

Abstract
The cerebellum is crucial for sensorimotor coordination. The cerebellar architecture not only requires proper development but also long-term integrity to ensure accurate functioning. Developmental defects such as impaired neuronal migration or neurodegeneration are thus detrimental to the cerebellum and can result in movement disorders including ataxias. In this study, we identify FBXO41 as a novel CNS-specific F-box protein that localizes to the centrosome and the cytoplasm of neurons and demonstrate that cytoplasmic FBXO41 promotes neuronal migration. Interestingly, deletion of the FBXO41 gene results in a severely ataxic gait in mice, which show delayed neuronal migration of granule neurons in the developing cerebellum in addition to deformities and degeneration of the mature cerebellum. We show that FBXO41 is a critical factor, not only for neuronal migration in the cerebellum, but also for its long-term integrity.
Keywords: ataxia, cerebellum, neuronal migration, F-box protein, degeneration
Introduction
The cerebellum is known for its crucial role in motor control and coordination. The adult cerebellar cortex exhibits a characteristic foliated morphology and well defined architecture consisting of three major layers: the molecular layer, the Purkinje cell layer, and the granule layer (Altman and Bayer, 1997). To establish proper lamination and circuitry, important events such as neuronal differentiation, morphogenesis, and migration need to be precisely regulated during cerebellar development (Altman and Bayer, 1997; Komuro and Yacubova, 2003; Sillitoe and Joyner, 2007). Defects in cerebellar development are often associated with various disorders including Dandy-Walker Malformations, Joubert Syndrome, and other congenital ataxias (Zoghbi, 2000; Millen and Gleeson, 2008; Basson and Wingate, 2013).
Neuronal migration of granule neurons is one of the critical developmental events in the cerebellum that ensures properly timed lamination. During embryonic development, the rhombic lip gives rise to cerebellar granule neuron progenitors, which migrate rostrally to settle in the cerebellar anlage (Wingate, 2001). After a phase of proliferation, which results in expansion of the external granular layer, granule neuron precursors start migrating inward through the developing molecular layer past the Purkinje cell layer to ultimately take up residence in the internal granular layer (Altman and Bayer, 1997). Needless to say, these events require thorough and precise orchestration. A growing number of factors that contribute to the successful migration of granule neurons have been identified (Komuro and Yacubova, 2003; Sillitoe and Joyner, 2007). Recent additions to the list of regulators, such as profilin1, adenomatous polyposis coli 2, or various E2 ubiquitin-conjugating enzymes, give an indication of how delicate and complex granule neuron migration really is (Kullmann et al., 2012; Shintani et al., 2012; Heng et al., 2015; Kumazawa et al., 2013; Huang et al., 2014; Jaarsma et al., 2014; Anckar and Bonni, 2015; Sathyamurthy et al., 2015; Yong et al., 2015).
Neuronal migration is intimately linked to the centrosome, a critical site for microtubule nucleation (Solecki et al., 2006; Higginbotham and Gleeson, 2007). Among the hundreds of centrosomal proteins (Andersen et al., 2003), a subset regulates neuronal migration and is also functionally intertwined (Wynshaw-Boris and Gambello, 2001; Tsai and Gleeson, 2005; Higginbotham and Gleeson, 2007; Kuijpers and Hoogenraad, 2011; Ramahi and Solecki, 2014). In addition to centrosomal proteins, microtubule-binding proteins have critical roles in neuronal migration because they control microtubule elongation and retraction (Tsai and Gleeson, 2005; Vallee et al., 2009; Falnikar et al., 2011). Not surprisingly, various small GTPases that control cytoskeletal dynamics have also been implicated in neuronal migration (Govek et al., 2011; Kawauchi, 2011). Even though recent studies have characterized E3 ligases that regulate neuronal migration by acting upstream of the centrosomal proteins Par6α or Pard3a, the Reelin-regulated signaling component Dab1, or the small GTPase RhoA (Feng et al., 2007; Famulski et al., 2010; Kannan et al., 2012a; Kannan et al., 2012b; Vadhvani et al., 2013), E3 ligases remain poorly explored regulators of neuronal migration.
In this study, we identified the F-box protein FBXO41, a potential E3 ubiquitin ligase, as a neuron-specific protein in the CNS that plays a role in neuronal migration. We also characterized the novel FBXO41 knock-out mouse that displays an ataxia-like motor phenotype.
Materials and Methods
Ethical statement.
All experiments involving live animals were conducted in accordance with animal protocols approved by the Niedersächsisches Landesamt für Verbraucherschutz und Lebensmittelsicherheit (Lower Saxony, Germany).
Constructs.
N-terminally myc- or GFP-tagged mouse FBXO41 was cloned into pCMVmyc or pEGFP vectors, respectively. A DNA-based template method was used to express short hairpin RNAs in the U6/(cytomegalovirus) cmv-EGFP vector. The sequences 5′-GGGTGCCGGCATCGAGAAGGT-3′ and 5′-AGCTGCTGCCGTGCCCT-3′ were used to construct the FBXO41 RNAi#5 and #8 plasmids, respectively. The rescue plasmids for FBXO41 and FBXO41-ΔCTR were generated by introducing silent mutations into the RNAi-targeting regions using site-directed mutagenesis. FBXO41 deletion mutants were generated by standard and fusion PCR. For the latter, two separate PCRs were performed to amplify fragments A and B omitting the region to be deleted but introducing compatible overhangs into each fragment. Subsequently, another PCR was performed to fuse both fragments resulting in a protein lacking the desired region.
Antibodies.
Antibodies used in this study include γ-tubulin (Sigma-Aldrich), pan 14-3-3 (Santa Cruz Biotechnology), pericentrin 2 (Abcam), mouse monoclonal GFP (Santa Cruz Biotechnology), rabbit polyclonal GFP (Invitrogen), β-Gal (Santa Cruz Biotechnology), SnoN (Santa Cruz Biotechnology), GFAP (Promega), PLP [gift from Prof. K.A. Nave, Max Planck Institute (MPI) of Experimental Medicine, Göttingen, Germany], GFAP (Nova Castra), Iba1 (WAKO), calbindin (Sigma-Aldrich), parvalbumin (Swant), NeuN (Millipore), PCNA (Abcam), β-gal antibody (Santa Cruz Biotechnology), and cleaved caspase 3 (Cell Signaling Technology).
FBXO41 antibodies were raised in rabbit against recombinant His6-tagged rat FBXO41 (aa124–184) isolated from bacteria using Nickel-NTA Sepharose (IBA). Polyclonal serum, obtained from Eurogentec, was then affinity purified using recombinant rat FBXO41 protein bound to amino link plus columns.
Primary neuron culture, transfections, and immunocytochemistry.
Murine astrocytic and oligodendroglial cultures were kindly provided by M. Simons (MPI of Experimental Medicine, Göttingen, Germany). Cerebellar granule neurons were cultured from postnatal day 6 (P6) Wistar rat murine cerebella as described previously (Bilimoria and Bonni, 2008).
Granule neurons were either transfected 48 h after plating (2 d in vitro or DIV2) using the modified calcium-phosphate method with the indicated plasmids together with plasmids expressing GFP-FBXO41 or GFP-FBXO41 deletion mutants to visualize the transfected neurons (Konishi et al., 2004). To rule out that the morphological effects observed due to genetic manipulations were not a result of their effect on cell survival, the anti-apoptotic protein Bcl-XL was coexpressed in all experiments.
Survival assay.
For survival assay in neurons, rat granule neurons were transfected at DIV2 with the required plasmids together with β-gal plasmid, which served as a transfection marker. The neurons were fixed at DIV6 and subjected to immunocytochemistry using the β-gal and cleaved-caspase-3 antibodies. In addition, the nuclei were stained with the DNA dye bisbenzimide Hoechst 33258. Cleaved caspase 3 staining was observed in apoptotic neurons. A minimum of 100 transfected neurons per condition were counted and included in the analysis. The percentage of dead and living cells was calculated. Three independent experiments were included in the analysis and statistical analysis was performed using GraphPad Prism 5.0 using ANOVA.
In vivo electroporation.
In vivo electroporation was performed as described previously (Konishi et al., 2004; Stegmüller et al., 2006; Kannan et al., 2012a). In brief, plasmid DNA diluted in PBS (3–4 μl/animal) was injected into the P4 Wistar rat cerebellar cortex together with 0.03% Fast Green using a Hamilton syringe with a 30 gauge needle. U6/cmvGFP or FBXO41 RNAi#5-cmvGFP and/or myc-CMV, FBXO41, FBXO1-Res, FBXO41ΔCTR, or FBXO41-Res ΔCTR expression plasmids together with the synapsin(prom)-EGFP plasmid at a concentration of 8 μg/μl were injected together with 1 μg/μl Bcl-XL expression plasmid. After DNA injection, the animals were subjected to five short electric pulses (160–170 V for 50 ms with intervals of 950 ms). Five days later, the cerebella were isolated and sectioned (40 μm) using a Leica cryostat and subjected to immunohistochemistry with a GFP antibody (Santa Cruz Biotechnology). In all control U6-cmv-GFP cerebella, we observed a 90% association of GFP-positive granule neurons with parallel fibers: the parallel fiber index (Konishi et al., 2004; Stegmüller et al., 2006). GraphPad Prism was used to perform statistical analysis of neuronal migration and distribution data using two-way ANOVA (Bonferroni correction) test. Sections were imaged using the Leica SP5.
Generation of knock-out mice.
FBXO41 knock-out mice embryonic stem (ES) cells (C57BL/6-FBXO41tm1(KOMP)Vlcg) were generated by and purchased from the KOMP Repository at the University of California–Davis. Gene ablation was achieved by replacing the entire FBXO41 gene by a ZenUB1 reporter/selection (LacZ/neomycin) cassette, flanked by loxP sites, via homologous recombination. The FBXO41 knock-out ES cells were injected into a blastocyst from C57BL/6 mice and the blastocyst then implanted into the uterus of the foster mother. The resulting heterozygous animals were mated to generate FBXO41 wild-type, heterozygous, and homozygous mice. The mice were genotyped using the following primers:
 |
Immunohistochemistry.
For LacZ staining, P30 adult heterozygous mice and wild-type littermates were subjected to transcardiac perfusion under deep anesthesia (ketamine/xylazine). After perfusion with PBS, pH 7.4, brains were fixed in situ with 4% (w/v) PFA in PBS. The brains were extracted from the cranium and postfixed in the same fixative for 3–4 h. After a brief rinse in PBS, brains were transferred to 30% sucrose in PBS for cryoprotection overnight. The brains were briefly washed and stored in PBS at 4°C. Vibratome sections of 30–50 μm were obtained with a Leica VT1000s Vibratome and collected in PBS. The free-floating sagittal brain sections were then incubated overnight at 37°C in the following solution: 2 mm MgCl2, 0.02% NP40, 0.01% sodium deoxycholate, 5 mm potassium ferrocyanide, 5 mm potassium ferricyanide, and 0.5 mg/ml X-gal in PBS. The sections were rinsed in PBS and mounted onto glass slides for imaging.
For paraffin sections, P12, P16, and P30 mice (FBXO41+/+ or FBXO41−/−) were subjected to transcardial perfusion with PBS followed by 4% PFA. The brains were extracted and postfixed in the same fixative overnight. After a brief rinse in PBS, the brains were either paraffin embedded and cut into 5 μm sagittal sections using a sliding microtome (Microm) or immersed in 30% sucrose for cryoprotection, embedded in OCT (Tissue Teck), and cut into 12-μm-thick sections using the Leica cryostat. The paraffin sections were stained with H&E or immunostained with antibodies against PCNA (Abcam), NeuN (Millipore), GFAP (Nova Castra), Iba1 (WAKO), and calbindin (Sigma-Aldrich) using the LSAB2 system (Dako). The TUNEL assay kit (Promega) was used to detect apoptotic neurons. DNase-digested tissue was used as positive control. Images were acquired using an Axiophot microsocope (Zeiss) and processed by ZEN software (Zeiss). For florescence staining on P16 FBXO41+/+ or FBXO41−/− cryosections, parvalbumin specific antibody was used and images were acquired using a Leica Sp5 confocal microscope.
The cellular density/distribution within the cerebellum was either counted manually or using a custom-designed ImageJ macro for DAB (GFAP) and H&E stainings, respectively. The sections were analyzed in a blinded manner with a least three sections per animal and three animals per genotype being included in the analysis. GraphPad Prism 5.0 was used to perform statistical analysis of cell migration and distribution data using paired t test and two-way ANOVA.
Mouse behavior.
The mice (P16: 5 males and 6 females, P30: 2 males and 2 females) were subjected to a previously established ataxia scoring protocol (Guyenet et al., 2010). To assess basic motor functions including balance and coordination, mice were subjected to four basic tests by a blinded investigator. The “clasping test” was used to assess whether the mice would clasp their forelimbs and hind limbs into their bodies or would spread their limbs when suspended by their tails (Lin et al., 2001). The ledge test, described previously (Garden et al., 2002), involved placing the P16 mouse on the 5-mm-wide and 30-cm-long ledge of a Plexiglas box and observing the response of the mouse. Although wild-type mice typically balanced themselves well and scurried along the bar of the cage, the knock-out mice with a neurological dysfunction typically froze in place or fell into the padded box. The animals were subjected to a gait analysis test as described previously (Guyenet et al., 2010) to evaluate coordination and muscle function. The mice were placed in a flat surface with their heads facing away from the investigator. The mice were scored on how well they supported their body weight and moved on four limbs. For the above tests, the mice were scored with a range of 0 to 3 (0 was for best performance and 3 for worst). All tests were repeated thrice for each animal and an average of the three trials was considered as the final score. The rotarod test, described previously (Radyushkin et al., 2009), was used to assess their balance and motor coordination. In brief, the rotarod is a rotating drum (Ugo Basile) at 12 rpm. The mice were placed individually on the rotarod and the latency of falling off was recorded using a stopwatch. The test period on the rotarod was 200 s.
Subcellular fractionation and centrosomal purification.
Tissue lysates of mice at different embryonic and postnatal stages, of granule neurons at various DIVs, and of cultured astrocytes and oligodendrocytes were subjected to SDS-PAGE and immunoblotting analyses as described previously (Stegmüller et al., 2006).
For subcellular fractionation, cultured cerebellar granule neurons at DIV8 were scraped into a detergent-free buffer A (10 mm HEPES, pH 7.9, 10 mm KCl, 0.1 mm EDTA, 0.1 mm EGTA, and protease inhibitors) and subjected to mechanical disruption using a 2 ml dounce homogenizer. While the nuclei were spun down at 500 g at 4°C, the supernatant was collected as the cytoplasmic fraction. Nuclei were washed once in 0.1% NP40-supplemented buffer A and then lysed in a buffer B (20 mm HEPES pH 7.9, 400 mm NaCl, 1 mm EDTA, 1 mm EGTA, and protease inhibitors) and pelleted at maximum speed (18,400 × g) at 4°C. The supernatant was collected as the nuclear fraction.
The centrosomal purification was performed as described previously (Vadhvani et al., 2013). Cultured granule neurons were exposed to cytochalasin D (1 μg/ml) and nocodazole (0.2 μm) for 1 h before lysis (1 mm HEPES, pH 7.2, 0.5% NP40, 0.5 mm MgCl2, and 0.1% β-mercaptoethanol supplemented with protease inhibitors 1 μg/ml pepstatin, 1 μg/ml aprotinin, 1 μg/ml leupeptin, and 1 mm PMSF). Cell lysates were then subjected to gradient centrifugation (60% sucrose, 10,000 × g, 30 min, 4°C). The resulting centrosomal fractions were purified by a discontinuous sucrose density gradient centrifugation (40%/50%/70% sucrose, 34,000 × g, 1 h, 4°C). Supernatant fractions were further subjected to immunoblotting analysis using the FBXO41 and γ-tubulin antibodies.
Results
FBXO41 is a neuron-specific F-box protein expressed in the CNS
To examine the role of FBXO41, we first generated and validated the affinity-purified antibody against FBXO41 (Fig. 1A). We then characterized the spatial and temporal expression of FBXO41 by immunoblotting murine tissue lysates obtained from various organs. We found FBXO41 to be expressed in CNS tissues including cortex, hippocampus, cerebellum, and spinal cord, but absent from the peripheral nervous system tissue such as sciatic nerve and from non-neural tissues (Fig. 1B,C). We also corroborated the CNS-specific expression of FBXO41 at the mRNA level with RT-PCR (Fig. 1D,E). Furthermore, we detected FBXO41 protein as early as embryonic day 10 (E10) in mouse (Fig. 1F) with abundant levels in postnatal stages in the cerebellum (Fig. 1G). Further immunoblotting analysis revealed that FBXO41 was expressed with increasing amounts in cerebellar granule neurons cultured for different DIV, but not in astrocytes or oligodendrocytes (Fig. 1H,I), thus confirming the neuron-specific expression. Last, on performing subcellular fractionation analyses of cultured granule neurons, we found that endogenous FBXO41 was localized in the cytoplasm and excluded from the nucleus (Fig. 1J).
Figure 1.

The F-box protein FBXO41 is exclusively expressed in CNS neurons. A, Cortical and splenic lysates from P20 mice were subjected to immunoblotting using the FBXO41 antibody alone or preabsorbed with FBXO41 antigen. Pan 14-3-3 antibody served as loading control. Arrow indicates specific bands. Asterisks indicate nonspecific bands. B, Lysates of indicated tissues isolated from P20 mice were subjected to immunoblotting using the FBXO41 and the pan 14-3-3 antibodies. The latter served as loading control. C, Lysates of indicated tissues were analyzed as described in B. D, E, RT-PCR of FBXO41 using RNA from indicated P20 mouse tissues. GAPDH served as loading control. F, Brain lysates from indicated embryonic ages were subjected to immunoblotting using the FBXO41 or pan 14-3-3 antibodies. The latter served as loading control. G, Lysates of mouse cerebella isolated at indicated ages were analyzed as described in B. H, Granule neuron lysates from indicated DIV were subjected to immunoblotting using the FBXO41 or pan 14-3-3 antibodies. The latter served as loading control. I, Lysates of cultured granule neurons, astrocytes, or oligodendrocytes were subjected to immunoblotting using the FBXO41, GFAP, or PLP antibodies. The γ-tubulin antibody served as loading control. J, Cultured granule neurons were subjected to subcellular fractionation analyses, followed by immunoblotting using the FBXO41 antibody or the FBXO41 antibody preabsorbed with FBXO41 antigen. The pan 14-3-3 and SnoN antibodies served as markers for the postnuclear supernatant (PNS) and the nuclear fraction (NF), respectively. Asterisks indicate nonspecific bands. K, Confocal image of cultured granule neurons transfected with a plasmid encoding GFP-FBXO41 and subjected to immunocytochemistry with the GFP antibody and the DNA dye 4′, 6-diamidino-2-phenylindole (DAPI). Arrowheads and arrow indicate axon and puncta-like structure near the nucleus, respectively. Inset is shown at lower exposure on the right. Scale bars, 20 μm. L, Confocal images of granule neurons transfected with the GFP-FBXO41 plasmid followed by immunocytochemistry with the GFP and pericentrin 2 antibodies together with the DNA dye DAPI. Arrows and arrowheads indicate FBXO41 and the centrosomal marker pericentrin 2, respectively. Scale bars, 10 μm. M, Cultured granule neurons were subjected to centrosomal purification using a sucrose gradient, followed by immunoblotting with the FBXO41 antibody. γ-tubulin served as marker for centrosomal enrichment in the bottom fractions. Cerebellar lysates from P16 mice served as a positive control.
Because our purified FBXO41 antibody failed to work in immunocytochemistry, we resorted to an overexpression analysis of GFP-tagged FBXO41 to support its cytoplasmic localization in neurons (Fig. 1K). In addition, we discovered a conspicuous dot-like accumulation of FBXO41 near the nucleus, which we later confirmed to be the centrosome because GFP-FBXO41 colocalized with the centrosomal marker pericentrin 2 (Fig. 1L). Consistent with these results, we found that endogenous FBXO41 also cosegregated with the centrosome-associated protein γ-tubulin in the bottom fractions of centrosome purification experiments using cultured granule neurons (Fig. 1M). These experiments identify FBXO41 as a neuron-specific protein in the CNS, which localizes to the centrosome and cytoplasm.
Cytoplasmic localization of FBXO41 is important for neuronal migration in the cerebellum
Having found that FBXO41 localized to different cellular regions, we generated GFP-tagged FBXO41 deletion mutants to identify the region that would target FBXO41 to the centrosome (Fig. 2A, B). Upon expression in HEK293T cells and in neurons, we identified a sequence that was required to direct FBXO41 to the centrosome (Fig. 2A,B); we refer to it as centrosomal targeting region (CTR, aa 179–354) hereafter. In contrast to wild-type FBXO41, FBXO41 ΔCTR failed to localize to the centrosome and dispersed in the cytoplasm in both HEK293T cells and granule neurons (Fig. 2A). Together, these experiments established the dual localization of FBXO41 at the centrosome and cytoplasm.
Figure 2.
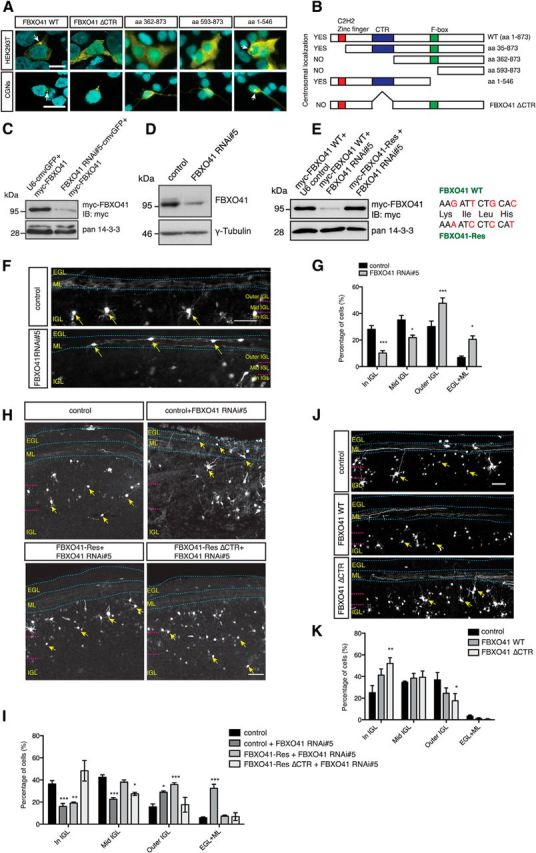
Cytoplasmic FBXO41 promotes neuronal migration. A, Confocal images of HEK293T cells and granule neurons (CGNs), transfected with plasmids encoding GFP-FBXO41, GFP-FBXO41 ΔCTR, or other GFP-FBXO41 mutants, followed by immunocytochemistry with the GFP antibody and the DNA dye DAPI. Arrows indicate centrosomes. Scale bars, 10 μm. B, Schematic summarizes localization analyses. C, HEK293T cells transfected with the myc-FBXO41 plasmid together with control bicistronic vector (U6-cmvGFP, which contains a U6 promoter and a GFP cassette) or the bicistronic FBXO41 RNAi#5-cmvGFP plasmid, were subjected to immunoblotting (IB) with the myc and pan 14-3-3 antibodies. The latter served as loading control. D, Granule neurons in suspension were mixed with indicated control vector or the FBXO41 RNAi#5 plasmid, electroporated with the Amaxa nucleofector, and cultured for 5 d. Lysates were immunoblotted with the FBXO41 and the γ-tubulin antibodies. The latter served as loading control. E, Lysates of HEK293T cells transfected with the plasmid encoding myc-FBXO41 together with control vector or FBXO41 RNAi#5 plasmid or the myc-FBXO41-Res plasmid together with FBXO41 RNAi#5 plasmid were subjected to immunoblotting using the myc and pan 14-3-3 antibodies. The latter served as loading control. Silent mutations in the FBXO41 RNAi targeting region are indicated in red. F, Cerebella of P4 rat pups were subjected to in vivo electroporation with bicistronic control vector (U6/cmv-GFP) or the bicistronic FBXO41 RNAi#5-cmvGFP plasmid. Five days later, coronal cerebellar sections were subjected to immunohistochemistry with the GFP antibody and the distribution of GFP+ neurons were analyzed within each of the cerebellar layers (EGL, ML, and IGL). The IGL was further divided into three equal parts: the outer IGL, middle IGL (Mid IGL), and the inner IGL (In IGL) for the analysis. Dotted lines show layers. Arrows indicate eletroporated neurons and scale bar equals 50 μm. G, Quantification of F. A total of 2346 neurons from four control and five FBXO41 RNAi#5 cerebella were analyzed (two-way ANOVA, ***p < 0.001, **p < 0.01, *p < 0.05, mean + SEM). H, Cerebella of P4 rat pups were subjected to in vivo electroporation with bicistronic control vector (U6/cmv-GFP) or the bicistronic FBXO41 RNAi#5-cmvGFP plasmid together with control, FBXO41-Res, or FBXO41-Res ΔCTR. Five days later, the cerebella were processed and analyzed as described in F. Arrows indicate electroporated neurons. Scale bar, 50 μm. I, Quantification of H. A total of 5515 neurons from five control, five FBXO41 RNAi#5, five FBXO41-Res, and five FBXO41-Res ΔCTR cerebella were analyzed (two-way ANOVA, ***p < 0.001, **p < 0.01, *p < 0.05, mean + SEM). J, Cerebella of P4 rat pups were injected with control vector, the FBXO41 plasmid, or the FBXO41 ΔCTR plasmid together with synapsin(prom)-GFP expression plasmid and subjected in vivo electroporation and processed as in F. Arrows indicate electroporated neurons and scale bar equals 50 μm. K, Quantification of J. A total of 4404 neurons from five control, five FBXO41 WT, and five FBXO41 ΔCTR cerebella were included in the analysis (two-way ANOVA, **p < 0.01, *p < 0.05, mean + SEM).
The centrosome and in particular centrosome-associated proteins have been the focus of many studies elucidating mechanisms of neuronal migration (Wynshaw-Boris and Gambello, 2001; Tsai and Gleeson, 2005; Higginbotham and Gleeson, 2007). Therefore, the centrosomal localization of FBXO41 prompted us to determine whether FBXO41 plays a role in neuronal migration using the in vivo electroporation technique (Konishi et al., 2004; Holubowska et al., 2014). We started out with a loss-of-function approach for which we generated several FBXO41 hairpin constructs and validated the efficient downregulation of exogenous FBXO41 in HEK293T cells and endogenous FBXO41 in granule neurons by the FBXO41 RNAi#5 plasmid (Fig. 2C,D). To establish the specificity of knock-down-induced phenotypes, we generated and validated an RNAi-resistant FBXO41-Rescue (Res) plasmid harboring four silent mutations in the targeting region (Fig. 2E).
We then injected the bicistronic FBXO41 RNAi#5-cmvGFP plasmid or control vector into the cerebella of P4 Wistar rat pups and subjected them to electrical pulses, followed by analyses of coronal sections at P9. Compared with control developing cerebella, in which neuronal precursors undergo radial migration from the external granule layer (EGL) to the internal granular layer (IGL) (Altman and Bayer, 1997), we found that significantly more FBXO41 knock-down neurons were stalled in the EGL/ML (Fig. 2F). We quantified the number of control-electroporated and FBXO41 RNAi neurons in four cerebellar regions (EGL/ML, inner, middle, and outer IGL) and found significantly more FBXO41 RNAi neurons in the EGL/ML (Fig. 2G). The remaining FBXO41 RNAi neurons were most abundant in the outer IGL, whereas significantly fewer FBXO41 RNAi neurons migrated into the middle and inner IGL, indicating a slower migration speed (Fig. 2G). These data suggest that FBXO41 promotes neuronal migration.
Next, we investigated whether the centrosomal or the cytoplasmic localization of FBXO41 is important for neuronal migration. We injected FBXO41 RNAi plasmid alone or together with the RNAi-resistant FBXO41-Res or FBXO41-Res ΔCTR plasmid or appropriate control vectors into the cerebella of P4 Wistar rat pups and subjected them to electrical pulses. Five days later, we analyzed electroporated cerebellar granule neurons in coronal sections. We found that, unlike FBXO41 RNAi neurons, the control neurons, FBXO41-Res neurons, and FBXO41-Res ΔCTR neurons managed to migrate efficiently away from the EGL/ML region (Fig. 2H). In the outer IGL, control neurons were indistinguishable from FBXO41-Res ΔCTR neurons, but significantly less numerous than FBXO41 RNAi or FBXO41-Res neurons (Fig. 2I). In the middle section of the IGL, we found expectedly less FBXO41 RNAi neurons, but an equal number of FBXO41-Res neurons and less FBXO41-Res ΔCTR neurons (Fig. 2I). In the inner IGL, both FBXO41 RNAi and FBXO41-Res neurons are significantly less numerous than control neurons (Fig. 2I). FBXO41-Res ΔCTR neurons, however, migrated equally efficient compared with control neurons. In addition, to underscore these findings, we examined FBXO41 gain-of-function in vivo. We electroporated P4 cerebella with control vector, the wild-type FBXO41, or FBXO41 ΔCTR expression plasmid and analyzed coronal sections from P9 pups (Fig. 2J). Quantification of the granule neurons that migrated into subdivided sections of the IGL demonstrated that control neurons are more numerous in the outer IGL and equally numerous in the middle IGL compared with wild-type FBXO41 and FBXO41 ΔCTR neurons (Fig. 2K). FBXO41 neurons migrated more efficiently into the inner IGL than control neurons and FBXO41 ΔCTR neurons were significantly more numerous in the inner IGL (Fig. 2K). Collectively, these data indicate that the FBXO41 RNAi-induced phenotype is specific to FBXO41 and suggest that cytoplasmic FBXO41 is crucial to stimulate the migration of cerebellar granule neurons.
FBXO41−/− mice display an ataxia-like phenotype and defects in cerebellar development
Having identified a role for FBXO41 in neuronal migration, we decided to take a mouse genetics approach to explore the effects of the loss of FBXO41 on neuronal migration. Therefore, we generated an FBXO41 knock-out (KO; C57BL/6-FBXO41tm1(KOMP)Vlcg) mouse in which the FBXO41 gene was replaced by a LacZ reporter cassette (for cloning strategy, see www.KOMP.org). After genotyping PCR to identify the integrated LacZ cassette (Fig. 3A), we performed immunoblotting analysis and determined the successful deletion of FBXO41 in the FBXO41−/− mice and observed approximately half the dosage in FBXO41+/− mice (Fig. 3B).
Figure 3.
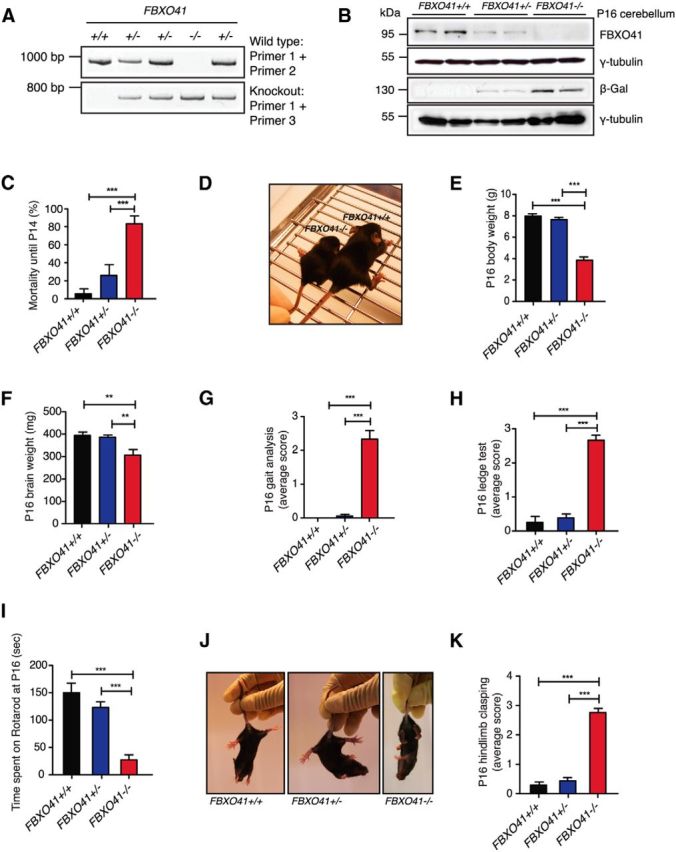
Phenotypical characterization of the FBXO41−/− mice. A, Representative genotyping PCR of FBXO41+/+, FBXO41+/−, or FBXO41−/− mice. B, Cerebellar lysates of P16 FBXO41+/+, FBXO41+/−, and FBXO41−/− mice were subjected to immunoblotting using the FBXO41, β-Gal, pan 14-3-3, or γ-tubulin antibodies. The latter two served as loading control. C, Mortality rate of FBXO41+/+, FBXO41+/−, and FBXO41−/− mice until P14 (ANOVA, ***p < 0.001, mean + SEM). D, Representative image of P16 FBXO41+/+ and FBXO41−/− littermates. E, Average body weight of P16 FBXO41+/+, FBXO41+/−, and FBXO41−/− mice. A total of 86 P16 mice were included in the analysis (ANOVA, **p < 0.01, ***p < 0.001, mean + SEM). F, Average brain weight of mice analyzed in E. G, P16 littermates were subjected to gait analysis. More than 10 mice of each genotype were included in the analysis (ANOVA, ***p < 0.001, mean + SEM); 0 = normal phenotype, 3 = worst manifestation of phenotype. H, P16 littermates were subjected to the ledge test. More than 10 mice of each genotype were included in the analysis (ANOVA, ***p < 0.001, mean + SEM); 0 = normal phenotype, 3 = worst manifestation of phenotype. I, P16 littermates were tested for time spent on rotarod with 240 s cutoff time and accelerating speed. More than 10 mice of each genotype were included in the analysis (ANOVA, ***p < 0.001, mean + SEM). J, Representative images of P16 FBXO41+/+, FBXO41+/−, and FBXO41−/− littermates. K, P16 littermates were tested for hindlimb clasping. More than 10 mice of each genotype were included in the analysis (ANOVA, ***p < 0.001, mean + SEM); 0 = normal phenotype, 3 = worst manifestation of phenotype.
We started out by examining the FBXO41−/− mice and observed a slight developmental delay already at P5, which became even more evident thereafter. Approximately 80% of homozygous mice died before P14 (Fig. 3C); the few surviving homozygous mice had a variable lifespan but never longer than 10 weeks. This compelled us to describe the strong phenotype displayed by FBXO41−/− mice at P16 (Fig. 3D). The most obvious characteristics of the P16 FBXO41−/− mice were severely reduced body and brain weight compared with their FBXO41+/− and FBXO41+/+ littermates (Fig. 3E,F).
In addition to reduced body size, FBXO41−/− mice exhibited motor deficits characterized by a severely ataxic gait. To quantify this aspect of the phenotype, we subjected FBXO41+/+, FBXO41+/−, and FBXO41−/− mice at P16 to an ataxia scoring protocol described previously by Guyenet et al. (2010), with 0 representing normalcy and 3 representing the strongest manifestation of the phenotype. We observed that FBXO41−/− mice displayed an abnormal gait characterized by dragging of the hind limbs, impaired balance, and tremors (Fig. 3G). We also tested the balance of FBXO41−/− mice along with their littermates using the ledge test (Fig. 3H) and their motor coordination on the rotarod test (Fig. 3I). We found that FBXO41−/− mice performed significantly worse than their littermates in all tested paradigms. FBXO41−/− mice also adopted a hindlimb clasping posture when suspended by the tail (Fig. 3J,K). The heterozygous FBXO41 mice were indistinguishable from wild-type littermates in all tests. These observations suggest that loss of FBXO41 in mice results in ataxia-like motor defects as a result of neurological deficits.
Abundant neuronal expression of FBXO41 in the brain
We next took advantage of the LacZ cassette driven by the FBXO41 promoter in the aforementioned mice to characterize the spatial expression of FBXO41 in the CNS by subjecting sagittal sections from P30 FBXO41+/− mice to LacZ staining. We observed a wide distribution of β-Galactosidase (β-Gal) throughout the brain (Fig. 4A), with abundant FBXO41 expression, for example, in various layers of the hippocampus and the cortex (Fig. 4B). We also detected a weaker expression in the spinal cord (Fig. 4C). In the cerebellum, FBXO41 was expressed in the granule cell layer and deep cerebellar nuclei, but was absent from Purkinje cells, interneurons of the molecular layer, and white matter (Fig. 4D). We excluded nonspecific signals by subjecting a brain section from an FBXO41+/+ littermate to LacZ staining together with the FBXO41−/− sections (Fig. 4E).
Figure 4.
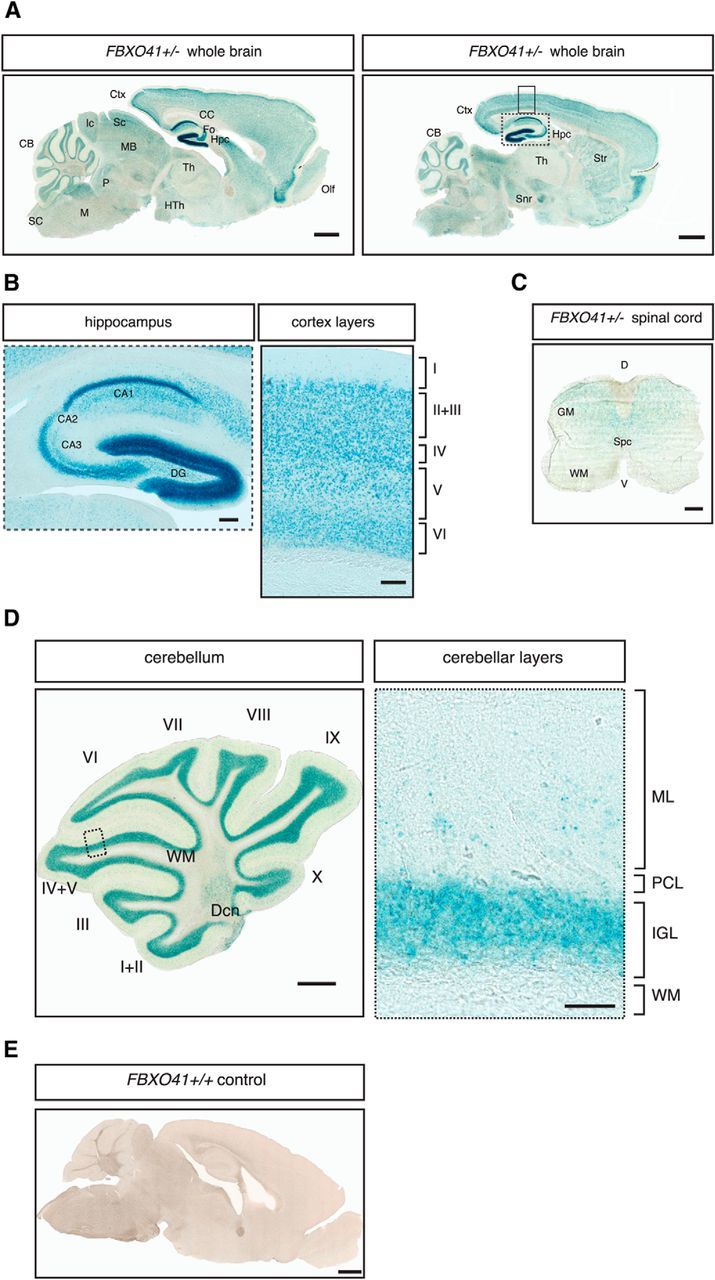
FBXO41 expression in the CNS. A, Forty-micrometer sagittal sections of the P30 FBXO41+/− mouse brain were subjected to LacZ staining. Ctx, Cortex; CB, cerebellum; CC, corpus callosum; Fo, cornix; Hpc, hippocampus; Th, thalamus; HTh, hypothalamus; Str, striatum; Snr, substantia nigra; Olf, olfactory bulb; MB, midbrain; Sc, superior colliculus; Ic, inferior colliculus; P, pons; M, medulla oblongata; SC, spinal cord. Scale bar, 1000 μm. B, Insets from A show higher magnification of the hippocampus and cortex, respectively. The roman numerals I-VI represent the cortical layers. CA1, Cornu ammonis 1; CA2, cornu ammonis 2; CA3, cornu ammonis 3; DG, dentate gyrus. Scale bars, 200 and 100 μm. C. Forty-micrometer sagittal section of the P30 FBXO41+/− mouse spinal cord was subjected to Lac Z staining. SC, Spinal cord; GM, gray matter; WM, white matter; SpC, spinal canal; D, dorsal; V, ventral. Scale bar, 200 μm. D, Forty-micrometer sagittal section of the P30 FBXO41+/− mouse cerebellum subjected to Lac Z staining. The roman numerals I-X represent cerebellar folia. Inset shows higher magnification of the various cerebellar layers. WM, White matter; Dcn, deep cerebellar nuclei; ML, molecular layer; PCL, Purkinje cell layer. Scale bars, 500 and 50 μm for the panel depicting the cerebellum and the inset with the higher magnification, respectively. E, The FBXO41+/+ mouse brain section served as a negative control. Scale bar, 1000 μm.
No signs of apoptosis or inflammation in the young FBXO41−/− mouse
Before investigating the potential role of FBXO41 in neuronal migration, we sought to establish whether FBXO41 had any effect on the overall integrity of the cells in the cerebellum as the in vivo electroporation experiments were performed in the presence of neuronal survival-promoting protein Bcl-XL. Therefore, we could not exclude a prosurvival function of FBXO41 in neurons and possible early effects on neuronal health. Compromised neuronal survival has been shown, for example, in the cerebellum of equally young repair-deficient Cockayne syndrome mice (Laposa et al., 2007). Therefore, we investigated whether the deletion of the FBXO41 gene would trigger any histopathological changes including neurodegeneration or inflammation in the young cerebellum. We examined the P16 cerebellum when neuronal migration has subsided and the cerebellar region that consists of precursor cells and postmitotic neurons (EGL) ceases to exist. We subjected sagittal paraffin sections from FBXO41+/+ and FBXO41−/− brains to immunohistochemistry. Using TUNEL staining to mark apoptotic cells, we could not find any increase in cell death in the cortex, hippocampus, or cerebellum at P16 (Fig. 5A–D). We also did not detect any activation of microglia in the P16 brain (Fig. 5E–H) or changes in astrogliosis (Fig. 5I,J). These results suggest that loss of FBXO41 does not trigger any degenerative changes in the adolescent brain at P16.
Figure 5.

FBXO41−/− mice show no signs of neurodegeneration or inflammation in the brain. A, E, I, Immunohistochemistry of paraffin sections from P16 FBXO41+/+ and FBXO41−/− cerebella using a TUNEL detection kit (A, apoptotic marker, DNAase-treated cerebellar tissue served as positive control), IBA1 antibodies (E, activated microglia, tissue from CNPaseCre/Cre mouse served as positive control; Lappe-Siefke et al., 2003), or the GFAP antibodies (I, astroglia). Scale bars: A, E, 20 μm; I, 50 μm. B–D, Quantification (cells/mm2) of TUNEL-positive cells in the cerebella, cortex, and hippocampus of FBXO41+/+ and FBXO41−/− mice. F–H, Quantification (cells/mm2) of IBA1-positive cells in the cerebella, cortex, and hippocampus of FBXO41+/+ and FBXO41−/− mice in I. B–D, F–H, Three independent FBXO41+/+ and FBXO41−/− littermates were included in the analyses (Student's t test, *p < 0.05, ns= nonsignificant, mean + SEM). J, Quantification (percentage of stained area/mm2) of GFAP-positive regions in the cortex, hippocampus, and cerebellum of FBXO41+/+ and FBXO41−/− mice in A (two-way ANOVA, mean + SEM).
Because we could not detect any degenerative events in the P16 FBXO41−/− brain, we explored whether loss of FBXO41 has any effects on the gross morphology of the brain possibly due to its role in migration. We thus analyzed H&E sections from P16 and included P12 and P30 in our analyses. The brain sizes of the FBXO41−/− mice were consistently smaller than the wild-type brains (Fig. 6A–C). Remarkably, we noticed that the P30 FBXO41−/− cerebellum adopted a distorted architecture with an unsual intralobular outfolding between lobules V and VI (Fig. 6C). We also quantified the area taken up by the cortex, hippocampus, and cerebellum with respect to the area of the entire brain. We found that, by P30, cortices and hippocampi from wild-type and homozygous FBXO41 mice occupied a comparable relative area (Fig. 6D,E). The cerebellum, however, occupied a significantly smaller area (Fig. 6F). These results indicate that the cerebellum displays the most obvious morphological defects of the FBXO41−/− brain at the ages analyzed.
Figure 6.

FBXO41−/− mice show reduced cerebellar size and abnormal architecture with age. A–C, Panels depict H&E-stained 5 μm sagittal brain sections of FBXO41+/+ and FBXO41−/− mice at P12, P16, and P30. Scale bars, 1000 μm. D–F, Graphs representing quantifications of relative sizes of the cortex, hippocampus, and the cerebellum, respectively, as observed in the FBXO41+/+ and −/− mice at the indicated ages (P12, P16, and P30). Areas are represented as percentage. Three wild-type and three knock-out littermate pairs were analyzed (two-way ANOVA, **p < 0.01, *p < 0.05, mean + SEM).
Loss of FBXO41 delays neuronal migration in the cerebellum
Due to the abnormal architecture of the P30 cerebellum in the FBXO41−/− mice and our findings that FBXO41 regulates neuronal migration, we decided to first examine the P16 cerebellum when neuronal migration events and thus lamination are completed. We observed abnormal cerebellar lamination in FBXO41−/− mice, which displayed a residual EGL and migrating cells of elongated shape in the ML (Fig. 7A). This was not the case in FBXO41+/+ cerebella (Fig. 7A). Quantification of this phenotype revealed a significant increase in the number of cells in the ML and residual EGL (Fig. 7B–D). We then monitored the development of the cerebellum of the KO mice before and after the completion of lamination. As a consequence of the delayed migration phenotype, we anticipated a thicker EGL in the younger cerebellum. This was indeed the case when we analyzed the P12 cerebella (Fig. 7E), where the EGL of FBXO41−/− cerebella took up a larger area relative to the total cerebellum (Fig. 7F), and we also found more cells in the ML (Fig. 7G). At P30, although there was no longer an EGL in the FBXO41−/− cerebella (Fig. 7H), the analysis of the ML still revealed a significantly larger number of cells in the knock-out ML compared with the wild-type condition (Fig. 7I,J). These findings indicate that, although the migrating cells eventually catch up and reach the IGL, FBXO41 is required to maintain a normal migration speed in the cerebellum.
Figure 7.
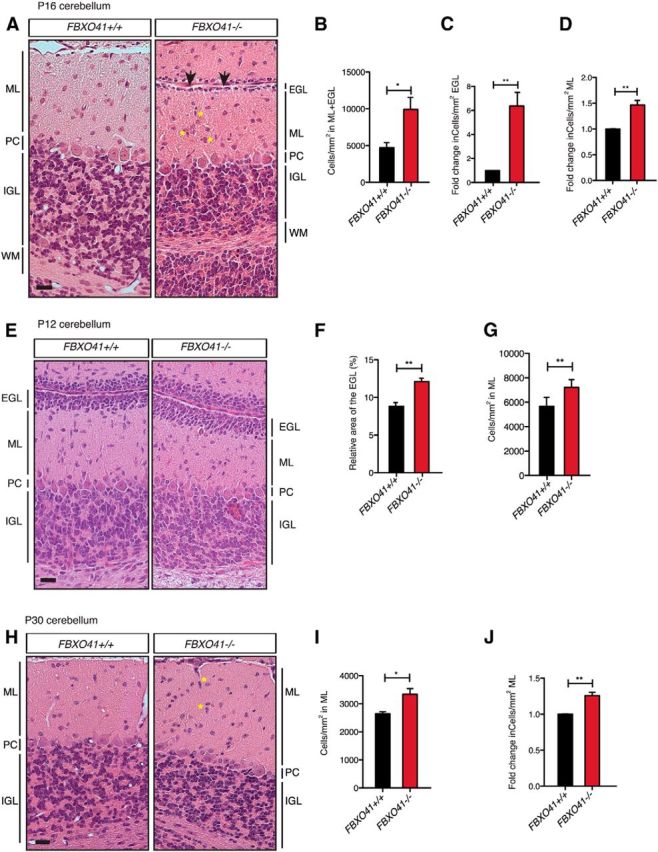
FBXO41−/− mice show developmental abnormalities in the cerebellum. A, E, H, Representative images of H&E-stained 5-μm-thick sagittal sections from P16, P12, and P30 FBXO41+/+ and FBXO41−/− cerebella, respectively. Arrowheads and asterisks in A and H indicate residual EGL and migrating neurons in the ML, respectively, in the FBXO41−/− cerebella at P16. PC, Purkinje cell layer. Scale bars, 20 μm. B–D, Quantification of the cell density in EGL+ML (B) and resulting fold change of cell number (cells/mm2) in EGL or ML (C, D) at P16. F, G, Quantification of the relative thickness of the EGL and cell density (cells/mm2) in ML at P12. I, J, Quantification of the cell density (cells/mm2) in ML and resulting fold change of cell number (cells/mm2) in ML at P30. B–D, F, G, I, J, Three independent FBXO41+/+ and FBXO41−/− littermates for all the indicated ages (P12, P16, P30) were included in the analyses (Student's t test, **p < 0.01, *p < 0.05, mean + SEM).
To characterize the nature of the supernumerous cells of the EGL and ML in more detail, we subjected sagittal paraffin sections form P12, P16, and P30 cerebella from wild-type and homozygous littermates to immunohistochemistry using NeuN, parvalbumin (PV), and PCNA antibodies to identify granule neurons, Purkinje cells, and mitotic precursors, respectively. While NeuN and PCNA gives insight into the composition of the EGL, PV shows the proper arrangement of Purkinje cells. NeuN staining revealed that the molecular layer of the FBXO41−/− cerebellum harbored at all analyzed ages significantly more NeuN+ cells compared with wild-type littermates (Fig. 8A,B). Interestingly, not all cells of the residual EGL of the P16 FBXO41−/− cerebellum were NeuN+. Interestingly, although at P12, PCNA+ cells were present in the outer EGL of both FBXO41+/+ and FBXO41−/− cerebella, we found mitotic PCNA+ cells in the residual EGL of the P16 FBXO41−/− cerebellum (Fig. 8C,D). At P30, there were no PCNA+ cells in the EGL or ML of either genotype. With the parvalbumin staining as a marker for Purkinje cells, we observed properly arranged Purkinje cells and the expansion of their dendrites into the ML of wild-type cerebella and all the way up to the residual EGL cells in the P16 FBXO41−/− cerebellum (Fig. 8E). In summary, our analyses indicate that the overall composition of the EGL is unaltered because it harbors both mitotic cells and postmitotic neurons. Our results also indicate that the more slowly subsiding EGL of the FBXO41−/− cerebellum still consists of granule neurons and mitotic precursor cells. Last, our analyses identify the supernumerous cells in the molecular layer as migrating granule neurons.
Figure 8.
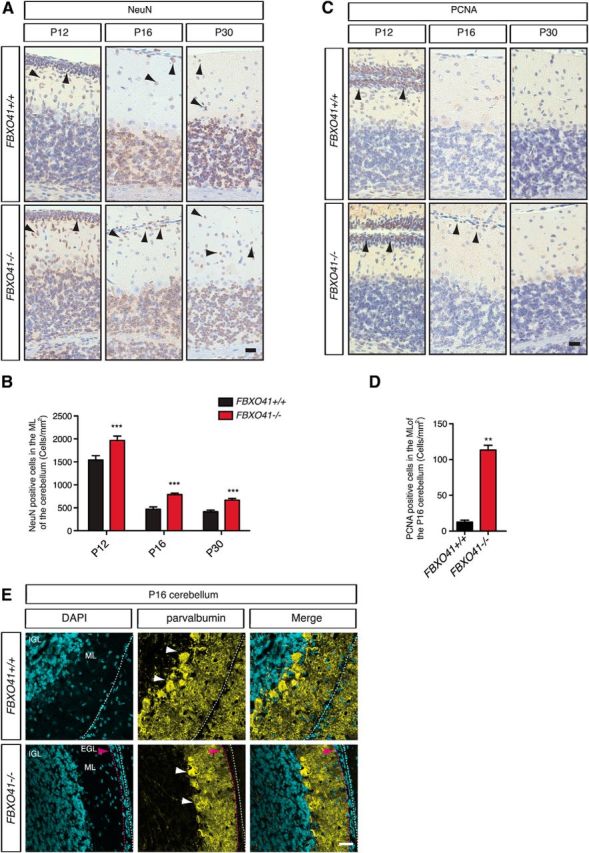
FBXO41−/− mice show delayed neuronal migration in the cerebellum. A, Representative images of 5-μm-thick sections from P12, P16, and P30 FBXO41+/+ and FBXO41−/− cerebella subjected to NeuN staining. Arrowheads indicate NeuN+ cells. Scale bar, 20 μm. B, Quantification of the density of NeuN+ cells (cells/mm2) in ML at the indicated ages. Three independent FBXO41+/+ and FBXO41−/− littermates for all the indicated ages (P12, P16, P30) were included in the analyses (two-way ANOVA, ***p < 0.001, mean + SEM). C, Representative images of 5-μm-thick sections from FBXO41+/+ and FBXO41−/− cerebella at P12, P16, and P30 subjected to PCNA staining. Arrowheads indicate PCNA+ cells. Scale bar, 20 μm. D, Quantification of PNCA+ cells in the ML of cerebella from three independent FBXO41+/+ and FBXO41−/− littermates (Student's t test, **p < 0.01, mean + SEM). E, Confocal images of cryosections from P16 FBXO41+/+ and FBXO41−/− cerebella subjected to immunohistochemistry using antibodies against parvalbumin (Purkinje cells and interneurons). Scale bar, 100 μm. Red arrowhead indicates residual EGL in the P16 FBXO41−/− cerebella. ML, Molecular layer.
In a final experiment, we addressed whether the delay in neuronal migration, and likely impairment of cerebellar wiring, affected not only the gross morphology, but also the integrity of the neurons in the cerebellum of the P30 FBXO41−/− mice. Although these mice gained weight, they were always significantly lighter than wild-type littermates (Fig. 9A) and were still motor impaired and unable to reach for the food tray and water bottle in the cage and thus required special supply of food and water. Analyses showed that the mice displayed an ataxic gait, had strong hindlimb clasping, and performed poorly on the ledge test (Fig. 9B–D). We also subjected FBXO41+/+ and FBXO41−/− brain sections to TUNEL staining and found little or no increase of apoptotic cells in the cortex but a significant increase in the hippocampus and the cerebellum (Fig. 9E–H). In the latter region, many of the apoptotic cells appeared clustered in the intralobular outfolding and collapsed lobule VI (Fig. 9E). These results suggest that the delay of cerebellar development as a result of loss of FBXO41 not only triggers changes in the morphology, but also affects the integrity of neurons.
Figure 9.

Motor deficits and neurodegeneration in the young adult FBXO41−/− mouse. A, Weight curve of FBXO41+/+ and FBXO41−/− mice over a period of 60 d. Four mice of each genotype were included in the analysis (two-way ANOVA, ***p < 0.001, **p < 0.01, mean + SEM). B, P30 littermates were tested for hindlimb clasping. Four mice of each genotype were included in the analysis (Student's t test, **p < 0.01, mean + SEM); 0 = normal phenotype, 3 = worst manifestation of phenotype. C, P30 littermates were subjected to gait analysis. Four mice of each genotype were included in the analysis (Student's t test, **p < 0.01, mean + SEM); 0 = normal phenotype, 3 = worst manifestation of phenotype. D, P30 littermates were subjected to the ledge test. Four mice of each genotype were included in the analysis (Student's t test, *p < 0.05, mean + SEM); 0 = normal phenotype, 3 = worst manifestation of phenotype. E, Immunohistochemistry of paraffin sections from P16 FBXO41+/+ and FBXO41−/− cerebella using a TUNEL detection kit. Red arrows indicate clusters of apoptotic cells and black arrows indicate individual apoptotic cells. Scale bars, 500 μm (panel 1), 100 μm (inset A) and 20 μm (inset B). F–H, Quantification of TUNEL+ cells. Three independent FBXO41+/+ and FBXO41−/− cerebella (F), cortices (G), and hippocampi (H) were included in the analyses (Student's t test, **p < 0.01, *p < 0.05, mean + SEM). I, Cultured cerebellar granule neurons were transfected at DIV2 with control vector, FBXO41 RNAi#5 (functional), or FBXO41 RNAi#8 (nonfunctional) plasmid together with the β-Gal reporter plasmid. Neurons were subjected to immunohistochemistry with the cleaved-caspase-3 antibody to mark apoptotic neurons and the DNA dye Hoechst at DIV6, followed by analysis. Scale bar, 50 μm. Arrowheads indicate transfected neurons. Insets show higher magnification. J, Quantification of apoptotic neurons in I. At least 100 neurons were counted per condition per experiment in three independent experiments (ANOVA, ***p < 0.001, mean + SEM). K, Granule neurons transfected with control plasmids (U6 and pCMVmyc), FBXO41 RNAi #5, and pCMVmyc or FBXO41 RNAi#5 and myc-FBXO41-Res expression plasmid were analyzed as in I. At least 100 neurons were counted per condition per experiment in three independent experiments (ANOVA, ***p < 0.001, mean + SEM).
To support the role of FBXO41 in apoptosis, we performed cell survival assays using the cerebellar granule neuron culture system. We examined control-transfected and FBXO41 RNAi neurons and found that knock-down of FBXO41 using a functional RNAi #5, but not the nonfunctional RNAi#8, leads to an increase in cleaved caspase 3+ neurons (Fig. 9I, J). To ensure the specificity of FBXO41 RNAi-induced cell death, we performed a rescue experiment and expressed the RNAi-resistant FBXO41 (FBXO41-Res) in the background of FBXO41 RNAi (Fig. 9K). We found that the number of apoptotic neurons in the rescue condition was reduced to control level. These results support a prosurvival role for FBXO41 in neurons.
Discussion
In this study, we have characterized the novel neuron-specific F-box protein FBXO41 in the establishment and maintenance of proper cerebellar architecture. FBXO41 belongs to the large family of F-box proteins that share the F-box domain. As a member of this family, FBXO41 is anticipated to be a subunit of an E3 ubiquitin ligase, which will be discussed in the follow-up paper (Mukherjee M., Holubowska A., Schwedhelm-Domeyer N., Gellerer A., Kannan M., Stegmüller J., unpublished data). FBXO41 has a striking neuron-specific expression in various areas of the CNS. At the subcellular level, FBXO41 localizes to the cytoplasm and the centrosome. In vivo electroporation experiments in the cerebellum revealed cytoplasmic FBXO41 as the crucial player. Further work will be necessary to identify mechanisms underlying FBXO41-mediated neuronal migration. Successful neuronal migration of the cerebellum is dependent on proper guidance by extrinsic cues, on cell-intrinsic mechanisms including cytoskeletal dynamics or transcription factors, and on the interplay of both (Solecki et al., 2006; Tiveron and Cremer, 2008; Romaniello et al., 2015). Because of FBXO41's abundant cytoplasmic expression in neurons, it could, for example, interfere intrinsically with the dynamics of the leading edge or intercept incoming extrinsic promigratory stimuli.
Upon systemic deletion of the FBXO41 gene in the mouse, we observed the most severe morphological changes in the cerebellum, characterized by the failure of migrating granule neurons to reach the IGL within a developmentally appropriate time window. A comparable delay in migration of granule neurons in the cerebellum has been described, for example, in mouse models lacking the actin-binding protein profilin1 or the microtubule-binding protein APC2 (Adenomatous Poliposis Coli 2; Kullmann et al., 2012; Shintani et al., 2012). Because we found obvious and persisting ataxia-like motor deficits in the cerebellum in the FBXO41−/− mouse, it is very likely that the major motor phenotype can be attributed to this phenotype. Cerebellar dysfunction has also been characterized in mice modeling human cerebellar ataxias, in which the development of the cerebellum has derailed. Deletion of the Ahi1 gene in mice for example results in abnormal cerebellar development and is thus viewed as a Joubert syndrome mouse model (Lancaster et al., 2011). In addition, conditional deletion of the Robo3 gene in the cerebellum, mutations of which are associated with horizontal gaze palsy with progressive scoliosis, recapitulate developmental deficits in the cerebellum that lead to severely ataxic mice (Renier et al., 2010; Badura et al., 2013).
Ataxia is often associated with degenerative events in the cerebellum (Zoghbi, 2000; Orr and Zoghbi, 2007; Klockgether and Paulson, 2011). Although we did not find any signs of apoptosis or inflammation at a very young age, we did observe apoptotic events in the young adult mice. This indicates that loss of FBXO41 triggers defects in neuronal migration in the developing cerebellum, which may contribute largely to the motor deficits in the young mouse. The onset of neurodegeneration in the young adult mouse could be a consequence of deferred wiring and faulty architecture in the cerebellum or the requirement of FBXO41 for long-term integrity and health of the neurons. Although migrating neurons of the cerebellum catch up in the older FBXO41−/− mouse and the animals gain weight, the impending neurodegeneration is likely also to contribute to the small-sized cerebellum and the persisting motor symptoms. Neurodegeneration in the cerebellum leading to motor dysfunction is causative in several human cerebellar disorders such as spinocerebellar ataxias (SCA), which have been successfully modeled (Grote and La Spada, 2003; Colomer Gould, 2012; Cendelin, 2014). Among these, the SCA7 mouse also displays early onset of ataxia and premature death (Yoo et al., 2003). Although other deficits contribute to the severity of the phenotype of this mouse, the ataxic phenotype can be ascribed to cerebellar degeneration.
We also find that FBXO41 is abundantly expressed in, for example, the cortex and the hippocampus. Therefore, it is conceivable that FBXO41 affects neuronal migration in these regions during development as well. The early postnatal cortex, however, does not appear to have obvious histopathological disturbances (unpublished data). This could mean that FBXO41 has only a migration-regulating role in the cerebellum or that migration deficits in the cortex have caught up in the course of development. A thorough examination of the lamination of the embryonic cortex is thus required to monitor the migration progress. The same holds true for other FBXO41-positive neurons that require migration to reach their final destination, including hippocampal neurons. FBXO41 is also expressed in adult cerebellar granule neurons when developmental events are completed. Given the high expression of FBXO41, not only in the adult cerebellum, but also several other areas of the brain including the hippocampus, a role in the development and maintenance of synapses is also possible and deserves further investigation.
Developmental delay of the cerebellum and cerebellar and hippocampal neurodegeneration upon loss of FBXO41 could also affect cognitive function. Although the hippocampus is famously associated with learning and memory, recent work has revealed that the cerebellum plays a significant role in cognition. Developmental deficiencies of the cerebellum have been linked to psychiatric disorders including autism spectrum disorders (Schmahmann and Sherman, 1998; Schmahmann et al., 2007; Fatemi et al., 2012; Basson and Wingate, 2013), so cognitive deficits may coexist along with motor impairment in the FBXO41−/− mice.
The ataxia-like phenotype of the knock-out mice supports the notion that FBXO41 is an important player in motor coordination and the FBXO41−/− mouse can thus be viewed as a novel model for congenital ataxia. Because ∼40% of ataxias are of unknown genetic causes (Sailer and Houlden, 2012), the search for causative genes is imperative and the discovery of new genes such as the FBXO41 gene and the recently identified TRCP3 gene as critical factors of cerebellar development and integrity are important because mutations and deletions in the these genes might be associated with unresolved forms of congenital ataxias (Becker et al., 2009; Becker, 2015). In addition, future elucidation of the FBXO41 pathway will shed more light onto the mechanisms that regulate cerebellar development and will contribute to our understanding of the role of FBXO41 in proper motor coordination and possibly disease.
Footnotes
The study was funded by the Max Planck Society, the Cluster of Excellence, the Deutsche Forschungsgemeinschaft Research Center for Nanoscale Microscopy and Molecular Physiology of the Brain, and the Junior Group Stipend of the University of Göttingen. We thank D. Brockelt, S. Vingill, and A. Matz for critical reading of the manuscript; U. Bode and M. Schindler for ES cell culture and ES cell injection, respectively; R. Bunse for help in the initial phase of the study; A. Gellerer for help in the later phase of the study; and S. Papiol for help with statistical analyses.
The authors declare no competing financial interests.
References
- Altman J, Bayer SA. Development of the cerebellar system: in relation to its evolution, structure, and functions. Boca Raton, FL: CRC; 1997. [Google Scholar]
- Anckar J, Bonni A. Regulation of neuronal morphogenesis and positioning by ubiquitin-specific proteases in the cerebellum. PLoS One. 2015;10:e0117076. doi: 10.1371/journal.pone.0117076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen JS, Wilkinson CJ, Mayor T, Mortensen P, Nigg EA, Mann M. Proteomic characterization of the human centrosome by protein correlation profiling. Nature. 2003;426:570–574. doi: 10.1038/nature02166. [DOI] [PubMed] [Google Scholar]
- Badura A, Schonewille M, Voges K, Galliano E, Renier N, Gao Z, Witter L, Hoebeek FE, Chédotal A, De Zeeuw CI. Climbing fiber input shapes reciprocity of Purkinje cell firing. Neuron. 2013;78:700–713. doi: 10.1016/j.neuron.2013.03.018. [DOI] [PubMed] [Google Scholar]
- Basson MA, Wingate RJ. Congenital hypoplasia of the cerebellum: developmental causes and behavioral consequences. Front Neuroanat. 2013;7:29. doi: 10.3389/fnana.2013.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker EB. From mice to men: TRPC3 in cerebellar ataxia. Cerebellum. 2015 doi: 10.1007/s12311-015-0663-y. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker EB, Oliver PL, Glitsch MD, Banks GT, Achilli F, Hardy A, Nolan PM, Fisher EM, Davies KE. A point mutation in TRPC3 causes abnormal Purkinje cell development and cerebellar ataxia in moonwalker mice. Proc Natl Acad Sci U S A. 2009;106:6706–6711. doi: 10.1073/pnas.0810599106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilimoria PM, Bonni A. Cultures of cerebellar granule neurons. CSH Protoc. 2008;2008 doi: 10.1101/pdb.prot5107. pdb.prot5107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cendelin J. From mice to men: lessons from mutant ataxic mice. Cerebellum and Ataxias. 2014;1:1–21. doi: 10.1186/2053-8871-1-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colomer Gould VF. Mouse models of spinocerebellar ataxia type 3 (Machado-Joseph disease) Neurotherapeutics. 2012;9:285–296. doi: 10.1007/s13311-012-0117-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falnikar A, Tole S, Baas PW. Kinesin-5, a mitotic microtubule-associated motor protein, modulates neuronal migration. Mol Biol Cell. 2011;22:1561–1574. doi: 10.1091/mbc.E10-11-0905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Famulski JK, Trivedi N, Howell D, Yang Y, Tong Y, Gilbertson R, Solecki DJ. Siah regulation of Pard3A controls neuronal cell adhesion during germinal zone exit. Science. 2010;330:1834–1838. doi: 10.1126/science.1198480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatemi SH, Aldinger KA, Ashwood P, Bauman ML, Blaha CD, Blatt GJ, Chauhan A, Chauhan V, Dager SR, Dickson PE, Estes AM, Goldowitz D, Heck DH, Kemper TL, King BH, Martin LA, Millen KJ, Mittleman G, Mosconi MW, Persico AM, et al. Consensus paper: pathological role of the cerebellum in autism. Cerebellum. 2012;11:777–807. doi: 10.1007/s12311-012-0355-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L, Allen NS, Simo S, Cooper JA. Cullin 5 regulates Dab1 protein levels and neuron positioning during cortical development. Genes Dev. 2007;21:2717–2730. doi: 10.1101/gad.1604207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garden GA, Libby RT, Fu YH, Kinoshita Y, Huang J, Possin DE, Smith AC, Martinez RA, Fine GC, Grote SK, Ware CB, Einum DD, Morrison RS, Ptacek LJ, Sopher BL, La Spada AR. Polyglutamine-expanded ataxin-7 promotes non-cell-autonomous purkinje cell degeneration and displays proteolytic cleavage in ataxic transgenic mice. J Neurosci. 2002;22:4897–4905. doi: 10.1523/JNEUROSCI.22-12-04897.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govek EE, Hatten ME, Van Aelst L. The role of Rho GTPase proteins in CNS neuronal migration. Dev Neurobiol. 2011;71:528–553. doi: 10.1002/dneu.20850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grote SK, La Spada AR. Insights into the molecular basis of polyglutamine neurodegeneration from studies of a spinocerebellar ataxia type 7 mouse model. Cytogenet Genome Res. 2003;100:164–174. doi: 10.1159/000072851. [DOI] [PubMed] [Google Scholar]
- Guyenet SJ, Furrer SA, Damian VM, Baughan TD, La Spada AR, Garden GA. A simple composite phenotype scoring system for evaluating mouse models of cerebellar ataxia. J Vis Exp. 2010:pii, 1787. doi: 10.3791/1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heng JI, Qu Z, Ohtaka-Maruyama C, Okado H, Kasai M, Castro D, Guillemot F, Tan SS. The zinc finger transcription factor RP58 negatively regulates Rnd2 for the control of neuronal migration during cerebral cortical development. Cereb Cortex. 2015;25:806–816. doi: 10.1093/cercor/bht277. [DOI] [PubMed] [Google Scholar]
- Higginbotham HR, Gleeson JG. The centrosome in neuronal development. Trends Neurosci. 2007;30:276–283. doi: 10.1016/j.tins.2007.04.001. [DOI] [PubMed] [Google Scholar]
- Holubowska A, Mukherjee C, Vadhvani M, Stegmüller J. Genetic manipulation of cerebellar granule neurons in vitro and in vivo to study neuronal morphology and migration. J Vis Exp. 2014 doi: 10.3791/51070. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang GJ, Edwards A, Tsai CY, Lee YS, Peng L, Era T, Hirabayashi Y, Tsai CY, Nishikawa S, Iwakura Y, Chen SJ, Flint J. Ectopic cerebellar cell migration causes maldevelopment of Purkinje cells and abnormal motor behaviour in Cxcr4 null mice. PLoS One. 2014;9:e86471. doi: 10.1371/journal.pone.0086471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaarsma D, van den Berg R, Wulf PS, van Erp S, Keijzer N, Schlager MA, de Graaff E, De Zeeuw CI, Pasterkamp RJ, Akhmanova A, Hoogenraad CC. A role for Bicaudal-D2 in radial cerebellar granule cell migration. Nat Commun. 2014;5:3411. doi: 10.1038/ncomms4411. [DOI] [PubMed] [Google Scholar]
- Kannan M, Lee SJ, Schwedhelm-Domeyer N, Stegmüller J. The E3 ligase Cdh1-anaphase promoting complex operates upstream of the E3 ligase Smurf1 in the control of axon growth. Development. 2012a;139:3600–3612. doi: 10.1242/dev.081786. [DOI] [PubMed] [Google Scholar]
- Kannan M, Lee SJ, Schwedhelm-Domeyer N, Nakazawa T, Stegmüller J. p250GAP is a novel player in the Cdh1-APC/Smurf1 pathway of axon growth regulation. PLoS One. 2012b;7:e50735. doi: 10.1371/journal.pone.0050735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawauchi T. Regulation of cell adhesion and migration in cortical neurons: not only Rho but also Rab family small GTPases. Small GTPases. 2011;2:36–40. doi: 10.4161/sgtp.2.1.15001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klockgether T, Paulson H. Milestones in ataxia. Mov Disord. 2011;26:1134–1141. doi: 10.1002/mds.23559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komuro H, Yacubova E. Recent advances in cerebellar granule cell migration. Cell Mol Life Sci. 2003;60:1084–1098. doi: 10.1007/s00018-003-2248-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi Y, Stegmüller J, Matsuda T, Bonni S, Bonni A. Cdh1-APC controls axonal growth and patterning in the mammalian brain. Science. 2004;303:1026–1030. doi: 10.1126/science.1093712. [DOI] [PubMed] [Google Scholar]
- Kuijpers M, Hoogenraad CC. Centrosomes, microtubules and neuronal development. Mol Cell Neurosci. 2011;48:349–358. doi: 10.1016/j.mcn.2011.05.004. [DOI] [PubMed] [Google Scholar]
- Kullmann JA, Neumeyer A, Gurniak CB, Friauf E, Witke W, Rust MB. Profilin1 is required for glial cell adhesion and radial migration of cerebellar granule neurons. EMBO Rep. 2012;13:75–82. doi: 10.1038/embor.2011.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumazawa A, Mita N, Hirasawa M, Adachi T, Suzuki H, Shafeghat N, Kulkarni AB, Mikoshiba K, Inoue T, Ohshima T. Cyclin-dependent kinase 5 is required for normal cerebellar development. Mol Cell Neurosci. 2013;52:97–105. doi: 10.1016/j.mcn.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster MA, Gopal DJ, Kim J, Saleem SN, Silhavy JL, Louie CM, Thacker BE, Williams Y, Zaki MS, Gleeson JG. Defective Wnt-dependent cerebellar midline fusion in a mouse model of Joubert syndrome. Nat Med. 2011;17:726–731. doi: 10.1038/nm.2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laposa RR, Huang EJ, Cleaver JE. Increased apoptosis, p53 up-regulation, and cerebellar neuronal degeneration in repair-deficient Cockayne syndrome mice. Proc Natl Acad Sci U S A. 2007;104:1389–1394. doi: 10.1073/pnas.0610619104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappe-Siefke C, Goebbels S, Gravel M, Nicksch E, Lee J, Braun PE, Griffiths IR, Nave KA. Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat Genet. 2003;33:366–374. doi: 10.1038/ng1095. [DOI] [PubMed] [Google Scholar]
- Lin CH, Tallaksen-Greene S, Chien WM, Cearley JA, Jackson WS, Crouse AB, Ren S, Li XJ, Albin RL, Detloff PJ. Neurological abnormalities in a knock-in mouse model of Huntington's disease. Hum Mol Genet. 2001;10:137–144. doi: 10.1093/hmg/10.2.137. [DOI] [PubMed] [Google Scholar]
- Millen KJ, Gleeson JG. Cerebellar development and disease. Curr Opin Neurobiol. 2008;18:12–19. doi: 10.1016/j.conb.2008.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr HT, Zoghbi HY. Trinucleotide repeat disorders. Annu Rev Neurosci. 2007;30:575–621. doi: 10.1146/annurev.neuro.29.051605.113042. [DOI] [PubMed] [Google Scholar]
- Radyushkin K, Hammerschmidt K, Boretius S, Varoqueaux F, El-Kordi A, Ronnenberg A, Winter D, Frahm J, Fischer J, Brose N, Ehrenreich H. Neuroligin-3-deficient mice: model of a monogenic heritable form of autism with an olfactory deficit. Genes Brain Behav. 2009;8:416–425. doi: 10.1111/j.1601-183X.2009.00487.x. [DOI] [PubMed] [Google Scholar]
- Ramahi JS, Solecki DJ. The PAR polarity complex and cerebellar granule neuron migration. Adv Exp Med Biol. 2014;800:113–131. doi: 10.1007/978-94-007-7687-6_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renier N, Schonewille M, Giraudet F, Badura A, Tessier-Lavigne M, Avan P, De Zeeuw CI, Chédotal A. Genetic dissection of the function of hindbrain axonal commissures. PLoS Biol. 2010;8:e1000325. doi: 10.1371/journal.pbio.1000325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romaniello R, Arrigoni F, Bassi MT, Borgatti R. Mutations in alpha- and beta-tubulin encoding genes: Implications in brain malformations. Brain Dev. 2015;37:273–280. doi: 10.1016/j.braindev.2014.06.002. [DOI] [PubMed] [Google Scholar]
- Sailer A, Houlden H. Recent advances in the genetics of cerebellar ataxias. Curr Neurol Neurosci Rep. 2012;12:227–236. doi: 10.1007/s11910-012-0267-6. [DOI] [PubMed] [Google Scholar]
- Sathyamurthy A, Yin DM, Barik A, Shen C, Bean JC, Figueiredo D, She JX, Xiong WC, Mei L. ERBB3-mediated regulation of Bergmann glia proliferation in cerebellar lamination. Development. 2015;142:522–532. doi: 10.1242/dev.115931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmahmann JD, Sherman JC. The cerebellar cognitive affective syndrome. Brain. 1998;121:561–579. doi: 10.1093/brain/121.4.561. [DOI] [PubMed] [Google Scholar]
- Schmahmann JD, Weilburg JB, Sherman JC. The neuropsychiatry of the cerebellum: insights from the clinic. Cerebellum. 2007;6:254–267. doi: 10.1080/14734220701490995. [DOI] [PubMed] [Google Scholar]
- Shintani T, Takeuchi Y, Fujikawa A, Noda M. Directional neuronal migration is impaired in mice lacking adenomatous polyposis coli 2. J Neurosci. 2012;32:6468–6484. doi: 10.1523/JNEUROSCI.0590-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sillitoe RV, Joyner AL. Morphology, molecular codes, and circuitry produce the three-dimensional complexity of the cerebellum. Annu Rev Cell Dev Biol. 2007;23:549–577. doi: 10.1146/annurev.cellbio.23.090506.123237. [DOI] [PubMed] [Google Scholar]
- Solecki DJ, Govek EE, Hatten ME. mPar6 alpha controls neuronal migration. J Neurosci. 2006;26:10624–10625. doi: 10.1523/JNEUROSCI.4060-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegmüller J, Konishi Y, Huynh MA, Yuan Z, Dibacco S, Bonni A. Cell-intrinsic regulation of axonal morphogenesis by the Cdh1-APC target SnoN. Neuron. 2006;50:389–400. doi: 10.1016/j.neuron.2006.03.034. [DOI] [PubMed] [Google Scholar]
- Tiveron MC, Cremer H. CXCL12/CXCR4 signalling in neuronal cell migration. Curr Opin Neurobiol. 2008;18:237–244. doi: 10.1016/j.conb.2008.06.004. [DOI] [PubMed] [Google Scholar]
- Tsai LH, Gleeson JG. Nucleokinesis in neuronal migration. Neuron. 2005;46:383–388. doi: 10.1016/j.neuron.2005.04.013. [DOI] [PubMed] [Google Scholar]
- Vadhvani M, Schwedhelm-Domeyer N, Mukherjee C, Stegmüller J. The centrosomal E3 ubiquitin ligase FBXO31-SCF regulates neuronal morphogenesis and migration. PLoS One. 2013;8:e57530. doi: 10.1371/journal.pone.0057530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallee RB, Seale GE, Tsai JW. Emerging roles for myosin II and cytoplasmic dynein in migrating neurons and growth cones. Trends Cell Biol. 2009;19:347–355. doi: 10.1016/j.tcb.2009.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingate RJ. The rhombic lip and early cerebellar development. Curr Opin Neurobiol. 2001;11:82–88. doi: 10.1016/S0959-4388(00)00177-X. [DOI] [PubMed] [Google Scholar]
- Wynshaw-Boris A, Gambello MJ. LIS1 and dynein motor function in neuronal migration and development. Genes Dev. 2001;15:639–651. doi: 10.1101/gad.886801. [DOI] [PubMed] [Google Scholar]
- Yong Y, Meng Y, Ding H, Fan Z, Tang Y, Zhou C, Luo J, Ke ZJ. PACT/RAX regulates the migration of cerebellar granule neurons in the developing cerebellum. Sci Rep. 2015;5:7961. doi: 10.1038/srep07961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo SY, Pennesi ME, Weeber EJ, Xu B, Atkinson R, Chen S, Armstrong DL, Wu SM, Sweatt JD, Zoghbi HY. SCA7 knockin mice model human SCA7 and reveal gradual accumulation of mutant ataxin-7 in neurons and abnormalities in short-term plasticity. Neuron. 2003;37:383–401. doi: 10.1016/S0896-6273(02)01190-X. [DOI] [PubMed] [Google Scholar]
- Zoghbi HY. Spinocerebellar ataxias. Neurobiol Dis. 2000;7:523–527. doi: 10.1006/nbdi.2000.0346. [DOI] [PubMed] [Google Scholar]