

Abstract
The major challenge for progressive multiple sclerosis therapy is the promotion of remyelination from inflammation-induced demyelination. A switch from an M1- to an M2-dominant polarization of microglia is critical in these repair processes. In this study, we identified the homeobox gene msh-like homeobox-3 (Msx3) as a new pivotal regulator for microglial polarization. MSX3 was induced during microglia M2 polarization and repressed in M1 cells. The expression of MSX3 in microglia was dynamically regulated during experimental autoimmune encephalomyelitis (EAE), which is an animal model of multiple sclerosis. The overexpression of MSX3 in microglia promoted M2 but impeded M1 polarization. Interrupting MSX3 expression in microglia accelerated inflammation-induced demyelination and neurodegeneration. The conditioned medium from MSX3-transduced microglia promoted oligodendrocyte progenitor survival, differentiation, and neurite outgrowth. The adoptive transfer of MSX3-transduced microglia suppressed EAE and facilitated remyelination within the murine CNS in EAE and the LPC model. Mechanically, chromatin immunoprecipitation assays also indicated that MSX3 directly regulated three key genes associated with microglia M2 polarization, including Pparg, Stat6, and Jak3. Importantly, we found that overexpression of MSX3 in human-derived microglia represents the M2 phenotype and ameliorated EAE after intraventricular injection. Our findings suggest a new homeobox protein-dependent mechanism for driving microglia M2 polarization and identify MSX3 as an attractive therapeutic approach for preventing inflammation-induced demyelination and promoting remyelination.
Keywords: EAE, microglia, MSX3, multiple sclerosis, polarization
Introduction
Multiple sclerosis (MS) is an autoimmune CNS demyelinating disease. Most patients with MS have an initial relapsing–remitting neurologic symptom that later develops into a secondary progressive course of irreversible disability (Trapp and Nave, 2008). Extensive remyelination that is observed only during the early stages of MS but that is impaired in the later stages might be the leading cause of this shift (Scolding et al., 1998). The failure of effective remyelination in progressive MS is considered to be a result of axonal degeneration (Zamvil and Steinman, 2003) and a differentiation block of oligodendrocyte progenitors (Kuhlmann et al., 2008; Shen et al., 2008) in chronic MS lesions.
Recently, microglia (MG) were demonstrated to be polarized to play different roles in regulating neuron and oligodendrocyte progenitor functions (Minghetti, 2005; Rasmussen et al., 2007; Peferoen et al., 2014). Microglia can be polarized into a proinflammatory and neurotoxic phenotype (M1) under stimulation with lipopolysaccharide, interferon-γ (IFNγ), or interleukin-17A (IL-17A), whereas IL-4 or IL-13 induces a state of immunological regulation phenotype (M2), which secretes anti-inflammatory cytokines and growth factors that promote oligodendrocyte progenitor differentiation and that protect neurons from damage (Butovsky et al., 2006; Kigerl et al., 2009; Liao et al., 2012; Miron et al., 2013). M1 microglia exist in all types of MS lesions (Miron et al., 2013). The number of M1 microglia correlates with the extent of axonal damage (Bitsch et al., 2000; Rasmussen et al., 2007; Miron et al., 2013), but M2 densities only increased in acute active lesions and in the rim of chronic active lesions where efficient remyelination occurs (Miron et al., 2013). A block in the M1 to M2 switch may contribute to remyelination failure in chronic inactive MS lesions (Miron et al., 2013).
Manipulating polarization phenotypes of microglia might be a promising therapeutic strategy for treating MS (Butovsky et al., 2006); however, our understanding of the transcriptional mechanism of the M2 polarization pathway remains limited. In this study, using comparative gene profiling of differentially activated microglia, we identified msh-like homeobox-3 (Msx3) as a particularly upregulated homeobox gene in M2 polarized microglia. MSX3 expression is reported to be highly restricted in the developing neural tube (Shimeld et al., 1996) and might be associated with generation of dorsal interneurons (Liu et al., 2004). However, whether this gene is involved in microglia polarization is unclear.
In the present study, MSX3 was found to drive microglia to adapt an M2 phenotype. MSX3-overexpressed microglia promoted the maturation of oligodendrocyte precursors and remyelination, protected neurons from injury, and prevented experimental autoimmune encephalomyelitis (EAE) progression, whereas the ablation of MSX3 in microglia induced the apoptosis of oligodendrocyte precursors, abrogated neuroprotection, and exacerbated EAE pathology. Mechanistically, MSX3 was found to control the expression of key proteins in M2 polarization, including JAK3, STAT6, and PPARγ. These findings established MSX3 as an important microglia polarization regulator, suggested a previously unknown signaling mechanism of MSX3 function, and provided a promising therapeutic approach for treating demyelinating diseases.
Materials and Methods
Animal experiments.
All animal experiments were performed in adherence with the National Institutes of Health Guidelines on the Care and Use of Laboratory Animals and approved by the Second Military Medical University Committee on Animal Care. For EAE induction, female C57BL/6 mice (8–10 weeks) were subcutaneously immunized with MOG35–55 (GL Biochem) in complete Freund's adjuvant containing heat-killed Mycobacterium tuberculosis (H37Ra strain; Difco). Pertussis toxin (Calbiochem-EMD Chemicals) in PBS was administered intraperitoneally on days 0 and 2. Mice were blindly examined daily for disease signs by independent researchers and were assigned scores on a scale of 0–5 as follows: 0, no clinical signs; 1, paralyzed tail; 2, paresis; 3, paraplegia; 4, paraplegia with forelimb weakness or paralysis; and 5, moribund state or death. For the focal demyelination model, a spinal cord lesion was induced as described previously (Arnett et al., 2004; Luo et al., 2014). Briefly, 1 μl of 1% l-a-lysophosphatidylcholine (LPC; lysolectin; Sigma-Aldrich) in 0.9% sodium chloride solution was microinjected into the dorsal column at T11–T12 spinal cord of C57BL/6 mice (8–10 weeks), followed by a laminectomy. The day of LPC injection was designated as day 0 (0 dpl). Lesion volumes were calculated by the lesion area from serial sections throughout the entire lesion based on the equation for volume of cylinder (volume = lesion area × length of lesion). For stereotactic transplantation of microglia in the EAE model, A small hole was drilled into the mouse skull, and a 10 μl Hamilton syringe with a 29 gauge needle was inserted into the right lateral ventricle (bregma, −0.7; lateral, 1.2; ventral, 2.4). For transplantation of microglia in the LPC-induced focal lesion model, a laminectomy was performed, and microglia were injected in the same area using the above paradigm at 3 dpl. For both models, microglia (1 × 106 cells in 10 μl) were injected at a flow rate of 1 μl/min.
Isolation of microglia derived from the spinal cord.
The spinal cords of normal or EAE mice were isolated, homogenized, filtrated, centrifuged, and suspended in 70% Percoll (GE Healthcare) and overlaid with 37 and 30% gradient. After centrifugation, the majority of mononuclear cells, found in the interface of 37 and 70% Percoll, were collected. These cells were incubated with a mouse CD16/CD32 antibody (BD Biosciences) for 5 min and surface stained with PE-labeled CD45 and FITC-labeled CD11b antibodies according to the manufacturer's instructions (BD Biosciences). CD45+/CD11b+ cells were then assessed and sorted on a Moflo XDP (Beckman Coulter).
Cell cultures.
CNS mixed glial cell cultures were generated and cultured in DMEM/F-12 containing 10% fetal bovine serum (D10; Invitrogen) and an antibiotic mixture (1% penicillin/streptomycin; Invitrogen) at 37°C and 5% CO2 for 10 d. For the purification of microglia, cultures were shaken for 6 h at 180 rpm at 37°C to collect microglia. For polarization, microglia were stimulated with IL-4 (10 ng/ml), transforming growth factor-β1 (TGF-β1; 5 ng/ml), IFNγ (100 ng/ml), or IL-17A (10 ng/ml; R&D Systems) for 24 h.
Primary human microglia (HM1900) were purchased from ScienCell Research Laboratories and cultured in D10 medium with 10 ng/ml macrophage-colony stimulation factor (Sigma-Aldrich).
Purified oligodendrocyte precursor cells (OPCs) were isolated by collecting the floating fraction of 10-d-old mixed glial cultures after the depletion of microglia as described above, with a 16 h incubation on a rotary shaker at 37°C at 210 rpm, and after the depletion of microglia and astrocytes by differential adhesion in uncoated Petri dishes for 1 h at 37°C and 5% CO2. The collected OPCs in D10 medium were then plated at densities between 50,000 and 5000 cells/cm2 and maintained at 37°C and 5% CO2. OPCs were grown in Neurobasal medium with 2% B27 (Invitrogen) and 10 ng/ml biotin (Sigma-Aldrich) with 5 μg/ml N-acetyl cysteine (Sigma-Aldrich). OPCs were treated with microglia conditioned medium (CM), which was added in a 1:1 ratio to OPC culture medium.
Hippocampi and cortices were dissected from E18 rat embryos. After digestion, neural cells were suspended and seeded at 5 × 104 cells per well. Neurons were cultured with Neurobasal medium/B27 and microglia conditioned medium in a 1:1 ratio for 24–72 h.
Cytotoxicity and apoptosis assay.
The viability of OPCs was evaluated by measuring lactate dehydrogenase (LDH) activity in the medium using a CytoTox96 nonradioactive assay (Promega) according to the manufacturer's protocol. Cells were stained with Annexin V-FITC and 7-AAD using a Cell Apoptosis Analysis kit (Sungene). The percentage of apoptotic cells was calculated from the proportion of 7-AAD−/Annexin V+ cells using a flow cytometer FC500 (Beckman Coulter).
Lentiviral transduction.
The coding sequence of MSX3 (MSX3OE) or GFP alone (CtrlOE) was ligated into the GV287 plasmid (GeneChem). The siRNAs for MSX3 (MSX3i) or GFP alone (Ctrli) were ligated into the pGCL-GFP plasmid (GeneChem). The sequence for the control siRNA was as follows: 5′-TTCTCCGAACGTGTCACGT-3′. The sequences for MSX3 siRNA were as follows: 5′-GAGCATCTCAGATCTCCCATT-3′, 5′-CAGGGTTAGGATGCTCTGGTT-3′, and 5′-AATCCGGACTTGTACCTTTAT-3′. Titers of concentrated viral particles ranged between 1 × 108 and 1 × 109 transducing units/ml. Lentiviral particles were added on day 2 to cultured microglia. The supernatant was removed 24 h after infection and replaced with D10 medium. Quantitative PCR (qPCR) analyses verified that lenti-MSX3i downregulated the level of MSX3 mRNA in microglia from nonstimulated or IL-4 treated groups (MGMSX3i, 0.14 ± 0.01 vs MGCtrli, 1 ± 0.00; one-way ANOVA with Tukey's post hoc test, p = 3.26E-4; MGIL-4 MSX3i, 0.42 ± 0.02 vs MGIL-4 Ctrli, 13.78 ± 2.45; p = 2.06E-5; df = 11) and that lenti-MSX3OE upregulated the level of MSX3 mRNA in microglia (MGMSX3OE/MGCtrlOE12.84 ± 2.63; t test, p = 0.004).
Microarray.
Total RNA was isolated from primary microglia using the RNeasy micro kit (QIAGEN), following the manufacturer's instructions, and examined for RNA integration using a Bioanalyzer 2100 (Agilent). The microarray analysis was performed using the Affymetrix GeneChip Mouse Genome 430 2.0 Arrays. The arrays were hybridized, washed, and scanned according to the standard Affymetrix protocol. The gene chip tests were performed by Shanghai Biochip Company. The raw data were normalized using the MAS 5.0 algorithm in GeneSpring software (version 11.0; Agilent). The logged expression values were adjusted by subtracting the raw-wise median and by subjecting to hierarchical clustering using TM4-MEV software (MultiExperiment Viewer).
RNA isolation and qPCR.
Total RNA was extracted from spinal cord or from the primary cell culture using Trizol (Invitrogen). First-strand cDNA was synthesized using a RevertAid First Strand cDNA Synthesis kit (Thermo Fisher Scientific, Fermentas). Quantitative PCR was performed on a MyiQ Real Time PCR system (Bio-Rad). Gene expression was expressed as the mRNA level, which was normalized to that of a standard housekeeping gene (Gapdh) using the ΔΔCT method.
Chromatin immunoprecipitation.
Chromatin immunoprecipitation (ChIP) was performed using an EZ ChIP Chromatin Immunoprecipitation kit (Millipore) according to the manufacturer's protocol. Briefly, 2 × 106 microglia were transduced with lenti-MSX3OE for 72 h and fixed with formaldehyde to a final concentration of 1%. Cross-linked chromatin was sonicated into 200–1000 bp fragments and immunoprecipitated using anti-Flag (Huabio) antibodies. Normal mouse IgG was used as a negative control. Quantitative PCR was conducted on an MyiQ Detection System.
Western blot analysis.
Primary cell cultures were homogenized in RIPA buffer that was supplemented with protease cocktail inhibitors (Roche). Cell lysates were subjected to Western blot analysis using anti-MSX3 (Abcam), anti-inducible nitric oxide (iNOS; Sigma-Aldrich), anti-phospho-STAT6 (phospho Y641; Abcam), anti-STAT6 (Abcam), Anti-JAK3 (Abgent), or anti-PPARγ (Santa Cruz Biotechnology) antibody. The protein bands were analyzed and quantified using densitometry and an Image-Pro Plus analysis (Media Cybernetics) normalizing phosphorylated or no modified protein to GAPDH bands.
ELISA.
The concentration of insulin-like growth factor-1 (IGF-1; R&D Systems), IL-1β (Neobioscience), TGF-β (Uscn Life Science), and activin-A (Uscn Life Science) were detected using an ELISA according to the manufacturer's instructions. Absorbance was read at 490 nm on a spectrophotometer, and sample concentrations were calculated using an equation generated from a standard curve.
Immunocytofluorescence staining.
Cells or tissue sections were fixed, permeabilized, and incubated with primary antibodies [MBP (Millipore), MAP2 (Sigma-Aldrich), or NF-H (Millipore Bioscience Research Reagents)] overnight at 4°C, followed by incubation with TRITC-conjugated or FITC-conjugated secondary antibody (Jackson ImmunoResearch) and counterstained with Hoechst33342 (Sigma-Aldrich). Fluorescence images were captured using fluorescence microscopy (DXM1200; Nikon) and quantified using Image-Pro Plus (Media Cybernetics).
Hematoxylin and eosin and Luxol fast blue staining.
Lumbar or thoracic spinal cords were isolated and cut into cryosections (14–20 μm thick) for immunohistochemistry. One of every six successive sections was collected from each animal. One pool of the sections was stained with Luxol fast blue (LFB) and counterstained with 0.5% neutral red. Another pool of the sections was stained with hematoxylin and eosin (H&E) solution and mounted in Permount (Fisher Scientific). Scoring for LFB or H&E sections was performed using blind examination by independent readers. At least four animals for each time point in different treated groups were included in one experiment, and 10 sections were examined per animal for each individual analysis. Three independent experiments were performed.
Transmission electron microscopy.
Briefly, spinal cords of mice were fixed in 2.5% glutaraldehyde for 2 h, postfixed in 1% osmium tetroxide for 45 min, dehydrated, and embedded in Araldite resin. Sections (1 μm) were stained with toluidine blue. Ultrathin sections (60 nm) were stained in uranyl acetate and lead citrate. Samples were visualized using a transmission electron microscope (H-7650; Hitachi) at 100 KV.
In situ hybridization.
Antisense and sense digoxigenin-labeled cRNA probes were synthesized with a DIG-RNA labeling kit (Roche) with linearized templates of MSX3 cDNA 90–704 (GenBank accession number NM_010836) containing T7 and T3 promoters. In situ hybridization was performing according to the previous report (Guo et al., 2008).
Statistical analysis.
The data were analyzed using one-way ANOVA with Tukey's post hoc test in multiple groups. The EAE model was analyzed using the nonparametric Mann–Whitney U test to compare two groups or the Kruskal–Wallis test with Dunn's post hoc test to compare three groups. The data are presented as mean ± SEM unless otherwise indicated. The value of p < 0.05 was considered statistically significant.
Results
The homeobox gene Msx3 is induced during microglia M2 polarization
To determine the transcriptional factors that are involved in microglia M2 polarization, we induced the primary microglia to M1 or M2 polarization and performed cDNA microarrays to compare gene expression patterns of differently polarized cells. The volcano plots show genes that are regulated (more than twofold and p < 0.05) after 10 ng/ml IL-4 or 100 ng/ml IFNγ treatment, including the M1 or M2 marker genes (Fig. 1A, arrows). The nucleus transcriptional factors that were selectively upregulated in M2 cells were analyzed (Fig. 1B). Notably, Msx3 is the only homeobox gene that is upregulated in M2 microglia (Fig. 1B). Western blot analysis consistently indicated MSX3 protein could hardly be detected in nonstimulated or IFNγ-stimulated microglia but was significantly increased after IL-4 stimulation (MSX3/GAPDH ratio: IL-4, 1.80 ± 0.18 vs IFNγ, 0.91 ± 0.04; one-way ANOVA with Tukey's post hoc test: IL-4, p = 4.2E-6; IFNγ, p = 0.001; df = 8; Fig. 1C). The level of MSX3 in microglia was further determined on different T-lymphocyte-associated cytokine stimulation. Quantitative PCR analyses verified that IL-4 enhanced MSX3 expression, whereas IFNγ decreased the MSX3 level. A slight increase in MSX3 expression was shown after TGF-β1 treatment, and strong downregulation was shown after exposure to IL-17A. Of these stimuli, IL-4 led to the most significant increase in mRNA levels of MSX3 in microglia in vitro (one-way ANOVA with Tukey's post hoc test: IL-4, p = 8.9E-7; TGF-β1, p = 0.002; IL-17A, p = 4.1E-5; IFNγ, p = 3.7E-6; df = 14; Fig. 1D). In contrast, neither IL-4 nor IFNγ induced robust changes in BMP2 mRNA (data not shown). Furthermore, we detected the time course expression of MSX3 after stimulation with IL-4. The level of MSX3 mRNA increased with a peak expression ∼24 h after IL-4 stimulation (fold change, 12.36 ± 1.21; unpaired t test, p = 1.9E-4; df = 4; Fig. 1E).
Figure 1.

Expression of MSX3 is dynamically regulated in microglia during EAE. A, Screening of microglia genes that are differentially expressed after IL-4 (10 ng/ml) or IFNγ (100 ng/ml) treatment. The volcano plots showing genes that are regulated (more than twofold and p < 0.05) after IL-4 or IFNγ treatment for 24 h. Probes for some M1 or M2 marker genes are shown (arrows). B, Heat map of the nuclear genes that are upregulated after IL-4 treatment (p < 0.05) and that are downregulated (p < 0.05) or unchanged after IFNγ treatment for 24 h. Untreated microglia (Ctrl) were used as the control. C, Western blot analysis of MSX3 expression in microglia after no stimulation, IFNγ stimulation, or IL-4 stimulation for 24 h. n = 3 blots. MSX3 is 22 kDa. D, MSX3 mRNA expression from microglia stimulated with TGF-β1 (5 ng/ml), IL-4 (10 ng/ml), IL-17A (10 ng/ml), and IFNγ (100 ng/ml) for 24 h. E, The time course of MSX3 mRNA expression on microglia stimulated with IL-4 (10 ng/ml). C–E, *p < 0.05; **p < 0.01; ***p < 0.005 versus MSX3 in naive microglia. F, Flow cytometry analysis of microglia isolated from the spinal cord of EAE mice. Mononuclear cells were isolated, stained for CD11b and CD45, and analyzed using FACS. CD11b+CD45low resting microglia (a), CD11b+CD45hi-activated microglia and peripheral macrophages (b), and CD11b−CD45hi lymphocytes (c) are shown. G, Relative MSX3 and FIZZ-1 mRNA expression in microglia from spinal cord of control mice (CFA without MOG) and EAE mice on day 11 (preclinic), day 16 (EAE peak), or day 40 (chronic EAE) after induction. *p < 0.05; **p < 0.01; ***p < 0.005 versus microglia from control mice. n = 8 mice per group. AU, Arbitrary units. The data are represented as the mean ± SEM.
MSX3 expression in microglia is dynamic regulated during EAE
The expression level of MSX3 in CD11b+CD45low microglia (Fig. 1F) was then analyzed at the mRNA level in the spinal cord tissue of mice with MOG-induced EAE at the preclinical phase (day 11 after immunization), at the peak of the diseases (day 16 after immunization), and at the chronic phase (day 40 after immunization). In control mice, there was a low level of MSX3 expression in microglia. In the EAE mice, MSX3 mRNA expression was unchanged at the preclinical phase, increased in microglia at the peak of disease (fold change: EAE/CFA, 12.29 ± 1.76; unpaired t test, p = 8.1E-4; df = 4), and persisted at lower levels during the chronic phase (fold change: EAE/CFA, 1.89 ± 0.37; unpaired t test, p = 0.027; df = 4). Consistent with the expression pattern of MSX3, mRNA levels of the M2 marker Retnla (also known as FIZZ-1) were also increased in microglia in mice with EAE at the peak of disease (fold change: EAE/CFA, 117.90 ± 18.10; unpaired t test, p = 8.0E-4; df = 4; Fig. 1G).
MSX3 regulates microglia polarization
Microglia de-ramification and attenuated process motility are correlative responses after microglia M1 polarization (Kettenmann et al., 2011). Primary microglia that were transduced with lentivirus encoding MSX3 RNA-interfering sequence (MGMSX3i) transformed to an amoeboid morphology and increased the cell area (MGMSX3i, 3.46 ± 1.03 vs MGCtrli, 1 ± 0.17; one-way ANOVA with Tukey's post hoc test, p = 8.5E-11; MGIL-4 MSX3i, 1.32 ± 0.13 vs MGIL-4 Ctrli, 0.47 ± 0.17; p = 0.002; df = 32) and the ramification index (MGMSX3i, 0.95 ± 0.12 vs MGCtrli, 0.44 ± 0.04; one-way ANOVA with Tukey's post hoc test, p = 4.1E-5; MGIL-4 MSX3i, 0.78 ± 0.13 vs MGIL-4 Ctrli, 0.42 ± 0.08; p = 0.002; df = 19). Consistently, microglia that were transduced with lentivirus encoding MSX3 CDS sequence (MGMSX3OE) displayed a branched, ramified morphology (data not shown).
These observations prompted us to explore whether the regulation of MSX3 expression in microglia resulted in phenotypic or in functional changes. IGF-1, MRC1 (also known as CD206), and FIZZ-1 were used as markers to define M2 polarization, whereas IL-1β, inducible nitric oxide (NOS2, also known as iNOS), and tumor necrosis factor-α (TNF-α) were used to define M1 polarization (Szanto et al., 2010). We found that interfering with MSX3 expression in microglia decreased IGF-1, CD206, and FIZZ-1 mRNA levels but increased IL-1β, iNOS, and TNF-α mRNA expression (Fig. 2A,B). In contrast, MGMSX3OE experienced an increase in IGF-1, CD206, and FIZZ-1 mRNA and a decrease in IL-1β, iNOS, and TNF-α mRNA (Fig. 2C,D). Accordingly, an ELISA showed that IGF-1 was increased in MGMSX3OE but decreased in MGMSX3i (MGMSX3i, 2.09 ± 0.19 ng/ml vs MGCtrli, 3.92 ± 0.62 ng/ml; one-way ANOVA with Tukey's post hoc test, p = 1.0E-6; MGIL-4 MSX3i, 4.17 ± 0.29 ng/ml vs MGIL-4 Ctrli, 5.92 ± 0.35 ng/ml; p = 2.5E-6; MGMSX3OE, 5.12 ± 0.44 ng/ml vs MGCtrlOE, 4.31 ± 0.43 ng/ml; p = 0.039; df = 35; Fig. 2E), whereas IL-1β was increased in MGMSX3i but decreased in MGMSX3OE (MGMSX3i, 15.52 ± 0.51 pg/ml vs MGCtrli, 12.01 ± 1.19 pg/ml; one-way ANOVA with Tukey's post hoc test, p = 0.008; MGIL-4 MSX3i, 13.60 ± 0.43 pg/ml vs MGIL-4 Ctrli, 10.84 ± 1.11 pg/ml; p = 0.040; MGMSX3OE, 11.82 ± 0.60 pg/ml vs MGCtrlOE, 13.62 ± 0.55 pg/ml; p = 0.043; df = 17; Fig. 2F). The results from Western blot analysis indicated that iNOS protein could not be detected in MGMSX3OE and its control but was increased in MGMSX3i with and without IL-4 stimulated (Fig. 2G), which was consistent with our mRNA results. In summary, MSX3 prompted M2 polarization and interfered with M1 polarization of microglia.
Figure 2.

MSX3 barricades the classical activation of microglia and plays a crucial role in the alternative activation of microglia. A, B, Effects of MSX3 RNAi on the polarization of nonstimulated or IL-4-stimulated microglia were determined using quantitative RT-PCR. C, D, Effects of MSX3 overexpression on the polarization of nonstimulated microglia were determined using quantitative RT-PCR. IGF-1, CD206, and FIZZ-1 were used as M2 markers (A, C), and IL-1β, iNOS, and TNF-α were used as M1 markers (B, D). E, F, ELISA was performed to measure IGF-1 and IL-1β secretion from culture supernatant harvested from MSX3 knock-down and MSX3 overexpressed microglia. G, Western blot analysis of iNOS expression in MSX3 knock-down or overexpressed microglia. Scanning densitometry was quantified and normalized to GAPDH. *p < 0.05; **p < 0.01; ***p < 0.005 versus control, as indicated. NS, Nonstimulated; MSX3i, MSX3 RNAi; Ctrli, RNAi control; MSX3OE, MSX3 overexpression; CtrlOE, overexpression control; AU, arbitrary units. Data are represented as the mean ± SEM of at least three independent experiments.
Regulating MSX3 in microglia controls the survival and maturation of oligodendrocyte precursor cells
We further explored whether regulating MSX3 expression in microglia affected the properties and functions of OPCs. First, OPCs were cultured in microglia CM and stained with annexin V and 7-AAD to assess apoptosis. Compared with control groups, the percentages of early apoptosis OPCs increased significantly in the MGMSX3i groups (one-way ANOVA with Tukey's post hoc test, p = 6.7E-5; df = 17). The CM from MGMSX3i also markedly induced cell death in OPCs in a time-dependent manner by LDH assay (MGMSX3i, 21.28 ± 0.63% vs MGCtrli, 11.54 ± 0.81%; one-way ANOVA with Tukey's post hoc test, p = 8.4E-12; MGIL-4 MSX3i, 19.34 ± 1.48% vs MGIL-4 Ctrli, 8.60 ± 0.36%; p = 3.38E-12; df = 15; Fig. 3A). These results indicated that loss function of MSX3 in microglia induced early apoptosis and accelerated cell death of OPCs.
Figure 3.
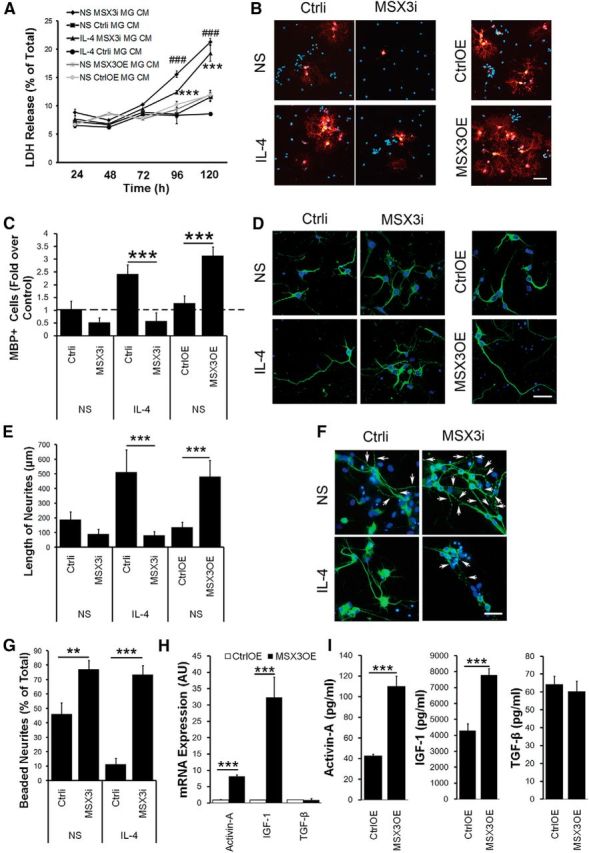
MSX3 is required both for microglia-induced differentiation of OPCs and neurite outgrowth. A, The influence of the CM from MSX3-transduced microglia on the cell viability of OPCs. OPCs were incubated with conditioned medium from MGMSX3i or MGMSX3OE pretreated with or without IL-4 for 24–120 h and analyzed using LDH assay. ###p < 0.005 versus CM of MGCtrli; ***p < 0.005 versus CM of MGCtrli pretreated with IL-4. B, C, The effects of MSX3-transduced microglia on OPC differentiation were determined using an anti-MBP antibody (red). D, F, Representative images of MAP2+ neurons (green) incubated with the CM from MGMSX3i with or without IL-4 stimulation for 72 h. E, Histogram showing the length of MAP2+ neurites (in micrometers). F, Arrows indicate neurite swelling and bead formation. G, Histogram showing the percentage of beaded per total neurites. H, I, Histogram showing factors secreted by MGMSX3OE in CM detected by Q-PCR (H) and ELISA (I). In A, C, E, G, H, and I, data are the mean ± SEM of three independent experiments. NS, Nonstimulation. *p < 0.05; **p < 0.01; ***p < 0.005. Scale bars, 50 μm.
We then explored whether regulating MSX3 expression in microglia could affect the differentiation of OPCs. The CM from MGMSX3OE promoted OPC differentiation by increasing MBP+ cells, whereas the CM from IL-4-stimulated MGMSX3i prohibited OPC differentiation (fold over control: MGIL-4 MSX3i, 0.58 ± 0.32 vs MGIL-4 Ctrli, 2.42 ± 0.37; one-way ANOVA with Tukey's post hoc test, p = 2.65E-9; MGMSX3OE, 3.14 ± 0.34 vs MGCtrlOE, 1.28 ± 0.28; p = 1.88E-9; df = 35; Fig. 3B,C).
Former studies have identified a number of M2-derived factors that have potential to promote the maturation and survival of OPCs, such as TGF-β (McKinnon et al., 1993), activin-A (Miron et al., 2013), and IGF-1 (Carson et al., 1993; Mason et al., 2003). To identify an M2 cell-derived regenerative factor, we took a candidate approach and investigated these factors in the MGMSX3OE.
Here, using qPCR analysis, we found the levels of activin-A and IGF-1 mRNAs were increased in the MGMSX3OE compared with that in MGCtrlOE (Fig. 3H). In contrast, we did not observe a significant difference on levels of TGF-β mRNA between these two groups. Accordingly, we found the protein levels of both activin-A and IGF-1 were increased in the conditioned medium of MGMSX3OE compared with that in MGCtrlOE (activin-A: MGMSX3OE, 110.34 ± 9.58 pg/ml vs MGCtrlOE, 42.89 ± 1.42 pg/ml; p = 1.97E-5; df = 6; IGF-1: MGMSX3OE, 7.79 ± 0.40 ng/ml vs MGCtrlOE, 4.31 ± 0.43 ng/ml; p = 1.13E-7; df = 10). We did not find a significant difference of TGF-β between these two groups at the protein level (MGMSX3OE, 60.42 ± 4.55 pg/ml vs MGCtrlOE, 64.34 ± 5.70 pg/ml; p = 0.388; df = 6; Fig. 3I).
Thus, MSX3 in microglia is crucial to the viability and maturation of OPCs in vitro, at least partly by secreting activin-A and IGF-1.
MSX3 controls microglia-mediated neurotoxicity and neurite outgrowth
To gain insight into the function of MSX3-transduced microglia, neurons were incubated with the CM from MGMSX3i or MGMSX3OE. After 72 h of incubation with the CM from MGMSX3i, the neurites shortened (MGIL-4 MSX3i, 80.70 ± 25.73 μm vs MGIL-4 Ctrli, 513.75 ± 151.51 μm; one-way ANOVA with Tukey's post hoc test, p = 1.59E-9; df = 33; Fig. 3D,E), and the neurons lost their intact shapes and began to form beads (MGMSX3i, 76.98 ± 6.32% vs MGCtrli, 46.11 ± 7.81%; one-way ANOVA with Tukey's post hoc test, p = 0.0084; MGIL-4 MSX3i, 73.33 ± 6.40% vs MGIL-4 Ctrli, 11.20 ± 4.15%; p = 4.12E-5; df = 42; Fig. 3F,G). In contrast, even after 72 h of incubation with the CM from MGMSX3OE, most of the neurons still displayed a normal morphology with integrated neurites (Fig. 3D). Notably, neurons appeared to have longer neurites after incubation with the CM from MGMSX3OE (MGMSX3OE, 481.56 ± 110.66 μm vs MGCtrlOE, 135.70 ± 34.26 μm; one-way ANOVA with Tukey's post hoc test, p = 3.98E-4; df = 33; Fig. 3D).
MSX3-transduced microglia regulate inflammation-induced demyelination in vivo
To determine the impact of MSX3 on inflammation-induced demyelination in vivo, we induced EAE in C57BL/6 mice and transplanted MSX3-modified microglia into the right cerebral ventricle (Fig. 4A) 8 d after immunization. The localization of the graft in the spinal cord was confirmed by anti-GFP immunohistochemistry at 30 d after immunization (data not shown). Neither naive murine MG nor naive human microglia (HM) transplantation groups showed any significant differences of EAE progression compared with the sham EAE group (data not shown). We found that IL-4-pretreated Ctrli microglia (MGIL-4 Ctrli) transplantation promoted EAE recovery compared with nonstimulated microglia (MGCtrli), whereas the transplantation of IL-4-pretreated MSX3 RNAi microglia (MGIL-4 MSX3i) exacerbated EAE (increased average daily score; Kruskal–Wallis test, p = 0.0055; df = 4; Fig. 4B) with an earlier onset. Although there were no significant changes in the average day of onset between the MGMSX3OE and the control MGCtrlOE transplantation groups, the average daily score in the MGMSX3OE group decreased after disease induction (Kruskal–Wallis test with Dunn's post hoc test: MGMSX3OE vs MGCtrlOE, p = 0.028; df = 4; Fig. 4B). The lumbar segment of the spinal cord from the MGMSX3OE transplantation group displayed mild demyelination (MGIL-4 MSX3i, 23.14 ± 2.24% vs MGIL-4 Ctrli, 4.22 ± 1.19%; one-way ANOVA with Tukey's post hoc test, p = 4.10E-12; MGMSX3OE, 3.18 ± 0.48% vs MGCtrlOE, 10.55 ± 1.47%; p = 2.23E-5; df = 24; Fig. 4C,D) and less infiltrating cells (MGIL-4 MSX3i, 633.3 ± 44.2 cells/mm2 vs MGIL-4 Ctrli, 90.2 ± 44.6 cells/mm2; one-way ANOVA with Tukey's post hoc test, p = 9.37E-13; MGMSX3OE, 70.5 ± 14.5 cells/mm2 vs MGCtrlOE, 185.2 ± 23.7 cells/mm2; p = 1.94E-4; df = 29; Fig. 4E,F) around the lesion sites. Immunohistochemistry using anti-MBP antibodies (MGIL-4 MSX3i, 1,123,887 ± 83,520 AU vs MGIL-4 Ctrli, 1,543,972 ± 70,939 AU; one-way ANOVA with Tukey's post hoc test, p = 1.05E-5; MGMSX3OE, 1,981,306 ± 91,120 AU vs MGCtrlOE, 1,355,158 ± 61,948 AU; p = 2.06E-8; df = 24; Fig. 5A,C) also confirmed the observation.
Figure 4.

Ablation of MSX3 in microglia exacerbates the progression of EAE, whereas overexpression of MSX3 ameliorates it. A, EAE mice were transplanted with IL-4-stimulated microglia transduced with MSX3i lentivirus (IL-4 MSX3i MG), IL-4 stimulated microglia transduced with RNAi control lentivirus (IL-4 Ctrli MG), or nontreated microglia transduced with RNAi control lentivirus (Ctrli MG) into the right lateral ventricle (day 8 after immunization). The diagram illustrates the experimental injection site. B, Average clinical score of EAE for each transplant group was evaluated from three experiments with five to eight mice per group. *p < 0.05; **p < 0.01. C–F, Histology analysis of the extent of demyelination and inflammation. C, E, Representative spinal cord sections of Luxol fast blue (C) and H&E staining (E) from EAE mice. Boxed areas in left columns are enlarged in the right columns. Asterisks and arrowheads indicate demyelination and cell infiltration, respectively. Scale bars = 200 μm. D, F, Histogram showing quantification of the percentage of demyelinated white matter (WM) in total WM (D) and the number of infiltrating cells around lesion sites of EAE mice (F). *p < 0.05; **p < 0.01; ***p < 0.005. Data are the mean ± SEM.
Figure 5.

MSX3-transduced microglia affects axon survival and the myelination of the spinal cord in EAE mice. A, Representative images of spinal cord sections of mice from microglia transduced with RNAi control lentivirus (Ctrli MG), IL-4-stimulated microglia transduced with RNAi control lentivirus (IL-4 Ctrli MG), IL-4-stimulated microglia transduced with MSX3 RNAi lentivirus (IL-4 MSX3i MG), microglia transduced with overexpression control lentivirus (CtrlOE MG), or microglia transduced with MSX3 overexpression lentivirus (MSX3OE MG). The sections were stained for neurofilaments using an anti-NF antibody (green) and for myelin using an anti-MBP antibody (red). The dashed line indicates the lesion boundary of demyelination, where the loss of NF staining (green) and the reconstruction of MBP (red) are accompanied by increased numbers of infiltrating cells (Hoechst, blue). In contrast, scarce infiltration is observed outside the dashed line with increased NF and normal MBP staining. B, Axon damage was shown by quantifying the intensity of NF staining (green). C, Demyelination was assessed by quantifying the intensity of the MBP staining (red). *p < 0.05; ***p < 0.005. Scar bar, 100 μm.
MSX3-transduced microglia supports axon survival during inflammation-induced demyelination
To explore the role of MSX3 in axonal degeneration during inflammation-induced demyelination, neurofilament (NF) immunofluorescent staining was used to visualize the surviving axons in lumbar spinal cord. We found that transplantation of MGIL-4 MSX3i decreased NF staining intensity compared with the MGIL-4 Ctrli group in the lumbar segment of the spinal cord, whereas the spinal cord from MGMSX3OE transplantation mice displayed much higher NF intensity compared with the control group (MGIL-4 MSX3i, 262,689 ± 95,980 AU vs MGIL-4 Ctrli, 801,448 ± 100,408 AU; one-way ANOVA with Tukey's post hoc test, p = 4.71E-6; MGMSX3OE, 1,147,594 ± 132,612 AU vs MGCtrlOE, 537,301 ± 97,081 AU; p = 7.19E-7; df = 24; Fig. 5A,B).
MSX3-transduced microglia promotes remyelination in vivo
Considering less demyelination (Fig. 4C,D) may be attributed to an effect of the MGMSX3OE on the immune reaction (Fig. 4E,F), to discern whether more myelinated nerve fibers remaining (Fig. 5) were the result of promoting the remyelination or just alleviating the demyelination, MGMSX3OE was transplanted in EAE mice after the peak phase (on day 22 after immunization), and the remyelination was determined at the chronic phase (on day 40 after immunization). We found transplantation of MGMSX3OE promoted EAE recovery and alleviated the clinical symptoms of ongoing EAE compared with transplantation of control microglia (MGCtrlOE; decreased average daily score from day 22 to day 40, Kruskal–Wallis test with Dunn's post hoc test: MGMSX3OE vs MGCtrlOE, p = 5.66E-11; df = 2; Fig. 6A). We then performed the electron microscopy (EM) analysis on day 40 after immunization. EM analysis showed that more thin myelin sheaths, a morphological hallmark of remyelination, were present in the ventral column of spinal cords in the MGMSX3OE group compared with that of the MGCtrlOE group (Fig. 6B). A higher percentage of myelinated axons (one-way ANOVA with Tukey's post hoc test, p = 4.60E-13; df = 165; Fig. 6C) and a lower g-ratio (axon diameter/fiber diameter; one-way ANOVA with Tukey's post hoc test, p = 4.68E-13; df = 133; Fig. 6D) were determined in MGMSX3OE-transplanted mice than in MGCtrlOE-transplanted mice.
Figure 6.

Ameliorating EAE progression and promoting remyelination by overexpressing MSX3 in microglia. EAE mice were given injections of PBS (Sham EAE), MSX3 overexpressing microglia (MSX3OE MG), or control lentivirus-transduced microglia (CtrlOE MG) into the right lateral ventricle (day 22 after immunization). A, The clinical score of EAE was shown for each transplantation group. The data represent average disease scores from five to eight mice per group, using Kruskal–Wallis test with Dunn's post hoc test. ***p < 0.005 versus CtrlOE MG or sham EAE group. B–D, Overexpression of MSX3 in microglia promoted remyelination in spine cords. Spine cords from each group (MSX3OE and CtrlOE) of EAE mice were harvested at 40 d after EAE induction. The ventral column of the L4 lumbar spine cords was examined. B, Representative electron micrographs of the demyelinated lesions of spinal cords at 40 d after EAE induction. The presence of thin myelin sheaths (remyelination, arrows) was observed in both the MSX3OE MG and CtrlOE MG groups. In contrast, the normal thick myelin sheaths (arrowheads) were observed in naive mice. Original magnification, ×700; high magnification, ×1500. Scale bars: top, 5 μm; bottom, 2 μm. C, Quantification of myelinated axons among total axons. D, Mean g-ratio (axon diameter divided by entire myelinated fiber diameter) was determined using ImageJ software. *p < 0.05; **p < 0.01; ***p < 0.005 versus control, as indicated. Data are the mean ± SEM, using one-way ANOVA with Tukey's post hoc test. Five to eight mice (40 d after EAE induction) from each group were analyzed.
To avoid the influences of MGMSX3OE on inflammation, the LPC-induced focal demyelination model was also performed. A local LPC lesion was generated in the dorsal spinal cord, followed by transplantation of microglia at 3 dpl. LFB staining illustrated that the volume of the demyelinated lesions in the MGMSX3OEgroup was significantly smaller than lesions in the MGCtrlOE group at 11 dpl, and a more extensive remyelination was observed at 15 dpl (Fig. 7A). Analysis of total lesion volume supported that, although no significant difference of lesions was observed in these two groups at 3 or 7 dpl (at 3 dpl: MGMSX3OE, 41.84 ± 1.84 vs MGCtrlOE, 39.72 ± 3.57; one-way ANOVA with Tukey's post hoc test, p = 0.962; at 7 dpl: MGMSX3OE, 51.82 ± 1.47 vs MGCtrlOE, 54.27 ± 4.24; p = 0.924; df = 31; Fig. 7B), more prominent remyelination occurred in the MGMSX3OE group compared with that in the MGCtrlOE group at 11 and 15 dpl (at 11 dpl: MGMSX3OE, 29.67 ± 1.79 vs MGCtrlOE, 39.32 ± 2.33; one-way ANOVA with Tukey's post hoc test, p = 0.003; at 15 dpl: MGMSX3OE, 3.34 ± 2.00 vs MGCtrlOE, 16.36 ± 0.99; p = 3.22E-5; df = 31; Fig. 7B). We then performed the EM analysis at 11 and 15 dpl. EM analysis showed that more thin myelin sheaths were present in the dorsal column of spinal cords in the MGMSX3OE group compared with that of the MGCtrlOE group (Fig. 7C). A higher percentage of myelinated axons (at 11 dpl: MGMSX3OE, 46.51 ± 3.54% vs MGCtrlOE, 15.41 ± 1.46%; one-way ANOVA with Tukey's post hoc test, p = 8.59E-7; at 15 dpl: MGMSX3OE, 72.06 ± 7.61% vs MGCtrlOE, 27.87 ± 5.21%; p = 6.24E-9; df = 19; Fig. 7D) and a lower g-ratio (axon diameter/fiber diameter; at 11 dpl: MGMSX3OE, 0.77 ± 0.03 vs MGCtrlOE, 0.83 ± 0.04; one-way ANOVA with Tukey's post hoc test, p = 0.012; at 15 dpl: MGMSX3OE, 0.74 ± 0.02 vs MGCtrlOE, 0.80 ± 0.04; p = 1.99E-5; df = 343; Fig. 7E–G) were determined in MGMSX3OE-transplanted mice than in MGCtrlOE-transplanted mice.
Figure 7.

Promoting remyelination in LPC-induced focal lesion model by overexpressing MSX3 in microglia. A, B, Histology analysis of the extent of remyelination in the MGMSX3OE and MGCtrlOE groups at 3, 7, 11, and 15 dpl. A, Representative dorsal column of spinal cord sections of Luxol fast blue from the MGMSX3OE (bottom row) and MGCtrlOE (top row) groups. Scale bar, 10 μm. B, Quantitative analysis of the lesion size of the dorsal column of spinal cords in the MGMSX3OE and MGCtrlOE groups. C–G, EM analysis of remyelination in the MGMSX3OE and MGCtrlOE groups at 11 and 15 dpl. C, Representative electron micrographs of the remyelinated area of spinal cords. The dorsal column of the lesioned spine cords were examined by EM, based on the 1 μm coronal sections of toluidine blue. The presence of thin myelin sheaths (remyelination) was observed in both the MGMSX3OE and MGCtrlOE groups. Scale bar, 4 μm. D, Quantification of myelinated axons among total axons. E, The mean g-ratio at 11 or 15 dpl was determined using ImageJ software. F, G, Analysis of the myelinated axons showed a reduction of the g-ratio in spinal cords in the MGMSX3OE groups (red) compared with the MGCtrlOE groups (blue) at 11 dpl (F) and 15 dpl (G). *p < 0.05; ***p < 0.005 versus control, as indicated. Data are the mean ± SEM, using one-way ANOVA with Tukey's post hoc test. Over 100 axons were counted per mouse. Five to eight mice from each group were analyzed.
Together, these results indicate the potent ability of MSX3-tranduced microglia to attenuate neural degeneration (Figs. 3, 5) and promote remyelination (Figs. 6, 7), thus preventing disease progression.
Overexpression of MSX3 in human-derived microglia represents the M2 phenotype and ameliorates EAE after intraventricular injection
To determine whether MSX3 could affect the polarization of human-derived microglia, the mRNA level of M2 markers (IGF-1 and CD206) and the M1 marker (TNF-α) were analyzed in human microglia transduced with lentivirus encoding the MSX3 CDS sequence (HMMSX3OE). Twenty-four hours after transduction, levels of both IGF-1 (unpaired t test, p = 0.021; df = 6) and CD206 (unpaired t test, p = 0.017; df = 6) were increased (Fig. 8A), whereas the level of TNF-α decreased (unpaired t test, p = 0.007; df = 10; Fig. 8B) compared with the control. Moreover, the progression of EAE was mitigated after transplanting HMMSX3OE 8 d after immunization (Fig. 8C). The average daily score in the HMMSX3OE group decreased significantly 16 d after disease induction (Mann–Whitney U test, p = 0.014; Fig. 8C). Consistently, the lumbar segment of the spinal cord of HMMSX3OE group mice displayed mild demyelination (MGMSX3OE, 1.15 ± 0.32% vs MGCtrlOE, 7.13 ± 1.05%; unpaired t test, p = 2.53E-7; df = 10; Fig. 8D,E) and less infiltrating cells around the lesion sites (MGMSX3OE, 84.3 ± 22.1 cells/mm2 vs MGCtrlOE, 202.9 ± 79.8 cells/mm2; unpaired t test, p = 0.003; df = 12; Fig. 8F,G).
Figure 8.

Human microglia is prone to M2 activation by overexpressing MSX3 and mitigating the progression of EAE. A, B, Effect of MSX3 overexpression on the activation of human microglia by qPCR for the M2 activation markers CD206 and IGF-1 (A) and for the M1 activation marker TNF-α (B). AU, Arbitrary units. *p < 0.05 versus the control indicated. C, Clinical score of EAE on human microglia transduced with overexpression control lentivirus (CtrlOE HM) and human microglia transduced with MSX3 overexpression lentivirus (MSX3OE HM) transplantation groups. The data represent average disease scores from three experiments with five to eight mice per group. *p < 0.05 versus CtrlOE HM. D–G, Histology analysis of the extent of demyelination and inflammation in the spinal cords of mice from the HMCtrlOE and HMMSX3OE transplantation groups. D, F, Luxol fast blue (D) and H&E (F) staining of representative spinal cord sections of EAE in groups as indicated. Boxed areas in the left columns are enlarged in the right columns. Asterisks and arrowheads indicate demyelination and cell infiltration, respectively. Scale bars, 200 μm. E, G, Quantification of the percentage of demyelinated white matter (WM) in total WM (E) and the number of infiltrating cells around the lesion site of EAE mice (G). *p < 0.05; **p < 0.01; ***p < 0.005.
MSX3 plays an essential role in the regulation of microglial polarization via PPARγ and the JAK/STAT6 pathway
To explore the mechanisms of MSX3 on microglia, we used ChIP analysis to investigate whether MSX3 could interact with the promoter areas of genes that are involved in M2 polarization, such as Pparg, Stat6, Cebpb, Jak1, Jak3, and Ptpn1 (also known as Ptp1b). MSX3 directly interacted with the Pparg, Stat6, and Jak3 promoters (Fig. 9A–C). Although STAT6 was a direct substrate of PTPN1 and was sensitive to JAK1's regulation (Lu et al., 2008), no direct MSX3 binding was found between these two genes promoters in ChIP analysis (data not shown). Moreover, we did not detect any interaction between MSX3 and the promoter of Cebpb (Ruffell et al., 2009; data not shown).
Figure 9.

MSX3 promotes PPARγ, STAT6, and JAK3 expression in microglia. A–C, MSX3 interacts with promoters of Pparg, Stat6, and Jak3 in primary cultured microglia. The enrichment of MSX3 on the promoter of the genes indicated was determined using ChIP qPCR in MSX3-overexpressed microglia 72 h after transduction, confirming a direct interaction between MSX3 and promoters of Pparg (A), Stat6 (B), and Jak3 (C). In contrast, no direct interaction between MSX3 and promoters of Jak1, Ptp1b, or Cebpb was detected. The data are shown as the means ± SEM of three independent experiments. **p < 0.01 versus the IgG control. D–F, qPCR analysis of PPARγ, STAT6, and JAK3 mRNA in microglia. G–I, Immunoblot analysis of the expression of PPARγ, phospho-STAT6, STAT6, and JAK3 in MSX3 RNAi microglia with or without IL-4 stimulation and their controls. J–L, Immunoblot analysis of the expression of PPARγ, phospho-STAT6, STAT6, and JAK3 in MSX3-overexpressed microglia and its control. Scanning densitometry was quantified and normalized to the control on the same Western blots. GAPDH expression was analyzed from the same samples as an internal control for all Western blots. Representative blots are shown from three independent experiments. The data are shown as means ± SEM of three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.005 versus control, as indicated. AU, Arbitrary units.
Interestingly, we found that PPARγ, JAK3, and STAT6 mRNA expression was repressed in MGMSX3i with or without IL-4 stimulation. Moreover, there were increased levels of JAK3, STAT6, and PPARγ mRNAs in MGMSX3OE (PPARγ: MGIL-4 MSX3i, 0.12 ± 0.17 vs MGIL-4 Ctrli, 4.04 ± 0.47; one-way ANOVA with Tukey's post hoc test, p = 0.003; MGMSX3OE, 7.46 ± 2.51 vs MGCtrlOE, 1.02 ± 0.07; p = 4.86E-5; df = 17; STAT6: MGIL-4 MSX3i, 0.51 ± 0.19 vs MGIL-4 Ctrli, 0.96 ± 0.02; p = 0.006; MGMSX3OE, 1.22 ± 0.08 vs MGCtrlOE, 0.95 ± 0.05; p = 0.032; df = 17; JAK3: MGIL-4 MSX3i, 0.72 ± 0.03 vs MGIL-4 Ctrli, 1.15 ± 0.06; p = 0.006; MGMSX3OE, 1.18 ± 0.20 vs MGCtrlOE, 0.86 ± 0.03; p = 0.039; df = 17; Fig. 9D–F). Consistently, we found a decrease in PPARγ, JAK3, and STAT6 protein levels in MGMSX3i and an augment of these molecules in MGMSX3OE (PPARγ/GAPDH ratio: MGIL-4 MSX3i, 6.03 ± 0.69 vs MGIL-4 Ctrli, 11.08 ± 1.69; one-way ANOVA with Tukey's post hoc test, p = 1.06E-5; MGMSX3OE, 3.69 ± 0.11 vs MGCtrlOE, 1.00 ± 0.06; p = 0.004; df = 17; JAK3/GAPDH ratio: MGIL-4 MSX3i, 1.00 ± 0.02 vs MGIL-4 Ctrli, 1.19 ± 0.04; p = 0.011; MGMSX3OE, 2.43 ± 0.22 vs MGCtrlOE, 0.93 ± 0.08; p = 1.68E-4; df = 17; STAT6/GAPDH ratio: MGIL-4 MSX3i, 1.75 ± 0.22 vs MGIL-4 Ctrli, 2.42 ± 0.11; p = 0.004; MGMSX3OE, 1.75 ± 0.04 vs MGCtrlOE, 0.93 ± 0.05; p = 6.86E-4; df = 17; Fig. 9G–L). We also observed a reduced phosphorylation level of STAT6 (P-STAT6) in MGMSX3i (Fig. 9H) and increased phosphorylation of STAT6 in MGMSX3OE (P-STAT6/GAPDH ratio: MGIL-4 MSX3i, 1.57 ± 0.11 vs MGIL-4 Ctrli, 3.16 ± 0.13; one-way ANOVA with Tukey's post hoc test: p = 9.42E-9; MGMSX3OE, 2.82 ± 0.04 vs MGCtrlOE, 0.93 ± 0.05; p = 1.30E-9; df = 17; Fig. 9K).
Discussion
MSX3 is a homeobox transcription factor affecting dorsal neuron generation during development (Liu et al., 2004). The biological functions of MSX3 in immune cells have not been studied. The present study revealed a novel function for MSX3 to enhance the M2 polarization of microglia through the direct gene induction of known M2 key transcription factors. We have also shown that MSX3-transduced microglia promoted the maturation of OPCs in vitro and remyelination in vivo, prevented neurons from damage, and ameliorated the clinical symptoms of EAE. The data suggested a new approach for treating demyelinating diseases by MSX3-transduced human microglia.
Msx3 is the third member of the murine Msx homeobox gene family, which is homologous to the msh gene of Drosophila (Shimeld et al., 1996). Previous studies illustrated that MSX3 expression is limited to the dorsal neuroepithelium of the developing CNS (Wang et al., 1996; Mehra-Chaudhary et al., 2001; Liu et al., 2004). Interestingly, the maximal expression of MSX3 has been shown to occur in embryos between 10.5 and 12.5 d postcoitus (Matsui et al., 2004), which coincides with the time when microglia are transferred into both the cephalic mesenchyme and the neuroepithelium of the CNS (Ginhoux et al., 2010). However, whether this molecule is expressed in microglia remains unknown. Our study demonstrated that MSX3 was expressed in CD11b+/CD45low naive adult microglia and that the expression level of MSX3 was enhanced in microglia after EAE. Although Msx1 and Msx2 belong to the same homeobox gene family, we could not detect significant expression levels of these two molecules in microglia (data not shown). Because microglia only constitute <10% of the CNS cells (Ponomarev et al., 2007), the low expression level of MSX3 by naive microglia might account for the extremely low expression level of MSX3 in the normal adult CNS that was reported in a previous study (Dillman et al., 2013). Notably, we found that MSX3 expression in primary cultured microglia was diversely changed under the stimulation of different cytokines. The level of MSX3 also changed dramatically in different phases of EAE, suggesting its regulable properties in the dynamic inflammatory environment.
Homeobox genes encode transcription factors that typically switch on cascades of other genes and that direct the formation of body structures during early embryonic development (Corsetti et al., 1992). In the present study, we found a new role of MSX3 in controlling microglia polarization. MSX3 promoted the M2 polarization of microglia by upregulating IGF-1, CD206, and FIZZ-1; however, MSX3 impeded M1 polarization by downregulating TNF-α, IL-1β, and iNOS. Moreover, the CM from MSX3-transduced microglia was shown to promote oligodendrocyte maturation and remyelination and support neuritis outgrowth.
Axonal degeneration and blockage of OPC differentiation are main characteristics of chronic MS lesions (Ruckh et al., 2012). Recent research has also shown that another important finding in chronic lesion is the lack of M2 polarized microglia (Miron et al., 2013). Importantly, manipulating the polarization phenotypes of microglia protects from inflammation-induced neurodegeneration and demyelination (Ponomarev et al., 2011). In the present study, we found that transplantation of MSX3-transduced mouse microglia into the lateral ventricles could alleviate inflammation-induced neurodegeneration and demyelination, ameliorate clinical symptoms, and induce remyelination from chronic EAE and that transplantation of MSX3-transduced microglia into the spinal cord could promote remyelination in the LPC-induced focal lesion. We also demonstrated that MSX3 could not only drive the M2 polarization of mouse microglia but also induce the M2 phenotype of human-derived microglia and that MSX3-transduced human microglia ameliorated EAE after intraventricular injection. These results were interesting because mouse M2 typically activates genes, including Arg1, Fizz-1, and Chil3 (also known as Ym-1; Lawrence and Natoli, 2011), which were not induced in human M2 cells (Lawrence and Natoli, 2011; Martinez et al., 2013), indicating a slight difference in the signaling pathway between human and mouse microglial polarization. The observation that MSX3 could promote the M2 polarization of both mouse and human microglia indicates that MSX3 serves as a key element in the microglia polarization pathway.
Together, this study highlights MSX3 delivery into localized microglia as a potential therapeutic approach for treating chronic demyelinating diseases. Our and others' recent studies also show that Trans-activation of transcription (TAT), a cell-penetrating peptide, could help the therapeutic molecules penetrating blood–brain barrier and cell membrane (Yu et al., 2010). So, it will be interesting to administer the TAT-MSX3 fusion peptide intravenously to explore whether this peptide could drive microglia in situ to M2 polarization and to promote remyelination in CNS demyelinated lesions.
Understanding the mechanism of the MSX3 functioning in microglia would advance our knowledge of the brain patrol cells and be valuable for MS treatment in the future. Our ChIP experiments demonstrated a direct interaction of MSX3 with the promoters of Jak3, Stat6, and Pparg. These molecules are key signal transducers that mediated M2 polarization. For example, IL-4 signaling is primarily mediated through the JAK–STAT6 pathway, which involves sequential phosphorylation of the IL-4 receptor complex, JAK1/3, and STAT6 (Martinez et al., 2009). We found that MSX3 increased JAK3 and STAT6 expression. MSX3 also induces the STAT6 activation by phosphorylation of STAT6. The increased phosphorylation of STAT6 may be attributable to increased STAT6 or JAK3 levels after MSX3 overexpression. IL-4 can also induce PPARγ expression through STAT6 (Huang et al., 1999), and transcriptional synergy between STAT6 and PPARγ is required for the full expression of M2 markers (Chawla, 2010). We found that MSX3 was not only essential but also sufficient in upregulating PPARγ expression. The direct regulation of PPARγ and indirect regulation through STAT6 may contribute to PPARγ upregulation after MSX3 overexpression. Previous studies also revealed PTPN1 and C/EBPβ as M2 regulators. PTPN1 was a negative regulator of IL-4-induced STAT6 signaling (Lu et al., 2008). C/EBPβ was induced by Toll-like receptor ligands or by CREB (cAMP-responsive element-binding protein) and was responsible for M2 gene expression (Ruffell et al., 2009). However, in the present study, the ChIP analysis did not show direct interactions of MSX3 with these two genes.
Our conditioned medium studies indicated that this M2 cell-driven regenerative response was mediated, at least in part, by secretion of the IGF-1 and activin-A. Though we cannot exclude the effects of other factors secreted by MSX3-overexpressed microglia, our finding highlights these two factors as potential critical secreted factors for the CNS myelin regeneration.
We conclude that Msx3, which is a particularly upregulated homeobox gene in M2 microglia, is a key element in microglia M2 polarization that is involved in the induction of key M2 pathway signal transducers, including Jak3, Stat6, and Pparg. MSX3-transduced microglia protects from inflammation-induced neurodegeneration and demyelination and facilitates remyelination. Here, we found that microglia, as a component of the innate immune response regardless of changes to the blood–brain barrier, was sufficient to polarize to the M2 phenotype. However, we do not exclude a possibility that MSX3 has a similar effect on the peripherally derived macrophages that enter the CNS after demyelination. In fact, we found the MSX3-transduced peritoneal macrophages could be polarized to the M2 phenotype in vitro, by upregulating the mRNA level of CD206 (Z. Yu, unpublished data). The effect of MSX3 on the macrophages needs to be further determined in vivo.
The results of our present study identify a new homeobox protein-dependent mechanism in microglia polarization and support the use of this homeobox transcription factor for therapeutic approaches targeting pathologies that are associated with microglia polarization, including MS, Alzheimer's and Parkinson's diseases, and CNS trauma.
Footnotes
This work was supported by the Key program of the National Natural Science Foundation of China (Grant 31130024), the National Key Basic Research Program (Grant 2011CB504401), the National Natural Science Foundation of China (Grants 31171030, 81371326, 31100765, and 31371068), and the Natural Science Foundation of Shanghai (Grant 11ZR1446600).
The authors declare no competing financial interests.
References
- Arnett HA, Fancy SP, Alberta JA, Zhao C, Plant SR, Kaing S, Raine CS, Rowitch DH, Franklin RJ, Stiles CD. bHLH transcription factor Olig1 is required to repair demyelinated lesions in the CNS. Science. 2004;306:2111–2115. doi: 10.1126/science.1103709. [DOI] [PubMed] [Google Scholar]
- Bitsch A, Schuchardt J, Bunkowski S, Kuhlmann T, Brück W. Acute axonal injury in multiple sclerosis. Correlation with demyelination and inflammation. Brain. 2000;123:1174–1183. doi: 10.1093/brain/123.6.1174. [DOI] [PubMed] [Google Scholar]
- Butovsky O, Landa G, Kunis G, Ziv Y, Avidan H, Greenberg N, Schwartz A, Smirnov I, Pollack A, Jung S, Schwartz M. Induction and blockage of oligodendrogenesis by differently activated microglia in an animal model of multiple sclerosis. J Clin Invest. 2006;116:905–915. doi: 10.1172/JCI26836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson MJ, Behringer RR, Brinster RL, McMorris FA. Insulin-like growth factor I increases brain growth and central nervous system myelination in transgenic mice. Neuron. 1993;10:729–740. doi: 10.1016/0896-6273(93)90173-O. [DOI] [PubMed] [Google Scholar]
- Chawla A. Control of macrophage activation and function by PPARs. Circ Res. 2010;106:1559–1569. doi: 10.1161/CIRCRESAHA.110.216523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corsetti MT, Briata P, Sanseverino L, Daga A, Airoldi I, Simeone A, Palmisano G, Angelini C, Boncinelli E, Corte G. Differential DNA binding properties of three human homeodomain proteins. Nucleic Acids Res. 1992;20:4465–4472. doi: 10.1093/nar/20.17.4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillman AA, Hauser DN, Gibbs JR, Nalls MA, McCoy MK, Rudenko IN, Galter D, Cookson MR. mRNA expression, splicing and editing in the embryonic and adult mouse cerebral cortex. Nat Neurosci. 2013;16:499–506. doi: 10.1038/nn.3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, Mehler MF, Conway SJ, Ng LG, Stanley ER, Samokhvalov IM, Merad M. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330:841–845. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Xu X, Gao X, Burnstock G, He C, Xiang Z. Expression of P2X5 receptors in the mouse CNS. Neuroscience. 2008;156:673–692. doi: 10.1016/j.neuroscience.2008.07.062. [DOI] [PubMed] [Google Scholar]
- Huang JT, Welch JS, Ricote M, Binder CJ, Willson TM, Kelly C, Witztum JL, Funk CD, Conrad D, Glass CK. Interleukin-4-dependent production of PPAR-gamma ligands in macrophages by 12/15-lipoxygenase. Nature. 1999;400:378–382. doi: 10.1038/22572. [DOI] [PubMed] [Google Scholar]
- Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev. 2011;91:461–553. doi: 10.1152/physrev.00011.2010. [DOI] [PubMed] [Google Scholar]
- Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci. 2009;29:13435–13444. doi: 10.1523/JNEUROSCI.3257-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlmann T, Miron V, Cui Q, Cuo Q, Wegner C, Antel J, Brück W. Differentiation block of oligodendroglial progenitor cells as a cause for remyelination failure in chronic multiple sclerosis. Brain. 2008;131:1749–1758. doi: 10.1093/brain/awn096. [DOI] [PubMed] [Google Scholar]
- Lawrence T, Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol. 2011;11:750–761. doi: 10.1038/nri3088. [DOI] [PubMed] [Google Scholar]
- Liao B, Zhao W, Beers DR, Henkel JS, Appel SH. Transformation from a neuroprotective to a neurotoxic microglial phenotype in a mouse model of ALS. Exp Neurol. 2012;237:147–152. doi: 10.1016/j.expneurol.2012.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Helms AW, Johnson JE. Distinct activities of Msx1 and Msx3 in dorsal neural tube development. Development. 2004;131:1017–1028. doi: 10.1242/dev.00994. [DOI] [PubMed] [Google Scholar]
- Lu X, Malumbres R, Shields B, Jiang X, Sarosiek KA, Natkunam Y, Tiganis T, Lossos IS. PTP1B is a negative regulator of interleukin 4-induced STAT6 signaling. Blood. 2008;112:4098–4108. doi: 10.1182/blood-2008-03-148726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo F, Burke K, Kantor C, Miller RH, Yang Y. Cyclin-dependent kinase 5 mediates adult OPC maturation and myelin repair through modulation of Akt and Gsk-3beta signaling. J Neurosci. 2014;34:10415–10429. doi: 10.1523/JNEUROSCI.0710-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Ann Rev Immunol. 2009;27:451–483. doi: 10.1146/annurev.immunol.021908.132532. [DOI] [PubMed] [Google Scholar]
- Martinez FO, Helming L, Milde R, Varin A, Melgert BN, Draijer C, Thomas B, Fabbri M, Crawshaw A, Ho LP, Ten Hacken NH, Cobos Jiménez V, Kootstra NA, Hamann J, Greaves DR, Locati M, Mantovani A, Gordon S. Genetic programs expressed in resting and IL-4 alternatively activated mouse and human macrophages: similarities and differences. Blood. 2013;121:e57–e69. doi: 10.1182/blood-2012-06-436212. [DOI] [PubMed] [Google Scholar]
- Mason JL, Xuan S, Dragatsis I, Efstratiadis A, Goldman JE. Insulin-like growth factor (IGF) signaling through type 1 IGF receptor plays an important role in remyelination. J Neurosci. 2003;23:7710–7718. doi: 10.1523/JNEUROSCI.23-20-07710.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui H, Takahashi T, Raghow R. The mechanism of biogenesis and potential function of the two alternatively spliced mRNAs encoded by the murine Msx3 gene. Mol Cell Biochem. 2004;267:1–11. doi: 10.1023/B:MCBI.0000049353.38098.51. [DOI] [PubMed] [Google Scholar]
- McKinnon RD, Piras G, Ida JA, Jr, Dubois-Dalcq M. A role for TGF-beta in oligodendrocyte differentiation. J Cell Biol. 1993;121:1397–1407. doi: 10.1083/jcb.121.6.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehra-Chaudhary R, Matsui H, Raghow R. MSX3 protein recruits histone deacetylase to down-regulate the Msx1 promoter. Biochem J. 2001;353:13–22. [PMC free article] [PubMed] [Google Scholar]
- Minghetti L. Role of inflammation in neurodegenerative diseases. Curr Opin Neurol. 2005;18:315–321. doi: 10.1097/01.wco.0000169752.54191.97. [DOI] [PubMed] [Google Scholar]
- Miron VE, Boyd A, Zhao JW, Yuen TJ, Ruckh JM, Shadrach JL, van Wijngaarden P, Wagers AJ, Williams A, Franklin RJ, ffrench-Constant C. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat Neurosci. 2013;16:1211–1218. doi: 10.1038/nn.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peferoen L, Kipp M, van der Valk P, van Noort JM, Amor S. Oligodendrocyte-microglia cross-talk in the central nervous system. Immunology. 2014;141:302–313. doi: 10.1111/imm.12163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponomarev ED, Maresz K, Tan Y, Dittel BN. CNS-derived interleukin-4 is essential for the regulation of autoimmune inflammation and induces a state of alternative activation in microglial cells. J Neurosci. 2007;27:10714–10721. doi: 10.1523/JNEUROSCI.1922-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponomarev ED, Veremeyko T, Barteneva N, Krichevsky AM, Weiner HL. MicroRNA-124 promotes microglia quiescence and suppresses EAE by deactivating macrophages via the C/EBP-alpha-PU.1 pathway. Nat Med. 2011;17:64–70. doi: 10.1038/nm.2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen S, Wang Y, Kivisäkk P, Bronson RT, Meyer M, Imitola J, Khoury SJ. Persistent activation of microglia is associated with neuronal dysfunction of callosal projecting pathways and multiple sclerosis-like lesions in relapsing–remitting experimental autoimmune encephalomyelitis. Brain. 2007;130:2816–2829. doi: 10.1093/brain/awm219. [DOI] [PubMed] [Google Scholar]
- Ruckh JM, Zhao JW, Shadrach JL, van Wijngaarden P, Rao TN, Wagers AJ, Franklin RJ. Rejuvenation of regeneration in the aging central nervous system. Cell Stem Cell. 2012;10:96–103. doi: 10.1016/j.stem.2011.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruffell D, Mourkioti F, Gambardella A, Kirstetter P, Lopez RG, Rosenthal N, Nerlov C. A CREB-C/EBPbeta cascade induces M2 macrophage-specific gene expression and promotes muscle injury repair. Proc Natl Acad Sci U S A. 2009;106:17475–17480. doi: 10.1073/pnas.0908641106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scolding N, Franklin R, Stevens S, Heldin CH, Compston A, Newcombe J. Oligodendrocyte progenitors are present in the normal adult human CNS and in the lesions of multiple sclerosis. Brain. 1998;121:2221–2228. doi: 10.1093/brain/121.12.2221. [DOI] [PubMed] [Google Scholar]
- Shen S, Sandoval J, Swiss VA, Li J, Dupree J, Franklin RJ, Casaccia-Bonnefil P. Age-dependent epigenetic control of differentiation inhibitors is critical for remyelination efficiency. Nat Neurosci. 2008;11:1024–1034. doi: 10.1038/nn.2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimeld SM, McKay IJ, Sharpe PT. The murine homeobox gene Msx-3 shows highly restricted expression in the developing neural tube. Mech Dev. 1996;55:201–210. doi: 10.1016/0925-4773(96)00505-9. [DOI] [PubMed] [Google Scholar]
- Szanto A, Balint BL, Nagy ZS, Barta E, Dezso B, Pap A, Szeles L, Poliska S, Oros M, Evans RM, Barak Y, Schwabe J, Nagy L. STAT6 transcription factor is a facilitator of the nuclear receptor PPARgamma-regulated gene expression in macrophages and dendritic cells. Immunity. 2010;33:699–712. doi: 10.1016/j.immuni.2010.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp BD, Nave KA. Multiple sclerosis: an immune or neurodegenerative disorder? Ann Rev Neurosci. 2008;31:247–269. doi: 10.1146/annurev.neuro.30.051606.094313. [DOI] [PubMed] [Google Scholar]
- Wang W, Chen X, Xu H, Lufkin T. Msx3: a novel murine homologue of the Drosophila msh homeobox gene restricted to the dorsal embryonic central nervous system. Mech Dev. 1996;58:203–215. doi: 10.1016/S0925-4773(96)00562-X. [DOI] [PubMed] [Google Scholar]
- Yu Z, Xu X, Xiang Z, Zhou J, Zhang Z, Hu C, He C. Nitrated alpha-synuclein induces the loss of dopaminergic neurons in the substantia nigra of rats. PloS One. 2010;5:e9956. doi: 10.1371/journal.pone.0009956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamvil SS, Steinman L. Diverse targets for intervention during inflammatory and neurodegenerative phases of multiple sclerosis. Neuron. 2003;38:685–688. doi: 10.1016/S0896-6273(03)00326-X. [DOI] [PubMed] [Google Scholar]