

Abstract
Amyloid β (Aβ) is thought to play an important role in the pathogenesis of Alzheimer's disease. Aβ may exert its neurotoxic effects via multiple mechanisms and in particular through degradation of excitatory synaptic transmission associated with impaired synaptic plasticity. In contrast, much less is known about Aβ effects at inhibitory synapses. This study investigates the impact of acute Aβ1-42 application on GABAergic synaptic transmission in rat somatosensory cortex in vitro. Whole-cell voltage-clamp recordings were obtained from layer V pyramidal cells, and monosynaptic GABAA receptor-mediated IPSCs were elicited. Bath-applied Aβ (1 μm) depressed the IPSCs on average to 60% of control, whereas a reversed sequence control peptide was ineffective. Paired-pulse stimuli indicated a postsynaptic site of action. This was further corroborated by a decreased postsynaptic responsiveness to local puffs of the GABAA receptor agonist isoguvacine. The Aβ-induced IPSC decline could be prevented with intracellular applications of p4, a blocker of GABAA receptor internalization. It is concluded that Aβ weakens synaptic inhibition via downregulation of GABAA receptors.
Keywords: Alzheimer, amyloid-β, cortex, GABA, IPSP, plasticity
Introduction
Alzheimer's disease is characterized by a steady cognitive decline and loss of memory function. A main pathophysiological agent is the protein β-amyloid (Aβ) that, although possibly playing a physiological role under normal conditions, gets transformed into a toxic substrate (Palop and Mucke, 2010). Aβ is known to increase Ca2+ influx into cells (Demuro et al., 2010) that may lead to hyperexcitability (Busche et al., 2008; Minkeviciene et al., 2009) and enhanced release of the neurotransmitter glutamate (Abramov et al., 2009). Both processes may interact in the subsequent removal of glutamatergic receptors from synapses (Hsieh et al., 2006). The loss of glutamate receptors is accompanied by an impairment of long-term potentiation, a long-lasting form of synaptic plasticity (Palop and Mucke, 2010; Walsh et al., 2002). It is believed that the decline in synaptic plasticity is responsible for the progressive loss of cognitive and memory functions in Alzheimer's patients (Selkoe, 2002). Although there is substantial evidence for an Aβ-associated pathophysiology at glutamatergic synapses, much less consistent data exist for GABA, the main inhibitory neurotransmitter in the forebrain. Early postmortem studies concluded that GABAergic cells were spared in Alzheimer's disease (for review, see Rissman et al., 2007). However, there is more recent evidence for differential upregulation and downregulation of particular GABAA receptor subunits in reconstituted human postmortem tissue of Alzheimer's patients, probably reflecting the coexistence of degenerative and compensatory processes (Limon et al., 2012). In animal models of Alzheimer's disease, tonic GABAergic inhibition was found to be upregulated in dentate granule cells of hippocampus (Wu et al., 2014). In contrast, phasic GABAergic synaptic transmission was found to be decreased (Busche et al., 2008), unaltered (Kamenetz et al., 2003), or enhanced (Palop et al., 2007). Part of these discrepancies may result from differences in the particular disease model investigated, the brain region examined, and/or experimental parameter assessed (e.g., morphological, neurochemical vs physiological) (Lanctôt et al., 2004). To better understand the role of synaptic inhibition in Alzheimer's disease, this study investigates the impact of Aβ on GABAergic synaptic transmission and describes an Aβ-mediated decline of IPSCs via GABAA receptor endocytosis.
Materials and Methods
Tissue preparation.
All experimental procedures were approved by the Bioresources Committee, Trinity College Dublin, and licensed by the Department of Health and Children, Ireland, in accordance with European Communities Council Directive (86/609/EEC). Wistar rats of either sex (P21-P25) were kept on a 12 h light/dark cycle with food and water ad libitum. Rats were killed by decapitation, and the brains were quickly removed. Each hemisphere was glued onto a specimen platform slanted at 10°. Parasagittal slices of 300 μm thickness containing somatosensory cortex were prepared on a vibratome (Microm HM650V) in 4°C cold standard ACSF. The ACSF contained the following (in mm): 125 NaCl, 1.25 NaH2PO4, 25 NaHCO3, 2.5 KCl, 1 MgCl2, 2 CaCl2, 10 glucose, pH 7.3, equilibrated with 5% CO2/95% O2. Slices were incubated in a submerged chamber at 34°C for 1 h and kept at room temperature thereafter.
Electrophysiology.
Individual slices were transferred into a submerged recording chamber and superfused at a rate of 6 ml/min with ACSF warmed to 34°C. Whole-cell patch-clamp recordings were performed under visual control with an infrared differential interference contrast video microscope (Olympus BX51WI) (Stuart et al., 1993). Layer V of the somatosensory cortex was identified at low magnification (4×), and individual pyramidal cells were visualized with a 40× water-immersion objective. Patch pipettes were pulled on a DMZ universal puller (Zeitz Instruments) from thick-walled glass capillaries (Hilgenberg). Pipettes were filled with a filtered solution containing the following (in mm): 125 Cs-gluconate, 5 NaCl, 0.1 EGTA, 10 HEPES, 4 ATP, 0.4 GTP, pH 7.3 (osmolarity = 290 mosmol). Whole-cell currents were recorded in continuous single-electrode voltage-clamp mode (BVC-700A, Dagan) and digitized at 3 kHz (Digidata 1440A, Molecular Devices). A liquid junction potential of −10 mV was left uncorrected. IPSCs were evoked locally with constant current pulses (0.1–0.3 ms, 50–200 μA) via insulated bipolar nickel-chromium wires (Good Fellow) that were connected to an isolated pulse stimulator (A-M Systems). Agonist-evoked responses were generated by applying isoguvacine puffs (0.5 mm in ACSF, Picospritzer III, Parker) of 2–4 ms duration and 2–10 PSI amplitude through a wide focal patch pipette that was positioned in the neighborhood of the cells.
Chemicals.
All drugs were stored as 1000× aliquots in distilled water and bath applied at final concentrations as indicated. Stock solutions of synthetic human β-amyloid 1-42 (shAβ1-42) and its reverse control 42-1 (Bachem) were prepared in 0.1% NH4OH (Teplow, 2006) at a concentration of 0.2 mm and kept at −20°C. Leupeptin and bestatin were from Sigma-Aldrich. All other drugs were from Tocris Bioscience. Aliquots of the dynamin inhibitory peptide (p4, QVPSRPNRAP) were dissolved in water at a concentration of 0.9 mm and kept at −20°C. A control peptide was generated by denaturating aliquots of p4 at 100°C for 10 min. p4 or its control was added to the pipette solution at the concentrations indicated.
Data analysis.
IPSC amplitudes were measured with 2 pairs of 1- to 3-ms-wide cursors for baseline and peak (pClamp 10, Molecular Devices). Paired stimuli were evoked at an interval of 50 ms, and the paired-pulse ratio was calculated as <IPSC2>/<IPSC1> (<> designates averages of 50 IPSC amplitudes). It was previously shown that calculating the paired-pulse ratio from averaged versus individual traces yields more accurate estimates (Kim and Alger, 2001). Baseline-subtracted, agonist-evoked responses were integrated to obtain an estimate of charge. Twenty consecutive amplitude or area values in control and Aβ were averaged and the ratio calculated to obtain an estimate of drug-induced changes. Data are presented as mean ± SEM, and n designates the number of cells. Statistical comparisons were done with the two-tailed, two-sample Student's t test.
Results
Aβ1-42 reduces GABAergic IPSCs
Layer V pyramidal cells were visually identified in the somatosensory cortex, and whole-cell patch-clamp recordings were established. To increase the electrochemical driving force for chloride, which had a Nernst equilibrium potential of −85 mV, individual neurons were voltage-clamped at 0 mV. Monosynaptic IPSCs were evoked focally with extracellular stimuli at 0.1–0.2 Hz after blocking ionotropic glutamatergic synaptic transmission with d-APV (50 μm) and DNQX (20 μm). QX314 (10 mm) was routinely added to the intracellular solution to block regenerative activity and GABAB receptors (Nathan et al., 1990). Evoked IPSCs in control had an average peak amplitude of 208 ± 39 pA (n = 14 cells). After recording a stable sequence of IPSCs in control, Aβ1-42 (1 μm) was added to the bath. Aβ application led to a decline of IPSC amplitudes to on average 61 ± 5% of control (n = 14 cells), which was statistically significant (p < 0.0005, paired t test) (Fig. 1A). In a subset of experiments, IPSCs were fully blocked by the selective antagonist bicuculline (10 μm) at the end of the recordings to verify that the IPSCs were exclusively mediated by GABAA receptors (e.g., see Fig. 4A). A similar series of experiments was subsequently undertaken with the control reverse sequence peptide Aβ42-1. Again, a stable series of IPSCs was recorded in normal ACSF. However, bath application of Aβ42-1 left IPSCs unaltered (Fig. 1B). Overall, the IPSC amplitudes were not significantly reduced by the control peptide to 93 ± 9% of control (n = 5, p > 0.1, paired t test), indicating that the observed depression of the IPSCs could be specifically attributed to Aβ1-42. Figure 1C shows a summary histogram of the net effects of Aβ1-42 and 42-1 on monosynaptic GABAA receptor-mediated IPSCs revealing a statistically significant difference between the two peptides (p < 0.01, unpaired t test).
Figure 1.
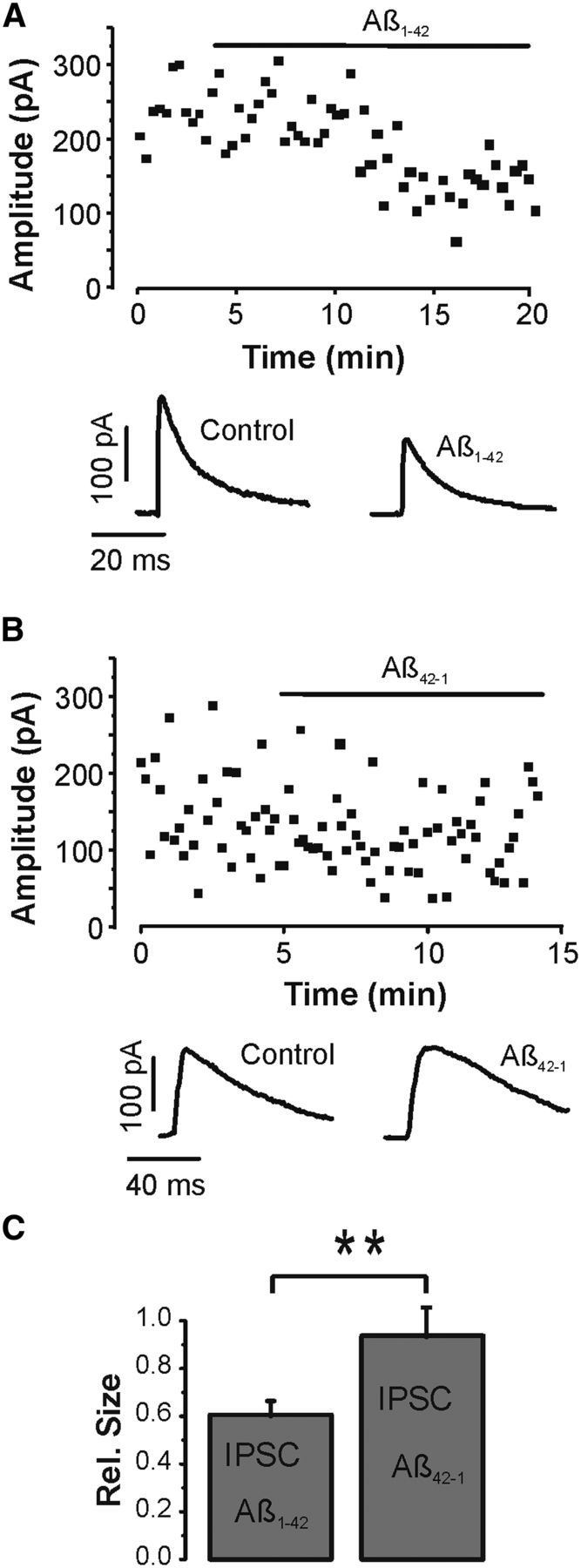
Aβ diminishes GABAergic synaptic transmission. A, IPSC amplitude time course (■) and sample IPSCs (bottom) recorded in whole-cell voltage-clamp mode at 0 mV. Aβ (1 μm) was bath applied as indicated by the horizontal bar. There is pronounced reduction of the IPSCs after the peptide was added. The glutamate receptor antagonists DNQX (20 μm) and APV (50 μm) were present throughout the experiment. B, Similar experiment to A with the reversed control peptide of Aβ. The control peptide had no significant effect on the IPSCs. C, Summary histograms (mean ± SEM) of all experiments with the active (n = 14) and control (n = 5) sequence of Aβ indicating a statistically significant difference. **p < 0.01 (unpaired t test).
Figure 4.
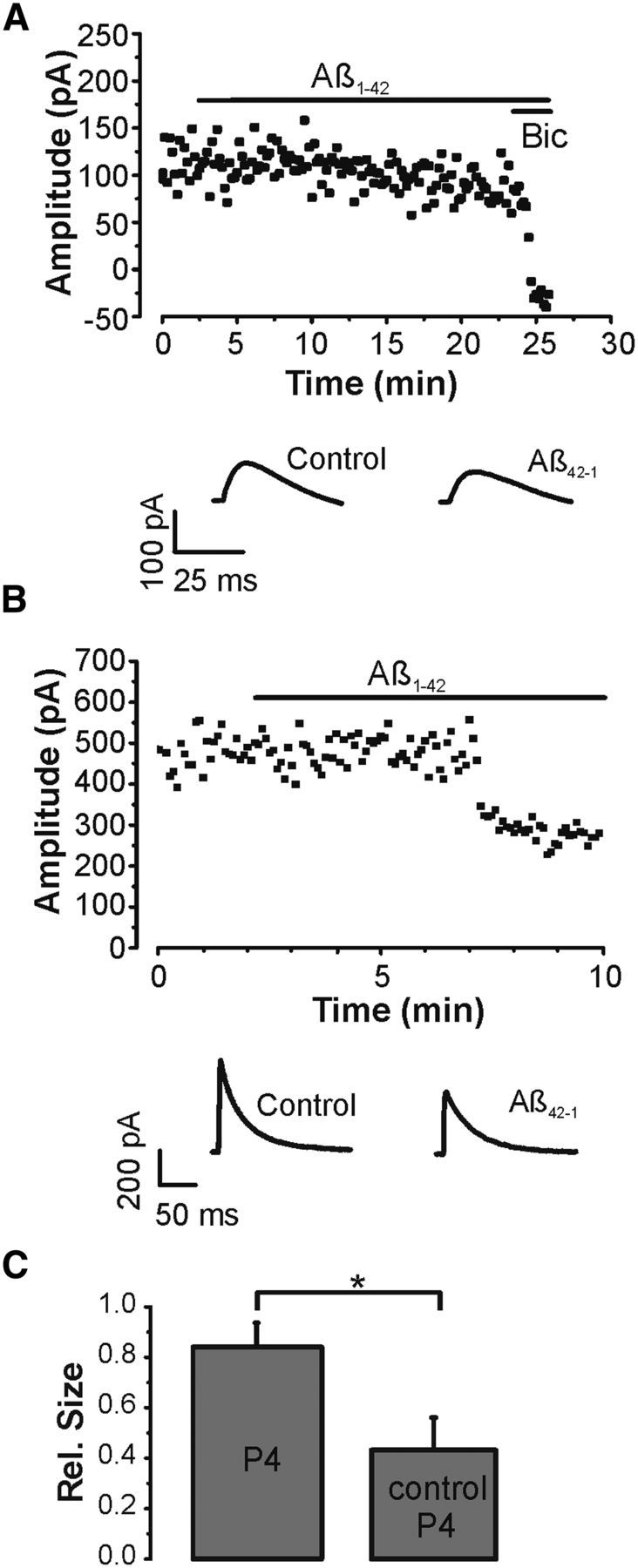
Aβ promotes GABAA receptor endocytosis. A, IPSC amplitude time course (■) and sample IPSCs (bottom) in an experiment where 50 μm p4 was added to the patch pipette solution together with the protease inhibitors leupeptin (0.1 mm) and bestatin (0.1 mm). Aβ (1 μm) was bath applied as indicated by the horizontal bar. IPSCs were completely blocked by bicuculline (10 μm) toward the end of the experiment. DNQX (20 μm), APV (50 μm), and 0.5 μm CGP52432 were present throughout the recordings. B, Amplitude time course (■) and sample IPSCs (bottom) in a similar recording with a control form of p4 applied intracellularly together with leupeptin (0.1 mm) and bestatin (0.1 mm). Aβ (1 μm) was added to the bath as indicated by the horizontal bar. DNQX (20 μm), APV (50 μm), and 0.5 μm CGP52432 were present throughout. C, Summary histogram (mean ± SEM) of all experiments involving p4 (n = 9) and its control (n = 4) shows a statistically significant difference. *p < 0.05 (unpaired t test).
Aβ1-42 acts postynaptically
At excitatory synapses, Aβ has been shown to affect neurotransmitter release as well as the number of neurotransmitter receptors in the postsynaptic membrane. A series of experiments was performed applying paired-pulse stimuli to determine the main site of action of Aβ at inhibitory synapses (Fig. 2A). Presynaptic GABAB receptors were routinely blocked in these experiments by adding 0.5 μm of the specific antagonist CGP52432 to the bath. Similar to previous paired-pulse experiments with GABAB receptors blocked (Pearce et al., 1995), the average paired-pulse ratio for IPSCs at 50 ms interstimulus interval showed a small depression of 0.88 ± 0.07. The paired-pulse ratio was not significantly altered by Aβ to 0.81 ± 0.07 (n = 14, p > 0.1, paired t test), indicating that the probability of GABA release is unlikely a main parameter affected by Aβ (Fig. 2B). To corroborate further a mainly postsynaptic site of Aβ action at inhibitory synapses, an additional set of recordings was performed by puffing the GABAA receptor agonist isoguvacine locally to neurons at an interpuff interval of 20 s (Fig. 3A). Glutamatergic synaptic transmission was again routinely blocked in these experiments by adding APV and DNQX to the bath. Repetitive isoguvacine puffs induced a series of stable transient outward currents in control that were reduced by Aβ and fully blocked by bicuculline (Fig. 3A). Overall, Aβ significantly reduced the total charge of the current from on average 516 ± 138 pC to 411 ± 143 pC (n = 6, p < 0.05, paired t test) (Fig. 3B). Together, the paired-pulse and isoguvacine data identify the postsynaptic membrane as the main target of Aβ actions in these experiments.
Figure 2.
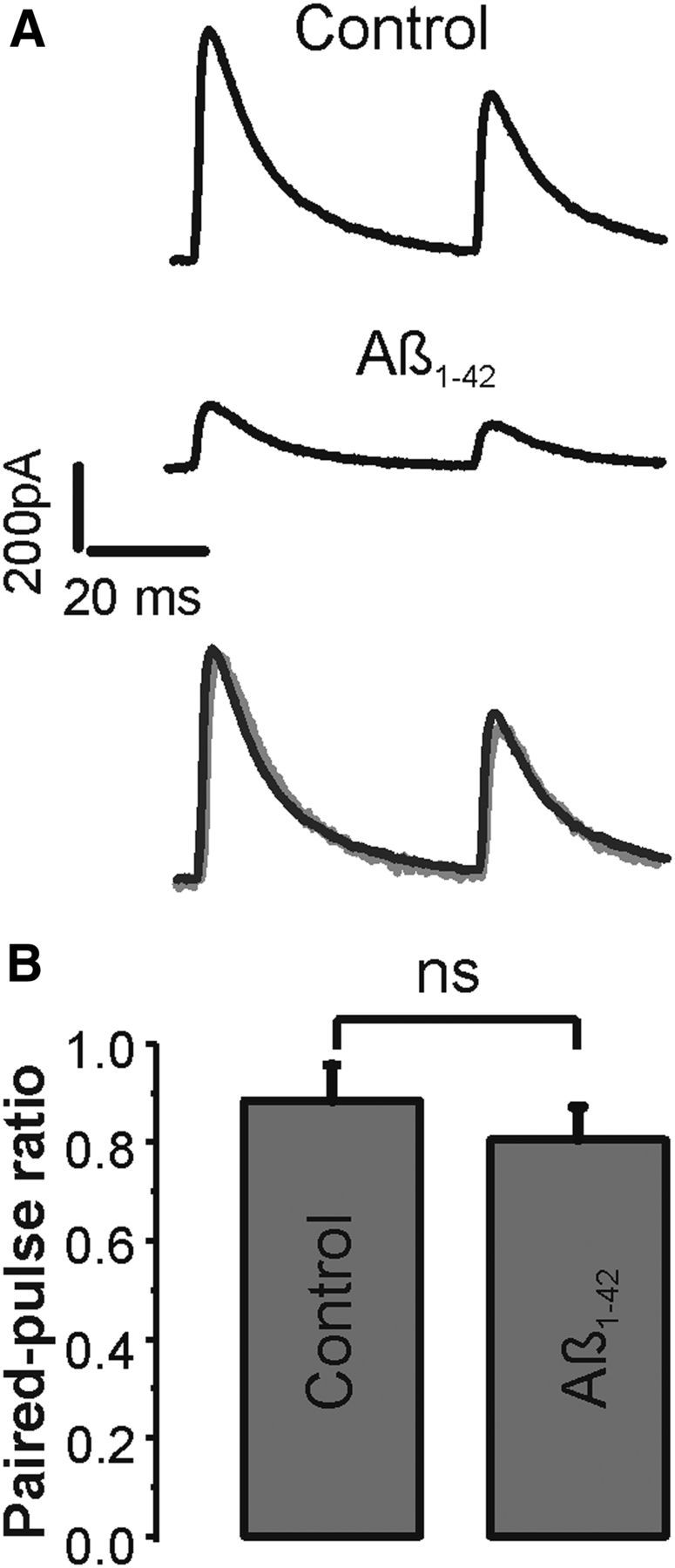
Short-term plasticity is unchanged by Aβ. A, Paired-pulse IPSCs elicited at a 50 ms interval in control and Aβ (1 μm). Bottom, Overlay of normalized control and Aβ traces for comparison. DNQX (20 μm), APV (50 μm), and CGP52432 (0.5 μm) were present throughout the recordings. B, Summary histograms (mean ± SEM) of all paired-pulse experiments in control and Aβ (n = 14) reveal no statistically significant difference. ns, Not significant (p > 0.1, paired t test).
Figure 3.
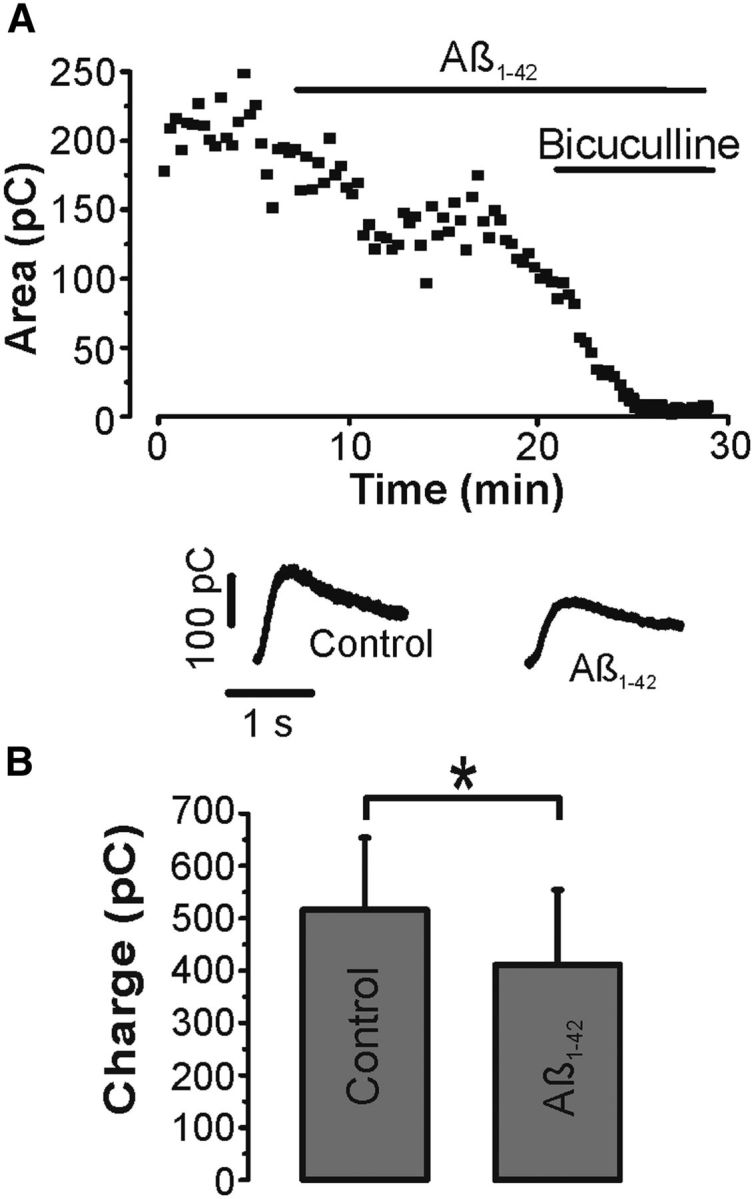
Aβ decreases agonist-evoked GABAA responses. A, Time course (■) and sample traces of transient outward currents generated by brief puffs of 0.5 mm isoguvacine repeated every 20 s in control, Aβ (1 μm), and bicuculline (10 μm). B, Summary histograms (mean ± SEM) of isoguvacine responses in Aβ versus control (n = 6) show a statistically significant difference. *p < 0.05 (paired t test).
Aβ1-42 downregulates GABA receptors
Endocytosis and exocytosis of GABAA receptors are important mechanisms through which neurons regulate the strength of synaptic inhibition (Vithlani et al., 2011). In particular, GABAA receptors are known to be removed from the plasma membrane by clathrin-mediated endocytosis, a process that requires interactions between dynamin and amphiphysin (Kittler et al., 2000). To investigate whether Aβ affects GABAA receptor endocytosis, IPSCs were elicited with the dynamin inhibitory peptide p4 (50 μm) intracellularly applied via the patch pipette in combination with the protease inhibitors leupeptin (0.1 mm) and bestatin (0.1 mm) (Fig. 4) (Kittler et al., 2000). When p4 was allowed to diffuse into cells, IPSCs were no longer significantly reduced by Aβ to 85 ± 9% of control (n = 9, p > 0.1, paired t test) (Fig. 4A). To verify that this effect was indeed attributable to p4, a similar series of experiments was performed with a heat-inactivated control version of p4. When the control form of p4 was included in the patch pipette solution together with leupeptin and bestatin, IPSCs were again significantly reduced by Aβ to 45 ± 13% of control (p < 0.05, n = 4, paired t test) (Fig. 4B). Comparison of all recordings with p4 and its control showed a statistically significant difference (p < 0.05, unpaired t test) (Fig. 4C). Overall, these experiments indicate that Aβ affects inhibitory synapses through GABAA receptor endocytosis.
Discussion
This study shows that Aβ, a main causative agent in the pathogenesis of Alzheimer's disease, directly interferes with synaptic inhibition in pyramidal cells of neocortex via downregulation of GABAA receptors. Controversial data exist on whether and to what degree Aβ affects synaptic inhibition in various experimental preparations. In acute applications, Aβ fragments 1-40 and 25-35 were shown to decrease agonist-evoked GABA responses in Aplysia neurons (Sawada and Ichinose, 1996). In contrast, GABA responses in acutely dissociated hippocampal neurons were potentiated by Aβ 25-35 and Aβ 31-35 (Zhang et al., 2009). This may indicate that the net effect of Aβ is fragment- and cell-type specific. In the present study, the Aβ 1-42 alloform was used because of its presence in Alzheimer's brains and its tendency to form oligomers, which are thought to be the main toxic agent in Alzheimer's disease. In line with the current findings, a comparable reduction of GABAergic inhibition was recently reported in the hippocampus under similar experimental conditions (Kurudenkandy et al., 2014). This indicates that acute Aβ effects on GABAergic synapses are not confined to neocortical areas but are also present in phylogenetically older parts of the brain. There is limited agreement on the adequate concentration of Aβ for experimental Alzheimer's studies. The concentration of Aβ1-42 in the interstitial fluid of patients with Alzheimer's disease can be in the range of picomolar to low nanomolar but is thought to be significantly higher in the vicinity of senile plaques (Mucke and Selkoe, 2012). Low micromolar concentrations of Aβ that are widely used in in vitro studies may therefore be of pathophysiological relevance, even more so because the actual Aβ concentration in the slice may be considerably lower than in the superfusing bath (Waters, 2010).
There are experimental data supporting presynaptic and postsynaptic sites of Aβ action at excitatory synapses. The paired-pulse data from this study show a reduction in IPSCs without concomitant change in the paired-pulse ratio. This is usually taken as an indicator for a postsynaptic site of action, although the paired-pulse ratio may not be fully conclusive (Kim and Alger, 2001). However, the similar Aβ-induced reduction of agonist-evoked GABA responses and IPSCs renders a major presynaptic contribution to the observed decline unlikely. Nevertheless, a presynaptic influence may become apparent with different concentrations of Aβ and/or stimulation patterns as seen at excitatory synapses (Ting et al., 2007; Abramov et al., 2009). Adding to the complexity of amyloid effects on GABAergic synaptic transmission is the recent observation that the amyloid precursor protein itself can limit the amount of GABA released through direct protein-protein interactions (Yang et al., 2009). This suggests that presynaptic as well as postsynaptic processes may contribute synaptic impairment at inhibitory synapses in Alzheimer's disease.
The number of GABAA receptors on the cell surface is controlled by receptor trafficking involving exocytosis and endocytosis (Vithlani et al., 2011). The prevention of the Aβ-induced decline of IPSCs by p4, a peptide that inhibits the dynamin-mediated removal of GABAA receptors from the plasma membrane (Kittler et al., 2000), indicates that Aβ exerts its effect via receptor endocytosis. A similar mechanism has previously been shown to underlie the Aβ-induced decrease of glutamatergic synaptic transmission (Hsieh et al., 2006). Of note, no p4-related upregulation of IPSCs was seen in the present study, as originally reported by Kittler et al. (2000). However, the absence of a p4-induced increase of GABA receptors is in line with previous data from cortical pyramidal neurons (Kurotani et al., 2008), indicating that this phenomenon may be cell-type specific. The detailed signaling pathways between Aβ and GABA receptor endocytosis remain to be determined. Synaptic excitotoxicity is unlikely involved because ionotropic glutamate receptors were routinely blocked in the experiments. Similarly, a role for metabotropic neurotransmitters is unlikely as the intracellularly added Cs+ and QX314 block several of their main effectors. Preliminary experiments with a high Ca2+ buffer did not prevent a decline of the IPSCs (data not shown), arguing against a role for Ca2+ triggered endocytosis. Direct interactions of Aβ with GABA receptors or Aβ-mediated intracellular signaling may provide alternative mechanisms to be tested. The current findings add to previous studies showing compromised GABAB receptor-dependent synaptic inhibition after Aβ application (Nava-Mesa et al., 2013). Therefore, Aβ appears to interfere with both ionotropic and metabotropic GABA responses. In addition, in animal models, nonsynaptic mechanisms have been reported through which inhibitory interneurons may be compromised (Verret et al., 2012). Thus, there is experimental evidence for the existence of multiple processes that could compromise inhibitory pathways in the etiology of Alzheimer's disease. An overall impairment of inhibitory circuits would be compatible with the occurrence of hyperexcitability that has been seen in animal models of Alzheimer's disease and Alzheimer's patients (Amatniek et al., 2006; Minkeviciene et al., 2009) and that may eventually lead to increased amyloid plaque formation (Bero et al., 2011).
Footnotes
This work was supported by Trinity Research and Innovation. I thank Prof. M.J. Rowan for insightful discussions and critical comments on the manuscript.
The authors declare no competing financial interests.
References
- Abramov E, Dolev I, Fogel H, Ciccotosto GD, Ruff E, Slutsky I. Amyloid-beta as a positive endogenous regulator of release probability at hippocampal synapses. Nat Neurosci. 2009;12:1567–1576. doi: 10.1038/nn.2433. [DOI] [PubMed] [Google Scholar]
- Amatniek JC, Hauser WA, DelCastillo-Castaneda C, Jacobs DM, Marder K, Bell K, Albert M, Brandt J, Stern Y. Incidence and predictors of seizures in patients with Alzheimer's disease. Epilepsia. 2006;47:867–872. doi: 10.1111/j.1528-1167.2006.00554.x. [DOI] [PubMed] [Google Scholar]
- Bero AW, Yan P, Roh JH, Cirrito JR, Stewart FR, Raichle ME, Lee JM, Holtzman DM. Neuronal activity regulates the regional vulnerability to amyloid-β deposition. Nat Neurosci. 2011;14:750–756. doi: 10.1038/nn.2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busche MA, Eichhoff G, Adelsberger H, Abramowski D, Wiederhold KH, Haass C, Staufenbiel M, Konnerth A, Garaschuk O. Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer's disease. Science. 2008;321:1686–1689. doi: 10.1126/science.1162844. [DOI] [PubMed] [Google Scholar]
- Demuro A, Parker I, Stutzmann GE. Calcium signaling and amyloid toxicity in Alzheimer disease. J Biol Chem. 2010;285:12463–12468. doi: 10.1074/jbc.R109.080895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S, Malinow R. AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron. 2006;52:831–843. doi: 10.1016/j.neuron.2006.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R. APP processing and synaptic function. Neuron. 2003;37:925–937. doi: 10.1016/S0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- Kim J, Alger BE. Random response fluctuations lead to spurious paired-pulse facilitation. J Neurosci. 2001;21:9608–9618. doi: 10.1523/JNEUROSCI.21-24-09608.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittler JT, Delmas P, Jovanovic JN, Brown DA, Smart TG, Moss SJ. Constitutive endocytosis of GABAA receptors by an association with the adaptin AP2 complex modulates inhibitory synaptic currents in hippocampal neurons. J Neurosci. 2000;20:7972–7977. doi: 10.1523/JNEUROSCI.20-21-07972.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurotani T, Yamada K, Yoshimura Y, Crair MC, Komatsu Y. State-dependent bidirectional modification of somatic inhibition in neocortical pyramidal cells. Neuron. 2008;57:905–916. doi: 10.1016/j.neuron.2008.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurudenkandy FR, Zilberter M, Biverstål H, Presto J, Honcharenko D, Strömberg R, Johansson J, Winblad B, Fisahn A. Amyloid-β-induced action potential desynchronization and degradation of hippocampal gamma oscillations is prevented by interference with peptide conformation change and aggregation. J Neurosci. 2014;34:11416–11425. doi: 10.1523/JNEUROSCI.1195-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanctôt KL, Herrmann N, Mazzotta P, Khan LR, Ingber N. GABAergic function in Alzheimer's disease: evidence for dysfunction and potential as a therapeutic target for the treatment of behavioural and psychological symptoms of dementia. Can J Psychiatry. 2004;49:439–453. doi: 10.1177/070674370404900705. [DOI] [PubMed] [Google Scholar]
- Limon A, Reyes-Ruiz JM, Miledi R. Loss of functional GABA(A) receptors in the Alzheimer diseased brain. Proc Natl Acad Sci U S A. 2012;109:10071–10076. doi: 10.1073/pnas.1204606109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minkeviciene R, Rheims S, Dobszay MB, Zilberter M, Hartikainen J, Fülöp L, Penke B, Zilberter Y, Harkany T, Pitkänen A, Tanila H. Amyloid beta-induced neuronal hyperexcitability triggers progressive epilepsy. J Neurosci. 2009;29:3453–3462. doi: 10.1523/JNEUROSCI.5215-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucke L, Selkoe DJ. Neurotoxicity of amyloid β-protein: synaptic and network dysfunction. Cold Spring Harb Perspect Med. 2012;2:a006338. doi: 10.1101/cshperspect.a006338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan T, Jensen MS, Lambert JD. The slow inhibitory postsynaptic potential in rat hippocampal CA1 neurones is blocked by intracellular injection of QX-314. Neurosci Lett. 1990;110:309–313. doi: 10.1016/0304-3940(90)90865-7. [DOI] [PubMed] [Google Scholar]
- Nava-Mesa MO, Jiménez-Díaz L, Yajeya J, Navarro-Lopez JD. Amyloid-β induces synaptic dysfunction through G protein-gated inwardly rectifying potassium channels in the fimbria-CA3 hippocampal synapse. Front Cell Neurosci. 2013;7:117. doi: 10.3389/fncel.2013.00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Mucke L. Amyloid-beta-induced neuronal dysfunction in Alzheimer's disease: from synapses toward neural networks. Nat Neurosci. 2010;13:812–818. doi: 10.1038/nn.2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO, Yu GQ, Kreitzer A, Finkbeiner S, Noebels JL, Mucke L. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease. Neuron. 2007;55:697–711. doi: 10.1016/j.neuron.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce RA, Grunder SD, Faucher LD. Different mechanisms for use-dependant depression of two GABAA-mediated IPSCs in rat hippocampus. J Physiol. 1995;484:425–435. doi: 10.1113/jphysiol.1995.sp020675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rissman RA, De Blas AL, Armstrong DM. GABA(A) receptors in aging and Alzheimer's disease. J Neurochem. 2007;103:1285–1292. doi: 10.1111/j.1471-4159.2007.04832.x. [DOI] [PubMed] [Google Scholar]
- Sawada M, Ichinose M. Amyloid beta proteins reduce the GABA-induced Cl-current in identified Aplysia neurons. Neurosci Lett. 1996;213:213–215. doi: 10.1016/0304-3940(96)12847-0. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Stuart GJ, Dodt HU, Sakmann B. Patch-clamp recordings from the soma and dendrites of neurons in brain slices using infrared video microscopy. Pflugers Arch. 1993;423:511–518. doi: 10.1007/BF00374949. [DOI] [PubMed] [Google Scholar]
- Teplow DB. Preparation of amyloid beta-protein for structural and functional studies. Methods Enzymol. 2006;413:20–33. doi: 10.1016/S0076-6879(06)13002-5. [DOI] [PubMed] [Google Scholar]
- Ting JT, Kelley BG, Lambert TJ, Cook DG, Sullivan JM. Amyloid precursor protein overexpression depresses excitatory transmission through both presynaptic and postsynaptic mechanisms. Proc Natl Acad Sci U S A. 2007;104:353–358. doi: 10.1073/pnas.0608807104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verret L, Mann EO, Hang GB, Barth AM, Cobos I, Ho K, Devidze N, Masliah E, Kreitzer AC, Mody I, Mucke L, Palop JJ. Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell. 2012;149:708–721. doi: 10.1016/j.cell.2012.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vithlani M, Terunuma M, Moss SJ. The dynamic modulation of GABA(A) receptor trafficking and its role in regulating the plasticity of inhibitory synapses. Physiol Rev. 2011;91:1009–1022. doi: 10.1152/physrev.00015.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Waters J. The concentration of soluble extracellular amyloid-β protein in acute brain slices from CRND8 mice. PLoS One. 2010;5:e15709. doi: 10.1371/journal.pone.0015709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Guo Z, Gearing M, Chen G. Tonic inhibition in dentate gyrus impairs long-term potentiation and memory in an Alzheimer's disease model. Nat Commun. 2014;5:4159. doi: 10.1038/ncomms5159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Wang Z, Wang B, Justice NJ, Zheng H. Amyloid precursor protein regulates Cav1.2 L-type calcium channel levels and function to influence GABAergic short-term plasticity. J Neurosci. 2009;29:15660–15668. doi: 10.1523/JNEUROSCI.4104-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Hou L, Gao X, Guo F, Jing W, Qi J, Qiao J. Amyloid β-protein differentially affects NMDA receptor and GABAA receptor-mediated current in rat hippocampal CA1 neurons. Prog Nat Sci. 2009;19:963–972. doi: 10.1016/j.pnsc.2008.11.006. [DOI] [Google Scholar]