20 μm), EPAC1/EPAC2 inhibition with ESI-09 (15 μm), and EPAC activation with 8-pCPT (10 μm) were performed on hippocampal cultures 16 h after plating. Evaluation of PKA inhibition was assessed 6 h after the addition of inhibitors. Four days in vitro (DIV) cortical neurons were used for biochemical assays with PKI (20 μm), 8-pCPT (10 μm), ESI-09 (15 μm), and ESI-05 (15 μm) treatments. Briefly, PKI, 8-pCPT, ESI-09, and ESI-05 were dissolved in plain neurobasal medium and incubated with the cultures for 30 min before Rap1B pull-down assays were performed. For experiments involving mature axonal markers, long-term neuronal cultures were treated 16 h after plating with 8-pCPT (10 μm) and maintaining the cells for 8 d to evaluate vGLUT and 10 d for synaptophysin and AnkG without changing the medium.
N2A cells were treated for 10 h with 8-pCPT (10 μm), forskolin (20 μm), or forskolin + PKI (20 μm). The final DMSO concentration in all experiments was maintained at <0.1%.
Immunofluorescence and image analysis.
Hippocampal neurons that were grown on coverslips were fixed at 18, 48, and 72 h after transfection with 4% (w/v) paraformaldehyde/4% (w/v) sucrose for 30 min at 37°C and washed with PBS 3 times for 5 min each. The cells were incubated with PBS containing 0.1% Triton X-100 for 5 min and then blocked with 5% (w/v) bovine serum albumin (BSA) in PBS for 1 h. After blocking, cells were incubated with primary antibodies diluted with 1% BSA in PBS overnight at 4°C, washed with PBS 3 times for 5 min each, incubated with fluorescent secondary antibody for 1 h, and washed with PBS 3 times for 5 min each. For AnkG staining, neurons were permeabilized with 0.1% Triton X-100 for 20 min at room temperature and blocked with 1% BSA for 1 h at room temperature. Neurons were incubated with antibodies against MAP-2 (1:500) and AnkG (1:150) overnight at 4°C. Later, secondary antibodies Alexa Fluor 546-conjugated anti-rabbit IgG and Alexa Fluor 488-conjugated anti-mouse IgG were added and incubated for 1 h at room temperature. Neurons were then washed for 1 h and Tau-1 antibody (1:500) was added and incubated overnight at 4°C. Secondary antibody Alexa Fluor 647-conjugated anti-mouse IgG was then added and incubated for 1 h at room temperature. Finally, coverslips were mounted on slides using FluorSave Reagent (Millipore, catalog #345789) and examined using a Zeiss LSM510 Meta Confocal Scanning Microscope equipped with Plan-Apochromat 40×/1.3 numerical aperture (NA) and Plan-Apochromat 63×/1.4 NA objective lenses. Digital images were quantified using the LSM 5 Image Browser (Zeiss) and ImageJ for axonal length, fluorescence intensity (values are expressed as arbitrary units of the relative fluorescent of EPAC1 and EPAC2 to β-III tubulin), and cell counting. Phenotypes were determined by measuring the total axon length and the percentage of polarized neurons. For 3D reconstruction of vGLUT1 axonal puncta with the Imaris Software package (Bitplane), z-stack images of 8 DIV neurons that had been transfected with Venus-VGLUT1 were acquired using the 63× objective at a resolution of 1024 × 1024 pixels. Three-dimensional images were generated with the Surpass tool for filaments (Tau-1-positive axons) and spots (Venus-VGLUT1-positive clusters). Automated quantification of the number of spots was performed with the MATLAB-based “find spots close to filaments” plugin. For the purposes of this study, we defined a polarized neuron as a neuron with an axon of >50 μm or more than twice the length of its soma that is positive for Tau-1 or SMI-31 staining localized in a proximal-distal gradient along the shaft and that is negative for MAP2 staining. EPAC1 KO neurons were examined using a HC PL APO CS2 40×/1.3 (oil) objective on a Leica SP8 Confocal Microscope (DMI 600). This experiment was performed at the University Medical Center Groningen Microscopy and Imaging Center, which is sponsored by the Netherlands Organization for Scientific Research (NOW) and 175-010-2009-023 (Zeiss 2P), University of Groningen Grants 40-00506-98-9021 (TissueGnostics TissueFAXS microscope).
GST fusion protein preparation and Rap1 activation pull-down assay.
Expression and purification of GST-conjugated proteins were performed as described previously (Henríquez et al., 2012). Briefly, BL21 (DE3) E. coli strains carrying GST-RalGDS-RBD plasmid were grown overnight in LB medium containing ampicillin at 37°C. The next day, cultures were diluted 1:100 and grown in fresh medium until they reached an OD600 of 0.6. Then, 1 mm isopropyl-β-d-thiogalactopyranoside (final concentration) was added. Cells were collected and lysed 2 h after induction by sonication in lysis buffer A (50 mm Tris-HCl, pH 8.0; 1% Triton X-100; 1 mm EDTA; 0.15 m NaCl; 25 mm NaF; 0.5 mm PMSF; 1× protease inhibitor complex, Roche). Cleared lysate was then incubated with glutathione-Sepharose beads (GE Healthcare). Loaded beads were washed 10 times with lysis buffer B (lysis buffer A plus 300 mm NaCl) at 4°C. The GST fusion proteins were quantified and visualized in SDS-PAGE gels stained with Coomassie brilliant blue.
For the Rap1 activation assay, beads loaded with RalGDS-RBD (Rap-binding domain of the Ral guanine nucleotide dissociation stimulator, which binds specifically to Rap1-GTP but not to the inactive Rap1-GDP form) were incubated for 1 h at 4°C with 1 mg of freshly made lysate from 3 DIV cortical neuron cultures or 1 mg of freshly made lysate from COS-7 cells expressing EPAC1 constructs. Cell lysates were produced using fishing buffer (50 mm Tris-HCl, pH 7.5; 10% glycerol; 1% Triton X-100; 150 mm NaCl; 10 mm MgCl2; 25 mm NaF; 1× protease inhibitor complex). The beads were washed three times with washing buffer (50 mm Tris-HCl, pH 7.5; 30 mm MgCl2; 40 mm NaCl) and resuspended in SDS-PAGE sampling buffer. The levels of Rap1B-GTP (presented as arbitrary units) were evaluated from Western blot analysis and normalized against total Rap1B with ImageJ (values are presented as arbitrary units).
Western blot analysis.
Neurons and cell lines grown on dishes were washed once with PBS and then incubated with RIPA (65 mm Tris, 155 mm NaCl, 1% Triton X-100, 0.25% sodium deoxycholate, 1 mm EDTA, pH 7.4, and a mixture of protease inhibitors: 5 μg/ml Na3VO4, 20 μm PMSF, 5 mm NaF). Then, cells were scraped from the plate, kept on ice for 15 minutes, and finally centrifuged for 20 min at 14,000 r.p.m. Supernatant fractions were denatured and subjected to SDS-PAGE, using 10% running gels for EPAC, 15% for Rap1B, and 12% for the PKA substrates Par6 and RhoA. Separated proteins were transferred to nitrocellulose membranes, which were then blocked with 5% BSA for 1 h at room temperature. The membranes were incubated with primary antibodies overnight at 4°C. Membranes were then washed using TBST (50 mm Tris-HCl, 150 mm NaCl, 0.05% w/v Tween 20, pH 7.4) four times for 10 min each and incubated with HRP-conjugated secondary antibody for 1 h at room temperature. Finally, the membrane blots were developed using Pierce ECL Western blotting substrate. Digital images of Western blots were quantified using ImageJ software and values are expressed as arbitrary units.
Statistical analysis.
All data represent the mean ± SEM of at least three independent experiments. Comparisons between two groups were made using the unpaired Student's t test. Comparisons between three or more groups were performed using a one-way ANOVA followed by Dunnett's or Tukey's post hoc test. A value of p < 0.05 was considered significant. Analyses were performed with GraphPad Prism software).
Results
Expression of EPAC1 and EPAC2 during neuronal polarization
Because Rap1 is the major downstream target of EPAC (Rehmann et al., 2006; Rehmann et al., 2008; Ponsioen et al., 2009), we first examined the expression and localization of both EPAC isoforms in stage II (unpolarized) and stage III (polarized) hippocampal neurons to assess whether the distribution of EPAC isoforms resembled that of active Rap1B. Both EPAC1 and EPAC2 were expressed at stages II and III, with no significant differences between the stages (Fig. 1a,d).
Figure 1.

EPAC 1 and EPAC2 are differentially expressed in cultured hippocampal neurons. a, EPAC1 immunoblot of lysates from cultured neurons at stages II and III normalized against actin (stage II, 0.86 ± 0.13; stage III, 0.98 ± 0.1; Student's t test, three independent experiments.) b, EPAC1 distribution in cultured hippocampal neurons at stages II and III. Tuj1 antibody against β-III-tubulin was used as neuron-specific marker. c, Quantitative analysis of EPAC1 distribution in stage II and III neurons (n = 20 neurons, one-way ANOVA with Tukey's post hoc test, three independent experiments). d, EPAC2 immunoblot analysis performed as in a (stage II, 0.10 ± 0.01; stage III, 0.07 ± 0.03; Student's t test, three independent experiments.) e, EPAC2 distribution in cultured hippocampal neurons at stages II and III. Cells were immunostained as in b. f, Quantitative analysis of EPAC2 distribution in stage II and III neurons (n = 30 neurons, one-way ANOVA with Tukey's post hoc test, three independent experiments). Data represent the mean ± SEM; n.s., not significant; *p < 0.05; ****p < 0.0001. Scale bars: B and E, top, 10 μm; B and E, bottom, 20 μm.
We then evaluated the subcellular distribution of EPAC1 and EPAC2 in unpolarized and polarized cultured neurons using semiquantitative immunofluorescence (Fig. 1b,e). Fluorescence intensities of EPAC1 (Fig. 1c) and EPAC2 (Fig. 1f) immunostaining (normalized to Tuj1 staining) were analyzed at stage II (18 h in culture). EPAC1 and EPAC2 were uniformly distributed in every neurite, but the fluorescence intensity of EPAC1 was significantly higher than that of EPAC2 (EPAC1: mean, 1.40 ± 0.03; n = 20 neurons, three experiments vs EPAC2: mean, 0.60 ± 0.04; n = 44 neurons; p < 0.0001; Fig. 1c,f). Next, we analyzed the fluorescence intensity at stage III (48 h in culture). EPAC1 was enriched at the tip of the longest neurite, which is the nascent axon (Fig. 1c). In contrast, EPAC2 distribution showed no significant differences among neurites (Fig. 1f).
These results suggest that EPAC1 may be important in promoting axon elongation in hippocampal neurons, similar to what has been described previously for DRG neurons (Shelly et al., 2007; Murray and Shewan, 2008).
EPAC induced multiples axons through Rap1B activation
To assess the role of EPAC activation in axonal elongation, we performed pull-down assays to evaluate pharmacological activation and inhibition of EPAC functions. We used a selective agonist, 8-pCPT, which activates EPAC but not PKA both in vitro and in vivo (Woolfrey et al., 2009; Lim et al., 2012), as well as a selective antagonist of EPAC1 and EPAC2 (ESI-09) and an antagonist of EPAC2 only (ESI-05; Tsalkova et al., 2012a; Tsalkova et al., 2012b). The immunoblot analysis showed a significant increase in the fraction of GTP-bound Rap1B in cortical neurons treated with 10 μm 8-pCPT and a significant reduction of Rap1B activity in cultures treated with 15 μm ESI-09 but not with 15 μm ESI-05 (Fig. 2a,b). Therefore, 8-pCPT stimulated Rap1B activity in neurons, whereas ESI-09 inhibited Rap1B activation. Interestingly, ESI-05, the selective EPAC2 antagonist, did not lead to reduced activation (relative to control) of Rap1B in cultured neurons, strongly suggesting that EPAC1 alone is involved in Rap1B activation in neurons.
Figure 2.
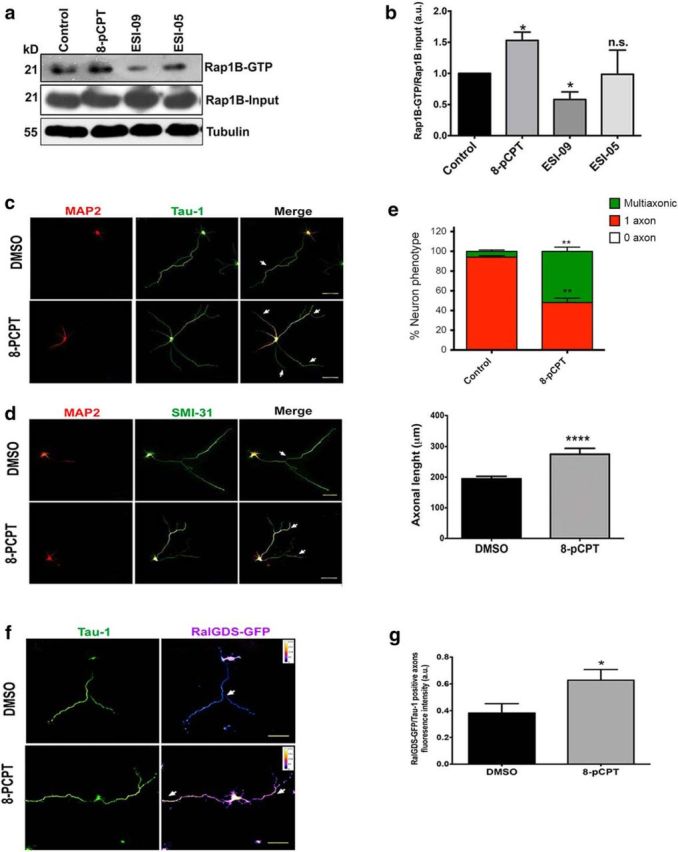
EPAC pharmacological activation results in multiaxonic neurons. a, 4 DIV cortical neurons were incubated with 8-pCPT, ESI-09, or ESI-05 for 30 minutes and then activated Rap1B was evaluated by pull down. Rap1B-GTP was normalized against Rap1B input and corrected against tubulin. b, Quantitative analyses of pull-down data as in a (Student's t test; three independent experiments). c, d Neurons cultured for 3 DIV with DMSO or 8-pCPT were immunostained for Tau-1 (c; axon, arrows) or phosphorylated MAP1B (d; SMI-31; axon, arrows) and MAP2 (somatodendritic compartment). e, Quantitative analysis of neurons stained as in c (n = 76 DMSO and n = 70 8-pCPT neurons, left; n = 70 DMSO and 63 8-pCPT neurons, right; Student's t tests; four independent experiments). f, Hippocampal neurons were transfected with the Ral-GDS-GFP construct, treated with DMSO or 8-pCPT, and stained for Tau-1 (arrows). g, Quantitative analysis of GFP fluorescence in axons (i.e., Tau-1-positive neurites) as in f (n = 36 neurons for DMSO and 55 neurons for 8-pCPT; Student's t tests, three independent experiments). Data represent the mean ± SEM; n.s., not significant; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. Scale bars: c, d, f, 50 μm.
We then investigated the functional role of EPAC proteins during in vitro culture of hippocampal neurons before polarization. Neurons were incubated with a single dose of 8-pCPT (10 μm) or a vehicle control for 3 DIV. Most vehicle-treated neurons formed one long Tau-1-positive axon and several shorter MAP2-positive minor neurites (Fig. 2c). In contrast, the 8-pCPT-treated neurons displayed multiple axonal processes that were positive for two axonal markers, hypophosphorylated Tau (epitope Tau-1; Fig. 2c,e) and mode I-phosphorylated MAP1B (epitope SMI-31; n = 50 neurons, three independent experiments; Fig. 2d). Moreover, neurons treated with 8-pCPT showed increased total axonal length compared with the control group (Fig. 2e). To confirm that 8-pCPT was indeed activating Rap1B protein, we transfected hippocampal neurons with a GFP-fused Ral-GDS-RBD construct, which is a reporter of active Rap1B that has been used previously to assess subcellular localization of active Rap1 in several cell types (Bivona et al., 2004; Jordan et al., 2005; Kortholt et al., 2010). Neurons were transfected upon plating and further incubated with 8-pCPT (10 μm) at 16 h after plating; at 3 DIV, they were stained with Tau-1. We quantified the fluorescence intensity of Ral-GDS-GFP in the last third of each Tau-1-positive axon. Control transfected neurons each had one Tau-1-positive axon that showed Ral-GDS-GFP fluorescence both along the axon shaft and within the cell body (Fig. 2f). In contrast, neurons treated with 8-pCPT each had multiple Tau-1-positive axons (Fig. 2f) and the Ral-GDS-GFP signal was significantly concentrated on each supernumerary axon (Fig. 2g). These data suggest that EPAC proteins mediate Rap1B activation in hippocampal neurons and this activation could be responsible for development of supernumerary axon.
Activation of EPAC by 8-pCPT induces multiple axons that are positive for mature axonal markers
We next investigated whether the short-term effects induced by EPAC activation, namely induction of supernumerary axons, were maintained in long-term neuronal cultures. For this, we evaluated the assembly of the axon initial segment (AIS) in long-term neurons treated with 8-pCPT. AIS is a physical and functional barrier between the somatodendritic and axonal compartments, where Na+ and K+ channels are recruited to trigger the action potential (Grubb and Burrone, 2010). The assembly of AIS represents the maturation and functional polarization of the axon (Rasband, 2010) We used 10 DIV neurons to determine whether neurons treated with 8-pCPT exhibited more than one Tau-1-positive process containing an assembled AIS. We evaluated the distribution of ankyrin G (AnkG) within the first 15–25 μm of the axon (Fig. 3a) because it is essential for the organization of the AIS (Kordeli et al., 1995; Galiano et al., 2012). 8-pCPT was added to the culture medium after 18 h in culture and the neurons were fixed at day 10. Of the DMSO-treated neurons, 94.1 ± 3.0% displayed only one Tau-1- and AnkG-positive axon, which was also negative for MAP2 staining (Fig. 3a,b). AnkG was concentrated in the proximal region of each axon (Fig. 3a), a pattern characteristic of AIS in cultured neurons (Kordeli et al., 1995; Galiano et al., 2012). In contrast, for neurons treated with 8-pCPT, 76.4 ± 11.5% had multiple axons that were positive for Tau-1 and AnkG and negative for MAP2 staining (Fig. 3a,b). Next, to determine whether these multiple axons were functional, we analyzed the distribution of the vesicular glutamate transporter vGLUT1 by transfecting a vGLUT1–Venus fusion construct into neurons that were then treated with 8-pCPT and cultured for 8 DIV before fixation. vGLUT1 is a reliable indicator of synaptic vesicle precursors loaded with glutamate (Bellocchio et al., 2000), even before synapses are formed (Sabo et al., 2006). In addition, vGLUT1 constructs that are overexpressed show the same subcellular distribution as the endogenous VGLUT1 protein, which is characterized by an obvious punctate pattern along the axon and, in particular, in presynaptic boutons (Wilson et al., 2005). Neurons were stained for Tau-1 and MAP2 and the distribution of vGLUT1-Venus was examined. As expected, transfected neurons that were treated with 8-pCPT had multiple Tau-1-positive axons (Fig. 3c). Venus immunofluorescence was then quantified in a 3D reconstruction of a 100 μm region along each axon (Fig. 3d). Control neurons had 11.4 ± 1.30 vGLUT1-Venus-positive spots per 100 μm of axonal length and axons from 8-pCPT-treated neurons had 11.5 ± 1.12 vGLUT1-Venus-positive spots per 100 μm of axonal length (Fig. 3e). In both cases, the distribution of vGLUT1-Venus in punctate structures was similar to those found in excitatory neurons (Wilson et al., 2005), suggesting that the multiple axons induced by EPAC activation might be functional.
Figure 3.

8-pCPT-treated multiaxonic neurons in long-term (10 DIV) culture display markers of mature axons. a, DMSO- and 8-pCPT-treated neurons were cultured for 10 DIV and stained for AnkG, MAP2, and Tau-1. Arrows indicate axons that are positive for AnkG and Tau-1 staining and negative for MAP2 staining. b, Quantitative analysis of neurons treated as in a (n = 60 neurons; ***p < 0.001; ****p < 0.0001; Student's t tests; four independent experiments). c, Neurons were transfected with vGLUT1-Venus (green) and then treated with DMSO or 8-pCPT for a total of 8 DIV. Fixed neurons were stained for Tau-1 (axons; white, arrows) and MAP2 (dendrites, red). d, Higher magnification of axons in DMSO- and 8-pCPT-treated neurons was used for z-stack reconstruction and semiautomated analyses of Venus-vGLUT1 puncta. Top, Raw images of neurons cultured for 10 DIV. Middle, Higher magnification of area indicated in white rectangle on top. Bottom, Filaments (Tau-1 staining in axons) and dots (Venus-vGLUT1 punctate staining). e, Quantification of data from neurons analyzed as in d (n = 12 neurons per treatment; n.s., not significant; Student's t tests; three independent experiments). f, Neurons were transfected with RFP (white), DMSO, or 8-pCPT and immunolabeled for endogenous synaptophysin (green) and MAP2 (red). Arrows indicate positive synapthophysin spots in axons. Data represent the mean ± SEM. Scale bars: a, 30 μm; c, top, 20 μm; c, bottom, 50 μm; d, top, 40 μm; d, bottom, 10 μm; f, bottom, 50 μm.
We investigated the expression and distribution of another axonal marker, synaptophysin, to confirm these results. Synaptophysin is a marker of synaptic density that predominantly shows granular or punctate staining along axons and accumulates in the axon, where it is involved in neurotransmitter release and is therefore a good indicator of axon functionality (Fletcher et al., 1991). We transfected neurons with RFP as a reporter gene to identify single neurons in confluent cultures. Next, cultures were treated with 8-pCPT and maintained for 10 DIV, fixed, and immunolabeled for synaptophysin and MAP2. Figure 3f shows representative images of a control neuron with a single axon and a neuron treated with 8-pCPT showing multiple axons that were also positive for synaptophysin (Fig. 3f, arrows). Synaptophysin immunostaining was very similar in both groups and showed canonical punctate structures widely distributed along the axons (Fletcher et al., 1991).
Gain of function of EPAC1 induces multiple axons similar to 8-pCPT-treated neurons
EPAC1 expression in brain tissue is higher at embryonic stages, whereas EPAC2 is barely detectable at this developmental stage (Murray and Shewan, 2008), similar to our findings in stage II and III hippocampal neurons and with ESI-05 in the biochemical analysis. We therefore tested whether EPAC1 is sufficient to modify neuronal polarity. We first performed pull-down assays to examine Rap1B activation in response to different forms of EPAC1 (WT, DN, and CA). COS-7 cells were transfected with HA-tagged EPAC1 constructs (as determined by Western blot analysis of transfected cells; Fig. 4a) and, as a negative control, an RFP-tagged Rap1 GTPase-activating protein (Rap1GAP), which leads to Rap1 inactivation. Pull-down results revealed that the amount of GTP-bound Rap1 increased in response to WT EPAC1 and CA EPAC1 and was no different from control levels in the presence of either the DN form of EPAC1 or that of Rap1GAP (Fig. 4a,b). As expected, results of this assay indicate that Rap1 activity changes in response to different EPAC1 constructs. To further confirm the contribution of EPAC1 on the multiple-axon phenotype in hippocampal neurons, we cotransfected neurons (2 h after plating) with the reporter vector RFP and WT, DN, or CA forms of EPAC1 or our negative control, RFP-Rap1GAP. Neuronal polarity was evaluated at 3 DIV by measuring the lengths of axons and Tau-1 and MAP2 immunostaining (Fig. 4c). Overexpression of WT and CA EPAC1 produced neurons with multiple axons (Fig. 4d). In contrast, overexpression of DN EPAC1 led to neurons that most often had only a single axon. Overexpression of CA EPAC1 also produced axons that were significantly longer than those of the RFP control, whereas DN EPAC1 and Rap1GAP overexpression induced a significant reduction in axonal length compared with the RFP control (Fig. 4e). Therefore, EPAC1 is sufficient to produce multiple axons during the development of neuronal polarity in vitro through Rap1B.
Figure 4.

EPAC1 genetic activation results in multiaxonic neurons. a, A Rap1B pull-down activation assay was used to evaluate the effect of WT, DN, and CA constructs of EPAC1 in COS7 cells. In addition, COS7 cells were transfected with a RAP1-GAP construct as a negative control. Only EPAC1 constructs have a HA-tag. b, Quantitative analysis of data shown in a (all compared with normalized control). c, Hippocampal neurons were transfected with the constructs described in a, with RFP as a volume marker. After 3 DIV, neurons were immunostained for Tau-1 (green, arrows) and MAP2 (red). d, e, Quantitative analysis of the percentage of neurons with multiple axons (d) and axonal length (e) from data shown in c. Data represent the mean ± SEM; n.s., not significant; *p < 0.05; **p < 0.01; ***p < 0.001. Scale bars: c, 50 μm.
Pharmacological inhibition of EPAC impairs axonal development
To clarify the role of EPAC in neuronal polarity, we used ESI-09, which inhibits cAMP-mediated activation of EPAC1 and EPAC2 but not PKA (Almahariq et al., 2013; Tsalkova et al., 2012b). Hippocampal neurons were treated at 16 h after plating with 15 μm ESI-09, incubated for 48 h, and immunostained for axonal and somatodendritic markers. ESI-09 disrupted the formation of axons, as characterized by weak Tau-1 staining and very short axons (Fig. 5a). Quantitative analyses revealed a significant reduction in the percentage of polarized neurons with ESI-09 compared with neurons treated with vehicle alone (Fig. 5b). Neurons treated with the EPAC activator 8-pCPT were all polarized (Fig. 5b). In the presence of ESI-09, neurons extended axons that were significantly shorter than the lengths of axons from control neurons (Fig. 5c). This effect did not occur with ESI-05 treatment (data not shown). These results suggest that EPAC is required for axon elongation and specification during the establishment of neuronal polarity and reinforce the idea that Rap1B activation during neuronal polarity is dependent on EPAC1 activation.
Figure 5.

EPAC inhibition results in decreased axonal elongation. a, Neurons were treated with control vehicle, EPAC agonist (8-pCPT), or antagonists of EPAC1 and EPAC2 (ESI-09) and then immunostained for Tau-1 (green, arrows) and MAP2 (red). b, c, Quantitative analysis of neurons treated as in a for the percentage of polarized (1 axon), no polarized (0 axon), and mutliaxonic neurons (b) and total axonal length (c; n = 50 neurons per treatment). Data represent the mean ± SEM; n.s., not significant compared with control; *p < 0.05; ****p < 0.001, one-way ANOVA with Dunnett's post hoc test; n = 4 (b) and n = 3 (c) independent experiments. Scale bars: a, 50 μm.
Axon specification regulated by EPAC is independent of PKA signaling
cAMP-PKA signaling is involved in establishing neuronal polarity through at least two different complementary mechanisms. Elevation of cAMP induces increased LKB1 phosphorylation by PKA, which promotes axon formation (Shelly et al., 2007; Shelly et al., 2010). In addition, phosphorylation of the E3-ligase Smurf1 by PKA switches its selectivity for ubiquitination of Par6 or RhoA, promoting the accumulation of Par6 and degradation of RhoA in the axonal compartment (Cheng et al., 2011a). In this context, we tested the influence of PKA inhibition in the presence of 8-pCPT. For this approach, we used the inhibitor PKI-(Myr-14–22)-amide, which binds to the free catalytic subunit of PKA and prevents phosphorylation of PKA targets (Dalton and Dewey, 2006) without the nonspecific effects that have been reported for other PKA inhibitors such as KT5720 and H89 (Davies et al., 2000; Murray, 2008).
To determine whether PKI can prevent phosphorylation of PKA targets, we added 20 μm PKI alone or in combination with 10 μm 8-pCPT to hippocampal neurons in culture. Western blotting of protein lysates was then performed using an antibody that specifically recognizes nuclear and cytoplasmic PKA substrates [i.e., PKA-specific phospho-motifs, (RR)-X-(S*/T*); Bacallao and Monje, 2013] to analyze the levels of PKA inhibition (Fig. 6a). At this concentration, PKI reduced the phosphorylation of PKA substrates either alone or in combination with 8-pCPT compared with cultures treated with vehicle alone (Fig. 6b). We then confirmed the effect of PKA inhibition on the establishment of neuronal polarity. We treated hippocampal neurons at 16 h after plating with DMSO alone (vehicle control), 20 μm PKI, or a combination of 20 μm PKI plus 10 μm 8-pCPT. The cultures were fixed at 2 DIV and analyzed by immunofluorescence for neuronal phenotype (Fig. 6c). Quantitative analyses revealed that PKI treatment did not affect neuronal polarization significantly (Fig. 6d). Interestingly, PKA inhibition did not affect the generation of multiple axons by EPAC activation with 8-pCPT treatments. In fact, the percentage of multiaxonic neurons was significantly higher in cultures treated with PKI plus 8-pCPT compared with the PKI and DMSO groups (Fig. 6d). In addition, a significant reduction in the length of the axons was observed in neurons treated with PKI compared with the control and PKI/8-pCPT groups (Fig. 6e). Neurons treated with PKI/8-pCPT showed axonal lengths that were significantly longer compared with the PKI and DMSO groups (Fig. 6e).
Figure 6.

EPAC-dependent axonal elongation is independent of PKA signaling. a, Whole protein extracts from 2 DIV cultured hippocampal neurons treated for 6 h with the vehicle (first lane), PKI (a PKA inhibitor; middle lane), and PKI in the presence of 8-pCPT (right lane) were analyzed by immunoblotting with an antibody that recognizes PKA-specific phosphorylation epitopes. Asterisks indicate epitopes that are less abundant in the presence of PKI. b, Quantitative analysis of data shown in a (****p < 0.0001; Student's t tests; three independent experiments). c, Neurons were treated with DMSO, PKI, or PKI + 8-pCPT and assessed for neuronal polarization. Neurons were immunolabeled for Tau-1 (green) and MAP2 (red). d, e, Quantitative analysis of neurons treated in c for the percentage of neuronal phenotypes (d) and axonal length (e; n = 50 neurons per analysis; **p < 0.01 and ***p < 0.001 versus control;#p < 0.0001 versus PKI; n.s., not significant; one-way ANOVA with Dunnett's (d) or Tukey's (e) post hoc test; three independent experiments). f, PKA signaling involved in neuronal polarization was evaluated in control and PKI + forskolin-treated, forskolin-treated, and 8-pCPT-treated N2A neuroblastoma cells as described previously (Cheng et al., 2011a). g, Quantitative analysis of cells treated in f (*p < 0.05, **p < 0.01; n.s., not significant; Student's t tests; three independent experiments). h-i, Cdc42-GTP levels were evaluated in neurons treated with 8-pCPT, 8-pCPT + PKI, and 8-pCPT + ESI-09, no significant differences was observed (three independent studies). A representative blot is presented. Data represent the mean ± SEM. Scale bars: c, 50 μm.
Moreover, we studied the effect of 8-pCPT on Par6 stabilization and RhoA degradation in neuro2A cells (N2A) to assess the role of PKA-dependent Smurf1 phosphorylation (Cheng et al., 2011a). We chose this experimental approach because it was demonstrated previously by Cheng et al. (2011a) that N2A cells treated with db-cAMP, an analog of cAMP, displayed reduced ubiquitination for Par6 and enhanced ubiquitination for RhoA, leading to differential ubiquitin-dependent degradation of these proteins by the proteasome (Cheng et al., 2011a).
Figure 6f shows that incubation of N2A cells with 10 μm 8-pCPT did not induce any obvious change in the overall levels of Par6 and RhoA (Fig. 6g). In contrast, 20 μm forskolin selectively increased the level of Par6 and decreased the level of RhoA (Fig. 6g). Forskolin effects on Par6 were abrogated when we used 20 μm PKI (Fig. 6g).
Furthermore, we evaluated whether Rap1B activation induced by the EPAC pharmacological agonist increased the activity of the small Rho GTPase Cdc42 because it has been reported previously that sequential activity of Rap1B and Cdc42 is essential for axon formation (Schwamborn and Püschel, 2004). Primary cultured neurons were incubated with 8-pCPT alone or in the presence of PKI or ESI-09 and active (GTP-bound) Cdc42 was evaluated by pull down. No differences were found among treatments, suggesting that Rap1B does not induce Cdc42 activation directly (Fig. 6h,i).
Loss of function and genetic deletion of EPAC1 reduce neuronal polarization in hippocampal neurons
Finally, we used two genetic approaches to validate results obtained with the EPAC1 pharmacological inhibitor ESI-09. First, we used an shRNA (pGIPZ-EPAC1-tGFP, shRNA-EPAC1) to suppress EPAC1 expression. Neurons were transfected with shRNA-Scramble and ShRNA-EPAC1 after 36 h of culture, immunostained for EPAC1, and analyzed in positive transfected neurons. Results shown in Figure 7a reveal a significant reduction of EPAC1 staining in the cell body of transfected neurons compared with the Scramble control. Moreover, EPAC1 staining in neurites was almost absent in shRNA-EPAC1-transfected neurons (Fig. 7a, inset, arrowhead), confirming knock-down. By visual inspection, we determined that neurons transfected with shRNA-EPAC1 on average displayed shorter axons (Fig. 7a, GFP panel).
Figure 7.
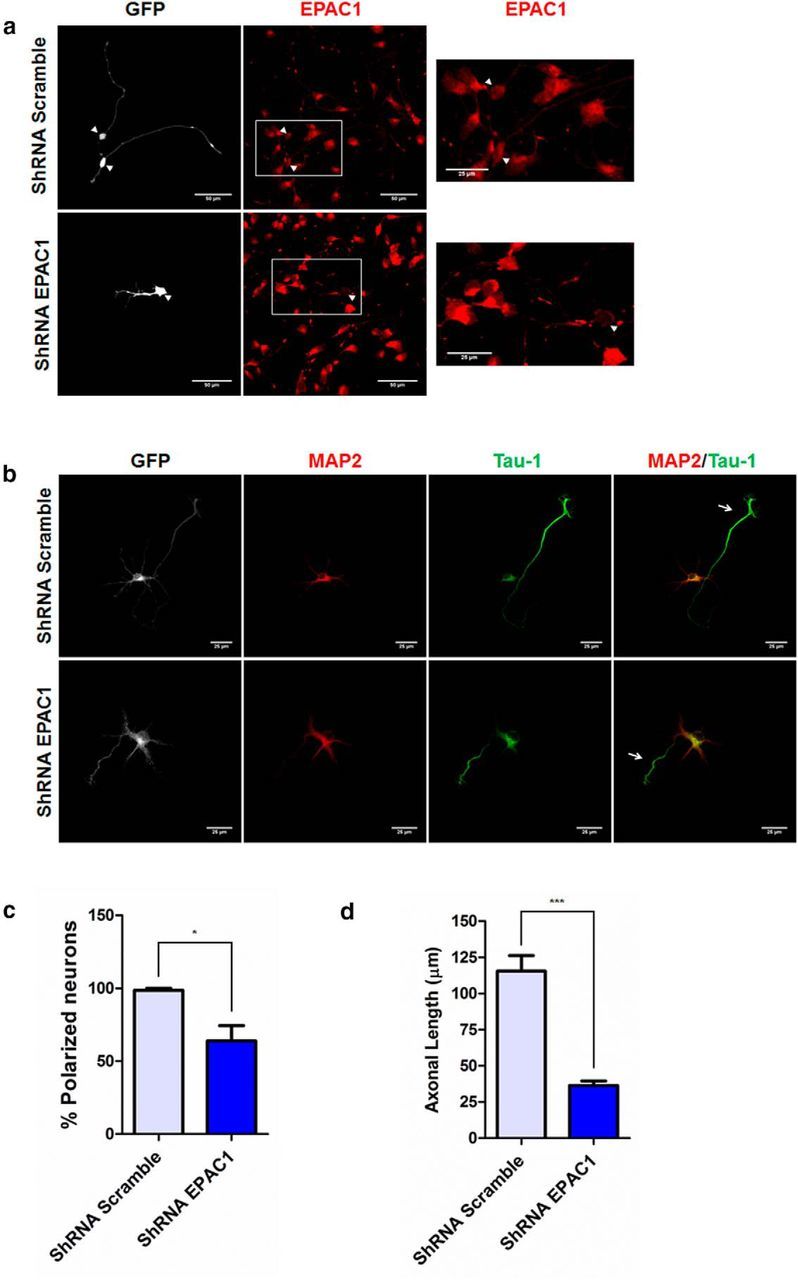
Knock-down of EPAC1 delays the polarization and decreases the length in hippocampal neurons. a, Neurons were transfected with ShRNA Scramble-GFP (top) and ShRNA EPAC1-GFP (bottom). After 36 h of culture, neurons were immunostained for EPAC1 (red). Transfected neurons were visualized with GFP marker (arrowhead). a, Right, Close-up view of the boxed areas. b, Neurons were transfected with shRNA as in a and assessed for neuronal polarization. Neurons were immunolabeled for Tau-1 (green, arrow) and MAP2 (red). Quantitative analysis of the percentage of polarized neurons (c, n = 31–40 cells, Student's t tests; *p < 0.05, three independent experiments) and axonal length (d, n = 31–40 cells, Student's t tests; p < 0.0001****, three independent experiments) from data shown in b. Data represent the mean ± SEM; n.s., not significant; *p < 0.05; ***p < 0.0001. Scale bars: a, 50 μm; a, close-up and b, 25 μm.
We used the same approach to analyze in detail the effect of shRNA-EPAC1 on the phenotype of hippocampal neurons. Knocked-down neurons were fixed and stained for MAP2 and Tau-1 antibodies (Fig. 7b). Knock-down of EPAC1 led to a reduction in the number of polarized neurons from 99 ± 1.3% in control cultures and 64 ± 11% of polarized neurons in EPAC1 knock-down (Fig. 7c). Moreover, the mean axonal length of neurons transfected with shRNA-EPAC1 was shorter compared with the control (shRNA-EPAC1: 36 ± 4 μm vs shRNA-Scrambled: 116 ± 11 μm; Fig. 7d).
In our last experiments, we cultured hippocampal neurons derived from EPAC1 KO mice that had been previously characterized by Laurent et al. (2015). We first determined that the EPAC1 protein is not expressed in the brain of mutant mice. Whole-brain extracts derived from control and EPAC1 KO mice were evaluated by immunoblot against anti-EPAC1 antibody. Figure 8a shows that null mutants lack EPAC1 protein. EPAC1 loss of function was indeed not compensated by increased EPAC2 expression because no changes in the overall amount of EPAC2 could be detected between WT and EPAC1 KO mice (Fig. 8b).
Figure 8.

EPAC1 KO hippocampal neurons show delayed polarization. a, Cerebral extracts (40 μg) from WT and EPAC1 KO E18 mice were analyzed for EPAC1 [a, WT brain = 1.4 ± 0.4 arbitrary units (a.u.)] and EPAC2 expression (b, WT brain = 0.37 ± 0.1 a.u.; EPAC1 KO brain = 0.35 ± 0.1 a.u, Student's t tests n.s.; n = 3 samples/genotype). c, Representative images from primary hippocampal neurons isolated from E18 WT and EPAC1 KO mice. Changes in neuronal differentiation at stages II and III were analyzed with anti β III tubulin, phalloidin (F-actin, gray), anti-Tau-1 (green, arrow), or anti-MAP2 (red) antibodies, respectively. d, Percentage of polarized neurons (blue) and nonpolarized neurons without an axon (white) after 3 DIV is shown. Neurons from EPAC KO mice show a significant reduction in the proportion of polarized neurons (n = 47–134 neurons per treatment). e, WT and EPAC1 KO neurons were cultured for 3 DIV and treated either with DMSO or 8-pCPT and immunostained for Tau-1 (green, arrows), MAP2 (red), phalloidin (F-actin, gray), respectively. Representative images show the formation of multiaxonic neurons only in WT neurons treated with 8-pCPT. EPAC1 KO neurons display polarized (white arrow) and no-polarized neurons. Data represent the mean ± SEM; n.s., not significant; *p < 0.05; **p < 0.001. Scale bars: c, 30 μm; e, 50 μm.
To study the morphology and axon development of the EPAC1 KO mice, hippocampal neurons were dissociated and maintained in culture for 1 DIV (stage II) and 2 DIV (stage III) and immunostained with the neuron-specific markers Tuj1, MAP2, and Tau-1, followed by staining of actin cytoskeleton with phalloidin-488. Figure 8b shows representative images of the neurons from WT and EPAC1 KO neurons. The majority of EPAC1 KO neurons cultured for 1 DIV show immature neurites typical for stage II, with no segregation of Tau-1 into the short neurite and normal MAP2 staining compared with WT cells. However, at 2 DIV, we observed that the culture of EPAC1 KO neurons shows a mix between nonpolarized (44 ± 9.7% of the cases) and polarized neurons positive for the axonal marker Tau-1 (56 ± 9.7%). In comparison, the population of polarized neuron in WT cultures was 77 ± 1%. To continue our study in these cultures, we analyzed EPAC1 KO neurons at 3 DIV. We found that some neurons in EPAC1 KO cultures extended processes normally, but at this stage, the rate of polarization became lower compared with WT neurons. Quantification of neurons at 3 DIV showed a significant population of unpolarized neurons in EPAC1 KO compared with WT (Fig. 8d; unpolarized: EPAC1 KO 68 ± 0%; WT 32.3 ± 8%). This suggests that the majority of the mutant neurons failed to generate a normal axon. This approach demonstrated that, whereas WT hippocampal neurons typically possessed a normal axon positive for Tau-1 (Fig. 8c), the mutant cells usually did not form an extended axon (Fig. 8c). Finally, we sought to determine whether the EPAC1 KO neurons were able to generate multiaxonic neurons in the presence of 10 μm 8-pCPT. Figure 8e shows representative neurons of WT and KO neurons treated with 10 μm 8-pCPT. The treatment with 8-pCPT in WT neurons led to multiaxonic neurons formation with axons positive for Tau-1 compared with EPAC1 KO neurons that only displayed one axon (Fig. 8e). Altogether, these results confirm the effect in axon formation obtained with the antagonist of EPAC1 (ESI-09) and suggest that EPAC1 is required for normal polarization of hippocampal neurons in vitro and is sufficient to produce multiple axons during the development of neuronal polarity.
Discussion
Our results indicate that EPAC is the GEF involved in Rap1B-dependent axon formation during neuronal polarization. Rap1B activation is an essential step in the determination of neuronal polarity (Schwamborn and Püschel, 2004).
Four lines of evidence support the role of EPAC1 in this process during neuronal polarization. First, similar to other proteins involved in neuronal polarization, EPAC1 is present at the growth cones of every neurite in stage II hippocampal neurons but becomes restricted to the axon alone in stage III neurons. Second, a specific agonist for EPAC resulted in the formation of neurons displaying multiple axons showing increased spatiotemporal activation of endogenous Rap1B-GTP (Bivona et al., 2004). Third, CA forms of EPAC1 also induced the formation of supernumerary axons in cultured neurons. Fourth, specific inhibition, loss of function, and genetic deletion of EPAC1 delay axon formation in cultured neurons. In this work, we used an improved form of an EPAC activator that drives Rap1B activation in vitro and in vivo efficiently without any detectable effect on PKA (Enserink et al., 2002; Rehmann et al., 2003). The presence of multiple neurites in individual neurons that were longer than their siblings and expressed Tau-1 and MAP1B but not MAP2 strongly suggests that these neurites are axons. Under our experimental conditions, neurons had more than one axon initial segment, as characterized by AnkG staining. AnkG clustering is required to maintain neuronal polarity and recruit other proteins to the AIS and to form a diffusion barrier (Rasband, 2010; Galiano et al., 2012) involved in the generation of action potentials. Axons from these multiaxon neurons expressed other markers of mature axons that are involved in synapse formation such as vGLUT1 and synaptophysin. vGLUT1 expression is observed in postnatal and hippocampal neurons at 7–14 DIV with a characteristic punctate distribution along the axon (Melo et al., 2013), which is similar to the expression pattern of the vGLUT1-Venus construct used here (Wilson et al., 2005). Synaptophysin is a synaptic vesicle protein that is distributed along the axon, forming large punctate structures that correspond to the dense accumulation of vesicles within presynaptic specializations (Fletcher et al., 1991). Synaptophysin clusters were present in supernumerary axons, suggesting that neurons with exacerbated polarity could be functional. These results are consistent with a previous analysis of the molecular identity of multiple axons that involved staining for synapsin I and recycling experiments using FM4–64 uptake (Schwamborn et al., 2007).
We also investigated the effects of EPAC loss of function during neuronal polarization. Pharmacological and genetic EPAC1 reduction and deletion lowered the incidence of neurons displaying multiples axons and the percentage of polarized neurons and total axonal length was reduced in neurons lacking EPAC1 expression. Interestingly, axon formation was not impaired when we used DN EPAC1 or Rap1GAP overexpression. These findings could be explained by residual Rap activity that supported normal polarization because of other Rap1B GEFs such as PDZ-GEF (Hisata et al., 2007), C3G (Hisata et al., 2007), or CalDAG-GEFI (Kawasaki et al., 1998b).
We used recently developed EPAC pharmacological inhibitors, the results from which reinforce the idea that EPAC1 is the GEF responsible for Rap1B activation. Our results are consistent with studies showing that ESI-09 is a potent EPAC inhibitor and is 100-fold more selective for EPAC than PKA (Almahariq et al., 2013). Inhibition of EPAC1 with ESI-09 suppresses pancreatic cancer cell migration, similar to genetic inactivation using siRNA against EPAC (Almahariq et al., 2013). In addition, in vivo pharmacological inhibition of EPAC using ESI-09 recapitulates the effects of rickettsial infection that are found in EPAC1 KO mice (Gong et al., 2013). Finally, ESI-09 antagonizes myelin sheath formation and Schwann cell differentiation, which are linked to EPAC functions (Bacallao and Monje, 2013). Although we cannot rule out the possibility that EPAC2 may also be involved in axonogenesis because both EPAC1 and EPAC2 are expressed throughout the brain, including the hippocampus and prefrontal cortex, our subcellular distribution experiments and the differential effects of ESI-09 and ESI-05 suggest that EPAC1 is responsible for Rap1B activation during neuronal polarity. Interestingly, our results show that treatment with 8-pCPT in EPAC1 KO neurons does not induce multiaxonic neurons, which suggests that EPAC2 may not be involved in the generation of multiple axons in hippocampal neurons cultured in vitro. Alternatively, lower relative EPAC2 levels found in embryonic neurons may not be enough to promote multiaxonic neurons upon 8-pCPT treatments.
We also studied the cross-talk between cAMP/PKA- and cAMP/EPAC-dependent pathways. PKA regulates neuronal polarity by two mechanisms. PKA phosphorylates Smurf1 in response to brain-derived neurotrophic factor (BDNF), reducing the degradation of Par6—a member of the polarity complex—and enhancing the degradation of the small GTPase RhoA that is an axonal growth-inhibitor protein (Cheng et al., 2011a; Cheng et al., 2011b). PKA also activates LKB1, which phosphorylates and activates SAD/MARKS kinase, promoting axonogenesis (Shelly et al., 2007). The EPAC agonist did not modify Par6 or RhoA levels, indicating that an additional cAMP-dependent mechanism is involved early during the acquisition of neuronal polarity. cAMP had been proposed as a key cytoplasmic factor involved in axonal outgrowth. EPAC1 and EPAC2 bind cAMP with similar affinity as the PKA holoenzyme, suggesting that both factors may respond to similar physiological concentrations of this second messenger (Dao et al., 2006). The concerted functions of EPAC and PKA signaling are dependent on cellular context and processes (Grandoch et al., 2010). Activation of the EPAC-dependent pathway may target several molecules that regulate axon formation and elongation, such as c-Jun N-terminal kinase (Hochbaum et al., 2003; Oliva et al., 2006) and the small GTPases Rit (Shi and Andres, 2005; Shi et al., 2006), Ras (Li et al., 2006; López De Jesús et al., 2006; Yoshimura et al., 2006), and Rho (Schwamborn and Püschel, 2004; Moon et al., 2013). An additional regulation point for such a complementary mechanism is related to the extracellular cues that trigger neuronal polarization. BDNF elevates cytoplasmic cAMP, leading to increased axonal elongation (Cheng et al., 2011b; Nakamuta et al., 2011). In contrast, nerve growth factor is not able to trigger axon elongation in cultured neurons even though it induces the activation of C3G, another Rap1B-GEF (Nakamuta et al., 2011), reinforcing the present findings that EPAC is the GEF involved in Rap1B activation during neuronal polarization. Finally, extrinsic or intrinsic mechanisms triggering neuronal polarity ultimately modify cytoskeleton dynamics. Our results suggest that Rap1B and Cdc42 activity may not be linked directly. Future studies should address the impact of EPAC functions on the molecular mechanisms downstream of Rap1B, which include actin dynamics, regulation of polarity, and membrane delivery. To our knowledge, this is the first identification of a molecular mechanism explaining Rap1B activation during neuronal polarity, which in addition links two complementary signaling cascades activated by cAMP.
The cAMP–PKA-dependent pathway involved in selective stabilization and degradation of proteins, which is relevant to axon formation, seems complementary to cAMP-EPAC-Rap1B signaling in neuronal polarity, strengthening the idea that both signaling pathways cooperate in mediating axon formation and elongation.
Footnotes
P.M.-L. was supported by Comisión Nacional de Investigación Científica y Tecnológica (CONICYT)-Chile (Graduate and Becas-Chile internship fellowship), an Abel Tasman program fellowship from University of Groningen, and a Universidad de Chile internship fellowship. D.R.H. was supported by Fondecyt (Postdoctoral Grant 3130316). C.G.-B. was supported by Fondecyt (Grant 1140325) and Research Ring ACT-1114 from CONICYT, Chile. We thank Michael Handford for proofreading the final version of the manuscript.
References
- Almahariq M, Tsalkova T, Mei FC, Chen H, Zhou J, Sastry SK, Schwede F, Cheng X. A novel EPAC-specific inhibitor suppresses pancreatic cancer cell migration and invasion. Mol Pharmacol. 2013;83:122–128. doi: 10.1124/mol.112.080689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacallao K, Monje PV. Opposing roles of PKA and EPAC in the cAMP-dependent regulation of schwann cell proliferation and differentiation [corrected] PLoS One. 2013;8:e82354. doi: 10.1371/journal.pone.0082354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellocchio EE, Reimer RJ, Fremeau RT, Jr, Edwards RH. Uptake of glutamate into synaptic vesicles by an inorganic phosphate transporter. Science. 2000;289:957–960. doi: 10.1126/science.289.5481.957. [DOI] [PubMed] [Google Scholar]
- Bivona TG, Wiener HH, Ahearn IM, Silletti J, Chiu VK, Philips MR. Rap1 up-regulation and activation on plasma membrane regulates T cell adhesion. J Cell Biol. 2004;164:461–470. doi: 10.1083/jcb.200311093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng PL, Poo MM. Early events in axon/dendrite polarization. Annu Rev Neurosci. 2012;35:181–201. doi: 10.1146/annurev-neuro-061010-113618. [DOI] [PubMed] [Google Scholar]
- Cheng PL, Lu H, Shelly M, Gao H, Poo MM. Phosphorylation of E3 ligase Smurf1 switches its substrate preference in support of axon development. Neuron. 2011a;69:231–243. doi: 10.1016/j.neuron.2010.12.021. [DOI] [PubMed] [Google Scholar]
- Cheng PL, Song AH, Wong YH, Wang S, Zhang X, Poo MM. Self-amplifying autocrine actions of BDNF in axon development. Proc Natl Acad Sci U S A. 2011b;108:18430–18435. doi: 10.1073/pnas.1115907108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conde C, Cáceres A. Microtubule assembly, organization and dynamics in axons and dendrites. Nat Rev Neurosci. 2009;10:319–332. doi: 10.1038/nrn2631. [DOI] [PubMed] [Google Scholar]
- Dalton GD, Dewey WL. Protein kinase inhibitor peptide (PKI): a family of endogenous neuropeptides that modulate neuronal cAMP-dependent protein kinase function. Neuropeptides. 2006;40:23–34. doi: 10.1016/j.npep.2005.10.002. [DOI] [PubMed] [Google Scholar]
- Dao KK, Teigen K, Kopperud R, Hodneland E, Schwede F, Christensen AE, Martinez A, Døskeland SO. Epac1 and cAMP-dependent protein kinase holoenzyme have similar cAMP affinity, but their cAMP domains have distinct structural features and cyclic nucleotide recognition. J Biol Chem. 2006;281:21500–21511. doi: 10.1074/jbc.M603116200. [DOI] [PubMed] [Google Scholar]
- Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Rooij J, Zwartkruis FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A, Bos JL. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature. 1998;396:474–477. doi: 10.1038/24884. [DOI] [PubMed] [Google Scholar]
- Enserink JM, Christensen AE, de Rooij J, van Triest M, Schwede F, Genieser HG, Døskeland SO, Blank JL, Bos JL. A novel Epac-specific cAMP analogue demonstrates independent regulation of Rap1 and ERK. Nat Cell Biol. 2002;4:901–906. doi: 10.1038/ncb874. [DOI] [PubMed] [Google Scholar]
- Fletcher TL, Cameron P, De Camilli P, Banker G. The distribution of synapsin I and synaptophysin in hippocampal neurons developing in culture. J Neurosci. 1991;11:1617–1626. doi: 10.1523/JNEUROSCI.11-06-01617.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galiano MR, Jha S, Ho TS, Zhang C, Ogawa Y, Chang KJ, Stankewich MC, Mohler PJ, Rasband MN. A distal axonal cytoskeleton forms an intra-axonal boundary that controls axon initial segment assembly. Cell. 2012;149:1125–1139. doi: 10.1016/j.cell.2012.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gärtner A, Huang X, Hall A. Neuronal polarity is regulated by glycogen synthase kinase-3 (GSK-3beta) independently of Akt/PKB serine phosphorylation. J Cell Sci. 2006;119:3927–3934. doi: 10.1242/jcs.03159. [DOI] [PubMed] [Google Scholar]
- Gekel I, Neher E. Application of an Epac activator enhances neurotransmitter release at excitatory central synapses. J Neurosci. 2008;28:7991–8002. doi: 10.1523/JNEUROSCI.0268-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong B, Shelite T, Mei FC, Ha T, Hu Y, Xu G, Chang Q, Wakamiya M, Ksiazek TG, Boor PJ, Bouyer DH, Popov VL, Chen J, Walker DH, Cheng X. Exchange protein directly activated by cAMP plays a critical role in bacterial invasion during fatal rickettsioses. Proc Natl Acad Sci U S A. 2013;110:19615–19620. doi: 10.1073/pnas.1314400110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Billault C, Muñoz-Llancao P, Henríquez DR, Wojnacki J, Conde C, Cáceres A. The role of small GTPases in neuronal morphogenesis and polarity. Cytoskeleton. 2012;69:464–485. doi: 10.1002/cm.21034. [DOI] [PubMed] [Google Scholar]
- Grandoch M, Roscioni SS, Schmidt M. The role of Epac proteins, novel cAMP mediators, in the regulation of immune, lung and neuronal function. Br J Pharmacol. 2010;159:265–284. doi: 10.1111/j.1476-5381.2009.00458.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubb MS, Burrone J. Building and maintaining the axon initial segment. Curr Opin Neurobiol. 2010;20:481–488. doi: 10.1016/j.conb.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henríquez DR, Bodaleo FJ, Montenegro-Venegas C, González-Billault C. The light chain 1 subunit of the microtubule-associated protein 1B (MAP1B) is responsible for Tiam1 binding and Rac1 activation in neuronal cells. PLoS One. 2012;7:e53123. doi: 10.1371/journal.pone.0053123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hisata S, Sakisaka T, Baba T, Yamada T, Aoki K, Matsuda M, Takai Y. Rap1-PDZ-GEF1 interacts with a neurotrophin receptor at late endosomes, leading to sustained activation of Rap1 and ERK and neurite outgrowth. J Cell Biol. 2007;178:843–860. doi: 10.1083/jcb.200610073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochbaum D, Tanos T, Ribeiro-Neto F, Altschuler D, Coso OA. Activation of JNK by Epac is independent of its activity as a Rap guanine nucleotide exchanger. J Biol Chem. 2003;278:33738–33746. doi: 10.1074/jbc.M305208200. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- Jordan JD, He JC, Eungdamrong NJ, Gomes I, Ali W, Nguyen T, Bivona TG, Philips MR, Devi LA, Iyengar R. Cannabinoid receptor-induced neurite outgrowth is mediated by Rap1 activation through G(alpha)o/i-triggered proteasomal degradation of Rap1GAPII. J Biol Chem. 2005;280:11413–11421. doi: 10.1074/jbc.M411521200. [DOI] [PubMed] [Google Scholar]
- Kaech S, Banker G. Culturing hippocampal neurons. Nat Protoc. 2006;1:2406–2415. doi: 10.1038/nprot.2006.356. [DOI] [PubMed] [Google Scholar]
- Kawasaki H, Springett GM, Mochizuki N, Toki S, Nakaya M, Matsuda M, Housman DE, Graybiel AM. A family of cAMP-binding proteins that directly activate Rap1. Science. 1998a;282:2275–2279. doi: 10.1126/science.282.5397.2275. [DOI] [PubMed] [Google Scholar]
- Kawasaki H, Springett GM, Toki S, Canales JJ, Harlan P, Blumenstiel JP, Chen EJ, Bany IA, Mochizuki N, Ashbacher A, Matsuda M, Housman DE, Graybiel AM. A Rap guanine nucleotide exchange factor enriched highly in the basal ganglia. Proc Natl Acad Sci U S A. 1998b;95:13278–13283. doi: 10.1073/pnas.95.22.13278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiermayer S, Biondi RM, Imig J, Plotz G, Haupenthal J, Zeuzem S, Piiper A. Epac activation converts cAMP from a proliferative into a differentiation signal in PC12 cells. Mol Biol Cell. 2005;16:5639–5648. doi: 10.1091/mbc.E05-05-0432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordeli E, Lambert S, Bennett V. AnkyrinG. A new ankyrin gene with neural-specific isoforms localized at the axonal initial segment and node of Ranvier. J Biol Chem. 1995;270:2352–2359. doi: 10.1074/jbc.270.5.2352. [DOI] [PubMed] [Google Scholar]
- Kortholt A, Bolourani P, Rehmann H, Keizer-Gunnink I, Weeks G, Wittinghofer A, Van Haastert PJ. A Rap/phosphatidylinositol 3-kinase pathway controls pseudopod formation [corrected] Mol Biol Cell. 2010;21:936–945. doi: 10.1091/mbc.E09-03-0177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent AC, Bisserier M, Lucas A, Tortosa F, Roumieux M, De Régibus A, Swiader A, Sainte-Marie Y, Heymes C, Vindis C, Lezoualc'h F. Exchange protein directly activated by cAMP 1 promotes autophagy during cardiomyocyte hypertrophy. Cardiovasc Res. 2015;105:55–64. doi: 10.1093/cvr/cvu242. [DOI] [PubMed] [Google Scholar]
- Li Y, Asuri S, Rebhun JF, Castro AF, Paranavitana NC, Quilliam LA. The RAP1 guanine nucleotide exchange factor Epac2 couples cyclic AMP and Ras signals at the plasma membrane. J Biol Chem. 2006;281:2506–2514. doi: 10.1074/jbc.M508165200. [DOI] [PubMed] [Google Scholar]
- Lim BK, Huang KW, Grueter BA, Rothwell PE, Malenka RC. Anhedonia requires MC4R-mediated synaptic adaptations in nucleus accumbens. Nature. 2012;487:183–189. doi: 10.1038/nature11160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin D, Edwards AS, Fawcett JP, Mbamalu G, Scott JD, Pawson T. A mammalian PAR-3-PAR-6 complex implicated in Cdc42/Rac1 and aPKC signalling and cell polarity. Nat Cell Biol. 2000;2:540–547. doi: 10.1038/35019582. [DOI] [PubMed] [Google Scholar]
- López De Jesús M, Stope MB, Oude Weernink PA, Mahlke Y, Börgermann C, Ananaba VN, Rimmbach C, Rosskopf D, Michel MC, Jakobs KH, Schmidt M. Cyclic AMP-dependent and Epac-mediated activation of R-Ras by G protein-coupled receptors leads to phospholipase D stimulation. J Biol Chem. 2006;281:21837–21847. doi: 10.1074/jbc.M604156200. [DOI] [PubMed] [Google Scholar]
- Melo CV, Mele M, Curcio M, Comprido D, Silva CG, Duarte CB. BDNF regulates the expression and distribution of vesicular glutamate transporters in cultured hippocampal neurons. PLoS One. 2013;8:e53793. doi: 10.1371/journal.pone.0053793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montenegro-Venegas C, Tortosa E, Rosso S, Peretti D, Bollati F, Bisbal M, Jausoro I, Avila J, Cáceres A, González-Billault C. MAP1B regulates axonal development by modulating Rho-GTPase Rac1 activity. Mol Biol Cell. 2010;21:3518–3528. doi: 10.1091/mbc.E09-08-0709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon MY, Kim HJ, Kim JG, Lee JY, Kim J, Kim SC, Choi IG, Kim PH, Park JB. Small GTPase Rap1 regulates cell migration through regulation of small GTPase RhoA activity in response to transforming growth factor-beta1. J Cell Physiol. 2013;228:2119–2126. doi: 10.1002/jcp.24383. [DOI] [PubMed] [Google Scholar]
- Murray AJ. Pharmacological PKA inhibition: all may not be what it seems. Sci Signal. 2008;1:re4. doi: 10.1126/scisignal.122re4. [DOI] [PubMed] [Google Scholar]
- Murray AJ, Shewan DA. Epac mediates cyclic AMP-dependent axon growth, guidance and regeneration. Mol Cell Neurosci. 2008;38:578–588. doi: 10.1016/j.mcn.2008.05.006. [DOI] [PubMed] [Google Scholar]
- Murray AJ, Tucker SJ, Shewan DA. cAMP-dependent axon guidance is distinctly regulated by Epac and protein kinase A. J Neurosci. 2009;29:15434–15444. doi: 10.1523/JNEUROSCI.3071-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawara A. Trk receptor tyrosine kinases: a bridge between cancer and neural development. Cancer Lett. 2001;169:107–114. doi: 10.1016/S0304-3835(01)00530-4. [DOI] [PubMed] [Google Scholar]
- Nakamuta S, Funahashi Y, Namba T, Arimura N, Picciotto MR, Tokumitsu H, Soderling TR, Sakakibara A, Miyata T, Kamiguchi H, Kaibuchi K. Local application of neurotrophins specifies axons through inositol 1,4,5-trisphosphate, calcium, and Ca2+/calmodulin-dependent protein kinases. Sci Signal. 2011;4:ra76. doi: 10.1126/scisignal.2002011. [DOI] [PubMed] [Google Scholar]
- Nishimura T, Yamaguchi T, Kato K, Yoshizawa M, Nabeshima Y, Ohno S, Hoshino M, Kaibuchi K. PAR-6-PAR-3 mediates Cdc42-induced Rac activation through the Rac GEFs STEF/Tiam1. Nat Cell Biol. 2005;7:270–277. doi: 10.1038/ncb1227. [DOI] [PubMed] [Google Scholar]
- Oldenburger A, Timens W, Bos S, Smit M, Smrcka AV, Laurent AC, Cao J, Hylkema M, Meurs H, Maarsingh H, Lezoualc'h F, Schmidt M. Epac1 and Epac2 are differentially involved in inflammatory and remodeling processes induced by cigarette smoke. FASEB J. 2014;28:4617–4628. doi: 10.1096/fj.13-248930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliva AA, Jr, Atkins CM, Copenagle L, Banker GA. Activated c-Jun N-terminal kinase is required for axon formation. J Neurosci. 2006;26:9462–9470. doi: 10.1523/JNEUROSCI.2625-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponsioen B, Gloerich M, Ritsma L, Rehmann H, Bos JL, Jalink K. Direct spatial control of Epac1 by cyclic AMP. Mol Cell Biol. 2009;29:2521–2531. doi: 10.1128/MCB.01630-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasband MN. The axon initial segment and the maintenance of neuronal polarity. Nat Rev Neurosci. 2010;11:552–562. doi: 10.1038/nrn2852. [DOI] [PubMed] [Google Scholar]
- Rehmann H, Schwede F, Døskeland SO, Wittinghofer A, Bos JL. Ligand-mediated activation of the cAMP-responsive guanine nucleotide exchange factor Epac. J Biol Chem. 2003;278:38548–38556. doi: 10.1074/jbc.M306292200. [DOI] [PubMed] [Google Scholar]
- Rehmann H, Das J, Knipscheer P, Wittinghofer A, Bos JL. Structure of the cyclic-AMP-responsive exchange factor Epac2 in its auto-inhibited state. Nature. 2006;439:625–628. doi: 10.1038/nature04468. [DOI] [PubMed] [Google Scholar]
- Rehmann H, Arias-Palomo E, Hadders MA, Schwede F, Llorca O, Bos JL. Structure of Epac2 in complex with a cyclic AMP analogue and RAP1B. Nature. 2008;455:124–127. doi: 10.1038/nature07187. [DOI] [PubMed] [Google Scholar]
- Sabo SL, Gomes RA, McAllister AK. Formation of presynaptic terminals at predefined sites along axons. J Neurosci. 2006;26:10813–10825. doi: 10.1523/JNEUROSCI.2052-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwamborn JC, Püschel AW. The sequential activity of the GTPases Rap1B and Cdc42 determines neuronal polarity. Nat Neurosci. 2004;7:923–929. doi: 10.1038/nn1295. [DOI] [PubMed] [Google Scholar]
- Schwamborn JC, Müller M, Becker AH, Püschel AW. Ubiquitination of the GTPase Rap1B by the ubiquitin ligase Smurf2 is required for the establishment of neuronal polarity. EMBO J. 2007;26:1410–1422. doi: 10.1038/sj.emboj.7601580. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Shelly M, Cancedda L, Heilshorn S, Sumbre G, Poo MM. LKB1/STRAD promotes axon initiation during neuronal polarization. Cell. 2007;129:565–577. doi: 10.1016/j.cell.2007.04.012. [DOI] [PubMed] [Google Scholar]
- Shelly M, Lim BK, Cancedda L, Heilshorn SC, Gao H, Poo MM. Local and long-range reciprocal regulation of cAMP and cGMP in axon/dendrite formation. Science. 2010;327:547–552. doi: 10.1126/science.1179735. [DOI] [PubMed] [Google Scholar]
- Shelly M, Cancedda L, Lim BK, Popescu AT, Cheng PL, Gao H, Poo MM. Semaphorin3A regulates neuronal polarization by suppressing axon formation and promoting dendrite growth. Neuron. 2011;71:433–446. doi: 10.1016/j.neuron.2011.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi GX, Andres DA. Rit contributes to nerve growth factor-induced neuronal differentiation via activation of B-Raf-extracellular signal-regulated kinase and p38 mitogen-activated protein kinase cascades. Mol Cell Biol. 2005;25:830–846. doi: 10.1128/MCB.25.2.830-846.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi GX, Rehmann H, Andres DA. A novel cyclic AMP-dependent Epac-Rit signaling pathway contributes to PACAP38-mediated neuronal differentiation. Mol Cell Biol. 2006;26:9136–9147. doi: 10.1128/MCB.00332-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi SH, Jan LY, Jan YN. Hippocampal neuronal polarity specified by spatially localized mPar3/mPar6 and PI 3-kinase activity. Cell. 2003;112:63–75. doi: 10.1016/S0092-8674(02)01249-7. [DOI] [PubMed] [Google Scholar]
- Suzuki S, Yokoyama U, Abe T, Kiyonari H, Yamashita N, Kato Y, Kurotani R, Sato M, Okumura S, Ishikawa Y. Differential roles of Epac in regulating cell death in neuronal and myocardial cells. J Biol Chem. 2010;285:24248–24259. doi: 10.1074/jbc.M109.094581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsalkova T, Mei FC, Cheng X. A fluorescence-based high-throughput assay for the discovery of exchange protein directly activated by cyclic AMP (EPAC) antagonists. PLoS One. 2012a;7:e30441. doi: 10.1371/journal.pone.0030441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsalkova T, Mei FC, Li S, Chepurny OG, Leech CA, Liu T, Holz GG, Woods VL, Jr, Cheng X. Isoform-specific antagonists of exchange proteins directly activated by cAMP. Proc Natl Acad Sci U S A. 2012b;109:18613–18618. doi: 10.1073/pnas.1210209109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson NR, Kang J, Hueske EV, Leung T, Varoqui H, Murnick JG, Erickson JD, Liu G. Presynaptic regulation of quantal size by the vesicular glutamate transporter VGLUT1. J Neurosci. 2005;25:6221–6234. doi: 10.1523/JNEUROSCI.3003-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolfrey KM, Srivastava DP, Photowala H, Yamashita M, Barbolina MV, Cahill ME, Xie Z, Jones KA, Quilliam LA, Prakriya M, Penzes P. Epac2 induces synapse remodeling and depression and its disease-associated forms alter spines. Nat Neurosci. 2009;12:1275–1284. doi: 10.1038/nn.2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura T, Arimura N, Kawano Y, Kawabata S, Wang S, Kaibuchi K. Ras regulates neuronal polarity via the PI3-kinase/Akt/GSK-3beta/CRMP-2 pathway. Biochem Biophys Res Commun. 2006;340:62–68. doi: 10.1016/j.bbrc.2005.11.147. [DOI] [PubMed] [Google Scholar]