

Abstract
Theoretical and animal work has proposed that prefrontal cortex (PFC) glutamate inhibits dopaminergic inputs to the ventral striatum (VS) indirectly, whereas direct VS glutamatergic afferents have been suggested to enhance dopaminergic inputs to the VS. In the present study, we aimed to investigate relationships of glutamate and dopamine measures in prefrontostriatal circuitries of healthy humans. We hypothesized that PFC and VS glutamate, as well as their balance, are differently associated with VS dopamine. Glutamate concentrations in the left lateral PFC and left striatum were assessed using 3-Tesla proton magnetic resonance spectroscopy. Striatal presynaptic dopamine synthesis capacity was measured by fluorine-18-l-dihydroxyphenylalanine (F-18-FDOPA) positron emission tomography. First, a negative relationship was observed between glutamate concentrations in lateral PFC and VS dopamine synthesis capacity (n = 28). Second, a positive relationship was revealed between striatal glutamate and VS dopamine synthesis capacity (n = 26). Additionally, the intraindividual difference between PFC and striatal glutamate concentrations correlated negatively with VS dopamine synthesis capacity (n = 24). The present results indicate an involvement of a balance in PFC and striatal glutamate in the regulation of VS dopamine synthesis capacity. This notion points toward a potential mechanism how VS presynaptic dopamine levels are kept in a fine-tuned range. A disruption of this mechanism may account for alterations in striatal dopamine turnover as observed in mental diseases (e.g., in schizophrenia).
SIGNIFICANCE STATEMENT The present work demonstrates complementary relationships between prefrontal and striatal glutamate and ventral striatal presynaptic dopamine using human imaging measures: a negative correlation between prefrontal glutamate and presynaptic dopamine and a positive relationship between striatal glutamate and presynaptic dopamine are revealed. The results may reflect a regulatory role of prefrontal and striatal glutamate for ventral striatal presynaptic dopamine levels. Such glutamate–dopamine relationships improve our understanding of neurochemical interactions in prefrontostriatal circuits and have implications for the neurobiology of mental disease.
Keywords: dopamine, FDOPA PET, glutamate, MRS, prefrontal cortex, striatum
Introduction
Dopamine is implicated in motivated behavior, reward learning, and cognitive control (Schultz, 1998; Bromberg-Martin et al., 2010; Cools, 2011). It further acts as neuromodulator that is assumed to be crucially involved in driving synaptic plasticity (Surmeier et al., 2007; Di Filippo et al., 2009; Lovinger, 2010; Schultz, 2013). Furthermore, dopaminergic signals exert their influence on different time scales (tonic and phasic) and on different neural systems [e.g., basal ganglia, prefrontal cortex (PFC)]. Theoretical accounts and empirical work clearly point to the idea that the dynamics of dopamine underlie complex regulatory mechanisms resulting in a fine-tuned, nonlinear system (Cools and D'Esposito, 2011). A disrupted balance of this system in any direction may result in alterations of the associated functions. In general, the aim to identify surrogate imaging markers of the regulation of presynaptic dopamine function is key to many questions in human neuroscience as well as a better understanding of the neurobiology of mental diseases.
One central concept regarding a potential regulation is that the regulation of striatal presynaptic dopamine is driven by a balanced engagement of excitatory (“accelerator”) and inhibitory (“brake”) glutamatergic inputs (Carlsson et al., 1999). This model has received tremendous attention, particularly with respect to the pathophysiology of schizophrenia (Carlsson et al., 1999; Laruelle et al., 2003; Stephan et al., 2006; Gonzalez-Burgos and Lewis, 2012; Schwartz et al., 2012) and neurochemical interactions in healthy individuals (Carlsson et al., 1999; Gleich et al., 2014). Specifically, the PFC has been proposed to inhibit striatal dopaminergic activity indirectly via GABAergic interneurons, ultimately influencing striatal dopamine activity (Carlsson et al., 1999; Sesack et al., 2003). This is supported by animal research in which the blockage of glutamate NMDA receptors in prefrontal regions resulted in increased dopamine release specifically in the ventral striatum (VS) (Del Arco et al., 2008; Usun et al., 2013). With respect to local glutamate, the VS receives direct excitatory glutamatergic input, particularly from the hippocampus and amygdala (Grace, 1991; Sesack et al., 2003; Schwartz et al., 2012). In accordance, animal research showed that increased striatal glutamate by reverse microdialysis resulted in higher availability of dopamine in the synaptic cleft, particularly in the VS (Segovia and Mora, 2001; Morales et al., 2012). The need for in vivo investigation of this mechanism in humans was formulated previously, and a close coupling between striatal glutamate and dopaminergic tone was proposed (de la Fuente-Sandoval et al., 2011). Yet, a direct in vivo relationship between proxy measures of striatal glutamate and ventral striatal presynaptic dopamine has not been examined in humans.
Here, we investigate this proposed complementary relationship between prefrontal and striatal glutamate and VS presynaptic dopamine using human multimodal imaging. Glutamate was measured using single voxel magnetic resonance spectroscopy (1H-MRS), and presynaptic dopamine was estimated as the dopamine synthesis capacity from fluorine-18-l-dihydroxyphenylalanine (FDOPA) positron emission tomography (PET) scans. We tested three hypotheses based on prior research: First, PFC glutamate concentrations correlate negatively with VS dopamine synthesis capacity. Second, striatal glutamate concentrations correlate positively with VS dopamine synthesis capacity. Third, the balance of the two glutamate measures (PFC minus striatal glutamate) is negatively related to VS dopamine synthesis capacity.
Materials and Methods
Participants
Thirty-three healthy adults were recruited via advertisements on Internet platforms and in local newspapers. Before invitation to the study, a telephone interview was conducted with each participant. During this interview, standardized questions regarding their history of medical and psychological diseases and treatments and magnetic resonance imaging (MRI) safety as well as short demographic questions were asked. Left-handed participants and participants with a prior neurological disease or a history of brain or head surgery, lifetime psychopharmacological treatment, or MRI contraindications (e.g., nonremovable ferromagnetic material) were excluded. Additionally, the Structured Clinical Interview for DSM-IV Axis I Disorders was assessed, and any Axis I disorder lead to exclusion from the study. The study was approved by the local ethics committee of the Charité–University Medicine Berlin, and participants received financial compensation for participation. All participants gave written informed consent. Besides the imaging measures described in the following, all participants were also cognitively characterized by their performance on the digit span test for working memory as well as the digit symbol substitution test for cognitive processing speed as part of the Wechsler Adult Intelligence Scale-III (Ryan and Paolo, 2001).
Magnetic resonance spectroscopy
Acquisition and analysis of MRS data.
MRS imaging was conducted at the Berlin Center for Advanced Neuroimaging of the Charité–University Medicine Berlin, Mitte campus. Absolute glutamate concentrations in the PFC and the striatum were acquired with 3-Tesla 1H-MRS using water-suppressed and unsuppressed spectra [point resolved spectroscopy; 128/8 averages; 90° flip angle; echo time (TE), 80 ms; repetition time (TR), 3 s; automatic shimming]. MRS data were collected in the same session after acquisition of a high-resolution T1 structural image (MPRAGE; 192 sagittal slices; TR, 1.9 s; TE, 2.52 ms; flip angle, 9°; FOV, 256 × 256; matrix size, 256 × 256; 1 × 1 × 1 mm resolution; axially oriented 3D sequence).
MRS voxel localization.
A 40 × 10 × 20 mm voxel was placed in the left PFC (Fig. 1). The PFC voxel was placed in the dorsolateral prefrontal cortex (on a lateral slice) anterior to the motor cortex, with the longer edge of the voxel parallel to the lateral sulcus. Then, on the same view, the voxel was located to be intermediate between the dorsal borders of the insula and the dorsal hemispheric midline lining. Then, on a coronal view, the voxel was rotated and tilted to be parallel to the frontal skull bones and, additionally, to contain as much gray matter (GM) as possible (Fig. 1).
Figure 1.
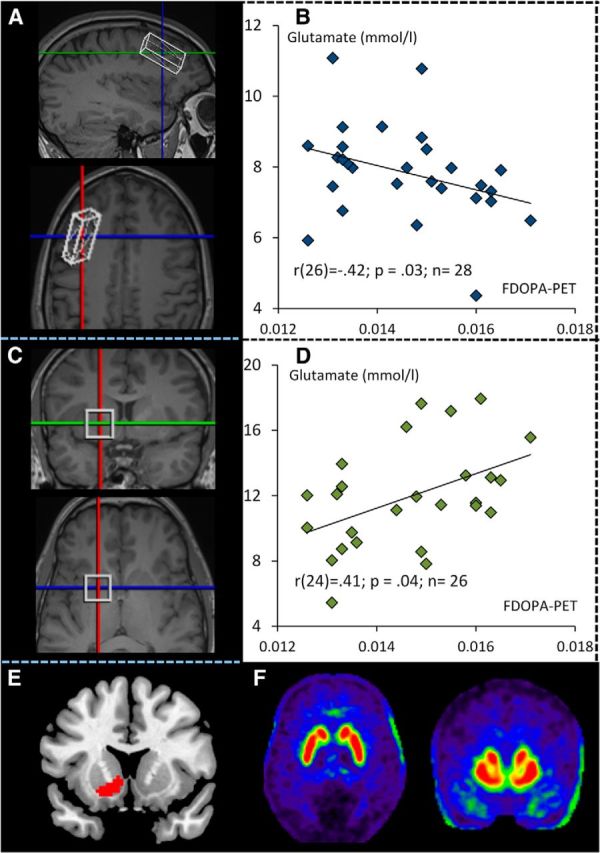
The relationship of the dopamine synthesis capacity in the ventral striatum with glutamate concentrations in the prefrontal cortex and ventral striatum. A, Position of the PFC MRS voxel. B, Scatter plot depicting the relationship of glutamate concentrations in the PFC and dopamine synthesis capacity in the ventral striatum. Spearman correlation coefficients are displayed. Glutamate is measured in millimoles per liter. C, Position of the MRS striatum voxel. D, Scatter plot depicting the relationship of striatal glutamate concentrations and ventral striatal dopamine synthesis capacity. Spearman correlation coefficients are displayed. Glutamate is measured in millimoles per liter. E, Ventral striatum region of interest used for extraction of FDOPA Ki values, displayed in red. F, Mean FDOPA PET dopamine synthesis capacity Ki map.
Furthermore, a 20 × 20 × 20 mm voxel was placed in the left striatum (Fig. 1). On a coronal plane, the voxel was first placed to contain the striatum in the center of the voxel. Then, due to individual differences in brain anatomy, the voxel was shifted dorsally and/or tilted counterclockwise on the coronal plane to include as many striatal and as few insular GM structures and as little CSF as possible. On the transversal and sagittal planes, voxels were individually shifted and tilted to contain as much GM as possible. All observers were trained on the localization of the voxels and were further guided by anatomical descriptions from textbooks.
Analysis of MRS data.
MRS data were analyzed using the Linear Combination of Model spectra (LCModel) commercial spectral-fitting package (Provencher, 2014), using water-suppressed and unsuppressed spectra. This method allows the quantification of many metabolites in the magnetic resonance spectrum in the region of interest (ROI); however, here we were only interested in glutamate concentrations. All MRS voxels were individually placed by anatomically trained MR operators, and all spectra in the PFC were reliably measured by definition of Cramér–Rao lower bounds of <20% fit deviation (mean fit deviation, 9.03%; SD, 2.80; range, 6–16%). We applied a more liberal criterion for the Cramér–Rao lower bounds in the VS (≤25%) due to lower MRS signal-to-noise ratio in deep brain structures (Schwerk et al., 2014; mean fit deviation, 17.31%; SD, 3.92; range, 7–25%).
Glutamate measured by MRS is considered to reflect the total content of glutamate in the region of interest (Rothman et al., 2011), independent of brain tissue compartments. Therefore, GM, white matter (WM), and CSF fractions within the MRS voxels were segmented using the unified segmentation approach (Ashburner and Friston, 2005) in Python software (Python Software Foundation; Python Language Reference available at https://docs.python.org/2/reference/index.html) based on the high-resolution T1 structural image. Subsequently, absolute glutamate concentrations were adjusted for GM and WM content within the voxel using the following formula: glutamate adjusted = glutamate absolute * (1/GM + WM). Throughout the rest of the present study, we report only adjusted glutamate concentrations. Glutamate concentrations have units of millimoles per liter.
FDOPA PET
Acquisition of PET data.
PET data were acquired at the department of nuclear medicine at the Rudolf Virchow Hospital in Berlin using a PET/CT scanner (Philips Gemini TF16) in 3D mode. After a low-dose transmission CT-scan, a dynamic “list-mode” emission recording lasting 60 min started simultaneously with intravenous bolus administration of 120–200 MBq FDOPA. List-mode data were framed (20 frames, 3 × 20 s, 3 × 1 min, 3 × 2 min, 3 × 3 min, 7 × 5 min, 1 × 6 min) and reconstructed iteratively with CT-based attenuation and scatter correction.
Analysis of PET data.
PET data were analyzed using Statistical Parametric Mapping 8 (Wellcome Department of Imaging Neuroscience, Institute of Neurology, London; http://www.fil.ion.ucl.ac.uk/spm/). The image frames were realigned to correct for head motion between frames. The individual mean images and individual T1 images were coregistered. Each participant's anatomical T1 image was spatially normalized using the unified segmentation approach (Ashburner and Friston, 2005). The computed normalization parameters were then applied to the coregistered PET frames. For statistical analysis, dopamine synthesis capacity was quantified as FDOPA Ki (minutes−1), which was estimated voxel by voxel using Gjedde–Patlak linear graphical analysis (Patlak and Blasberg, 1985). Radioactivity time curves in a standard cerebellum mask as defined in WFU PickAtlas excluding vermis (Tzourio-Mazoyer et al., 2002) were used as the input function. The linear fit was restricted to the time interval 20–60 min after injection. As the ROI, we selected the left ventral (limbic) striatum (Martinez et al., 2003; Howes et al., 2012), analogous to left hemisphere MRS voxel placement for both regions, and limbic/ventral because it matched the localization of the striatal MRS voxel most closely. Mean Ki values were extracted for the ventral striatal ROI from voxelwise map of each individual.
Statistical analysis of dopamine–glutamate relationships
Absolute glutamate concentrations in the PFCs of four participants could not be fitted due to bad signal-to-noise ratios. In the striatum, absolute glutamate concentrations could also not be fitted in five participants. These data were excluded from further analyses. For statistical analyses, we first tested whether dependent variables were normally distributed using the Shapiro–Wilk test. This test showed that the glutamate concentrations in the PFC (W(29) = 0.85; p < 0.01) and the striatum (W(28) = 0.77; p < 0.001) were not normally distributed. Furthermore, Ki values were distributed normally in both samples (PFC glutamate–VS Ki relationship, W(28) = 0.95, p = 0.15; striatal glutamate–VS Ki relationship, W(28)= 0.94, p = 0.10). Due to nonnormal distributions of glutamate concentrations, Spearman correlation coefficients were used for all analyses. We further tested for the presence of outliers by standardizing glutamate concentrations and FDOPA Ki values into z-scores and subsequently excluding subjects with a z-score above 3 SDs (Field, 2009). This led to exclusion of one outlier regarding PFC glutamate and two outliers for striatal glutamate concentrations. Thus, the final correlation analyses comprised 28 participants regarding the relationship between PFC glutamate concentrations and ventral striatal dopamine synthesis capacity (age range, 20–43 years; mean age, 28.9 years; SD, 5.53 years; 12 females) and 26 participants for the relationship between VS glutamate concentrations and ventral striatal dopamine synthesis capacity (age range, 20–39 years; mean age, 26.31 years; SD, 4.89 years; 11 females). Additionally, to index the balance of PFC and VS glutamate, we subtracted glutamate concentrations in the striatum from glutamate concentrations in the PFC to compute “ΔGlu”. This could only be computed when glutamate concentrations were available for both regions, which led to an overlapping sample of 24 participants. Then, we tested whether ΔGlu was related to VS dopamine synthesis capacity using Spearman correlation coefficient. We also explored correlations of all neurochemical imaging measures with the two collected cognitive tests, the digit span and digit symbol substitution tests.
Results
Glutamate concentration and tissue composition within voxels
GM- and WM-adjusted mean glutamate concentrations were 7.85 mmol/L in PFC (n = 28; min = 4.36 mmol/L; max = 11.08 mmol/L; SD, 1.34) and 11.94 mmol/L in the striatum (n = 26; min = 5.44 mmol/L; max = 17.94 mmol/L; SD, 3.16 mmol/L). GM, WM, and CSF fractions within PFC and striatal voxels (after exclusion of outliers) are reported in Table 1; please note the relatively large contribution of WM.
Table 1.
Tissue fractions in PFC and striatum voxels
| GM |
WM |
CSF |
||||
|---|---|---|---|---|---|---|
| Fraction | SD | Fraction | SD | Fraction | SD | |
| PFC voxel (n = 28) | 0.54 | 0.09 | 0.37 | 0.10 | 0.08 | 0.04 |
| Striatal voxel (n = 26) | 0.41 | 0.10 | 0.58 | 0.10 | 0.01 | 0.01 |
Please note the relatively large contribution of WM.
Dopamine synthesis capacity
The mean Ki in the left ventral striatum was 0.0147 (n = 33; min = 0.0126; max = 0.0174; SD, 0.0014).
Glutamate–dopamine relationships
First, a negative relationship was observed between left PFC glutamate concentrations and left VS dopamine synthesis capacity (n = 28; Spearman's r(26) = −0.42; p = 0.03; Fig. 1). Second, we found a positive relationship between left striatal glutamate concentrations and left VS dopamine synthesis capacity Ki (n = 26; Spearman's r(24) = 0.41; p = 0.04; Fig. 1). Third, ΔGlu showed a significant negative correlation with left VS dopamine synthesis capacity Ki (n = 24; Spearman's r(22) = −0.46; p = 0.02; Fig. 2). PFC and striatal glutamate concentrations were not correlated significantly (n = 24; Spearman's r(22) = −0.11; p = 0.58).
Figure 2.

Relationship between ΔGlu and dopamine synthesis capacity in the VS. ΔGlu is the difference score of glutamate concentrations (PFC and VS). Displayed is the negative association between ventral striatal FDOPA PET Ki values and ΔGlu.
We additionally tested whether the observed correlations were influenced by gray matter fractions within the MRS voxels using nonparametric partial correlations (Conover, 1999). The correlations between PFC glutamate and ventral striatal FDOPA Ki (n = 28; r(25) = −0.373; p = .054) and between striatal glutamate and ventral striatal FDOPA Ki (n = 26; r(23)= 0.397; p = .049) remained at the border of significance when controlling for gray matter fractions.
Correlations between neurochemical measures and cognitive tests
When exploring associations between all neurochemical measures (VS dopamine synthesis capacity, striatal and PFC glutamate concentrations, and ΔGlu) and the two cognitive tests acquired (digit span for working memory and digit symbol substitution for cognitive processing speed), no significant correlations were observed (all r values between 0.15 and −0.34; all p > 0.07).
Discussion
Using multimodal imaging, we provide two main results: a negative correlation between PFC glutamate and VS dopamine synthesis capacity and a positive relationship between striatal glutamate and VS dopamine synthesis capacity. These results point toward a potential mechanism of how PFC and striatal glutamate might be complementarily involved in keeping subcortical presynaptic dopamine within a fine-tuned range. In support, we also show that the balance between PFC glutamate and striatal glutamate is negatively related to VS dopamine synthesis capacity. These findings suggest that the applied neuroimaging measures may serve as a surrogate of a neurochemical balance in prefrontostriatal circuits and may thus have important implications for the pathophysiology of psychiatric disorders, in particular, schizophrenia.
Inverse coupling between prefrontal glutamate and ventral striatal dopamine
First, PFC glutamate concentrations were negatively related to VS dopamine synthesis capacity. In line, animal research demonstrated that PFC glutamate may act on VS dopamine via inhibitory GABAergic interneurons (Balla et al., 2009). The observed negative correlation of PFC glutamate and VS dopamine also resonates with pharmacological challenge studies; after NMDA receptor blockage by ketamine, increased VS dopamine release was observed in human and animal research (Rowland et al., 2005; Del Arco et al., 2008; Stone et al., 2012; Usun et al., 2013). Still, it remains unclear how dopamine release exactly relates to dopamine synthesis capacity as measured via PET. In animal research, the specific NMDA antagonist 3-[(R)-2-carboxypiperazin-4-yl]-propyl-1-phophonic acid (CPP) was injected in the PFC, and increased extracellular concentrations of VS dopamine were observed subsequently (Del Arco et al., 2008). Thus, the present result fits with findings from animal research regarding the regulation of VS dopamine levels.
Positive coupling between ventral striatal glutamate and ventral striatal dopamine
The second finding, a positive relationship between VS glutamate concentrations and VS dopamine synthesis capacity, is also supported by animal research. In animals, glutamate reuptake in the striatum was first blocked by the reuptake inhibitor l-trans-pyrrolidine-2,4-dicarboxilic acid, leading to an increase in extracellular dopamine and glutamate (among others). Subsequently, CPP was administered in the VS, which resulted in attenuation of extracellular dopamine (Segovia and Mora, 2001). Thus, these pharmacological effects in animals show the same direction as the positive correlation observed here. Furthermore, it was suggested that direct excitatory limbic afferents to the VS mainly stem from the hippocampus (Grace et al., 2007; Goto and Grace, 2008). However, it is impossible to distinguish between excitatory and inhibitory glutamatergic afferents to the VS using MRS imaging, because MRS measures the total glutamate concentration.
The balance of prefrontal vs striatal glutamate relates positively to ventral striatal dopamine
Third, we show that interindividual differences between PFC and VS glutamate concentrations correlated negatively with VS dopamine synthesis capacity. Thus, a balance of PFC–VS glutamate may play a role for fine-tuned regulation of VS presynaptic dopamine. This notion is supported by research proposing complementary glutamatergic inputs to the striatum from the “inhibitory” prefrontal and “excitatory” limbic system (Carlsson et al., 1999). Our observation fits to developmental animal models of schizophrenia, in which a disruption of prefrontal–hippocampal inputs to the VS resulted in aberrant VS plasticity (Belujon et al., 2014). Further studies helped to identify the ventral hippocampus as one important player for elevated presynaptic dopamine function (Blaha et al., 1997; Legault and Wise, 1999; Grace et al., 2007). With regard to regulation of VS presynaptic dopamine, it remains an open question whether interindividual variation arises from PFC or VS glutamate alone or whether the interaction might be decisive. This question may be more appropriately addressed in future studies measuring the entire trio of PFC and VS glutamate and striatal presynaptic dopamine combined with pharmacological challenges of dopamine and glutamate in animals and, if possible, humans.
Implications for mental diseases
The presented results are of interest regarding the “dopamine-glutamate hypothesis of schizophrenia” (Laruelle et al., 2003). From meta-analyses, increased striatal presynaptic dopamine is well known in schizophrenia, with the most convincing evidence for the more dorsal or associative parts of the striatum (Howes et al., 2012; Fusar-Poli and Meyer-Lindenberg, 2013), though it has also been shown in ventral striatal locations (McGowan et al., 2004; Kumakura et al., 2007). However, the origin of elevated striatal presynaptic dopamine remains unclear. It was proposed that a hypofunction of prefrontal NMDA receptors could be key and lead to elevated striatal presynaptic dopamine levels in patients (Laruelle et al., 2003). This idea resonates with the negative correlation between PFC glutamate and presynaptic VS dopamine synthesis capacity in the present study. Regarding PFC glutamate concentrations in schizophrenia, inconsistencies were present across studies with respect to medication (Poels et al., 2014). However, a recent meta-analysis showed decreased frontal glutamate in schizophrenia patients, but medication status could not be taken into account (Marsman et al., 2013). Disrupted interactions between dopamine and glutamate may be involved in aberrant modulation of synaptic plasticity (Surmeier et al., 2007; Di Filippo et al., 2009). So-called “dysconnectivity” was proposed as a common biological characteristic of schizophrenia (Heinz et al., 2003; Stephan et al., 2006, 2009), e.g., between PFC and hippocampus (Meyer-Lindenberg et al., 2005) or PFC and parietal regions (Deserno et al., 2012). This may represent a potential intermediate endophenotype of schizophrenia.
The presented findings may also be relevant for other mental diseases. For instance, addiction disorders were proposed to be associated with disrupted synaptic plasticity (Lüthi and Lüscher, 2014), and glutamate-associated plasticity is mediated by neuromodulators like dopamine and serotonin (Stephan et al., 2006; Heinz et al., 2011). Blunted presynaptic and postsynaptic striatal dopamine function was repeatedly observed in addiction (Volkow et al., 1996; Heinz et al., 2004; Martinez et al., 2005). Additionally, disrupted modulation of plasticity-related learning signals by dopamine was shown in alcohol dependence (Deserno et al., 2015a). Studies of glutamate concentrations in frontal lobe structures were more heterogeneous (Thoma et al., 2011; Mon et al., 2012; Abé et al., 2013; Ende et al., 2013). Most distinct abnormalities were observed in the anterior cingulate cortex, whereas no differences were reported in the dorsolateral PFC (Mon et al., 2012). Furthermore, ACC glutamate concentrations were found to vary as function of abstinence (Mon et al., 2012; Abé et al., 2013). Another study indicated that frontal WM glutamate may be reduced in alcohol dependence (Ende et al., 2013). Whereas interactions of glutamate and dopamine have been a target in animal models (Adrover et al., 2014; Nimitvilai et al., 2014), studies in humans are lacking so far. Thus, our findings provide a platform for studying glutamate–dopamine interactions in humans across mental diseases.
Limitations
First, glutamate concentrations measured by MRS reflect the total content of glutamate in a region, rather than a direct neurotransmitter contribution, limiting biological plausibility. However, glutamate measured by MRS and also dopamine synthesis capacity measured via FDOPA PET relate to important behavioral and neural signatures in healthy participants (Gallinat et al., 2007; Cools, 2008; Jocham et al., 2012; Schlagenhauf et al., 2012; Schmaal et al., 2012; Gleich et al., 2014; Deserno et al., 2015b) and in schizophrenia patients (Meyer-Lindenberg et al., 2002; Fusar-Poli et al., 2011; Fusar-Poli and Meyer-Lindenberg, 2013).
Second, specific neurophysiological mechanisms of how PFC and striatal glutamate regulate striatal presynaptic dopamine turnover remain to be elucidated. There has been substantial progress in translating animal models to human research focusing on schizophrenia (Modinos et al., 2015). It was shown that hippocampal electrophysiological activity enhances phasic firing of midbrain dopamine neurons (Grace et al., 2007), indicating a potential excitatory effect of glutamatergic input on midbrain dopamine firing via the hippocampus. Such glutamatergic input was shown to act locally at striatal presynaptic dopamine terminals via ionotropic (e.g., NMDA) receptors to facilitate tonic and impulse-independent phasic dopamine release (Borland and Michael, 2004), but glutamate may also indirectly enhance striatal dopamine via reuptake inhibition (Whitton, 1997). Regarding prefrontal glutamate, there is support that glutamatergic projections from the PFC influence dopaminergic projections to the striatum via GABA interneurons (Mora et al., 2002). Interestingly, infusion of the GABA(B) receptor agonists C4H12NO2P and baclofen into the PFC and striatum reduced dopamine levels, and this effect was reversed by a GABA antagonist (Balla et al., 2009). However, more research regarding specific receptor interactions potentially mediating the presented findings is needed.
Third, all presented findings are based on correlations and therefore preclude conclusions regarding causality. Future research should aim to investigate causal relationships more directly, e.g., by using multimodal imaging in combination with pharmacological interventions.
Conclusion
To our knowledge, this study provides first time in vivo evidence for an inverse coupling between prefrontal glutamate and striatal presynaptic dopamine function and a positive coupling between striatal glutamate and striatal presynaptic dopamine function in healthy human participants. Furthermore, we show that the balance between PFC and striatal glutamate also relates to VS presynaptic dopamine. These findings support theoretical assumptions regarding glutamate–dopamine interactions and point toward human imaging surrogate markers regarding the regulation of presynaptic dopamine function.
Footnotes
This work was supported by grants from the German Ministry for Education and Research to (BMBF 01GQ0914; to J.G.) and the German Research Foundation (DFG GA707/6-1, DFG SCHL1969/1-1, DFG SCHL 1969/2-1; to J.G., F.S.). R.C.L. was supported by the German National Academic Foundation, and R. Boehme was supported by the German Research Foundation (GRK 1123/2). L.D. and F.S. were supported by the Max Planck Society. We thank Stephan Lücke, Teresa Katthagen, Yu Fukuda, Sarah Diner, Jakob Kaminski, Saineb Alaa-Eddine, and Charlotte Witt for help with data acquisition and recruitment of the participants.
The authors declare no competing financial interests.
References
- Abé C, Mon A, Durazzo TC, Pennington DL, Schmidt TP, Meyerhoff DJ. Polysubstance and alcohol dependence: unique abnormalities of magnetic resonance-derived brain metabolite levels. Drug Alcohol Depend. 2013;130:30–37. doi: 10.1016/j.drugalcdep.2012.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adrover MF, Shin JH, Alvarez VA. Glutamate and dopamine transmission from midbrain dopamine neurons share similar release properties but are differentially affected by cocaine. J Neurosci. 2014;34:3183–3192. doi: 10.1523/JNEUROSCI.4958-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner J, Friston KJ. Unified segmentation. Neuroimage. 2005;26:839–851. doi: 10.1016/j.neuroimage.2005.02.018. [DOI] [PubMed] [Google Scholar]
- Balla A, Nattini ME, Sershen H, Lajtha A, Dunlop DS, Javitt DC. GABAB/NMDA receptor interaction in the regulation of extracellular dopamine levels in rodent prefrontal cortex and striatum. Neuropharmacology. 2009;56:915–921. doi: 10.1016/j.neuropharm.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belujon P, Patton MH, Grace AA. Role of the prefrontal cortex in altered hippocampal-accumbens synaptic plasticity in a developmental animal model of schizophrenia. Cereb Cortex. 2014;24:968–977. doi: 10.1093/cercor/bhs380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaha CD, Yang CR, Floresco SB, Barr AM, Phillips AG. Stimulation of the ventral subiculum of the hippocampus evokes glutamate receptor-mediated changes in dopamine efflux in the rat nucleus accumbens. Eur J Neurosci. 1997;9:902–911. doi: 10.1111/j.1460-9568.1997.tb01441.x. [DOI] [PubMed] [Google Scholar]
- Borland LM, Michael AC. Voltammetric study of the control of striatal dopamine release by glutamate. J Neurochem. 2004;91:220–229. doi: 10.1111/j.1471-4159.2004.02708.x. [DOI] [PubMed] [Google Scholar]
- Bromberg-Martin ES, Matsumoto M, Hikosaka O. Dopamine in motivational control: rewarding, aversive, and alerting. Neuron. 2010;68:815–834. doi: 10.1016/j.neuron.2010.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson A, Waters N, Carlsson ML. Neurotransmitter interactions in schizophrenia–therapeutic implications. Biol Psychiatry. 1999;46:1388–1395. doi: 10.1016/S0006-3223(99)00117-1. [DOI] [PubMed] [Google Scholar]
- Conover WJ. Practical nonparametric statistics. Ed 3. New York: Wiley; 1999. [Google Scholar]
- Cools R. Role of dopamine in the motivational and cognitive control of behavior. Neuroscientist. 2008;14:381–395. doi: 10.1177/1073858408317009. [DOI] [PubMed] [Google Scholar]
- Cools R. Dopaminergic control of the striatum for high-level cognition. Curr Opin Neurobiol. 2011;21:402–407. doi: 10.1016/j.conb.2011.04.002. [DOI] [PubMed] [Google Scholar]
- Cools R, D'Esposito M. Inverted-U-shaped dopamine actions on human working memory and cognitive control. Biol Psychiatry. 2011;69:e113–e125. doi: 10.1016/j.biopsych.2011.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Fuente-Sandoval C, León-Ortiz P, Favila R, Stephano S, Mamo D, Ramírez-Bermúdez J, Graff-Guerrero A. Higher levels of glutamate in the associative-striatum of subjects with prodromal symptoms of schizophrenia and patients with first-episode psychosis. Neuropsychopharmacology. 2011;36:1781–1791. doi: 10.1038/npp.2011.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Arco A, Segovia G, Mora F. Blockade of NMDA receptors in the prefrontal cortex increases dopamine and acetylcholine release in the nucleus accumbens and motor activity. Psychopharmacology (Berl) 2008;201:325–338. doi: 10.1007/s00213-008-1288-3. [DOI] [PubMed] [Google Scholar]
- Deserno L, Sterzer P, Wüstenberg T, Heinz A, Schlagenhauf F. Reduced prefrontal-parietal effective connectivity and working memory deficits in schizophrenia. J Neurosci. 2012;32:12–20. doi: 10.1523/JNEUROSCI.3405-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deserno L, Beck A, Huys QJ, Lorenz RC, Buchert R, Buchholz HG, Plotkin M, Kumakara Y, Cumming P, Heinze HJ, Grace AA, Rapp MA, Schlagenhauf F, Heinz A. Chronic alcohol intake abolishes the relationship between dopamine synthesis capacity and learning signals in the ventral striatum. Eur J Neurosci. 2015a;41:477–486. doi: 10.1111/ejn.12802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deserno L, Huys QJM, Boehme R, Buchert R, Heinze HJ, Grace AA, Dolan RJ, Heinz A, Schlagenhauf F. Ventral striatal dopamine reflects behavioral and neural signatures of model-based control during sequential decision making. Proc Natl Acad Sci U S A. 2015b;112:1595–1600. doi: 10.1073/pnas.1417219112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Filippo M, Picconi B, Tantucci M, Ghiglieri V, Bagetta V, Sgobio C, Tozzi A, Parnetti L, Calabresi P. Short-term and long-term plasticity at corticostriatal synapses: implications for learning and memory. Behav Brain Res. 2009;199:108–118. doi: 10.1016/j.bbr.2008.09.025. [DOI] [PubMed] [Google Scholar]
- Ende G, Hermann D, Demirakca T, Hoerst M, Tunc-Skarka N, Weber-Fahr W, Wichert S, Rabinstein J, Frischknecht U, Mann K, Vollstädt-Klein S. Loss of control of alcohol use and severity of alcohol dependence in non-treatment-seeking heavy drinkers are related to lower glutamate in frontal white matter. Alcohol Clin Exp Res. 2013;37:1643–1649. doi: 10.1111/acer.12149. [DOI] [PubMed] [Google Scholar]
- Field AP. Discovering statistics using SPSS (and sex and drugs and rock “n” roll) Ed 3. Thousand Oaks, CA: Sage; 2009. [Google Scholar]
- Fusar-Poli P, Meyer-Lindenberg A. Striatal presynaptic dopamine in schizophrenia, part II: meta-analysis of [(18)F/(11)C]-DOPA PET studies. Schizophr Bull. 2013;39:33–42. doi: 10.1093/schbul/sbr180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusar-Poli P, Stone JM, Broome MR, Valli I, Mechelli A, McLean MA, Lythgoe DJ, O'Gorman RL, Barker GJ, McGuire PK. Thalamic glutamate levels as a predictor of cortical response during executive functioning in subjects at high risk for psychosis. Arch Gen Psychiatry. 2011;68:881–890. doi: 10.1001/archgenpsychiatry.2011.46. [DOI] [PubMed] [Google Scholar]
- Gallinat J, Kunz D, Lang UE, Neu P, Kassim N, Kienast T, Seifert F, Schubert F, Bajbouj M. Association between cerebral glutamate and human behaviour: the sensation seeking personality trait. Neuroimage. 2007;34:671–678. doi: 10.1016/j.neuroimage.2006.10.004. [DOI] [PubMed] [Google Scholar]
- Gleich T, Lorenz RC, Pöhland L, Raufelder D, Deserno L, Beck A, Heinz A, Kühn S, Gallinat J. Frontal glutamate and reward processing in adolescence and adulthood. Brain Struct Funct. 2014 doi: 10.1007/s00429-014-0844-3. Advance online publication. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Burgos G, Lewis DA. NMDA receptor hypofunction, parvalbumin-positive neurons and cortical gamma oscillations in schizophrenia. Schizophr Bull. 2012;38:950–957. doi: 10.1093/schbul/sbs010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto Y, Grace AA. Limbic and cortical information processing in the nucleus accumbens. Trends Neurosci. 2008;31:552–558. doi: 10.1016/j.tins.2008.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace AA. Phasic versus tonic dopamine release and the modulation of dopamine system responsivity: a hypothesis for the etiology of schizophrenia. Neuroscience. 1991;41:1–24. doi: 10.1016/0306-4522(91)90196-U. [DOI] [PubMed] [Google Scholar]
- Grace AA, Floresco SB, Goto Y, Lodge DJ. Regulation of firing of dopaminergic neurons and control of goal-directed behaviors. Trends Neurosci. 2007;30:220–227. doi: 10.1016/j.tins.2007.03.003. [DOI] [PubMed] [Google Scholar]
- Heinz A, Romero B, Gallinat J, Juckel G, Weinberger DR. Molecular brain imaging and the neurobiology and genetics of schizophrenia. Pharmacopsychiatry. 2003;36(Suppl 3):S152–S157. doi: 10.1055/s-2003-45123. [DOI] [PubMed] [Google Scholar]
- Heinz A, Siessmeier T, Wrase J, Hermann D, Klein S, Grüsser SM, Grüsser-Sinopoli SM, Flor H, Braus DF, Buchholz HG, Gründer G, Schreckenberger M, Smolka MN, Rösch F, Mann K, Bartenstein P. Correlation between dopamine D(2) receptors in the ventral striatum and central processing of alcohol cues and craving. Am J Psychiatry. 2004;161:1783–1789. doi: 10.1176/appi.ajp.161.10.1783. [DOI] [PubMed] [Google Scholar]
- Heinz AJ, Beck A, Meyer-Lindenberg A, Sterzer P, Heinz A. Cognitive and neurobiological mechanisms of alcohol-related aggression. Nat Rev Neurosci. 2011;12:400–413. doi: 10.1038/nrn3042. [DOI] [PubMed] [Google Scholar]
- Howes OD, Kambeitz J, Kim E, Stahl D, Slifstein M, Abi-Dargham A, Kapur S. The nature of dopamine dysfunction in schizophrenia and what this means for treatment. Arch Gen Psychiatry. 2012;69:776–786. doi: 10.1001/archgenpsychiatry.2012.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jocham G, Hunt LT, Near J, Behrens TEJ. A mechanism for value-guided choice based on the excitation-inhibition balance in prefrontal cortex. Nat Neurosci. 2012;15:960–961. doi: 10.1038/nn.3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumakura Y, Cumming P, Vernaleken I, Buchholz H-G, Siessmeier T, Heinz A, Kienast T, Bartenstein P, Gründer G. Elevated [18F]fluorodopamine turnover in brain of patients with schizophrenia: an [18F]fluorodopa/positron emission tomography study. J Neurosci. 2007;27:8080–8087. doi: 10.1523/JNEUROSCI.0805-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laruelle M, Kegeles LS, Abi-Dargham A. Glutamate, dopamine, and schizophrenia: from pathophysiology to treatment. Ann N Y Acad Sci. 2003;1003:138–158. doi: 10.1196/annals.1300.063. [DOI] [PubMed] [Google Scholar]
- Legault M, Wise RA. Injections of N-methyl-d-aspartate into the ventral hippocampus increase extracellular dopamine in the ventral tegmental area and nucleus accumbens. Synapse. 1999;31:241–249. doi: 10.1002/(SICI)1098-2396(19990315)31:4<241::AID-SYN1>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Lovinger DM. Neurotransmitter roles in synaptic modulation, plasticity and learning in the dorsal striatum. Neuropharmacology. 2010;58:951–961. doi: 10.1016/j.neuropharm.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüthi A, Lüscher C. Pathological circuit function underlying addiction and anxiety disorders. Nat Neurosci. 2014;17:1635–1643. doi: 10.1038/nn.3849. [DOI] [PubMed] [Google Scholar]
- Marsman A, van den Heuvel MP, Klomp DW, Kahn RS, Luijten PR, Hulshoff Pol HE. Glutamate in schizophrenia: a focused review and meta-analysis of 1H-MRS studies. Schizophr Bull. 2013;39:120–129. doi: 10.1093/schbul/sbr069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez D, Slifstein M, Broft A, Mawlawi O, Hwang DR, Huang Y, Cooper T, Kegeles L, Zarahn E, Abi-Dargham A, Haber SN, Laruelle M. Imaging human mesolimbic dopamine transmission with positron emission tomography. Part II: Amphetamine-induced dopamine release in the functional subdivisions of the striatum. J Cereb Blood Flow Metab. 2003;23:285–300. doi: 10.1097/01.WCB.0000048520.34839.1A. [DOI] [PubMed] [Google Scholar]
- Martinez D, Gil R, Slifstein M, Hwang DR, Huang Y, Perez A, Kegeles L, Talbot P, Evans S, Krystal J, Laruelle M, Abi-Dargham A. Alcohol dependence is associated with blunted dopamine transmission in the ventral striatum. Biol Psychiatry. 2005;58:779–786. doi: 10.1016/j.biopsych.2005.04.044. [DOI] [PubMed] [Google Scholar]
- McGowan S, Lawrence AD, Sales T, Quested D, Grasby P. Presynaptic dopaminergic dysfunction in schizophrenia: a positron emission tomographic [18F]fluorodopa study. Arch Gen Psychiatry. 2004;61:134–142. doi: 10.1001/archpsyc.61.2.134. [DOI] [PubMed] [Google Scholar]
- Meyer-Lindenberg A, Miletich RS, Kohn PD, Esposito G, Carson RE, Quarantelli M, Weinberger DR, Berman KF. Reduced prefrontal activity predicts exaggerated striatal dopaminergic function in schizophrenia. Nat Neurosci. 2002;5:267–271. doi: 10.1038/nn804. [DOI] [PubMed] [Google Scholar]
- Meyer-Lindenberg A, Kohn PD, Kolachana B, Kippenhan S, McInerney-Leo A, Nussbaum R, Weinberger DR, Berman KF. Midbrain dopamine and prefrontal function in humans: interaction and modulation by COMT genotype. Nat Neurosci. 2005;8:594–596. doi: 10.1038/nn1438. [DOI] [PubMed] [Google Scholar]
- Modinos G, Allen P, Grace AA, McGuire P. Translating the MAM model of psychosis to humans. Trends Neurosci. 2015;38:129–138. doi: 10.1016/j.tins.2014.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mon A, Durazzo TC, Meyerhoff DJ. Glutamate, GABA, and other cortical metabolite concentrations during early abstinence from alcohol and their associations with neurocognitive changes. Drug Alcohol Depend. 2012;125:27–36. doi: 10.1016/j.drugalcdep.2012.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mora F, Arco AD, Segovia G. Glutamate-dopamine interactions in striatum and nucleus accumbens of the conscious rat during aging. In: Graybiel AM, Delong MR, Kitai ST, editors. The basal ganglia VI, Advances in behavioral biology. Vol 54. New York: Springer; 2002. pp. 615–622. [Google Scholar]
- Morales I, Fuentes A, Ballaz S, Obeso JA, Rodriguez M. Striatal interaction among dopamine, glutamate and ascorbate. Neuropharmacology. 2012;63:1308–1314. doi: 10.1016/j.neuropharm.2012.08.007. [DOI] [PubMed] [Google Scholar]
- Nimitvilai S, Herman M, You C, Arora DS, McElvain MA, Roberto M, Brodie MS. Dopamine D2 receptor desensitization by dopamine or corticotropin releasing factor in ventral tegmental area neurons is associated with increased glutamate release. Neuropharmacology. 2014;82:28–40. doi: 10.1016/j.neuropharm.2014.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patlak CS, Blasberg RG. Graphical evaluation of blood-to-brain transfer constants from multiple-time uptake data. Generalizations. J Cereb Blood Flow Metab. 1985;5:584–590. doi: 10.1038/jcbfm.1985.87. [DOI] [PubMed] [Google Scholar]
- Poels EM, Kegeles LS, Kantrowitz JT, Javitt DC, Lieberman JA, Abi-Dargham A, Girgis RR. Glutamatergic abnormalities in schizophrenia: a review of proton MRS findings. Schizophr Res. 2014;152:325–332. doi: 10.1016/j.schres.2013.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provencher S. LCModel & LCMgui user's manual. 2014. http://s-provencher.com/pub/LCModel/manual/manual.pdf.
- Rothman DL, De Feyter HM, de Graaf RA, Mason GF, Behar KL. 13C MRS studies of neuroenergetics and neurotransmitter cycling in humans. NMR Biomed. 2011;24:943–957. doi: 10.1002/nbm.1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowland LM, Bustillo JR, Mullins PG, Jung RE, Lenroot R, Landgraf E, Barrow R, Yeo R, Lauriello J, Brooks WM. Effects of ketamine on anterior cingulate glutamate metabolism in healthy humans: a 4-T proton MRS study. Am J Psychiatry. 2005;162:394–396. doi: 10.1176/appi.ajp.162.2.394. [DOI] [PubMed] [Google Scholar]
- Ryan JJ, Paolo AM. Exploratory factor analysis of the WAIS-III in a mixed patient sample. Arch Clin Neuropsychol. 2001;16:151–156. [PubMed] [Google Scholar]
- Schlagenhauf F, Rapp MA, Huys QJM, Beck A, Wüstenberg T, Deserno L, Buchholz HG, Kalbitzer J, Buchert R, Bauer M, Kienast T, Cumming P, Plotkin M, Kumakura Y, Grace AA, Dolan RJ, Heinz A. Ventral striatal prediction error signaling is associated with dopamine synthesis capacity and fluid intelligence. Hum Brain Mapp. 2012;34:1490–1499. doi: 10.1002/hbm.22000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmaal L, Goudriaan AE, van der Meer J, van den Brink W, Veltman DJ. The association between cingulate cortex glutamate concentration and delay discounting is mediated by resting state functional connectivity. Brain Behav. 2012;2:553–562. doi: 10.1002/brb3.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz W. Predictive reward signal of dopamine neurons. J Neurophysiol. 1998;80:1–27. doi: 10.1152/jn.1998.80.1.1. [DOI] [PubMed] [Google Scholar]
- Schultz W. Updating dopamine reward signals. Curr Opin Neurobiol. 2013;23:229–238. doi: 10.1016/j.conb.2012.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz TL, Sachdeva S, Stahl SM. Glutamate neurocircuitry: theoretical underpinnings in schizophrenia. Front Pharmacol. 2012;3:195. doi: 10.3389/fphar.2012.00195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwerk A, Alves FD, Pouwels PJ, van Amelsvoort T. Metabolic alterations associated with schizophrenia: a critical evaluation of proton magnetic resonance spectroscopy studies. J Neurochem. 2014;128:1–87. doi: 10.1111/jnc.12398. [DOI] [PubMed] [Google Scholar]
- Segovia G, Mora F. Involvement of NMDA and AMPA/kainate receptors in the effects of endogenous glutamate on extracellular concentrations of dopamine and GABA in the nucleus accumbens of the awake rat. Brain Res Bull. 2001;54:153–157. doi: 10.1016/s0361-9230(00)00432-9. [DOI] [PubMed] [Google Scholar]
- Sesack SR, Carr DB, Omelchenko N, Pinto A. Anatomical substrates for glutamate-dopamine interactions: evidence for specificity of connections and extrasynaptic actions. Ann N Y Acad Sci. 2003;1003:36–52. doi: 10.1196/annals.1300.066. [DOI] [PubMed] [Google Scholar]
- Stephan KE, Baldeweg T, Friston KJ. Synaptic plasticity and dysconnection in schizophrenia. Biol Psychiatry. 2006;59:929–939. doi: 10.1016/j.biopsych.2005.10.005. [DOI] [PubMed] [Google Scholar]
- Stephan KE, Friston KJ, Frith CD. Dysconnection in schizophrenia: from abnormal synaptic plasticity to failures of self-monitoring. Schizophr Bull. 2009;35:509–527. doi: 10.1093/schbul/sbn176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone JM, Dietrich C, Edden R, Mehta MA, De Simoni S, Reed LJ, Krystal JH, Nutt D, Barker GJ. Ketamine effects on brain GABA and glutamate levels with 1H-MRS: relationship to ketamine-induced psychopathology. Mol Psychiatry. 2012;17:664–665. doi: 10.1038/mp.2011.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier DJ, Ding J, Day M, Wang Z, Shen W. D1 and D2 dopamine-receptor modulation of striatal glutamatergic signaling in striatal medium spiny neurons. Trends Neurosci. 2007;30:228–235. doi: 10.1016/j.tins.2007.03.008. [DOI] [PubMed] [Google Scholar]
- Thoma R, Mullins P, Ruhl D, Monnig M, Yeo RA, Caprihan A, Bogenschutz M, Lysne P, Tonigan S, Kalyanam R, Gasparovic C. Perturbation of the glutamate-glutamine system in alcohol dependence and remission. Neuropsychopharmacology. 2011;36:1359–1365. doi: 10.1038/npp.2011.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzourio-Mazoyer N, Landeau B, Papathanassiou D, Crivello F, Etard O, Delcroix N, Mazoyer B, Joliot M. Automated anatomical labeling of activations in SPM using a macroscopic anatomical parcellation of the MNI MRI single-subject brain. Neuroimage. 2002;15:273–289. doi: 10.1006/nimg.2001.0978. [DOI] [PubMed] [Google Scholar]
- Usun Y, Eybrard S, Meyer F, Louilot A. Ketamine increases striatal dopamine release and hyperlocomotion in adult rats after postnatal functional blockade of the prefrontal cortex. Behav Brain Res. 2013;256:229–237. doi: 10.1016/j.bbr.2013.08.017. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Fowler JS, Logan J, Hitzemann R, Ding YS, Pappas N, Shea C, Piscani K. Decreases in dopamine receptors but not in dopamine transporters in alcoholics. Alcohol Clin Exp Res. 1996;20:1594–1598. doi: 10.1111/j.1530-0277.1996.tb05936.x. [DOI] [PubMed] [Google Scholar]
- Whitton PS. Glutamatergic control over brain dopamine release in vivo and in vitro. Neurosci Biobehav Rev. 1997;21:481–488. doi: 10.1016/S0149-7634(96)00034-6. [DOI] [PubMed] [Google Scholar]