

Abstract
Anxiety-related psychiatric disorders represent one of the largest health burdens worldwide. Single nucleotide polymorphisms of the FK506 binding protein 51 (FKBP51) gene have been repeatedly associated with anxiety-related disorders and stress sensitivity. Given the intimate relationship of stress and anxiety, we hypothesized that amygdala FKBP51 may mediate anxiety-related behaviors. Mimicking the stress effect by specifically overexpressing FKBP51 in the basolateral amygdala (BLA) or central amygdala resulted in increased anxiety-related behavior, respectively. In contrast, application of a highly selective FKBP51 point mutant antagonist, following FKBP51mut BLA-overexpression, reduced the anxiogenic phenotype. We subsequently tested a novel FKBP51 antagonist, SAFit2, in wild-type mice via BLA microinjections, which reduced anxiety-related behavior. Remarkably, the same effect was observed following peripheral administration of SAFit2. To our knowledge, this is the first in vivo study using a specific FKBP51 antagonist, thereby unraveling the role of FKBP51 and its potential as a novel drug target for the improved treatment of anxiety-related disorders.
Keywords: amygdala, antidepressants, anxiety, FKBP51, PTSD
Introduction
Anxiety-related behaviors are highly prevalent in stress-induced psychiatric disorders, such as major depression, posttraumatic stress disorder (PTSD), bipolar disorder, and schizophrenia (Chrousos, 2009; Lupien et al., 2009). Despite that these disorders contribute significantly to disease burden worldwide, their underlying molecular mechanisms are still poorly understood (Tennant, 2001; Krishnan and Nestler, 2008; Nestler and Hyman, 2010). In addition, a substantial proportion of patients do not benefit from the currently available treatment options (Binder and Holsboer, 2006; Thase, 2006); thus, there is a desperate need to increase our understanding of the mechanisms involved in the etiology of these disorders, so that novel therapeutics can be developed (Porcelli et al., 2011).
Human genetic studies have repeatedly implicated the FKBP5 gene, encoding FK506 binding protein 51 (FKBP51), in numerous psychiatric disorders (Binder et al., 2004, 2008; Koenen et al., 2005; Binder, 2009; Brent et al., 2010; Lavebratt et al., 2010; Ellsworth et al., 2013; Klengel et al., 2013). FKBP51 is an imunophilin best known to alter glucocorticoid receptor (GR) sensitivity (Riggs et al., 2003; Binder et al., 2004). When FKBP51 acts on the GR, the affinity of GR for cortisol, the main hormone produced in response to stress, and GR nuclear translocation is reduced (Wochnik et al., 2005). As a recurring characteristic of mood and anxiety disorders is altered GR expression and signaling (Holsboer, 2000; Pariante and Miller, 2001; Yehuda, 2009), FKBP51 is an attractive target to reverse deficits in the GR system, at least in a subset of patients (Schmidt et al., 2012).
Associations of single nucleotide polymorphisms (SNPs) in FKBP5 have repeatedly been found in various psychiatric case-control populations, including major depression (MD) (Appel et al., 2011; Menke et al., 2013), bipolar disorder (Binder et al., 2004; Tatro et al., 2009; Willour et al., 2009; Lavebratt et al., 2010), and PTSD (Binder et al., 2008; Mehta et al., 2011). In addition, FKBP51 variants also have been linked with suicidal behavior (Willour et al., 2009; Brent et al., 2010; Roy et al., 2010; Supriyanto et al., 2011). Interestingly, alterations in FKBP51 expression in the amygdala have been reported in postmortem samples from suicide victims (Pérez-Ortiz et al., 2013). The amygdala plays a pivotal role in the neurocircuitry of fear and anxiety disorders (LeDoux, 2007; Shin and Liberzon, 2010; Dias et al., 2013), and was also shown to modulate the hypothalamic-pituitary-adrenal (HPA) axis through negative feedback mediated by the GR (Gray, 1993; Davis, 1994; McEwen and Sapolsky, 1995).
We have previously shown that FKBP51 mRNA expression is strongly upregulated in the amygdala following acute stress (Scharf et al., 2011), and that FKBP51 knock-out mice are less affected by chronic social defeat stress, likely mediated by increased GR sensitivity (Hartmann et al., 2012b). However, a causal link of FKBP51 to anxiety-related behavior is still missing. We approached this challenge by manipulating FKBP51 expression in the basolateral amygdala (BLA) and central amygdala (CeA), respectively, demonstrating that FKBP51 most notably in the BLA enhances anxiety. Next, we took advantage of the recent development of the first selective and potent FKBP51 antagonists (Gopalakrishnan et al., 2012a,b; Gaali et al., 2015) and could show that these ligands applied directly in the amygdala or via the periphery have profound anxiolytic properties. Pharmacological inhibition of FKBP51 might thus constitute a mechanistically novel therapeutic approach for the prevention and treatment of stress-related anxiety disorders.
Materials and Methods
Experimental design
Experiment 1.
As FKBP51 expression has previously been shown to be upregulated following acute stress (Scharf et al., 2011), we mimicked this effect by overexpressing FKBP51 in the BLA or CeA. Anxiety-related behavior was subsequently confirmed for the BLA (n = 12–14 per group) and partially for the CeA (n = 9–10 per group). Experiments were additionally repeated following FKBP51 overexpression in the dorsal hippocampus (dHc), as a negative control (n = 12–13 per group). The testing order was as follows: open field (OF), elevated plus maze (EPM), dark-light box (DaLi), and forced swim test (FST). All animals underwent the same testing battery in the same order of the tests.
Experiment 2.
Ligand2, an FKBP51 antagonist that specifically binds to the mutated FKBP51F67V but not wild-type FKBP51, was analyzed for its potential to penetrate the blood–brain barrier. FKBP51F67V preserves the repressing activity of wild-type FKBP51 on the GR, but can specifically be inhibited by the FKBP51 antagonist Ligand2. Animals received a single injection of either 0.1 mg/kg body weight (BW), 1.0 mg/kg BW, 10.0 mg/kg BW Ligand2, or vehicle solution (n = 4 per group). Next, we tested the ability of Ligand2 to rescue the anxiogenic effects induced by FKBP51F67V overexpression in the BLA (n = 11–13 per group). The testing order was as follows: OF, EPM, DaLi, and FST. All animals underwent the same testing battery in the same order of the tests. To confirm the specificity and to rule out any potential drug side effects of the antagonist in the control group, we performed an additional control experiment, in which naive C57BL/6N mice received a single injection of 10.0 mg/kg BW Ligand2 or vehicle solution and were subsequently tested with regard to anxiety-related behavior (n = 8 per group).
Experiment 3.
Wild-type FKBP51 was blocked via peripheral administration of SAFit2 in naive mice and anxiety-related behavior was analyzed in the EPM and DaLi 1 and 16 h after the injection (2 separate cohorts of mice; n = 10–11 per group).
Experiment 4.
Wild-type FKBP51 was blocked in the BLA via micro-injections of SAFit2 (0.5 μl per injection) in mice. The effects on anxiety-related behavior was analyzed in the DaLi (n = 14 per group) 16 h after the application.
Animals
For all experiments, male C57BL/6N mice at the age of 12–15 weeks were used (Charles River Laboratories). The mice were held under standard conditions (12 h light/dark cycle, lights on at 08:00 A.M.; temperature 23 ± 2°C), were single housed and acclimatized to the room for 2 weeks before the commencing experiments. Food (Altromin 1324, Altromin) and tap water were available ad libitum. A separate batch of animals was used for each experiment. Experiments were performed in the animal facilities of the Max Planck Institute of Psychiatry in Munich (Germany) in accordance with the European Communities' Council Directive 2010/63/EU. Protocols were approved by the committee for the Care and Use of Laboratory animals of the Government of Upper Bavaria, Germany. All efforts were made to minimize animal suffering.
Viral overexpression of FKBP51
Viral overexpression was performed using an adeno-associated bicistronic AAV1/2 vector (GeneDetect) as described previously (Schmidt et al., 2011). In Experiment 1, the vector contained a CAG-HA-tagged-FKBP51-WPRE-BGH-polyA expression cassette (containing the coding sequence of human Fkbp51 NCBI CCDS ID CCDS4808.1). For Experiment 2, the vector contained a CAG-HA-tagged-FKBP51-F67V-mutant-WPRE-BGH-polyA expression cassette (containing the coding sequence of the human Fkbp51 NCBI CCDS ID CCDS4808.1) with a F67V point mutation (Gaali et al., 2015). For the control group, we used the same vector construct without expression of FKBP51 (CAG-Null/Empty-WPRE-BGH-polyA). Virus production, amplification, and purification were performed by GeneDetect. Mice were anesthetized with isoflurane, and 0.5 μl (BLA, dHc) or 0.025 μl (CeA) of either AAV-FKBP51, AAV-FKBP51F67V or AAV-Empty [titers: 1.3 × 1012 genomic particles/ml (Experiment 1), 2.6 × 1012 genomic particles/ml (Experiment 2)] were bilaterally injected in the BLA, CeA, or hippocampus at 0.06 μl/min by glass capillaries with tip resistance of 2–4 MΩ in a stereotactic apparatus. The following coordinates were used: (1) BLA: 1.0 mm posterior to bregma, 3.5 mm lateral from midline, and 3.8 mm below the surface of the skull; (2) CeA: 1.2 mm posterior to bregma, 2.6 mm lateral from midline, and 4.5 mm below the surface of the skull; and (3) hippocampus [CA1 and dentate gyrus (DG) region of the dorsal hippocampus]: 1.9 mm posterior to bregma, 1.3 mm lateral from midline, and 1.3/1.8 mm below the surface of the skull. After surgery, mice were treated for 5 d with Metacam via drinking water. Behavioral testing started 4 weeks after virus injection. Successful overexpression of the FKBP51-forms was verified by in situ hybridization and immunofluorescence. Animals that were not infected bilaterally in the dorsal hippocampus, BLA or CeA were excluded from the analysis. Quantification of overexpression was achieved by in situ hybridization using the riboprobe described below.
Implantation of guiding cannulas
For Experiment 4, stainless steel cannulas (10 mm, 26 G) were implanted above the BLA during anesthesia using the same setup as in Experiments 1 and 2. A hole was drilled to implant each cannula ventrally into the brain. The cannulas were fixed in the skull by two screws (DIN 84 MS S1*3 screws, stainless steel, Schrauben Presiniger) and dental cement (Paladur, Heraeus Kulzer). After surgery, the animals were singly housed and allowed to recover for at least 7 d. Injection cannulas (Hamilton, 30 G) that extended 1 mm beyond the tip of the guiding cannula were used for the microinjections.
Behavioral testing
All behavioral tests were recorded using a videotracking system (Anymaze 4.20; Stoelting). The following behavioral tests were performed: OF, EPM, DaLi. Only test-naive mice were used for each experiment, unless specified otherwise (Experiments 1 and 2). The testing procedures were performed as described below. The behavioral tests were performed between 08:30 A.M. and 12:30 P.M. in the same room in which the mice were housed.
Open field
The OF test was performed to investigate general locomotor activity in a novel environment, which could be a confounding factor in other exploration-based tests as the EPM or DaLi. Testing was performed in an empty OF arena (50 × 50 × 50 cm) made of gray polyvinyl chloride (PVC), which was evenly illuminated with 15 Lux. The low illumination of the OF arena was specifically chosen to investigate locomotion behavior and not create an aversive center region that may induce anxiety-related behavior. Testing time was 15 min and parameter of interest was the total distance traveled.
Elevated plus maze
The EPM was conducted to display changes in anxiety-related behavior. The device consisted of a plus-shaped platform with two opposing open arms (30 × 5 × 0.5 cm) and two opposing enclosed arms (30 × 5 × 15 cm), made of gray PVC, which were connected by a central area (5 × 5 cm). The whole device was elevated 50 cm above the floor. The illumination was 25 Lux in the open arms and <10 Lux in the closed arms. Testing duration was 10 min in Experiments 1 and 3, and 5 min in Experiment 2. Mice were placed into the center zone facing one of the enclosed arms at the start of the test. The time spent in the open arms compared with the total arm time, as well as the number of open arm entries were analyzed. Animals that fell off the open arm of the apparatus during testing were excluded from the analysis.
Dark-light box
The DaLi test was used as another paradigm to measure anxiety-related behavior. The apparatus consisted of a rectangular box with two compartments, the dark compartment (15 × 20 × 25 cm), with <10 lux and the light compartment (30 × 20 × 25 cm), lit with 700 lux. Both compartments were connected by a 4-cm-long tunnel. At the beginning of the test, each mouse was placed in the center of the dark compartment facing the back wall of the apparatus. Testing duration was 7 min in Experiment 1 and 10 min in Experiments 2 and 3. The time spent in the lit compartment, as well as the number of lit compartment entries, the latency to the first entry to the lit compartment and the distance traveled in the lit compartment were analyzed.
Forced swim test
The FST was used to assess stress-coping behavior. Each mouse was placed into a 2 L glass beaker (diameter: 13 cm, height: 24 cm) filled with tap water (21 ± 1°C) to a height of 15 cm, so that the mouse could not touch the bottom with its hindpaws or tail. Testing time was 6 min. The parameters struggling, swimming, floating, and latency to first floating were scored by an experienced observer, blind to condition or treatment of the animals.
Drugs
Both FKBP51 antagonists (Ligand2 and SAFit2) were solubilized in 4% EtOH, 5% Tween80 and 5% PEG400 in 0.9% saline (Experiments 2 and 3) or in artificial CSF (Experiment 4). In Experiment 2, animals received an intraperitoneal (i.p.) injection of either vehicle (4% EtOH, 5% Tween80, and 5% PEG400 in 0.9% saline) or Ligand2 (0.1 mg/kg BW, 1.0 mg/kg BW or 10 mg/kg BW) solution 1 h prior perfusion to assess the blood–brain barrier penetration. Mice that underwent surgeries and behavioral analysis in Experiment 2 received an intraperitoneal injection of vehicle or Ligand2 (10 mg/kg BW) on 10 consecutive days. On testing days, the injection was applied 1 h before the start of the test. Naive mice (without surgery) received a single intraperitoneal injection of vehicle or Ligand2 (10 mg/kg BW) 1 h before the start of the behavioral test. In Experiment 3, animals received an intraperitoneal injection of vehicle or SAFit2 (Gaali et al., 2015; 20 mg/kg BW) 1 or 16 h before the start of the test. In Experiment 4, during a quick anesthesia of isoflurane, animals received a bilateral microinjection into the BLA (0.5 μl per side) of vehicle or SAFit2 (20 mg/kg BW) 16 h before the start of the test.
Sampling procedure
At the day of kill, animals were deeply anesthetized with isoflurane. For animals that were used to assess the pharmacokinetics of Ligand2 in Experiment 2, blood samples were taken by cardiac puncture, kept on ice, and later centrifuged at 8000 rpm at 4°C for 15 min. Plasma was transferred to new, labeled tubes and stored at −20°C. These mice were perfused intracardially with saline; brains were removed and immediately stored at −80°C. The concentration of the FKBP51 antagonist in plasma and brain was quantified by LC-MS/MS. All other animals of Experiments 1 and 2 were perfused intracardially with 4% paraformaldehyde. Brains were removed, postfixed overnight in 4% paraformaldehyde, followed by subsequent overnight incubation in 30% sucrose solution at 4°C, and then stored at −80°C until processing for in situ hybridization and immunohistochemistry. Brains of Experiment 4 were removed and snap-frozen on dry ice. Correct cannula placement was examined by postmortem histological verification.
In situ hybridization and immunohistochemistry
For in situ hybridization and immunohistochemistry, frozen brains were coronally sectioned in a cryostat microtome at 25 μm and kept at −80°C. In situ hybridization using a 35S UTP-labeled ribonucleotide probe for Fkbp51 (forward primer: 5′-CTTGGACCACGCTATGGTTT; reverse primer: 5′-GGATTGACTGCCAACACCTT) was performed as described previously (Schmidt et al., 2007). Immunofluorescence (mouse anti-Ha, clone Ha-7, no. H9658; 1:1000, Sigma-Aldrich; Donkey anti-mouse AlexaFluor 488, 1:500; Invitrogen) was performed on free-floating sections (n = 3 per mouse) as described previously (Wang et al., 2011). Images of autoradiographs and immunohistochemistry were obtained with an Axioplan 2 microscope (Zeiss) and an Olympus Fluoview1000 confocal microscope (Olympus), respectively.
Ligand2 in vitro characterization
N2a neuroblastoma cells were transfected with a plasmid encoding myr-Venus together with plasmids encoding FKBP51wt, FKBP51F67V, or an empty plasmid (control). Cells were treated with or without 20 μm Ligand2 for 24 h, followed by fixation, fluorescence imaging, and morphological analysis using ImageJ. Twenty-five to 40 cells per group were analyzed. Ligand2 binding affinities for FKBPs were determined in a biochemical fluorescence polarization assay using 3 nm of the fluorescent tracers C40-(Fluoresceinyl-Gly)-Rapamycin and 2 nm of purified human FKBP51.
Statistical analysis
The data presented are shown as mean + SEM and were analyzed by the commercially available software SPSS 17.0 and Sigma-Aldrich Plot 11.0. All statistical analyses were performed two-sided. Datasets were tested for normal distribution. When two groups were compared, the unpaired Student's t test was applied. In case data were not normally distributed the nonparametric Mann–Whitney U test (MW test) was used. For four group comparisons, two-way ANOVA was performed, followed by Tukey's post hoc test, as appropriate. P values <0.05 were considered significant. Investigators were blinded to the experimental groups during the experiments and data analyses.
Results
FKBP51 overexpression in the amygdala induces anxiogenic behavior
We overexpressed FKBP51 in the BLA by viral mediated gene transfer and subsequently investigated the anxiety-related and stress-coping behavior in these mice. Correct and stable FKBP51 overexpression in the BLA was quantified by in situ hybridization (t(26) = 105, p < 0.001, t test) and was additionally validated by immunofluorescence (Fig. 1A–D).
Figure 1.
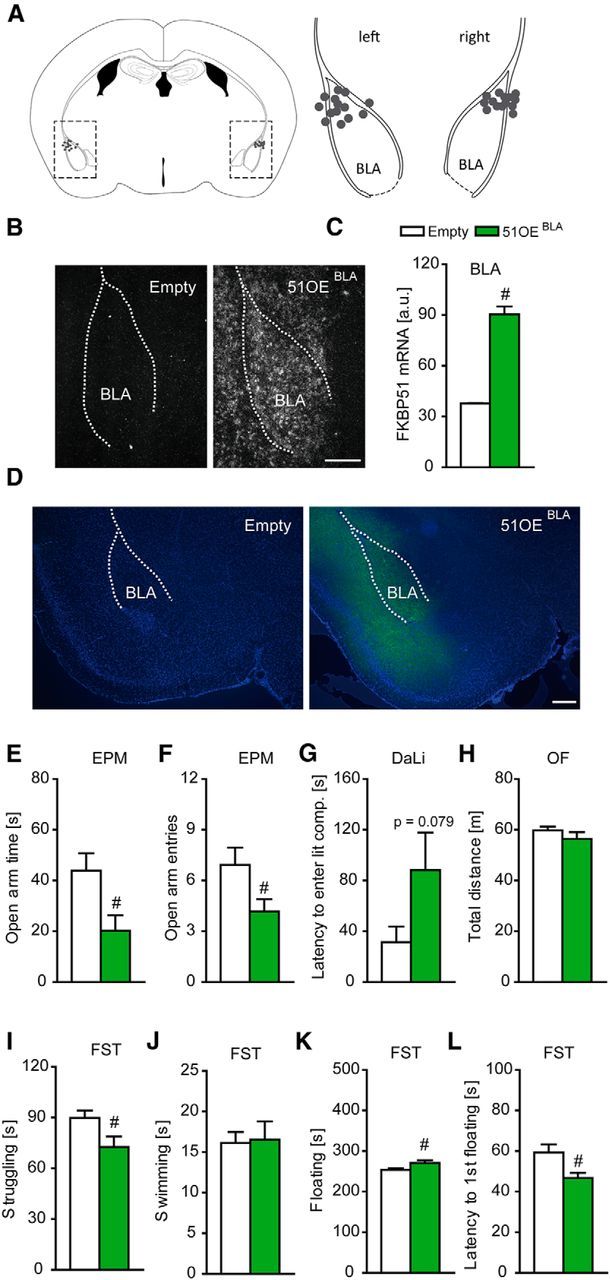
Overexpression of FKBP51 in the BLA induces anxiety-related behavior. A, Schematic representation of the viral injection into the BLA. Gray dots represent the location of the injection tip. B, Representative autoradiographs of viral FKBP51 mRNA expression in the BLA 7 weeks after injection. C, Quantification of FKBP51 mRNA levels in the BLA. Infection with the viral construct induced a strong increase. D, Representative immunohistochemistry images of viral FKBP51 expression (Ha-tag Immuno, green; DAPI, blue) in the BLA region 7 weeks after injection. E, F, FKBP51 overexpression in the BLA leads to significantly reduced open arm time and open arm entries in the EPM. G, 51OEBLA mice show a trend toward increased latency to enter the lit compartment in the DaLi. H, General locomotion as assessed in the OF is not affected by FKBP51 overexpression in the BLA. I–L, 51OEBLA mice show reduced stress-coping behavior in the FST, reflected in decreased time struggling, increased time floating and decreased latency to first floating, whereas time swimming remained unaffected; n = 12–14 per group; #p < 0.05. Data are expressed as mean + SEM. Scale bars, 250 μm.
Mice with overexpression of FKBP51 in the BLA (51OEBLA mice) showed increased anxiety-like behavior in the EPM, which was reflected by significantly reduced open arm time (t(22) = 2.593, p < 0.05, t test) and significantly decreased open arm entries (t(22) = 2.198, p < 0.05, t test) compared with control mice (Fig. 1E,F). These findings were further supported by a trend toward increased latency to enter the aversive lit compartment (z = −1.778, p = 0.079, MW test) in the DaLi test (Fig. 1G). This anxiogenic phenotype of 51OEBLA mice was independent of general locomotor activity, which did not differ between the groups (Fig. 1H). Moreover, 51OEBLA mice showed reduced active stress-coping behavior in the FST, which was reflected in significantly reduced time struggling (t(24) = 2.263, p < 0.05, t test), increased time floating (t(24) = −2.142, p < 0.05, t test) and a decreased latency to first floating (t(24) = 2.643, p < 0.05, t test; Fig. 1I–L).
In addition, we overexpressed FKBP51 in the CeA (51OECeA mice). Correct and stable FKBP51 overexpression in the CeA was quantified by in situ hybridization (t(19) = 19.240, p < 0.001, t test) and was additionally validated by immunofluorescence (Fig. 2A–D). 51OECeA mice displayed a mild anxiogenic phenotype with significantly reduced open arm time (t(16) = 2.29, p < 0.05, t test) and a trend toward decreased open arm entries (t(16) = 1.971, p = 0.066, t test) in the EPM (Fig. 2E,F). However, we did not observe any anxiogenic behavior in the DaLi, nor did we see any changes in general locomotion in the OF or altered stress-coping behavior in the FST (Fig. 2G–L).
Figure 2.
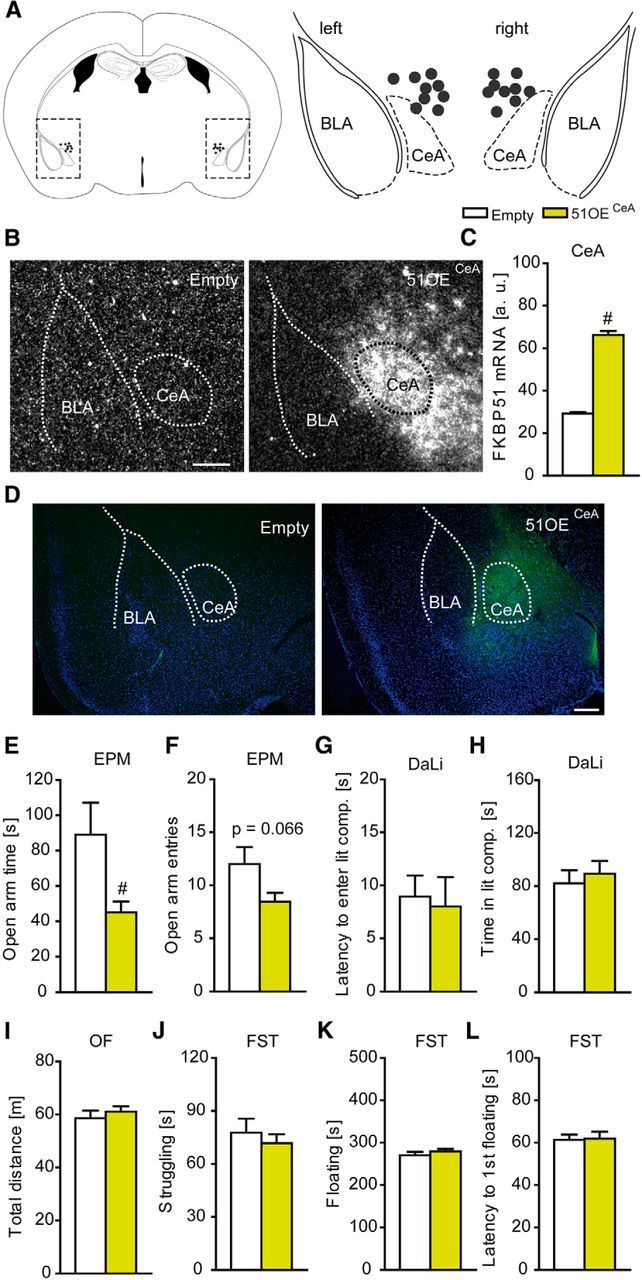
Overexpression of FKBP51 in the CeA. A, Schematic representation of the viral injection into the CeA. Gray dots represent the location of the injection tip. B, Representative autoradiographs of viral FKBP51 mRNA expression in the CeA 7 weeks after injection .C, Quantification of FKBP51 mRNA levels in the CeA. Infection with the viral construct induced a strong increase. D, Representative immunohistochemistry images of viral FKBP51 expression (Ha-tag Immuno, green; DAPI, blue) in the CeA region 7 weeks after injection. E, F, FKBP51 overexpression in the CeA leads to significantly reduced open arm time and a trend toward decreased open arm entries in the EPM. G, H, 51OECeA mice show no alterations in the DaLi. I–L, General locomotion as assessed in the OF and stress-coping behavior in the FST are not affected by FKBP51 overexpression in the CeA; n = 9–10; #p < 0.05. Data are expressed as mean + SEM. Scale bars, 250 μm.
Furthermore, the effects were amygdala specific, as we did not observe any differences in anxiety-related behavior in mice with overexpression of FKBP51 in the dorsal hippocampus (Fig. 3). Correct and stable FKBP51 overexpression in the dorsal hippocampus was quantified by in situ hybridization (CA1, z = −4.333; CA3, z = −4.179; DG, z = −4.333; p < 0.001, MW test) and was additionally validated by immunofluorescence (Fig. 3A–D).
Figure 3.

Overexpression of FKBP51 in the dHc. A, Schematic representation of the viral injection sites into the CA1 and DG of the dHc. B, Representative immunohistochemistry images of viral FKBP51 expression (Ha-tag Immuno, green; DAPI, blue) in the dHc 7 weeks after injection. C, Representative autoradiographs of viral FKBP51 mRNA expression in the dHc 7 weeks after injection. D, Quantification of FKBP51 mRNA levels in the CA1, CA3, and DG of the dHc. Infection with the viral construct induced a strong increase. E–H, No differences between 51OEdHc and Empty groups were found in the EPM and DaLi; n = 12–13, #p < 0.05. Data are expressed as mean + SEM. Scale bars, 500 μm.
Specific pharmacological blockade of a fully functional FKBP51 mutant overexpressed in the BLA reduces the anxiogenic phenotype
We subsequently examined whether the anxiogenic phenotype induced by BLA-restricted FKBP51 overexpression could be rescued with an FKBP51 antagonist. To avoid confounding effects by inhibition of endogenous FKBP51 elsewhere in the brain or side effects due to inhibition of the closely related FKBP52, we overexpressed a mutant form of FKBP51 with a single point mutation (51OEF67V-BLA mice) in the BLA. The FKBP51F67V mutant was chosen because it retains the anti-neuritotrophic of wild-type FKBP51WT in N2a neuroblastoma cells (two-way ANOVA: genotype × treatment interaction: F(2,200) = 135,862, p < 0.001; Fig. 4A; Quintá et al., 2010). Unlike FKBP51WT, however, FKBP51F67V can be selectively inhibited by the engineered Ligand2, which has been designed to complement the F67V mutation (Fig. 4B). Moreover, Ligand2 also discriminated against FKBP52, and it dose-dependently accumulated in the plasma and the brain 1 h after the injection (Fig. 4C,D), thereby allowing for the first time to pharmacologically interrogate FKBP51 in the presence of FKBP52 in vivo.
Figure 4.
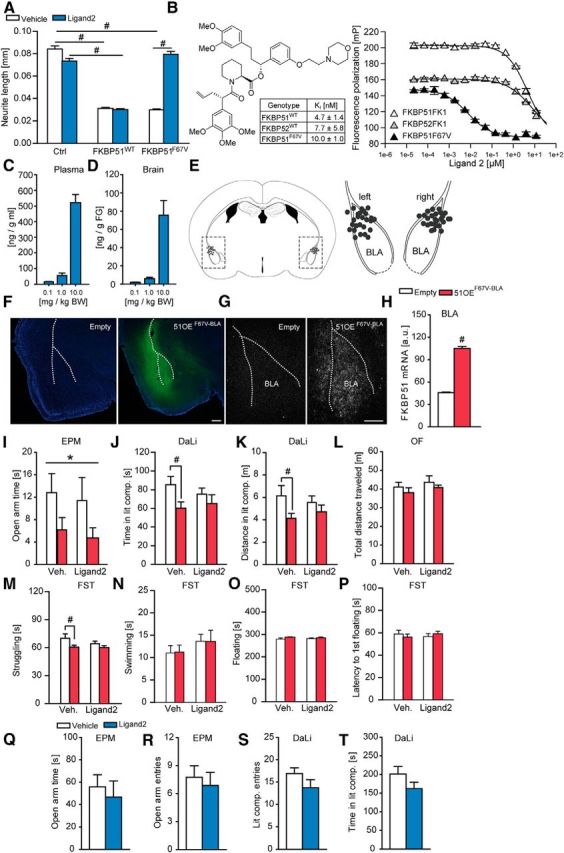
The FKBP51F67V antagonist Ligand2 reverses the anxiogenic phenotype induced by overexpression of FKBP51F67V in the amygdala. A, The anti-neuritotrophic effect of mutant FKBP51F67V, but not of FKBP51wt, can be fully blocked by Ligand2. N2a neuroblastoma cells were transfected with a plasmid encoding myr-Venus together with plasmids encoding FKBP51wt, FKBP51F67V or an empty plasmid (control). Cells were treated with or without 20 μm Ligand2 for 24 h, followed by fixation, fluorescence imaging, and morphological analysis using ImageJ. Data represent mean + SEM of 25–40 analyzed cells, post hoc tukey: #p < 0.001. B, Ligand2 binds to FKBP51F67V, but not to wild-type FKBP51 or FKBP52. Chemical structure of the FKBP51F67V-selective antagonist Ligand2 and binding affinities for FKBPs determined in a biochemical fluorescence polarization assay using 3 nm of the fluorescent tracers C40-(Fluoresceinyl-Gly)-Rapamycin and 2 nm of purified human FKBP51. C, D, Ligand2 pharmacokinetics. The compound was administered intraperitoneally at the indicated dose and the plasma and brain concentrations were determined after 1 h. Ligand2 accumulates in the brain in a dose-dependent manner (n = 4). E, Schematic representation of the viral injection into the BLA. Gray dots represent the location of the injection tip. F, Representative immunohistochemistry images of viral FKBP51F67V expression (Ha-tag Immuno, green; DAPI, blue) in the BLA 7 weeks after injection. G, Representative autoradiographs of viral FKBP51F67V mRNA expression in the BLA 7 weeks after injection. H, Quantification of FKBP51 mRNA levels in the BLA. Infection with the viral construct induced a strong increase. I, FKBP51F67V overexpression in the BLA leads to significantly reduced open arm time, independent of treatment group in the EPM. J, K, Vehicle treated 51OEF67V-BLA mice spend significantly less time and travel less distance in the lit compartment of the DaLi compared with vehicle treated control animals. This effect is abolished in the Ligand2 treated groups. L, General locomotion in the OF does not differ between the experimental groups. M, Vehicle treated 51OEF67V-BLA mice spend significantly less time struggling in the FST compared with vehicle treated control animals. This effect is abolished in the Ligand2 treated groups. N–P, No differences were found for the parameters time swimming, floating and latency to first floating between the groups in the FST; n = 11–13; *p < 0.05 (ANOVA genotype effect); #p < 0.05 (Tukey post hoc test); Q–T, Ligand2 did not show any overt behavioral side effects in the EPM and DaLi in naive mice (n = 8). Data are expressed as mean + SEM. Scale bars, 250 μm.
Similar to FKBP51wt, stable overexpression of FKBP51F67V in the BLA (z = −6.186; p < 0.001, MW test; Fig. 44E–H) induced an anxiogenic phenotype in the EPM, which was reflected in a significantly reduced open arm time (two-way ANOVA: main genotype effect: F(1,41) = 4.696, p < 0.05; Fig. 4I). This finding was independent of the treatment groups. In the DaLi test, 51OEF67V-BLA mice spent significantly less time (two-way ANOVA: main genotype effect: F(1,45) = 4.671, p < 0.05) and traveled less (two-way ANOVA: main genotype effect:, F(1,45) = 4.514, p < 0.05) in the lit compartment than the empty control groups (Fig. 4J,K). Intriguingly, this genotype effect was only significant between the vehicle treated groups but not in the ligand 2-treated group, suggesting an anxiolytic effect by FKBP51F67V-BLA inhibition (Tukey's post hoc test: p < 0.05). The OF)test served again as control, and revealed that the anxiety-related phenotype of 51OEF67V-BLA mice is independent of general locomotion, which did not differ between the experimental groups (Fig. 4L). In addition, 51OEF67V-BLA mice spent significantly less time struggling (two-way ANOVA: main genotype effect: F(1,45) = 4.863, p < 0.05) in the FST than the empty control groups (Fig. 4M). Interestingly, this genotype effect was only significant between the vehicle treated groups but not under FKBP51 antagonist conditions (Tukey's post hoc test: p < 0.05), which may implicate a protective effect by FKBP51F67V-BLA blockade. However, we did not observe any significant differences for the other parameters assessed in the FST (Fig. 4N–P). Moreover, we found no differences in anxiety-related behavior in naive mice treated with Ligand 2 compared with the vehicle treated group (Fig. 4Q–T), thereby arguing against potential off-target effects of the drug.
Peripheral administration of SAFit2 induces anxiolytic behavior in naive mice
In a next step, we explored the consequences inhibiting total FKBP51 after peripheral administration of SAFit2, the first potent and selective antagonist for wild-type FKBP51 (Gaali et al., 2015). When SAFit2 was applied 1 h before testing, we did not observe any alterations in anxiety-related behavior (Fig. 5A–D). However, SAFit2 treatment induced an anxiolytic phenotype in mice injected 16 h prior testing, which was reflected in a significantly increased open arm time in the EPM (z = −2.183, p < 0.05, MW test; Fig. 5E). This finding was further supported by an increase in the number of entries to the lit compartment (t(20) = 2.593, p < 0.01, t test) in combination with a significant increase in the time spent in the lit compartment (t(20) = −2.275, p < 0.05, t test) in the DaLi (Fig. 5F,G). Moreover, SAFit2 treatment led to a significantly reduced latency to enter the lit compartment (z = −2 to 265, p < 0.05, MW test), as well as a significantly increased distance traveled (t(20) = −2.371, p < 0.05, t test) in the lit compartment (Fig. 5H,I).
Figure 5.
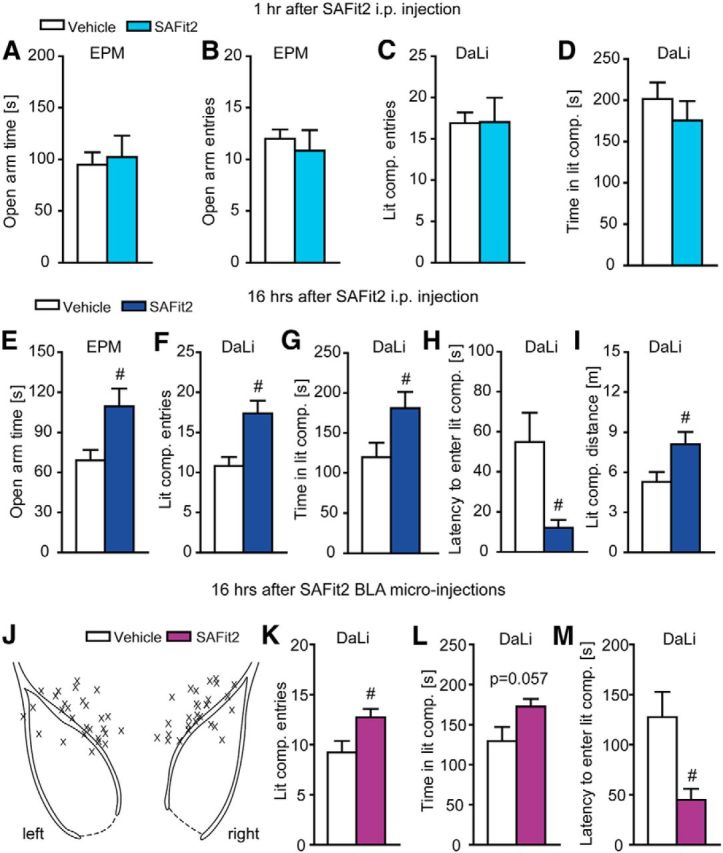
Inhibition of wild-type FKBP51 by SAFit2 induces anxiolytic behavior in a time-dependent manner. A–D, Mice tested 1 h after peripheral SAFit2 injection (i.p.) shown no alterations in anxiety-related behavior in the EPM or DaLi. E, Mice show a significant increase in the open arm time of the EPM 16 h after peripheral administration of SAFit2. F, G, SAFit2 leads to significantly increased entries to the lit compartment and significantly more time spent in the lit compartment of the DaLi 16 h after peripheral application. H, I, Mice treated with SAFit2 showed a significantly decreased latency to enter and a significantly increased distance traveled in the lit compartment of the DaLi 16 h after the intraperitoneal injection; n = 10–11. J, Schematic representation of the cannula placement. X represents the tip of the guiding cannula. K, mice treated with SAFit2 via BLA microinjections show a significant increase in the number of lit compartment entries in the DaLi. L, M, SAFit2 BLA microinjections lead toward a trend to increased time spent in the lit compartment and a significantly decreased latency to enter the lit compartment of the DaLi; n = 14, #p < 0.05. Data are expressed as mean + SEM.
BLA microinjections of the selective FKBP51 antagonist SAFit2 induce anxiolytic behavior in mice
Encouraged by these findings and to investigate a genuine relationship between FKBP51 in the amygdala and anxiety, we administered SAFit2, into the BLA via micro-injections 16 h before testing (Fig. 5J). Directly opposite to the effect of FKBP51 overexpression in the amygdala, SAFit2 treatment induced an anxiolytic phenotype, which was reflected in a significantly increased number of lit compartment entries (t(26) = −2.438, p < 0.05, t test), an increased time spent in the lit compartment (z = −1.930, p = 0.057, MW test) and a significantly reduced latency to enter the lit compartment (z = −2.895, p < 0.01, MW test; Fig. 5K–M).
Discussion
The amygdala is an important brain region for the mediation of stress responses, fear, and anxiety (Joëls and Baram, 2009), and is thus highly implicated in mood and anxiety disorders (Etkin and Wager, 2007; Siegle et al., 2007; Drevets et al., 2008). FKBP51 mRNA levels are significantly upregulated in the amygdala of mice after acute stressors or dexamethasone treatment (Scharf et al., 2011). Interestingly, altered amygdala activity, connectivity, and volume have been associated with FKBP51 variants and depression or childhood adversity (Holz et al., 2015). In addition, alterations in FKBP51 mRNA or protein levels have also been observed in autopsy samples from the amygdala of suicide victims (Pérez-Ortiz et al., 2013). In-line with these studies, we found that mice with FKBP51 overexpression in the BLA have increased anxiety-related behavior, suggesting that the natural induction of amygdala FKBP51 expression following stress functionally mediates stress-induced anxiety. Moreover, this anxiogenic phenotype was independent of locomotion, as we did not find any differences in the OF test. In addition, 51OEBLA mice also revealed reduced active stress-coping behavior in the FST. Interestingly, overexpression of FKBP51 in the CeA also led to a mild anxiogenic effect in the EPM. Although we cannot completely rule out that adjacent amygdala nuclei were affected by our AAV overexpression strategy, the neuroanatomical analyses supported a high degree of regional specificity of the viral spread. Furthermore, the increased anxiety-related effects were amygdala-specific, as FKBP51 overexpression in the dHc did not induce any changes in anxiety-like behavior.
FKBP52 is a close homolog of FKBP51 that shares >75% sequence homology, but acts as a functional opponent; whereas, FKBP51 decreases GR nucleus translocation and signaling, FKBP52 enhances GR translocation to the nucleus (Wochnik et al., 2005). We have previously reported that FKBP51 knock-out mice have a reduced corticosterone response to forced swim stress, whereas mice lacking one allele of FKBP52 display increased corticosterone levels to this stressor and an anxiogenic phenotype (Hartmann et al., 2012a,b). Thus, the selectivity of an FKBP51 antagonist is critical. We approached this challenge by using a bump-and-hole approach to first overexpress a functionally fully active FKBP51 variant carrying a point mutation (FKBP51F67V), which can selectively be blocked by the specifically designed FKBP51F67V-antagonist, Ligand2. We demonstrated that overexpression of FKBP51F67V in the BLA induced a similar anxiogenic phenotype in mice as overexpression of wild-type FKBP51, thereby confirming our initial results. When these mice were subchronically treated with Ligand2, the increase in anxiety-related behavior in the DaLi box was rescued and the stress-coping phenotype in the FST was reduced. However, we were not able to fully reverse the induced phenotype, especially in the EPM, indicating that the daily treatment regimen may not have been sufficient to continuously block FKBP51F67V activity in the BLA, potentially due to an unfavorable pharmacokinetics of Ligand2 following repeated injections, in contrast to the robust AAV-mediated FKBP51 F67V overexpression. To work under more naturalistic settings, we continued to work with a selective antagonist (SAFit2) against native FKBP51.
As microinjections of SAFit2 (the selective and potent novel antagonist for wild-type FKBP51) into the BLA induced a significant anxiolytic phenotype, we were further able to link amygdala FKBP51 expression to anxiety-like behavior. These results are in-line with a previous study demonstrating that knockdown of FKBP51 in the BLA reduces stress-induced anxiety in mice (Attwood et al., 2011). Intriguingly, peripheral (i.p.) injection of SAFit2 replicated the anxiolytic effect of direct amygdala administration in a time-dependent manner, suggesting applicability in a clinical setting.
In summary, our results provide the proof-of-concept for FKBP51 inhibitors to modulate anxiety in vivo. These results pave the way for the development of FKBP51 antagonists and their transition into the clinic for the improved treatment of stress-related psychiatric diseases.
Footnotes
This study was supported by the Max Planck Society. We thank Daniela Harbich, Bianca Schmid, and Anna Mederer for their excellent technical assistance, and Natalie Matosin for proofreading the paper.
A.K., S.G., G.R. and F.H. have filed patent applications for FKBP51 inhibitors. The remaining authors declare no competing financial interests.
References
- Appel K, Schwahn C, Mahler J, Schulz A, Spitzer C, Fenske K, Stender J, Barnow S, John U, Teumer A, Biffar R, Nauck M, Völzke H, Freyberger HJ, Grabe HJ. Moderation of adult depression by a polymorphism in the FKBP5 gene and childhood physical abuse in the general population. Neuropsychopharmacology. 2011;36:1982–1991. doi: 10.1038/npp.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwood BK, Bourgognon JM, Patel S, Mucha M, Schiavon E, Skrzypiec AE, Young KW, Shiosaka S, Korostynski M, Piechota M, Przewlocki R, Pawlak R. Neuropsin cleaves EphB2 in the amygdala to control anxiety. Nature. 2011;473:372–375. doi: 10.1038/nature09938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder EB. The role of FKBP5, a co-chaperone of the glucocorticoid receptor in the pathogenesis and therapy of affective and anxiety disorders. Psychoneuroendocrinology. 2009;34:S186–S195. doi: 10.1016/j.psyneuen.2009.05.021. [DOI] [PubMed] [Google Scholar]
- Binder EB, Holsboer F. Pharmacogenomics and antidepressant drugs. Ann Med. 2006;38:82–94. doi: 10.1080/07853890600551045. [DOI] [PubMed] [Google Scholar]
- Binder EB, Salyakina D, Lichtner P, Wochnik GM, Ising M, Pütz B, Papiol S, Seaman S, Lucae S, Kohli MA, Nickel T, Künzel HE, Fuchs B, Majer M, Pfennig A, Kern N, Brunner J, Modell S, Baghai T, Deiml T, et al. Polymorphisms in FKBP5 are associated with increased recurrence of depressive episodes and rapid response to antidepressant treatment. Nat Genet. 2004;36:1319–1325. doi: 10.1038/ng1479. [DOI] [PubMed] [Google Scholar]
- Binder EB, Bradley RG, Liu W, Epstein MP, Deveau TC, Mercer KB, Tang Y, Gillespie CF, Heim CM, Nemeroff CB, Schwartz AC, Cubells JF, Ressler KJ. Association of FKBP5 polymorphisms and childhood abuse with risk of posttraumatic stress disorder symptoms in adults. JAMA. 2008;299:1291–1305. doi: 10.1001/jama.299.11.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brent D, Melhem N, Ferrell R, Emslie G, Wagner KD, Ryan N, Vitiello B, Birmaher B, Mayes T, Zelazny J, Onorato M, Devlin B, Clarke G, DeBar L, Keller M. Association of FKBP5 polymorphisms with suicidal events in the treatment of resistant depression in adolescents (TORDIA) study. Am J Psychiatry. 2010;167:190–197. doi: 10.1176/appi.ajp.2009.09040576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrousos GP. Stress and disorders of the stress system. Nat Rev Endocrinol. 2009;5:374–381. doi: 10.1038/nrendo.2009.106. [DOI] [PubMed] [Google Scholar]
- Davis M. The role of the amygdala in emotional learning. Int Rev Neurobiol. 1994;36:225–266. doi: 10.1016/S0074-7742(08)60305-0. [DOI] [PubMed] [Google Scholar]
- Dias BG, Banerjee SB, Goodman JV, Ressler KJ. Towards new approaches to disorders of fear and anxiety. Curr Opin Neurobiol. 2013;23:346–352. doi: 10.1016/j.conb.2013.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drevets WC, Price JL, Furey ML. Brain structural and functional abnormalities in mood disorders: implications for neurocircuitry models of depression. Brain Struct Funct. 2008;213:93–118. doi: 10.1007/s00429-008-0189-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellsworth KA, Moon I, Eckloff BW, Fridley BL, Jenkins GD, Batzler A, Biernacka JM, Abo R, Brisbin A, Ji Y, Hebbring S, Wieben ED, Mrazek DA, Weinshilboum RM, Wang L. FKBP5 genetic variation: association with selective serotonin reuptake inhibitor treatment outcomes in major depressive disorder. Pharmacogenet Genomics. 2013;23:156–166. doi: 10.1097/FPC.0b013e32835dc133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etkin A, Wager TD. Functional neuroimaging of anxiety: a meta-analysis of emotional processing in PTSD, social anxiety disorder, and specific phobia. Am J Psychiatry. 2007;164:1476–1488. doi: 10.1176/appi.ajp.2007.07030504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaali S, Kirschner A, Cuboni S, Hartmann J, Kozany C, Balsevich G, Namendorf C, Fernandez-Vizarra P, Sippel C, Zannas AS, Draenert R, Binder EB, Almeida OF, Rühter G, Uhr M, Schmidt MV, Touma C, Bracher A, Hausch F. Selective inhibitors of the FK506-binding protein 51 by induced fit. Nat Chem Biol. 2015;11:33–37. doi: 10.1038/nchembio.1699. [DOI] [PubMed] [Google Scholar]
- Gopalakrishnan R, Kozany C, Gaali S, Kress C, Hoogeland B, Bracher A, Hausch F. Evaluation of synthetic FK506 analogues as ligands for the FK506-binding proteins 51 and 52. J Med Chem. 2012a;55:4114–4122. doi: 10.1021/jm201746x. [DOI] [PubMed] [Google Scholar]
- Gopalakrishnan R, Kozany C, Wang Y, Schneider S, Hoogeland B, Bracher A, Hausch F. Exploration of pipecolate sulfonamides as binders of the FK506-binding proteins 51 and 52. J Med Chem. 2012b;55:4123–4131. doi: 10.1021/jm201747c. [DOI] [PubMed] [Google Scholar]
- Gray TS. Amygdaloid CRF pathways: role in autonomic, neuroendocrine, and behavioral responses to stress. Ann N Y Acad Sci. 1993;697:53–60. doi: 10.1111/j.1749-6632.1993.tb49922.x. [DOI] [PubMed] [Google Scholar]
- Hartmann J, Wagner KV, Dedic N, Marinescu D, Scharf SH, Wang XD, Deussing JM, Hausch F, Rein T, Schmidt U, Holsboer F, Müller MB, Schmidt MV. Fkbp52 heterozygosity alters behavioral, endocrine and neurogenetic parameters under basal and chronic stress conditions in mice. Psychoneuroendocrinology. 2012a;37:2009–2021. doi: 10.1016/j.psyneuen.2012.04.017. [DOI] [PubMed] [Google Scholar]
- Hartmann J, Wagner KV, Liebl C, Scharf SH, Wang XD, Wolf M, Hausch F, Rein T, Schmidt U, Touma C, Cheung-Flynn J, Cox MB, Smith DF, Holsboer F, Müller MB, Schmidt MV. The involvement of FK506-binding protein 51 (FKBP5) in the behavioral and neuroendocrine effects of chronic social defeat stress. Neuropharmacology. 2012b;62:332–339. doi: 10.1016/j.neuropharm.2011.07.041. [DOI] [PubMed] [Google Scholar]
- Holsboer F. The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology. 2000;23:477–501. doi: 10.1016/S0893-133X(00)00159-7. [DOI] [PubMed] [Google Scholar]
- Holz NE, Buchmann AF, Boecker R, Blomeyer D, Baumeister S, Wolf I, Rietschel M, Witt SH, Plichta MM, Meyer-Lindenberg A, Banaschewski T, Brandeis D, Laucht M. Role of FKBP5 in emotion processing: results on amygdala activity, connectivity and volume. Brain Struct Funct. 2015;220:1355–1368. doi: 10.1007/s00429-014-0729-5. [DOI] [PubMed] [Google Scholar]
- Joëls M, Baram TZ. The neuro-symphony of stress. Nat Rev Neurosci. 2009;10:459–466. doi: 10.1038/nrn2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klengel T, Mehta D, Anacker C, Rex-Haffner M, Pruessner JC, Pariante CM, Pace TW, Mercer KB, Mayberg HS, Bradley B, Nemeroff CB, Holsboer F, Heim CM, Ressler KJ, Rein T, Binder EB. Allele-specific FKBP5 DNA demethylation mediates gene-childhood trauma interactions. Nat Neurosci. 2013;16:33–41. doi: 10.1038/nn.3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenen KC, Saxe G, Purcell S, Smoller JW, Bartholomew D, Miller A, Hall E, Kaplow J, Bosquet M, Moulton S, Baldwin C. Polymorphisms in FKBP5 are associated with peritraumatic dissociation in medically injured children. Mol Psychiatry. 2005;10:1058–1059. doi: 10.1038/sj.mp.4001727. [DOI] [PubMed] [Google Scholar]
- Krishnan V, Nestler EJ. The molecular neurobiology of depression. Nature. 2008;455:894–902. doi: 10.1038/nature07455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavebratt C, Aberg E, Sjöholm LK, Forsell Y. Variations in FKBP5 and BDNF genes are suggestively associated with depression in a Swedish population-based cohort. J Affect Disord. 2010;125:249–255. doi: 10.1016/j.jad.2010.02.113. [DOI] [PubMed] [Google Scholar]
- LeDoux J. The amygdala. Curr Biol. 2007;17:R868–R874. doi: 10.1016/j.cub.2007.08.005. [DOI] [PubMed] [Google Scholar]
- Lupien SJ, McEwen BS, Gunnar MR, Heim C. Effects of stress throughout the lifespan on the brain, behaviour and cognition. Nat Rev Neurosci. 2009;10:434–445. doi: 10.1038/nrn2639. [DOI] [PubMed] [Google Scholar]
- McEwen BS, Sapolsky RM. Stress and cognitive function. Curr Opin Neurobiol. 1995;5:205–216. doi: 10.1016/0959-4388(95)80028-X. [DOI] [PubMed] [Google Scholar]
- Mehta D, Gonik M, Klengel T, Rex-Haffner M, Menke A, Rubel J, Mercer KB, Pütz B, Bradley B, Holsboer F, Ressler KJ, Müller-Myhsok B, Binder EB. Using polymorphisms in FKBP5 to define biologically distinct subtypes of posttraumatic stress disorder: evidence from endocrine and gene expression studies. Arch Gen Psychiatry. 2011;68:901–910. doi: 10.1001/archgenpsychiatry.2011.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menke A, Klengel T, Rubel J, Brückl T, Pfister H, Lucae S, Uhr M, Holsboer F, Binder EB. Genetic variation in FKBP5 associated with the extent of stress hormone dysregulation in major depression. Genes Brain Behav. 2013;12:289–296. doi: 10.1111/gbb.12026. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, Hyman SE. Animal models of neuropsychiatric disorders. Nat Neurosci. 2010;13:1161–1169. doi: 10.1038/nn.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pariante CM, Miller AH. Glucocorticoid receptors in major depression: relevance to pathophysiology and treatment. Biol Psychiatry. 2001;49:391–404. doi: 10.1016/S0006-3223(00)01088-X. [DOI] [PubMed] [Google Scholar]
- Pérez-Ortiz JM, García-Gutiérrez MS, Navarrete F, Giner S, Manzanares J. Gene and protein alterations of FKBP5 and glucocorticoid receptor in the amygdala of suicide victims. Psychoneuroendocrinology. 2013;38:1251–1258. doi: 10.1016/j.psyneuen.2012.11.008. [DOI] [PubMed] [Google Scholar]
- Porcelli S, Drago A, Fabbri C, Gibiino S, Calati R, Serretti A. Pharmacogenetics of antidepressant response. J Psychiatry Neurosci. 2011;36:87–113. doi: 10.1503/jpn.100059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintá HR, Maschi D, Gomez-Sanchez C, Piwien-Pilipuk G, Galigniana MD. Subcellular rearrangement of hsp90-binding immunophilins accompanies neuronal differentiation and neurite outgrowth. J Neurochem. 2010;115:716–734. doi: 10.1111/j.1471-4159.2010.06970.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riggs DL, Roberts PJ, Chirillo SC, Cheung-Flynn J, Prapapanich V, Ratajczak T, Gaber R, Picard D, Smith DF. The Hsp90-binding peptidylprolyl isomerase FKBP52 potentiates glucocorticoid signaling in vivo. EMBO J. 2003;22:1158–1167. doi: 10.1093/emboj/cdg108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A, Gorodetsky E, Yuan Q, Goldman D, Enoch MA. Interaction of FKBP5, a stress-related gene, with childhood trauma increases the risk for attempting suicide. Neuropsychopharmacology. 2010;35:1674–1683. doi: 10.1038/npp.2009.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharf SH, Liebl C, Binder EB, Schmidt MV, Müller MB. Expression and regulation of the Fkbp5 gene in the adult mouse brain. PloS One. 2011;6:e16883. doi: 10.1371/journal.pone.0016883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt MV, Sterlemann V, Ganea K, Liebl C, Alam S, Harbich D, Greetfeld M, Uhr M, Holsboer F, Müller MB. Persistent neuroendocrine and behavioral effects of a novel, etiologically relevant mouse paradigm for chronic social stress during adolescence. Psychoneuroendocrinology. 2007;32:417–429. doi: 10.1016/j.psyneuen.2007.02.011. [DOI] [PubMed] [Google Scholar]
- Schmidt MV, Schülke JP, Liebl C, Stiess M, Avrabos C, Bock J, Wochnik GM, Davies HA, Zimmermann N, Scharf SH, Trümbach D, Wurst W, Zieglgänsberger W, Turck C, Holsboer F, Stewart MG, Bradke F, Eder M, Müller MB, Rein T. Tumor suppressor down-regulated in renal cell carcinoma 1 (DRR1) is a stress-induced actin bundling factor that modulates synaptic efficacy and cognition. Proc Natl Acad Sci U S A. 2011;108:17213–17218. doi: 10.1073/pnas.1103318108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt MV, Paez-Pereda M, Holsboer F, Hausch F. The prospect of FKBP51 as a drug target. ChemMedChem. 2012;7:1351–1359. doi: 10.1002/cmdc.201200137. [DOI] [PubMed] [Google Scholar]
- Shin LM, Liberzon I. The neurocircuitry of fear, stress, and anxiety disorders. Neuropsychopharmacology. 2010;35:169–191. doi: 10.1038/npp.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegle GJ, Thompson W, Carter CS, Steinhauer SR, Thase ME. Increased amygdala and decreased dorsolateral prefrontal BOLD responses in unipolar depression: related and independent features. Biol Psychiatry. 2007;61:198–209. doi: 10.1016/j.biopsych.2006.05.048. [DOI] [PubMed] [Google Scholar]
- Supriyanto I, Sasada T, Fukutake M, Asano M, Ueno Y, Nagasaki Y, Shirakawa O, Hishimoto A. Association of FKBP5 gene haplotypes with completed suicide in the Japanese population. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35:252–256. doi: 10.1016/j.pnpbp.2010.11.019. [DOI] [PubMed] [Google Scholar]
- Tatro ET, Everall IP, Masliah E, Hult BJ, Lucero G, Chana G, Soontornniyomkij V, Achim CL, HIV Neurobehavioral Research Center Differential expression of immunophilins FKBP51 and FKBP52 in the frontal cortex of HIV-infected patients with major depressive disorder. J Neuroimmune Pharmacol. 2009;4:218–226. doi: 10.1007/s11481-009-9146-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennant C. Work-related stress and depressive disorders. J Psychosom Res. 2001;51:697–704. doi: 10.1016/S0022-3999(01)00255-0. [DOI] [PubMed] [Google Scholar]
- Thase ME. Preventing relapse and recurrence of depression: a brief review of therapeutic options. CNS Spectr. 2006;11:12–21. doi: 10.1017/s1092852900015212. [DOI] [PubMed] [Google Scholar]
- Wang XD, Chen Y, Wolf M, Wagner KV, Liebl C, Scharf SH, Harbich D, Mayer B, Wurst W, Holsboer F, Deussing JM, Baram TZ, Müller MB, Schmidt MV. Forebrain CRHR1 deficiency attenuates chronic stress-induced cognitive deficits and dendritic remodeling. Neurobiol Dis. 2011;42:300–310. doi: 10.1016/j.nbd.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willour VL, Chen H, Toolan J, Belmonte P, Cutler DJ, Goes FS, Zandi PP, Lee RS, MacKinnon DF, Mondimore FM, Schweizer B, DePaulo JR, Jr, Gershon ES, McMahon FJ, Potash JB, Bipolar Disorder Phenome Group, NIMH Genetics Initiative Bipolar Disorder Consortium Family-based association of FKBP5 in bipolar disorder. Mol Psychiatry. 2009;14:261–268. doi: 10.1038/sj.mp.4002141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wochnik GM, Rüegg J, Abel GA, Schmidt U, Holsboer F, Rein T. FK506-binding proteins 51 and 52 differentially regulate dynein interaction and nuclear translocation of the glucocorticoid receptor in mammalian cells. J Biol Chem. 2005;280:4609–4616. doi: 10.1074/jbc.M407498200. [DOI] [PubMed] [Google Scholar]
- Yehuda R. Status of glucocorticoid alterations in post-traumatic stress disorder. Ann N Y Acad Sci. 2009;1179:56–69. doi: 10.1111/j.1749-6632.2009.04979.x. [DOI] [PubMed] [Google Scholar]