

Abstract
Multiple small-molecule inhibitors of the β-secretase enzyme (BACE1) are under preclinical or clinical investigation for Alzheimer's disease (AD). Prior work has illustrated robust lowering of central amyloid β (Aβ) after acute administration of BACE1 inhibitors. However, very few studies have assessed the overall impact of chronically administered BACE1 inhibitors on brain amyloid burden, neuropathology, and behavioral function in aged preclinical models. We investigated the effects of a potent nonbrain-penetrant BACE1 inhibitor, delivered directly to the brain using intracerebroventricular infusion in an aged transgenic mouse model. Intracerebroventricular infusion of the BACE1 inhibitor (0.3–23.5 μg/d) for 8 weeks, initiated in 17-month-old Tg2576 mice, produced dose-dependent increases in brain inhibitor concentrations (0.2–13 μm). BACE1 inhibition significantly reversed the behavioral deficit in contextual fear conditioning, and reduced brain Aβ levels, plaque burden, and associated pathology (e.g., dystrophic neurites), with maximal effects attained with ∼1 μg/d dose. Strikingly, the BACE1 inhibitor also reversed amyloid pathology below baseline levels (amyloid burden at the start of treatment), without adversely affecting cerebral amyloid angiopathy, microhemorrhages, myelination, or neuromuscular function. Inhibitor-mediated decline in brain amyloid pathology was associated with an increase in microglial ramification. This is the first demonstration of chronically administered BACE1 inhibitor to activate microglia, reverse brain amyloid pathology, and elicit functional improvement in an aged transgenic mouse model. Thus, engagement of novel glial-mediated clearance mechanisms may drive disease-modifying therapeutic benefit with BACE1 inhibition in AD.
Introduction
Alzheimer's disease (AD) is a progressive neurodegenerative disorder characterized by a decline in cognitive and memory functions and by an aberrant accumulation of brain amyloid plaques derived from amyloid β (Aβ) peptides. Consistent with the hypothesis that accumulation of Aβ contributes to disease pathogenesis, Aβ-lowering agents and antibodies have presented promising therapeutic avenues for AD. Several late-stage trials with anti-Aβ antibodies and γ-secretase inhibitors recently failed to meet primary clinical endpoints due to lack of target engagement and/or side effects that prevented testing of therapeutically relevant doses (Aisen et al., 2013; Toyn and Ahlijanian, 2014). However, inhibitors of the β-secretase (BACE1) enzyme have shown robust lowering of CSF Aβ levels in young healthy subjects, and are in late-stage clinical trials for AD (Yan and Vassar, 2014), raising interest in elucidating underlying mechanisms of BACE1 inhibition in aging models.
BACE1 is a promising drug target, since mutations of the amyloid precursor protein (APP) that increase its cleavage by BACE1 increase the risk for AD, while others that inhibit cleavage by BACE1 significantly reduce the risk for AD and age-related cognitive decline (Vassar et al., 2009; Jonsson et al., 2012). BACE1 knock-outs rescue amyloid pathology and behavioral deficits in mutant APP-overexpressing mice (for review, see Vassar et al., 2009). Whereas altered processing of neuregulin and myelination are evident in young mice lacking BACE1 (Hu et al., 2006; Willem et al., 2006), they appear to be unaltered in adult BACE1 knock-out mice or with subchronic BACE1 inhibition in APP-overexpressing mice (Sankaranarayanan et al., 2008). BACE1 inhibitors can reduce brain Aβ levels, prevent amyloid pathology, or improve behavioral function when tested in young animals before development of extensive pathology (for review, see Ghosh et al., 2012). However, studies assessing prolonged exposures in aged preclinical models are necessary to predict treatment effects in clinical AD.
We sought to determine whether chronic exposure to a potent BACE1 inhibitor could affect brain amyloid pathology and behavioral deficit in aged Tg2576 mice, which have significant pathology before onset of treatment. Our data provide the first evidence for behavioral improvement, reduction in amyloid pathology below pretreatment levels, and engagement of microglia as a potential underlying mechanism following BACE1 inhibition in an aged APP transgenic mouse model. These findings offer novel insights supporting BACE1 inhibition as a viable therapeutic with potential for disease-modifying effects in AD.
Materials and Methods
Mice and experimental design.
The Animal Care and Use Committee at Medtronic's Physiological Research Laboratory, accredited by the Association for Assessment and Accreditation of Laboratory Animal Care, approved all procedures. Male Tg2576, overexpressing APPSWE, the Swedish β-site mutant form of APP, and corresponding wild-type (WT) mice (B6/SJL background; Taconic), aged 16.5–17.7 months, were uniformly distributed across treatment groups to implant osmotic minipumps (Alzet model 2006; Durect) for subcutaneous or intracerebroventricular infusion of the BACE1 inhibitor (compound C; in vitro IC50, 2–8 nm; Meredith et al., 2008).
Tg2576 mice received intracerebroventricular infusions of compound (0.3 μg/d, n = 12; 1.1 μg/d, n = 13; 23.5 μg/d, n = 12) or vehicle alone (n = 14). As previously described for intracerebroventricular infusion (Thakker et al., 2009), osmotic minipumps were connected via catheters to brain-infusion cannulae and placed subcutaneously, before stereotaxically implanting the cannulae on the skull. PBS, pH 7.2, provided the vehicle for 0.3 and 1.1 μg/d doses, while the 23.5 μg/d dose was formulated in 1:1 PEG300/DMSO solution due to limited solubility. The two intracerebroventricular vehicle groups revealed no significant differences for any study outcome, and were combined into a single vehicle group. Other transgenic mice received subcutaneous infusions of compound C (1.1 μg/d, n = 9) or PBS vehicle (n = 10). WT mice provided phenotype controls (intracerebroventricular vehicle, n = 10; intracerebroventricular compound C, 1.1 μg/d, n = 10; subcutaneous vehicle, n = 10; subcutaneous compound C, 1.1 μg/d, n = 10). Infusion of test articles, with endotoxin levels <0.03 endotoxin units/ml, was performed for 8 weeks, starting at age ∼17.2 months and terminating at ∼19.1 months.
At termination, CSF samples were withdrawn, and mice transcardially perfused with ice-cold saline to harvest tissue. Ipsilateral and contralateral brain hemispheres, relative to the infused right lateral ventricle, were randomly assigned for biochemical and histological analyses. Similar to prior work with larger molecules (Thakker et al., 2009), data from both pilot and present studies revealed no significant differences between the two brain hemispheres for any outcomes.
Behavioral assessment.
A week before termination, mice were tested for contextual and cued memory, as illustrated in Figure 1A and previously described (Thakker et al., 2009). Locomotor activity was assessed in a cubical arena (40 cm3) for ≤1 h using the Limelight video tracking system (Coulbourn Instruments). Grip strength for all limbs was measured using the grip strength meter (Columbus Instruments).
Figure 1.

Chronic intracerebroventricular infusion with BACE1 inhibitor, compound C, rescued behavioral deficit and reversed amyloid burden in aged Tg2576 mice. A, Fear-conditioning paradigm: mice were trained on day 1 in a specific context with a series of conditioning stimuli (tone cue followed by mild foot-shock). On day 2, freezing was evaluated in the same context (contextual fear memory), novel context (baseline), or cue alone (cued fear memory). B, Vehicle-treated Tg2576 mice show a deficit in contextual memory, compared with WT mice. A dose-dependent improvement in contextual fear memory was evident with intracerebroventricularly infused compound C. C, Low-magnification (×4) images illustrate amyloid plaques stained with Campbell–Switzer stain in cortex and hippocampus of Tg2576 mice at baseline and 8 weeks after intracerebroventricular infusion with vehicle or compound C. D, Percentage area occupied by amyloid plaques in vehicle-treated and compound C-treated mice. Significance versus vehicle-treated mice (**p < 0.01, ***p < 0.001) and versus baseline (#p < 0.05, ##p < 0.01, ###p < 0.001).
Histology.
Brain hemispheres were fixed in paraformaldehyde (4% in saline) and, using MultiBrain Technology (NeuroScience Associates), coronally sectioned at 35 μm. For each stain, we used a series of 18–20 sections, equally spaced at 350 μm intervals, across the entire cerebrum. Accordingly, analyses involved an average from multiple observations per hemisphere per animal, as follows: cortex (18–20), hippocampus (7–9), striatum (9–11), corpus callosum (14). We quantified parenchymal amyloid plaques, dense-core plaques, cerebral amyloid angiopathy (CAA), cerebral microhemorrhages, dystrophic neurites, and activated microglia, using protocols described previously (Thakker et al., 2009). We evaluated brain sAPPβ (secreted form of β-APP) using an antibody specific to the neoepitope ISEVNL of sAPPβ derived from BACE1 cleavage of APPSWE (1:1500, clone 6A1, IBL International; Fukumoto et al., 2010). Sciatic nerves were individually embedded in blocks of glycol methacrylate, coronally sectioned at 2 μm, and visualized with modified Lee's stain. G-ratios were quantified as a ratio of inner circumference of the axon (without myelin) over the outer circumference of the nerve fiber (including myelin), for ≥32 axons from four nerve segments collected per animal (Midwest ToxPath Sciences).
Brain extractions and Aβ ELISA.
Brain samples were extracted using 0.2% diethylamine for soluble Aβ (Sankaranarayanan et al., 2008), followed by 70% formic acid for insoluble Aβ as previously described (Kawarabayashi et al., 2001). We used previously published methods to measure brain compound levels (Meredith et al., 2008), and sandwich ELISAs to measure brain and CSF Aβ levels (Easton et al., 2013).
Statistical analyses.
All analyses were performed blind to treatment. An average score was used when two individuals scored the outcomes; the interscorer reliability ranged from 0.955 to 0.989 (p < 0.0001). Data are expressed as mean ± SEM. Statistical analyses used GraphPad Prism 4.0 (GraphPad) to perform one-way ANOVA followed by Tukey's post hoc test for all analyses, except microhemorrhage severity, which was analyzed using Kruskal–Wallis followed by Dunn's multiple-comparison test. Differences were considered significant at p < 0.05.
Results
Dose-dependent increases in compound levels within the brain parenchyma were observed in ∼19.1-month-old Tg2576 mice following intracerebroventricular infusion of the BACE1 inhibitor (compound C) over 8 weeks: 0.25 ± 0.06, 0.71 ± 0.26, and 13.21 ± 5.15 μm, for infusions maintained at 0.3, 1.1, and 23.5 μg/d, respectively. In contrast, brain exposures were negligible in the subcutaneous infusion group: 0.01 ± 0.01 μm with 1.1 μg/d infusion. Robust brain compound exposures with intracerebroventricular delivery allowed testing hypotheses on multiple functional, biochemical, and pathological endpoints following chronic BACE1 inhibition.
Aged Tg2576 mice exhibit substantial deficit in contextual fear conditioning compared with WT littermates (Jacobsen et al., 2006). Compound C elicited a significant dose-dependent reversal of this deficit, with complete reversal noted at 1.1 μg/d infused intracerebroventricularly (Fig. 1B). In contrast, the same dose failed to alter behavioral responses after subcutaneous infusion in transgenic mice (p = 0.8), or with either intracerebroventricular (Fig. 1B) or subcutaneous delivery in WT mice (p = 0.5). The effects of intracerebroventricularly infused compound C were specific to contextual memory deficits, with cued-conditioning behavior (p = 0.3) and general locomotion (p = 0.5) being unchanged. Vehicle-infused transgenics exhibited hyperlocomotion (p < 0.01), but no changes in cued fear conditioning relative to vehicle-infused WTs (p = 0.67). The brain compound C EC50 for rescue of contextual fear memory was ∼0.25 ± 0.06 μm. These results demonstrate that chronic BACE1 inhibition can reverse behavioral deficits in aged Tg2576 mice.
Using histology and biochemical endpoints, we evaluated the impact of chronic BACE1 inhibition on amyloid burden. Tg2576 mice show robust levels of amyloid burden at ≥16 months of age (Kawarabayashi et al., 2001). Consistent with this finding, we observed extensive plaque burden in the cortex and hippocampus of our cohort at baseline (age, ∼17.2 months), with insignificant increases in the plaque burden in vehicle-treated mice at end of study (age, ∼19.1 months; Fig. 1C,D). Whereas subcutaneous infusion of compound C failed to alter cortical (p = 0.9) or hippocampal (p = 0.8) plaque load, intracerebroventricular infusion produced a dose-dependent reduction of plaques in both brain regions (Fig. 1C,D), with maximal effects obtained with the 1.1 μg/d dose. In particular, intracerebroventricular doses of 1.1 and 23.5 μg/d significantly reduced the plaque burden by >45%, compared with not only vehicle-treated animals but also baseline animals (Fig. 1D). Similarly, dense-core plaques decreased by ≤52% in mice treated with intracerebroventricular BACE1 inhibitor, relative to the plaques present in vehicle-treated (p < 0.01) or baseline (p < 0.05) animals (data not shown). Subcutaneously delivered inhibitor had no effects (p = 0.9). CAA or microhemorrhages were unaltered with intracerebroventricular (Table 1) or subcutaneous infusions (p = 0.9, CAA; p = 0.6, microhemorrhages).
Table 1.
Chronic intracerebroventricular infusion with compound C does not alter CAA, microhemorrhages, integrity of myelin, or neuromuscular function in aged Tg2576 micea
| Treatment | Daily dose (μg/d) | CAA (% area) | Microhemorrhages |
Grip strength (g) | Myelin integrity |
|||
|---|---|---|---|---|---|---|---|---|
| Frequency | Severity | Corpus callosum (mm3) | Striatum (% area) | Sciatic nerve (g-ratio) | ||||
| Compound C | 0.3 | 0.75 ± 0.09 | 3.4 ± 0.5 | 1.2 ± 0.1 | 205.1 ± 8.6 | 0.35 ± 0.01 | 25.9 ± 2.5 | 0.61 ± 0.01 |
| 1.1 | 0.67 ± 0.08 | 3.5 ± 0.7 | 1.2 ± 0.1 | 200.5 ± 8.1 | 0.38 ± 0.01 | 23.4 ± 1.7 | 0.59 ± 0.01 | |
| 23.5 | 0.89 ± 0.17 | 4.1 ± 0.5 | 1.5 ± 0.2 | 221.5 ± 10.1 | 0.40 ± 0.02 | 26.6 ± 4.0 | 0.59 ± 0.01 | |
| Vehicle | — | 0.83 ± 0.11 | 3.9 ± 0.4 | 1.2 ± 0.1 | 213.4 ± 6.6 | 0.38 ± 0.01 | 20.4 ± 1.0 | 0.59 ± 0.01 |
aQuantitation of CAA in leptomeninges and vasculature within brain cortices and hippocampi. Frequency of cerebral microhemorrhages analyzed by counting the number of perivascular hemosiderin-positive cell clusters per section (average of 18–20 sections per brain). Microhemorrhage severity estimated by assigning a grade of 1–4 for clusters of 1–4, 5–10, 11–20, or >20 hemosiderin-positive cells, respectively. Grip-strength recordings taken for all limbs and averaged over five trials per mouse. Myelin integrity measured using g-ratios, Caveliari's volumetric method, and percentage area occupied by myelin-positive signal, for peripheral (sciatic nerves), and central (corpus callosum, striatum) nerve fibers.
In agreement with its effects on amyloid plaques, intracerebroventricularly delivered compound C significantly decreased both soluble and insoluble Aβ42 levels in the brain by ≤47 and ≤41%, respectively (Fig. 2A,B). A trend for reduction in soluble and insoluble Aβ40 in the brain was also evident with a significant decline (∼35%) in the 23.5 μg/d dose (Fig. 2D,E). Unlike brain Aβ, CSF Aβ40 or Aβ42 remained unchanged with treatment (Fig. 2C,F). Subcutaneously delivered compound had no significant impact on biochemical outcomes (p = 0.8, soluble Aβ42; p = 0.9, insoluble Aβ42; p = 0.1, CSF Aβ42; p = 0.7, soluble Aβ40; p = 0.9, insoluble Aβ40; p = 0.2, CSF Aβ40). Consistent with the ring-like staining of BACE1 observed around clear cores of unstained amyloid plaques in aged Tg2576 mice (Zhao et al., 2007), we noted a robust ring-like sAPPβ immunostaining in Tg2576, but not in WT, mice (Fig. 2G). Brain sAPPβ immunostaining was reduced in mice infused with compound C (Fig. 2G), providing direct evidence for BACE1 inhibition in aged Tg2576 mice.
Figure 2.

Chronic intracerebroventricular infusion with compound C reduces brain Aβ42 and Aβ40 levels, but not CSF Aβ, in aged Tg2576 mice. A, B, Dose-dependent decline in soluble (A) and insoluble (B) brain Aβ42, 8 weeks after intracerebroventricular infusion of vehicle or compound C. C, No significant changes in CSF Aβ42. D, E, Significant reduction in brain Aβ40 in soluble (D) and insoluble (E) pools at the highest dose, 23.5 μg/d. F, No significant changes in CSF Aβ40. Significance versus vehicle-treated mice (*p < 0.05, **p < 0.01). G, Brain sAPPβ immunostaining in WT or Tg2576 mice infused intracerebroventricularly with vehicle or compound C. Scale bar, 100 μm.
Dystrophic neurites were visualized, using the deOlmos silver stain, as black rings of swollen distorted axons or dendrites surrounding dense-core amyloid plaques (Fig. 3A). A significant decline in dystrophic neurites was evident with ∼53% reduction with the 1.1 μg/d dose of the intracerebroventricularly infused compound (Fig. 3B). Subcutaneously infused compound did not elicit any effects (p = 0.9). GFAP-positive astrocytotic clusters also showed a dose-dependent reduction by ≤49% upon chronic intracerebroventricular, but not subcutaneous (p = 0.6), infusion of the BACE1 inhibitor (Fig. 3C,D).
Figure 3.
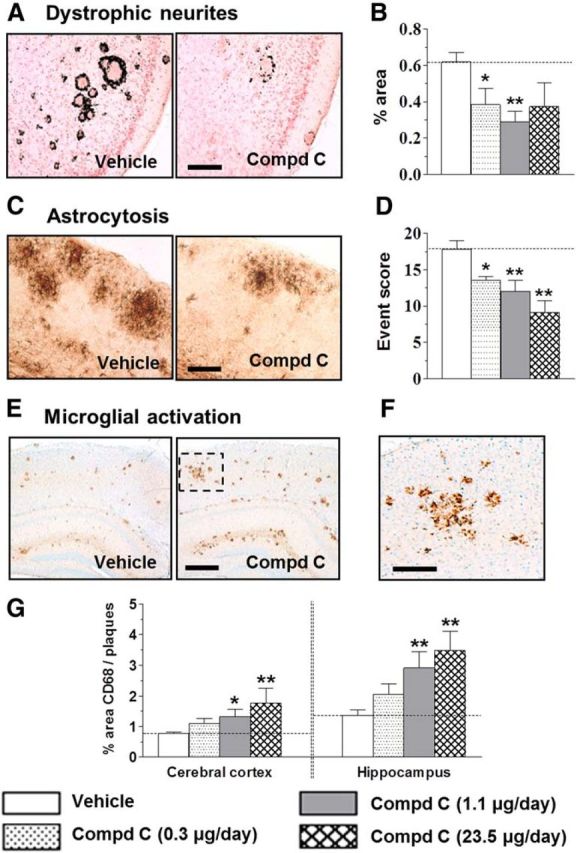
Chronic BACE1 inhibition led to reduction in dystrophic neurites and astrocytosis and to enhanced microglial activation in the brains of aged Tg2576 mice. A, Dystrophic neurites revealed using deOlmos silver stain in vehicle and 1.1 μg/d compound C-treated mice. B, Dose-dependent reduction in percentage area occupied by dystrophic neurites after intracerebroventricular infusion with compound C. C, Reactive astrocytes stained for GFAP in cortex from vehicle-treated and 1.1 μg/d compound C-treated mice. D, Compound C dose-dependently reduced astrocytosis, scored as the number of clusters (events) per section. Data are average from 18 to 20 sections per brain. E, CD68-labeled microglia in cortex and hippocampus of vehicle-treated or compound C (1.1 μg/d)-treated Tg2576 mice. F, Magnified image inset from E. G, Dose-dependent increase in microglial activation around plaques with compound C treatment. Microglial activation measured as percentage CD68-positive area relative to the area occupied by amyloid plaques (Thakker et al., 2009). *p < 0.05, **p < 0.01, significantly different from vehicle-treated mice. Scale bars: A, C, F, 200 μm; E, 500 μm.
Staining for CD68, a marker for phagocytic activity, revealed clusters of intensely stained microglia with relatively short processes surrounding the deposited amyloid (Fig. 3E,F). Whereas subcutaneous delivery had no effects (p = 0.7), intracerebroventricular infusion of the compound augmented microglial activation ∼2-fold in both cortex and hippocampus (Fig. 3G). A significant inverse correlation was apparent between activated microglia and amyloid burden in the cortex (R2 = 0.902, p < 0.0001) and hippocampus (R2 = 0.704, p < 0.0001), indicating a strong association between the two outcomes after chronic BACE1 inhibition.
Pilot work, assessing effects on brain Aβ after 2 week intracerebroventricular infusion in the Tg2576 mice, helped define compound doses (0.3 and 1.1 μg/d) for chronic evaluation in this study. A feasibly high dose of 23.5 μg/d was incorporated to assess maximal efficacy in different endpoints and any dose-dependent toxicity with BACE1 inhibition in the study. We observed maximal reversal of the behavioral deficit and amyloid pathology in aged Tg2576 mice at 1.1 μg/d infusions, and no additional statistically significant benefit at 23.5 μg/d for any outcomes. Overall mortality (11.8%) during blind execution of the study was within expected limits for aged mice. At study completion, the groups associated with mortality were uncovered as follows: Tg2576 mice infused intracerebroventricularly with vehicle (1 of 14), compound C at 1.11 μg/d (1 of 13); or 23.52 μg/d (6 of 12); WT mice infused intracerebroventricularly with vehicle (1 of 10) or subcutaneously with 1.11 μg/d compound C (4 of 10). Higher mortality was evident in transgenic mice receiving intracerebroventricular dose of 23.5 μg/d and in WT mice receiving subcutaneous dose at 1.11 μg/d.
We evaluated the consequences of chronic compound C dosing on central and peripheral myelination and neuromuscular function, since BACE1 knock-down was associated with myelin abnormalities (Hu et al., 2006; Willem et al., 2006). Staining for myelin basic protein revealed no significant changes in myelin integrity in the corpus callosum or striatum. G-ratio measurements revealed no treatment effects on sciatic nerve myelination (Table 1). Neuromuscular function remained intact, as evidenced by lack of significant differences in grip strength between vehicle-treated and compound C-treated animals (Table 1).
Discussion
In this study, we demonstrate for the first time that chronic BACE1 inhibition in aged Tg2576 mice can reduce brain amyloid to levels below that at start of treatment and rescue deficits in contextual fear memory. A robust increase in microglial ramification, associated with brain amyloid reduction, suggests recruitment of novel clearance pathways following BACE1 inhibition in aged transgenic mice.
We sought proof-of-concept of whether chronic intracerebroventricular infusion of a potent BACE1 inhibitor can reduce brain amyloid and/or improve function in aged Tg2576 mice, despite high P-glycoprotein-mediated efflux from the brain (Meredith et al., 2008). Intracerebroventricular doses ≤1.1 μg/d produced robust brain exposures that were well tolerated and revealed maximal efficacy. In contrast, intracerebroventricular infusion at 23.5 μg/d or subcutaneous infusion at 1.1 μg/d led to high mortality. Deaths were likely due to high compound exposures leading to non-BACE1-mediated toxicity, given lack of any changes in myelin integrity or neuromuscular function unlike BACE1 knock-out mice (Hu et al., 2006; Willem et al., 2006). A recent trial with an orally bioavailable BACE1 inhibitor was terminated due to nonspecific retinal degeneration, with similar toxicity observed in BACE1 knock-out mice (May et al., 2011). In light of these data, we illustrate that low-dose intracerebroventricular BACE1 inhibitor can improve function with lower risk of adverse effects.
The BACE1 inhibitor elicited a significant improvement in contextual fear memory as well as attenuation of dystrophic neurites and astrocytosis in the brain of aged transgenics. Although pathogenesis is unclear, age-dependent increase in brain Aβ may drive soluble oligomeric species to alter synaptic transmission and calcium handling in neurons, ultimately leading to structural and behavioral deficits (Cleary et al., 2005; Venkitaramani et al., 2007; Shankar et al., 2008). We hypothesize that such deficits may be mitigated following robust reduction in brain Aβ42 as well as amyloid plaques after intracerebroventricular BACE1 inhibition. Further, BACE1 inhibition may ameliorate such deficits via modulation of cAMP-dependent CREB pathways, a critical component of long-term synaptic plasticity (Chen et al., 2012).
Previous studies have shown modest reductions in brain Aβ after 6 months of intraperitoneal injections with a BACE1 inhibitor (Fukumoto et al., 2010), while others have demonstrated lack of efficacy with secretase inhibition after significant amyloid has deposited in Tg2576 mice (Das et al., 2012). Particularly for BACE1 inhibitors, low efficacy may also be due to the enzyme's high affinity for APPSWE expressed in Tg2576 mice (Eketjäll et al., 2013). Together, our data of robust amyloid reduction below baseline suggest that, in addition to lowering Aβ synthesis, the intracerebroventricularly delivered BACE1 inhibitor may reduce brain amyloid via yet-unknown mechanisms.
Intriguingly, the BACE1 inhibitor produced a dose-dependent increase in microglial ramification that inversely correlated with amyloid burden. Microglia play a key role in amyloid clearance by phagocytosing brain Aβ (Koenigsknecht-Talboo et al., 2008) but also in neuroinflammation (Weitz and Town, 2012). Pulse-chase studies to evaluate the synthesis and clearance rates for CSF Aβ have uncovered a significant impairment of Aβ clearance in AD (Mawuenyega et al., 2010), advocating for CNS amyloid clearance as a plausible therapy for AD. Further, studies demonstrate that enhanced amyloid clearance, when combined with reduced Aβ synthesis, effectively attenuates brain amyloid burden (Wang et al., 2011). Accordingly, we hypothesize that an increase in microglia-mediated clearance together with reduced Aβ synthesis, ensuing from chronic BACE1 inhibition, results in robust brain amyloid reduction in aged Tg2576 mice. Our hypothesis is supported by several recent studies implicating microglial pathways, such as pathways involving TREM2 (the triggering receptor expressed on myeloid cells-2), CD33, the scavenger receptor Scara1, and autophagy in Aβ clearance and AD pathogenesis (Jiang et al., 2013; Griciuc et al., 2013; Frenkel et al., 2013; Lucin et al., 2013).
In summary, we demonstrate functional improvement, along with significant brain amyloid reduction and enhanced microglial ramification, with a potent BACE1 inhibitor in aged Tg2576 mice. Our results provide novel insights in aged transgenics, and support evaluation of potent BACE1 inhibitors in AD subjects.
Footnotes
This work was supported by Medtronic and Bristol-Myers Squibb. We sincerely acknowledge the meticulous data analyses support from Bradley Nix.
All authors were, at the time of the work, employees of Medtronic or Bristol-Myers Squibb as indicated.
References
- Aisen PS, Vellas B, Hampel H. Moving towards early clinical trials for amyloid-targeted therapy in Alzheimer's disease. Nat Rev Drug Discov. 2013;12:324–325. doi: 10.1038/nrd3842-c1. [DOI] [PubMed] [Google Scholar]
- Chen Y, Huang X, Zhang YW, Rockenstein E, Bu G, Golde TE, Masliah E, Xu H. Alzheimer's β-secretase (BACE1) regulates the cAMP/PKA/CREB pathway independently of β-amyloid. J Neurosci. 2012;32:11390–11395. doi: 10.1523/JNEUROSCI.0757-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Natural oligomers of the amyloid-β protein specifically disrupt cognitive function. Nat Neurosci. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- Das P, Verbeeck C, Minter L, Chakrabarty P, Felsenstein K, Kukar T, Maharvi G, Fauq A, Osborne BA, Golde TE. Transient pharmacologic lowering of Aβ production prior to deposition results in sustained reduction of amyloid plaque pathology. Mol Neurodegener. 2012;7:39. doi: 10.1186/1750-1326-7-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easton A, Sankaranarayanan S, Tanghe A, Terwel D, Lin AX, Hoque N, Bourin C, Gu H, Ahlijanian M, Bristow L. Effects of sub-chronic donepezil on brain Abeta and cognition in a mouse model of Alzheimer's disease. Psychopharmacology. 2013;230:279–289. doi: 10.1007/s00213-013-3152-3. [DOI] [PubMed] [Google Scholar]
- Eketjäll S, Janson J, Jeppsson F, Svanhagen A, Kolmodin K, Gustavsson S, Radesäter AC, Eliason K, Briem S, Appelkvist P, Niva C, Berg AL, Karlström S, Swahn BM, Fälting J. AZ-4217: a high potency BACE inhibitor displaying acute central efficacy in different in vivo models and reduced amyloid deposition in Tg2576 mice. J Neurosci. 2013;33:10075–10084. doi: 10.1523/JNEUROSCI.1165-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenkel D, Wilkinson K, Zhao L, Hickman SE, Means TK, Puckett L, Farfara D, Kingery ND, Weiner HL, El Khoury J. Scara1 deficiency impairs clearance of soluble amyloid-β by mononuclear phagocytes and accelerates Alzheimer's-like disease progression. Nat Commun. 2013;4:2030. doi: 10.1038/ncomms3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumoto H, Takahashi H, Tarui N, Matsui J, Tomita T, Hirode M, Sagayama M, Maeda R, Kawamoto M, Hirai K, Terauchi J, Sakura Y, Kakihana M, Kato K, Iwatsubo T, Miyamoto M. A noncompetitive BACE1 inhibitor TAK-070 ameliorates Aβ pathology and behavioral deficits in a mouse model of Alzheimer's disease. J Neurosci. 2010;30:11157–11166. doi: 10.1523/JNEUROSCI.2884-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh AK, Brindisi M, Tang J. Developing β-secretase inhibitors for treatment of Alzheimer's disease. J Neurochem. 2012;120:71–83. doi: 10.1111/j.1471-4159.2011.07476.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griciuc A, Serrano-Pozo A, Parrado AR, Lesinski AN, Asselin CN, Mullin K, Hooli B, Choi SH, Hyman BT, Tanzi RE. Alzheimer's disease risk gene CD33 inhibits microglial uptake of amyloid β. Neuron. 2013;78:631–643. doi: 10.1016/j.neuron.2013.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Hicks CW, He W, Wong P, Macklin WB, Trapp BD, Yan R. Bace1 modulates myelination in the central and peripheral nervous system. Nat Neurosci. 2006;9:1520–1525. doi: 10.1038/nn1797. [DOI] [PubMed] [Google Scholar]
- Jacobsen JS, Wu CC, Redwine JM, Comery TA, Arias R, Bowlby M, Martone R, Morrison JH, Pangalos MN, Reinhart PH, Bloom FE. Early-onset behavioral and synaptic deficits in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2006;103:5161–5166. doi: 10.1073/pnas.0600948103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang T, Yu JT, Zhu XC, Tan L. TREM2 in Alzheimer's disease. Mol Neurobiol. 2013;48:180–185. doi: 10.1007/s12035-013-8424-8. [DOI] [PubMed] [Google Scholar]
- Jonsson T, Atwal JK, Steinberg S, Snaedal J, Jonsson PV, Bjornsson S, Stefansson H, Sulem P, Gudbjartsson D, Maloney J, Hoyte K, Gustafson A, Liu Y, Lu Y, Bhangale T, Graham RR, Huttenlocher J, Bjornsdottir G, Andreassen OA, Jönsson EG, et al. A mutation in APP protects against Alzheimer's disease and age-related cognitive decline. Nature. 2012;488:96–99. doi: 10.1038/nature11283. [DOI] [PubMed] [Google Scholar]
- Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Ashe KH, Younkin SG. Age-dependent changes in brain, CSF, and plasma amyloid-β protein in the Tg2576 transgenic mouse model of Alzheimer's disease. J Neurosci. 2001;21:372–381. doi: 10.1523/JNEUROSCI.21-02-00372.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenigsknecht-Talboo J, Meyer-Luehmann M, Parsadanian M, Garcia-Alloza M, Finn MB, Hyman BT, Bacskai BJ, Holtzman DM. Rapid microglial response around amyloid pathology after systemic anti-Aβ antibody administration in PDAPP mice. J Neurosci. 2008;28:14156–14164. doi: 10.1523/JNEUROSCI.4147-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucin KM, O'Brien CE, Bieri G, Czirr E, Mosher KI, Abbey RJ, Mastroeni DF, Rogers J, Spencer B, Masliah E, Wyss-Coray T. Microglial beclin 1 regulates retromer trafficking and phagocytosis and is impaired in Alzheimer's disease. Neuron. 2013;79:873–886. doi: 10.1016/j.neuron.2013.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, Yarasheski KE, Bateman RJ. Decreased clearance of CNS β-amyloid in Alzheimer's disease. Science. 2010;330:1774. doi: 10.1126/science.1197623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May PC, Dean RA, Lowe SL, Martenyi F, Sheehan SM, Boggs LN, Monk SA, Mathes BM, Mergott DJ, Watson BM, Stout SL, Timm DE, Smith Labell E, Gonzales CR, Nakano M, Jhee SS, Yen M, Ereshefsky L, Lindstrom TD, Calligaro DO, et al. Robust central reduction of amyloid-β in humans with an orally available, non-peptidic β-BACE1 inhibitor. J Neurosci. 2011;31:16507–16516. doi: 10.1523/JNEUROSCI.3647-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meredith JE, Jr, Thompson LA, Toyn JH, Marcin L, Barten DM, Marcinkeviciene J, Kopcho L, Kim Y, Lin A, Guss V, Burton C, Iben L, Polson C, Cantone J, Ford M, Drexler D, Fiedler T, Lentz KA, Grace JE, Jr, Kolb J, et al. P-glycoprotein efflux and other factors limit brain amyloid beta reduction by beta-site amyloid precursor protein-cleaving enzyme 1 inhibitors in mice. J Pharmacol Exp Ther. 2008;326:502–513. doi: 10.1124/jpet.108.138974. [DOI] [PubMed] [Google Scholar]
- Sankaranarayanan S, Price EA, Wu G, Crouthamel MC, Shi XP, Tugusheva K, Tyler KX, Kahana J, Ellis J, Jin L, Steele T, Stachel S, Coburn C, Simon AJ. In vivo beta-BACE1–1 inhibition leads to brain Abeta lowering and increased alpha-BACE1 processing of amyloid precursor protein without effect on neuregulin-1. J Pharmacol Exp Ther. 2008;324:957–969. doi: 10.1124/jpet.107.130039. [DOI] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-β protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakker DR, Weatherspoon MR, Harrison J, Keene TE, Lane DS, Kaemmerer WF, Stewart GR, Shafer LL. Intracerebroventricular amyloid-β antibodies reduce cerebral amyloid angiopathy and associated micro-hemorrhages in aged Tg2576 mice. Proc Natl Acad Sci U S A. 2009;106:4501–4506. doi: 10.1073/pnas.0813404106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyn JH, Ahlijanian MK. Interpreting Alzheimer's disease clinical trials in light of the effects on amyloid-β. Alzheimers Res Ther. 2014;6:14. doi: 10.1186/alzrt244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassar R, Kovacs DM, Yan R, Wong PC. The β-secretase enzyme BACE in health and Alzheimer's disease: regulation, cell biology, function, and therapeutic potential. J Neurosci. 2009;29:12787–12794. doi: 10.1523/JNEUROSCI.3657-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkitaramani DV, Chin J, Netzer WJ, Gouras GK, Lesne S, Malinow R, Lombroso PJ. β-Amyloid modulation of synaptic transmission and plasticity. J Neurosci. 2007;27:11832–11837. doi: 10.1523/JNEUROSCI.3478-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang A, Das P, Switzer RC, 3rd, Golde TE, Jankowsky JL. Robust amyloid clearance in a mouse model of Alzheimer's disease provides novel insights into the mechanism of amyloid-immunotherapy. J Neurosci. 2011;31:4124–4136. doi: 10.1523/JNEUROSCI.5077-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitz TM, Town T. Microglia in Alzheimer's disease: it's all about context. Int J Alzheimers Dis. 2012;2012:314185. doi: 10.1155/2012/314185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willem M, Garratt AN, Novak B, Citron M, Kaufmann S, Rittger A, DeStrooper B, Saftig P, Birchmeier C, Haass C. Control of peripheral nerve myelination by the β-secretase BACE1. Science. 2006;314:664–666. doi: 10.1126/science.1132341. [DOI] [PubMed] [Google Scholar]
- Yan R, Vassar R. Targeting the β secretase BACE1 for Alzheimer's disease therapy. Lancet Neurol. 2014;13:319–329. doi: 10.1016/S1474-4422(13)70276-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Fu Y, Yasvoina M, Shao P, Hitt B, O'Connor T, Logan S, Maus E, Citron M, Berry R, Binder L, Vassar R. β-Site amyloid precursor protein cleaving enzyme1 levels become elevated in neurons around amyloid plaques: implications for Alzheimer's disease pathogenesis. J Neurosci. 2007;27:3639–3649. doi: 10.1523/JNEUROSCI.4396-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]