

Abstract
Parvalbumin (Pv)-positive inhibitory interneurons effectively control network excitability, and their optogenetic activation has been reported to block epileptic seizures. An intense activity in GABAergic interneurons, including Pv interneurons, before seizures has been described in different experimental models of epilepsy, raising the hypothesis that an increased GABAergic inhibitory signal may, under certain conditions, initiate seizures. It is therefore unclear whether the activity of Pv interneurons enhances or opposes epileptiform activities. Here we use a mouse cortical slice model of focal epilepsy in which the epileptogenic focus can be identified and the role of Pv interneurons in the generation and propagation of seizure-like ictal events is accurately analyzed by a combination of optogenetic, electrophysiological, and imaging techniques. We found that a selective activation of Pv interneurons at the focus failed to block ictal generation and induced postinhibitory rebound spiking in pyramidal neurons, enhancing neuronal synchrony and promoting ictal generation. In contrast, a selective activation of Pv interneurons distant from the focus blocked ictal propagation and shortened ictal duration at the focus. We revealed that the reduced ictal duration was a direct consequence of the ictal propagation block, probably by preventing newly generated afterdischarges to travel backwards to the original focus of ictal initiation. Similar results were obtained upon individual Pv interneuron activation by intracellular depolarizing current pulses. The functional dichotomy of Pv interneurons here described opens new perspectives to our understanding of how local inhibitory circuits govern generation and spread of focal epileptiform activities.
Keywords: channelrhodopsin-2, cortex, epileptiform activity, optogenetics, parvalbumin, rebound spiking
Introduction
Within the brain, inhibitory signals that originate from different GABAergic interneurons play fundamental roles in establishing proper operation of neural circuits (Markram et al., 2004; Monyer and Markram, 2004; Kepecs and Fishell, 2014). GABA-mediated inhibition shapes the response of principal neurons to synaptic inputs, controls local circuit excitability, and generates network oscillations by synchronizing the firing in large neuronal ensembles (Bartos et al., 2007; Buzsáki and Wang, 2012). GABAergic interneurons are also involved in the pathophysiology of brain disorders, including epilepsy, where they may, under certain conditions, favor seizure initiation (Avoli and de Curtis, 2011; Jiruska et al., 2013). However, GABA antagonists are ictogenic (Gutnick et al., 1982; Miles and Wong, 1983; Connors, 1984), and inhibition is commonly thought to counteract seizures. Indeed, GABAergic interneurons, and in particular parvalbumin (Pv)-expressing interneurons, can generate a feedforward inhibition that opposes seizure spread in both experimental models (Trevelyan et al., 2006; Cammarota et al., 2013) and patients (Schevon et al., 2012). Moreover, whereas studies in brain slices suggest that optogenetic activation of GABAergic interneurons can enhance epileptiform activity (Shiri et al., 2014; Yekhlef et al., 2014), optogenetic in vivo studies reveal that Pv interneuron activation interrupts spontaneous ongoing seizures (Krook-Magnuson et al., 2013; Paz et al., 2013). Optogenetics was thus proposed as a promising approach for those epilepsies, such as temporal lobe epilepsy, that have typical activity arising from restricted epileptogenic sites. Increasing inhibition at this site by Pv interneuron optogenetic activation may be technically feasible and represent a potential alternative to resective surgery commonly used in untreatable temporal lobe epilepsy forms. Most importantly, it has the potential not only to interrupt ongoing seizures, but also to suppress the abnormal activity that precedes seizures preventing seizure generation. To test this hypothesis, light has to be delivered at the epileptogenic focus. However, the unpredictable nature of focal seizures renders the identification of the epileptogenic focus in the intact brain a daunting challenge. We thus used a focal epilepsy model in temporal cortex (TCx) slices (Losi et al., 2010) that gives the unique opportunity to identify the precise site of seizure initiation and to generate an epileptic event at any given time. The hallmark of this in vitro model is that it allows precisely controlled experiments that cannot be accomplished in currently available in vivo models, where the seizure initiation site is not identifiable in advance. The fact that in our model we know where and when the seizure will be generated allowed us to investigate whether and how Pv interneurons differentially control initiation and propagation of focal epileptiform activities. We addressed two main questions that will clarify the role of these cells in epileptiform activities. First, does Pv interneuron activation suppress the ictogenic activity that, at the epileptogenic focus, contributes to ictal event generation? Second, does Pv interneuron activation out of the focus (i.e., the ictal propagating region) oppose ictal event spread? To answer these questions, we combined single/dual cell patch-clamp and local field potential recordings with optogenetics and Ca2+ imaging in cortical slice preparations.
Materials and Methods
Animals.
All experiments involving animals were performed in strict accordance with the guidelines established by the European Communities Council Directive and approved by the National Council on Animal Care of the Italian Ministry of Health. The PV-IRES-Cre (parvalbumin internal ribosomal entry site (IRES) Cre-recombinase) mouse strain (B6;129P2-Pvalbtm1(cre)Arbr/J, id #017320) was purchased from The Jackson Laboratory. The G42 mouse strain (Chattopadhyaya et al., 2004) was donated by Alberto Bacci (ICM- Institut du Cerveau et de la Moelle épinière, Paris, France). Control slices were obtained from littermates mice of the corresponding mouse strain.
Viral injections.
Stereotaxic injections were performed on postnatal day 0–2 (P0-P2) PV-IRES-Cre mice. Pups were deeply anesthetized by hypothermia and placed on a custom neonatal stereotaxic apparatus maintained at 4°C during surgery; 100–200 nl of AAV2/1.EF1.dflox.hChR2(H134R)-mCherry.WPRE.hGH (Addgene 20297) or AAV1.EF1a. DIO.hChR2(H134R)-eYFP.WPRE.hGH (Addgene 20298, University of Pennsylvania Viral Vector Core) was injected at stereotaxic coordinates of 0.85 mm anterior from the λsuture, 2.5 mm lateral from midline, and 0.15 mm depth, by means of a glass micropipette. After injections, the skin was sutured and pups were revitalized under a heat lamp and returned to the dam.
Slice preparations and Ca2+ dye loading.
Coronal slices were prepared from G42 mice, and Pv-Cre mice of either sex injected with saline solution or with AAV between postnatal day 15 and 23. Briefly, mice were deeply anesthetized with intraperitoneal injected Zoletil (40 mg/kg) and decapitated; the brain was quickly removed and transferred to ice-cold standard ACSF, sACSF (in mm as follows: 125 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 25 glucose, pH 7.4, with 5% CO2/95% O2). After brain dissection on the coronal plane, 350-μm-thick slices were cut with a vibratome (VT1000S, Leica Microsystems) in the solution described previously (Dugué et al., 2005). Slices were transferred for 1 min in a 95% O2 and 5% CO2 saturated solution containing (in mm) the following: 225 d-mannitol, 2.5 KCl, 1.25 NaH2PO4, 26 NaHCO3, 25 glucose, 0.8 CaCl2, 8 MgCl2, 2 kynurenic acid with 5% CO2/95% O2. Slices were finally transferred in sACSF at 30°C for 20 min and then maintained at room temperature.
Slice loading with the fluorescence Ca2+ indicator Rhod-2 AM (10 μm; Invitrogen) was performed for 50–60 min at 30°C. Although Rhod-2 is known to accumulate in mitochondria (Kovács et al., 2005), with this loading protocol, it was highly present in the cytosol. Dye loading was performed in the sACSF solution containing sulfinpyrazone (200 μm, Sigma-Aldrich), pluronic (0.12%, Sigma-Aldrich), and kynurenic acid (1 mm, Sigma-Aldrich). After loading, slices were recovered and kept at room temperature.
Ca2+ imaging.
Images were acquired with a single-photon upright laser-scanning microscope (TCS-SP5-RS, Leica Microsystems) with an acquisition time frame from 351 to 491 ms (five to seven line averaging). Rhod-2 AM was excited at 543 nm. The microscope was also equipped with a CCD camera for differential interference contrast image acquisition.
Electrophysiology and induction of focal ictal discharges.
Brain slices were continuously perfused in a submerged chamber (Warner Instruments) at a rate of 3–4 ml/min with (in mm) the following: 120 NaCl, 3.2 KCl, 1 KH2PO4, 26 NaHCO3, 0.5 MgCl2, 2 CaCl2, 10 glucose, pH 7.4 (with 5% CO2/95% O2). Single- and dual-cell recordings were performed in current-clamp and voltage-clamp configuration using a multiclamp-700B amplifier (Molecular Devices). Signals were filtered at 1 kHz and sampled at 10 kHz with a Digidata 1440s interface and pClamp10 software (Molecular Devices). Typical pipette resistance was 3–4 mΩ. Access resistance was monitored throughout the recordings and was typically <25 mΩ. Whole-cell intracellular pipette solution was (in mm) as follows: 145 K-gluconate, 2 MgCl2, 0.5 EGTA, 2 Na2ATP, 0.2 Na2GTP, 10 HEPES, pH 7.2, with KOH, osmolarity, 305–315 mOsm (calculated liquid junction potential: −14 mV). Data were not corrected for the liquid junction potential.
Pyramidal cells were identified on the basis of their distinct morphology characterized by the triangular shape of the soma, a main apical dendrite pointing toward the pia and the absence of a main dendrite in the opposite direction. Their biophysical identity was confirmed by their response to hyperpolarizing and depolarizing 750 ms current steps. In particular, regular spiking pyramidal neurons showed a firing discharge with no spike amplitude accommodation (except for the second action potential in some cells), small afterhyperpolarization, and low steady-state frequency (15–23 Hz with 200 pA current injection). ChR2-positive Pv-fast spiking (FS) cells were identified by fluorescence and their biophysical identity was confirmed by high steady-state firing frequency (50 – 90 Hz with 400 pA current injection), no spike amplitude accommodation or frequency adaptation and large afterhyperpolarization. The following parameters were measured to characterize passive membrane properties: resting membrane potential (Vrest) was recorded immediately after the rupture of the neuronal membrane; input resistance (Rin) was determined by measuring the voltage change in response to a small hyperpolarizing and depolarizing current pulses (±50 pA, 750 ms) at resting potential.
All patched neurons were from cortical layer V-VI. All pyramidal neurons were voltage-clamped at −50 mV. In Pv-FS interneurons, we performed whole-cell current-clamp recordings at resting potential or juxtasomal recordings without rupture of the membrane patch using pipettes filled with the bath perfusing solution. Ictal event induction experiments were performed in the presence of 4-aminopyridine (4-AP, 50–100 μm) and bath temperature was maintained at 30–32°C by an inline solution heater and temperature controller (TC-324B, Warner Instruments). A pressure ejection unit (PDES, NPI Electronics) was used to apply a single or double pulse to NMDA (1 mm, Sigma-Aldrich)-containing pipettes with a 3 s interval, a pressure of 4–10 psi, and a duration of 200–600 ms. The NMDA pipette was positioned in vicinity of the rhinal fissure. The epileptogenic focus is defined as the region up to 300 μm from the pipette tip where neurons are directly activated by NMDA (Gómez-Gonzalo et al., 2010), whereas the ictal propagating region is the region distant at least 400 μm from the NMDA pipette tip. In most experiments, the NMDA glass pipette included an AgCl2 electrode for extracellular local field potential recordings. Field potential signals were filtered at 1 kHz, amplified by an AM-amplifier (AM Systems), and sampled at 10 kHz.
Optical stimulation.
Photostimulation was performed with a 473 nm continuous wave, solid-state laser source (Cobolt). The laser was directly controlled by a command voltage with a TTL signal. Light was delivered to brain slices via an optical fiber (ThorLabs) with 50 or 300 μm diameter positioned close to the surface of the slice. Blue light intensity was adjusted with neutral density filters (ThorLabs) to obtain a power between 1 and 3 mW (measured at the fiber tip). The photostimulated area for the 300 μm and for the 50 μm diameter fiber positioned at ∼300 μm from the slice surface was evaluated to be ∼0.9 mm2 and 0.048 mm2, respectively.
Data analysis.
Data analysis was performed with Clampfit 10, Origin 8.0 (Microcal Software), Microsoft Office and MATLAB 7.6.0 R2008A (MathWorks).
Ictal events analysis.
In voltage-clamp recordings, the recruitment of principal neurons into propagating ictal events was marked by the transition from the predominant inhibitory to the predominant excitatory phase (tIE), which is defined as the timing in which the ratio between the inhibition and the excitation index is for the first time <0.1. Inhibition and excitation indices were calculated, respectively, as the part of the positive and the negative component of the time derivative of the trace that exceeded 5× the mean standard deviation (SD) evaluated during the baseline (Cammarota et al., 2013). Ictal event duration in voltage-clamp recordings was calculated from the tIE to the end of the last afterdischarge (see Afterdischarge analysis, below) recorded with an instant frequency >0.1 Hz.
Local field potential recordings were filtered with low-pass Gaussian filter. The ictal discharge onset at the focus was considered as the first detectable event in the field potential recordings larger than 4× the mean SD. The end of the ictal discharge at the focus was determined as the last epileptic event detectable in the field potential recordings. Action potentials in juxtasomal recordings from Pv interneurons were identified as spikes in voltage-clamp at 0 mV that exceeded 5× the mean SD evaluated during the baseline.
Afterdischarge analysis.
The afterdischarges typically characterize the clonic activity that follows the tonic phase of long-lasting ictal events and were identified in voltage-clamp recordings as large excitatory bursts of <3 s duration. Afterdischarge onsets were set by the amplitude of the event that exceeded 3× the mean SD evaluated during the baseline. To evaluate the delay between excitatory bursts in voltage-clamp recordings from two pyramidal neurons located at different distances from the epileptogenic focus, we calculated the cross-correlation between the time derivative of both signals, in time bins of 1000 ms separated by 30 ms. We finally represented the pseudo-colors plot of the cross-correlation. We measured the lag corresponding to the maximal value of the cross-correlation before and after the change in direction of the afterdischarge.
Synchrony of pyramidal neurons.
Spikes were recorded in current-clamp mode and the spike timing was obtained by thresholding the traces in Clampfit. Files holding the spike timing were analyzed by custom software to compute the nearest neighbor (NN) distance of each spike. The distance was always computed from the spikes recorded in the cell that had a lower frequency of firing. A simulated spike train was obtained by randomly placing spikes on a 2 s time window respecting the basic statistics of the original data: the randomized data had the same spike density and similar distribution of distance from the preceding spike. The simulation of the experiments that included optogenetic activation included a refractory window, determined for each simulated cell, in which spike density was zero.
Ca2+ imaging experiments.
The Ca2+ signal is reported as ΔF/F0, where F0 is the baseline fluorescence. Significant Ca2+ events were considered when exceeding 3× SD of baseline. No background subtraction or other manipulations were applied to digitized Ca2+ signal images that are reported as raw data. Ca2+ signals from neurons were obtained using Leica Applicaction Suite (Leica Microsystems).
Statistical analysis.
Data are shown as mean ± SEM. Quantitative results were analyzed using t test and paired t test setting the statistical significance at p < 0.05. Wilcoxon signed-rank test was used to evaluate the statistical significance of the firing rate change in pyramidal neurons (see Fig. 5). Cumulative probability distributions of Figure 6 were compared using Kolmogorov–Smirnov test.
Figure 5.

Optogenetic activation of Pv interneurons induce postinhibitory rebound spiking in pyramidal neurons. A, B, Representative simultaneous current-clamp recordings from a pyramidal neuron, depolarized to action potential threshold by steady-state current injection, and a Pv interneuron in the absence (A, left panel) and presence of pulsed light stimulation at 0.5 Hz (B, left panels). Recordings were performed in the presence of 4-AP and 0.5 mm Mg2+. Histograms of the relative firing rate as a function of time, calculated over 50 ms time bins, obtained by the analysis of 300 consecutive sweeps (A, B, right panels). Open arrowheads indicate the main spike clustering. Red line indicates the multipeak fitting of the firing rate over time (reduced χ2 = 0.99). Calibration: 200 ms, 20 mV. C, Distribution of normalized firing rate changes during rebound spiking both in basal conditions (empty circles, n = 10) and in the presence of 4-AP (full circles, n = 14). The mean value of both datasets is represented by the empty square (284 ± 28.1%, n = 24 cells, 14 slices, 6 mice). ***p < 0.001 (Wilcoxon signed-rank test).
Figure 6.
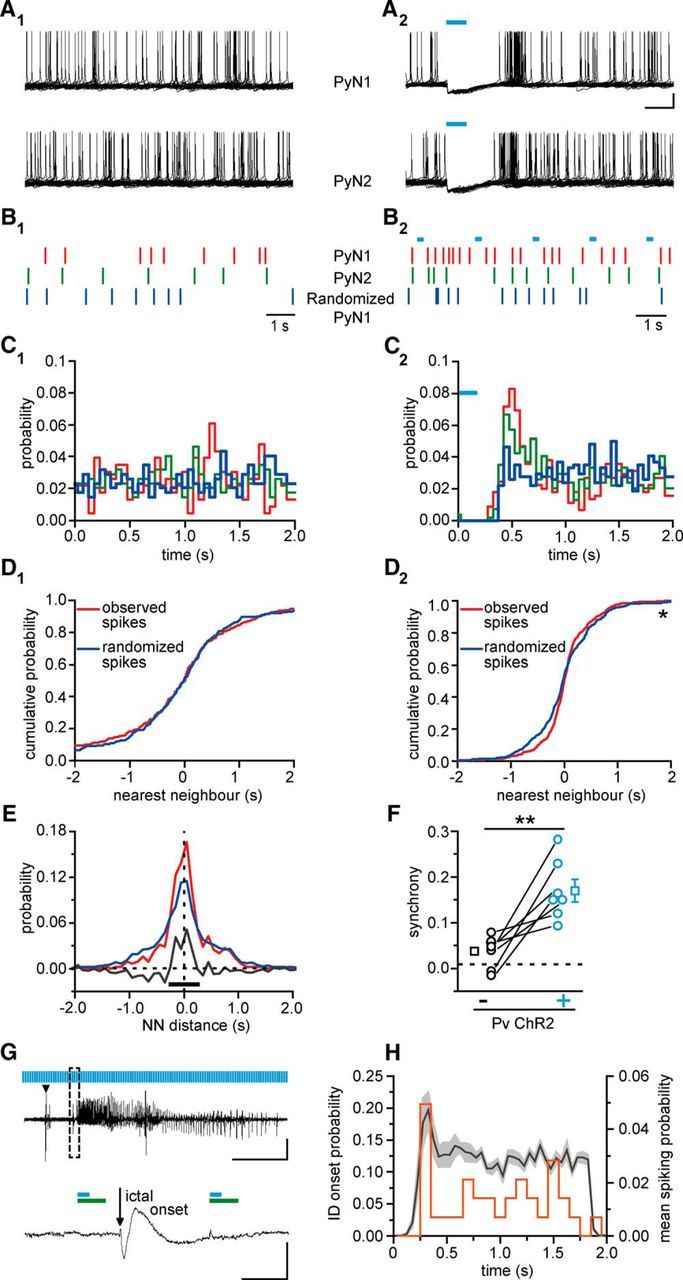
Optogenetic activation of Pv interneurons promotes synchronous firing in pyramidal neurons. A1, A2, Spike trains recorded in 2-s-long sweeps from two neighboring pyramidal neurons in the absence (A1) and in the presence (A2) of light pulse stimulation at 0.5 Hz. Cyan bars represent 150 ms light pulses. The rhythmic activation of Pv interneurons causes a temporal modulation of spikes in both pyramidal neurons. Calibration: 200 ms, 20 mV. B1, B2, Raster plots showing the spike timing of the two pyramidal neurons (red and green bars) and a randomized trial (blue bars). C1, C2, Probability distribution in the 2 s period in the absence (C1) and in the presence (C2) of rhythmic optogenetic activation of Pv interneurons. D1, D2, For each spike produced by the pyramidal neuron 2 (PyN2), the distance from the closest spike produced by PyN1 was computed. The observed cumulative distribution of the nearest neighbor distances is plotted in red (n = 230 spikes). The distribution was computed again after randomization of the spike train of PyN1 (blue trace, n = 230 spikes). In the presence of optogenetic stimulation (D2), the difference between the observed cumulative distribution and the cumulative distribution obtained after randomization is statistically significant (n = 447 spikes, *p < 0.05, Kolomogorov–Smirnov test). E, The integral of the difference between observed and simulated distributions has been computed in an interval centered ∼0, returning a measure of the synchronization between the two pyramidal neurons. F, Summary report for the increase in pyramidal neuron synchrony induced by Pv interneuron activation (n = 7 pairs, 5 mice) in the presence (black circles) and in the absence (cyan circles) of pulsed light stimulation both in basal ACSF and 4-AP. **p < 0.01. G, Representative local field potential recording of an NMDA-evoked ictal event in the presence of light pulse stimulation. The ictal event onset between two light pulses is shown at enlarged scales. Green bars represent the firing suppression period in pyramidal neurons induced by light stimulation of ChR2-expressing Pv interneurons (cyan bars, 150 ms at 0.5 Hz). Calibration: 40 s, 0.01 mV; inset, 500 ms, 0.01 mV. H, Distribution of both the ictal onset probability (orange line, n = 31 ictal events in 100 ms time bins, 17 mice) and the pyramidal neuron mean spiking probability in 4-AP (light gray represents SEM; n = 24 cells, 6 mice; 50 ms time bins) as a function of time during pulsed light stimulation of Pv interneurons. The 0 value is set as the end of the light stimulus.
Tissue processing and immunohistochemical procedures.
Postnatal day 21 (P21) mice were deeply anesthetized with an intraperitoneal injection of Zoletil (40 mg/kg) and perfused transcardially with 0.9% saline, followed by 4% PFA in 0.1 m phosphate buffer (PB), pH 7.4. Brains were removed from the skull, postfixed for 6 h in the same solution, cryoprotected in a 30% sucrose solution in 0.1 m PB, pH 7.4, frozen, and cryostat sectioned (Leica Microsystems). Free-floating coronal serial sections (30 μm) were collected in multiwell dishes. For immunofluorescence, sections were incubated overnight at 4°C in primary antibody diluted in 0.01 m PBS, pH 7.4, 0.5% Triton X-100, and 1% normal serum of the same species of the secondary antibody, and then incubated for 1 h at room temperature in the appropriate secondary antibody. Sections were mounted and coverslipped with a DABCO [1,4-diazobicyclo-(2,2,2)octane]-based antifade mounting medium. The following primary antibodies were used: anti-GABA (1:1000 rabbit, Sigma, A2052) and anti-paravalbumin (PV) (1:1000; mouse; Sigma, P3088). Secondary antibodies were as follows: goat anti-rabbit Alexa-488 (1:800, Invitrogen, A11034) and goat anti-mouse Alexa-488 (1:800, Invitrogen, A11029).
Confocal single image and z-stacks (1 μm optical steps) were acquired with a Leica SP5 inverted confocal microscope (Leica Microsystems). The percentage of mCherry-positive cells in temporal cortex (3 slices for each animal, range −2.92 to −4.04 mm from bregma) expressing either GABA or paravalbumin was evaluated on confocal z-stacks captured through the thickness of the slice using ImageJ/Fiji software. Data are mean ± SEM.
Results
The expression of the light-gated cation channel channelrhodopsin-2 (ChR2) in Pv interneurons was induced by injecting AAV vectors carrying the doublefloxed ChR2 sequence in Pv-Cre mouse pups (ChR2 Pv mice). ChR2 was tagged with the red fluorescent reporter mCherry. Immunohistological experiments in TCx slice preparations obtained from postnatal day 18–23 ChR2 Pv mice showed that a large fraction of ChR2-positive cells stained for GABA (96.7 ± 0.97%, n = 319 cells, 3 mice; Fig. 1Aa2–a5) and for Pv (96.8 ± 1.00%, n = 1006 cells, 6 mice; Fig. 1Aa6–a9). By using Pv-Cre mice crossed with a red fluorescence reporter line (floxed td-Tomato) and injected with AAV carrying the ChR2 sequence tagged with the enhanced yellow fluorescent protein (eYFP), we quantified that 61.3 ± 5.80% (n = 1456 cells, 6 mice) of PV cells expressed ChR2. All recorded ChR2-positive cells displayed a fast-spiking discharge pattern compatible with those of Pv-positive fast spiking interneurons (Levy and Reyes, 2012; Schiff and Reyes, 2012). Single-cell patch-clamp recordings from ChR2-positive cells revealed that blue light illumination (λ = 473 nm) reliably lead to membrane depolarization and action potential firing (Fig. 1B). The rate of action potential induced by light could be reliably mimicked by applying intracellular current pulses of different amplitudes (mean current injection, 320 ± 33.3 pA, ChR2-mediated mean firing rate, 73 ± 2.8 Hz; n = 8 cells, 6 mice; Fig. 1B). In the presence of the proconvulsant agent 4-AP, the firing rate evoked by light pulses was similar to control conditions (p = 0.26; n = 5, 5 mice, paired t test; Fig. 1C). Light-induced action potential exhibited unchanged mean amplitude and half-width, and a reduced threshold (amplitude, 69.3 ± 2.46 vs 68.6 ± 4.14 mV, p = 0.84; half-width, 1.07 ± 0.081 vs 0.95 ± 0.101 ms, p = 0.41; threshold, −59.7 ± 0.40 mV vs −56.7 ± 1.08, p = 0.03; n = 5 cells, 5 mice, paired t test).
Figure 1.
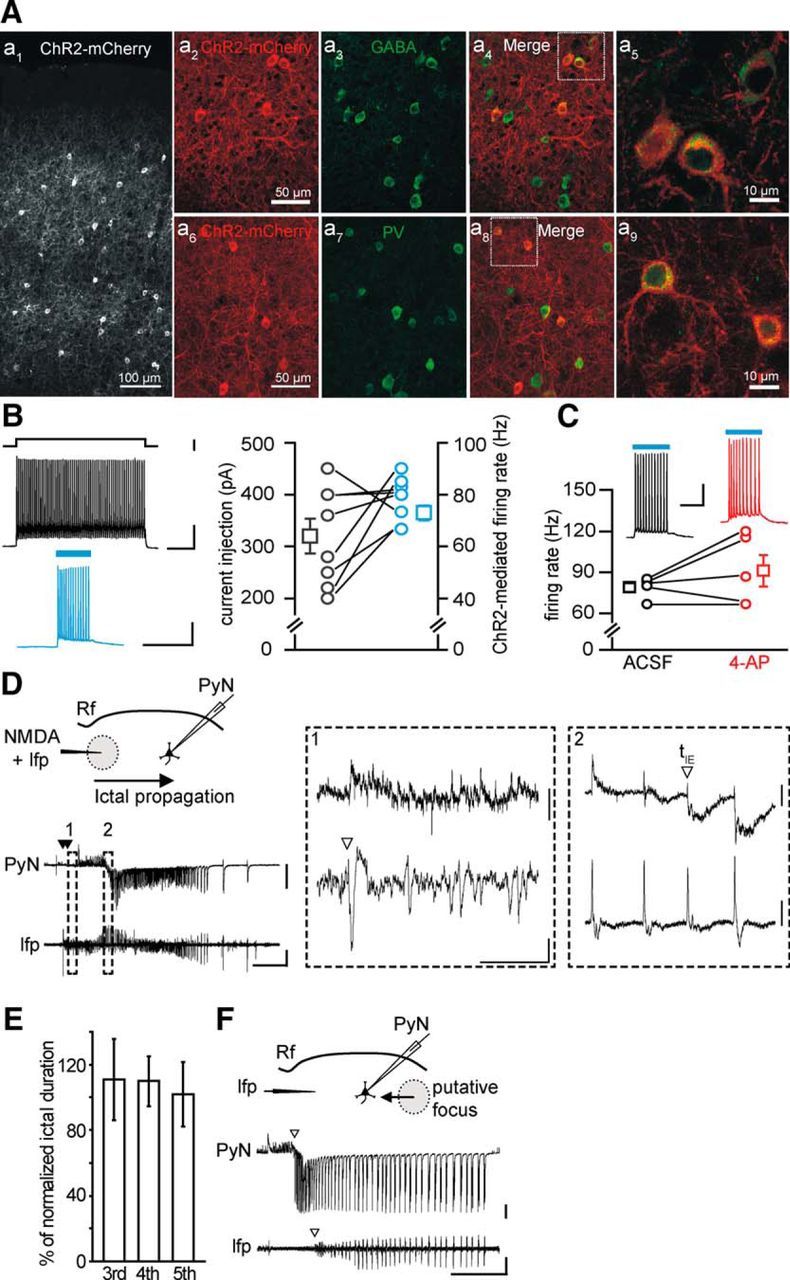
Characterization of ChR2-expressing neurons in Pv-Cre injected mice. A, Immunohistological characterization of ChR2-expressing neurons. Aa1, Confocal image of a cortical section from Pv-Cre mice injected with AAV-transducing ChR2-mCherry. Aa2–a5, A large fraction of ChR2-mCherry-positive cells (a2) stains for GABA (a3, n = 325 cells, 3 mice). The ChR2-mCherry and GABA fluorescence signals are shown merged in a4 (multiconfocal stack). A higher-magnification image of two ChR2-mCherry and Pv-positive cells is shown in a5 (single confocal image). Aa6–a9, Same as in a2–a5 for cortical sections that were stained against PV (n = 319 cells, 3 mice). The large majority of cells expressing ChR2-mCherry also stain for GABA and Pv, confirming that transgenes are expressed selectively in inhibitory neurons expressing parvalbumin. Scale bars in a2 and a6 apply to a3, a4, a7, and a8. B, Representative traces of the action potential firing in a ChR2-expressing Pv interneuron in response to an intracellular depolarizing current pulse (top left) and to a 473 nm light stimulation of 150 ms duration (bottom left). Right, Amount of injected current required to evoke a firing rate (in the first 150 ms, black circles) similar to that elicited by light stimulation (cyan circles, n = 8 cells, 6 mice). Calibration: top, 200 ms, 500 pA, 20 mV; bottom, 200 ms, 20 mV. C, Action potential firing rate in response to 473 nm light stimulation of ChR2-expressing Pv interneurons in the absence (black, ACSF) and in the presence of 100 μm 4-AP and Mg2+ 0.5 mm (red, n = 5, 5 mice). Calibration: 100 ms, 20 mV. D, Schematic of the experiment performed in a TCx slice from a Pv-Cre mouse injected with AAV-transducing ChR2, and representative traces of voltage-clamp recordings (Vh = −50 mV) from a pyramidal neuron located 1 mm from the NMDA application site (PyN, top) and of local field potentials (lfp, bottom) recorded from the focus. A double NMDA pulse (closed arrowheads) evoked an ictal event that propagated from the focal site of NMDA application to the distant pyramidal neuron. Calibration: 40 s, 500 pA, 0.01 mV. Rf, Rhinal fissure. Insets illustrate at expanded time scales the ictal event onsets (open arrowheads) at the focal area (1) and at the propagating region (2), as revealed by lfp change and voltage-clamp recording from a pyramidal neuron (see Materials and Methods). Calibration: 2 s; inset 1, 40 pA, 0.002 mV; inset 2, 100 pA, 0.004 mV. E, Mean duration of the third, fourth, and fifth ictals normalized to the averaged duration of the first and second ictal (n = 30 ictals, 5 mice, p = 0.97 between third and fourth, p = 0.62 between third and fifth, paired t test). F, Spontaneous ictal event arising from an unpredictable focus. As revealed by the different ictal onset in the PyN and the field potential recordings (open arrowheads), the epileptiform activity propagated first to the pyramidal neuron and second to the site of field potential recording. Calibration: 40 s, 500 pA, 0.01 mV. In this and the other figures, traces may report unclamped action potentials, closed arrowheads indicate NMDA pulses, and cyan bars represent 473 nm light pulses. Empty squares represent mean values. Error bars indicate SEM.
In our focal epilepsy model using TCx slice preparations, a seizure-like ictal discharge could be generated by a double application of NMDA (100–500 ms of duration) delivered to layer V-VI neurons, in the presence of 50–100 μm 4-AP and 0.5 mm Mg2+ (Losi et al., 2010) (Fig. 1D). The NMDA application represents an experimental tool that we used to drive normal networks to seizure-like ictal discharges. However, the seizure-like ictal discharges evoked in brain slice preparations closely mimic in vivo recorded seizures, including activation of feedforward inhibition, rapid propagation across the cortex, transitions from tonic to clonic phase, and interictal discharges (Prince and Wilder, 1967; Anderson et al., 1986; Trevelyan et al., 2006, 2007a, b). Indeed, the epileptic discharge escaped from the epileptogenic focus and propagated to distant regions where it evoked a series of outward inhibitory currents reflecting the feedforward inhibition activated by focal seizure events, followed by inward currents in pyramidal neurons (Trevelyan et al., 2006; Cammarota et al., 2013) (Fig. 1D). In no circumstances were NMDA-evoked ictal events observed to cross the rhinal fissure. Successive NMDA stimulations triggered repetitive ictal events of comparable duration (n = 30 ictal events, 5 mice; Fig. 1E) provided that a time interval of at least 5 min was applied to overcome the postictal refractory period. Spontaneous ictal events did not occur between successive NMDA applications. Only in specifically designed experiments, in which no NMDA challenges were applied, were ictal events observed to arise spontaneously from unpredictable foci, and they were similar to NMDA-evoked ictal events (Fig. 1F). These important control experiments demonstrate that ChR2 expression in Pv interneurons does not, per se, impair the potential of the neural network to generate epileptiform activities.
Optogenetic activation of Pv interneurons at the epileptogenic focus fails to block the generation and prolongs the duration of ictal events
Previous in vivo studies showed that spontaneous ongoing seizures can be aborted by optogenetic activation of Pv interneurons (Krook-Magnuson et al., 2013; Paz et al., 2013). We hypothesized that optogenetic activation of Pv interneurons, which in our model can be performed before ictal event onset, could prevent seizure generation. We delivered light pulses at 0.5 Hz or higher frequency (2 and 4 Hz; pulse duration, 150 ms) at the site where NMDA will be subsequently applied (called from here on, focus) by using a large fiber optic (diameter, 300 μm) positioned between the focus and the patched pyramidal neuron (located at ∼400 μm from the focus; Fig. 2A,B). We found that a light pulse activation of ChR2-expressing Pv interneurons initiated 30 s before the ictogenic double NMDA pulse failed to inhibit ictal event generation in all experiments (n = 24 ictal events, 11 mice; Fig. 2C,D).
Figure 2.

Optogenetic activation of Pv interneurons does not prevent ictal event generation. A, Schematic of the experimental configuration also showing the typical firing of patched pyramidal neurons. Calibration: 0.2 s, 30 mV, 200 pA. B, Differential interference contrast image of the cortical region showing the NMDA-containing pipette that is used to both apply NMDA pulses and record extracellular local field potentials, the optical fiber used to activate ChR2 Pv interneurons, and the patch pipette onto a pyramidal neuron located at ∼400 μm from the ictal initiation site. A confocal z-stack maximal projection showing fluorescent ChR2-mCherry expressing Pv interneurons from the same region is also reported. C, Representative voltage-clamp recordings (Vh = −50 mV) from a pyramidal neuron and simultaneous local field potential recordings of ictal events in the absence (left) and in the presence (right) of pulsed light stimulation. A light-evoked IPSC is reported at enlarged scale. Calibration: 40 s, 500 pA, 0.01 mV; inset, 1 s, 100 pA. D, Percentage of ictal events evoked by double NMDA pulses in the presence and in the absence of pulsed stimulation applied at 0.5 Hz (n = 16, 10 mice), 2 Hz and 4 Hz (n = 8, 4 mice). E, Representative voltage-clamp recordings from a pyramidal neuron and simultaneous field potential recordings of ictal events in the absence (left) and in the presence (right) of a pulsed light stimulation at 0.5 Hz (n = 30 ictals, 13 mice) or 2 Hz (n = 5 ictals, 3 mice) that initiated after ictal onset. Calibration: 40 s, 0.01 mV, 500 pA. F, Ictal duration distribution in the absence (black) and in the presence (cyan) of pulsed light stimulation (n = 16, 10 mice). ***p < 0.001.
We next asked whether an optogenetic activation of Pv interneurons, which started 5–15 s after the initiation of an ictal event, could interrupt the epileptic discharge, as it was reported for spontaneously occurring ongoing seizures in the living brain (Krook-Magnuson et al., 2013; Paz et al., 2013; Ledri et al., 2014). Surprisingly, we found that optogenetic stimulation at 0.5 or 2 Hz invariably failed to stop both ictal events evoked by the NMDA challenge (n = 35, 16 mice; Fig. 2E) and those occurring spontaneously (n = 9, 5 mice, data not shown). In contrast, upon a continuous light pulse activation of Pv cells (150 ms at 0.5 Hz), the majority of ictal events (9 of 16) showed an increased duration. As a mean, ictal event duration (n = 16) was significantly increased with respect to controls (from 144 ± 17.0 s to 194 ± 18.1 s, n = 16 ictal events, 10 mice, p = 0.00031, paired t test; Fig. 2E,F).
Activation of Pv interneurons at the epileptogenic focus facilitates ictal event generation
The prolongation of ictal event duration by optogenetic light stimulation suggests that activation of Pv interneurons might have a pro- rather than an anti-epileptic action. We thus tested the hypothesis that Pv interneuron activity can lower focal ictal event threshold. To this aim, we used slices from ChR2 Pv mice and a single NMDA pulse, which has been reported to be a subthreshold stimulus for ictal event generation (Gómez-Gonzalo et al., 2010; Losi et al., 2010). In each experiment, we first controlled the efficacy of NMDA stimulation and the slice integrity by applying a double NMDA pulse that reliably evoked an ictal event. We then applied a single NMDA pulse and found that, as previously reported (Gómez-Gonzalo et al., 2010; Losi et al., 2010), it induced a short-lasting change in the local field potential and no response in pyramidal neurons distant from the focus in 31 of 33 single NMDA pulses (Fig. 3B, left, right, Fig. 3F; n = 33, 9 mice). Surprisingly, when single NMDA pulses were coupled with light stimulation at 0.5 Hz, they reliably evoked an ictal event (Fig. 3B, middle), which escaped from the focus and rapidly recruited distal pyramidal neurons in 22 of 23 single NMDA pulses (n = 23, 9 mice; Fig. 3F). As a further control, we tested whether, in slices from Pv-Cre mice injected with saline solution, light pulse stimulation at the focus could affect per se seizure threshold. Under these conditions, a single NMDA pulse remained ineffective (WT, n = 9 single NMDA pulses, 3 mice; Fig. 3F). The duration of ictal events evoked by single NMDA pulses in the presence of light stimulation (n = 6 ictals, 4 mice) was comparable with that of ictal events evoked by double NMDA pulses (n = 6 ictals, 4 mice, p = 0.46, t test; Fig. 3C), and it was significantly longer than the response evoked by single NMDA pulses in the absence of light stimulation, as measured by field potential changes (n = 12 single NMDA pulses, 4 mice, p = 0.0058, t test). Furthermore, the latency of ictal event onset after single NMDA pulses in the presence of light, as measured by field potential recordings at the focus, was also significantly shorter than that of the events generated by double NMDA pulses (n = 6 ictals, 4 mice, p = 0.0054, paired t test; Fig. 3D). Consistent with these results, in additional experiments we found that the recruitment of pyramidal neurons distant from the focus into the ictal event evoked by a single NMDA pulse (n = 20 ictals, 7 mice) was also significantly faster than that evoked by double NMDA pulses (n = 14 ictals, 7 mice, p = 0.00023, t test; Fig. 3E; see Materials and Methods). These latter observations suggest that, upon light stimulation, the neuronal network of the focus more readily respond to the ictogenic stimulus. We then performed some experiments restricting illumination to a smaller area by using a 50-μm-diameter optical fiber. Under these conditions, a single NMDA pulse coupled with the optogenetic stimulation also successfully evoked an ictal event (n = 3 ictals, 2 mice; Fig. 3G,H). Moving the optical fiber to a region distant ∼1 mm from the focus failed to elicit an ictal event (n = 3 single NMDA pulses, 2 mice; Fig. 3I), indicating that the optogenetic stimulation needs to recruit Pv interneurons at the focus to significantly lower ictal event threshold. A contribution to the prompt response of the neuronal network to NMDA stimulation could potentially derive from an impairment of inhibition, possibly due to a depolarization block of Pv interneurons that, at the focus, are overstimulated by the synergic action of NMDA and light pulses. This possibility was ruled out by juxtasomal recordings of firing activity from Pv interneurons at the focus. These recordings revealed that light pulses applied during the NMDA stimulation did not cause a depolarization block in any of the cells studied (n = 13, 2 mice; mean distance from the NMDA pipette tip, 210.2 ± 1.24 μm), rather inducing in these cells an intense firing, which showed unchanged mean action potential frequency before and during NMDA stimulation (mean ± SEM, 67.7 ± 5.27 Hz before and 67.8 ± 7.72 Hz during NMDA stimulation). Because each cortical Pv interneuron is synaptically connected to hundreds of nearby pyramidal neurons (Wang et al., 2002; Packer and Yuste, 2011) and local optical stimulation of Pv cells is effective in decreasing the focal seizure threshold, we hypothesized that the activity of a single interneuron might be enough to mediate this phenomenon. To address this question, we activated individual Pv interneurons using G42 mice, which express GFP in a subpopulation of Pv interneurons (Chattopadhyaya et al., 2004). After patching GFP-positive cells that were classified as FS interneurons (Fig. 4A), we applied a single NMDA pulse that, as expected, failed to evoke an ictal event and only evoked a short-lasting change in the local field potential trace and a transient firing discharge in the Pv interneuron (n = 11 single NMDA pulses, 3 mice; Fig. 4B, left, right). We then applied a sequence of depolarizing current pulses (1 s steps at 0.5 Hz for 60 s) that triggered in the Pv interneuron a sustained, rhythmic firing discharge. Under these conditions, a single NMDA pulse evoked an ictal event as revealed by the local field potential and the intracellular recordings (Fig. 4B, middle) in five out of ten single NMDA pulses (n = 10, 3 mice; Fig. 4C). As observed in the previous optogenetic experiments (see Fig. 3C), the duration of the ictal events (n = 5, 3 mice) was strikingly longer than the duration of the field potential change evoked by single NMDA pulses in the absence of single-cell stimulation (n = 11 single NMDA pulses, 3 mice, p = 0.0027, t test; Fig. 4D). The duration of these ictal events was comparable with the duration of the ictal events evoked by double NMDA pulses in the same experiments (n = 5 ictals, 3 mice, p = 0.73, t test; Fig. 5D). Furthermore, the latency of ictal event onset after single NMDA pulses paired with individual Pv cell stimulation was also significantly shorter than that of the events elicited by double NMDA pulses (n = 5 ictals, 3 mice, p = 0.042, paired t test; Fig. 4E). This reduction was similar, although less marked, to what observed in optogenetic experiments (Fig. 3D).
Figure 3.

Optogenetic activation of Pv interneurons at the epileptogenic focus promotes ictal event generation. A, Schematic of the experiment and (B) representative voltage-clamp recording (Vh = −50 mV) from a pyramidal neuron 700 μm from the focus and simultaneous local field potential recording in response to a single NMDA pulse in the absence (left, right) and in the presence (middle) of light stimulation at 0.5 Hz, which was initiated 30 s before the NMDA challenge. Note the ictal response evoked by a single NMDA pulse in the presence of optogenetic stimulation. Calibration: 40 s, 0.01 mV, 500 pA. C, Distribution of normalized event duration in double NMDA pulses (black circles, n = 6, 4 mice) and single NMDA pulse experiments in the absence (green circles, n = 12, 4 mice) and in the presence (cyan, n = 6, 4 mice) of light stimulation. As in the representative experiment reported in B, in the six experiments the effect of a single NMDA pulse was checked twice, both before and after the ictal event evoked by the single NMDA pulse coupled with light stimulation. All data points are normalized to the mean duration of double NMDA-evoked events. D, Paired distribution of the delay in ictal onset measured from local field potentials recorded at the focus in a subset of the experiments (n = 6 ictals, 4 mice) that are reported in E. E, Distribution of the delay between the first NMDA pulse and the recruitment of the pyramidal neuron, as measured by tIE (see Materials and Methods), in ictal events evoked by double NMDA pulses in the absence of light stimulation (black, n = 14, 7 mice), and single NMDA pulses in the presence of light stimulation (cyan, n = 20, 7 mice). F, Bar histogram of the percentage of ictal events evoked by a single NMDA pulse in slices from Pv-Cre mice injected with saline solution (n = 9 trials, 3 mice) and from ChR2 Pv mice in the absence (−, green bar, n = 33 trials, 9 mice) and in the presence (+, cyan, n = 23 trials, 9 mice) of pulsed light stimulation. G–I, Schematic of the experiment (top) and representative voltage-clamp (Vh = −50 mV) recordings (bottom) of the response to a single NMDA pulse in a pyramidal neuron located 250 μm from the focus in the absence of light stimulation (G), in the presence of light stimulation at the focus (H), and in the presence of light stimulation in distant regions 1 mm from the focus (I) (n = 3 ictal events, 2 mice). **p < 0.01. ***p < 0.001.
Figure 4.

Individual Pv interneuron activation at the epileptogenic focus promotes ictal event generation. A, Schematic of the experiment also showing the typical firing discharge of an FS interneuron expressing GFP in a TCx slice from a G42 mouse. Calibration: 0.5 s, 20 mV, 500 pA. B, Representative current-clamp recordings at resting potential from a Pv interneuron located at the focus and simultaneous local field potential recordings in response to a single NMDA pulse in the absence (left, right) and in the presence (middle) of 1 s intracellular current pulses at 0.5 Hz (dashed line). The intense firing activity in the interneuron facilitates the generation of a full ictal event (middle). Calibration: 40 s, 30 mV, 0.01 mV. C, Percentage of ictal events successfully evoked by a single NMDA pulse in the absence (−, n = 11, 3 mice) and in the presence (+, n = 10, 3 mice) of current pulse stimulation. D, Distribution of event duration evoked by double NMDA pulse (black circles, n = 5, 3 mice) and by single NMDA pulse in the absence (green, n = 11, 3 mice) and in the presence (cyan, n = 5, 3 mice) of evoked Pv interneuron firing activity. In each experiment, single NMDA pulses without light stimulation were performed at least twice as in the representative experiment reported in B. All data points are normalized to the mean duration of double NMDA pulse-evoked ictal events. E, Paired distribution of ictal onset delay measured from local field potential recorded at the focus (n = 5, 3 mice). *p < 0.05. **p < 0.01.
Together, these results demonstrate that rhythmic Pv interneuron activity at the epileptogenic focus, obtained either through wide field optogenetic stimulation or depolarizing current pulses applied to individual cells, contributes to lower the threshold for ictogenesis and turns a subthreshold stimulus into a reliable ictogenic trigger.
Optogenetic activation of Pv interneurons induces postinhibitory rebound spiking and promotes coordinated firing in pyramidal neurons
Activation of GABAergic interneuron inhibitory signals may enhance seizure generation by synchronizing neural excitatory networks (for review, see Avoli and de Curtis, 2011). We therefore tested whether Pv interneuron activation enhances coordinated firing in pyramidal neurons. To answer this question, we first performed current-clamp recordings from ChR2-expressing Pv interneuron-pyramidal neuron pairs during 0.5 Hz light pulse stimulation (Fig. 5). Whereas Pv interneurons were activated by blue light pulses, tonic spiking of the pyramidal cell (induced by holding the cell at depolarized potential; Fig. 5A) was largely suppressed (Fig. 5B). Moreover, both in the presence (n = 14 cells, 6 mice; Fig. 5B) and in the absence of 4-AP (n = 10 cells, 6 mice), pyramidal neurons also exhibited marked postinhibitory rebound spiking after the end of the light stimulus and successive spike clusterings. This rebound spiking period was accompanied by a significant increase in the firing rate in 24 of the 35 pyramidal neurons recorded. On average, during the rebound, a threefold increase of the firing rate was observed in these neurons (n = 24 cells, 6 mice, p = 1.19E-7, Wilcoxon signed-rank test; Fig. 5C). In paired recordings from pyramidal neurons, we then explored the hypothesis that the spike clustering during the rebound period caused by the rhythmic Pv interneuron activation could increase the degree of synchronization in pyramidal neurons (Fig. 6A1,A2). Under control conditions, two neighboring pyramidal neurons (mean distance, 52.6 ± 11.05 μm, 7 pairs, 5 mice) generated action potentials that fell randomly within the 2 s window (Fig. 6A1,B1), as also shown by the probability distribution (Fig. 6C1, red and green traces). To evaluate the degree of correlation between the two spike trains, we computed the NN distance between the spikes from the two neurons. The cumulative distribution of all NN distances is shown by the red curve (observed spikes) in Figure 6D1. Next, we shuffled the spike timing of one neuron and recomputed the NN distances (Fig. 6B1,C1, randomized PyN1). When spikes were shuffled, we found that the cumulative distribution of NN distances (blue curve, randomized spikes) was not significantly different from the distribution of observed spikes (red curve) demonstrating that the spiking of the two pyramidal neurons was not correlated (n = 230 spikes, p = 0.96, Kolmogorov–Smirnov test; Fig. 6D1). During blue light illumination delivered to activate Pv cells, we observed the suppression of firing and a rebound spiking in both neurons immediately after the light stimulus (Fig. 6A2). We generated a spike train by shuffling the spikes in the nonsilenced window (randomized PyN1; Fig. 6B2,C2). The comparison of the cumulative distributions of the NN distances of the observed spikes (red trace) and randomized spikes (blue trace) showed that the randomized spikes had a wider distribution with respect to that of the recorded spikes, indicating that after light pulses the spikes of the two neurons tended to group together (n = 447 spikes, p = 0.015, Kolmogorov–Smirnov test; Fig. 6D2). This effect is quantified in Figure 6E where the NN distance distributions for the observed (red curve) and the randomized spike train (blue curve) are reported. The difference between the two distributions (gray trace) indicates that Pv interneurons induced in pyramidal neurons an excess of synchronous spikes in a time window of 500 ms after release of inhibition. Similar results were obtained in the absence of 4-AP and the increase in synchrony for all pairs, both in 4-AP and in basal conditions, is reported in Figure 6F (n = 7 pairs, 5 mice, p = 0.0054, paired t test).
These windows of enhanced synchrony generated in the pyramidal neuron network by the rhythmic inhibition of Pv interneurons may be at the basis of the ictogenic effect induced by light pulse stimulation at the epileptogenic focus. If this were the case, we would expect the ictal events to initiate after the recovery from inhibition, and not during the firing suppression period (354 ± 19.0 ms, n = 24, 6 mice) in pyramidal neurons, as measured in each cell by the minimal time between the onset of the light pulse and the first recorded action potential. Consistent with this prediction, we found that all ictal events (n = 35, 19 mice), with one exception (representing 2.86% of all ictal events, opposed to an expected 17.7%), initiated during these windows of enhanced synchronous excitation in pyramidal neurons (Fig. 6G). Furthermore, the distribution of the ictal event onset probability exhibited a peak that coincides with the rebound peak in the mean spiking probability distribution of pyramidal neurons (Fig. 6H).
Activation of Pv interneurons in regions distant from the focus blocks the propagation and shortens the duration of ictal events
We next investigated the role of Pv interneurons in the regulation of ictal event propagation. To this aim, we specifically stimulated Pv interneurons in the propagating region by positioning the optical fiber (50 μm diameter) at ∼1 mm from the focus (Fig. 7A). In the presence of Pv interneuron stimulation, the recruitment of the pyramidal neuron into the propagating ictal event was prevented in six of eight double NMDA pulses (n = 8, 3 mice), whereas in the absence of Pv interneuron stimulation, a double NMDA pulse evoked an ictal event that always recruited the pyramidal neuron (n = 14 double NMDA pulses, 3 mice; Fig. 7B,C). Interestingly, in the six experiments in which the recruitment of the pyramidal neuron was prevented (Fig. 7D, cyan closed circles), ictal event duration at the focus was reduced to 33 ± 4% of paired controls (n = 6 ictal events, 3 mice, p = 0.009, paired t test; Fig. 7D). In the two unsuccessful experiments (Fig. 7D, open cyan circles), ictal event duration at the focus was not affected.
Figure 7.

Optogenetic activation of Pv interneurons distant from the focus blocks ictal event propagation and shortens ictal event duration at the focus. A, Schematic of the experiment. B, Representative voltage-clamp recordings (Vh = −50 mV) from a pyramidal neuron located >1 mm from the focus and simultaneous local field potential recordings at the focus in the absence (left and right) and in the presence (middle) of light stimulation starting 60 s before the double NMDA pulse. Calibration: 40 s, 0.01 mV, 500 pA. C, Percentage of propagating ictal events in the absence (n = 14, 3 mice) and in the presence of light stimulation (n = 8, 3 mice). D, Paired distribution of ictal event duration evoked by double NMDA pulses in the absence (empty and full black circles, n = 8, 3 mice) and in the presence (empty and full cyan circles, n = 8, 3 mice) of light stimulation, as measured from local field potentials recorded at the focus. The duration of the two ictal events that were not blocked by local optogenetic activation of Pv interneurons was not reduced. Mean values of black and cyan full circles are represented by full squares. Paired sample t test was performed on the values obtained from the six successful experiments (3 mice). **p < 0.01.
Given that the activation of single Pv interneurons is sufficient to mimic the effect of optogenetic Pv interneuron activation at the focus (Fig. 4), we tested the hypothesis that activation of a single Pv interneuron is also sufficient to block the recruitment of neighboring pyramidal neurons into the propagating ictal event. The progression of ictal events was monitored by current-clamp recordings from GFP-positive Pv FS interneurons in slices from G42 mice and, in a subset of experiments, also by simultaneous Ca2+ imaging from neurons loaded with the Ca2+ indicator Rhod-2 AM. As reported previously (Trevelyan et al., 2006; Cammarota et al., 2013), spatially distinct clusters of neurons were recruited into the propagating ictal event at different times (Fig. 8A, red and blue spots, B, red and blue traces). Notably, the recruitment of the second cluster of neurons (Fig. 8A,B, blue traces and spots), occurred shortly after firing impairment of the Pv interneuron (Fig. 8B, black trace). We then applied a sequence of 1 s intracellular depolarizing current pulses at 0.5 Hz that evoked in the patched Pv interneuron a firing discharge similar to that induced by the optogenetic stimulation, starting 30 s before the double NMDA pulse. Under these conditions, and in all but one of the performed experiments (n = 12 double NMDA pulses, 8 mice; Fig. 8C), the firing impairment in the patched Pv interneuron was prevented (Fig. 8B, middle, inset). As a consequence, only part of the neurons that belong to the first cluster (Fig. 8B, red trace) closer to the focus, but not neurons of the second cluster (Fig. 8B, blue traces) surrounding the patched Pv interneuron, were recruited into the ictal event. In the presence of Pv interneuron stimulation, the mean number of neurons recruited into the propagating ictal event was significantly reduced with respect to controls (n = 3 double NMDA pulses, 2 mice, p = 0.02, paired t test; Fig. 8D). The recruited neurons exhibited a drastic shortening of the ictal event, as measured from the Ca2+ signal change (n = 29 neurons; Fig. 8D) with respect to controls (n = 175 neurons, 3 ictals, 2 mice, p = 1.95E-60, t test; Fig. 8D). The shortening of ictal events was similar to that observed in the optogenetic experiments where ictal event duration at the focus was significantly reduced after the propagation was blocked by optogenetic Pv interneuron activation (Fig. 7D).
Figure 8.

Individual Pv interneuron activation in penumbra regions blocks ictal event propagation. A, Schematic of the experiment and differential interference contrast image illustrating the neuronal clusters progressively recruited into the ictal event in Ca2+ imaging experiments. B, Representative single current-clamp recordings from a Pv interneuron and simultaneous mean Ca2+ signal from two neuronal clusters (red and blue traces; soma position of these neurons is reported in A according to their different time of recruitment into the propagating ictal event. Steps of current were repeatedly injected to evoke firing activity (inset) that prevented the occurrence of a full ictal event. Calibration: 40 s, ΔF/F0 40%, 40 mV; inset, 5 s, 20 mV. C, Bar histogram of the percentage of NMDA evoked ictal events without (−, n = 23, 8 mice) or during (+, n = 12, 8 mice) stimulation of individual Pv interneurons. D, Bar histograms reporting the number of recruited neurons (left) and ictal event duration (right) measured in red neuron Ca2+ signals, in the absence and in the presence of current pulse stimulation of Pv interneurons (n = 3 ictal events, 2 mice). *p < 0.05. ***p < 0.001.
The afterdischarges travel backwards to the focus and affect ictal event duration
What process can underlie the ictal event shortening at the focus following Pv interneuron stimulation in the propagating region? The ictal event wave front generates new sources of the afterdischarges as it advances across the cortex (Trevelyan et al., 2007a). If the afterdischarges emerging from recently recruited territories travel back with respect to that of the propagating ictal event, as it was described for spontaneous ictal events in slices of occipital cortex (Trevelyan et al., 2007a), they could sustain and prolong the clonic phase of the ictal event. To address this hypothesis, we analyzed the timing of the afterdischarges in voltage-clamp paired recordings from two pyramidal neurons located at different distances from the focus (Fig. 9A). We found that the ictal event wavefront propagated first to the neuron closer to the focus (Fig. 9B, top) and then to the more distant neuron (Fig. 9B, bottom). During the clonic phase, however, the afterdischarges that initially propagated in the same direction as the ictal event wavefront changed direction (Fig. 9C). As revealed by the cross-correlation diagram, after the switch in direction, all the afterdischarges propagated toward the original site of ictal event generation (Fig. 9B,C, bottom). Similar results were obtained in four ictal events from three pyramidal neuron pairs (3 mice; Fig. 9D; see Materials and Methods).
Figure 9.

Afterdischarges invert their propagation direction during ictal events. A, Schematic of the experiment also illustrating the switch in the direction of ictal propagation. B, Representative traces of a dual voltage-clamp (Vh = −50 mV) recording from two pyramidal neurons 1 mm apart and pseudocolor plot of the cross-correlation (see Materials and Methods) during a focal ictal event showing the switch in the afterdischarge direction (n = 4, 3 mice). Calibration: 10 s, 500 pA. C, Two afterdischarges at enlarged time scale with cross-correlation diagrams of the relative bin. Calibration: 200 ms, 200 pA. D, Mean lag before and after the switch in the afterdischarge direction (n = 4, 3 mice).
Discussion
Using a temporal cortex slice model, we investigated the contribution of Pv-expressing GABAergic interneurons to the different stages that characterize a focal seizure-like ictal discharge. Our results show that, at the epileptogenic focus, a rhythmic activity in Pv interneurons, induced by optogenetic stimulation or current pulses to individual cells, enhances the generation of focal ictal events, whereas in regions distant from the focus the same interneuron activity blocks ictal event spread.
It is widely recognized that the role of GABAergic interneurons on brain networks goes far beyond the general depressant inhibitory action originally described (Freund and Katona, 2007; Bonifazi et al., 2009; Hu et al., 2014; Somogyi et al., 2014). Regarding epilepsy, data favoring either anticonvulsant or proconvulsant role are reported. Indeed, although an effective control of seizure propagation by feedforward inhibition was described previously (Prince and Wilder, 1967; Schwartz and Bonhoeffer, 2001; Trevelyan et al., 2006, 2007b; Schevon et al., 2012; Cammarota et al., 2013), increased GABAergic inhibitory signals were reported to favor, under certain conditions, seizure initiation (Avoli et al., 1996; Cossart et al., 2005; Derchansky et al., 2008; Gnatkovsky et al., 2008; Mann and Mody, 2008; Avoli and de Curtis, 2011; Grasse et al., 2013; Jiruska et al., 2013). Recent optogenetic studies also lead to conflicting views on the role of inhibition in epilepsy. In seizure models in vivo (Krook-Magnuson et al., 2013; Paz et al., 2013), optogenetic activation of GABAergic interneurons was found to suppress ongoing epileptiform activities, whereas in other models in vitro optogenetic activation of Pv interneurons and, although less efficiently, somatostatin interneurons enhanced spontaneous ictal events (Shiri et al., 2014; Yekhlef et al., 2014). Our study reconciles these conflicting views by revealing that, in the same model of epileptiform activity, Pv interneurons play a dual opposing role, either favoring the generation or opposing the propagation of ictal events. The ultimate nature of their action appears to be determined by the spatial and temporal relationship between their rhythmic inhibitory activity and the emergent network dynamics that at the focus generate an ictal event and in distant regions govern ictal event propagation.
We found that an optogenetic activation of Pv interneurons at the focus fails to block NMDA-evoked ictal events. The same stimulation initiated a few seconds after the onset of epileptiform activity also failed to interrupt ongoing ictal events and significantly prolonged their duration. Previous optogenetic studies in hippocampal slices revealed that coactivation of several interneuron subclasses impacts epileptiform activity by prolonging the interval between successive interictal-like events more effectively than activation of Pv or somatostatin interneurons alone (Ledri et al., 2014). It cannot therefore be excluded that a similar approach could also affect the generation of NMDA-evoked ictal events.
We also found that a subthreshold stimulus for ictal event generation became suprathreshold if coupled with optogenetic Pv interneuron activation. Intracellular current pulses to individual Pv interneurons also favored ictal event generation, demonstrating that this effect is independent on the tool used to activate Pv interneurons (optogenetics vs electrophysiology) and that single-cell activation is sufficient to mediate this phenomenon.
To explain this result, we hypothesized that at the focus an enhanced coordinated spiking in pyramidal neurons contributes to ictal event initiation. In this framework, and consistent with previous studies (Cobb et al., 1995; Stark et al., 2013), we observed that the rhythmic inhibition originating from Pv interneurons was followed by a postinhibitory rebound spiking that boosted excitation and drove coordinated activity in pyramidal neurons. We suggest that these windows of enhanced coordinated firing contribute to the increased local circuit excitability that, at the focus, initiates an ictal discharge in response to subthreshold excitation. In accordance with this view, NMDA-evoked ictal events were generated in these time windows, immediately after the firing suppression period induced in pyramidal neurons by Pv interneuron activation. These observations render unlikely that GABAergic signaling by Pv interneurons favored ictal event generation by directly depolarizing pyramidal neurons. Because of intracellular Cl− accumulation in pyramidal neurons upon intense interneuron activity (Fujiwara-Tsukamoto et al., 2004; Rivera et al., 2004), GABA signaling may become excitatory (Staley et al., 1995; Kaila et al., 1997), contributing to both seizure onset (Pathak et al., 2007) and afterdischarge generation (Ellender et al., 2014). If this were the case, however, ictal events are expected to initiate in coincidence with the Pv interneuron-mediated GABaergic inhibition onto pyramidal neurons and not during the rebound spiking that follows the recovery from inhibition, as we observed.
Our results also showed that, when an ictal event was evoked at the focus, its propagation was arrested by increasing inhibition in the propagating region through either an optogenetic stimulation restricted to Pv interneurons of this region or intracellular current pulse to individual interneurons. This finding is consistent with previous in vivo observations revealing that optogenetic GABAergic interneuron activation can suppress ongoing epileptiform activities (Krook-Magnuson et al., 2013; Paz et al., 2013; Ledri et al., 2014). Indeed, the epileptogenic focus could not be precisely identified in these studies. Therefore, the optogenetic stimulation most likely did not include the Pv interneurons at the focus and it arrested ictal event propagation, in accordance with our study.
The cellular events that underlined the block of ictal event propagation might involve the impact in propagating regions of two different patterns of network activity: the successive waves of excitation of the propagating ictal discharge and the rhythmic inhibition generated locally by Pv interneurons. It is noteworthy that the same rhythmic activity induced by current pulses in individual Pv interneurons that in propagating regions blocked the ictal discharge at the focus enhanced ictal event generation. This result may reflect the reduced connectivity in brain slices. However, it cannot be excluded that a similar action can be also exerted in the intact brain given that inhibition provided by a single cortical Pv interneuron can be extended to hundreds of synaptically connected pyramidal neurons (Wang et al., 2002; Packer and Yuste, 2011).
The potential of Pv interneurons to interrupt ictal event propagation is further supported by results from our Ca2+ imaging experiments. We observed that, upon activation of intense rhythmic firing in individual Pv interneurons, neurons surrounding the stimulated Pv interneurons were not recruited into propagating ictal discharge. However, neurons that were closer to the focal area were still recruited, but they exhibited a drastic ictal discharge shortening similar to that observed after ictal spread was blocked by optogenetic Pv interneuron activation in the propagating region. A possible cellular event that may account for ictal event shortening at the focus comes from afterdischarge analysis in pair recordings from pyramidal neurons at different distances from the focus. Obtained data suggest that the reduced ictal event duration could be a direct consequence of ictal event propagation block that prevented newly generated afterdischarges to travel backwards to the original focus. Notably, the afterdischarges showed only one switch in the direction of propagation suggesting that ictal events propagated unidirectionally from the site of NMDA application (in proximity to the rhinal fissure) across the temporal cortex. Consistent with this view, afterdischarges in spontaneous ictal events, probably arising from multiple foci, have been reported to frequently switch the direction of propagation (Trevelyan et al., 2007a). Given the limits of brain slice preparations, these results need to be validated in the living brain. However, they suggest that halting seizure propagation in patients may result in seizure shortening in the entire neural network, including the foci. This may represent a promising tool to prevent neurological damages frequently associated with long-lasting seizures.
We identified the action of Pv interneurons in a brain slice model in which the distinct dynamics of ictal event initiation and propagation could be investigated separately. This preparation has known limitations with respect to the living brain state, such as reduced long-range axonal connections and the removal of cortico-thalamo-cortical projections. It should also be noted that the local NMDA excitation represents an experimental tool that we used to drive normal networks to ictal events. Caution is, therefore, warranted before extrapolating our data to the epileptic brain. Nevertheless, it must be also underlined that NMDA-induced ictal events were similar to those arising spontaneously from unpredictable foci. Most importantly, our cortical slice model of focal ictal events allows experiments that can be hardly performed in the living brain. For example, the optogenetic stimulation restricted to Pv interneurons of the focus that we performed in our model is unfeasible in currently available in vivo models of seizures where seizure initiation site is not identifiable in advance.
In conclusion, the functional dichotomy of Pv interneurons that we here describe extends our understanding of the cellular mechanisms that in local circuits control the generation and the propagation of focal epileptiform discharges. Results obtained validate the efficacy of an optogenetic activation of Pv interneurons in opposing seizure propagation, but, in parallel, they also show that GABAergic inhibition can enhance seizure generation by promoting synchronized network activity. The promising use of optogenetics in seizure control (Tye and Deisseroth, 2012; Kokaia et al., 2013) must therefore be carefully weighted to avoid its potential negative consequences.
Footnotes
This work was supported by Telethon Italy Grant GGP10138/GGP12265, Cariparo Foundation, National Research Council Aging Project, and Fondo per gli Investimenti della Ricerca di Base Grant RBAP11X42L to G.C. and San Paolo Programma in Neuroscienze, Ministero dell'Istruzione, dell'Università e della Ricerca Fondo per Gli Investimenti Della Ricerca di Base Grant RBAP11X42L, Telethon-Italy Grant GGP10138, and a FP7 DESIRE project to T.F. We thank Alberto Bacci for critical reading of a previous version of the manuscript; and Karl Deisseroth for AAV2/1.EF1.dflox.hChR2(H134R)-mCherry.WPRE.hGH.
The authors declare no competing financial interests.
References
- Anderson WW, Lewis DV, Swartzwelder HS, Wilson WA. Magnesium-free medium activates seizure-like events in the rat hippocampal slice. Brain Res. 1986;398:215–219. doi: 10.1016/0006-8993(86)91274-6. [DOI] [PubMed] [Google Scholar]
- Avoli M, de Curtis M. GABAergic synchronization in the limbic system and its role in the generation of epileptiform activity. Prog Neurobiol. 2011;95:104–132. doi: 10.1016/j.pneurobio.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avoli M, Barbarosie M, Lücke A, Nagao T, Lopantsev V, Köhling R. Synchronous GABA-mediated potentials and epileptiform discharges in the rat limbic system in vitro. J Neurosci. 1996;16:3912–3924. doi: 10.1523/JNEUROSCI.16-12-03912.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartos M, Vida I, Jonas P. Synaptic mechanisms of synchronized gamma oscillations in inhibitory interneuron networks. Nat Rev Neurosci. 2007;8:45–56. doi: 10.1038/nrn2044. [DOI] [PubMed] [Google Scholar]
- Bonifazi P, Goldin M, Picardo MA, Jorquera I, Cattani A, Bianconi G, Represa A, Ben-Ari Y, Cossart R. GABAergic hub neurons orchestrate synchrony in developing hippocampal networks. Science. 2009;326:1419–1424. doi: 10.1126/science.1175509. [DOI] [PubMed] [Google Scholar]
- Buzsáki G, Wang XJ. Mechanisms of gamma oscillations. Annu Rev Neurosci. 2012;35:203–225. doi: 10.1146/annurev-neuro-062111-150444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cammarota M, Losi G, Chiavegato A, Zonta M, Carmignoto G. Fast spiking interneuron control of seizure propagation in a cortical slice model of focal epilepsy. J Physiol. 2013;591:807–822. doi: 10.1113/jphysiol.2012.238154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyaya B, Di Cristo G, Higashiyama H, Knott GW, Kuhlman SJ, Welker E, Huang ZJ. Experience and activity-dependent maturation of perisomatic GABAergic innervation in primary visual cortex during a postnatal critical period. J Neurosci. 2004;24:9598–9611. doi: 10.1523/JNEUROSCI.1851-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb SR, Buhl EH, Halasy K, Paulsen O, Somogyi P. Synchronization of neuronal activity in hippocampus by individual GABAergic interneurons. Nature. 1995;378:75–78. doi: 10.1038/378075a0. [DOI] [PubMed] [Google Scholar]
- Connors BW. Initiation of synchronized neuronal bursting in neocortex. Nature. 1984;310:685–687. doi: 10.1038/310685a0. [DOI] [PubMed] [Google Scholar]
- Cossart R, Bernard C, Ben-Ari Y. Multiple facets of GABAergic neurons and synapses: multiple fates of GABA signalling in epilepsies. Trends Neurosci. 2005;28:108–115. doi: 10.1016/j.tins.2004.11.011. [DOI] [PubMed] [Google Scholar]
- Derchansky M, Jahromi SS, Mamani M, Shin DS, Sik A, Carlen PL. Transition to seizures in the isolated immature mouse hippocampus: a switch from dominant phasic inhibition to dominant phasic excitation. J Physiol. 2008;586:477–494. doi: 10.1113/jphysiol.2007.143065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugué GP, Dumoulin A, Triller A, Dieudonné S. Target-dependent use of co-released inhibitory transmitters at central synapses. J Neurosci. 2005;25:6490–6498. doi: 10.1523/JNEUROSCI.1500-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellender TJ, Raimondo JV, Irkle A, Lamsa KP, Akerman CJ. Excitatory effects of parvalbumin-expressing interneurons maintain hippocampal epileptiform activity via synchronous afterdischarges. J Neurosci. 2014;34:15208–15222. doi: 10.1523/JNEUROSCI.1747-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund TF, Katona I. Perisomatic inhibition. Neuron. 2007;56:33–42. doi: 10.1016/j.neuron.2007.09.012. [DOI] [PubMed] [Google Scholar]
- Fujiwara-Tsukamoto Y, Isomura Y, Kaneda K, Takada M. Synaptic interactions between pyramidal cells and interneurone subtypes during seizure-like activity in the rat hippocampus. J Physiol. 2004;557:961–979. doi: 10.1113/jphysiol.2003.059915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnatkovsky V, Librizzi L, Trombin F, de Curtis M. Fast activity at seizure onset is mediated by inhibitory circuits in the entorhinal cortex in vitro. Ann Neurol. 2008;64:674–686. doi: 10.1002/ana.21519. [DOI] [PubMed] [Google Scholar]
- Gómez-Gonzalo M, Losi G, Chiavegato A, Zonta M, Cammarota M, Brondi M, Vetri F, Uva L, Pozzan T, de Curtis M, Ratto GM, Carmignoto G. An excitatory loop with astrocytes contributes to drive neurons to seizure threshold. PLoS Biol. 2010;8:e1000352. doi: 10.1371/journal.pbio.1000352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grasse DW, Karunakaran S, Moxon KA. Neuronal synchrony and the transition to spontaneous seizures. Exp Neurol. 2013;248:72–84. doi: 10.1016/j.expneurol.2013.05.004. [DOI] [PubMed] [Google Scholar]
- Gutnick MJ, Connors BW, Prince DA. Mechanisms of neocortical epileptogenesis in vitro. J Neurophysiol. 1982;48:1321–1335. doi: 10.1152/jn.1982.48.6.1321. [DOI] [PubMed] [Google Scholar]
- Hu H, Gan J, Jonas P. Interneurons. Fast-spiking, parvalbumin+ GABAergic interneurons: from cellular design to microcircuit function. Science. 2014;345:1255–1263. doi: 10.1126/science.345.6202.1255. [DOI] [PubMed] [Google Scholar]
- Jiruska P, de Curtis M, Jefferys JG, Schevon CA, Schiff SJ, Schindler K. Synchronization and desynchronization in epilepsy: controversies and hypotheses. J Physiol. 2013;591:787–797. doi: 10.1113/jphysiol.2012.239590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaila K, Lamsa K, Smirnov S, Taira T, Voipio J. Long-lasting GABA-mediated depolarization evoked by high-frequency stimulation in pyramidal neurons of rat hippocampal slice is attributable to a network-driven, bicarbonate-dependent K+ transient. J Neurosci. 1997;17:7662–7672. doi: 10.1523/JNEUROSCI.17-20-07662.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kepecs A, Fishell G. Interneuron cell types are fit to function. Nature. 2014;505:318–326. doi: 10.1038/nature12983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokaia M, Andersson M, Ledri M. An optogenetic approach in epilepsy. Neuropharmacology. 2013;69:89–95. doi: 10.1016/j.neuropharm.2012.05.049. [DOI] [PubMed] [Google Scholar]
- Kovács R, Kardos J, Heinemann U, Kann O. Mitochondrial calcium ion and membrane potential transients follow the pattern of epileptiform discharges in hippocampal slice cultures. J Neurosci. 2005;25:4260–4269. doi: 10.1523/JNEUROSCI.4000-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krook-Magnuson E, Armstrong C, Oijala M, Soltesz I. On-demand optogenetic control of spontaneous seizures in temporal lobe epilepsy. Nat Commun. 2013;4:1376. doi: 10.1038/ncomms2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledri M, Madsen MG, Nikitidou L, Kirik D, Kokaia M. Global optogenetic activation of inhibitory interneurons during epileptiform activity. J Neurosci. 2014;34:3364–3377. doi: 10.1523/JNEUROSCI.2734-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy RB, Reyes AD. Spatial profile of excitatory and inhibitory synaptic connectivity in mouse primary auditory cortex. J Neurosci. 2012;32:5609–5619. doi: 10.1523/JNEUROSCI.5158-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losi G, Cammarota M, Chiavegato A, Gómez-Gonzalo M, Carmignoto G. A new experimental model of focal seizures in the entorhinal cortex. Epilepsia. 2010;51:1493–1502. doi: 10.1111/j.1528-1167.2009.02472.x. [DOI] [PubMed] [Google Scholar]
- Mann EO, Mody I. The multifaceted role of inhibition in epilepsy: seizure-genesis through excessive GABAergic inhibition in autosomal dominant nocturnal frontal lobe epilepsy. Curr Opin Neurol. 2008;21:155–160. doi: 10.1097/WCO.0b013e3282f52f5f. [DOI] [PubMed] [Google Scholar]
- Markram H, Toledo-Rodriguez M, Wang Y, Gupta A, Silberberg G, Wu C. Interneurons of the neocortical inhibitory system. Nat Rev Neurosci. 2004;5:793–807. doi: 10.1038/nrn1519. [DOI] [PubMed] [Google Scholar]
- Miles R, Wong RK. Single neurones can initiate synchronized population discharge in the hippocampus. Nature. 1983;306:371–373. doi: 10.1038/306371a0. [DOI] [PubMed] [Google Scholar]
- Monyer H, Markram H. Interneuron Diversity series: molecular and genetic tools to study GABAergic interneuron diversity and function. Trends Neurosci. 2004;27:90–97. doi: 10.1016/j.tins.2003.12.008. [DOI] [PubMed] [Google Scholar]
- Packer AM, Yuste R. Dense, unspecific connectivity of neocortical parvalbumin-positive interneurons: a canonical microcircuit for inhibition? J Neurosci. 2011;31:13260–13271. doi: 10.1523/JNEUROSCI.3131-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak HR, Weissinger F, Terunuma M, Carlson GC, Hsu FC, Moss SJ, Coulter DA. Disrupted dentate granule cell chloride regulation enhances synaptic excitability during development of temporal lobe epilepsy. J Neurosci. 2007;27:14012–14022. doi: 10.1523/JNEUROSCI.4390-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz JT, Davidson TJ, Frechette ES, Delord B, Parada I, Peng K, Deisseroth K, Huguenard JR. Closed-loop optogenetic control of thalamus as a tool for interrupting seizures after cortical injury. Nat Neurosci. 2013;16:64–70. doi: 10.1038/nn.3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prince DA, Wilder BJ. Control mechanisms in cortical epileptogenic foci: “surround” inhibition. Arch Neurol. 1967;16:194–202. doi: 10.1001/archneur.1967.00470200082007. [DOI] [PubMed] [Google Scholar]
- Rivera C, Voipio J, Thomas-Crusells J, Li H, Emri Z, Sipilä S, Payne JA, Minichiello L, Saarma M, Kaila K. Mechanism of activity-dependent downregulation of the neuron-specific K-Cl cotransporter KCC2. J Neurosci. 2004;24:4683–4691. doi: 10.1523/JNEUROSCI.5265-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schevon CA, Weiss SA, McKhann G, Jr, Goodman RR, Yuste R, Emerson RG, Trevelyan AJ. Evidence of an inhibitory restraint of seizure activity in humans. Nat Commun. 2012;3:1060. doi: 10.1038/ncomms2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiff ML, Reyes AD. Characterization of thalamocortical responses of regular-spiking and fast-spiking neurons of the mouse auditory cortex in vitro and in silico. J Neurophysiol. 2012;107:1476–1488. doi: 10.1152/jn.00208.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz TH, Bonhoeffer T. In vivo optical mapping of epileptic foci and surround inhibition in ferret cerebral cortex. Nat Med. 2001;7:1063–1067. doi: 10.1038/nm0901-1063. [DOI] [PubMed] [Google Scholar]
- Shiri Z, Manseau F, Levesque M, Williams S, Avoli M. Interneuron activity leads to initiation of low-voltage fast-onset seizures. Ann Neurol. 2014 doi: 10.1002/ana.24342. doi: 10.1002/ana.24342. Advance online publication. Retrieved Dec. 27, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somogyi P, Katona L, Klausberger T, Lasztóczi B, Viney TJ. Temporal redistribution of inhibition over neuronal subcellular domains underlies state-dependent rhythmic change of excitability in the hippocampus. Philos Trans R Soc Lond B Biol Sci. 2014;369:20120518. doi: 10.1098/rstb.2012.0518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley KJ, Soldo BL, Proctor WR. Ionic mechanisms of neuronal excitation by inhibitory GABAA receptors. Science. 1995;269:977–981. doi: 10.1126/science.7638623. [DOI] [PubMed] [Google Scholar]
- Stark E, Eichler R, Roux L, Fujisawa S, Rotstein HG, Buzsáki G. Inhibition-induced theta resonance in cortical circuits. Neuron. 2013;80:1263–1276. doi: 10.1016/j.neuron.2013.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trevelyan AJ, Sussillo D, Watson BO, Yuste R. Modular propagation of epileptiform activity: evidence for an inhibitory veto in neocortex. J Neurosci. 2006;26:12447–12455. doi: 10.1523/JNEUROSCI.2787-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trevelyan AJ, Baldeweg T, van Drongelen W, Yuste R, Whittington M. The source of afterdischarge activity in neocortical tonic-clonic epilepsy. J Neurosci. 2007a;27:13513–13519. doi: 10.1523/JNEUROSCI.3005-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trevelyan AJ, Sussillo D, Yuste R. Feedforward inhibition contributes to the control of epileptiform propagation speed. J Neurosci. 2007b;27:3383–3387. doi: 10.1523/JNEUROSCI.0145-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tye KM, Deisseroth K. Optogenetic investigation of neural circuits underlying brain disease in animal models. Nat Rev Neurosci. 2012;13:251–266. doi: 10.1038/nrn3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Gupta A, Toledo-Rodriguez M, Wu CZ, Markram H. Anatomical, physiological, molecular and circuit properties of nest basket cells in the developing somatosensory cortex. Cereb Cortex. 2002;12:395–410. doi: 10.1093/cercor/12.4.395. [DOI] [PubMed] [Google Scholar]
- Yekhlef L, Breschi GL, Lagostena L, Russo G, Taverna S. Selective activation of parvalbumin- or somatostatin-expressing interneurons triggers epileptic seizure-like activity in the mouse medial entorhinal cortex. J Neurophysiol. 2014 doi: 10.1152/jn.00841.2014. doi: 10.1152/jn.00841.2014. Advance online publication. Retrieved Dec. 10, 2014. [DOI] [PubMed] [Google Scholar]