

Abstract
The survival of postmitotic neurons needs continuous degradation of cyclin B1, a mitotic protein accumulated aberrantly in the damaged brain areas of Alzheimer's disease and stroked patients. Degradation of cyclin B1 takes place in the proteasome after ubiquitylation by the anaphase-promoting complex/cyclosome (APC/C)–cadherin 1 (Cdh1), an E3 ubiquitin ligase that is highly active in neurons. However, during excitotoxic damage—a hallmark of neurological disorders—APC/C–Cdh1 is inactivated, causing cyclin B1 stabilization and neuronal death through an unknown mechanism. Here, we show that an excitotoxic stimulus in rat cortical neurons in primary culture promotes cyclin B1 accumulation in the mitochondria, in which it binds to, and activates, cyclin-dependent kinase-1 (Cdk1). The cyclin B1–Cdk1 complex in the mitochondria phosphorylates the anti-apoptotic protein B-cell lymphoma extra-large (Bcl-xL), leading to its dissociation from the β subunit of F1Fo–ATP synthase. The subsequent inhibition of ATP synthase activity causes complex I oxidative damage, mitochondrial inner membrane depolarization, and apoptotic neuronal death. These results unveil a previously unrecognized role for mitochondrial cyclin B1 in the oxidative damage associated with neurological disorders.
Keywords: Bcl-xL, cyclin B1, excitotoxicity, F1Fo-ATP synthase, mitochondria, neurodegeneration
Introduction
An increasing body of evidence indicates that the progressive neuronal death associated with neurodegenerative diseases is a consequence of a failed attempt of postmitotic neurons to aberrantly reenter the cell cycle (Herrup, 2013). Thus, in patients suffering from Alzheimer's disease (Vincent et al., 1997; Yang et al., 2003) and stroke (Love, 2003), as well as in experimental models of cerebral ischemia (Erdö et al., 2004; Wen et al., 2004; Rashidian et al., 2007), the affected brain areas aberrantly accumulate cyclin B1. This is compatible with the notion that cyclin B1 may be involved in the neuronal death associated with those pathologies. In healthy neurons, cyclin B1 is degraded persistently after ubiquitylation by the E3 ubiquitin ligase anaphase-promoting complex/cyclosome (APC/C)–cadherin 1 (Cdh1; Almeida et al., 2005). However, during excitotoxic damage—a hallmark of neurological disorders (Bolaños et al., 2009; Wang and Qin, 2010)—APC/C–Cdh1 is inactivated, causing aberrant stabilization of cyclin B1 (Maestre et al., 2008), which mediates neuronal death (Almeida et al., 2005; Maestre et al., 2008) through a yet elusive molecular mechanism.
Excitotoxicity is one of the most common phenomena that underlie the pathophysiology of neurological disorders (Wang and Qin, 2010) and involves overactivation of glutamatergic receptors triggering massive Ca2+ entry into the postsynaptic neuron. After an excitotoxic insult, mitochondria are thus overloaded of Ca2+, resulting in inner membrane potential (Δψm) disruption, which precedes neuronal apoptotic death (Bolaños et al., 2009). Accordingly, keeping Δψm intact represents an interesting therapeutic strategy against neurodegeneration (Smith et al., 2008). However, besides mitochondrial Ca2+ (Duchen, 2012), there are other yet unknown factor(s) responsible for the Δψm loss and neuronal death. Here, we show that, during an excitotoxic stimulus, cyclin B1 is accumulated in the mitochondria, accounting for the Δψm loss and neuronal death. Furthermore, we found that this effect occurred through cyclin B1-mediated activation of cyclin-dependent kinase-1 (Cdk1), which phosphorylated B-cell lymphoma extra-large (Bcl-xL), resulting in its dissociation from the β subunit of F1Fo–ATP synthase. The subsequent inhibition in ATP synthase activity caused increased mitochondrial superoxide anion (O2·−) that damaged complex I, leading to the Δψm loss and bioenergetic crisis responsible for apoptotic neuronal death.
Materials and Methods
Ethical statement regarding the use of animals.
All animals (pregnant Wistar female rats) used in this work were obtained from the Animal Experimentation Service of the University of Salamanca, in accordance with Spanish legislation (RD 1201/2005) under license from the Spanish government. Protocols were approved by the Bioethics Committee of the University of Salamanca.
Cell cultures.
Primary cultures of rat cortical neurons were prepared from fetal Wistar rats (of either sex) of 16 d of gestation (Maestre et al., 2008), seeded at 2.5 × 105 cells/cm2 in different-sized plastic plates coated with poly-d-lysine (15 μg/ml) and incubated in DMEM (Sigma-Aldrich) supplemented with 10% fetal calf serum (FCS; Roche Diagnostics). Cells were incubated at 37°C in a humidified 5% CO2-containing atmosphere. At 48 h after plating, the medium was replaced with DMEM supplemented with 5% horse serum (Sigma-Aldrich), 20 mm d-glucose, and, on day 4, cytosine arabinoside (10 μm) to prevent non-neuronal proliferation. Cells were used for the experiments on day 6–7 in vitro. Human embryonic kidney-293T (HEK293T) cells were maintained in DMEM supplemented with 10% (v/v) FCS. Twenty-four hours before the experiment, cells were reseeded at 1.8 × 105 cells/cm2 in plates coated previously with poly-d-lysine (15 μg/ml).
Plasmid constructions and site-directed mutagenesis.
The following plasmid constructions were used: (1) pIRES2–EGFP (Invitrogen), either empty [control; expressing green fluorescent protein (GFP)] or containing the full-length cDNA of human Cyclin B1 (Cyclin B1; expressing both cyclin B1 and GFP; Maestre et al., 2008); (2) pcDNA3–GFP (expressing GFP) or pcDNA3–cyclin B1–GFP, encoding human cyclin B1 fused to GFP (from J. Pines, Gurdon Institute, University of Cambridge, Cambridge, UK; Almeida et al., 2005); (3) pSuper–neo.gfp (Oligoengine), including the small hairpin sequences for luciferase (control; 5′-CTGACGCGGAATACTTCGA-3′) or cyclin B1 (shCyclin B1; 5′-GATGGAGCTGATCCAAACC-3′; nucleotides 478–496, GenBank accession number AY338491; Almeida et al., 2005; Maestre et al., 2008); (4) pMitoDsRed2 plasmid vector (MitoRed; Clontech), expressing red fluorescent protein in mitocondria; and (5) p8–Bcl-xL (from F. Pimentel, Cancer Research Institute, University of Salamanca, Spanish National Research Council, Salamanca, Spain) was subjected to site-directed mutagenesis on Ser42, which was replaced by Ala or Asp residues to obtain the phosphodefective [Bcl-xL(A); to block Cdk1 phosphorylation] and phosphomimetic [Bcl-xL(D)] forms of Bcl-xL (GenBank accession number NM_138578.1), respectively, using the QuikChange XL kit (Stratagene), followed by DpnI digestion. The forward and reverse oligonucleotides designed were as follows, respectively: 5′-TGGCACCTGGCAGACGCCCCCGCGGTGAATGGA-3′ and 5′-TCCATTCACCGCGGGGGCGTCTGCCAGGTGCCA-3′ for Ser42Ala; and 5′-TGGCACCTGGCAGACGACCCCGCGGTGAATGGA-3′ and 5′-TCCATTCACCGCGGGGTCGTCTGCCAGGTGCCA-3′ for Ser42Asp.
Small interfering RNA.
Specific depletion of Cdk5 was achieved by using small (21 bp) interfering double-stranded ribonucleotides (siRNA) designed specifically to target the coding sequence of the rat Cdk5 mRNA (Maestre et al., 2008). We used the following siRNA (only the forward strand shown): 5′-AAGCCGUACCCGAUGUAUC-3′ (nucleotides 859–877, GenBank accession number NM_080885). An siRNA against luciferase (5′-CUGACGCGGAAUACUUCGAUU-3′) was used as control siRNA (siControl; Maestre et al., 2008). Annealed siRNAs were purchased from Dharmacon (Abgene, Thermo Fisher Scientific).
Cell transfections and treatments.
All transfections with plasmid constructions were performed using Lipofectamine 2000 (Invitrogen; Maestre et al., 2008), following the instructions of the manufacturer. After transfections, cells were incubated further for 4–24 h until the experiments and cell collection were performed. Transfections of neurons with siRNAs were performed using Lipofectamine RNAiMAX (Invitrogen) following the instructions of the manufacturer and used after 72 h.
To promote an excitotoxic insult, neurons were incubated with 100 μm glutamate plus 10 μm glycine in HBSS (in mm: 134.2 NaCl, 5.26 KCl, 0.43 KH2PO4, 4.09 NaHCO3, 0.33 Na2HPO4, 5.44 glucose, 20 HEPES, and 4 CaCl2, pH 7.4) for 5 min and incubated further in culture medium for the indicated time period (Almeida and Bolaños, 2001). When indicated, incubations were performed in the presence of 100 ng/ml actinomycin D (ActD; Sigma), 1 μg/ml cycloheximide (CHX; Sigma), 10 μm ProTAME (Boston Biochem, R&D Systems), and 1 mm glutathione ethyl ester (GSH-EE; Sigma) or with 10 μm roscovitine (Rosc; Sigma).
Subcellular fractionation.
Cells were washed with cold PBS containing 1 mm MgCl2, harvested with cytosolic buffer (10 mm HEPES, 1.5 mm MgCl2, 10 mm KCl, 1 mm EDTA, 0.1% NP-40, v/v, 1.5 m sucrose, and protease and phosphatase inhibitors mixture, pH 7.9), triturated with a micropipette to promote cell lysis, left on ice for 30 min, and vortexed for 10 s. After checking cell lysis under a light microscope, extracts were centrifuged at 830 × g for 10 min. Lysis of the nuclei was performed by resuspending the nuclear pellet in nuclear buffer (50 mm HEPES, 1.5 mm MgCl2, 10 mm KCl mm, 0.5 mm NaCl, 1 mm EDTA, 1% NP-40, v/v, and protease and phosphatase inhibitor mixture, pH 7.9), triturated with a micropipette, left on ice for 2 h, vortexed (10 s), boiled (5 min), and sonicated (5 min). The supernatant (mitochondrial and cytosolic fractions) was then centrifuged at 17,000 × g for 12 min (4°C), and the cytosolic fraction (supernatant) was lysed in 2× RIPA buffer (2% sodium dodecylsulphate, 2 mm EDTA, 2 mm EGTA, and 50 mm Tris, pH 7.5), supplemented with phosphatase inhibitors (1 mm Na3VO4 and 50 mm NaF) and protease inhibitors [100 μm phenylmethylsulfonyl fluoride (PMSF), 50 μg/ml anti-papain, 50 μg/ml pepstatin, 50 μg/ml amastatin, 50 μg/ml leupeptin, 50 μg/ml bestatin, and 50 μg/ml soybean trypsin inhibitor], and boiled for 5 min (Maestre et al., 2008). The mitochondrial fraction (pellet) was resuspended in isolation medium (in mm: 320 sucrose, 1 potassium EDTA, and 10 Tris-HCl, pH 7.4) and was homogenized in a tight-fitting glass–Teflon homogenizer (20 strokes; Almeida and Medina, 1998). Mitochondrial fraction was either lysed with 2× RIPA buffer for protein analysis by immunoblotting or used for the identification of intramitochondrial localization of cyclin B1, as described below.
Mitochondrial fraction was incubated in buffer (in mm: 1 EDTA and 10 MOPS-KOH, pH 7.2) containing 320 or 70 mm sucrose on ice for 30 min and was centrifuged at 16,000 × g at 4°C for 10 min. Pellets were resuspended in RIPA buffer, and all fractions were analyzed by immunoblotting.
The protease protection assay (Wang et al., 2014 1) was performed by incubating mitochondrial fraction in buffer (in mm: 1 EDTA, 10 MOPS-KOH, and 320 sucrose, pH 7.2) on ice for 30 min with or without 25 or 50 μg/ml trypsin. Proteolysis was stopped by addition of 1 mm PMSF, and samples were centrifuged 16,000 × g at 4°C for 10 min. Pellet were resuspended in RIPA buffer, and proteins were analyzed by immunoblotting.
Western blot.
Cells were lysed in 2% sodium dodecylsulphate, 2 mm EDTA, 2 mm EGTA, and 50 mm Tris, pH 7.5, supplemented with phosphatase inhibitors (1 mm Na3VO4, 50 mm NaF) and protease inhibitors (100 μm phenylmethylsulfonyl fluoride, 50 μg/ml anti-papain, 50 μg/ml pepstatin, 50 μg/ml amastatin, 50 μg/ml leupeptin, 50 μg/ml bestatin, and 50 μg/ml soybean trypsin inhibitor), stored on ice for 30 min, and boiled for 10 min. Aliquots of cell extracts were subjected to SDS polyacrylamide gel (MiniProtean; Bio-Rad) and blotted with primary antibodies at dilutions ranging from 1:200 to 1:1000 overnight at 4°C. Antibodies used were anti-cyclin B1 (BD Biosciences Pharmingen), anti-Cdk1 (Santa Cruz Biotechnology) anti-voltage-dependent anion channel (VDAC; Merck Millipore), anti-proliferating cell nuclear antigen (BD Biosciences Pharmingen), anti-GAPDH (Ambion), anti-Bcl-xL (BD Biosciences), anti-phosphoserine (Zymed, Invitrogen), anti-β-ATPase (Abcam), anti-Hsp60 (Abcam), anti-translocase of the outer mitochondrial membrane 20 (TOM20; Abcam), and anti-Ndusf1 [NADH dehydrogenase (ubiquinone) Fe-S protein 1, 75kDa (NADH–coenzyme Q reductase); Santa Cruz Biotechnology]. After incubation with horseradish peroxidase-conjugated goat anti-rabbit IgG (Santa Cruz Biotechnology) or goat anti-mouse IgG (1:10,000 dilution; Santa Cruz Biotechnology), membranes were incubated immediately with the enhanced chemiluminescence kit SuperSignal West Dura (Pierce, Thermo Fisher Scientific) for 5 min or Immobilon Western Chemiluminiscent HRP Substrate (Merck Millipore) for 1 min, before exposure to Kodak XAR-5 film for 1–5 min, and the autoradiograms were scanned. Biologically independent replicates (three to four different independent cell cultures) were always performed, and a representative Western blot is shown. The protein abundances were measured by densitometry of the bands on the films using NIH ImageJ version 1.48u4 software and were normalized against the corresponding loading control.
RT-PCR.
Total RNA was purified from neurons using a commercially available kit (Sigma). Cyclin B1 and GAPDH mRNA expression was analyzed by 3% agarose electrophoresis after RT-PCR using the following forward and reverse oligonucleotides, respectively: 5′-CAACTGGAGGAAGAGCAGTCA-3′and 5′-CATCTGAACCTGTATTAGCCA-3′ for Cyclin B1; and 5′- GGGTGTGAACCACGAGAAAT-3′ and 5′- GACTGTGGTCATGAGCCCTT-3′ for GAPDH. RT was performed at 48°C for 50 min, and PCR conditions were 10 min at 95°C, 35 cycles of 30 s at 95°C, 30 s at 52°C, and 1 min at 72°C. Final extension was performed for 10 min at 72°C. In no case was a band detected by PCR without RT.
Cdk1 kinase assay.
Cells were lysed in ice-cold buffer containing 50 mm Tris, pH 7.5, 150 mm NaCl, 2 mm EDTA, and 1% NP-40, supplemented with the phosphatase and protease inhibitors cited above. After clearing debris by centrifugation, extracts (200 μg protein) were incubated with anti-Cdk1 (2 μg) for 4 h at 4°C, followed by the addition of 30 μl of protein A-Sepharose (GE Healthcare) for 2 h at 4°C. Immunoprecipitates were washed four times in lysis buffer and resuspended in kinase buffer (in mm: 50 HEPES, pH 7.5, 10 MgCl2, 1 EDTA, and 0.1 dithiothreitol) containing 20 μm ATP, 2 μCi of [γ-32P]ATP, and histone H1 (1 mg/ml; Sigma-Aldrich). Samples were subjected to SDS-PAGE (12%), and transferred proteins were visualized by autoradiography or blotted with anti-Cdk1.
Coimmunoprecipitation assay.
Cells were lysed in ice-cold buffer containing 50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 2 mm EDTA, and 1% NP-40, supplemented with the phosphatase and protease inhibitors cited in Western blot analysis. Cell extracts were clarified by centrifugation, and supernatants (500 μg of protein) were incubated with 2 μg of anti-Bcl-xL for 4 h at 4°C, followed by the addition of 30 μl of protein A-Sepharose (GE Healthcare) for 2 h at 4°C. Immunoprecipitates were washed extensively with lysis buffer and detected by Western blot analysis (Gomez-Sanchez et al., 2011).
Isolation of mitochondria.
Mitochondrial fractions were isolated as described by Almeida and Medina (1998), which provides a rapid method for isolation of intact functional mitochondria from cultured cells. In brief, cells were washed twice with cold PBS and collected in isolation medium (in mm: 320 sucrose, 1 potassium EDTA, and 10 Tris-HCl, pH 7.4), centrifuged at 600 × g for 5 min (4°C), and resuspended in isolation medium. Cells were homogenized in a tight-fitting glass–Teflon homogenizer (20 strokes), and the nuclei and lysed membranes were removed by centrifugation at 1500 × g for 10 min (4°C). We then centrifuged the supernatant at 17,000 × g for 11 min (4°C), and pellet (mitochondrial fraction) was suspended in isolation buffer. The supernatant contained the cytosolic fraction.
Blue native PAGE and immunoblotting for oxidized protein detection.
Isolated mitochondria (0.5 mg of protein) were treated with 50 mm N-ethylmaleimide (NEM) for 10 min at 37°C, pelleted, and washed with isolation medium (in mm: 320 sucrose, 1 potassium EDTA, and 10 Tris-HCl, pH 7.4). Then, mitochondria were treated with 2 mm dithiothreitol for 10 min and washed with isolation medium. After treating mitochondria with 25 mm iodoacetyl-LC-biotin for 10 min in the dark, mitochondria were incubated in ice-cold digitonin buffer [1% (w/v) digitonin, 1 m 6-aminohexanoic acid, and 50 mm Bis-Tris HCl, pH 7.0] for 5 min in ice. After centrifugation for 30 min at 13,000 × g, the supernatant was collected, and 5% (v/v) Coomassie Brilliant Blue G-250 in 1 m 6-aminohexanoic acid was added to the supernatant. Samples were resolved on a 1-mm-thick 3–12% polyacrylamide gradient gel. Gel was dyed with a nitroblue tetrazolium test to remark the situation of complex I, and the band was excised with a razor blade and incubated for 5 min at room temperature in 125 mm Tris-HCl, pH 7.0, containing 1% SDS and 50 mm NEM before insertion over the 1.5-mm-thick SDS–polyacrylamide gel. Stacking gel was polymerized around the excised bands, and proteins were separated by electrophoresis in a MiniProtean system (Bio-Rad). Oxidized proteins were detected after blotting onto nitrocellulose membranes with streptavidin–horseradish peroxidase conjugate (1:1000; Thermo Fisher Scientific) to visualize biotinylated reagents. Signal detection was performed with an enhanced chemiluminescence kit (Hurd et al., 2008).
Determination of mitochondrial enzyme activities.
Cells were collected by trypsinization, centrifuged, and resuspended in 300 μl of 0.1 m potassium phosphate buffer, pH 7.0. Samples were freeze–thawed three times to ensure cellular disruption. Enzyme activities were measured in cell lysates (80–100 μg of protein) at 30°C using a Kontron spectrophotometer (Uvikon XL; NorthStar Scientific). Nicotinamide adenine dinucleotide (NADH)–CoQ1 reductase (complex I) activity was measured as the rotenone-sensitive rate of NADH oxidation (at 340 nm; Ragan et al., 1987). Cell lysates was incubated in 25 mm potassium phosphate, pH 7.2, containing 0.2 mm NADH, 10 mm MgCl2, 1 mm KCN, and 2.5 mg of fat-free bovine serum albumin (BSA), and the reaction was initiated with 50 μm ubiquinone-1. For measurement of the rotenone-insensitive NADH oxidation activity, the assay was also performed in the presence of 10 μm rotenone.
Succinate–cytochrome c reductase (complex II–III) activity was measured as the antimycin A-sensitive rate of cytochrome c reduction (at 550 nm; King, 1967). The reaction mixture contained 100 mm potassium phosphate, pH 7.4, 0.3 mm EDTA, 1 mm KCN, and 0.1 mm cytochrome c and was initiated with 20 mm succinate. The assay was also performed in the presence of 20 μg/ml antimycin A to determine the actimycin A-insensitive complex II–III activity.
Cytochome c oxidase (complex IV) was measured as the rate of cytochrome c oxidation (at 550 nm; Wharton and Tzagoloff, 1967). The test cuvette contained 10 mm potassium phosphate, pH 7.0, and 50 μm reduced cytochrome c. The reference cuvette also contained 1 mm K3Fe(CN)6. The reaction was initiated by adding cell lysate to the test cuvette, and the first-order velocity constant (k) was calculated.
The ATPase activity was measured in a linked enzyme system as the oligomycin-sensitive rate of NADH oxidation (at 340 nm), using the method of Soper and Pedersen (1979). The oxidation of NADH to NAD+ results from a coupled reaction catalyzed by pyruvate kinase and lactate dehydrogenase in the presence of phosphoenolpyruvate, dependent on ADP production by the ATPase in the presence of ATP. The reaction mixture contained 50 mm Tris-HCl, pH 8.0, 5 mm phosphoenolpyruvate, 2 mm KCN, 0.3 mm NADH, 100 mm HCl, 6 mm MgCl2, 10 μm rotenone, 25 U/ml lactate dehydrogenase, 25 U/ml pyruvate kinase, and 6 mm ATP. The rate inhibited by the addition of 5 μg/ml olygomycin was taken to be the ATPase activity.
Citrate synthase activity was measured as in the study by Shepherd and Garland (1966) (at 412 nm). Cell lysates were incubated in 100 mm Tris-HCl, pH 8.0, 0.1 mm acetyl-CoA, 0.2 mm 5,5′-dithiobis(2-nitrobenzoic acid), 0.1% (v/v) Triton X-100, and 200 μm oxaloacetate.
All enzyme activities were expressed as nanomoles per minute per milligram of protein, except for cytochrome c oxidase, which was expressed as the first-order rate constant (k) per minute per milligram of protein.
Oxygen consumption assay.
Cells were removed from the flasks by mild trypsinization. The rate of oxygen consumption was measured in 106 cells suspended in HBSS (containing 5.5 mm glucose) at 30°C, using a Clark-type dissolved oxygen electrode (Digital model 20; Rank Brothers). Results are expressed as nanomoles of O2 consumed per minute per 106 cells (Almeida et al., 2001).
Immunocytochemistry.
Neurons grown on glass coverslips were fixed with 4% (v/v, in PBS) paraformaldehyde for 30 min and immunostained with mouse anti-Cyclin B1 (1:100) and rabbit anti-Bcl-xL (1:100; BD Biosciences) antibodies (Gomez-Sanchez et al., 2011). Immunolabeling was detected using anti-mouse IgG–Cy3 (1:500) or anti-rabbit IgG–Cy5 (1:500; Jackson ImmunoResearch). Coverslips were washed, mounted in SlowFade light antifade reagent (Invitrogen) on glass slides, and examined using a scanning laser confocal microscope (TSC SL; Leica) with three lasers [multiline argon (458, 488 nm), green helium–neon (543 nm), and red helium–neon (633 nm)] and equipped with 40×, 63× (1.4 numerical aperture) HCX PL Apo oil-immersion objectives for high-resolution imaging.
Flow cytometric detection of apoptotic cell death.
Neurons were detached carefully from the plates using 1 mm EDTA (tetrasodium salt) in PBS, pH 7.4, and were stained with annexin V–adenomatous polyposis coli (APC) and 7-aminoactinomycin D (7-AAD) in binding buffer (in mm: 100 HEPES, 140 NaCl, and 2.5 CaCl2) to determine quantitatively the percentage of apoptotic neurons by flow cytometry. Cells were stained with annexin V–APC and 7-AAD in binding buffer (in mm: 100 HEPES, 140 NaCl, and 2.5 CaCl2), according to the instructions of the manufacturer, and 3 × 105 cells were analyzed, in four replicates per condition, on a FACScalibur flow cytometer (15 mW argon ion laser tuned at 488 nm; CellQuest software; BD Biosciences). Transfected (identified by GFP fluorescence) cell populations were analyzed, and the annexin V–APC-stained cells that were 7-AAD-negative were considered to be apoptotic (Gomez-Sanchez et al., 2011).
Flow cytometric detection of active caspase-3.
Active caspase-3 was detected in the GFP-expressing (GFP+) neurons using the ApoActive3 kit (Bachem). After detaching cells with 1 mm EDTA (tetrasodium salt) and centrifuging, cell pellets were fixed during 20 min, resuspended in PBS plus 2% BSA, and incubated for 1 h with 1× rabbit anti-caspase 3. Cells were then incubated with 1:500 anti-rabbit Cy3 (Jackson ImmunoResearch) for 1 h. Between each step, cells were washed with either PBS (until labeling of samples) or PBS plus 1% BSA and resuspended in PBS plus 1% BSA before analysis by flow cytometry (tuned at 488 nm; CellQuest software; BD Biosciences).
Flow cytometric detection of Δψm and mitochondrial superoxide generation.
Δψm was assessed using the MitoProbe DilC1(5) Assay Kit for Flow Cytometry (Invitrogen), and stained cells were analyzed on the FL1 and FL4 channels of a FACScalibur flow cytometer (15 mW argon ion laser tuned at 488 nm; CellQuest software; BD Biosciences). Δψm values were expressed as percentages, using carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (10 μm) to define the 0% Δψm values. Mitochondrial superoxide production was assessed using the fluorescent MitoSox probe (Invitrogen) by flow cytometry, as described previously (Quintana-Cabrera et al., 2012).
Protein determinations.
Protein concentrations were determined in the cell suspensions, lysates, or in parallel cell culture incubations after solubilization with 0.1 m NaOH. Protein concentrations were determined as described previously (Lowry et al., 1951), using BSA as standard.
Statistical analysis.
All measurements in cell culture were performed, at least, in triplicate, and the results are expressed as the mean ± SEM values from at least three different culture preparations. For the comparisons between two groups of values, the statistical analysis of the results was performed by the Student's t test. For multiple values comparisons, we used one-way ANOVA, followed by Bonferroni's test. The statistical analysis was performed using the SPSS 16.0 software for Macintosh. In all cases, p < 0.05 was considered significant.
Results
Cyclin B1–Cdk1 activity mediates neuronal apoptotic death via the mitochondrial pathway on an excitotoxic stimulus
To investigate the molecular mechanism responsible for cyclin B1-mediated neurodegeneration, neurons were incubated with 100 μm glutamate for 5 min, followed by harvesting at different time points, a previously validated excitotoxic stimulus known to cause cyclin B1 stabilization (Maestre et al., 2008). As shown in Figure 1A, glutamate triggered a time-dependent accumulation of endogenous cyclin B1 in whole-cell extracts, confirming our previous findings (Maestre et al., 2008).
Figure 1.

Cyclin B1–Cdk1 activity mediates neuronal apoptotic death on an excitotoxic stimulus. Rat cortical neurons were treated with glutamate (100 μm, 5 min) and were further incubated in culture medium for 1–20 h. A, Western blots of neuronal extracts were probed sequentially with antibodies to cyclin B1 and Cdk1, followed by GAPDH as loading control. B, CyclinB1 and GAPDH mRNA expression was analyzed by 3% agarose electrophoresis after RT-PCR. GAPDH is a housekeeping gene and used as loading control in PCR. One representative gel is shown of three. Shown is the relative cyclin B1 mRNA levels, as normalized with GAPDH, averaged from three independent neuronal cultures (n = 3). After glutamate stimulation (100 μm, 5 min), neurons were incubated in culture medium containing the transcriptional inhibitor ActD (100 ng/ml), the protein synthesis inhibitor CHX (1 μg/ml), or the APC/C inhibitor ProTAME (10 μm) for 20 h. C, Treatment with ActD or CHX did not prevent glutamate-induced cyclin B1 accumulation in neurons. D, ProTAME promoted neuronal cyclin B1 stabilization. E, Neurons on day 4 in vitro were transfected with an siRNA against luciferase (siControl; 100 nm) or with siRNA against Cdk5 (siCdk5; 100 nm) for 3 d. Knockdown of Cdk5 (siCdk5-treated neurons) prevented cyclin B1 accumulation in glutamate-treated neurons. In A, C, D, and E, a representative Western blot is shown of three. Bar graphs represent the relative cyclin B1 protein abundance, as normalized with GAPDH, averaged from at least three independent neuronal cultures. In all cases, the represented values are means ± SEM (n = 3–4 independent neuronal cultures). *p < 0.05 versus untreated (−Glu) neurons. F, Glutamate triggered a time-dependent stimulation of Cdk1 activity, as assessed by the ability of the protein extracts to phosphorylate, in vitro, histone H1. Transfection with siCdk5 abrogated glutamate-caused Cdk1 activation. Each bar represents the mean ± SEM of three independent neuronal cultures. *p < 0.05 versus siControl 0 h. G, Neurons on day 4 in vitro were transfected with an shRNA against luciferase (Control) or with an shRNA against cyclin B1 (shCyclin B1) for 2 d. Then, neurons were incubated (or not in the Control condition) with glutamate (100 μm, 5 min; Glutamate condition) after incubation with culture medium for the indicated time points. When indicated, Rosc (10 μm) was added to the culture medium. Both shCyclin B1 and Rosc abrogated neuronal apoptotic death caused by glutamate but did not modify apoptosis in control (untreated) neurons (20 h: shCyclin B1, 13.52 ± 0.89%; Rosc, 12.77 ± 0.79%), as assessed by annexin V+/7-AAD− quantification by flow cytometry. Representative flow cytometric dot plots are shown of four independent experiments. Data are the mean ± SEM from four independent neuronal cultures (n = 4). *p < 0.05 versus control; #p < 0.05 versus glutamate. AD, Actinomycin D; MW, molecular weight.
Cyclin B1 mRNA was unaltered (Fig. 1B), and its protein accumulation was not prevented by the transcription inhibitor ActD and the protein synthesis inhibitor CHX (Fig. 1C), indicating that glutamate induced cyclin B1 protein stabilization by a posttranslational mechanism. We reported that APC/C–Cdh1 activity destabilizes cyclin B1 protein as an essential survival mechanism for neurons (Almeida et al., 2005). Moreover, excitotoxic stimulus inactivates APC/C–Cdh1, causing aberrant cyclin B1 stabilization that leads to apoptotic neuronal death (Maestre et al., 2008). Here, we show that the APC/C inhibitor ProTAME mimicked glutamate-induced cyclin B1 accumulation (Fig. 1D). In accordance with the finding that the inactivation of APC/C–Cdh1 during excitotoxicity requires Cdk5 (Maestre et al., 2008), this protein was knocked down by RNA interference using a previously validated siRNA against Cdk5 (siCdk5; Maestre et al., 2008), which resulted in the abolishment of glutamate-induced cyclin B1 accumulation (Fig. 1E). Altogether, these results confirm our previous findings (Maestre et al., 2008) that an excitotoxic stimulus activates Cdk5, which inhibits APC/C, leading to aberrant cyclin B1 accumulation.
During the excitotoxic insult, the increase in cyclin B1 protein paralleled that of Cdk1 activity, as assessed by its ability to phosphorylate histone H1 (Fig. 1F), suggesting functional activation of the cyclin B1–Cdk1 complex. Furthermore, such Cdk1 activation was abolished by siRNA-mediated knockdown of Cdk5 (Fig. 1F), confirming the involvement of Cdk5 in this process. To assess whether the cyclin B1–Cdk1 complex was responsible for neuronal death after the excitotoxic stimulus, we next knocked down cyclin B1 using a previously validated shRNA (shCyclin B1; Almeida et al., 2005) or inhibited Cdk(s) with Rosc. As shown in Figure 1G, both treatments mostly attenuated the time-dependent neuronal apoptotic death triggered by glutamate. Conversely, ectopic expression of cyclin B1 triggered a time-dependent increase in apoptotic neuronal death (Fig. 2A) and loss of cyclin B1-containing (GFP+) neurons (Fig. 2B) that reached the statistical significance after 18 h of transfection. Axonal disruption, characteristic of apoptotic death, was also observed by epifluorescence after 24 h of the ectopic expression of a cyclin B1 (Fig. 2B). Moreover, active caspase-3 was increased in cyclin B1-expressing neurons as assessed by flow cytometry (Fig. 2C), confirming apoptotic death. Cyclin B1-mediated apoptotic death was abolished by the caspase inhibitors zVAD-fmk and zDEVD-fmk, as well as by the caspase-3-specific inhibitor zDQMD-fmk (Fig. 2D). However, inhibiting caspase-8 with zIETD-fmk or caspase-2 with zVDVAD-fmk failed to prevent cyclin B1-mediated apoptotic death (Fig. 2D), ruling out the non-mitochondrial apoptotic pathway in the process. In contrast, inhibition of mitochondrial-dependent caspase-9 with zLEHD-fmk fully prevented cyclin B1-mediated apoptotic death (Fig. 2D). Thus, cyclin B1 induces neuronal apoptotic death via the intrinsic mitochondrial pathway through caspase-3/caspase-9 activation.
Figure 2.

Cyclin B1 induces neuronal apoptotic death via the intrinsic mitochondrial pathway activation. A, Rat cortical neurons were transfected with 0.8 μg/106 cells pIRES2–EGFP, either empty (Control; expressing GFP) or containing the full-length cDNA of human cyclin B1 (Cyclin B1; expressing both cyclin B1 and GFP) for 6–24 h. cyclin B1 expression time dependently induced neuronal apoptotic death, as assessed by annexin V+/7-AAD− quantification by flow cytometry. *p < 0.05 versus control. B, Epifluorescence microphotographs revealed the typical axonal disruption of apoptosis in cyclin B1-transfected neurons (24 h). Scale bar, 50 μm. Transfection rate (percentage of GFP+ neurons) was determined by flow cytometry. *p < 0.05 versus control. C, Cyclin B1 induced caspase 3 cleavage and activation by 24 h after transfection of neurons, as showed by quantitative analyses of GFP+ (transfected) neurons by flow cytometry. *p < 0.05 versus control. D, Rosc (10 μm) or caspase inhibitors (zVAD-fmk and zDEVD-fmk; 100 μm) prevented cyclin B1-mediated neuronal death at 24 h of transfection. Inhibition of caspases-8 (zIETD-fmk; 100 μm) or caspase-2 (zVDVAD-fmk, 100 μm) was ineffective. Caspase-3 (zDQMD-fmk; 100 μm) or caspase-9 inhibition (zLEHD-fmk; 50 μm) abrogated cyclin B1-mediated neuronal death. *p < 0.05 versus control. In all cases, data are the mean ± SEM of four independent neuronal cultures (n = 4).
Cyclin B1–Cdk1 accumulates in mitochondria on the excitotoxic stimulus, leading to oxidative stress and energy deficiency
In view of the evidence for a mitochondrial involvement in cyclin B1-mediated neuronal apoptotic death, we next sought to investigate whether cyclin B1, and its catalytic partner Cdk1, occurred in this organelle. Interestingly, glutamate treatment promoted the accumulation of endogenous cyclin B1 and Cdk1 in mitochondria purified from neurons, as revealed by Western blotting (Fig. 3A). This was confirmed in HEK293T cells transfected with the full-length cDNA encoding cyclin B1 (Fig. 3B).
Figure 3.
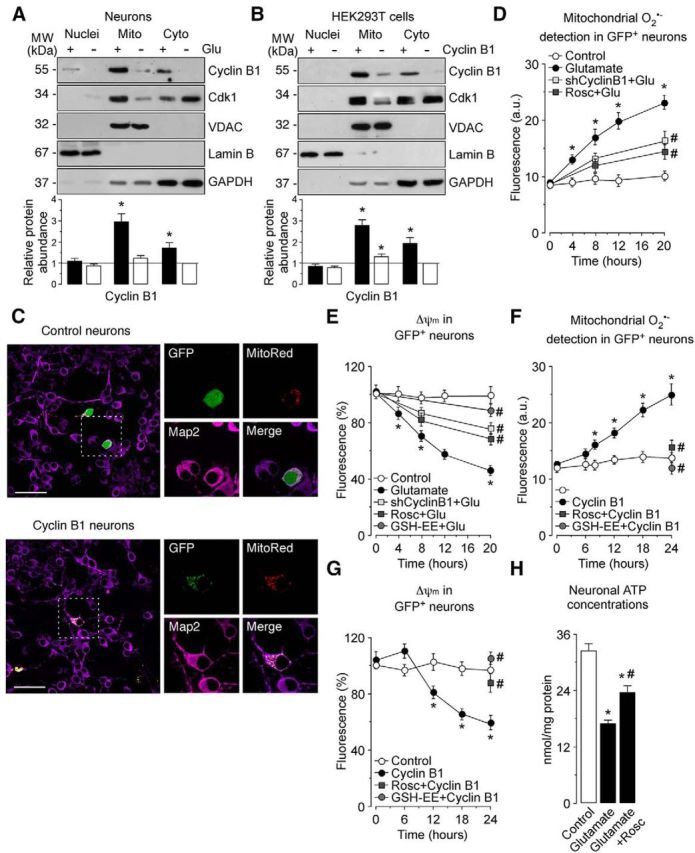
cyclin B1–Cdk1 accumulates in mitochondria on the excitotoxic stimulus, leading to oxidative stress and energy deficiency. A, Neurons were treated with 100 μm glutamate for 5 min and were further incubated in culture medium for 20 h. Western blots of nuclear (Nuclei), mitochondrial (Mito), and cytosolic (Cyto) extracts were probed sequentially with antibodies to cyclin B1 and Cdk1, followed by VDAC as mitochondrial marker, Lamin B as nuclear marker, and GAPDH as cytosolic marker. A representative Western blot is shown of three. Bar graphs represent the relative cyclin B1 protein abundance compared with the cytosolic fraction from untreated (−Glu) cells. Each bar represents the mean ± SEM of three independent neuronal cultures (n = 3). *p < 0.05 versus untreated neurons. B, HEK293T cells were transfected with 0.8 μg/106 cells pIRES2–EGFP, either empty (expressing GFP) or containing the full-length cDNA of human Cyclin B1 (expressing both cyclin B1 and GFP) for 24 h. Western blot were performed as in A. Bar graphs represent the relative cyclin B1 protein abundance compared with cytosolic fraction from control (−Cyclin B1) cells. Each bar represents the mean ± SEM of three independent cell cultures (n = 3). *p < 0.05 versus control cells. C, Neurons were cotransfected with plasmid encoding either GFP (0.8 μm/106 cells pcDNA3–GFP; control) or cyclin B1 fused with GFP (0.8 μm/106 cells pcDNA3–cyclin B1-GFP; cyclin B1-GFP) and the mitochondrial marker MitoRed (0.8 μm/106 cells pMitoDsRed2) for 12–14 h. Microphotograph reveals colocalization of cyclin B1–GFP, but not GFP (control), with MitoRed in neurons. Neurons were immunostained with the neuronal marker Map2 (violet). Scale bar, 50 μm. D, Neurons on day 4 in vitro were transfected with an shRNA against luciferase (Control) or with an shRNA against cyclin B1 (shCyclin B1) for 2 d. Then, neurons were treated with 100 μm glutamate for 5 min and were further incubated in culture medium for 4–20 h. When indicated, Rosc (10 μm) was added to the culture medium. Glutamate time dependently induced mitochondrial O2·− generation in neurons. Both shCyclin B1 and Rosc partially abrogated glutamate-caused mitochondrial O2·− generation in neurons but did not modify values in control (untreated) neurons (20 h: shCyclin B1, 10.39 ± 0.67%; Rosc, 9.33 ± 0.54%). *p < 0.05 versus control; #p < 0.05 versus glutamate. E, Neurons were transfected and treated as in D. Glutamate time dependently induced mitochondrial depolarization in neurons. Treatment with shCyclin B1, Rosc (10 μm), and GSH-EE (1 mm) prevented glutamate-caused Δψm collapse in neurons. However, these treatments did not modify values in control (untreated) neurons (20 h: shCyclin B1, 105.4 ± 8.79%; Rosc, 109.3 ± 9.25%; GSH-EE, 112.7 ± 7.74). *p < 0.05 versus control; #p < 0.05 versus glutamate. F, Neurons were transfected with 0.8 μg/106 cells pIRES2–EGFP, either empty (Control; expressing GFP) or containing the full-length cDNA of human cyclin B1 (Cyclin B1; expressing both cyclin B1 and GFP) for 4–24 h. When indicated, neurons were treated with Rosc (10 μm) and GSH-EE (1 mm). Cyclin B1 expression time dependently induced mitochondrial O2·− generation in neurons, as assessed by flow cytometry. Both Rosc and GSH-EE fully prevented cyclin B1-mediated mitochondrial O2·− generation in neurons but did not modify values in control neurons (24 h: Rosc, 11.3 ± 0.87%; GSH-EE, 7.8 ± 0.64). G, Neurons were transfected and treated as in F. Expression of cyclin B1 time dependently induced mitochondrial depolarization in neurons. Treatments with Rosc (10 μm) and GSH-EE (1 mm) abrogated glutamate-caused Δψm collapse in neurons. However, these treatments did not modify values in control neurons (24 h: Rosc, 102.3 ± 8.49%; GSH-EE, 109.7 ± 9.21). *p < 0.05 versus control. H, Neurons were transfected and treated as in D. Rosc (10 μm) partially prevented ATP depletion caused by glutamate. *p < 0.05 versus control; #p < 0.05 versus glutamate. In D–H, data are the mean ± SEM from four independent neuronal cultures (n = 4). MW, Molecular weight.
To further corroborate the occurrence of cyclin B1 in mitochondria, neurons were cotransfected with MitoRed—which delivers the red fluorescent protein specifically into mitochondria—and cyclin B1–GFP (or GFP alone as control) cDNA constructs. Confocal images showed that MitoRed fluorescence colocalized with that of GFP in neurons transfected with the cyclin B1–GFP fusion cDNA (cyclin B1 neurons) but not with those transfected with GFP alone (control neurons; Fig. 3C). Thus, the accumulation of cyclin B1 protein during either glutamate treatment or exogenous cyclin B1 expression occurs primarily in mitochondria. Because glutamate-mediated excitotoxicity is known to increase mitochondrial superoxide (O2·−) production and inner membrane depolarization, we next investigated the possible involvement of mitochondrial cyclin B1 in these phenomena. As shown in Figure 3D, mitochondrial O2·− time dependently increased from 4 h after the excitotoxic stimulus, an effect that was mostly attenuated but not abolished, by knocking down cyclin B1 with shCyclin B1, or by inhibiting Cdk1 with Rosc. Moreover, glutamate time dependently decreased Δψm, which was prevented partially by shCyclin B1 or Rosc (Fig. 3E), mirroring the effects on mitochondrial O2·− (Fig. 3D). Interestingly, incubation of neurons with the free radical scavenger GSH-EE nearly abolished glutamate-induced Δψm collapse (Fig. 3E), suggesting that increased O2·− preceded Δψm loss. Likewise, the ectopic expression of cyclin B1 mimicked glutamate treatment, because neurons displayed a time-dependent increase in O2·− (Fig. 3F) and Δψm loss (Fig. 3G). Furthermore, these effects were rescued completely by either Rosc (Fig. 3F) or GSH-EE (Fig. 3G). Thus, our time course study shows that Δψm loss precedes apoptotic death on the excitotoxic stimulus, indicating that mitochondrial dysfunction is a cause, not a consequence, of the neuronal apoptotic process, thus confirming the importance for Δψm maintenance in neuronal survival (White and Reynolds, 1996; Almeida and Bolaños, 2001). Because Δψm loss on excitotoxic damage is often associated with energy depletion, we determined the concentration of neuronal ATP, which was decreased (Fig. 3H); this effect was prevented partially by Rosc, indicating the contribution of Cdk activity in energy depletion. Thus, glutamate-mediated cyclin B1–Cdk1 activation in mitochondria induces oxidative stress, leading to mitochondrial dysfunction and energy deficiency.
Cyclin B1–Cdk1 activation inhibits ATP synthase, leading to oxidative damage to complex I
Next, we aimed to understand the molecular mechanism responsible for mitochondrial dysfunction by cyclin B1–Cdk1 activity. To do so, we first analyzed the mitochondrial respiratory chain in HEK293T cells transfected with the full-length cDNA encoding cyclin B1. As expected, cyclin B1 protein levels time dependently increased in the transfected cells (Fig. 4A). As shown in Figure 4B, cyclin B1 induced a time-dependent inhibition in mitochondrial complex I activity; however, complexes II–III and IV were unaffected (complex II–III: control, 2.73 ± 0.11 nmol · min−1 · mg−1 protein; cyclin B1, 2.60 ± 0.13 nmol · min−1 · mg−1 protein; complex IV: 0.33 ± 0.03, 0.27 ± 0.03 k · min−1 · mg−1 protein; citrate synthase: control, 135 ± 10.3; cyclin B1, 141 ± 11.9). Interestingly, we also found that ATP synthase activity was decreased on cyclin B1 expression (Fig. 3C). Notably, inhibition of ATP synthase activity by cyclin B1 preceded (12 h; Fig. 3C) that of complex I (18 h; Fig. 3B). In view of the inhibitory effect of cyclin B1 on complex I, we sought to investigate whether the mitochondrial respiration was impaired. As shown in Figure 4D, the rate of O2 consumption showed a biphasic effect on cyclin B1 expression, being increased at 12 h and decreased at 24 h. It is noteworthy that the increase in O2 respiration (12 h; Fig. 4D) occurs at a time point in which complex I was unaffected (Fig. 4B) and ATP synthase was inhibited (Fig. 4C). In view of this intriguing phenomenon, we were prompted to investigate the mechanism responsible for this effect.
Figure 4.

Cyclin B1–Cdk1 activation inhibits ATP synthase, leading to oxidative damage to complex I. HEK293T cells were transfected with 0.8 μg/106 cells pIRES2–EGFP, either empty (Control; expressing GFP) or containing the full-length cDNA of human cyclin B1 (Cyclin B1; expressing both cyclin B1 and GFP). A, Western blots of cell extracts were probed sequentially with antibodies to cyclin B1, followed by GAPDH as loading control. A representative Western blot is shown of three. Bar graphs represent the relative cyclin B1 protein abundance compared with 0 h (immediately after transfection). Each bar represents the mean ± SEM of three independent cell cultures (n = 3). *p < 0.05 versus 0 h. B, Mitochondrial complex I activity was measured by spectrometry at different times after transfections. *p < 0.05 versus control. C, ATP synthase activity was measured by spectrometry at different times after transfections. *p < 0.05 versus control. D, HEK293T cells were suspended in HBSS, and the rate of oxygen consumption was measured at different times after transfections by using with a Clark-type dissolved oxygen electrode. *p < 0.05 versus control. E, Δψm was assessed by flow cytometry in HEK293T at different times after transfections. *p < 0.05 versus control. F, HEK293T cells were transfected with 0.8 μg/106 cells pIRES2–EGFP (control) or pIRES2–cyclin B1–EGFP (cyclin B1) either in the absence of presence of Rosc (10 μm) and GSH-EE (1 mm) for 24 h. Cyclin B1 induced complex I oxidation, as judged by the increase in oxidized proteins detected by immunoblotting from isolated biotin–NEM-labeled mitochondrial complex I subunits, which was abrogated by Rosc and GSH-EE. A representative Western blot is shown of three. Bar graphs represent the relative band intensity compared with control. Each bar represents the mean ± SEM of three independent Western blots (n = 3). *p < 0.05 versus control; #p < 0.05 versus cyclin B1. G, H, HEK293T cells were transfected and treated as in F. Mitochondrial complex I activity (G) and ATP synthase activity (H) were measured by spectrometry at 24 h after transfections. *p < 0.05 versus control; #p < 0.05 versus cyclin B1. In B–E, G, and H, data are expressed as the mean ± SEM, from three or four independent cell cultures (n = 3–4). MW, Molecular weight.
Because ATP synthase inhibition is known to induce transient mitochondrial inner membrane hyperpolarization (Brand et al., 2004; Sánchez-Cenizo et al., 2010; Formentini et al., 2014), we next assessed Δψm in cells expressing cyclin B1. As shown in Figure 4E, Δψm increased at 12 h, and it was followed by a decrease at 24 h of transfection, thus mimicking the same time-dependent changes of cell O2 consumption (Fig. 4D). Thus, it is tempting to suggest that cyclin B1 primarily inhibited ATP synthase, leading to increased mitochondrial O2 consumption, Δψm and O2·−, which secondarily will lead to damage to complex I. In fact, increased Δψm enhances mitochondrial O2·− generation (Brand et al., 2004; Sánchez-Cenizo et al., 2010; Formentini et al., 2014), and, in neurons, cyclin B1 increased Δψm (although it did not reach statistical significance; Fig. 3G) and mitochondrial O2·− (Fig. 3F). Moreover, because mitochondrial O2·− causes primarily complex I oxidative damage (Bolaños et al., 2009), we then assessed whether cyclin B1–Cdk1 activity causes oxidative damage to complex. As shown in Figure 4F, cyclin B1 induced complex I oxidation, as judged by the increase in oxidized proteins detected by immunoblotting from isolated biotin–NEM-labeled mitochondrial complex I subunits. Complex I oxidation was prevented by GSH-EE (Fig. 4F) and Rosc (Fig. 4F). Furthermore, complex I inhibition caused by cyclin B1 expression in HEK293T cells was rescued by Rosc and GSH-EE (Fig. 4G). These results strongly suggest complex I oxidative damage by cyclin B1–Cdk1 activity. Moreover, ATP synthase inhibition caused by cyclin B1 expression in HEK293T cells was rescued by Rosc but not by GSH-EE, suggesting that cyclin B1–Cdk1-mediated inhibition of ATP synthase was not oxidative (Fig. 4H). Thus, we propose that the sequence of events occurring by cyclin B1–Cdk1 activation first starts by inhibiting ATP synthase, which leads to enhanced O2 consumption and Δψm, which stimulates O2·−. Such an increase in O2·− causes complex I oxidative damage, leading to the delayed impairment of cell O2 consumption.
Cyclin B1–Cdk1 phosphorylates Bcl-xL, leading to its dissociation from β-F1Fo–ATP synthase and causing ATP synthase and complex I inhibition on the excitotoxic stimulus
We next sought to investigate the molecular mechanism whereby cyclin B1–Cdk1 inhibited ATP synthase. According to a recent study, the enzymatic activity of ATP synthase can be activated by direct interaction of its β-subunit with Bcl-xL in neurons (Alavian et al., 2011). Conversely, in tumor cells, cyclin B1–Cdk1 phosphorylates and inactivates some members of the Bcl-2 protein family (Harley et al., 2010; Terrano et al., 2010; Sakurikar et al., 2012). Thus, we hypothesized whether the inhibition of ATP synthase activity on glutamate excitotoxicity was the consequence of a putative interference of cyclin B1–Cdk1 with the interaction between Bcl-xL and β-F1Fo–ATP synthase. In fact, confocal imaging revealed that endogenous cyclin B1 and Bcl-xL colocalized in neurons after the excitotoxic stimulus (Fig. 5A). In good agreement with our Western blot analyses (Fig. 1A), cyclin B1 was absent in untreated neurons as assessed by confocal microscopy (Fig. 5A). To further confirm cyclin B1 and Bcl-xL colocalization, neurons were cotransfected with cDNA constructs encoding either GFP (control) or cyclin B1–GFP, with Bcl-xL. The results show that, whereas GFP showed a spread subcellular localization, cyclin B1–GFP colocalized with Bcl-xL (Fig. 5B). Furthermore, Bcl-xL coimmunoprecipitated with both expressed cyclin B1 (Fig. 5C) or endogenously accumulated cyclin B1 during glutamate treatment (Fig. 5D). Rosc, which binds to the ATP binding site of Cdk1 catalytic domain (De Azevedo et al., 1997), abolished such interactions (Fig. 5C, D), suggesting Bcl-xL phosphorylation by cyclin B1–Cdk1. To test this possibility directly, protein extracts obtained from either HEK293T cells expressing cyclin B1 or glutamate-treated neurons were immunoprecipitated with anti-Bcl-xL, followed by anti-phospho-Ser Western blotting. The results showed that either exogenously expressed (Fig. 6A) or endogenously accumulated (Fig. 6B) cyclin B1 phosphorylated Bcl-xL, an effect that was abrogated by Rosc (Fig. 6A,B). Next, we determined whether mitochondrial cyclin B1–Cdk1-mediated phosphorylation of Bcl-xL accounts for ATP synthase inhibition. Bcl-xL may occur at the outer mitochondrial membrane (Vander Heiden et al., 1997; Billen et al., 2008; Ding et al., 2014); however, other authors have described its localization at either the inner membrane or matrix, in which it would interact with the β-F1Fo–ATP synthase to regulate neuronal bioenergetic efficiency of neurons (Alavian et al., 2011; Chen et al., 2011). Here, we aimed to determine the submitochondrial fraction in which cyclin B1, Bcl-xL, and β-F1Fo–ATP synthase colocalized in neurons on the excitotoxic stimulus. To do this, mitoplasts were prepared by incubating mitochondria in hypotonic buffer (70 mm sucrose), and then proteins were analyzed by Western blot. In good agreement with the rupture of the outer mitochondrial membrane occurring at 70 mm sucrose, Tom20, an outer mitochondrial membrane protein marker, was lost (Fig. 6C). However, cyclin B1, Bcl-xL, and β-F1Fo–ATP synthase proteins were still present in these mitoplast extracts, similar to the mitochondrial matrix protein Hsp60 and the inner membrane protein Ndusf1, a complex I subunit (Fig. 6C). To further corroborate the submitochondrial localization of cyclin B1, a protease-protection assay was performed in mitochondria isolated from glutamate-treated neurons using trypsin to digest exposed proteins. As showed in Figure 6D, similarly to Hsp60 and Ndusf1, but unlike Tom20, cyclin B1, Bcl-xL, and β-F1Fo–ATP synthase were protected from the trypsin digestion. These data demonstrate that cyclin B1, Bcl-xL, and β-F1Fo–ATP synthase colocalize in the mitochondrial compartment, either inner membrane or matrix, on a neuronal excitotoxic stimulus.
Figure 5.
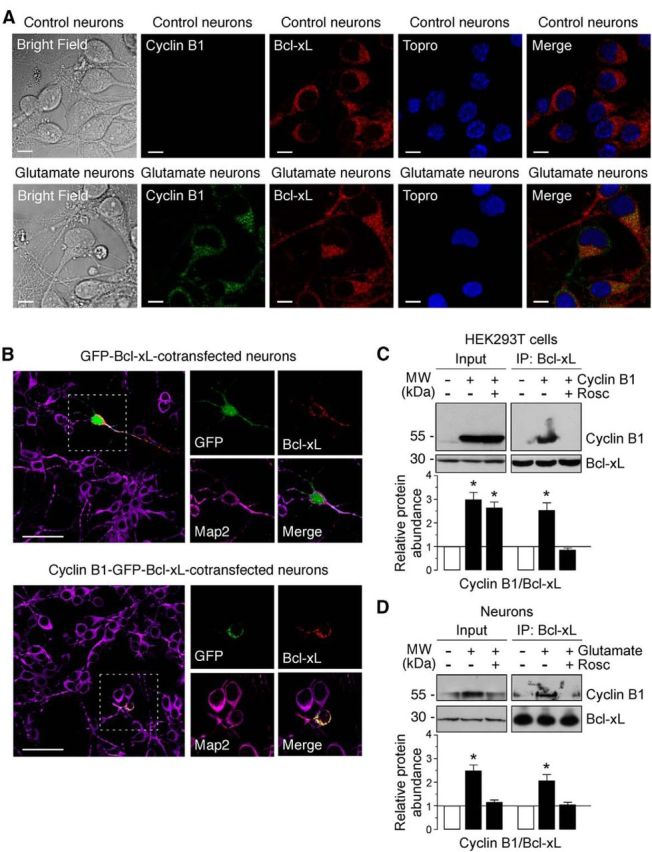
Cyclin B1–Cdk1 localizes to mitochondria in neurons on excitotoxic stimulus. A, Neurons were treated or not (control) with 100 μm glutamate for 5 min and were further incubated in culture medium for 12–14 h. Cyclin B1 and Bcl-xL were detected by immunocytochemistry. Scale bars, 10 μm. Microphotograph reveals colocalization of cyclin B1 with Bcl-xL in neurons. B, Neurons were cotransfected with plasmids encoding either GFP (0.8 μm/106 cells pcDNA3–GFP; control) or cyclin B1 fused with GFP (0.8 μm/106 cells pcDNA3–cyclin B1–GFP; cyclin B1–GFP) and Bcl-xL (0.8 μm/106 cells p8–Bcl-xL) for 12–14 h. Microphotograph reveals that cyclin B1–GFP, but not GFP (control), colocalizes with Bcl-xL. Neurons were immunostained with the neuronal marker Map2 (violet). Scale bars, 50 μm. C, HEK293T cells were transfected with 0.8 μg/106 cells pIRES2–EGFP, either empty (−Cyclin B1) or containing the full-length cDNA of human cyclin B1 (Cyclin B1), in either the absence or presence of Rosc (10 μm). At 24 h after transfections, cellular extracts were obtained and immunoprecipitated with anti-Bcl-xL antibody and analyzed by Western blot for cyclin B1 and Bcl-xL. Of the whole cellular extracts used for immunoprecipitation, 10% were loaded on SDS-PAGE as an input control. Coimmunoprecipitation assays revealed that cyclin B1 coprecipitated with Bcl-xL. A representative Western blot is shown of three. Bar graphs represent the relative band intensity compared with control. Each bar represents the mean ± SEM of three independent cell cultures (n = 3). *p < 0.05 versus control (−Cyclin B1). D, Neurons were treated or not (−Glu) with 100 μm glutamate for 5 min and were further incubated in culture medium for 20 h, in either the absence or presence of Rosc (10 μm). Coimmunoprecipitation assay was performed as indicated in C. Cyclin B1 coprecipitated with Bcl-xL in neurons on excitotoxic stimulus. A representative Western blot is shown of three. Bar graphs represent the relative band intensity compared with control. Each bar represents the mean ± SEM of three independent neuronal cultures (n = 3). *p < 0.05 versus untreated (−Glu) neurons. MW, Molecular weight.
Figure 6.

cyclin B1–Cdk1 phosphorylates Bcl-xL, leading to its dissociation from β-F1Fo–ATP synthase on the excitotoxic stimulus. A, HEK293T cells were transfected with 0.8 μg/106 cells pIRES2–EGFP, either empty or containing the full-length cDNA of human cyclin B1, in either the absence of presence of Rosc (10 μm). At 24 h after transfections, cellular extracts were obtained and immunoprecipitated with anti-Bcl-xL antibody and analyzed by Western blot for phospho-Serine (PSer) and Bcl-xL. Expression levels of cyclin B1 (Input) is shown in Figure 5C. B, Neurons were treated or not with 100 μm glutamate for 5 min and were further incubated in culture medium for 20 h. When indicated, Rosc (10 μm) was added to the culture medium. Neuronal extracts were immunoprecipitated with anti-Bcl-xL antibody and analyzed by Western blot for phospho-Serine (PSer) and Bcl-xL. C, D, Neurons were exposed to glutamate as in B, and submitochondrial localization of cyclin B1, Bcl-xL, and β-F1Fo–ATP synthase was detected by mitoplasting (C) and protease protection assay (D). C, Mitochondria isolated from untreated (−Glu) or glutamate-treated (+Glu) neurons were incubated in isosmotic (320 mm) or hypotonic (70 mm) sucrose buffer, and extracts were subjected to Western blot analysis with cyclin B1, Bcl-xL, β-ATPase, Hsp60 (a matrix protein), Ndusf1 (and inner membrane protein), and Tom20 (an outer membrane protein). D, Mitochondria isolated from glutamate-treated neurons were incubated in mitochondrial buffer with or without 25 or 50 μg/ml soybean trypsin. Extracts were analyzed by immunoblotting with indicated antibodies. E, HEK293T cells were transfected as described in A. Cellular extracts were immunoprecipitated with anti-Bcl-xL antibody and analyzed by Western blot for β-F1Fo–ATP synthase (βATPase) and Bcl-xL. Of the whole cellular extracts used for immunoprecipitation, 10% were loaded on SDS-PAGE as an input control. Coimmunoprecipitation assays revealed that β-F1Fo–ATP synthase failed to coprecipitate with Bcl-xL in cyclin B1-transfected cells, which was prevented by Rosc. F, Neurons were exposed to glutamate as in B, and neuronal extracts were immunoprecipitated as in E. Coimmunoprecipitation assays revealed that glutamate avoided β-F1Fo–ATP synthase and Bcl-xL interaction in neurons, which was prevented by Rosc. In A, B, E, and F, a representative Western blot is shown of three. Bar graphs represent the relative band intensity compared with −Cyclin B1 (A, E) or −Glu (B, F). Each bar represents the mean ± SEM of three independent cell (A, E) or neuronal (B, F) cultures (n = 3). *p < 0.05 versus −Cyclin B1 (A, E) or − Glu (B, F). MW, Molecular weight.
Next, the interaction between Bcl-xL and β-F1Fo–ATP synthase (βATPase) was analyzed in neurons during an excitotoxic stress. The interaction of Bcl-xL with βATPase was disrupted by either exogenously expressed cyclin B1 in HEK293T cells (Fig. 6E) or by endogenous cyclin B1, accumulated during excitotoxic stress (Fig. 6F). Rosc rescued the cyclin B1–Cdk1-mediated Bcl-xL–βATPase interaction (Fig. 6E,F). These data indicate that glutamate-mediated cyclin B1–Cdk1 activation promotes Bcl-xL phosphorylation, resulting in its dissociation from β-F1Fo–ATP synthase. To elucidate whether such a mechanism elicits any functional consequence on ATP synthase activity, we performed site-directed mutagenesis to generate Bcl-xL(D) and Bcl-xL(A) (Abbott et al., 1986). Because Ser-62 in Bcl-xL is a high score putative phosphorylation target of Cdk1 (Terrano et al., 2010), we decided to mutate this residue. HEK293T cells were cotransfected with cyclin B1 together with either Bcl-xL(A) or Bcl-xL(D). We first confirmed that the phosphodefective Bcl-xL mutant Bcl-xL(A) was not phosphorylated by cyclin B1–Cdk1 (Fig. 7A). As revealed by the coimmunoprecipitation assay, βATPase interacted with the phosphodefective, but not with the phosphomimetic, Bcl-xL mutant (Fig. 7B). Furthermore, the expression of the phosphodefective, but not the phosphomimetic, Bcl-xL mutant in neurons restored the interaction of Bcl-xL with the β-F1Fo–ATP synthase on the excitotoxic stimulus (Fig. 7C). These results demonstrate that glutamate-induced Bcl-xL phosphorylation at Ser42 disrupts the Bcl-xL–β-subunit interaction, hence explaining the mechanism of the previously reported inhibition of ATP synthase activity (Alavian et al., 2011). In fact, expression of the phosphodefective Bcl-xL (Fig. 7D) mutant form exerted a dominant-negative effect, because it was unable to inhibit ATP synthase activity during cyclin B1 expression (Fig. 7E). However, expression of the phosphomimetic form (Fig. 7D) imitated the effect of endogenous Bcl-xL at inhibiting ATP synthase activity (Fig. 7E). These results indicate that cyclin B1–Cdk1 mediated phosphorylation of Bcl-xL at Ser62, triggering its dissociation from βATPase, is sufficient to inhibit ATP synthase activity. Finally, we aimed to elucidate whether inhibition of ATP synthase activity in this manner accounted for complex I inactivation. As shown in Figure 7F, the inhibition of complex I activity caused by cyclin B1 was abolished by coexpressing the phosphodefective, but not the phosphomimetic, form of Bcl-xL. Thus, ATP synthase inhibition by cyclin B1–Cdk1 is sufficient to promote complex I impairment.
Figure 7.

Cyclin B1–Cdk1-induced Bcl-xL phosphorylation leading to its dissociation from β-F1Fo–ATP synthase causes ATP synthase and complex I inhibition on the excitotoxic stimulus. A, HEK293T cells were cotransfected with 0.8 μg/106 cells pIRES2–EGFP, either empty (Control) or containing the full-length cDNA of human cyclin B1, together with 0.8 μg/106 cells P8, either empty (Control) or containing the full-length cDNA of human Bcl-xL, the phosphodefective Bcl-xL(A) or the phosphomimetic Bcl-xL(D) Ser62–Bcl-xL mutants. Cell extracts were immunoprecipitated with anti-Bcl-xL antibody and analyzed by Western blot for phospho-Serine (PSer) and Bcl-xL. B, HEK293T cells were cotransfected as in B. Coimmunoprecipitation assay revealed that βATPase coprecipitated with Bcl-xL(A) but not with Bcl-xL(D). C, Neurons were transfected with 0.8 μg/106 cells P8, either empty or containing the full-length cDNA of human phosphodefective Bcl-xL(A) or phosphomimetic Bcl-xL(D) Ser62-Bcl-xL mutants. Neurons were then treated or not (−Glu) with 100 μm glutamate for 5 min and were further incubated in culture medium for 20 h. Coimmunoprecipitation assay reveals that the Bcl-xL(A), but not the Bcl-xL(D), mutant restored the interaction of Bcl-xL with the β-F1Fo–ATP synthase in neurons on the excitotoxic stimulus. In B and C, a representative Western blot is shown of three. Bar graphs represent the relative band intensity compared with −Cyclin B1 (B) or −Glu (C). Each bar represents the mean ± SEM of three independent cell (B) or neuronal (C) cultures (n = 3). *p < 0.05 versus −Cyclin B1 (B) or − Glu (C). E, F, HEK293T cells were cotransfected as in B. The expression of Bcl-xL(A), but not Bcl-xL(D), fully prevented inhibition of ATP synthase (E) and complex I (F) activities in cyclin B1-expressing cells. *p < 0.05 versus −Cyclin B1. Data are expressed as the mean ± SEM of three independent cell cultures (n = 3). MW, Molecular weight.
Cyclin B1–Cdk1-induced Bcl-xL phosphorylation accounts for oxidative stress and mitochondrial dysfunction on excitotoxic stimulus
We finally sought to ascertain whether Bcl-xL phosphorylation is essential in the signaling cascade leading to oxidative stress, Δψm collapse, and neuronal apoptotic death during glutamate excitotoxicity. To do so, we used either exogenously expressed cyclin B1 or endogenously accumulated cyclin B1 during glutamate treatment, both in neurons. We found that expression of the phosphodefective mutant form of Bcl-xL fully prevented the increase in mitochondrial O2·− (Fig. 8A), Δψm collapse (Fig. 8B), and apoptotic death (Fig. 8C) triggered by cyclin B1. However, these phenomena were unaffected by expression of the phosphomimetic form of Bcl-xL (Fig. 8A–C). Furthermore, glutamate-mediated increase in mitochondrial O2·− (Fig. 8D), Δψm collapse (Fig. 8E), and apoptotic death (Fig. 8F) were prevented by expressing the phosphodefective Bcl-xL mutant but not by its phosphomimetic form (Fig. 8D–F). These results indicate that Ser62 phosphorylation of Bcl-xL accounts for oxidative stress, mitochondrial dysfunction, and the proapoptotic effect of cyclin B1–Cdk1 in excitotoxicity. To further confirm this, we assessed whether Ser62 mutants affected the anti-apoptotic function of Bcl-xL. To do so, we expressed its phosphodefective (A) mutant, which fully prevented etoposide-induced neuronal apoptosis (Fig. 8G). Conversely, expression of its phosphomimetic (D) form failed to protect neurons from etoposide-mediated neurotoxicity. These data are in good agreement with previous findings showing that Bcl-xL is phosphorylated at residue Ser62, hence abolishing its anti-apoptotic function, in response to anti-mitotic drugs in cancer cells (Schmitt et al., 2007; Upreti et al., 2008; Antony et al., 2010).
Figure 8.

Cyclin B1–Cdk1-induced Bcl-xL phosphorylation accounts for oxidative stress and mitochondrial dysfunction on the excitotoxic stimulus. A–C, Neurons cells were cotransfected with 0.8 μg/106 cells pIRES2–EGFP, either empty (Control) or containing the full-length cDNA of human cyclin B1, together with 0.8 μg/106 cells P8, either empty (Control) or containing the full-length cDNA of human phosphodefective Bcl-xL(A) or phosphomimetic, Bcl-xL(D) Ser62-Bcl-xL mutants. Mitochondrial O2·− generation (A), mitochondrial membrane potential (Δψm) (B), and neuronal apoptotic death (annexin V+/7-AAD− neurons) (C) were assayed by flow cytometry in transfected (GFP+) neurons. The expression of Bcl-xL(A), but not Bcl-xL(D), prevented cyclin B1-induced oxidative stress (A), Δψm loss (B), and apoptosis (C) in neurons. *p < 0.05 versus −Cyclin B1 (control). D–F, Neurons were transfected with 0.8 μg/106 cells P8, either empty (control) or containing human phosphodefective Bcl-xL(A) or phosphomimetic Bcl-xL(D) Ser62–Bcl-xL mutants. Neurons were then treated or not (−Glu) with 100 μm glutamate for 5 min and were further incubated in culture medium for 20 h. The expression of Bcl-xL(A), but not Bcl-xL(D), prevented glutamate-induced oxidative stress (D), Δψm loss (E), and apoptosis (F) in neurons. *p < 0.05 versus −Glu (control); #p < 0.05 versus glutamate. G, Neurons were transfected as in D and then were exposed to etoposide (10 μm) for 24 h. The expression of Bcl-xL(A), but not Bcl-xL(D), prevented glutamate-induced neuronal apoptosis. *p < 0.05 versus −Etoposide (control). In all cases, data are expressed as the mean ± SEM of three or four independent neuronal cultures (n = 3–4).
Discussion
We have shown previously that an excitotoxic challenge by overactivation of NMDA receptors triggers the stabilization of cyclin B1 because of a cascade of events involving Ca2+-mediated activation of calpain to breakdown p35 into p25, which strongly activated Cdk5 activity in the nucleus (Maestre et al., 2008). Active Cdk5 hyperphosphorylated Cdh1, leading to its dissociation from APC/C, which causes the inhibition of Cdh1 ability to coactivate APC/C (Jaquenoud et al., 2002; Maestre et al., 2008). Here, we now demonstrate that the stabilization of cyclin B1 after this excitotoxic challenge occurs selectively in mitochondria. In a previous work, forced expression of a mutant form of cyclin B1 harboring a mitochondrial leading sequence in dividing cells exerted the coordination of the mitochondrial respiration with cell-cycle progression, especially the G2/M transition (Wang et al., 2014). However, recruitment of endogenous cyclin B1, as we herein show, has not been reported so far. This result is intriguing, because naive cyclin B1 apparently lacks a mitochondrial leading sequence. In addition, postmitotic neurons do not need to exert this coordination (Wang et al., 2014), which is an exclusive function of proliferating cells. Thus, the mechanism whereby cyclin B1 specifically recruits to mitochondria and the consequence(s) of such a subcellular localization remain unknown.
We show that Cdk1 is responsible for the recruitment of cyclin B1 to mitochondria, because both proteins colocalize in the organelle. Furthermore, because we found that Bcl-xL phosphorylated at Ser62, i.e., its Cdk1-cognate phosphorylating residue (Terrano et al., 2010), the formation of a cyclin B1–Cdk1 complex in mitochondria is highly likely. In good agreement with previous studies performed in cancer cells (Schmitt et al., 2007; Upreti et al., 2008; Antony et al., 2010), phosphorylation at Ser62 lost the anti-apoptotic activity of Bcl-xL in neurons, suggesting its involvement in mitochondrial dysfunction during glutamate treatment. However, because its phosphomimetic form failed to affect mitochondrial membrane potential and oxidative stress, additional mechanisms cannot be disregarded. Bcl-xL is known to interact with the β subunit of the F1Fo–ATP synthase to form a stable complex and maximize the efficiency of production of ATP (Alavian et al., 2011). Here, we show that phosphorylation of Ser62 in Bcl-xL is sufficient to promote its dissociation from the β subunit of the F1Fo–ATP synthase, leading to inhibition of ATP synthase enzymatic activity. Inhibition of ATP synthase activity is known to affect the Δψm (Brand et al., 2004; Sánchez-Cenizo et al., 2010; Formentini et al., 2014). In fact, in our hands, glutamate exerted a cyclin B1-mediated biphasic effect on Δψm, namely a transient increase followed by a delayed collapse. During the increased Δψm phase, the mitochondrial respiratory chain is known to stimulate O2·− (Sánchez-Cenizo et al., 2010; Formentini et al., 2014), which we found in neurons on the excitotoxic stimulus. Mitochondrial complex I contains O2·−-sensitive Fe-S clusters (Sazanov, 2014) and specific cysteine residues (Chouchani et al., 2013) that are essential for optimal electron transfer toward ubiquinone. Accordingly, an increased production of O2·−, by either complex I or complex III (Murphy, 2009), caused both delayed oxidative damage to complex I and impairment in the electron transfer likely responsible for the inhibition of its activity. This effect had functional consequences for the intact cell, because both Δψm was lost in a delayed manner, and the rate of O2 consumption was impaired dramatically.
The delayed collapse in Δψm observed under our conditions is likely attributable to the inefficacy of complex I at pumping protons into the intermembrane space, a trigger of the mitochondrial transition pore. It has been shown recently that this pore is formed by dimerization of F1Fo–ATP synthase β subunits (Carraro et al., 2014). Thus, dissociation of phosphorylated Bcl-xL from the F1Fo–ATP synthase β subunits that we herein describe might be an important step in this process by allowing free F1Fo–ATP synthase β subunits to dimerize. Subsequent opening of the mitochondrial transition pore will initiate mitochondrial outer membrane permeabilization to promote the release of proapoptotic factors (Galluzzi et al., 2009; Bernardi, 2013), shown previously to be sensitive to Bcl-xL binding to the F1Fo–ATP synthase β subunit (Chen et al., 2011; Alavian et al., 2014).
In conclusion, here we decipher a cascade of events triggered by the overactivation of glutamate receptors in neurons, an underlying feature of several neurological diseases. This transduction pathway requires mitochondrial cyclin B1 to activate Cdk1 to promote phosphorylation of anti-apoptotic Bcl-xL. Phosphorylated Bcl-xL releases the β subunit of F1Fo–ATP synthase, likely forming the transition pore that promotes apoptosis. This mechanism can account for the known deleterious effects of cyclin B1 accumulation in the degenerating brain areas of neurological diseases (Vincent et al., 1997; Love, 2003; Yang et al., 2003). Furthermore, the negative regulation of mitochondrial function by cyclin B1–Cdk1 activity that we describe here may have implications beyond the neural tissue. For instance, it is known that cancer cells upregulate aerobic glycolytic metabolism to support aberrant cell proliferation (Vander Heiden et al., 2009), which is coupled with decreased APC/C–Cdh1 activity (Almeida et al., 2010; Moncada et al., 2012). The aberrantly accumulated cyclin B1 in mitochondria causing inhibition of complex I impairs the energetic efficiency of mitochondria, and this would likely stimulate glycolysis in proliferating cells. The combination of such a metabolic switch with the abnormal ability of cancer cells to form the transition pore (Brenner and Grimm, 2006; Javadov et al., 2011) would represent a previously unrecognized mechanism whereby cyclin B1–Cdk1 promotes cancer cell progression.
Footnotes
This work was funded by Instituto de Salud Carlos III Grants PI12/00685 and RD12/0014/0007 (A.A.P.), RD12/0043/0021 (J.P.B.), FI10/00492 (M.V.-P.d.T.), and CP14/00010 (M.D.-E.), Ministerio de Economia y Competitividad Grant SAF2013-41177-R (J.P.B.), SP3-People-MC-ITN programme of the European Commission Grant 608381 (J.P.B.), and the European Regional Development Fund. The technical assistances of Monica Carabias and Monica Resch are acknowledged.
The authors declare no competing financial interests.
References
- Abbott WA, Griffith OW, Meister A. Gamma-glutamyl-glutathione. Natural occurrence and enzymology. J Biol Chem. 1986;261:13657–13661. [PubMed] [Google Scholar]
- Alavian KN, Li H, Collis L, Bonanni L, Zeng L, Sacchetti S, Lazrove E, Nabili P, Flaherty B, Graham M, Chen Y, Messerli SM, Mariggio MA, Rahner C, McNay E, Shore GC, Smith PJ, Hardwick JM, Jonas EA. Bcl-xL regulates metabolic efficiency of neurons through interaction with the mitochondrial F1FO ATP synthase. Nat Cell Biol. 2011;13:1224–1233. doi: 10.1038/ncb2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alavian KN, Beutner G, Lazrove E, Sacchetti S, Park HA, Licznerski P, Li H, Nabili P, Hockensmith K, Graham M, Porter GA, Jr, Jonas EA. An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc Natl Acad Sci U S A. 2014;111:10580–10585. doi: 10.1073/pnas.1401591111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida A, Bolaños JP. A transient inhibition of mitochondrial ATP synthesis by nitric oxide synthase activation triggered apoptosis in primary cortical neurons. J Neurochem. 2001;77:676–690. doi: 10.1046/j.1471-4159.2001.00276.x. [DOI] [PubMed] [Google Scholar]
- Almeida A, Medina JM. A rapid method for the isolation of metabolically active mitochondria from rat neurons and astrocytes in primary culture. Brain Res Brain Res Prot. 1998;2:209–214. doi: 10.1016/S1385-299X(97)00044-5. [DOI] [PubMed] [Google Scholar]
- Almeida A, Almeida J, Bolaños JP, Moncada S. Different responses of astrocytes and neurons to nitric oxide: the role of glycolytically-generated ATP in astrocyte protection. Proc Natl Acad Sci U S A. 2001;98:15294–15299. doi: 10.1073/pnas.261560998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida A, Bolaños JP, Moreno S. Cdh1/Hct1-APC is essential for the survival of postmitotic neurons. J Neurosci. 2005;25:8115–8121. doi: 10.1523/JNEUROSCI.1143-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida A, Bolaños JP, Moncada S. E3 ubiquitin ligase APC/C-Cdh1 accounts for the Warburg effect by linking glycolysis to cell proliferation. Proc Natl Acad Sci U S A. 2010;107:738–741. doi: 10.1073/pnas.0913668107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antony R, Lukiw WJ, Bazan NG. Neuroprotectin D1 induces dephosphorylation of Bcl-xL in a PP2A-dependent manner during oxidative stress and promotes retinal pigment epithelial cell survival. J Biol Chem. 2010;285:18301–18308. doi: 10.1074/jbc.M109.095232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi P. The mitochondrial permeability transition pore: a mystery solved? Front Physiol. 2013;4:95. doi: 10.3389/fphys.2013.00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billen LP, Kokoski CL, Lovell JF, Leber B, Andrews DW. Bcl-XL inhibits membrane permeabilization by competing with Bax. PLoS Biol. 2008;6:e147. doi: 10.1371/journal.pbio.0060147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolaños JP, Moro MA, Lizasoain I, Almeida A. Mitochondria and reactive oxygen and nitrogen species in neurological disorders and stroke: therapeutic implications. Adv Drug Deliv Rev. 2009;61:1299–1315. doi: 10.1016/j.addr.2009.05.009. [DOI] [PubMed] [Google Scholar]
- Brand MD, Affourtit C, Esteves TC, Green K, Lambert AJ, Miwa S, Pakay JL, Parker N. Mitochondrial superoxide: production, biological effects, and activation of uncoupling proteins. Free Radic Biol Med. 2004;37:755–767. doi: 10.1016/j.freeradbiomed.2004.05.034. [DOI] [PubMed] [Google Scholar]
- Brenner C, Grimm S. The permeability transition pore complex in cancer cell death. Oncogene. 2006;25:4744–4756. doi: 10.1038/sj.onc.1209609. [DOI] [PubMed] [Google Scholar]
- Carraro M, Giorgio V, Šileikyte J, Sartori G, Forte M, Lippe G, Zoratti M, Szabò I, Bernardi P. Channel formation by yeast F-ATP synthase and the role of dimerization in the mitochondrial permeability transition. J Biol Chem. 2014;289:15980–15985. doi: 10.1074/jbc.C114.559633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YB, Aon MA, Hsu YT, Soane L, Teng X, McCaffery JM, Cheng WC, Qi B, Li H, Alavian KN, Dayhoff-Brannigan M, Zou S, Pineda FJ, O'Rourke B, Ko YH, Pedersen PL, Kaczmarek LK, Jonas EA, Hardwick JM. Bcl-xL regulates mitochondrial energetics by stabilizing the inner membrane potential. J Cell Biol. 2011;195:263–276. doi: 10.1083/jcb.201108059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouchani ET, Methner C, Nadtochiy SM, Logan A, Pell VR, Ding S, James AM, Cochemé HM, Reinhold J, Lilley KS, Partridge L, Fearnley IM, Robinson AJ, Hartley RC, Smith RA, Krieg T, Brookes PS, Murphy MP. Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I. Nat Med. 2013;19:753–759. doi: 10.1038/nm.3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Azevedo WF, Leclerc S, Meijer L, Havlicek L, Strnad M, Kim SH. Inhibition of cyclin-dependent kinases by purine analogues: crystal structure of human cdk2 complexed with roscovitine. Eur J Biochem. 1997;243:518–526. doi: 10.1111/j.1432-1033.1997.0518a.x. [DOI] [PubMed] [Google Scholar]
- Ding J, Mooers BH, Zhang Z, Kale J, Falcone D, McNichol J, Huang B, Zhang XC, Xing C, Andrews DW, Lin J. After embedding in membranes antiapoptotic Bcl-XL protein binds both Bcl-2 homology region 3 and helix 1 of proapoptotic Bax protein to inhibit apoptotic mitochondrial permeabilization. J Biol Chem. 2014;289:11873–11896. doi: 10.1074/jbc.M114.552562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen MR. Mitochondria, calcium-dependent neuronal death and neurodegenerative disease. Pflugers Arch. 2012;464:111–121. doi: 10.1007/s00424-012-1112-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdö F, Trapp T, Mies G, Hossmann KA. Immunohistochemical analysis of protein expression after middle cerebral artery occlusion in mice. Acta Neuropathol. 2004;107:127–136. doi: 10.1007/s00401-003-0789-8. [DOI] [PubMed] [Google Scholar]
- Formentini L, Pereira MP, Sánchez-Cenizo L, Santacatterina F, Lucas JJ, Navarro C, Martínez-Serrano A, Cuezva JM. In vivo inhibition of the mitochondrial H+-ATP synthase in neurons promotes metabolic preconditioning. EMBO J. 2014;33:762–778. doi: 10.1002/embj.201386392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Blomgren K, Kroemer G. Mitochondrial membrane permeabilization in neuronal injury. Nat Rev Neurosci. 2009;10:481–494. doi: 10.1038/nrn2665. [DOI] [PubMed] [Google Scholar]
- Gomez-Sanchez JC, Delgado-Esteban M, Rodriguez-Hernandez I, Sobrino T, Perez de la Ossa N, Reverte S, Bolaños JP, Gonzalez-Sarmiento R, Castillo J, Almeida A. The human Tp53 Arg72Pro polymorphism explains different functional prognosis in stroke. J Exp Med. 2011;208:429–437. doi: 10.1084/jem.20101523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harley ME, Allan LA, Sanderson HS, Clarke PR. Phosphorylation of Mcl-1 by CDK1-cyclin B1 initiates its Cdc20-dependent destruction during mitotic arrest. EMBO J. 2010;29:2407–2420. doi: 10.1038/emboj.2010.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrup K. Post-mitotic role of the cell cycle machinery. Curr Opin Cell Biol. 2013;25:711–716. doi: 10.1016/j.ceb.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurd TR, Requejo R, Filipovska A, Brown S, Prime TA, Robinson AJ, Fearnley IM, Murphy MP. Complex I within oxidatively stressed bovine heart mitochondria is glutathionylated on Cys-531 and Cys-704 of the 75-kDa subunit: potential role of CYS residues in decreasing oxidative damage. J Biol Chem. 2008;283:24801–24815. doi: 10.1074/jbc.M803432200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaquenoud M, van Drogen F, Peter M. Cell cycle-dependent nuclear export of Cdh1p may contribute to the inactivation of APC/C(Cdh1) EMBO J. 2002;21:6515–6526. doi: 10.1093/emboj/cdf634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javadov S, Hunter JC, Barreto-Torres G, Parodi-Rullan R. Targeting the mitochondrial permeability transition: cardiac ischemia-reperfusion versus carcinogenesis. Cell Physiol Biochem. 2011;27:179–190. doi: 10.1159/000327943. [DOI] [PubMed] [Google Scholar]
- King TE. Preparation of succinate cytochrome c reductase and the cytochrome b-c1 particle, and reconstitution of succinate cytochrome c reductase. Methods Enzymol. 1967;10:216–225. doi: 10.1016/0076-6879(67)10043-8. [DOI] [Google Scholar]
- Love S. Neuronal expression of cell cycle-related proteins after brain ischaemia in man. Neurosci Lett. 2003;353:29–32. doi: 10.1016/j.neulet.2003.09.004. [DOI] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Lewis-Farr A, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- Maestre C, Delgado-Esteban M, Gomez-Sanchez JC, Bolaños JP, Almeida A. Cdk5 phosphorylates Cdh1 and modulates cyclin B1 stability in excitotoxicity. EMBO J. 2008;27:2736–2745. doi: 10.1038/emboj.2008.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moncada S, Higgs EA, Colombo SL. Fulfilling the metabolic requirements for cell proliferation. Biochem J. 2012;446:1–7. doi: 10.1042/BJ20120427. [DOI] [PubMed] [Google Scholar]
- Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana-Cabrera R, Fernandez-Fernandez S, Bobo-Jimenez V, Escobar J, Sastre J, Almeida A, Bolaños JP. gamma-Glutamylcysteine detoxifies reactive oxygen species by acting as glutathione peroxidase-1 cofactor. Nat Commun. 2012;3:718. doi: 10.1038/ncomms1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragan CI, Wilson MT, Darley-Usmar VM, Lowe PN. Subfractionation of mitochondria and isolation of the proteins of oxidative phosphorylation. In: Darley-Usmar VM, Rickwood D, Wilson TM, editors. Mitochondria: a practical approach. London: IRL; 1987. pp. 79–112. [Google Scholar]
- Rashidian J, Iyirhiaro GO, Park DS. Cell cycle machinery and stroke. Biochim Biophys Acta. 2007;1772:484–493. doi: 10.1016/j.bbadis.2006.11.009. [DOI] [PubMed] [Google Scholar]
- Sakurikar N, Eichhorn JM, Chambers TC. Cyclin-dependent kinase-1 (Cdk1)/cyclin B1 dictates cell fate after mitotic arrest via phosphoregulation of antiapoptotic Bcl-2 proteins. J Biol Chem. 2012;287:39193–39204. doi: 10.1074/jbc.M112.391854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Cenizo L, Formentini L, Aldea M, Ortega AD, García-Huerta P, Sánchez-Aragó M, Cuezva JM. Up-regulation of the ATPase inhibitory factor 1 (IF1) of the mitochondrial H+-ATP synthase in human tumors mediates the metabolic shift of cancer cells to a Warburg phenotype. J Biol Chem. 2010;285:25308–25313. doi: 10.1074/jbc.M110.146480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sazanov LA. The mechanism of coupling between electron transfer and proton translocation in respiratory complex I. J Bioenerg Biomembr. 2014;46:247–253. doi: 10.1007/s10863-014-9554-z. [DOI] [PubMed] [Google Scholar]
- Schmitt E, Beauchemin M, Bertrand R. Nuclear colocalization and interaction between bcl-xL and cdk1(cdc2) during G2/M cell-cycle checkpoint. Oncogene. 2007;26:5851–5865. doi: 10.1038/sj.onc.1210396. [DOI] [PubMed] [Google Scholar]
- Shepherd D, Garland PB. ATP controlled acetoacetate and citrate synthesis by rat liver mitochondria oxidising palmitoyl-carnitine, and the inhibition of citrate synthase by ATP. Biochem Biophys Res Commun. 1966;22:89–93. doi: 10.1016/0006-291X(66)90607-3. [DOI] [PubMed] [Google Scholar]
- Smith RA, Adlam VJ, Blaikie FH, Manas AR, Porteous CM, James AM, Ross MF, Logan A, Cochemé HM, Trnka J, Prime TA, Abakumova I, Jones BA, Filipovska A, Murphy MP. Mitochondria-targeted antioxidants in the treatment of disease. Ann N Y Acad Sci. 2008;1147:105–111. doi: 10.1196/annals.1427.003. [DOI] [PubMed] [Google Scholar]
- Soper JW, Pedersen PL. Isolation of an oligomycin-sensitive ATPase complex from rat liver mitochondria. Methods Enzymol. 1979;55:328–333. doi: 10.1016/0076-6879(79)55040-X. [DOI] [PubMed] [Google Scholar]
- Terrano DT, Upreti M, Chambers TC. Cyclin-dependent kinase 1-mediated Bcl-xL/Bcl-2 phosphorylation acts as a functional link coupling mitotic arrest and apoptosis. Mol Cell Biol. 2010;30:640–656. doi: 10.1128/MCB.00882-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upreti M, Galitovskaya EN, Chu R, Tackett AJ, Terrano DT, Granell S, Chambers TC. Identification of the major phosphorylation site in Bcl-xL induced by microtubule inhibitors and analysis of its functional significance. J Biol Chem. 2008;283:35517–35525. doi: 10.1074/jbc.M805019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, Chandel NS, Williamson EK, Schumacker PT, Thompson CB. Bcl-xL regulates the membrane potential and volume homeostasis of mitochondria. Cell. 1997;91:627–637. doi: 10.1016/S0092-8674(00)80450-X. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent I, Jicha G, Rosado M, Dickson DW. Aberrant expression of mitotic cdc2/cyclin B1 kinase in degenerating neurons of Alzheimer's disease brain. J Neurosci. 1997;17:3588–3598. doi: 10.1523/JNEUROSCI.17-10-03588.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Qin ZH. Molecular and cellular mechanisms of excitotoxic neuronal death. Apoptosis. 2010;15:1382–1402. doi: 10.1007/s10495-010-0481-0. [DOI] [PubMed] [Google Scholar]
- Wang Z, Fan M, Candas D, Zhang TQ, Qin L, Eldridge A, Wachsmann-Hogiu S, Ahmed KM, Chromy BA, Nantajit D, Duru N, He F, Chen M, Finkel T, Weinstein LS, Li JJ. Cyclin B1/Cdk1 coordinates mitochondrial respiration for cell-cycle G2/M progression. Dev Cell. 2014;29:217–232. doi: 10.1016/j.devcel.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Y, Yang S, Liu R, Brun-Zinkernagel AM, Koulen P, Simpkins JW. Transient cerebral ischemia induces aberrant neuronal cell cycle re-rentry and Alzheimer's disease-like tauopathy in female rats. J Biol Chem. 2004;279:22684–22692. doi: 10.1074/jbc.M311768200. [DOI] [PubMed] [Google Scholar]
- Wharton DC, Tzagoloff A. Cytochrome oxidase from beef heart mitochondria. Methods Enzymol. 1967;10:245–250. doi: 10.1016/0076-6879(67)10048-7. [DOI] [Google Scholar]
- White RJ, Reynolds IJ. Mitochondrial depolarization in glutamate-stimulated neurons: an early signal specific to excitotoxic exposure. J Neurosci. 1996;16:5688–5697. doi: 10.1523/JNEUROSCI.16-18-05688.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Mufson EJ, Herrup K. Neuronal cell death is preceded by cell cycle events at all stages of Alzheimer's disease. J Neurosci. 2003;23:2557–2563. doi: 10.1523/JNEUROSCI.23-07-02557.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]