

Abstract
Polyglutamine (polyQ) expansion of the androgen receptor (AR) causes Kennedy's disease/spinobulbar muscular atrophy (KD/SBMA) through poorly defined cellular mechanisms. Although KD/SBMA has been thought of as a motor neuron disease, recent evidence indicates a key role for skeletal muscle. To resolve which early aspects of the disease can be caused by neurogenic or myogenic mechanisms, we made use of the tet-On and Cre-loxP genetic systems to selectively and acutely express polyQ AR in either motor neurons (NeuroAR) or myocytes (MyoAR) of transgenic mice. After 4 weeks of transgene induction in adulthood, deficits in gross motor function were seen in NeuroAR mice, but not MyoAR mice. Conversely, reduced size of fast glycolytic fibers and alterations in expression of candidate genes were observed only in MyoAR mice. Both NeuroAR and MyoAR mice exhibited reduced oxidative capacity in skeletal muscles, as well as a shift in fast fibers from oxidative to glycolytic. Markers of oxidative stress were increased in the muscle of NeuroAR mice and were reduced in motor neurons of both NeuroAR and MyoAR mice. Despite secondary pathology in skeletal muscle and behavioral deficits, no pathological signs were observed in motor neurons of NeuroAR mice, possibly due to relatively low levels of polyQ AR expression. These results indicate that polyQ AR in motor neurons can produce secondary pathology in muscle. Results also support both neurogenic and myogenic contributions of polyQ AR to several acute aspects of pathology and provide further evidence for disordered cellular respiration in KD/SBMA skeletal muscle.
Keywords: androgen receptor, neuromuscular, polyglutamine, SBMA, skeletal muscle, transgenic mice
Introduction
Kennedy's disease (also known as spinobulbar muscular atrophy, hereafter referred to as KD/SBMA), is a degenerative neuromuscular disease caused by CAG trinucleotide repeat expansion mutations of AR (Kennedy et al., 1968; La Spada et al., 1991). KD/SBMA is one of several CAG repeat expansion diseases, including Huntington's disease, the spinocerebellar ataxias, and dentatorubral-pallidoluysian atrophy (Zoghbi and Orr, 2000; Orr and Zoghbi, 2007), in which the resulting polyglutamine expansion of the associated gene product causes neurodegeneration. The cellular and molecular etiology of KD/SBMA remains poorly understood and there is currently no treatment or cure.
Until mouse models of KD/SBMA were generated, this disease was generally thought to be a primary motor neuron disease (i.e., neurogenic in origin) based on clinical and histopathological observations of patients. Cellular models of this disease have therefore focused on modeling actions in motor neurons. However, recent studies in mouse models of KD/SBMA have increasingly pointed to contributions of polyglutamine (polyQ) expansion of the androgen receptor (AR) in skeletal muscle (Monks et al., 2007, Yu el al 2006, Palazzolo et al., 2009), which are necessary for many chronic aspects of the disease (Cortes et al., 2014; Lieberman et al., 2014). In the absence of models with selective polyQ AR expression in myocytes or motor neurons, it remains unclear whether polyQ AR in these cells is sufficient to induce pathology. Furthermore, given that the mouse models used to date express polyQ AR throughout development, it is unclear which aspects of the disease process result from the acute actions of polyQ AR rather than less direct mechanisms such as latent developmental effects or compensatory actions within cells and tissues.
The current study therefore evaluates acute pathology in a novel mouse model in which expression of polyQ AR is induced in adulthood for 4 weeks and expression of polyQ AR is limited to either skeletal muscle cells (myocytes) or to motor neurons. We find evidence for neurogenic contributions to motor dysfunction and some forms of muscle pathology, whereas myogenic contributions to muscle pathology and alterations in muscle gene expression are more prominent.
Materials and Methods
Transgene engineering
HB9-rtTA/tetO-Cre.
A 1 kb AgeI/FseI fragment of rtTA coding sequence (cds) from a modified pTet-On plasmid (Le et al., 2008) was inserted between the 9.4 kb promoter of HB9 and IRES-EGFP of the plasmid pHB9-MCS-IRES-EGFP (kind gift from T. Jessel, Columbia University) to give the recombinant plasmid pHB9-rtTA-IRES-EGFP (Fig. 1A; abbreviated hereafter to pHB9-rtTA). The construction of the tetO-Cre transgene has been described previously (Rao and Monks, 2009). A 12 kb fragment of the HB9-rtTA-IRES-EGFP transgene was excised from the plasmid pHB9-rtTA with XhoI. The 3.5 kb tetO-Cre fragment was isolated from ptetO-Cre with HindIII and XhoI as described previously (Rao and Monks, 2009).
Figure 1.
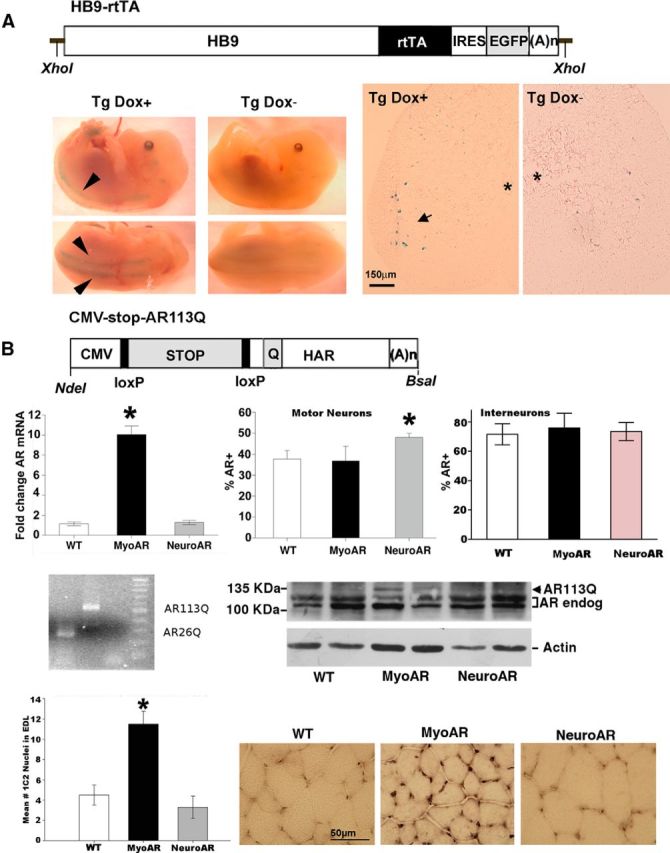
A, Top, Diagram of HB9-rtTA-IRES-EGFP transgene. HB9, Ppromoter of homeobox9 gene; rtTA, reverse tet-controlled transactivator; IRES, internal ribosomal entry site; EGFP, enhanced green fluorescent protein; (A)n, mouse metallothionein polyadenylation signal. Bottom, Photomicrographs of crosses of LacZ reporter mice with HB9-rtTA-IRES-EGFP/tetO-Cre mice. Bottom left, E14.5 Tg embryos gestating in dams treated with the tetracycline analog Dox for 3 d before dissection (Dox+) or untreated dams (Dox−). Arrows indicate staining along spinal column corresponding to ventral roots. Bottom right, Sections from adult Tg mice either treated with Dox or untreated demonstrating staining in motor neurons after Dox administration. Asterisk indicates the position of central canal. Arrow indicates stained motor neurons after Dox treatment. Scale bar, 150 μm. B, Top, Diagram of CMV-AR113Q transgene. CMV, mini cytomegalovirus promoter; STOP, floxed NEO-stop cassette; HAR, human AR clone with expanded CAG repeat (Q). loxP sites and restriction sites used to excise the constructs from plasmid before microinjection are also indicated. Second row, AR measurement in MyoAR and NeuroAR mice. As expected, AR was elevated in muscle only in MyoAR mice and in motor neurons only in NeuroAR mice. Left, qPCR measurement of AR in anterior tibialis muscle indicates AR mRNA is elevated in myoAR mice ∼10× relative to WT (n, 6/group). Middle, the percentage of motor neurons immunoreactive for AR in the retrodorsolateral nucleus is elevated relative to WT (n = 6–11 per group). Right, No differences in the number of AR+ interneurons in lamina X (n = 3–5 per group). *p < 0.05 for indicated comparison. Third row, PolyQ-expanded AR is expressed selectively in muscle. Left, PCR products obtained using primers that flank the CAG repeat region of the transgene on genomic DNA from CMV-stop-AR113Q mice demonstrate expanded region relative to DNA from an AR26Q clone. No template control and 100 bp ladder are also shown. Right, Western blot for AR in muscle of two mice of each genotype showing that polyQ AR (AR113Q) is detectable only in MyoAR mice. Actin Western blot used as a loading control is shown at bottom. Fourth row, 1C2-immunoreactive nuclei are observed in muscle of MyoAR mice. Left, Graph indicating that the number of 1C2+ nuclei is increased selectively in MyoAR mice (n = 3/group). *p < 0.05 for indicated comparison. Right, Photomicrographs illustrating increased 1C2 staining in EDL muscle of MyoAR mice. Scale bar, 50 μm.
CMV-stop-AR113Q.
A floxed stop sequence was obtained from pBS-Neostop (kind gift from Scott Zeitlin, University of Virginia; Dragatsis and Zeitlin, 2001). An NheI site was removed from within this sequence via blunting before the addition of flanking NheI sites, which were introduced at existing XbaI and KpnI sites using linkers. An NheI fragment containing the loxP Neostop sequence was then ligated between promoter and coding sequences of the p5HBhAR113QcDNA transgene (kind gift from Andrew Lieberman, University of Michigan; Yu et al., 2006), yielding CMV-stop-AR113Q (Fig. 1B).
Animals
Transgenes (either CMV-stop-AR113Q or both HB9-rtTA and tetO-Cre) were injected (or coinjected) into pronuclei of (C57BL/6 × C3H)F2 zygotes in the transgenic facility of the Van Andel Research Institute (Grand Rapids, MI). Zygotes were then implanted into pseudopregnant foster mothers. Genomic DNA from resulting founder mice were then genotyped by quantitative PCR (qPCR) with rtTA-specific and Cre-specific primers, as described previously (Rao and Monks, 2009), and with primers complementary to sequences in the neomycin resistance cassette within the floxed STOP sequence (primers = AGG ATC TCC TGT CAT CTC ACC TTG CTC CTG, AAG AAC TCG TCA AGA AGG CGA TAG AAG GCG). We verified that the CMV-stop-AR113Q transgene integrated into the genome of transgenic (Tg) mice with an intact CAG expansion using qPCR with transgene-specific primers flanking the CAG repeat region (F = ACC GAG GAG CTT TCC AGA AT, R = CTC ATC CAG GAC CAG GTA GC) on samples of genomic DNA (Fig. 1B).
HB9-rtTA/tetO-Cre double-Tg and CMV-stop-AR113Q single-Tg founders were used for further analyses. Founder lines were maintained by back-crossing Tg mice to wild-type (WT) C57BL/6 mice. For all studies, WT controls are siblings negative for both inducer and AR transgenes. Unless otherwise noted, all experimental animals were adult male mice. Histological and gene expression studies were only performed on a subset of animals in which behavior was measured.
Animals were housed in standard conditions at the University of Toronto–Mississauga. All procedures were approved by the University Animal Care Committee in accordance with the Canadian Council on Animal Care and National Institutes of Health guidelines. To induce transgene expression, the tetracycline analog doxycycline (Dox) (D9891 doxycycline hyclate; Sigma) was administered in drinking water at a concentration of 2 mg/ml supplemented with 5% sucrose and provided ad libitum to Dox-treated (Dox+) animals in light-proofed water bottles for 3 d as described previously (Rao and Monks, 2009).
Analysis of HB9-rtTA/tetO-cre transgene expression using loxP LacZ reporter mice
Mice from each founding line were crossed with LacZ reporter mice to characterize transgene expression as described previously (Rao and Monks, 2009). Briefly, to localize Cre activity, HB9-rtTA/tetO-Cre double-Tg founders were crossed with homozygous B6.129S4-Gt(ROSA)26Sortm1Sor/J reporter mice obtained from The Jackson Laboratory. In the resulting progeny, Cre expression results in a blue color in a β-galactosidase assay (X-gal staining). Resulting triple-Tg offspring were genotyped from yolk sac, placenta, or pup tail genomic DNA and embryos were aged according to the day when vaginal plugs were observed, which was counted as embryonic day 0.5 (E0.5).
Embryonic pups were induced by delivering Dox to the dam. Control (Dox−) mice received standard drinking water. A minimum of three adult mice of each sex in each group (Tg Dox+, Tg Dox−, and WT Dox+) were examined. A total of 15 Tg and 16 WT embryos were examined, but these were not sexed.
Embryos were dissected in ice-cold 1× PBS and placed in fixative solution (2% paraformaldehyde/0.2% glutaraldehyde). Fixed embryos were then stained with 0.1% X-gal (5-bromo-4-chloro-3-indolyl-β-d-galactoside) for 24 h. After staining, embryos were further fixed in 4% paraformaldehyde and then photographed using a Nikon CoolPix 995 camera attached to a Nikon SMZ800 dissection microscope.
Cryostat sections (12 μm) of spinal cords were prepared and kept frozen at −80°C until X-gal staining. Slides were then placed in the fixative solution (described above) before incubation in X-gal solution (described above) for 24 h. Slides were stained with 0.05% safranin before dehydration with graded ethanol and clearing with xylene. Slides were coverslipped using Permount.
Measurement of motor function
Body weight and gross motor function of adult male mice was assessed 4 weeks after Dox treatment using a battery of tests as described previously (Monks et al., 2007). At least 6 animals per group were measured, all of which were, ∼4 months of age at Dox treatment (range 114–124). The testing session included three trials of hang test, gait, grip strength, and constant-speed rotorod, with the best performance being recorded for each day.
Measurement of mRNA using qPCR
qPCR for AchRa, AchRb, Ankrd1, Hsp7, Fbxo32 and Vegfa was performed as described in Mo et al., 2010. Briefly, DNase-treated mRNA was reverse transcribed using dT20VN primer (Sigma) with SuperScript II before ampification with SYBR Green JumpStart Taq ReadyMix (Sigma). The Mx4000 Multiplex Quantitative PCR System (Stratagene) was used for data acquisition and analysis according to the instructions of the manufacturer. Samples were incubated at 95°C for 10 min before thermal cycling (40 cycles of: 95°C for 30 s, 57°C for 30 s, and 72°C for 30 s). qPCR for AR was performed in the same way with the following primers: F = AGG AAG CAG TAT CCG AAG GCA, R = GGA CAC CGA CAC TGC CTT ACA. All samples were run in duplicate and with a no reverse transcription condition as technical controls for reproducibility and to rule out genomic DNA contamination. A melting curve was also performed to assess product quality. Efficiency of reaction was then determined for ROX-normalized fluorescence measurements and a final calculation of the fold induction from the Ct values and the expression of each test gene was normalized to the level of GAPDH.
Measurement of AR by Western blotting
Quadriceps muscles were dissected and frozen at −80°C until use. Samples were homogenized with a glass tissue grinder and then sonicated in ice-cold RIPA lysis buffer supplemented with protease inhibitor mixture (Sigma-Aldrich). Protein was quantified through the Bradford assay (Thermo Fisher Scientific) and 50 μg of total protein per sample was resolved by 7.5% Tris-glycine SDS-PAGE and wet transferred onto a PVDF membrane (Immobilon-P; Thermo Fisher Scientific). After blocking with 5% skim milk in TBS/Triton X-100 (0.05%), the membrane was then incubated with primary antibody overnight at 4°C. Polyclonal rabbit anti-AR (1:50; Santa Cruz Biotechnology) and monoclonal rabbit anti-actin (1:10000; Sigma-Aldrich) primary antibodies were used. The next day, the membrane was rinsed with TBS/Triton X-100 and incubated with HRP-conjugated goat anti-rabbit (Thermo Fisher Scientific) for 2 h at room temperature. Chemiluminescent signal was detected using the SuperSignal West Femto Substrate (Thermo Fisher Scientific).
Histology
Analysis of cell number and size in muscle with hemotoxylin and eosin, as well as spinal cords with thionin, were performed as described previously (Monks et al., 2007; Niel et al., 2009). Succinate dehydrogenase (SDH) and NADH staining was performed as described previously (Monks et al., 2007; Musa et al., 2011). Fiber typing was performed using SDH in combination with anti-fast myosin heavy chain (My32; Sigma) on alternate sections as described previously (Fernando et al., 2010). AR immunohistochemistry on spinal cord sections was performed using a rabbit monoclonal anti-AR antibody (1:100, 3165-1; Epitomics) as described previously (Swift-Gallant and Monks, 2013). The percentage of AR-positive cells was determined by counting AR+ cells in the retrodorsolateral nucleus (RDLN) manually at 400× magnification and then correcting to total cell number by restaining with neutral red as described previously (Zuloaga et al., 2007). For lamina X neurons, a similar approach was taken, but cell counts were performed automatically using ImageJ on photomicrographs of a single section corresponding to the middle of the rostrocaudal extent of the RDLN.
Staining for PolyQ AR was performed using the 1C2 antibody (clone MAB1574, 1:10,000; Millipore). Quantification of 1C2 staining was performed manually on two nonoverlapping photomicrographs taken at 1000× magnification from an extensor digitorum longus (EDL) muscle section for each subject. Counts were then averaged for each subject before statistical analysis.
Staining for motor neurons was performed using an anti-choline acetyltransferase (ChAT) antibody (1:100; polyclonal MAB144P-200UL; Millipore) The total number of neurons in the RDLN was estimated by live counting the ChAT-stained motor neurons at 400× magnification in every fourth section of its rostrocaudal extent.
Staining for oxidative stress markers (8-OHdG and nitrotyrosine) were performed using immunohistochemistry largely as described above, with the following modifications. Tissue was incubated in 0.05% sodium citrate buffer at 85°C for 30 min before blocking in 10% normal horse serum and incubating with primary antibodies overnight at 4°C (1:100, 8-OHdG SC-66036; Santa Cruz Biotechnology; 1:100, anti-nitrotyrosine, MAB5404; EMD-Millipore). Quantification of relative optical density (ROD) was performed on images for two soleus (SOL) and spinal cord sections. Images were captured at 200× magnification and ROD was calculated using ImageJ. Values obtained from no primary control sections were subtracted from matched primary sections. The entire image was measured for SOL sections and three motor neurons per subject were outlined in ImageJ for cord sections.
For statistical analysis, data were analyzed using ANOVA for genotype, followed by Tukey's tests for pairwise comparisons. In all cases, α was set at 0.05.
Results
A summary of the results is presented in Table 1.
Table 1.
Summary of phenotypes of MyoAR and NeuroAR mice
| Motor | Gene expression | Fiber type | Oxidative stress | Inclusions | |
|---|---|---|---|---|---|
| MyoAR | Normal | Dysregulated | Shift to glycolytic | Normal in muscle, reduced in cord | +Muscle, −cord |
| NeuroAR | Mild deficit | Normal | Shift to glycolytic | Increased in muscle, reduced in cord | −Muscle, −cord |
Tg mice
Characterization of the ACTA-rtTA/tetO-Cre mouse has been described previously (Rao and Monks, 2009). We verified tissue- and cell-type specificity of HB9-rtTA/tetO-Cre using a similar strategy of crossing with a LacZ reporter mouse (the HB9-rtTA transgene is depicted in Fig. 1A, the coinjected tetO-Cre transgene was identical to that used in Rao and Monks, 2009). β-galactosidase activity in reporter mice (Fig. 1A) was restricted to motor neuron cell bodies in the spinal cord and processes in ventral roots and β-galactosidase was readily detected only after treatment with the tetracyline analog Dox.
To generate mice that selectively express polyQ AR in myocytes (referred to hereafter as MyoAR), we crossed ACTA-rtTA/tetO-Cre and CMV-stop-AR113Q mice. Selective transgene expression in MyoAR mice was verified using qPCR for AR in skeletal muscle. As expected, AR mRNA and protein were elevated in the skeletal muscle of Tg MyoAR mice relative to WT littermate controls (Fig. 1B), but not in crosses between HB9-rtTA/tetO-Cre and CMV-stop-AR113Q (referred to hereafter as NeuroAR), which were engineered to express polyQ AR in motor neurons. Interestingly, the protein expression in muscle was considerably lower than predicted by mRNA (Fig. 1B). This observation may be explained by the technical difficulty in solubilizing polyQ AR aggregates. Consistent with this hypothesis, abundant 1C2-immunoreactive nuclei were observed in MyoAR muscle (Fig. 1B). To resolve the cell types within the spinal cord resulting from NeuroAR, we further validated specificity of overexpression using immunohistochemistry for AR. As expected, NeuroAR mice had an increased percentage of AR+ motor neurons in the RDLN relative to both MyoAR mice and WT mice, but no increase in AR+ neurons in lamina X (Fig. 1B). Together, these results indicate that specificity in polyQ AR expression had been achieved, with MyoAR and NeuroAR mice expressing polyQ AR in a muscle-specific and motor-neuron-specific manner, respectively.
Behavioral analysis
In contrast to HSA-AR mice (Monks et al., 2007), no overt atrophic phenotype was observed in either MyoAR or NeuroAR mice. Both NeuroAR and MyoAR mice appeared grossly normal after acute (4 weeks) transgene induction. No overt postural (e.g., kyphosis) or behavioral signs (e.g., lethargy, joint contracture) of neuromuscular atrophy were present nor was there a reduction in body weight (Fig. 2). However, evidence of motor dysfunction on endurance tasks was found in NeuroAR mice (Fig. 2). Although muscle-specific wild-type AR overexpression can result in profound muscular atrophy (Monks et al., 2007), these results suggest that other sites of polyQ AR expression, longer periods of transgene induction, and/or higher levels of transgene overexpression would be required for more overt signs of pathology in CMV-stop-AR113Q mice. Certainly, the level of overexpression of AR in skeletal muscle of MyoAR mice is orders of magnitude smaller than that in HSA-AR mice (Monks et al., 2007). Because we are primarily interested in the site(s) which initiate(s) the early stages of pathology within KD/SBMA, we did not attempt to determine the end stages of disease progress in these mice.
Figure 2.

Motor deficits in endurance-related tests in NeuroAR but not MyoAR mice. Mice were tested 4 weeks after induction with Dox and reduced performance was observed only in NeuroAR mice. Differences in motor function were not secondary to altered body weight (n = 6–11 per group). *p < 0.05 relative to MyoAR; +p < 0.10 relative to WT.
Gene expression
We measured expression of several candidate genes (Ankrd1, Chrna1, Chrnb1, Fbxo32, Hspb7, and Vegfa), which were previously identified as being dysregulated in a microarray study of gene expression in skeletal muscle of mouse models of KD/SBMA (Mo et al., 2010). Here, we found that most of these genes (Ankrd1, Chrna1, Chrnb1, and Hspb7) were similarly altered in MyoAR mice (Fig. 3). Interestingly, we found no alteration of any of these candidate genes in the muscle of NeuroAR mice, although it remains possible that motor neuron expression of polyQ AR affects expression of other KD/SBMA candidate genes in muscle. These results indicate that alterations in skeletal muscle gene expression typical of KD/SBMA can be reproduced solely with acute expression of polyQ AR in myocytes.
Figure 3.

Alterations in gene expression in anterior tibialis muscle of MyoAR mice. qPCR was used to estimate mRNA abundance of Ankrd1, Chrna1, Chrnb1, Fbxo32, Hspb7, and Vegfa (n = 6 per group). Alterations in Ankrd1, Chrna1, Chrnb1, and Hspb7 similar to those seen in other mouse models of KD/SBMA were observed in MyoAR but not NeuroAR mice. *p < 0.05 relative to all other groups.
Histopathology
We assessed potential histopathology in skeletal muscle and spinal cord sections (Fig. 4) and found no obvious disorganization of these tissues. We also found neither a loss of motor neurons nor a reduction in their size in either NeuroAR or MyoAR mice. We confirmed that there was no obvious loss in motor neuron functionality using a ChAT stain (Fig. 4). Similarly, we found no alterations in number of EDL or SOL fibers. Together, these results indicate that neither acute myocyte nor acute motor neuron expression of polyQ AR is sufficient to cause motor neuron loss or obvious muscular pathology. These features of KD/SBMA may require other loci and/or longer periods of polyQ AR expression to manifest.
Figure 4.

Absence of gross histopathology in skeletal muscle or spinal cord of MyoAR or NeuroAR mice. Hematoxylin and eosin stained muscle sections from extensor digitorum longus (EDL) and soleus (SOL) from WT, MyoAR, and NeuroAR mice (Top). Thionin-stained lumbar 5 spinal cord sections (cord) with associated motor neuron pools (MN) are also presented (middle and bottom, respectively). Scale bars, 50 μm for muscle and MN photomicrographs and 150 μm for spinal cord. Note: No difference in fiber number in EDL and SOL muscles was observed for WT, MyoAR, and NeuroAR mice (n = 11–21 per group). In addition, no difference in number or functionality (as shown by ChAT staining) of lumbar segment 5 and 6 retrodorsolateral (RDLN) motor neurons was observed (n = 5–9 per group).
Diaphorase staining
Because diaphorase staining is altered in other mouse models of KD/SBMA (Monks et al., 2007; Musa et al., 2011; Rinaldi et al., 2012), we examined NADH (corresponding to complex I of the electron transport chain) and SDH (corresponding to complex II of the electron transport chain). Although we did not observe disorganization in diaphorase staining such as that exhibited by HSA-AR mice (Monks et al., 2007), we found a global reduction in NADH and SDH staining in both MyoAR and NeuroAR mice (Fig. 5A,B), indicating that deficits in oxidative metabolism in muscle can result from acute polyQ AR toxicity either directly in myocytes or indirectly via motor neurons.
Figure 5.

Reduced diaphorase staining in both MyoAR and NeuroAR mice. A, Photographs of SDH-stained extensor digitorum longus (EDL) and soleus (SOL) from WT, MyoAR, and NeuroAR mice indicate dramatically reduced oxidative capacity. Scale bar, 50 μm. B, Quantification of the percentage of fibers staining darkly for SDH and NADH (n = 5–10 per group) confirms that both MyoAR and NeuroAR mice have fewer fibers with high oxidative metabolic capacity in EDL and SOL. C, Fiber typing indicates a shift in fast fibers in the EDL from oxidative to glycolytic in both MyoAR and NeuroAR mice (n = 4–9). The size of the minor ellipse (measured in micrometers) of fast glycolytic fibers was reduced in the EDL of MyoAR but not NeuroAR mice. *p < 0.05 relative to WT.
Fiber typing
We found a shift in proportion of fast fibers in EDL but not SOL in both NeuroAR and MyoAR mice such that fewer fast oxidative fibers, but more fast glycolytic (FG) fibers, are observed (Fig. 5C, top). A reduction in size of fast glycolytic fibers was also observed in EDL of MyoAR but not NeuroAR mice, consistent with mild muscular atrophy (Fig. 5C, bottom). These results indicate that polyQ AR action in either muscle or motor neuron is sufficient to alter FG muscle fibers, but this effect is limited by muscle type, as has been reported for contraction dynamics in the HSA-AR model (Oki et al., 2013).
Oxidative stress
8-OHdG and nitrotyrosine are markers of oxidative stress that are increased in tissues of a mouse model of KD/SBMA (Iida et al., 2014). We found that 8-OHdG staining of the SOL muscle was significantly increased in NeuroAR Tg mice relative to WT and MyoAR mice (Fig. 6). In contrast, nitrotyrosine staining was significantly reduced in motor neurons of both MyoAR and NeuroAR mice (Fig. 6). No significant differences were found between genotypes after 8-OHdG staining of motor neurons or after nitrotyrosine staining in muscle. These results suggest a complex relationship between polyQ AR expression and oxidative stress in muscle and motor neurons, which depends on tissue type and marker of stress. The results are also consistent with the notion that oxidative stress in muscle participates in motor dysfunction, but are inconsistent with its participation in gene dysregulation or reduced oxidative metabolism.
Figure 6.

Oxidative stress markers in muscle and motor neurons of MyoAR and NeuroAR mice. Top, Photomicrographs of 8-OHdG staining in soleus (SOL) muscle (top row) and nitrotyrosine staining in spinal cord sections (bottom row) of WT, MyoAR, and NeuroAR mice. Scale bar, 100 μm. Bottom, Graphs representing mean ± SEM ROD (measured in arbitrary units) of 8-OHdG staining in SOL (right) and nitrotrosine staining in motor neurons (left). 8-OHdG is elevated in SOL of NeuroAR mice relative to both WT and MyoAR mice (n = 3–4 per group). Nitrotyrosine is reduced in both MyoAR and NeuroAR mice relative to WT. *p < 0.05 for indicated comparison.
Discussion
The present study indicates that acute polyQ AR overexpression restricted to either skeletal myocytes or to motor neurons can produce features of pathology associated with more global and chronic expression of polyQ AR in other mouse models. This is consistent with the necessity of polyQ AR in myocytes for many, but not all, aspects of pathology (Cortes et al., 2014). MyoAR mice exhibited alterations in gene expression in skeletal muscle typical of KD/SBMA, along with some reduction in fiber size. NeuroAR mice displayed some gross motor deficits. Interestingly, both MyoAR and NeuroAR mice exhibited reduced diaphorase staining in skeletal muscle and associated fiber type switching. Importantly, neither MyoAR nor NeuroAR produced loss of motor neurons or myocytes, supporting our attempt to model early stages of disease progression.
Although we found many pathological features in NeuroAR and MyoAR mice that were similar to those reported in other mouse models of KD/SBMA, in some cases, pathology manifested differently. Notably, although increased diaphorase staining has been reported in HSA-AR mice (Monks et al., 2007) and AR97Q mice (Rinaldi et al., 2012), we found that it was reduced in MyoAR and NeuroAR mice. Similarly, alterations in fiber type were more pronounced in the EDL rather than SOL in the present study, whereas contraction dynamics were more severely affected in SOL in HSA-AR mice (Oki et al., 2013). This puzzling finding might reflect a nonlinear relationship between AR pathology and oxidative metabolism of muscle, such that aberrant AR produces dysregulation of mitochondrial function rather than a simple increase or decrease. Alternatively, because our model involves a relatively acute transgene induction, it may be that the increase in diaphorase staining in chronic mouse models is preceded by reduced diaphorase staining. Nonetheless, the present findings support the notion that altered AR signaling affects oxidative metabolism and/or mitochondria in skeletal muscle in a variety of contexts (Monks et al., 2007; Musa et al., 2011; Fernando et al., 2010; Rinaldi et al., 2012; Ranganathan et al., 2009) and also indicate that these alterations may have both myogenic and neurogenic origins.
Despite the disturbance in oxidative metabolism indicated by diaphorase staining, we found no clear association between reduced diaphorase staining in muscle and alterations in markers of oxidative stress (8-OHdG and nitrotyrosine). Although we did not measure diaphorase staining in spinal cord, nitrotyrosine (but not 8-OHdG) was decreased in both MyoAR and NeuroAR mice, which would be expected if oxidative metabolism is reduced. However, nitrotyrosine staining was unaffected in muscle of MyoAR and NeuroAR mice, which show decreased diaphorase staining. This observation argues against a simple scenario in which oxidative stress and oxidative metabolism change in concert with one another. An alternative explanation may be that the oxidative stress markers instead track ongoing energetic demands rather than oxidative capacity. According to this view, oxidative stress in motor neurons might be reduced as a consequence of reduced spontaneous activity, for example, whereas 8-OHdG staining in SOL was increased in NeuroAR mice, possibly reflecting increased task demands of motor testing due to endurance deficits in these mice.
That MyoAR and NeuroAR can manifest similar pathology raises the question of how these sites of action can converge through necessarily divergent mechanisms. Cell autonomous effects, such as the alterations in gene expression in skeletal muscle observed in MyoAR mice, are easily understood, but more complex conceptual models are required to explain cell nonautonomous effects. Muscle–nerve interactions are well studied and mechanisms that are thought to mediate normal development, plasticity, and repair of neuromuscular systems (e.g., release of growth factors by musculature and regulation of muscle physiology via alterations in synaptic transmission) are likely to play a role as well. Indeed, several theories of KD/SBMA center on dysregulation of muscle-nerve interactions (Rocchi and Pennuto, 2013; Katsuno et al., 2006).
Importantly, neither MyoAR nor NeuroAR produced a full spectrum of KD/SBMA symptoms. This may be explained in several ways. It remains possible that longer treatments or higher expression levels might produce more pathology. Although MyoAR mice have relatively strong expression of polyQ AR, relatively low levels of polyQ AR expression in motor neurons in NeuroAR mice are likely because we saw no obvious increase in 1C2 immunoreactivity or AR-immunoreactive nuclear inclusions in the spinal cords of NeuroAR mice. AR-immunoreactive inclusions are a hallmark pathological sign in KD/SBMA, but they are inconsistently present in cell and mouse models. Higher expression levels and/or longer periods of expression may be necessary for their manifestation. It seems likely that the length of time of polyQ AR expression is a more decisive factor for inclusions because insoluble aggregates of polyQ AR occur late in the cytotoxic process (Takahashi et al., 2008) and we did not observe inclusions in muscle of MyoAR mice (which have relatively strong expression) in the present study. Aside from expression level and length of time of polyQ AR expression, endogenous AR produced by the mice may protect against KD/SBMA symptoms (Thomas et al., 2006). More to the point, it remains possible that expression is required in more than one cell type and that expression of polyQ AR in other cell types in muscle, the nervous system, or elsewhere are pivotal in KD/SBMA etiology. Finally, complex interactions between several sites of polyQ AR action may be required.
Footnotes
This work was supported by funding from the Canadian Institutes of Health Research (Operating Grant JNM-116637 to D.A.M. and Operating Grants MOP-126090 and MOP-86549 to H.-Y.M.C.) and the Natural Sciences and Engineering Research Council of Canada (Discovery Grants RGPIN 312458 to D.A.M. and RGPIN 386495 to H-Y.M.C.). We thank Paulina Phan for technical assistance.
The authors declare no competing financial interests.
References
- Cortes CJ, Ling SC, Guo LT, Hung G, Tsunemi T, Ly L, Tokunaga S, Lopez E, Sopher BL, Bennett CF, Shelton GD, Cleveland DW, La Spada AR. Muscle expression of mutant androgen receptor accounts for systemic and motor neuron disease phenotypes in spinal and bulbar muscular atrophy. Neuron. 2014;82:295–307. doi: 10.1016/j.neuron.2014.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragatsis I, Zeitlin S. A method for the generation of conditional gene repair mutations in mice. Nucleic Acids Res. 2001;29:E10. doi: 10.1093/nar/29.3.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernando SM, Rao P, Niel L, Chatterjee D, Stagljar M, Monks DA. Myocyte androgen receptors increase metabolic rate and improve body composition by reducing fat mass. Endocrinology. 2010;151:3125–3132. doi: 10.1210/en.2010-0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iida M, Katsuno M, Nakatsuji H, Adachi H, Kondo N, Miyazaki Y, Tohnai G, Ikenaka K, Watanabe H, Yamamoto M, Kishida K, Sobue G. Pioglitazone suppresses neuronal and muscular degeneration caused by polyglutamine-expanded androgen receptors. Hum Mol Genet. 2015;24:314–329. doi: 10.1093/hmg/ddu445. [DOI] [PubMed] [Google Scholar]
- Katsuno M, Adachi H, Waza M, Banno H, Suzuki K, Tanaka F, Doyu M, Sobue G. Pathogenesis, animal models and therapeutics in spinal and bulbar muscular atrophy (SBMA) Exp Neurol. 2006;200:8–18. doi: 10.1016/j.expneurol.2006.01.021. [DOI] [PubMed] [Google Scholar]
- Kennedy WR, Alter M, Sung JH. Progressive proximal spinal and bulbar muscular atrophy of late onset: a sex-linked recessive trait. Neurology. 1968;18:671–680. doi: 10.1212/WNL.18.7.671. [DOI] [PubMed] [Google Scholar]
- La Spada AR, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature. 1991;352:77–79. doi: 10.1038/352077a0. [DOI] [PubMed] [Google Scholar]
- Le YZ, Zheng W, Rao PC, Zheng L, Anderson RE, Esumi N, Zack DJ, Zhu M. Inducible expression of cre recombinase in the retinal pigmented epithelium. Invest Ophthalmol Vis Sci. 2008;49:1248–1253. doi: 10.1167/iovs.07-1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman AP, Yu Z, Murray S, Peralta R, Low A, Guo S, Yu XX, Cortes CJ, Bennett CF, Monia BP, La Spada AR, Hung G. Peripheral androgen receptor gene suppression rescues disease in mouse models of spinal and bulbar muscular atrophy. Cell Rep. 2014;7:774–784. doi: 10.1016/j.celrep.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monks DA, Johansen JA, Mo K, Rao P, Eagleson B, Yu Z, Lieberman AP, Breedlove SM, Jordan CL. Overexpression of wild-type androgen receptor in muscle recapitulates polyglutamine disease. Proc Natl Acad Sci U S A. 2007;104:18259–18264. doi: 10.1073/pnas.0705501104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo K, Razak Z, Rao P, Yu Z, Adachi H, Katsuno M, Sobue G, Lieberman AP, Westwood JT, Monks DA. Microarray analysis of gene expression by skeletal muscle of three mouse models of Kennedy disease/spinal bulbar muscular atrophy. PLoS One. 2010;5:e12922. doi: 10.1371/journal.pone.0012922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musa M, Fernando SM, Chatterjee D, Monks DA. Subcellular effects of myocyte-specific androgen receptor overexpression in mice. J Endocrinol. 2011;210:93–104. doi: 10.1530/JOE-11-0071. [DOI] [PubMed] [Google Scholar]
- Niel L, Shah AH, Lewis GA, Mo K, Chatterjee D, Fernando SM, Hong MH, Chang WY, Vollmayr P, Rosen J, Miner JN, Monks DA. Sexual differentiation of the spinal nucleus of the bulbocavernosus is not mediated solely by androgen receptors in muscle fibers. Endocrinology. 2009;150:3207–3213. doi: 10.1210/en.2008-1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oki K, Wiseman RW, Breedlove SM, Jordan CL. Androgen receptors in muscle fibers induce rapid loss of force but not mass: implications for spinal bulbar muscular atrophy. Muscle Nerve. 2013;47:823–834. doi: 10.1002/mus.23813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr HT, Zoghbi HY. Trinucleotide repeat disorders. Annu Rev Neurosci. 2007;30:575–621. doi: 10.1146/annurev.neuro.29.051605.113042. [DOI] [PubMed] [Google Scholar]
- Palazzolo I, Stack C, Kong L, Musaro A, Adachi H, Katsuno M, Sobue G, Taylor JP, Sumner CJ, Fischbeck KH, Pennuto M. Overexpression of IGF-1 in muscle attenuates disease in a mouse model of spinal and bulbar muscular atrophy. Neuron. 2009;63:316–328. doi: 10.1016/j.neuron.2009.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranganathan S, Harmison GG, Meyertholen K, Pennuto M, Burnett BG, Fischbeck KH. Mitochondrial abnormalities in spinal and bulbar muscular atrophy. Hum Mol Genet. 2009;18:27–42. doi: 10.1093/hmg/ddn310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao P, Monks DA. A tetracycline-inducible and skeletal muscle-specific Cre recombinase transgenic mouse. Dev Neurobiol. 2009;69:401–406. doi: 10.1002/dneu.20714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinaldi C, Bott LC, Chen KL, Harmison GG, Katsuno M, Sobue G, Pennuto M, Fischbeck KH. Insulinlike growth factor (IGF)-1 administration ameliorates disease manifestations in a mouse model of spinal and bulbar muscular atrophy. Mol Med. 2012;18:1261–1268. doi: 10.2119/molmed.2012.00271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocchi A, Pennuto M. New routes to therapy for spinal and bulbar muscular atrophy. J Mol Neurosci. 2013;50:514–523. doi: 10.1007/s12031-013-9978-7. [DOI] [PubMed] [Google Scholar]
- Swift-Gallant A, Monks DA. Androgen receptor expression in satellite cells of the neonatal levator ani of the rat. Dev Neurobiol. 2013;73:448–454. doi: 10.1002/dneu.22066. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Kikuchi S, Katada S, Nagai Y, Nishizawa M, Onodera O. Soluble polyglutamine oligomers formed before inclusion body formation are cytotoxic. Hum Mol Genet. 2008;17:345–356. doi: 10.1093/hmg/ddm311. [DOI] [PubMed] [Google Scholar]
- Thomas PS, Jr, Fraley GS, Damien V, Woodke LB, Zapata F, Sopher BL, Plymate SR, La Spada AR. Loss of endogenous androgen receptor protein accelerates motor neuron degeneration and accentuates androgen insensitivity in a mouse model of X-linked spinal and bulbar muscular atrophy. Hum Mol Genet. 2006;15:2225–2238. doi: 10.1093/hmg/ddl148. [DOI] [PubMed] [Google Scholar]
- Yu Z, Dadgar N, Albertelli M, Gruis K, Jordan C, Robins DM, Lieberman AP. Androgen-dependent pathology demonstrates myopathic contribution to the Kennedy disease phenotype in a mouse knock-in model. J Clin Invest. 2006;116:2663–2672. doi: 10.1172/JCI28773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoghbi HY, Orr HT. Glutamine repeats and neurodegeneration. Annu Rev Neurosci. 2000;23:217–247. doi: 10.1146/annurev.neuro.23.1.217. [DOI] [PubMed] [Google Scholar]
- Zuloaga DG, Morris JA, Monks DA, Breedlove SM, Jordan CL. Androgen-sensitivity of somata and dendrites of spinal nucleus of the bulbocavernosus (SNB) motoneurons in male C57BL6J mice. Horm Behav. 2007;51:207–212. doi: 10.1016/j.yhbeh.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]