

Abstract
PET imaging of nicotinic acetylcholine receptors (nAChRs) could become an effective tool for the diagnosis and therapy evaluation of neurologic diseases. Despite this, the role of nAChRs α4β2 receptors after brain diseases such as cerebral ischemia and its involvement in inflammatory reaction is still largely unknown. To investigate this, we performed in parallel in vivo magnetic resonance imaging (MRI) and positron emission tomography (PET) with 2[18F]-fluoro-A85380 and [11C]PK11195 at 1, 3, 7, 14, 21, and 28 d after middle cerebral artery occlusion (MCAO) in rats. In the ischemic territory, PET with 2[18F]-fluoro-A85380 and [11C]PK11195 showed a progressive binding increase from days 3–7, followed by a progressive decrease from days 14–28 after cerebral ischemia onset. Ex vivo immunohistochemistry for the nicotinic α4β2 receptor and the mitochondrial translocator protein (18 kDa) (TSPO) confirmed the PET findings and demonstrated the overexpression of α4β2 receptors in both microglia/macrophages and astrocytes from days 7–28 after experimental ischemic stroke. Likewise, the role played by α4β2 receptors on neuroinflammation was supported by the increase of [11C]PK11195 binding in ischemic rats treated with the α4β2 antagonist dihydro-β-erythroidine hydrobromide (DHBE) at day 7 after MCAO. Finally, both functional and behavioral testing showed major impaired outcome at day 1 after ischemia onset, followed by a recovery of the sensorimotor function and dexterity from days 21–28 after experimental stroke. Together, these results suggest that the nicotinic α4β2 receptor could have a key role in the inflammatory reaction underlying cerebral ischemia in rats.
Keywords: α4β2, [11C]PK11195, 2[18F]-fluoro-A85380, cerebral ischemia, PET, TSPO
Introduction
Nicotinic acetylcholine receptors (nAChRs) are ion-gated channels composed of five subunits (α2–α10 and β2–β4) that are widely expressed in the skeletal neuromuscular junction and throughout both the peripheral nervous system and the CNS (Hogg et al., 2003). The most abundant subtypes of nAChRs in mammalian brain are heteromeric receptors containing α4 and β2 subunits and homomeric α7 (Mukhin et al., 2000). The nicotinic acetylcholine system plays a crucial role in the mediation of memory learning, drug addiction, and control of pain (Gotti et al., 1997). Furthermore, nAChRs play important roles in neurodegenerative diseases (O'Neill et al., 2002). Postmortem brains from patients with Alzheimer's disease (AD) (Burghaus et al., 2000) and Parkinson's disease (PD) (Guan et al., 2002) showed a decrease of the density of nAChRs. Likewise, the stimulation of nAChRs protects neurons from insults associated with neurodegenerative disorders (Mudo et al., 2007), ischemic damage (Shimohama et al., 1998), and intracerebral hemorrhage (Hijioka et al., 2011). This physiological mechanism responds to the cholinergic modulation of macrophage/microglia activation by nAChRs receptors and its activation has been ultimately related to the suppression of the inflammation (Wang et al., 2003, Shytle et al., 2004, Nizri et al., 2008). Therefore, the in vivo imaging of nAChRs with PET might be crucial to further understanding the role of these receptors on inflammation underlying brain diseases (Kimes et al., 2003). Such promising PET radiotracers as the 2-18F-fluoro-3-[2(S)-2-azetidinylmethoxy]pyridine (2[18F]-fluoro-A85380) have appeared for the study of central nAChR (Bottlaender et al., 2003, Kimes et al., 2003, Ding et al., 2004, Zanotti-Fregonara et al., 2012). 2[18F]-fluoro-A85380 is a radioligand with high affinity to the α4β2 receptor that has shown a binding decrease in human brain sections of AD (Schmaljohann et al., 2004) and PD (Schmaljohann et al., 2006). Therefore, PET 2[18F]-fluoro-A85380 has been suggested as a promising radiotracer with which to evaluate the loss of nicotinic neurons in neurodegenerative diseases. Nevertheless, although nAChRs are also expressed in microglia (Furukawa et al., 2013), the use of 2[18F]-fluoro-A85380 to image brain inflammation has been little explored to date. The purpose of the present study was to investigate changes in the levels of α4β2 nAChRs in the rat brain after cerebral ischemia using PET with 2[18F]-fluoro-A85380 and immnuhistochemistry. In particular, we were interested in clarifying the relationship of the nAChRs expression with the activation of glial cells after cerebral ischemia in rats. Ischemic rats were subjected to PET studies with [11C]PK11195, a specific radioligand for the translocator protein (18 kDa) (TSPO), to image brain inflammation (Rojas et al., 2007). Likewise, [11C]PK11195 binding was used to evaluate the effect of a selective antagonist for α4β2 receptors [dihydro-β-erythroidine hydrobromide (DHβE)] on the neuroinflammatory reaction after ischemic stroke in rats. The results reported here may have a significant practical importance because they can provide novel information about the role of nAChRs on the inflammatory reaction after cerebral ischemia and might ultimately contribute to a better design of anti-inflammatory strategies for the treatment of ischemic stroke.
Materials and Methods
Cerebral ischemia and treatment.
Adult male Sprague Dawley rats (300 g body weight; Janvier) (n = 35) were used. Animal studies were approved by the animal ethics committee of Center for Cooperative Research in Biomaterials CIC biomaGUNE and local authorities and were conducted in accordance with the Directives of the European Union on animal ethics and welfare. Transient focal ischemia was produced by a 2 h intraluminal occlusion of the middle cerebral artery (MCA) followed by reperfusion as described previously (Justicia et al., 2001). Briefly, rats were anesthetized with 4% isoflurane in 100% O2 and a 2.6 cm length of 4-0 monofilament nylon suture was introduced into the right external carotid artery up to the level where the MCA branches out. Animals were sutured and placed in their cages with free access to water and food. After 2 h, the animals were reanesthetized and the filament was removed to allow reperfusion. Six rats were repeatedly examined before (day 0) and at 1, 3, 7, 14, 21, and 28 d after ischemia to evaluate the temporal PET binding of both α4β2 and TSPO. The animals studied at day 0 were considered the baseline control group.
A group of six rats were inoculated daily for a total of 7 d from 1 h afater MCAO with 0.5 ml of DHβE (3 mg/kg, i.p.). A control ischemic group of eight rats received daily the same volume of vehicle (normal saline). At day 7, treated and control rats were imaged with PET to determine the effect of DHβE on the TSPO expression. Finally, a total of 15 rats were used to perform ex vivo studies (immnuhistochemistry) at 0, 7, and 28 d after cerebral ischemia. Therefore, MCAO was induced to 30 rats.
MRI.
T2-weighting (T2W) MRI scans were performed in ischemic animals at 24 h after reperfusion to select the rats (n = 6) presenting corticostriatal lesions to be included in the PET studies. Before the scans, anesthesia was induced with 4% isoflurane and maintained by 2–2.5% of isofluorane in 100% O2 during the scan. Animals were placed into a rat holder compatible with MRI acquisition systems and maintained normothermic using a water-based heating blanket at 37°C. Measurements were performed using an 11.7 T Bruker Biospec system with a 72 mm volumetric quadrature coil for excitation and a 20 mm surface coil for reception. Acquisition parameters for the T2-weighted spin echo images were as follows: TR/TE = 3300/30 ms, FOV = 1.8 × 1.8 cm2, matrix = 200 × 200, number of excitations = 3, slice thickness = 0.8 mm. Contiguous slices covering all the infarcted volume were acquired and fat suppression was used.
MRI analysis.
MRI (T2W) images were used to calculate the lesion volume. ROIs were defined manually using open source 3D Slicer image analysis software (version 3.6.3; www.slicer.org) for each rat on the region of increased signal in the ipsilateral hemisphere. The total lesion volume was calculated by summing the area of the infarcted regions of all slices affected by the lesion.
Radiochemistry.
For the production of [11C]PK11195, [11C]CH4 was directly generated in an IBA Cyclone 18/9 cyclotron and transferred to a TRACERlab FXC Pro synthesis module (GE Healthcare), where [11C]CH3I was generated. At the end of the process, [11C]CH3I was distilled under continuous helium flow (20 ml/min) and introduced in a 2 ml stainless steel reaction loop, precharged with a solution of N-methyl-2-(2-amino-4-cyanophenylthio)-benzylamine (MASB, 1 mg; ABX) in dimethylsulfoxide (80 μl). The reaction mixture was purified with HPLC. The collected fraction was reformulated by dilution with water (20 ml), retention on a C-18 cartridge (Sep-Pak Light; Waters) and elution with ethanol (1 ml) and saline (9 ml). Filtration through a 0.22 μm sterile filter yielded the final solution. Typical radiochemical yields and specific activities were 33 ± 5% (end of bombardment) and 135 ± 18 GBq/μmol (end of synthesis), respectively. Radiochemical purity was >98% in all cases.
2-[18F]-fluoro-A85380 was produced using a TRACERlab FXFN synthesis module (GE Healthcare). Briefly, the [18F]F− was trapped in a preconditioned QMA cartridge and transferred to the reactor by sequential elution with a solution of K2CO3 (3.5 mg) in water (0.5 ml) and a solution of Kryptofix K2.2.2 (15 mg) in acetonitrile (1 ml). After evaporation to dryness, a solution containing 3 mg of nitro-AP (ABX) in 1 ml of dry acetonitrile was added. The reaction was performed at 85°C for 15 min. The reactor was then cooled at room temperature and dichloromethane (0.5 ml), trifluoroacetic acid (0.5 ml), and purified water (2.5 ml) were sequentially added. After stirring for 15 s, the mixture was injected into a semipreparative HPLC system. A mixture of 0.2% trifluoroacetic acid in water/methanol/acetonitrile (85/4/11) was used as mobile phase and a Nucleosil 100–7 250 mm column (Macherey Nagel) was used as a stationary phase. The collected fraction was formulated by dilution with sodium hydroxide aqueous solution (30 ml of water + 2 ml of 0.5 m NaOH), retention on a C-18 cartridge (Sep-Pak Plus; Waters) and elution with ethanol (2 ml) and phosphate buffer solution. Filtration through 0.22 μm sterile filters yielded the final 2-[18F]-fluoro-A85380 solution. Average radiochemical yield was 7.8 ± 1.2% (end of synthesis) and the radiochemical purity was > 98% in all cases. The specific activity of 2-[18F]-fluoro-A85380 was 150 ± 30 GBq/μmol and the mass dose was 0.0665 ± 0.011 μg.
PET scans and data acquisition.
PET scans were repeatedly performed before (day 0) and at 1, 3, 7, 14, 21, and 28 d after reperfusion using a General Electric eXplore Vista CT camera. Scans were performed in rats anesthetized with 4% isofluorane and maintained by 2–2.5% of isofluorane in 100% O2.The tail vein was catheterized with a 24-gauge catheter for intravenous administration of the radiotracer. Animals were placed into a rat holder compatible with PET acquisition system and maintained normothermic using a water-based heating blanket. Animals were subjected to two PET scans to assess TSPO binding ([11C]PK11195) and α4β2 receptor binding (2-[18F]-fluoro-A85380) at every time point before and after ischemia onset. First, ∼30 MBq of [11C]PK11195 were injected concomitantly with the start of the PET acquisition. Brain dynamic images were acquired (34 frames: 6 × 5, 6 × 15, 6 × 60, 8 × 120, and 8 × 300 s) in the 400–700 keV energetic window, with a total acquisition time of 64 min, providing a 175 × 175 matrix with a pixel size of 0.887 mm and 61 slices. Second, after at least 180 min (∼9 half-lives of 11C), animals were reanesthetized and placed on the PET camera and ∼30 MBq of 2-[18F]-fluoro-A85380 was injected concomitantly with the start of the PET acquisition. The acquisition protocol was the same as for [11C]PK11195. After each PET scan, CT acquisitions were also performed (140 μA intensity, 40 kV voltage), providing anatomical information of each animal as well as the attenuation map for the later image reconstruction. Dynamic acquisitions were reconstructed (decay and CT-based attenuation corrected) with filtered back projection using a Ramp filter with a cutoff frequency of 0.5 mm−1.
PET image analysis.
PET images were analyzed using PMOD image analysis software version 3.506 (PMOD Technologies). To verify the anatomical location of the signal, PET images were coregistered to the anatomical data of a MRI rat brain template. Two types of volumes of interest (VOIs) were established as follows. A first set of VOIs was defined to study the whole brain [11C]PK11195 and 2-[18F]-fluoro-A85380 binding potential nondisplaceable (PBND). Whole-brain VOIs were manually drawn in both the entire ipsilateral and contralateral hemispheres containing the territory irrigated by the middle cerebral artery on slices of a MRI (T2W) rat brain template from the PMOD software. A second set of VOIs was automatically generated in cortex, striatum, and thalamus using the regions proposed by the PMOD rat brain template to study the evolution of BPND of the radiotracers to the specific regions in both ipsilateral and contralateral cerebral hemispheres. The simplified reference tissue model (Lammertsma and Hume, 1996) from the PMOD software was used to assess BPND. This model relies on a two-tissue reversible compartment for a reference region (cerebellum).
Immunohistochemistry.
Immunohistochemistry staining was performed at day 0 (control) and days 7 and 28 after reperfusion. Animals were terminally anesthetized and killed by decapitation. The brain was removed, frozen, and cut in 5-μm-thick sections in a cryostat. Sections were fixed in acetone (−20°C) for 2 minutes, washed with PBS, saturated with a solution of 5% BSA/0.5% Tween in PBS for 15 minutes at room temperature, and incubated for 1 h at room temperature with primary antibodies BSA (5%)/Tween (0.5%) in PBS. The first set of sections were stained for α4β2 with rabbit anti-rat α4β2 (1:500; Abcam), for CD11b with mouse anti-rat CD11b (1:300; Serotec), and for the glial fibrillary acidic protein (GFAP) with chicken anti-rat GFAP (1:500; Abcam). The second set of sections was stained for TSPO with a rabbit anti-rat TSPO (NP155, 1:1000), for CD11b, and for GFAP. Sections were washed (3 × 10 minutes) in PBS and incubated for 1 h at room temperature with secondary antibodies Alexa Fluor 350 goat anti-rabbit IgG, Alexa Fluor 594 goat anti-mouse IgG, and Alexa Fluor 488 goat anti-chicken IgG (Invitrogen, 1:1000) in 5% BSA/0.5% Tween in PBS, washed again (3 × 10 minutes) in PBS, and mounted with a prolong antifade kit in slices (Invitrogen, Madrid). Standardized images acquisition was performed with an Axio Observer Z1 (Zeiss) equipped with a motorized stage.
Cell counts and immunoreactivity quantification.
The number of α4β2/CD11b and α4β2/GFAP-immunopositive cells and microglial/astrocytic immunoreactivity for α4β2 receptors within the ischemic area was assessed at 0, 7, and 28 d after ischemia. Cells were counted in 10 different fields at 40× magnification. Representative images of areas showing the highest staining density were acquired and cells were counted manually. Immunoreactivity was analyzed using ImageJ version 1.48 software. Microglial/α4β2 receptor immunoreactivity was calculated using the mean intensity value of α4β2 receptor-immunostained pixels in the CD11b staining-positive area. For that, the CD11b-staining-positive ROIs were defined by thresholding and converting into a binary mask (Jo et al., 2014). Then, the CD11b-positive ROIs were analyzed for α4β2 receptor intensity and the mean intensity value of pixels was used to define microglial/α4β2 immunoreactivity. Data represent the mean value of 150 ROIs obtained from four animals, two slides per animal, and five different fields per slide. The mean intensity value of astrocytic α4β2-positive pixels was measured similarly in GFAP-positive areas.
Neurological and behavioral testing.
Two main neurological tests were used to assess neurological and behavioral deficits after cerebral ischemia in rats. The assessment of neurological outcome induced by cerebral ischemia was based on a previously reported 9-neuroscore test (Menzies et al., 1992). Four consecutive tests were performed on every ischemic animal before (day 0) and at 1, 3, 7, 14, 21, and 28 d after MCAO as follows: (1) spontaneous activity (moving and exploring = 0, moving without exploring = 1, no moving = 2); (2) left drifting during displacement (none = 0, drifting only when elevated by the tail and pushed or pulled = 1, spontaneous drifting = 2, circling without displacement or spinning = 3); (3) parachute reflex (symmetrical = 0, asymmetrical = 1, contralateral forelimb retracted = 2); and (4) resistance to left forepaw stretching (stretching not allowed = 0, stretching allowed after some attempts = 1, no resistance = 2). Total score could range from 0 (normal) to a 9 (highest handicap) point-scale.
The adhesion/removal tape test was performed as described previously (Bouet et al., 2009) to evaluate the deficits and behavioral recovery on every ischemic animal before (day 0) and at 1, 3, 7, 14, 21, and 28 d after MCAO. In brief, it consists of applying adhesive tape on each forepaw of the animal and measuring the time to contact (the time until contact with the mouth) and the time to remove them up to a maximum of 2 minutes. Animals were trained in this task 7 d before ischemia. This behavior implies correct paw and mouth sensitivity (time to contact) and correct dexterity (time to remove).
Statistical analyses.
For PET binding values, the statistical analysis was performed as follows: BPND values for each animal, brain region (whole brain, cortex, striatum and thalamus), and brain hemisphere (ipsilateral and contralateral) were calculated at each time point. Binding values within each region, time point, and hemisphere were averaged and compared with the averaged baseline control values (before MCAO) using one-way ANOVA followed by Dunnett's multiple-comparison tests for post hoc analysis. Likewise, cellular expression and immunoreactivity of both microglial/α4β2 and astrocytic/α4β2 receptors at days 7 and 28 after ischemia were compared with control values (day 0) using the same statistical analysis as for PET imaging. The effect of DHβE in ischemic rats was compared with control infarcted rats using an unpaired t test. Neurological outcome comparisons were performed as follows: animals were subjected to the 9-neuroscore test before MCAO and at 1, 3, 7, 14, 21, and 28 d after cerebral ischemia. The results within each time point were averaged and compared with baseline average values using Mann–Whitney U tests. Behavioral outcome comparisons by forepaw (ipsilateral and contralateral) were then performed. Animals were subjected to the adhesion/removal tape test before MCAO and at different time points after cerebral ischemia. The results within each time point and forepaw (ipsilateral and contralateral) were averaged and compared with baseline values using Mann–Whitney U tests. To assess the differences between forepaws (ipsilateral vs contralateral), behavioral outcome averaged values at each time point and forepaw were compared using two-way ANOVA. The level of significance was regularly set at p < 0.05. Statistical analyses were performed with GraphPad Prism version 4.02 software.
Results
The levels and distribution of nicotinic α4β2 and TSPO receptors were explored by PET imaging after a 2 h MCAO and 1–28 d reperfusion in rats. All images were quantified in standard units; that is, BPND for both 2-[18F]-fluoro-A85380 and [11C]PK11195. The images with normalized color scale illustrate the evolution of the PET signals at control and at 1, 7, 14, and 28 d after reperfusion (Fig. 1). The extent of brain damage after cerebral ischemia was assessed using T2W MRI at 1 d after ischemia onset. Hyperintensities of T2W images showed similar infarct extents as well as locations affected. All ischemic rats subjected to nuclear studies showed cortical and striatal MRI alterations (mean ± SD = 306.16 ± 37.78 mm3, n = 6).
Figure 1.
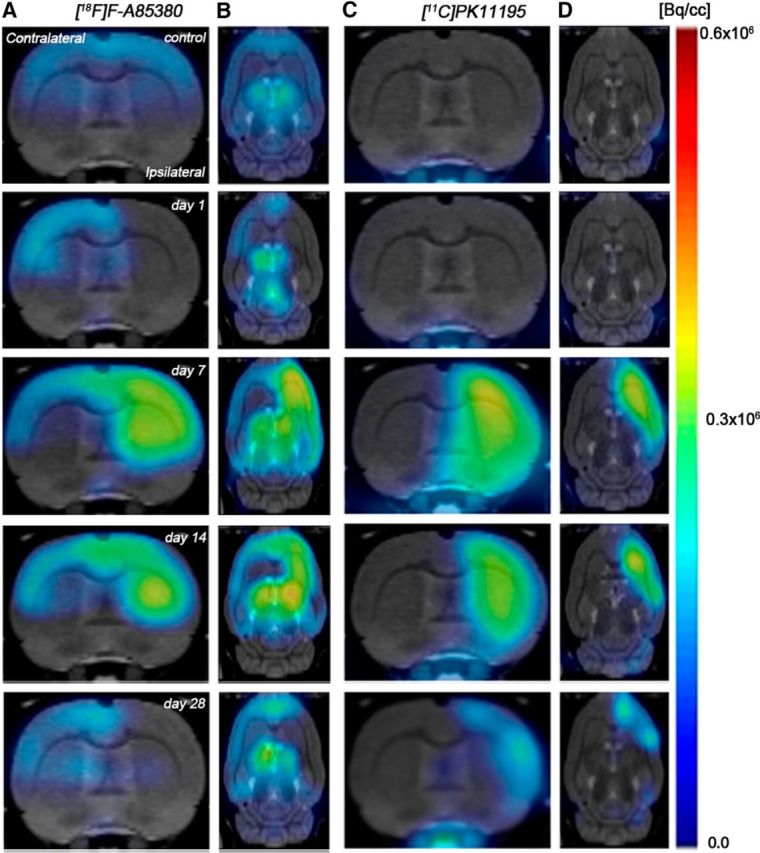
Serial images of [11C]PK11195 and 2-[18F]-fluoro-A85380 PET at day 0 (control), days 1, 7, 14, and 28 after MCAO. Normalized coronal and axial PET images of [11C]PK11195 (A, B) and 2-[18F]-fluoro-A85380 (C, D) signals before and after cerebral ischemia are coregistered with a MRI (T2W) rat template to localize anatomically the PET signal from left to right. Images correspond to the same representative animal for each time and condition and radiotracer.
2-[18F]-fluoro-A85380 PET after cerebral ischemia
The time course of nicotinic receptor α4β2 was evaluated using 2-[18F]-fluoro-A85380 in both the ipsilateral and contralateral cerebral cortex, striatum, thalamus, and whole brain at day 0 (control) and 1, 3, 7, 14, 21, and 28 d after MCAO (Fig. 2, n = 6). All studied regions showed a similar 2-[18F]-fluoro-A85380-binding evolution after long-term focal cerebral ischemia. In the ipsilateral whole brain (cerebrum), the binding values for 2-[18F]-fluoro-A85380 presented a slight nonsignificant decrease at day 1, followed by a progressive PET signal increase later on. In fact, the highest binding value was reached at 7 d after reperfusion compared with day 0 (control) values (F(6,41) = 3.42, p < 0.05; Fig. 2A). Subsequently, the PET signal showed a progressive decrease from days 14–28 compared with day 7. In the contralateral whole brain, 2-[18F]-fluoro-A85380 PET binding reached pseudocontrol values during the first 3 d, followed by a nonstatistically mild increase at day 7 compared with control values. This was followed by a progressive decline on days 14–28 after cerebral ischemia (Fig. 2B). The cerebral cortex in the ipsilateral hemisphere showed a statistically increase of 2-[18F]-fluoro-A85380 PET signal at day 7 in relation to control, followed by a progressive decrease from day 14 on (F(6,41) = 4.30, p < 0.05; Fig. 2C). In contrast, nonstatistically significant differences were observed in the contralateral hemisphere despite the weak increase of PET signal observed from day 7 to day 14 after reperfusion (Fig. 2D). In the ipsilateral (occluded MCA) striatum, PET-binding levels found from day 7 to day 21 after reperfusion were higher than those observed in the cerebral cortex, showing an increase of nicotinic receptor expression under this neuropathologic situation (F(6,41) = 12.54, p < 0.01; p < 0.05 with respect to control animals; Fig. 2E). In addition, contralateral striatum exhibited lower 2-[18F]-fluoro-A85380 PET-binding levels than that observed in the cerebral cortex and thalamus, showing a lower density of nicotinic α4β2 receptors in the nonischemic striatum in relation to other brain regions (Fig. 2F). Likewise, thalamus at day 0 (control) showed the highest 2-[18F]-fluoro-A85380-binding potential values (Fig. 2G,H) compared with other rat brain regions evaluated at the same time point (F(6,41) = 2.30, p < 0.001; p < 0.01 with respect to striatum and cortex, data not shown). These findings are consistent with the following rank order: thalamus > cortex > striatum of the nicotinic α4β2 receptors distribution in rodent brain (Nirogi et al., 2012). Moreover, the nicotinic PET values increased in both ipsilateral (F(6,41) = 2.30, p < 0.05, with respect to day 0; Fig. 2G) and contralateral (F(6,41) = 1.74, p < 0.05, with respect to day 0; Fig. 2H) thalamus after reperfusion, which is consistent with the other cerebral regions evaluated in the present study.
Figure 2.
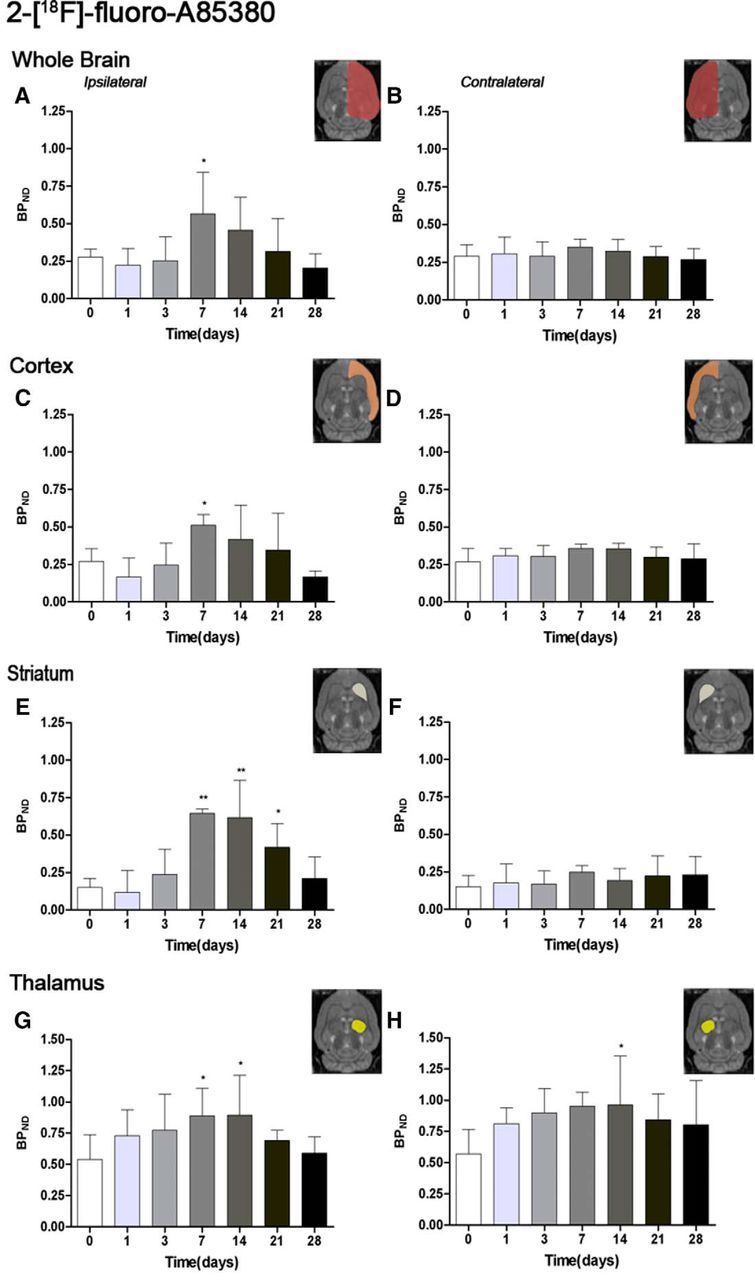
Time course of the progression of the 2-[18F]-fluoro-A85380 PET signal before and after cerebral ischemia. BPND (mean ± SD) of 2-[18F]-fluoro-A85380 was quantified in eight VOIs. The entire ipsilateral cerebral hemisphere (A), contralateral cerebral hemisphere (B), ipsilateral cortex (C), contralateral cortex (D), ipsilateral striatum (E), contralateral striatum (F), ipsilateral thalamus (G), and contralateral thalamus (H) are shown. The upper right panels of each figure show the selected brain ROIs for the quantification defined on a slice of a MRI (T2W) template. Rats (n = 6) were repeatedly examined by PET before (day 0) and at 1, 3, 7, 14, 21, and 28 d after ischemia. *p < 0.05 and **p < 0.01 compared with control.
[11C]PK11195 PET after cerebral ischemia
The time course of TSPO receptor was evaluated using [11C]PK11195 in both the ipsilateral and contralateral cortex, striatum, thalamus, and whole brain at day 0 (control) and 1, 3, 7, 14, 21, and 28 d after MCAO (Fig. 3, n = 6). All studied regions exhibited a similar [11C]PK11195-binding evolution after long-term focal cerebral ischemia. The intact or noninflamed cerebral tissue shows a very low TSPO receptor expression, followed by a dramatic upregulation due to the inflammatory reaction, suggesting its usefulness in imaging neuroinflammatory processes (Chen and Guilarte, 2008). This fact has been supported by the very low [11C]PK11195-binding level observed at control and at day 1 after cerebral ischemia. Likewise, all studied brain regions have exhibited a similar TSPO overexpression after cerebral ischemia (Fig. 3). The ischemic whole-brain hemisphere presented a dramatic increase of PET [11C]PK11195 values at day 3, followed by the highest increase at day 7 and a subsequently progressive decrease from days 14–28 after MCAO (F(6,41) = 20.07, p < 0.05; p < 0.01with respect to the control animals; Fig. 3A). In the contralateral hemisphere, the PET signal showed a nonstatistically significant increase the first and second week after reperfusion (Fig. 3B). Cerebral cortex showed a very similar binding uptake as that observed in the whole brain over the following month after cerebral ischemia (F(6,41) = 15.40, p < 0.05; p < 0.01with respect to the control animals; Figs. 3C,D). In the ipsilateral striatum, BPND increased sharply from day 0–3, peaking at day 7 and followed by a progressive decrease from day 14–28 after stroke (F(6,41) = 24.86, p < 0.05; p < 0.01 vs control values; Fig. 3E). Likewise, striatum experienced the highest [11C]PK11195-binding uptake with respect to the other cerebral regions evaluated, showing a greater increase of TSPO expression in this region after MCAO in rats. The nonischemic striatum showed a weak nonstatistically significant increase from days 7–14 compared with values at day 0 (Fig. 3F). Thalamic regions also suffered an increase of inflammatory reaction underlying cerebral ischemia. The ipsilateral thalamic region showed a significant increase of the PET signal in the ipsilateral thalamus at days 7, 14, and 21 in relation to the control animals (F(6,41) = 7.40, p < 0.01, p < 0.05; Fig. 3G). Despite this, a lower increase of the PET [11C]PK11195 signal was observed in the ipsilateral thalamus in relation to areas irrigated by the middle cerebral artery, such as as cortex and striatum. Likewise, PET with [11C]PK11195 displayed a nonsignificant increase of the inflammatory process in the contralateral thalamus at days 7 and 14 after ischemia (Fig. 3H).
Figure 3.
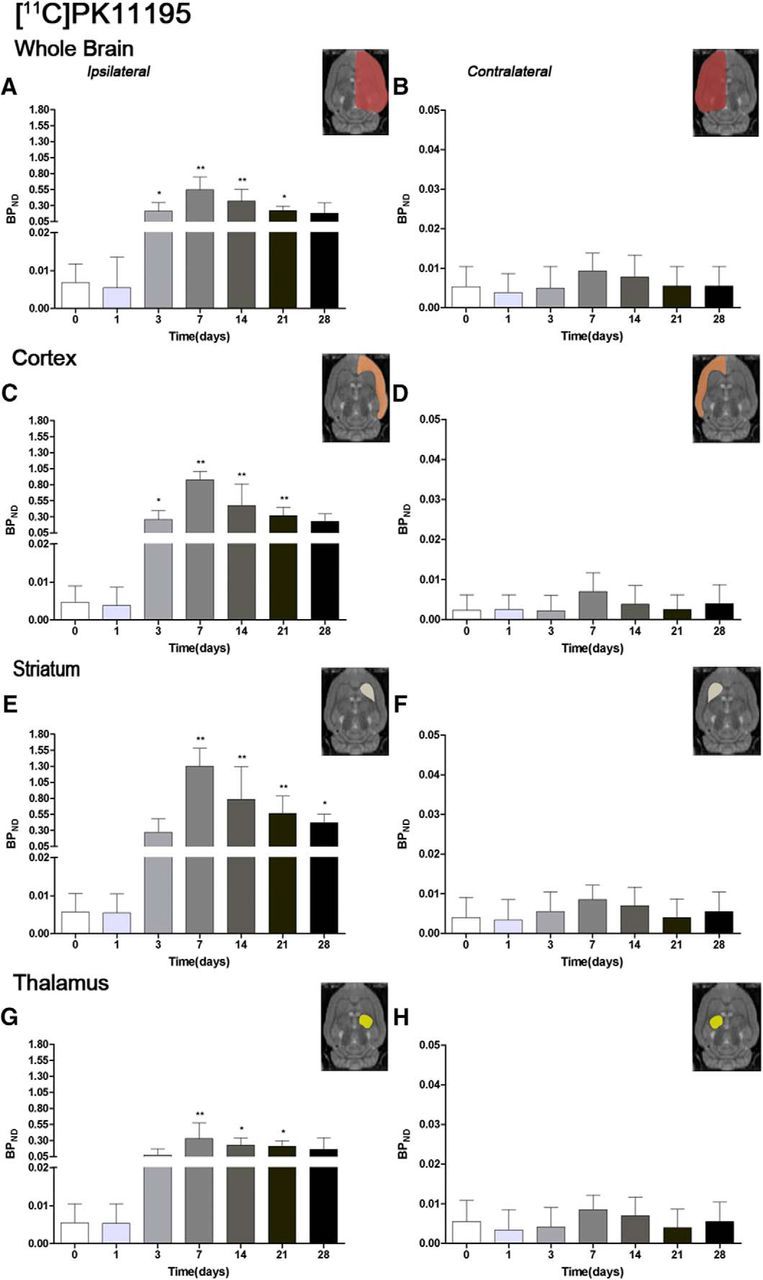
Time course of the progression of the [11C]PK11195 PET signal before and after cerebral ischemia. The BPND (mean ± SD) of [11C]PK11195 was quantified in eight VOIs. The entire ipsilateral cerebral hemisphere (A), contralateral cerebral hemisphere (B), ipsilateral cortex (C), contralateral cortex (D), ipsilateral striatum (E), contralateral striatum (F), ipsilateral thalamus (G), and contralateral thalamus (H) are shown. The upper right panels of each figure show the selected brain ROIs for the quantification defined on a slice of a MRI (T2W) template. Rats (n = 6) were repeatedly examined by PET before (day 0) and at 1, 3, 7, 14, 21, and 28 d after ischemia. *p < 0.05 and **p < 0.01 compared with control.
Expression of α4β2 and TSPO after ischemia
Immunofluorescence staining exhibited α4β2 and TSPO expression in two glial subpopulations, microglia and astrocytes after ischemia (Fig. 4, n = 3 (1 rat per time point considered)). At day 7, cells with the morphology of amoeboid-reactive microglia/macrophages showed intense CD11b immunoreactivity in the lesion (Fig. 4B,H, green), followed by a decrease at day 28 (Fig. 4C,I, green). The overreactivity of microglia colocalized with the cellular expression of both nicotinic α4β2 (Fig. 4B,C, blue and green) and TSPO (Fig. 4H,I, red and green) at days 7 and 28 after ischemia. Likewise, astrocytes displayed an increase of the GFAP immunoreactivity from days 7 (Fig. 4E,K, white) to 28 (Fig. 4F,L, white) after ischemia, forming a thin astrocytic rim in the vicinity of the lesion. In addition, reactive astrocytes showed colocalization with nicotinic α4β2 expression (Fig. 4E,F, blue and white) and TSPO (Fig. 4K,L, red and white) after cerebral ischemia (7 and 28 d).
Figure 4.

Immunofluorescent labeling of α4β2 (blue), CD11b (green), GFAP (white), and TSPO (red) in the ischemic area. The data show temporal evolution of both α4β2 and TSPO expression in microglial and astrocytic cells at day 0 (control, n = 1, left column), day 7 (n = 1, middle column), and day 28 (n = 1, right column) after cerebral ischemia. A–C, G–I, Number of CD11b-reactive microglia/macrophages (arrowheads) increase at 7 d. D–F, J–L, Number of GFAP-positive astrocytes (arrows) increase in the ischemic area over time. α4β2 (A–F) and TSPO (G–L) immunoreactivities colocalize with the temporal activation of CD11b and GFAP after cerebral ischemia. Scale bars, 5 μm.
Cellular expression and immunoreactivity of nicotinic receptors in microglia and astrocytes
Immunohistochemistry for microglia and astrocytes illustrated the expression of nicotinic α4β2 receptor in glial suptypes after cerebral ischemia (Fig. 5A) time course of α4β2+/CD11b+ microglia/macrophages and α4β2+/GFAP+ astrocytes (n = 15, 5 rats per time point considered) and microglial and astrocytic/α4β2 receptor signal intensity (n = 12, 4 rats per time point; 3 rats were discarded from the analysis because of technical problems) was performed in the ischemic lesion before (day 0) and at days 7 and 28 after ischemia. An overall increase in α4β2+/CD11b+ cells and intensity of microglial/α4β2 receptors was found at day 7, followed by a sharp decrease at day 28 (F(2,14) = 67.27, p < 0.01 with respect to day 0; Fig. 5A; F(2,11) = 125.2, p < 0.01; p < 0.05 with respect to day 0; Fig. 5B). In addition, both the number of α4β2+/GFAP+ cells and the signal intensity of astrocytic/α4β2 receptors showed a progressive increase from day 7 to day 28 (F(2,14) = 120.7, p < 0.05 with respect to day 0; Fig. 5C; F(2,11) = 10.17, p < 0.01 with respect to day 0; Fig. 5D).
Figure 5.

Temporal profiles of α4β2+/CD11b+ microglial cells (A), microglial/α4β2 immunoreactivity (B), α4β2+/GFAP+ astrocytic cells (C), and astrocytic/α4β2 immunoreactivity (D) at day 0, 7, and 28 after cerebral ischemia. α4β2+/CD11b+ cells increase versus control at day 7 followed by a dramatic decline at day 28 (A, n = 15, 5 rats/time point). Intensity of microglial/α4β2 receptors increases versus control at day 7, followed by a dramatic decline at day 28 (B, n = 12, 4 rats/time point). The number of α4β2+/GFAP+ cells increases versus control at days 7 and 28 after ischemia (C, n = 15, 5 rats/time point). Finally, the intensity of astrocytic α4β2 receptors increases versus control at days 7 and 28 (D, n = 12, 4 rats/time point). *p < 0.05 and **p < 0.01 compared with control.
Effect of DHβE on neuroinflammation after MCAO
The levels and distribution of TSPO receptors were explored by PET imaging after the chronic treatment with the α4 receptor antagonist DHβE and vehicle 7 d after MCAO (Fig. 6, n = 14). All images were quantified in standard units, BPND. The images with normalized color scale illustrate the evolution of the [11C]PK11195 PET signal in control (Fig. 6A, n = 8) and DHβE-treated (Fig. 6B, n = 6) ischemic rats. DHβE treatment caused a significant increase of [11C]PK11195 binding in the treated ischemic cerebral hemisphere in relation to nontreated ischemic control rats (t(12) = 2.20, p < 0.05; Fig. 6C).
Figure 6.
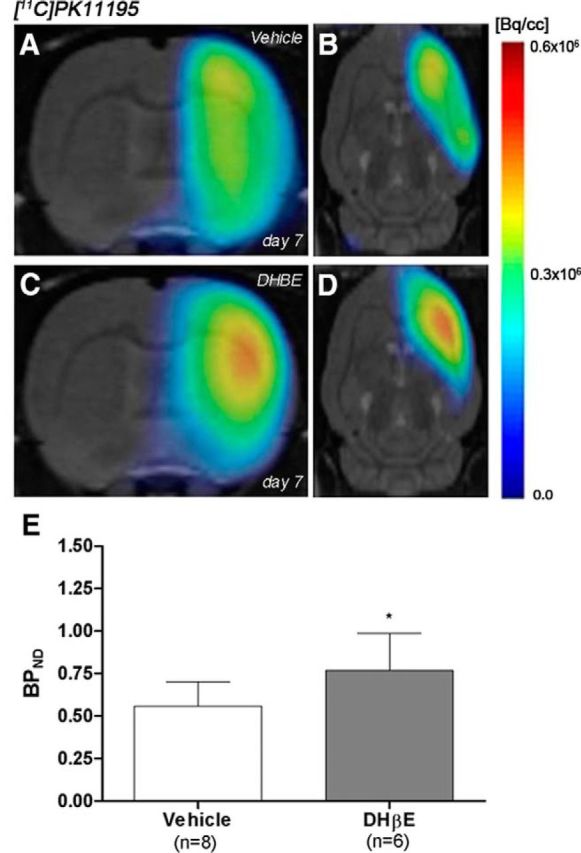
Normalized coronal and axial PET images of [11C]PK11195 for control (A, B) and DHβE-treated (C, D) rats at day 7 after cerebral ischemia are coregistered with a MRI (T2W) rat template to localize anatomically the PET signal from left to right. The BPND (mean ± SD) of [11C]PK11195 was quantified in the entire ipsilateral cerebral hemisphere. Vehicle (n = 8) and DHβE-treated (n = 6) rats were examined by PET 7 d after ischemia (E). *p < 0.05 compared with control.
Time course neurologic score and behavioral test after cerebral ischemia
Ischemic animals (n = 6) presented a major impairment from days 1–3 after MCAO in relation to day 0 (control). The neurologic impairment showed a significant increase versus that in the control at days 1, 3, 7, 14, 21, and 28 (t(10) = 13.86, p < 0.01). After day 3, rats exhibited a trend to a progressive functional recovery over time at days 7, 14, 21, and 28 (t(12) = 13.86, p < 0.01) in relation to day 1 after ischemia (Fig. 7A). These results showed a neurologic recovery of the animals over time after cerebral ischemia. After the neurologic score, the animals were subjected to the adhesion/removal tape test. Animals showed a worst taping contact (Fig. 7B) and remove (Fig. 7C) performances of the contralateral in relation to the ipsilateral forepaw. Statistical analyses using two-way ANOVA by forepaw (ipsilateral and contralateral) and time followed by the Bonferroni test showed a significant increase in contact time in the contralateral forepaw at 1, 3, and 7 d (F(6,83) = 4.32, p < 0.05) with respect to corresponding times in the ipsilateral forepaw (Fig. 7B). Statistical analysis by using Mann–Whitney U tests within the contralateral forelimb demonstrated a worst contact performance at 1 and 3 d (t(10) = 2.02, p < 0.001), followed by an improvement of paw and mouth sensitivity from days 7–28 (t(10) = 2.02, p < 0.01) with respect to day 0 (Fig. 7B). In the ipsilateral forepaw, ischemic animals exhibited an increase in the time to contact at days 1 and 3 (t(10) = 44.58, p < 0.01 vs control values), followed by a behavioral recovery from day 7 on (Fig. 7B). Statistical analyses using two-way ANOVA show a significant increase in remove time in the contralateral forepaw at day 7 (F(6,83) = 4.43, p < 0.05) with respect to corresponding times in the ipsilateral forepaw (Fig. 7C). Ischemic animals showed the worst performance of the taping remove test from day 1 to day 3 (t(10) = 57.18, p < 0.001), followed by a dexterity recovery 7–28 d after ischemia (t(10) = 57.18, p < 0.01) in the contralateral forepaw. Subsequently, the ipsilateral forepaw showed the uppermost time to remove at days 1 and 3 (t(10) = 2.70, p < 0.01) in relation to the day 0, followed by a fast improvement from day 7 to day 28 after MCAO.
Figure 7.
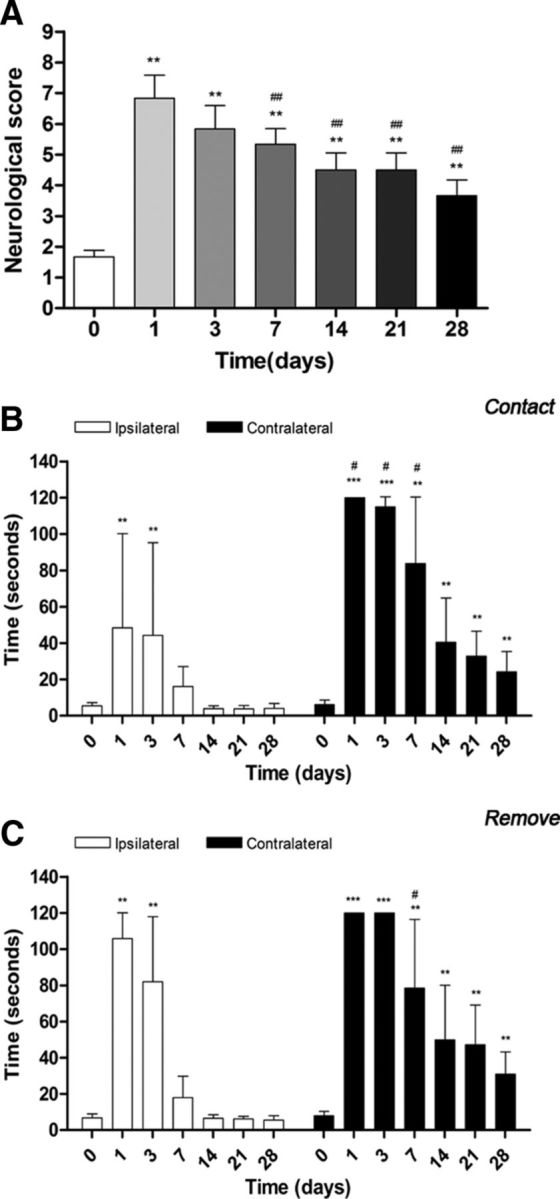
Neurologic and behavioral outcomes before (day 0) and 1, 3, 7, 14, 21, and 28 d after cerebral ischemia. The neurologic score (A), the time to contact (B), and the time to remove (C) tape tests showed an improvement over time. For A, **p < 0.01 compared with control; ##p < 0.01 compared with day 1. For B and C, #p < 0.05 compared with corresponding time to ipsilateral and **p < 0.01 and ***p < 0.001 compared with control.
Discussion
PET imaging of TSPO with [11C]PK11195
After brain injury, glial cells (microglia/macrophages and astrocytes) become reactive, overexpressing TSPO receptors in response to the inflammatory environment (Rojas et al., 2007, Martín et al., 2010, Winkeler et al., 2010). For this reason, these receptors have become a potential target for the development of radioligands to study the neuroinflammatory processes with PET (Chauveau et al., 2008, Damont et al., 2013). [11C]PK11195, a selective TSPO radioligand, has been widely used to evaluate the expression of this receptor in rodents (Rojas et al., 2007, Schroeter et al., 2009, Hughes et al., 2012) and in human brain (Pappata et al., 2000, Gerhard et al., 2005, Price et al., 2006) after cerebral ischemia. In addition, [11C]PK11195 has been used extensively to study such neurodegenerative diseases as multiple sclerosis (Banati et al., 2000), AD (Cagnin et al., 2001), and PD (Bartels and Leenders, 2007). Recently, an impressive number of selective radioligands for TSPO have been generated (Damont et al., 2013), limiting research on neuroinflammation by PET imaging almost exclusively to the study of this receptor. Therefore, the identification of new upregulated receptors in reactive glial cells as nAChRs is of great interest to the in vivo imaging research on neuroinflammation. Because of this, we have assessed in parallel the in vivo expression of both nAChRs (α4β2) and TSPO in reactive glial cells using PET-imaging procedures in combination with immunochemistry and neurofunctional evaluation after long-term transient MCAO in rats.
PET imaging of α4β2 with 2[18F]-fluoro-A85380
Previous in vivo PET studies performed with 2[18F]-fluoro-A85380 have shown a good penetration in the rat brain (Valette et al., 1999). In the present study, the PET 2[18F]-fluoro-A85380-binding uptake in the healthy rat brain was mainly concentrated in the cerebral cortex, striatum, and thalamus (Figs. 1; 2), which was consistent with the well known distribution of α4β2 in both human and rat healthy brains (Kimes et al., 2003, Nirogi et al., 2012). In the pathologic brain, 2[18F]-fluoro-A85380 has been used to determine the degeneration of neural cells containing nAChRs after neurodegenerative diseases such as AD and PD (Schmaljohann et al., 2004, Schmaljohann et al., 2006). Nevertheless, nAChRs are not only expressed in neurons, but also in microglia/macrophages, and the modulation of these receptors might be related to different inflammatory responses (Wang et al., 2003, Shytle et al., 2004). Despite this, the present study is the first time that 2[18F]-fluoro-A85380 has been evaluated as a potential biomarker for in vivo PET imaging of neuroinflammation.
In vivo and ex vivo evidence of α4β2 receptor overexpression after ischemia
After cerebral ischemia, 2[18F]-fluoro-A85380 binding increased in different brain regions in the injured hemisphere. The increase of α4β2 receptor availability ran in parallel with the [11C]PK11195 binding over the following month to ischemia onset (Fig. 1), showing a hypothetical role of α4β2 on neuroinflammation after stroke. One day after MCAO, 2[18F]-fluoro-A85380 binding experienced a slight decline due to either the decrease of the blood flow and perfusion (Martín et al., 2012a, Martín et al., 2012b) or the loss of nicotinic neurons as a consequence of the ischemic process (Fig. 2). This decrease was followed by a parallel increase of 2[18F]-fluoro-A85380 and [11C]PK11195 binding from day 3 in the different brain regions considered, which peaked at day 7 in the ipsilateral hemisphere and showed a progressive decline to pseudocontrol values from day 14 to day 28 after ischemia onset (Fig. 2; 3). These results are in agreement with Martín et al. (2010), who showed, after monitoring the in vivo postischemic TSPO receptor expression by PET, a gradual increase of TSPO receptor availability from days 4–7, which peaked at day 11 and was followed by a slight decline afterward. In our study, the same ROIs used to calculate the PET signal in the ipsilateral hemisphere were used to study PET binding in the nonischemic side. PET binding for both radiotracers in the contralateral hemisphere showed a nonsignificant increase over the following week after MCAO onset. A similar increase of TSPO expression has been observed at day 11 after cerebral ischemia with [18F]DPA-714-PET in rats (Martín et al., 2010). Therefore, in vivo PET imaging has showed a large concordance between [11C]PK11195 and 2[18F]-fluoro-A85380 binding uptake profiles, showing an overexpression of α4β2 receptors in the inflammatory brain regions after long-term cerebral ischemia in rats. (Figs. 1, 2, 3).
To confirm these findings, both α4β2 and TSPO expression were characterized by ex vivo immunohistochemistry at days 1, 7, and 28 after reperfusion (Fig. 4). These results verified the expression of α4β2 receptors in both microglia/macrophages and astrocytes after cerebral ischemia that ran in parallel with the expression pattern showed by TSPO. In addition, immunohistochemistry showed an overall increase of α4β2+/CD11b+ cells and microglial/α4β2 receptors immunoreactivity at day 7, followed by a dramatic decrease at day 28 after cerebral ischemia. Likewise, both α4β2+/GFAP+ cells and astrocytic/α4β2 receptors signal intensity experienced a progressive increase of reactive atrocytic cells from day 7 on (Fig. 5). Therefore, the present results support the previous PET imaging results obtained with 2[18F]-fluoro-A85380, showing that the α4β2 receptor overexpression observed at day 7 after MCAO was promoted by the α4β2 receptor expression increase by microglial and astrocytic cells. Despite these findings, little is known concerning the role of nicotinic receptors on inflammation underlying experimental ischemic stroke.
Modulation of the neuroinflammatory response using DHβE
The expression of nAChR α7 has been found in microglia/macrophages and its presence is required for acetylcholine inhibition of macrophage TNF release, becoming an essential key for the regulation of inflammation (Wang et al., 2003, Shytle et al., 2004). In addition, the stimulation of α4β2 receptors by nicotine has promoted inhibition of β-amyloid toxicity to cortical neurons and its activity has been reversed by the specific α4β2 nicotinic receptor antagonist DHβE (Kihara et al., 1998). For this reason, a daily treatment after cerebral ischemia with DHβE was performed to explore the effect of α4β2 receptors on the neuroinflammatory reaction using [11C]PK11195 PET (Fig. 6). DHβE-treated ischemic rats showed a significant increase of [11C]PK11195 PET binding in the ischemic hemisphere in relation to control ischemic rats, confirming the role exerted by α4β2 on inflammatory reaction after experimental ischemic stroke (Fig. 6). Likewise, these results stand in agreement with the anti-inflammatory and neuroprotective activity effects performed by such nicotinic receptor activators as nicotine and acetylcholinesterase inhibitors in animal models of cerebral ischemia (Chen et al., 2013), intracerebral hemorrhage (Hijioka et al., 2011), and neurodegeneration (Nizri et al., 2006, Park et al., 2007, Nizri et al., 2008).
Neurofunctional outcome after cerebral ischemia
The peak of the inflammatory reaction observed by PET imaging from day 7 to day 14 was consistent with the functional recovery observed in the ischemic rats. Both neurological and behavioral tests performed in the present study showed similar results during the first week after ischemia, followed by a gradual recovery of neurologic handicap and a faster improvement of contact/removal patch test from the second to the third week after reperfusion (Fig. 7). Therefore, animals experienced a quasi-recovery of functional outcome 1 month after stroke. Likewise, such capacity to undergo motor and cognitive recovery has been monitored in adult human brain after stroke (Rossini et al., 2003).
Summary and conclusions
We report here the PET imaging of 2[18F]-fluoro-A85380 to assess α4β2 bioavailability and its relationship with the neuroinflammatory response after long-term focal cerebral ischemia in rats. These results confirmed a binding increase of 2[18F]-fluoro-A85380 in the ipsilateral hemisphere during the first week after cerebral ischemia, followed by a progressive PET-binding decrease later on. These results are consistent with the binding profile of the radiotracer for neuroinflammation [11C]PK11195 after experimental stroke in rats. In addition, ex vivo immunohistochemistry showed for the first time an overexpression of the α4β2 in both microglia/macrophages and astrocytes, and treatment with DHBE was able to promote an increase of inflammatory reaction after MCAO. Therefore, these results provide novel information about the role of the α4β2 receptor on the inflammatory reaction underlying cerebral ischemia in rats. Finally, these findings might contribute to the discovery of novel biomarkers that might guide the synthesis of new imaging contrast agents and open new avenues into the diagnosis, evaluation, and therapy of all spectra of neurologic diseases.
Footnotes
We thank M. González, A. Leukona, M. Errasti, and A. Arrieta for technical support with radiosynthesis and technical assistance in the PET studies and the Department of Industry of the Basque Government for financial support.
The authors declare no competing financial interests.
References
- Banati RB, Newcombe J, Gunn RN, Cagnin A, Turkheimer F, Heppner F, Price G, Wegner F, Giovannoni G, Miller DH, Perkin GD, Smith T, Hewson AK, Bydder G, Kreutzberg GW, Jones T, Cuzner ML, Myers R. The peripheral benzodiazepine binding site in the brain in multiple sclerosis: quantitative in vivo imaging of microglia as a measure of disease activity. Brain. 2000;123:2321–2337. doi: 10.1093/brain/123.11.2321. [DOI] [PubMed] [Google Scholar]
- Bartels AL, Leenders KL. Neuroinflammation in the pathophysiology of Parkinson's disease: evidence from animal models to human in vivo studies with [11C]-PK11195 PET. Mov Disord. 2007;22:1852–1856. doi: 10.1002/mds.21552. [DOI] [PubMed] [Google Scholar]
- Bottlaender M, Valette H, Roumenov D, Dollé F, Coulon C, Ottaviani M, Hinnen F, Ricard M. Biodistribution and radiation dosimetry of 18F-fluoro-A-85380 in healthy volunteers. J Nucl Med. 2003;44:596–601. [PubMed] [Google Scholar]
- Bouet V, Boulouard M, Toutain J, Divoux D, Bernaudin M, Schumann-Bard P, Freret T. The adhesive removal test: a sensitive method to assess sensorimotor deficits in mice. Nat Protoc. 2009;4:1560–1564. doi: 10.1038/nprot.2009.125. [DOI] [PubMed] [Google Scholar]
- Burghaus L, Schütz U, Krempel U, de Vos RA, Jansen Steur EN, Wevers A, Lindstrom J, Schröder H. Quantitative assessment of nicotinic acetylcholine receptor proteins in the cerebral cortex of Alzheimer patients. Brain Res Mol Brain Res. 2000;76:385–388. doi: 10.1016/S0169-328X(00)00031-0. [DOI] [PubMed] [Google Scholar]
- Cagnin A, Brooks DJ, Kennedy AM, Gunn RN, Myers R, Turkheimer FE, Jones T, Banati RB. In-vivo measurement of activated microglia in dementia. Lancet. 2001;358:461–467. doi: 10.1016/S0140-6736(01)05625-2. [DOI] [PubMed] [Google Scholar]
- Chauveau F, Boutin H, Van Camp N, Dollé F, Tavitian B. Nuclear imaging of neuroinflammation: a comprehensive review of [11C]PK11195 challengers. Eur J Nucl Med Mol Imaging. 2008;35:2304–2319. doi: 10.1007/s00259-008-0908-9. [DOI] [PubMed] [Google Scholar]
- Chen MK, Guilarte TR. Translocator protein 18 kDa (TSPO): molecular sensor of brain injury and repair. Pharmacol Ther. 2008;118:1–17. doi: 10.1016/j.pharmthera.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Nie H, Tian L, Tong L, Yang L, Lao N, Dong H, Sang H, Xiong L. Nicotine-induced neuroprotection against ischemic injury involves activation of endocannabinoid system in rats. Neurochem Res. 2013;38:364–370. doi: 10.1007/s11064-012-0927-6. [DOI] [PubMed] [Google Scholar]
- Damont A, Roeda D, Dollé F. The potential of carbon-11 and fluorine-18 chemistry: illustration through the development of positron emission tomography radioligands targeting the translocator protein 18 kDa. J Labelled Comp Radiopharm. 2013;56:96–104. doi: 10.1002/jlcr.2992. [DOI] [PubMed] [Google Scholar]
- Ding YS, Fowler JS, Logan J, Wang GJ, Telang F, Garza V, Biegon A, Pareto D, Rooney W, Shea C, Alexoff D, Volkow ND, Vocci F. 6-[18F]Fluoro-A-85380, a new PET tracer for the nicotinic acetylcholine receptor: studies in the human brain and in vivo demonstration of specific binding in white matter. Synapse. 2004;53:184–189. doi: 10.1002/syn.20051. [DOI] [PubMed] [Google Scholar]
- Furukawa S, Sameshima H, Yang L, Ikenoue T. Activation of acetylcholine receptors and microglia in hypoxic-ischemic brain damage in newborn rats. Brain Dev. 2013;35:607–613. doi: 10.1016/j.braindev.2012.10.006. [DOI] [PubMed] [Google Scholar]
- Gerhard A, Schwarz J, Myers R, Wise R, Banati RB. Evolution of microglial activation in patients after ischemic stroke: a [11C](R)-PK11195 PET study. Neuroimage. 2005;24:591–595. doi: 10.1016/j.neuroimage.2004.09.034. [DOI] [PubMed] [Google Scholar]
- Gotti C, Fornasari D, Clementi F. Human neuronal nicotinic receptors. Prog Neurobiol. 1997;53:199–237. doi: 10.1016/S0301-0082(97)00034-8. [DOI] [PubMed] [Google Scholar]
- Guan ZZ, Nordberg A, Mousavi M, Rinne JO, Hellström-Lindahl E. Selective changes in the levels of nicotinic acetylcholine receptor protein and of corresponding mRNA species in the brains of patients with Parkinson's disease. Brain Res. 2002;956:358–366. doi: 10.1016/S0006-8993(02)03571-0. [DOI] [PubMed] [Google Scholar]
- Hijioka M, Matsushita H, Hisatsune A, Isohama Y, Katsuki H. Therapeutic effect of nicotine in a mouse model of intracerebral hemorrhage. J Pharmacol Exp Ther. 2011;338:741–749. doi: 10.1124/jpet.111.182519. [DOI] [PubMed] [Google Scholar]
- Hogg RC, Raggenbass M, Bertrand D. Nicotinic acetylcholine receptors: from structure to brain function. Rev Physiol Biochem Pharmacol. 2003;147:1–46. doi: 10.1007/s10254-003-0005-1. [DOI] [PubMed] [Google Scholar]
- Hughes JL, Jones PS, Beech JS, Wang D, Menon DK, Aigbirhio FI, Fryer TD, Baron JC. A microPET study of the regional distribution of [11C]-PK11195 binding following temporary focal cerebral ischemia in the rat. Correlation with post mortem mapping of microglia activation. Neuroimage. 2012;59:2007–2016. doi: 10.1016/j.neuroimage.2011.10.060. [DOI] [PubMed] [Google Scholar]
- Jo S, Yarishkin O, Hwang YJ, Chun YE, Park M, Woo DH, Bae JY, Kim T, Lee J, Chun H, Park HJ, Lee da Y, Hong J, Kim HY, Oh SJ, Park SJ, Lee H, Yoon BE, Kim Y, Jeong Y, Shim I, Bae YC, Cho J, Kowall NW, Ryu H, Hwang E, Kim D, Lee CJ. GABA from reactive astrocytes impairs memory in mouse models of Alzheimer's disease. Nat Med. 2014;20:886–896. doi: 10.1038/nm.3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justicia C, Pérez-Asensio FJ, Burguete MC, Salom JB, Planas AM. Administration of transforming growth factor-alpha reduces infarct volume after transient focal cerebral ischemia in the rat. J Cereb Blood Flow Metab. 2001;21:1097–1104. doi: 10.1097/00004647-200109000-00007. [DOI] [PubMed] [Google Scholar]
- Kihara T, Shimohama S, Urushitani M, Sawada H, Kimura J, Kume T, Maeda T, Akaike A. Stimulation of alpha4beta2 nicotinic acetylcholine receptors inhibits beta-amyloid toxicity. Brain Res. 1998;792:331–334. doi: 10.1016/S0006-8993(98)00138-3. [DOI] [PubMed] [Google Scholar]
- Kimes AS, Horti AG, London ED, Chefer SI, Contoreggi C, Ernst M, Friello P, Koren AO, Kurian V, Matochik JA, Pavlova O, Vaupel DB, Mukhin AG. 2-[18F]F-A-85380: PET imaging of brain nicotinic acetylcholine receptors and whole body distribution in humans. FASEB J. 2003;17:1331–1333. doi: 10.1096/fj.02-0492fje. [DOI] [PubMed] [Google Scholar]
- Lammertsma AA, Hume SP. Simplified reference tissue model for PET receptor studies. Neuroimage. 1996;4:153–158. doi: 10.1006/nimg.1996.0066. [DOI] [PubMed] [Google Scholar]
- Martín A, Boisgard R, Thézé B, Van Camp N, Kuhnast B, Damont A, Kassiou M, Dollé F, Tavitian B. Evaluation of the PBR/TSPO radioligand [(18)F]DPA-714 in a rat model of focal cerebral ischemia. J Cereb Blood Flow Metab. 2010;30:230–241. doi: 10.1038/jcbfm.2009.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martín A, Macé E, Boisgard R, Montaldo G, Thézé B, Tanter M, Tavitian B. Imaging of perfusion, angiogenesis, and tissue elasticity after stroke. J Cereb Blood Flow Metab. 2012a;32:1496–1507. doi: 10.1038/jcbfm.2012.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martín A, San Sebastián E, Gómez-Vallejo V, Llop J. Positron emission tomograghy with [(1)(3)N]ammonia evidences long-term cerebral hyperperfusion after 2h-transient focal ischemia. Neuroscience. 2012b;213:47–53. doi: 10.1016/j.neuroscience.2012.03.050. [DOI] [PubMed] [Google Scholar]
- Menzies SA, Hoff JT, Betz AL. Middle cerebral artery occlusion in rats: a neurological and pathological evaluation of a reproducible model. Neurosurgery. 1992;31:100–106. doi: 10.1227/00006123-199207000-00014. discussion 106–107. [DOI] [PubMed] [Google Scholar]
- Mudo G, Belluardo N, Fuxe K. Nicotinic receptor agonists as neuroprotective/neurotrophic drugs: progress in molecular mechanisms. J Neural Transm. 2007;114:135–147. doi: 10.1007/s00702-006-0561-z. [DOI] [PubMed] [Google Scholar]
- Mukhin AG, Gündisch D, Horti AG, Koren AO, Tamagnan G, Kimes AS, Chambers J, Vaupel DB, King SL, Picciotto MR, Innis RB, London ED. 5-Iodo-A-85380, an alpha4beta2 subtype-selective ligand for nicotinic acetylcholine receptors. Mol Pharmacol. 2000;57:642–649. doi: 10.1124/mol.57.3.642. [DOI] [PubMed] [Google Scholar]
- Nirogi R, Kandikere V, Bhyrapuneni G, Saralaya R, Muddana N, Ajjala DR. Rat thalamic alpha(4)beta(2) neuronal nicotinic acetylcholine receptor occupancy assay using LC-MS/MS. J Pharmacol Toxicol Methods. 2012;65:136–141. doi: 10.1016/j.vascn.2012.04.007. [DOI] [PubMed] [Google Scholar]
- Nizri E, Hamra-Amitay Y, Sicsic C, Lavon I, Brenner T. Anti-inflammatory properties of cholinergic up-regulation: A new role for acetylcholinesterase inhibitors. Neuropharmacology. 2006;50:540–547. doi: 10.1016/j.neuropharm.2005.10.013. [DOI] [PubMed] [Google Scholar]
- Nizri E, Irony-Tur-Sinai M, Faranesh N, Lavon I, Lavi E, Weinstock M, Brenner T. Suppression of neuroinflammation and immunomodulation by the acetylcholinesterase inhibitor rivastigmine. J Neuroimmunol. 2008;203:12–22. doi: 10.1016/j.jneuroim.2008.06.018. [DOI] [PubMed] [Google Scholar]
- O'Neill MJ, Murray TK, Lakics V, Visanji NP, Duty S. The role of neuronal nicotinic acetylcholine receptors in acute and chronic neurodegeneration. Curr Drug Targets CNS Neurol Disord. 2002;1:399–411. doi: 10.2174/1568007023339166. [DOI] [PubMed] [Google Scholar]
- Pappata S, Levasseur M, Gunn RN, Myers R, Crouzel C, Syrota A, Jones T, Kreutzberg GW, Banati RB. Thalamic microglial activation in ischemic stroke detected in vivo by PET and [11C]PK1195. Neurology. 2000;55:1052–1054. doi: 10.1212/WNL.55.7.1052. [DOI] [PubMed] [Google Scholar]
- Park HJ, Lee PH, Ahn YW, Choi YJ, Lee G, Lee DY, Chung ES, Jin BK. Neuroprotective effect of nicotine on dopaminergic neurons by anti-inflammatory action. Eur J Neurosci. 2007;26:79–89. doi: 10.1111/j.1460-9568.2007.05636.x. [DOI] [PubMed] [Google Scholar]
- Price CJ, Wang D, Menon DK, Guadagno JV, Cleij M, Fryer T, Aigbirhio F, Baron JC, Warburton EA. Intrinsic activated microglia map to the peri-infarct zone in the subacute phase of ischemic stroke. Stroke. 2006;37:1749–1753. doi: 10.1161/01.STR.0000226980.95389.0b. [DOI] [PubMed] [Google Scholar]
- Rojas S, Martín A, Arranz MJ, Pareto D, Purroy J, Verdaguer E, Llop J, Gómez V, Gispert JD, Millán O, Chamorro A, Planas AM. Imaging brain inflammation with [(11)C]PK11195 by PET and induction of the peripheral-type benzodiazepine receptor after transient focal ischemia in rats. J Cereb Blood Flow Metab. 2007;27:1975–1986. doi: 10.1038/sj.jcbfm.9600500. [DOI] [PubMed] [Google Scholar]
- Rossini PM, Calautti C, Pauri F, Baron JC. Post-stroke plastic reorganisation in the adult brain. Lancet Neurol. 2003;2:493–502. doi: 10.1016/S1474-4422(03)00485-X. [DOI] [PubMed] [Google Scholar]
- Schmaljohann J, Minnerop M, Karwath P, Gündisch D, Falkai P, Guhlke S, Wüllner U. Imaging of central nAChReceptors with 2-[18F]F-A85380: optimized synthesis and in vitro evaluation in Alzheimer's disease. Appl Radiat Isot. 2004;61:1235–1240. doi: 10.1016/j.apradiso.2004.02.026. [DOI] [PubMed] [Google Scholar]
- Schmaljohann J, Gündisch D, Minnerop M, Bucerius J, Joe A, Reinhardt M, Guhlke S, Biersack HJ, Wüllner U. In vitro evaluation of nicotinic acetylcholine receptors with 2-[18F]F-A85380 in Parkinson's disease. Nucl Med Biol. 2006;33:305–309. doi: 10.1016/j.nucmedbio.2005.12.012. [DOI] [PubMed] [Google Scholar]
- Schroeter M, Dennin MA, Walberer M, Backes H, Neumaier B, Fink GR, Graf R. Neuroinflammation extends brain tissue at risk to vital peri-infarct tissue: a double tracer [11C]PK11195- and [18F]FDG-PET study. J Cereb Blood Flow Metab. 2009;29:1216–1225. doi: 10.1038/jcbfm.2009.36. [DOI] [PubMed] [Google Scholar]
- Shimohama S, Greenwald DL, Shafron DH, Akaika A, Maeda T, Kaneko S, Kimura J, Simpkins CE, Day AL, Meyer EM. Nicotinic alpha 7 receptors protect against glutamate neurotoxicity and neuronal ischemic damage. Brain Res. 1998;779:359–363. doi: 10.1016/S0006-8993(97)00194-7. [DOI] [PubMed] [Google Scholar]
- Shytle RD, Mori T, Townsend K, Vendrame M, Sun N, Zeng J, Ehrhart J, Silver AA, Sanberg PR, Tan J. Cholinergic modulation of microglial activation by alpha 7 nicotinic receptors. J Neurochem. 2004;89:337–343. doi: 10.1046/j.1471-4159.2004.02347.x. [DOI] [PubMed] [Google Scholar]
- Valette H, Bottlaender M, Dollé F, Guenther I, Fuseau C, Coulon C, Ottaviani M, Crouzel C. Imaging central nicotinic acetylcholine receptors in baboons with [18F]fluoro-A-85380. J Nucl Med. 1999;40:1374–1380. [PubMed] [Google Scholar]
- Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, Li JH, Wang H, Yang H, Ulloa L, Al-Abed Y, Czura CJ, Tracey KJ. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature. 2003;421:384–388. doi: 10.1038/nature01339. [DOI] [PubMed] [Google Scholar]
- Winkeler A, Boisgard R, Martín A, Tavitian B. Radioisotopic imaging of neuroinflammation. J Nucl Med. 2010;51:1–4. doi: 10.2967/jnumed.109.065680. [DOI] [PubMed] [Google Scholar]
- Zanotti-Fregonara P, Maroy R, Peyronneau MA, Trebossen R, Bottlaender M. Minimally invasive input function for 2-18F-fluoro-A-85380 brain PET studies. Eur J Nucl Med Mol Imaging. 2012;39:651–659. doi: 10.1007/s00259-011-2004-9. [DOI] [PubMed] [Google Scholar]