

Abstract
Protein interacting with C-kinase 1 (PICK1) has received considerable attention, because it interacts with a broad range of neurotransmitter receptors, transporters, and enzymes and thereby influences their localization and function in the CNS. Although it is suggested that putative partners of PICK1 are involved in neurological diseases such as schizophrenia, Parkinson's disease, chronic pain, and amyotrophic lateral sclerosis, the functions of PICK1 in neurological disorders are not clear. Here, we show that oxidative stress, which is tightly associated with neurological diseases, occurs in PICK1−/− mice. The oxidation in PICK1−/− mice was found selectively in neurons and was age dependent, leading to microglial activation and the release of inflammatory factors. Neurons in the cortex and hippocampus from PICK1−/− mice showed increased vulnerability to oxidants and reduced capacity to metabolize reactive oxygen species (ROS); this was caused by reduced glutathione content and impaired cysteine transport. The dysregulated expression of glutathione was attributed to a decrease of the surface glutamate transporter excitatory amino acid carrier 1 (EAAC1). Overexpression of PICK1 could rescue the surface expression of EAAC1 and ameliorate the glutathione deficit in PICK1−/− neurons. Finally, reduced surface EAAC1 was associated with defective Rab11 activity. Transfection with dominant-negative Rab11 effectively suppressed surface EAAC1 and increased ROS production. Together, these results indicate that PICK1 is a crucial regulator in glutathione homeostasis and may play important roles in oxidative stress and its associated neurodegenerative diseases.
Keywords: EAAC1, glutathione, oxidant, oxidative stress, PICK1, Rab11
Introduction
Protein interacting with C-kinase 1 (PICK1) is first cloned as a protein kinase C (PKC)-binding protein by yeast two-hybrid screening (Staudinger et al., 1995). Over the past two decades, PICK1 has received considerable attention because it is the only known protein that possesses a PSD-95/DlgA/ZO-1 (PDZ) domain and a Bin–Amphiphysin–Rvs (BAR) domain and thereby participates in many aspects of brain function (Xu and Xia, 2006). Through the PDZ domain, PICK1 interacts with a broad range of neurotransmitter receptors, transporters, and enzymes (Deken et al., 2001; Xu and Xia, 2006; Hanley, 2008). Through the BAR domain, PICK1 anchors its partners and influences their synaptic localization and function (Xu and Xia, 2006; Hanley, 2008). The best characterized function of PICK1 is that it regulates trafficking of the GluA2 subunit of AMPA receptor during long-term depression and long-term potentiation in the hippocampus and the cerebellum (Xia et al., 2000; Hanley and Henley, 2005; Steinberg et al., 2006; Terashima et al., 2008). In summary, a wealth of information has revealed important physiological functions of PICK1 in the activity-dependent modulation of synaptic transmission (Hanley, 2008).
Recent evidence implies that PICK1 is involved in neurological disorders, such as schizophrenia, Parkinson's disease (PD), chronic pain, amyotrophic lateral sclerosis (ALS), epilepsy, and cocaine-seeking behavior (Focant and Hermans, 2013). It is suggestive that the involvement of PICK1 in neurological diseases is relevant to its interactions with putative partners (Focant and Hermans, 2013), such as PKC, GluA2, metabotropic glutamate receptor subunit mGluR7, acid-sensing ion channel (ASIC), and enzyme serine racemase (Xu and Xia, 2006; Focant and Hermans, 2013). Notably, dysregulations of PICK1 with GluA2 and ASIC are hypothesized to be the main mechanisms underlying multiple disorders (Focant and Hermans, 2013). For example, a lack of PICK1 abrogates the development of chronic pain in complete Freund's adjuvant-injected mice (Atianjoh et al., 2010). In comparison, the evidence for the function of PICK1–ASIC interaction in disorders is only from in vitro studies, which show increased ASIC-mediated acidotoxicity and enhanced surface expression of ASIC2a by PICK1 in cultured cells (Joch et al., 2007; Jin et al., 2010). However, these findings are insufficient to illustrate roles of PICK1 in neurological disorders. Additional studies are needed to understand functions of PICK1 in neurological disorders.
Oxidative stress is defined by an imbalance between reactive oxygen species (ROS) and antioxidants (Preiser, 2012). Excessive oxidative stress, shown as increased levels of oxidation biomarkers, has been incriminated in several pathological conditions, including neurodegenerative diseases (Preiser, 2012). Here we showed increased oxidative stress in the brain of PICK1−/− mice and in the serum of a PICK1-mutant patient. The age-dependent oxidation was caused by reduced glutathione (GSH) content in neurons. The GSH dysregulation was mediated by the decrease of surface type 3 glutamate transporter excitatory amino acid carrier 1 (EAAC1), which was associated with defective activity of Rab11. Our results suggest that PICK1 is a crucial regulator in oxidative stress and plays important roles in neurodegenerative diseases.
Materials and Methods
Animals.
All experiments were approved by the Animal Experimentation Ethics Committee of Zhejiang University and were designed specifically to minimize the number of animals used. Original breeding pairs of PICK1−/− mice were obtained from Jun Xia (Hong Kong University of Science and Technology, Hong Kong, China) and maintained at the Experimental Animal Center of Zhejiang University. Mice were kept under temperature-controlled conditions on a 12 h light/dark cycle with food and water available ad libitum. In vivo experiments were done in a batch of mice of the same sex.
Antibodies and reagents.
Antibody against 4-hydroxy-2-nonenal (4-HNE) was from Abcam. Antibodies to AMP-activated protein kinase (AMPK), phosphorylated AMPK at Thr172 (pAMPK), voltage-dependent anion channel (VDAC), myc tag, and Rab11 were from Cell Signaling Technology. Antibody to EAAC1 (for Western blotting) was from Alpha Diagnostic. Antibodies to nitrotyrosine, microtubule-associated protein-2 (MAP-2), EAAC1 (for immunostaining), glial fibrillary acid protein (GFAP), and β-actin were from Millipore. Antibody to flotillin-1 was from BD Biosciences. Antibody to ionized Ca2+-binding adapter molecule 1 (Iba1) was from Wako. Mouse anti-PICK1 antibody was from NeuroMab. Rabbit polyclonal anti-PICK1 antibody, which was against the N-terminal 29 aa of mouse PICK1, was homemade according to Cao et al. (2007). Anti-GluA2 antibody was a gift from Richard Huganir (Johns Hopkins University, Baltimore, MD). Horseradish peroxidase-conjugated secondary antibodies were from Thermo Fisher Scientific. The terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) kit was from Roche. IgG antibody, Alexa Fluor-conjugated secondary antibodies, 4′,6-diamidino-2-phenylindole (DAPI), 5,6-carboxy-2′,7′-dichlorofluorescein diacetate (DCFDA), Neurobasal medium, Alexa Fluor 488 C5-maleimide (maleimide), and B27 supplements were from Invitrogen. The protease inhibitor mixture was from Merck. Other chemicals were from Sigma unless stated otherwise.
Neuronal culture.
Primary neuronal culture was performed based on our previous work (Zhu et al., 2012). In brief, cortices were dissected from Sprague Dawley rats (embryonic day 18) or mice (embryonic day 16). Dissociated neurons were plated onto dishes at a density of 2000–3000 cells/cm2 for immunostaining or 7000–10,000 cells/cm2 for biochemical experiments. Cell density was measured using an inverted phase-contrast microscope (Olympus) before experiments. Neurons were cultured in Neurobasal medium supplemented with 2% B-27 and 2 mm l-alanyl-glutamine (NM medium). At 48 h after plating, primary cultures were treated with 2.5 μm AraC for 24 h and replaced with fresh NM medium. Cultures were maintained at 37°C in a humidified incubator gassed with 95% O2 and 5% CO2. The culture medium was renewed every 3 d.
Lentivirus transfection.
Lentivirus encoding scrambled shRNA, PICK1–shRNA, PICK1, or dominant-negative Rab11 (Rab11-DN, Rab11-S25N; Ren et al., 1998) was prepared by GenePharma. The sequence for PICK1 shRNA was 5′-GCC TCA CCA TCA AGA AGT ACC-3′ (Anggono et al., 2011). Overexpressed PICK1 was based on the coding sequence of rat PICK1 gene (GenBank accession number AF542094.1). The cDNA encoding Rab11 (GenBank accession number BC010722) was amplified by PCR. Rab11-DN was generated by PCR-based site-specific mutagenesis and fused with a myc tag for detection. All sequences were verified by DNA sequencing. PICK1 shRNA and scrambled shRNA were driven by the U6 promoter, whereas overexpressed PICK1 and Rab11-DN were driven by the elongation factor 1α promoter. pVSV-G plasmid driven by the CMV promoter expressed the envelope glycoprotein of vesicular stomatitis virus and thus replaced the lentiviral env gene. All virus-packed plasmids were transduced into cultures at 20 multiplicities 4 d before experiments. Only when >90% of cultured neurons were transfected, which was confirmed by mCherry fluorescence, experiments were continued.
Preparation of plasma membrane fractions.
Mouse cortices were homogenized gently in 1 ml of buffer (in mm: 250 sucrose, 3.3 CaCl2, 10 KCl, 5 MgCl2, 1 EDTA, 1 dithiothreitol, and 50 Tris-HCl, pH 7.2) supplemented with the protease inhibitor. The homogenate was centrifuged at 1200 × g for 10 min at 4°C, and the supernatant was centrifuged again at 8000 × g for 10 min at 4°C. The supernatant was further centrifuged at 105,000 × g for 1 h at 4°C, and the precipitant was used for Western blotting.
Surface biotinylation.
Cultured neurons were rinsed with cold PBS containing 1 mm MgCl2 and 0.5 mm CaCl2 and then incubated with 1 mg/ml sulfo-NHS-LC-biotin (Thermo Fisher Scientific) for 30 min at 4°C. Cells were quenched with 100 mm glycine for 15 min at 4°C, lysed in ice-cold RIPA buffer (50 mm Tris-HCl, 0.1% SDS, 0.5% sodium deoxycholate, and 1% Triton X-100), and centrifuged for 20 min at 4°C. The BCA assay (Thermo Fisher Scientific) was used to measure the concentration of proteins in the supernatant, which was next incubated with NeutrAvidin beads (Thermo Fisher Scientific) for 2 h at 4°C. Biotinylated proteins were extracted using 2× SDS sample buffer supplemented with 50 mm dithiothreitol for 20 min at 50°C before Western blotting.
Coimmunoprecipitation.
The coimmunoprecipitation (Co-IP) experiments were performed according to previous work (Wang et al., 2013). Mouse cortices were lysed in RIPA buffer plus the protease inhibitor. The protein concentration was measured using the BCA assay after centrifugation at 16,000 × g at 4°C for 10 min. Decimus supernatant was used for input, and the remainder was for IP. Precleared solubilized preparations were incubated with mouse anti-PICK1 antibody, which was precoupled to protein A-Sepharose beads (GE Healthcare) at 2–4 μg antibody/1 ml beads for 2 h in 50 mm Tris-HCl. Proteins on the beads were extracted with 2× SDS sample buffer and boiled for 5 min before Western blotting.
Western blotting.
Western blotting was performed as reported previously (Wang et al., 2013). In brief, equal amounts of proteins were loaded and fractionated on 10% SDS-polyacrylamide gels, transferred to PVDF membrane (Millipore), immunoblotted with antibodies, and visualized by enhanced chemiluminescence (Pierce Biotechnology). The primary antibody dilutions used were nitrotyrosine (1:1000), 4-HNE (1:1000), PICK1 (rabbit antibody; 1:2000), pAMPK (1:1000), AMPK (1:1000), VDAC (1:1000), GluA2 (1:2000), EAAC1 (1:2000), Rab11 (1:1000), flotillin-1 (1:10,000), myc (1:1000), β-actin (1:10,000), and secondary antibodies (1:10,000). Film signals were scanned digitally and quantitated using NIH ImageJ 1.42q.
Real-time PCR.
The mRNA levels were assessed by RT-PCR using Bio-Rad CFX96 real-time detection system. In brief, cDNA was synthesized by reverse transcription using oligo-dT18 as the primer and proceeded to real-time PCR with gene-specific primers in the presence of SYBR Premix Ex Taq (Takara). Quantification was performed using the comparative cycle threshold method with β-actin as the internal control. The forward and reverse primers were used: interleukin 1β (IL1β), forward, 5′-CTG GTG TGT GAC GTT CCC ATTA-3′ and reverse, 5′-CCG ACA GCA CGA GGC TTT-3′; IL6, forward, 5′-TTC CAT CCA GTT GCC TTC TTG-3′ and reverse, 5′-TTG GGA GTG GTA TCC TCT GTG A-3′; interferon γ (IFNγ), forward, 5′-TGC TGA TGG GAG GAG AGA TGT CT-3′ and reverse, 5′-TTT CTT TCA GGG ACA GCC TGT T-3′; and β-actin, forward, 5′-GGC TGT ATT CCC CTC CAT CG-3′ and reverse, 5′- CCA GTT GGT AAC AAT GCC ATG T-3′. The amplification efficiency and specificity were confirmed before RT-PCR assay.
TUNEL assay.
TUNEL assays were performed according to the instructions of the manufacturer (Roche). In brief, cultured neurons (DIV9) were exposed to H2O2 (200 μm) or glutamate (50 μm) for 30 min and incubated in fresh medium for 3.5 h. Cells were subsequently fixed in 4% paraformaldehyde for 30 min at room temperature and then permeabilized with 0.1% Triton X-100 and 0.1% sodium citrate for 2 min on ice. After washing with PBS, samples were incubated with TUNEL reaction mixture containing TdT in a humidified 37°C chamber for 1 h, mounted using ProLong Gold Antifade Reagent with DAPI (Invitrogen), and photographed using a confocal microscope (Olympus).
ROS measurement and DCF imaging.
Neurons (DIV9) were used for measuring ROS as described previously (Valencia and Morán, 2001; Li et al., 2010). Neurons were incubated with 10 μm DCFDA at 37°C for 30 min. After incubation, the neurons were washed in prewarmed PBS and photographed using a confocal microscope (Olympus) immediately. Images were acquired through a 40× water-immersion objective (numerical aperture, 0.8) using 488 nm excitation and 515 nm emission for the green signal, and 543 nm excitation and 572 nm emission for the red signal. All parameters used in confocal microscopy, including the laser excitation power, detector and offset gains, and the pinhole diameter, were consistent in each experiment. The measurement of DCF imaging was according to Li et al. (2010). In brief, each field with at least two neurons was taken for imaging. After adjustment of the focus on cell body or neurite, images were immediately captured. At least four different fields were measured per culture dish and used for quantification with NIH ImageJ. Somata and neurites were manually tracked by tracing their edges. A neurite was determined by criteria including the explicit focus, a clear origin point from the soma, and separation from other processes. The pixels in the region of interest (ROI) and the area adjacent to the ROI were measured in somata or neurites. The fluorescence intensity of each ROI was calculated by subtracting the intensity in the adjacent area. The imaging data analysts were blind to experimental conditions until the data were integrated.
Manipulation of GSH content in mice.
Buthionine sulfoximine (BSO; 0.66 mg/g body weight) was injected intraperitoneally into 2-month-old wild-type (WT) and PICK1−/− mice twice a day for 4 d. N-Acetylcysteine (NAC; 0.15 mg/g body weight) was injected intraperitoneally into 4-month-old WT and PICK1−/− mice as a single daily dose for 4 consecutive weeks. In some mice, NAC injection was accompanied by BSO to investigate their combined action.
GSH measurement.
The GSH levels were measured using the GSH reductase method and maleimide imaging (Aoyama et al., 2006). In the GSH reductase method, mouse cortices were homogenized with ice-cold 5% sulfosalicylic acid and centrifuged at 1200 × g for 15 min. The supernatant was diluted with PBS containing 1 mm EDTA, pH 7.0, and supplemented with the same volume of reaction mixture (in mm: 1 EDTA, 0.3 5,5′-dithiobis-(2-nitrobenzoic acid), and 0.4 NADPH) plus 2 IU/ml GSH reductases to measure GSH content. Fluorescent maleimide imaging was performed to assess GSH content in situ, because GSH is the dominant species of reactive thiols (Aoyama et al., 2006). In brief, sections were washed with PBS, fixed in 4% paraformaldehyde, and incubated overnight at 4°C with 2.5 μm maleimide in PBS containing 2% goat serum, 0.2% Triton X-100, and 0.1% bovine serum albumin (BSA). The sections were photographed using a confocal microscope (Olympus). The signal intensity was measured in the cell-body layer of each section using an ROI of constant size. Fluorescent intensity was analyzed using NIH ImageJ.
Immunohistochemistry and immunocytochemistry.
The acquirement and analysis of immunostaining in cultured cells and slices were according to our previous work (Zhu et al., 2012; Xie et al., 2015). Coronal sections were cut at 30 μm and placed in blocking solution (1% BSA, 0.3% Triton X-100, and 10% normal goat serum) for 1 h at room temperature. After washing with PBS, sections were incubated with primary antibodies overnight at 4°C and then with secondary antibodies for 1 h at room temperature. The secondary antibodies Alexa Fluor 488-conjugated goat anti-rabbit IgG and/or Alexa Fluor 594-conjugated goat anti-mouse IgG were diluted at 1:1000. The sections were mounted using ProLong Gold Antifade Reagent with DAPI (Invitrogen). Cultured cells were fixed with 4% paraformaldehyde for 15 min at room temperature, washed with PBS and permeabilized in 0.2% Triton X-100 for 10 min, blocked in 10% BSA for 1 h, and labeled with primary antibodies overnight at 4°C. Cells were then incubated with a 1:1000 dilution of Alexa Fluor 488-conjugated goat anti-rabbit IgG and/or a 1:1000 dilution of Alexa Fluor 594-conjugated goat anti-mouse IgG for 1 h at room temperature. The primary antibody dilutions used for immunohistochemistry and immunocytochemistry were nitrotyrosine (1:200), MAP-2 (1:1000), Iba1 (1:1000), GFAP (1:1000), EAAC1 (1:200), and Rab11 (1:100). All antibodies were diluted in PBS containing 1% BSA and 1% normal goat serum. Microscopy was performed using 20× (numerical aperture, 0.75 for sections) or 60× (numerical aperture, 1.35 for cells) oil-immersion objectives.
Cysteine uptake assay.
l-[14C]cysteine (20 μCi/ml; PerkinElmer Life and Analytical Sciences) uptake was performed at room temperature for 30 s in PBS containing 1% BSA according to previous work (Li et al., 2010). The final concentrations of l-[14C]cysteine were set as 0.1, 1, 5, 10, and 50 μm to study the uptake efficiency curve. Dithiothreitol (25 mm) was added to work solutions for the radioactivity detection in cell lysates. After uptake experiments, neurons were washed three times in cold PBS and lysed in 0.2 ml of cold 50 mm HEPES, pH 7.4, 100 mm KCl, 2 mm EDTA, and 1 mm dithiothreitol containing 1% Triton X-100 on ice for 15 min. Lysates (20 μl) were used for scintillation count in triplicate.
Mitochondrial respiration assay.
Real-time oxygen consumption rate (OCR) was measured on a Seahorse XF96 extracellular flux analyzer (Seahorse Bioscience) according to the instructions of the manufacturer and a previous report (Yu et al., 2014). Cultured cortical neurons were seeded in 96-well FluxPaks (Seahorse Bioscience) at a density of 10,000 cells per well. Cells (DIV9) were first incubated in Seahorse assay medium supplemented with 25 mm glucose, 1 mm sodium pyruvate, and 2 mm l-alanyl-glutamine, and we adjusted the pH to 7.4. Plates were equilibrated in this medium for 30 min without CO2. OCR measurements were then taken under a basal condition, and, after the addition of oligomycin (1 μm), fluoro-carbonyl cyanide phenylhydrazone (FCCP; 0.5 μm), rotenone (1 μm), and antimycin A (5 μm) sequentially. Non-mitochondrial OCR was determined as the OCR after antimycin A and rotenone treatment. Basal OCR was determined as OCR before oligomycin minus non-mitochondrial OCR. ATP-lined OCR was determined as OCR before oligomycin minus OCR after oligomycin. Maximal OCR was determined as the OCR after FCCP minus non-mitochondrial OCR (Yu et al., 2014). OCR values were normalized to the protein level in each well. The mean of six replicate wells per experiment was obtained across individual experiments.
Clinical information, blood sampling, and ELISA measurement of nitrotyrosine.
The human study was approved by the Ethics Committees of the Hospital of Central South University (Changsha, China) and Zhejiang Provincial People's Hospital (Hangzhou, China) and was conducted in conformance with the policies and principles contained in the Federal Policy for the Protection of Human Subjects and in the Declaration of Helsinki. Informed consents were given by all participants. The patient with a homozygous PICK1 mutation (33 years old) was the same Chinese man reported previously, who carries a missense mutation (G393R) in the C terminal of PICK1 (Liu et al., 2010). This homozygous patient does not show any evident physiological defects or inflammatory disease other than globozoospermia (Liu et al., 2010). As controls, four unrelated and healthy Chinese men (30–35 years old) were genotyped by sequencing exon 13 of the PICK1 gene. Blood samples (5 ml taken into EDTA by venipuncture) were obtained from the PICK1-mutant patient in Xiangya Hospital and from controls in Zhejiang Provincial People's Hospital at ∼10:00 A.M. Samples were collected from the PICK1-mutant patient once a week, four times to minimize systematic errors. Immediately after collection, the blood was centrifuged and the serum was collected and stored at −80°C until ELISA. The levels of nitrotyrosine were measured using an ELISA kit (Cell Biolabs), according to the instructions of the manufacturer.
Statistical analysis.
Data analysis was performed using Excel 2003 (Microsoft), Igor Pro 6.0 (Wavemetrics), and SPSS 16.0 statistical program (SPSS). SDs for control groups were calculated according to the average of all control data points (Lim et al., 2014). Statistical differences were determined using unpaired two-sided Student's t test for two group comparison or one-way ANOVA test followed by Tukey's post hoc test for multiple comparisons. The accepted level of significance was p < 0.05. n represents the number of animals or cultures tested. Data in the text and figures are presented as mean ± SEM.
Results
Increased oxidative stress in PICK1−/− mice
Neural tissue oxidation was examined in 4-month-old WT and PICK1−/− mice by Western blotting using antibodies against nitrotyrosine and 4-HNE, which are formed by oxidant interactions with proteins and lipids, respectively (Zigdon et al., 2013). We found that levels of nitrotyrosine and 4-HNE were increased markedly in multiple regions of PICK1−/− mouse brain, such as the cortex, hippocampus, and cerebellum (Fig. 1A), implying that the level of oxidative stress proteins related to nitrotyrosine or 4-HNE is increased. The increments appeared similar for nitrotyrosine and 4-HNE in the cortex, hippocampus, and cerebellum (Fig. 1B). Immunocytochemical staining showed an increased nitrotyrosine signal in the hippocampal CA3 and forebrain of PICK1−/− mice, but this signal was negligible in the CA1 area (Fig. 1C). These results clearly indicated increased oxidation in PICK1−/− mice, which has not been reported yet. We next examined the age dependence of this oxidation because previous work has shown that oxidative stress is age dependent (Preiser, 2012). Using Western blotting, we found that both nitrotyrosine and 4-HNE were barely detectable in WT and PICK1−/− mice at 1 month (Fig. 1D). However, PICK1−/− mice of all ages displayed significantly increased nitrotyrosine and 4-HNE compared with the corresponding WT mice at the same age from 2 to 12 months, and the increments reached a maximum at 12 months (Fig. 1D). These data indicated that the increased oxidation in PICK1−/− mice is age dependent.
Figure 1.
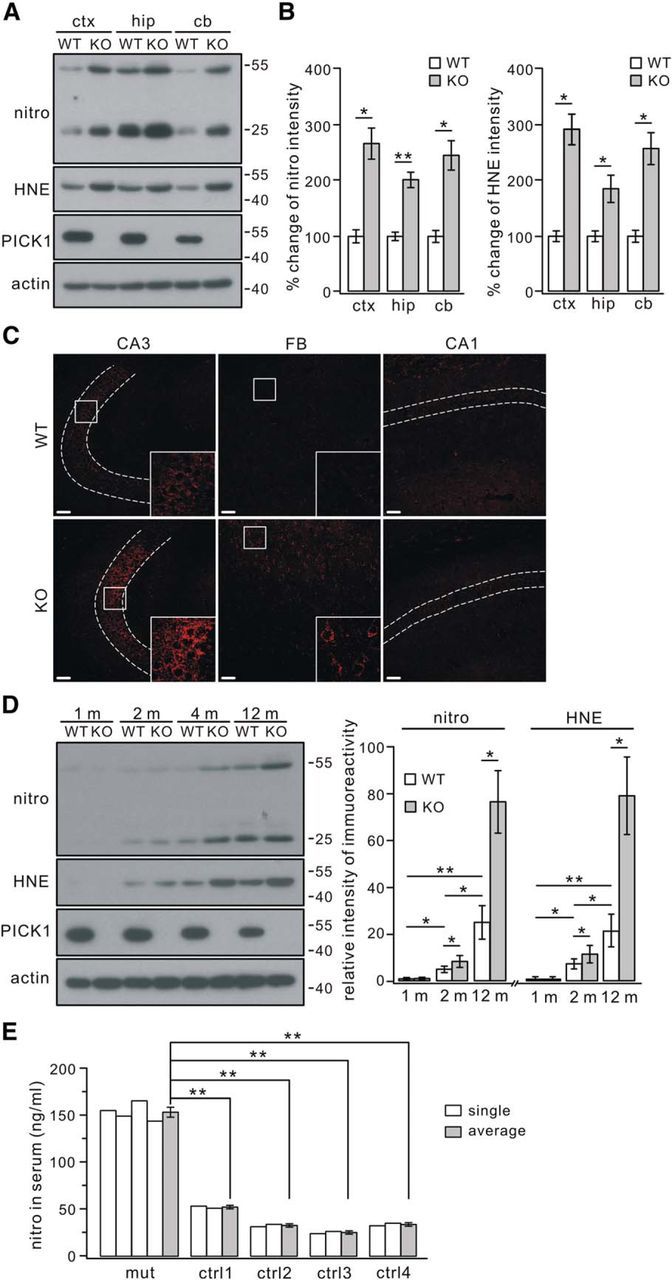
Increased oxidative stress in PICK1−/− mouse brain. A, Total proteins extracted from 4-month-old WT and PICK1−/− (KO) mouse cortex (ctx), hippocampus (hip), and cerebellum (cb) were probed with antibodies to nitrotyrosine (nitro), 4-HNE (HNE), PICK1, and β-actin (actin). B, Levels of nitrotyrosine and 4-HNE intensity were normalized to the corresponding β-actin level, and percentage changes relative to corresponding WT are shown in the histograms: nitro, 100 ± 10% (WT, ctx), 270 ± 26% (KO, ctx), 100 ± 6% (WT, hip), 201 ± 12% (KO, hip), 100 ± 9% (WT, cb), 248 ± 25% (KO, cb); HNE, 100 ± 8% (WT, ctx), 295 ± 25% (KO, ctx), 100 ± 8% (WT, hip), 186 ± 22% (KO, hip), 100 ± 9% (WT, cb), 249 ± 27% (KO, cb). n = 4 pairs. C, Immunoreactivity for nitrotyrosine in the CA3, forebrain (FB), and CA1 from 4-month-old WT and KO mice. Dashed lines show pyramidal cell layers. The higher magnification of areas in the white boxes shows the different staining intensity in CA3 and FB areas between WT and KO mice, but no difference was found in CA1 pyramidal layers. Scale bars, 50 μm. D, Expression of nitrotyrosine and 4-HNE in WT and KO cortices at 1, 2, 4, and 12 months. Signal intensity ratios (nitrotyrosine/actin) were as follows: WT, 1.4 ± 0.5% (1 month), 5.5 ± 1.4% (2 months), and 25.4 ± 7.1% (12 months); KO, 1.5 ± 0.5% (1 month), 8.8 ± 2.5% (2 months), and 76.8 ± 13.3% (12 months). Signal intensity ratios (4-HNE/actin) were as follows: WT, 1.2 ± 0.4% (1 month), 7.8 ± 1.8% (2 months), and 21.7 ± 6.6% (12 months); KO, 1.2 ± 0.4% (1 month), 11.9 ± 3.4% (2 months), and 79.4 ± 16.2% (12 months). n = 4 pairs. Note that levels of both nitrotyrosine and 4-HNE in WT mice were significantly increased with age. E, Concentrations of nitrotyrosine in serum samples from controls (ctrl) and PICK1-mutant patient (mut). “single” indicates each ELISA test performed in one subject. All tests from the same subject were averaged and labeled as “average.” Averaged concentrations of nitrotyrosine were 154 ± 5 ng/ml (mut, n = 4), 53 ± 1 ng/ml (ctrl1, n = 2), 34 ± 1 ng/ml (ctrl2, n = 2), 26 ± 1 ng/ml (ctrl3, n = 2), and 35 ± 1 ng/ml (ctrl4, n = 2). *p < 0.05, **p < 0.01.
Previous work reported a globozoospermia-suffering Chinese patient who carries a homozygotic mutation in the PICK1 gene (Liu et al., 2010). Therefore, we investigated whether the serum level of nitrotyrosine is changed in this patient based on previous work demonstrating that the presence of nitrotyrosine in plasma and serum is a good indicator of oxidative stress induced by PD and inflammatory diseases (Seet et al., 2010; Codoñer-Franch et al., 2011; Fernández et al., 2013). ELISA tests showed that the nitrotyrosine concentration in the serum from the homozygous patient were significantly higher than controls (Fig. 1E), suggesting that this patient may suffer from increased oxidation. However, it should be noted that more tests are needed for an accurate evaluation of oxidative damage in this patient.
Oxidation is localized in neurons and accompanied by microglial activation in PICK1−/− mice
We next examined the cellular localization of nitrotyrosine using the neuronal marker MAP-2, the microglial marker Iba1, and the astrocytic marker GFAP. We found that the immunostaining of nitrotyrosine was localized mainly to CA3 neurons of 4-month-old PICK1−/− mice (Fig. 2A). In contrast, there was almost no colocalization of nitrotyrosine with Iba1 or GFAP (Fig. 2A). Thus, it was concluded that the oxidative stress occurs in neurons.
Figure 2.

Oxidative stress in neurons and activated microglia in PICK1−/− mice. A, Representative nitrotyrosine immunostaining (nitro) in the CA3 area of 4-month-old PICK1−/− (KO) mice, showing that it only colocalized with MAP-2. White arrows indicate the colocalization of nitrotyrosine and MAP-2 (n = 4 pairs). Scale bars, 50 μm. B, Representative staining with DAPI and TUNEL in cortices from 12-month-old WT and KO mice. No apparent TUNEL signal was found in these cortices (n = 4 pairs). Scale bars, 50 μm. C, Iba1 immunostaining shows that microglia were much activated in 12-month-old KO cortex. The higher magnification of areas in the white boxes shows the distinct morphology of microglia between WT and KO cortices. Scale bars, 50 μm. D, The percentage changes of Iba1-positive cell numbers relative to WT were 100 ± 8% (WT) and 264 ± 51% (KO) (n = 4 pairs). E, Ratios of IL1β, IL6, and IFNγ mRNA to β-actin mRNA from WT and KO mouse cortex were calculated, and the percentage changes relative to corresponding WT are shown in the histograms: 100 ± 7% (WT, IL1β), 286 ± 14% (KO, IL1β), 100 ± 5% (WT, IL6), 190 ± 5% (KO, IL6), 100 ± 11% (WT, IFNγ), and 201 ± 21% (KO, IFNγ) (n = 4 pairs). *p < 0.05, **p < 0.01.
Oxidative stress is known to be an early indication of neuronal degeneration and dysfunction (Moreira et al., 2005; Preiser, 2012). To assess potential neurodegeneration, cryosections of cortices from 12-month-old WT and PICK1−/− mice were subjected to TUNEL tests. Neurons in sections from PICK1−/− mice did not display obvious TUNEL signal (Fig. 2B), suggesting that oxidative stress does not result in gross neurodegeneration at this stage. Injured neurons cause resting microglia to become activated (Streit et al., 1999). Thus, the activation of microglia was examined using Iba1 staining in the cortices of WT and PICK1−/− mice of 12 months. Our results indicated that microglia were activated in these mice, as shown by increased somal size and hypertrophic processes (Fig. 2C; Mosher and Wyss-Coray, 2014) and increased numbers of Iba1-stained cells (Fig. 2D). Quantitative RT-PCR indicated that the inflammatory factors IL1β, IL6, and IFNγ, which are released along with microglial activation (Mosher and Wyss-Coray, 2014), were increased remarkably (Fig. 2E). Despite these observations, no obvious phagocytic microglial cells with rounded macrophage-like morphology were found at this stage, indicating that PICK1−/− mice are not undergoing neuronal death at the stage we examined (Streit et al., 1999).
Increased ROS and vulnerability to oxidants in PICK1−/− neurons
Oxidative stress is caused by excessive production of ROS (Noh and Ha, 2011). Using DCFDA, a cell-permeable nonfluorescent dye that is oxidized into green fluorescent DCF in the presence of oxidants (Kirkland et al., 2007; Li et al., 2010), two experiments were undertaken to measure ROS level in PICK1-deficient neurons. First, cortical neurons from WT and PICK1−/− mice were cultured and labeled with DCFDA at DIV9. We found that DCF signals in the somata and neurites were increased remarkably in PICK1−/− neurons (Fig. 3A,B). Second, cultured rat cortical neurons (DIV5) were transduced with scrambled shRNA or PICK1–shRNA and stained with DCFDA 4 d later. Although PICK1–shRNA knockdown effectively suppressed the expression of PICK1 in cultured neurons (Fig. 3C), quantification of the fluorescence intensity revealed that DCF signals in somata and neurites were both increased compared with controls (Fig. 3D,E). These experiments suggested that redox imbalance occurs when PICK1 expression is suppressed.
Figure 3.
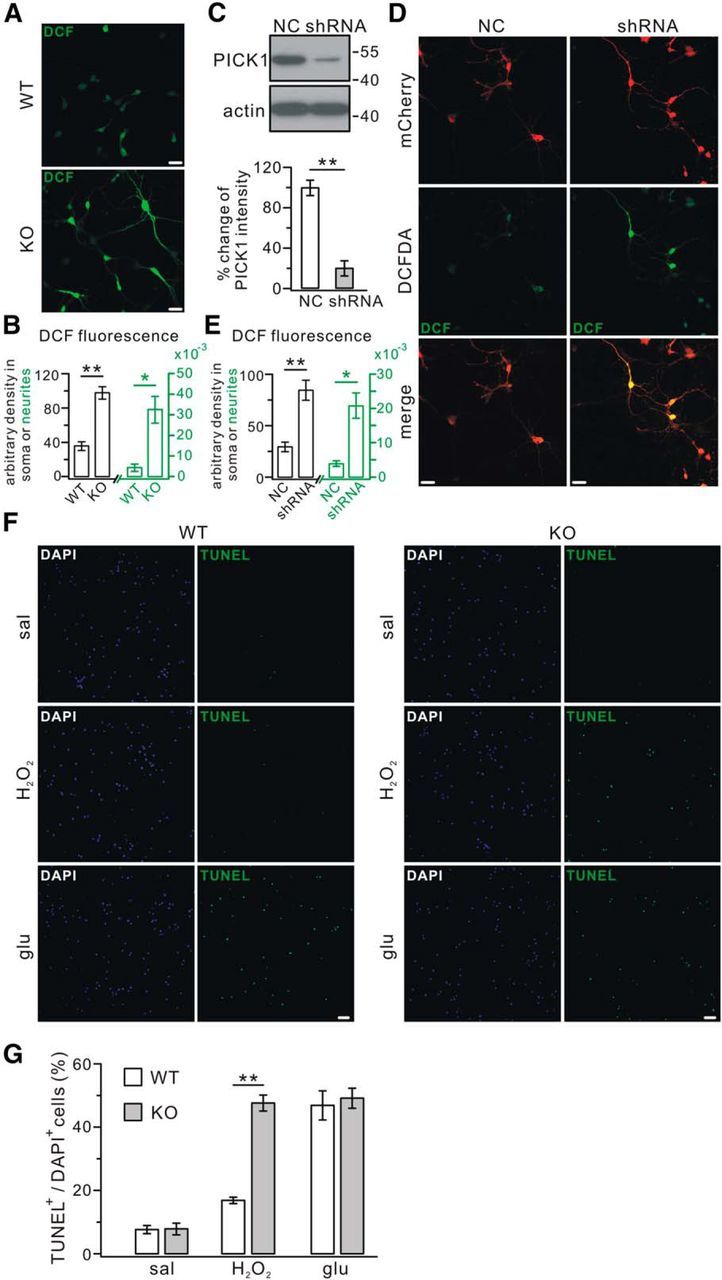
Assessment of ROS and cell vulnerability. A, Cortical neurons (DIV9) from embryonic WT and PICK1−/− (KO) mice were cultured and stained with DCFDA. Confocal imaging showed that KO neurons showed significantly stronger DCF signals. Scale bars, 25 μm. B, Average fluorescent DCF intensity in the soma was 36.4 ± 5.0 (WT, n = 28) versus 97.7 ± 7.2 (KO, n = 24). Average fluorescence intensity in the processes (per micrometers) was 0.0047 ± 0.0018 (WT, n = 28) versus 0.033 ± 0.0065 (KO, n = 24). Note that axes and labels for soma and neurites are distinguished in black and green, respectively. C, Cortical neuronal cultures (DIV5) were transfected with mCherry-tagged scrambled shRNA (NC) or mCherry-tagged PICK1 shRNA (shRNA), and lysates were probed with antibodies against PICK1 and β-actin (actin) 4 d after the transfection (DIV9). PICK1 levels were normalized to β-actin levels, and the percentage changes relative to NC are plotted in the bottom (NC, 100 ± 7%; shRNA, 20 ± 7%; n = 5). D, Virus-transfected neurons (DIV9) were treated with DCFDA to assess ROS level. NC neurons had low DCF fluorescence, whereas shRNA neurons had much brighter DCF fluorescence. Scale bars, 25 μm. E, Average fluorescence intensity in the soma was 30.1 ± 4.6 (NC, n = 28) versus 85.2 ± 9.8 (shRNA, n = 24). Average fluorescence intensity in the processes (per micrometers) was 0.0041 ± 0.0008 (NC, n = 28) versus 0.021 ± 0.0037 (shRNA, n = 24). Note that axes and labels for soma and neurites are distinguished in black and green, respectively. F, Cell vulnerability was identified by TUNEL staining in cultured cortical WT and KO neurons (DIV9). Staining with DAPI and TUNEL was performed at 4 h with saline (sal) or 3.5 h after 30 min incubation with H2O2 (200 μm) or glutamate (50 μm; glu). Scale bars, 50 μm. G, Ratios of TUNEL-positive/DAPI-positive cells were as follows: sal, 8.0 ± 1.0% (WT) and 8.2 ± 1.6% (KO); H2O2, 17.2 ± 0.7% (WT) and 47.6 ± 2.3% (KO); glu, 47.3 ± 4.3% (WT) and 49.5 ± 2.9% (KO) (n = 5). *p < 0.05, **p < 0.01.
To evaluate the vulnerability of neurons, cultured cortical neurons from WT and PICK1−/− mice were exposed to the oxidant H2O2, and TUNEL assays were performed. Our results showed that PICK1−/− neurons were several times more sensitive to H2O2 than WT neurons (Fig. 3F,G), suggesting that neurons are more vulnerable in the condition of PICK1 deficiency. To further evaluate the vulnerability, WT and PICK1−/− neurons were also exposed to glutamate, which increases cell vulnerability via neurotoxicity (Trotti et al., 1998). Our results showed that the application of glutamate induced a comparable increase of TUNEL signal in both WT and PICK1−/− neurons (Fig. 3F,G), suggesting that the change in surface glutamate receptors or scaled synaptic transmission caused by PICK1 deficiency (Anggono et al., 2011) does not result in excitotoxicity. Together, these data indicated that the vulnerability specific to oxidants is increased in PICK1−/− neurons.
GSH is reduced in PICK1−/− mice
We next studied how ROS was increased in PICK1−/− mice. Several experiments were used to examine the functions of mitochondria, which is involved in ROS production (Starkov, 2008). First, the total expression of AMPK and its phosphorylation at Thr172 (pAMPK) were examined because the phosphorylation of AMPK is an energy indicator that senses the change of the AMP/ATP ratio (Hardie et al., 2012). Western blots showed that pAMPK was not changed in 4-month-old PICK1−/− mice (Fig. 4A). Second, the expression of VDAC, which is critical to mitochondrial function (De Stefani et al., 2012), was not altered in 4-month-old PICK1−/− mice (Fig. 4B). Third, mitochondrial respiration function was assessed in cultured WT and KO cells (DIV9) by measuring OCR parameters under different respiratory conditions. Our results showed that the progress curves for WT and knock-out (KO) cells were very similar (Fig. 4C), and no difference was found in the values of basal OCR, ATP-linked OCR, and maximal OCR between WT and KO cells (Fig. 4D), implying that the respiratory function is not influenced by the loss of PICK1. Together, these data suggested that the increment of ROS may not be related to mitochondria-mediated energy metabolism.
Figure 4.
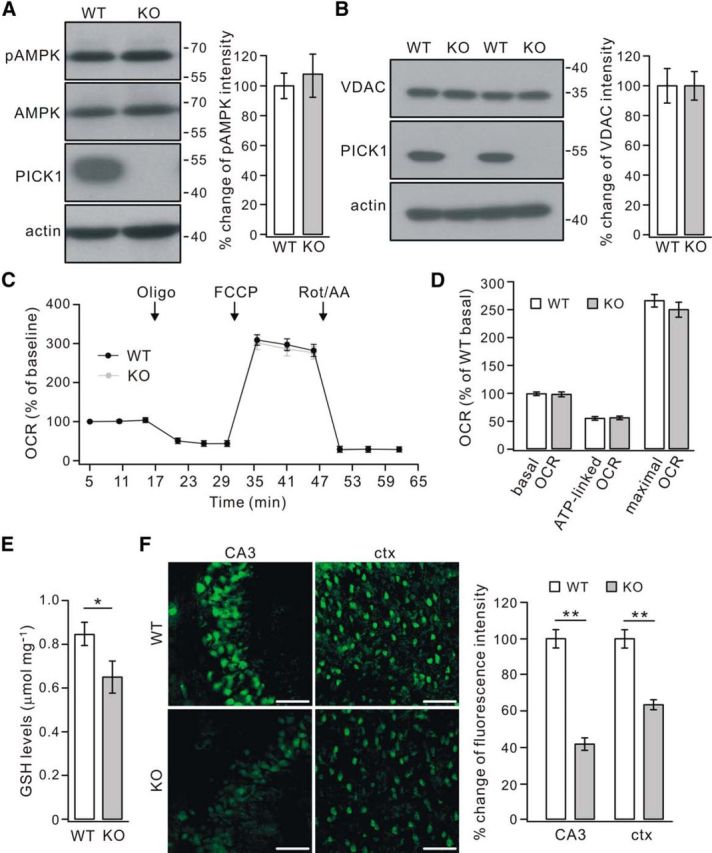
Reduced GSH in PICK1−/− mice. A, Representative bands immunoblotted with antibodies against pAMPK, AMPK, PICK1, and β-actin (actin) in 4-month-old WT and PICK1−/− (KO) mice. Levels of pAMPK and AMPK intensity were normalized to β-actin. The percentage changes of pAMPK/AMPK ratio relative to WT are shown in the histogram: 100 ± 8% (WT) and 106 ± 14% (KO) (n = 5 pairs; p > 0.05). B, Representative bands immunoblotted with antibodies against VDAC, PICK1, and β-actin (actin). Levels of VDAC intensity were normalized to β-actin, and the percentage changes relative to WT are shown in the histogram: 100 ± 11% (WT) and 100 ± 9% (KO) (n = 5 pairs; p > 0.05). C, Representative OCR measurement in WT and KO cortical cultures (DIV9) under the basal condition followed by sequential addition of oligomycin (Oligo), FCCP, and rotenone plus antimycin A (Rot/AA). All OCR values were normalized to protein level and shown as the percentage changes of WT and KO baseline, respectively. Error bars represent averages of six replicate wells. D, Basal OCR, ATP-linked OCR, and maximal OCR values in WT and KO neurons were expressed as percentage changes of averaged WT basal OCR value (n = 4 experiments). The percentage changes were as follows: basal OCR, 100 ± 2% (WT), 99 ± 3% (KO); ATP-linked OCR, 56 ± 2% (WT), 57 ± 2% (KO); maximal OCR values, 267 ± 10% (WT), 251 ± 12% (KO) (n = 4). E, GSH levels in the cortex from 4-month-old WT and KO mice. The average concentrations of GSH in micromoles per milligram protein were 0.85 ± 0.05 (WT) and 0.65 ± 0.07 (KO) (n = 5 pairs). F, Reactive thios were evaluated in the CA3 and cortex (ctx) using maleimide fluorescence. Fluorescence was reduced in the neurons in KO sections relative to those in WT sections. Scale bars, 50 μm. Percentage changes of maleimide fluorescent intensity relative to WT were as follows: 100 ± 3% (WT, CA3), 42 ± 2% (KO, CA3), 100 ± 3% (WT, ctx), and 63 ± 3% (KO, ctx) (n = 5 pairs). *p < 0.05, **p < 0.01.
Alternatively, the ROS accumulation might be attributable to the depletion of antioxidants, which quickly remove ROS (Birben et al., 2012). Therefore, we measured the levels of GSH, a major antioxidant in the brain (Dringen, 2000; Birben et al., 2012), in WT and PICK1−/− mice at 4 months. Our results showed that PICK1−/− mice had significantly lower levels of GSH than WT mice (Fig. 4E). To confirm the altered GSH homeostasis, staining with maleimide was used to assess the reactive GSH in hippocampal and cortical sections from 4-month-old WT and PICK1−/− mice, according to previous work (Aoyama et al., 2006). The results showed that maleimide fluorescent signal was reduced markedly in PICK1−/− neurons located in the CA3 and cortex (Fig. 4F). Together, these results indicated that GSH is reduced in PICK1−/− mice.
Distinct effects of BSO and NAS in WT and PICK1−/− mice
BSO has been shown to inhibit the formation of GSH (Griffith and Meister, 1979; Aoyama et al., 2006). Given that PICK1−/− mice had reduced GSH (Fig. 4), cell oxidation would be more severe if they were challenged with BSO. Such a study was performed using 4 d injection of BSO in 2-month-old WT and PICK1−/− mice, in which the oxidation was rather mild (Fig. 1D). Western blotting in harvested cortices revealed that nitrotyrosine and 4-HNE were increased in WT and PICK1−/− mice after BSO treatment (Fig. 5A). Nevertheless, the increments were much greater in PICK1−/− mice, as shown by 2.9-fold (nitrotyrosine) and 1.8-fold (4-HNE) increases in WT mice and 3.9-fold (nitrotyrosine) and 2.7-fold (4-HNE) increases in PICK1−/− mice (p < 0.01 for the increments of both nitrotyrosine and 4-HNE in PICK1−/− mice compared with WT mice; Fig. 5A). Likewise, immunohistochemical staining showed that the increase of nitrotyrosine was much greater in the CA3 region from PICK1−/− mice, as shown by a 1.3-fold increase in WT mice and a 2.1-fold increase in PICK1−/− mice (p < 0.01 for the increment of nitrotyrosine fluorescence in PICK1−/− mice compared with WT mice; Fig. 5B). In a parallel study, GSH content was assessed using maleimide staining in cortical slices. We found a strong decrease of maleimide signal in both WT and PICK1−/− sections from mice treated with BSO (Fig. 5C). However, statistics showed that the decrease of fluorescence was greater in PICK1−/− sections (p < 0.05; Fig. 5C). Together, these data again supported that loss of PICK1 leads to more vulnerability to oxidation.
Figure 5.

Opposite roles of BSO and NAC in oxidation and GSH content. A, Saline (sal) or BSO was administered to 2-month-old WT or PICK1−/− (KO) mice twice a day for 4 consecutive d. Lysates were probed with antibodies to nitrotyrosine (nitro), 4-HNE (HNE), PICK1, and β-actin (actin). All protein levels were normalized to actin, and the percentage changes are shown in the histograms: nitro, 100 ± 8% (WT + sal), 161 ± 15% (KO + sal), 293 ± 34% (WT + BSO), and 621 ± 36% (KO + BSO); HNE, 100 ± 6% (WT + sal), 149 ± 11% (KO + sal), 177 ± 17% (WT + BSO), and 399 ± 37% (KO + BSO) (n = 4 pairs; ANOVA test). B, Representative images of nitrotyrosine staining in the CA3 area of 2-month-old WT and PICK1−/− mice treated with either saline or BSO. Scale bars, 50 μm. Percentage changes of nitrotyrosine fluorescence were as follows: 100 ± 5% (WT + sal), 119 ± 9% (KO + sal), 126 ± 14% (WT + BSO), and 248 ± 15% (KO + BSO) (n = 4 pairs; ANOVA test). C, Reactive thios were evaluated using maleimide staining in cortical sections from 2-month-old WT and KO mice. The maleimide signal was decreased in sections from both WT and KO mice treated with BSO (right column). Scale bars, 50 μm. Percentage changes of fluorescent intensities were as follows: 100 ± 4% (WT + sal), 40 ± 4% (WT + BSO), 90 ± 3% (KO + sal), and 22 ± 2% (KO + BSO) (n = 4 pairs; ANOVA test). D, NAC was administered to 4-month-old WT or KO mice. Lysates were probed with antibodies to nitrotyrosine (nitro), 4-HNE (HNE), PICK1, and β-actin (actin). All protein levels were normalized to actin, and percentage changes are shown in the histograms: nitro, 100 ± 6% (WT + sal), 75 ± 7% (WT + NAC), 272 ± 20% (KO + sal), 104 ± 17% (KO + NAC); HNE, 100 ± 2% (WT + sal), 93 ± 3% (WT + NAC), 282 ± 11% (KO + sal), 134 ± 9% (KO + NAC) (n = 4 pairs; ANOVA test). E, Biochemical determination of GSH in cortices from 4-month-old KO mice treated with saline, NAC, BSO, or BSO plus NAC. GSH concentrations were (in μmol/mg protein) 0.63 ± 0.08 (sal), 1.33 ± 0.12 (NAC), 0.35 ± 0.06 (BSO), and 0.53 ± 0.05 (BSO+NAC), indicating that the effect of NAC is attenuated by the concomitant administration of BSO (n = 4 pairs; ANOVA test). *p < 0.05, **p < 0.01.
Cysteine is the rate-limiting substrate for the synthesis of GSH (Dringen, 2000). NAC, a cysteine precursor, passively crosses lipid membranes and thereby provides cysteine to cells for GSH synthesis (Mazor et al., 1996; Parsons and Chipman, 2000). To investigate whether NAC induces an increase in GSH and relieves oxidative stress in PICK1−/− mice, a single dose of NAC was administered daily to 4-month-old mice for 4 weeks, and the expression of nitrotyrosine and 4-HNE was measured afterward. Although nitrotyrosine and 4-HNE were reduced in both WT and PICK1−/− cortex, the degree of reduction was more prominent in PICK1−/− mice (Fig. 5D), indicating that the oxidation in PICK1−/− neurons is primarily attributable to the lack of cysteine.
To ensure that BSO and NAC both acted on GSH, we administered NAC and BSO alone or simultaneously for 4 d in 2-month-old PICK1−/− mice. The GSH content of the cortex showed that the effects of NAC and GSH were opposite: NAC significantly increased GSH but BSO decreased it (Fig. 5E). Furthermore, the NAC-induced increase in GSH content was reversed primarily by BSO when NAC and BSO were coadministered (Fig. 5E).
Surface EAAC1 is reduced in PICK1−/− mice
It has been shown that EAAC1 is the primary route for cysteine uptake (Zerangue and Kavanaugh, 1996), and inhibitors of EAAC1 prevent neuronal GSH synthesis in the presence of extracellular cysteine (Chen and Swanson, 2003; Himi et al., 2003). Therefore, we investigated whether EAAC1 expression is influenced in PICK1−/− mice. Western blots showed that total expression of EAAC1 was not changed in 4-month-old PICK1−/− mice (Fig. 6A). However, the enriched EAAC1 in the purified membrane fraction was decreased significantly in PICK1−/− mice (Fig. 6A). Next, rat cortical neurons were cultured and transduced with scrambled shRNA or PICK1–shRNA (Fig. 6B). Surface expression of EAAC1 was measured at DIV9 using biotinylation, and the results showed that it was much reduced (Fig. 6B). Meanwhile, the surface expression of GluA2 was increased (Fig. 6B), consistent with previous work (Anggono et al., 2011). As in the in vivo study, total EAAC1 was not changed in PICK1-knockdown neurons (Fig. 6B).
Figure 6.
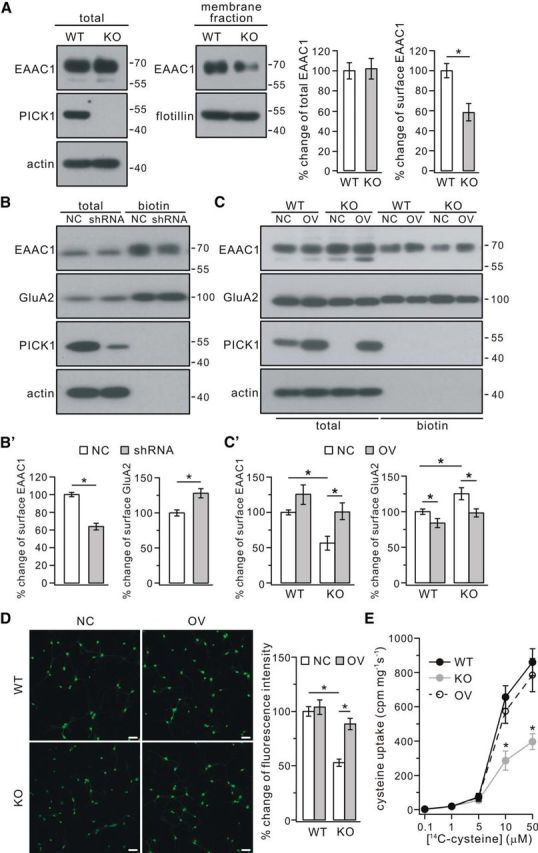
Surface EAAC1 was reduced in PICK1−/− mice. A, EAAC1 in the membrane fraction decreased in PICK1−/− mice (KO) at 4 months. Actin and flotillin were used as loading controls of total and membrane fractions, respectively. Percentage changes of EAAC1 expression were 100 ± 8% (WT, total), 102 ± 10% (KO, total), 100 ± 7% (WT, surface), and 59 ± 9% (KO, surface) (n = 4 pairs; *p < 0.05). B, Cultured cortical neurons (DIV9) were transfected with mCherry-tagged scrambled shRNA (NC) or mCherry-tagged PICK1 shRNA (shRNA). Surface (biotin) EAAC1 and GluA2 were isolated by biotinylation and detected by antibodies to EAAC1, GluA2, PICK1, and β-actin (actin). B′, Percentage changes of surface signal intensities were 100 ± 2% (EAAC1, NC), 65 ± 3% (EAAC1, shRNA), 100 ± 4% (GluA2, NC), and 130 ± 6% (GluA2, shRNA) (n = 4; *p < 0.05). C, Cultured cortical neurons (DIV9) from WT and KO mice were treated with control lentivirus (NC) or lentivirus encoding PICK1 (OV). Surface (biotin) EAAC1 and GluA2 were isolated by biotinylation and detected by antibodies to EAAC1, GluA2, PICK1, and actin. C′, Percentage changes of surface signal intensities were as follows: EAAC1, 100 ± 3% (WT + NC), 126 ± 13% (WT + OV), 55 ± 9% (KO + NC), 101 ± 12% (KO + OV); GluA2, 100 ± 3% (WT + NC), 85 ± 5% (WT + OV), 126 ± 8% (KO + NC), 98 ± 5% (KO + OV) (n = 4; *p < 0.05, ANOVA test). D, Effects of overexpressing PICK1 on GSH content using maleimide staining in cultured cortical neurons from WT and KO mice. Neurons (DIV9) from WT and KO mice were treated with control lentivirus or lentivirus encoding PICK1. The maleimide signal was significantly lower in KO neurons than WT neurons, and expression of PICK1 significantly increased the signal intensity in KO neurons. Scale bars, 50 μm. Percentage changes of fluorescent intensities were as follows: 100 ± 4% (WT + NC), 104 ± 6% (WT + OV), 55 ± 3% (KO + NC), and 89 ± 5% (KO + OV) (n = 4; *p < 0.05, ANOVA test). E, WT, KO, and OV cultured neurons (DIV9) were used for performing cysteine uptake at room temperature for 30 s with l-[14C]cysteine concentrations of 0.1, 1, 5, 10, and 50 μm. The uptaken l-[14C]cysteine in harvested cell lysates (cpm/mg protein/s) with cysteine concentration of 10 and 50 μm were as follows: 10 μm, 657 ± 66 (WT), 286 ± 56 (KO), 574 ± 70 (OV); 50 μm, 860 ± 78 (WT), 397 ± 46 (KO), 785 ± 96 (OV) (n = 4; *p < 0.05 when KO was compared with WT or OV, ANOVA test).
We next investigated whether overexpression of PICK1 rescues the surface expression of EAAC1. Cultured cortical neurons from WT and PICK1−/− mice were transduced with control lentivirus or lentiviral plasmid encoding PICK1. Our results showed that the amount of EAAC1 on the cell surface was decreased significantly, and the surface expression of GluA2 was increased in the PICK1−/− neurons (Fig. 6C), similar to PICK1-knockdown experiments in neuronal cultures (Fig. 6B). The altered expression of EAAC1 and GluA2 was rescued effectively by the overexpression of PICK1 (Fig. 6C). To determine whether PICK1 overexpression is sufficient to rescue the GSH content, plasmids encoding PICK1 were lentiviral transduced into cultured PICK1−/− neurons, and maleimide staining was examined. Our results showed that the fluorescence intensity of maleimide was attenuated in PICK1−/− neurons, and the overexpression of PICK1 effectively rescued the maleimide signal in these neurons (Fig. 6D).
To detect directly the cysteine transport, the uptake of radiolabeled cysteine was undertaken according to previous work (Li et al., 2010). As shown in Figure 6E, cultured cortical neurons (DIV9) from WT and PICK1−/− mice took up cysteine with similar rates in the presence of low concentrations of l-cysteine (0.1, 1, or 5 μm). Nevertheless, PICK1−/− neurons took up markedly less cysteine than WT neurons did with concentrations of l-cysteine at 10 and 50 μm (Fig. 6E). Overexpression of PICK1 in PICK1−/− neurons effectively rescued the uptake of cysteine (Fig. 6E). These results, combining with surface EAAC1 measurements and maleimide staining experiments, demonstrated that the downregulation of GSH in PICK1−/− mice is mediated by decreased surface EAAC1.
Rab11 accumulates in the cytoplasm of PICK1−/− neurons
EAAC1 is recycled through Rab11-positive recycling endosomes (González et al., 2007), and Rab11 dysfunction decreases the trafficking of EAAC1 to the cell surface and impairs cysteine uptake (Li et al., 2010). Thus, it was of interest to investigate the interaction between EAAC1 and Rab11 in PICK1−/− mice. Co-IP experiments showed that Rab11 was precipitated robustly by PICK1 in vivo (Fig. 7A), suggesting that PICK1 interacts with Rab11. Because Rab11 requires secure association with membranes to execute its functions (Li et al., 2009), the expression level of Rab11 enriched in the membrane was assessed in PICK1−/− mice. We found that Rab11 enriched in the purified membrane fraction was reduced significantly, but its total expression was not altered in 4-month-old PICK1−/− mice (Fig. 7B,B′). To examine whether the interaction between EAAC1 and Rab11 is changed, coimmunostaining with their antibodies was performed in cultured cortical neurons. In agreement with previous observations (Coco et al., 1997; Li et al., 2010), our results showed that, in WT neurons, immunoreactivity of EAAC1 occurred mainly at punctate structures in the cytoplasm and neurites, and EAAC1 and Rab11 colocalized in some structures (Fig. 7C). Interestingly, immunoreactive EAAC1 and Rab11 were both more concentrated in the cytoplasm of PICK1−/− neurons than WT neurons (Fig. 7C). Quantification of fluorescence signals revealed a significant increase in the somata of PICK1−/− neurons compared with WT neurons (n = 30 WT and 30 PICK1−/− neurons; EAAC1 fluorescence, 25.7 ± 1.5 for WT vs 34.7 ± 1.8 for PICK1−/−, p < 0.01; Rab11 fluorescence, 57.6 ± 3.9 for WT vs 75.6 ± 2.8 for PICK1−/−, p < 0.05). Meanwhile, the fluorescence of EAAC1 and Rab11 were both decreased in neurites of PICK1−/− neurons, accompanied by reduced loci along neurites in which EAAC1 and Rab11 were colabeled (Fig. 7C). Together, these data suggested that loss of PICK1 results in more accumulation of Rab11 in the cytoplasm and less EAAC1 on the cell surface.
Figure 7.

Dissociation between EAAC1 and Rab11. A, Precleared brain lysates from WT mice were immunoprecipitated with mouse anti-PICK1 antibody. The immunoprecipitates were probed with antibodies to PICK1 (rabbit polyclonal antibody), Rab11, and β-actin (actin). The experiment was performed three times. Mouse IgG was used as negative control. B, Rab11 expression in the membrane fraction was decreased in 4-month-old PICK1−/− mice. β-Actin (actin) and flotillin were used as loading controls of total and membrane fractions, respectively. B′, The percentage changes of Rab11 in membrane fraction were 100 ± 7% (WT) and 49 ± 4% (KO) (n = 4 pairs; **p < 0.01). C, Primary cortical neurons (DIV9) from WT and KO mouse embryos were fixed and processed for immunolabeling with antibodies against Rab11 (green) and EAAC1 (red). The merged signals (yellow) show areas of colocalization of Rab11 and EAAC1. The boxed regions contain part of neurites and are enlarged below. White arrows indicate loci along neurites in which EAAC1 and Rab11 were colabeled. Yellow arrows point to accumulation of immunoreactive Rab11 and EAAC1 in the soma of one KO neuron. Scale bars, 20 μm. D, Cultured cortical neurons (DIV5) were transfected with myc-tagged Rab11-DN or control (NC). At 4 d after transfection, surface (biotin) EAAC1 was isolated by biotinylation and detected by antibodies to EAAC1, PICK1, myc, Rab11, and β-actin (actin). The white arrowhead indicates the band of myc-tagged Rab11-DN. E, Levels of total EAAC1 and PICK1 were normalized to β-actin. Surface EAAC1 was normalized to total EAAC1. Percentage changes are shown in the histograms: 100 ± 4% (total EAAC1, NC), 96 ± 4% (total EAAC1, DN), 100 ± 7% (surface EAAC1, NC), 50 ± 8% (surface EAAC1, DN), 100 ± 8% (total PICK1, NC), and 100 ± 7% (total PICK1, DN) (n = 4; *p < 0.05). F, Rab11-DN-transfected neurons (DIV9) were treated with DCFDA to assess ROS level. NC neurons had low DCF fluorescence, whereas mutant neurons had brighter DCF fluorescence. Scale bars, 25 μm. G, Average fluorescence intensity in the soma was 11.3 ± 1.8 (NC, n = 25) versus 41.0 ± 3.4 (Rab11-DN, n = 25). Average fluorescence intensity in the processes (per micrometers) was 0.0020 ± 0.0003 (NC, n = 25) versus 0.0108 ± 0.0014 (Rab11-DN, n = 25). **p < 0.01.
To investigate whether Rab11 is causally related to EAAC1 surface expression and oxidative stress, cultured rat cortical neurons (DIV5) were transduced with control lentivirus or Rab11-DN, and two subsequent experiments were performed. First, surface EAAC1 was measured using biotinylation. Our results showed that Rab11-DN effectively suppressed the level of surface EAAC1 in these neurons (DIV9; Fig. 7D,E). Second, DCFDA staining was observed in these cells. Our results showed that DCF signals in somata and neurites were significantly increased 4 d after transfection (Fig. 7F,G). These data suggested a causal relationship between Rab11 and surface EAAC1 and ROS production. A similar conclusion was drawn by Li et al. (2010), in which the overexpression of active Rab11 improves cysteine uptake and elevates GSH levels.
Discussion
The main finding of this study is that PICK1 deficiency causes a defect in the constitutive targeting of neuronal glutamate/cysteine transporter EAAC1 on the cell surface, which in turn impairs the generation of GSH to handle ROS in neurons. Our results also suggest that the impaired surface expression of EAAC1 is attributable to defective Rab11 activity. These findings suggest a novel concept that PICK1 is essential in resistance to oxidative stress and may thereby play crucial roles in oxidative stress-related chronic diseases.
Genetic or pharmacological manipulations of PICK1 expression or function have been suggested to influence neurological disorders, such as PD, ALS, schizophrenia, chronic pain, and epilepsy, through putative partners of PICK1 (Focant and Hermans, 2013). However, the mechanisms underlying the roles of PICK1 in these diseases are not clear. For example, enhanced excitotoxicity by increased d-serine and defects in glutamate uptake are implicated in the motor neuron death in patients suffering from ALS (Rothstein et al., 1995; Sasabe et al., 2007). Because PICK1 interacts with serine racemase (Fujii et al., 2006) and glutamate transporter GLT1b (Bassan et al., 2008), it is suggested that PICK1 participates in ALS by controlling serine synthesis and glutamate uptake (Focant and Hermans, 2013). Nevertheless, this speculation is not persuasive because there is no evidence for an altered interaction between PICK1 and serine racemase or GLT1b in the progression of ALS. Here, we propose an alternative possibility for the involvement of PICK1 in neurological disorders, that is, PICK1 dysfunction leads to worsened oxidative stress, which in turn results in the pathogenesis of degenerative disease. In fact, it has been shown that oxidative stress is associated tightly with neurodegeneration in Alzheimer's disease, PD, and ALS (Schulz et al., 2000; Nunomura et al., 2006; Preiser, 2012), and the inhibition of oxidative stress inhibits the progression of these diseases (Preiser, 2012). Therefore, PICK1 may constitute an interesting candidate for the understanding of oxidation-related brain disorders and deserves additional attention. Our ELISA results showed that the level of nitrotyrosine was increased in the serum of one patient who carries a homozygous mutation in the PICK1 gene (Liu et al., 2010). Because the patient and controls had no inflammatory disease during the period of blood collection, this finding implicated a correlation between PICK1 mutation and oxidative stress. However, there are two caveats that should be noted regarding to this result. First, it is unclear whether the missense mutation (G393R) located in the C-terminal region of PICK1, which antagonizes the lipid-binding capability of PICK1 (Xu and Xia, 2006), leads to the dysfunction of PICK1. Second, the evidence from the serum assay is not sufficient, because another report argues that oxidation biomarkers in the serum may not be the faithful indices of oxidative stress status (Korolainen and Pirttilä, 2009). A complete profile of measurements of oxidative damage is needed to investigate whether this phenomenon is consistent in all members of the patient's family and whether they or their ancestors suffer from a certain neurodegenerative disease.
It has been shown that PICK1 is expressed broadly in the brain (Xu and Xia, 2006). However, we found that PICK1 deficiency causes the accumulation of nitrotyrosine and HNE only in neurons (Fig. 2A). High level of oxidation has also been reported in neurons rather than glial cells in EAAC1−/− mice (Aoyama et al., 2006). The selective neuronal oxidation in PICK1−/− mice might be attributable to the downregulation of membrane EAAC1, which is mainly expressed in neurons (Rothstein et al., 1994). Another possibility is that the ability to clear ROS is much greater in astrocytes, because they contain more GSH and associated enzymes for GSH metabolism and thereby are less vulnerable to oxidant challenge (Wilson, 1997). In addition, Aoyama et al. (2006) found that both CA1 and CA3 neurons display significant loss of GSH and increase of ROS in EAAC1−/− mice. Differently, oxidative stress was not observed in CA1 pyramidal neurons from PICK1−/− mice (Fig. 1C). Although this discrepancy may be explained by different vulnerability between CA3 and CA1 neurons in response to stress (Wilde et al., 1997; Kadar et al., 1998; Mao et al., 2013) or by different severity induced by EAAC1 or PICK1 deficiency, the precise underlying mechanism remains to be explored.
The regulatory mechanisms for EAAC1 are essentially unknown, despite limited observations on the modulation of EAAC1 activity and trafficking. For instance, the adaptor protein GTRAP3–18 has been shown to prevent EAAC1 maturation by retaining EAAC1 at the endoplasmic reticulum as a regulator of trafficking (Ruggiero et al., 2008; Maier et al., 2009). EAAC1 is distributed mainly in the cytoplasm of neurons (He et al., 2000), and its trafficking is modulated by PKC and platelet-derived growth factor (Fournier et al., 2004). It has also been demonstrated that the trafficking of EAAC1 from recycling endosomes relies on active Rab11 (Li et al., 2010). Here, we added an important signaling to the mechanisms of EAAC1 regulation by showing that loss of PICK1 resulted in reduced targeting of EAAC1 to the cell surface, which was mediated suggestively by reduced active Rab11. Aberrant Rab11 in PICK1−/− mice was confirmed by its decreased expression in the membrane fraction and the impaired colocalization between EAAC1 and Rab11 in neurites (Fig. 7B,C). Although we showed that PICK1 is coimmunoprecipitated with Rab11 in vivo (Fig. 7A), the mechanism for the dysregulation of Rab11 by PICK1 deficiency needs to be explored further. Although PICK1 is required for synaptic plasticity, infusion of PICK1 in cerebellar and hippocampal neurons has no effect on synaptic currents, suggesting that the regulation of GluA2 by PICK1 is activity dependent (Xia et al., 2000; Jin et al., 2006). In this sense, our work first implied a constitutive regulation of Rab11-containing vesicle trafficking by PICK1 in neurons.
Rab11 plays important roles in the trafficking of GluA1 to spines during LTP through the myosin-V–Rab11 complex (Park et al., 2004; Lisé et al., 2006; Correia et al., 2008; Wang et al., 2008). Currently no evidence is present for the relationship between Rab11 and GluA2 trafficking. Distinctly, PICK1 is critical for the activity-dependent and -independent trafficking of GluA2. PICK1 can keep GluA2 in a cytosolic pool and affects its synaptic targeting (Jin et al., 2006; Xu et al., 2014). Loss of PICK1 causes a constitutive upregulation of surface GluA2 (Anggono et al., 2011; Fig. 6). It has been shown that the myosin–Rab11 complex is not required for the constitutive trafficking of GluA1 (Correia et al., 2008). Whether the effects of PICK1 on GluA2 trafficking is mediated by Rab11 needs to investigated.
Our pilot study showed that PICK1 did not interact directly with EAAC1. Thus, EAAC1 is distinct from the type 2 glutamate transporter GLT1, because the latter interacts with PICK1 in hippocampal neurons (Bassan et al., 2008). Although GLT1 also mediates cysteine uptake (Chen et al., 2002; Chen and Swanson, 2003), its inhibition only affects ∼10% of cysteine uptake (Li et al., 2010), and its affinity for cysteine is >10-fold lower than that of EAAC1 (Zerangue and Kavanaugh, 1996). Thus, it is unlikely that GLT1 makes a significant contribution to defective GSH generation in PICK1−/− mice.
We considered that reduced surface expression of EAAC1 and GSH decrease were responsible for oxidative stress in PICK1−/− mice. This consideration was from the comparisons with previous reports. First, it has been shown that GSH content is reduced in EAAC1−/− mice (Aoyama et al., 2006), Huntington's disease (HD) transgenic mice (Li et al., 2010), and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-treated mice (Aoyama et al., 2008). The reduction of GSH was ∼36% in EAAC1−/− mice (Aoyama et al., 2006), ∼30% in MPTP-treated mice (Aoyama et al., 2008), or ∼28% in HD neurons (Li et al., 2010), which was close to GSH loss in the present work (∼24%; Fig. 4E). Second, the surface expression of EAAC1 was downregulated, but the total EAAC1 was not changed in HD neurons (Li et al., 2010) and MPTP-treated mice (Aoyama et al., 2008). Because that GSH is a key regulator of oxidation and EAAC1 is a primary route for neuronal cysteine uptake, it was concluded that reduced surface EAAC1 and subsequent defects in GSH content are responsible for oxidative stress and neurodegeneration in their models (Aoyama et al., 2006, 2008; Li et al., 2010). However, it should be noted that other possible mechanisms may also participate in the oxidative stress in PICK1−/− mice, although we showed that mitochondrial function was basically normal in this mice.
A notable feature of PICK1−/− mice was that the abnormality of oxidative stress was age dependent, despite the finding that brain sections from young PICK1−/− mice had displayed weak expression of nitrotyrosine and 4-HNE. We failed to find apparent neuronal degeneration in 12-month-old PICK1−/− mice, albeit they displayed microglia-mediated inflammatory responses (Fig. 2C–E). The delayed onset of neuronal degeneration might be because the surface EAAC1 was only reduced (but not eliminated) in PICK1−/− mice, or because the cumulative effect of prolonged impairment of neuronal GSH homeostasis was not sufficient to induce cell death. Similarly, Aoyama et al. (2006) also did not find neuronal degeneration in EAAC1−/− mice until 11 months after birth. The delayed onset of neurodegeneration in PICK1−/− mice may be in parallel to the delayed onset of human neurodegenerative disorders.
Footnotes
This work was supported by National Basic Research Program of the Ministry of Science and Technology of China Grant 2011CB504400, National Natural Science Foundation of China Grants 31460257, 31471024, 31271148, and 31200818, and National Fund for Talent Training in Basic Science Grant J1103603. We thank Dr. Jun Xia (Hong Kong University of Science and Technology, Hong Kong, China) for providing PICK1−/− mice and Dr. Richard Huganir (Johns Hopkins University, Baltimore, MD) for providing GluA2 antibody. We thank Drs. Minxin Guan and Pingping Jiang (College of Life Science, Zhejiang University, Hangzhou, China) for help with Seahorse experiments. We are grateful for helpful advice from Dr. Junyu Xu and members of Shen laboratory. We thank the Core Facilities of Zhejiang University Institute of Neuroscience for technical assistance.
All authors declare no competing financial interests.
References
- Anggono V, Clem RL, Huganir RL. PICK1 loss of function occludes homeostatic synaptic scaling. J Neurosci. 2011;31:2188–2196. doi: 10.1523/JNEUROSCI.5633-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoyama K, Suh SW, Hamby AM, Liu J, Chan WY, Chen Y, Swanson RA. Neuronal glutathione deficiency and age-dependent neurodegeneration in the EAAC1 deficient mouse. Nat Neurosci. 2006;9:119–126. doi: 10.1038/nn1609. [DOI] [PubMed] [Google Scholar]
- Aoyama K, Matsumura N, Watabe M, Nakaki T. Oxidative stress on EAAC1 is involved in MPTP-induced glutathione depletion and motor dysfunction. Eur J Neurosci. 2008;27:20–30. doi: 10.1111/j.1460-9568.2007.05979.x. [DOI] [PubMed] [Google Scholar]
- Atianjoh FE, Yaster M, Zhao X, Takamiya K, Xia J, Gauda EB, Huganir RL, Tao YX. Spinal cord protein interacting with C kinase 1 is required for the maintenance of complete Freund's adjuvant-induced inflammatory pain but not for incision-induced post-operative pain. Pain. 2010;151:226–234. doi: 10.1016/j.pain.2010.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassan M, Liu H, Madsen KL, Armsen W, Zhou J, Desilva T, Chen W, Paradise A, Brasch MA, Staudinger J, Gether U, Irwin N, Rosenberg PA. Interaction between the glutamate transporter GLT1b and the synaptic PDZ domain protein PICK1. Eur J Neurosci. 2008;27:66–82. doi: 10.1111/j.1460-9568.2007.05986.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birben E, Sahiner UM, Sackesen C, Erzurum S, Kalayci O. Oxidative stress and antioxidant defense. World Allergy Organ J. 2012;5:9–19. doi: 10.1097/WOX.0b013e3182439613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao M, Xu J, Shen C, Kam C, Huganir RL, Xia J. PICK1-ICA69 heteromeric BAR domain complex regulates synaptic targeting and surface expression of AMPA receptors. J Neurosci. 2007;27:12945–12956. doi: 10.1523/JNEUROSCI.2040-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Aoki C, Mahadomrongkul V, Gruber CE, Wang GJ, Blitzblau R, Irwin N, Rosenberg PA. Expression of a variant form of the glutamate transporter GLT1 in neuronal cultures and in neurons and astrocytes in the rat brain. J Neurosci. 2002;22:2142–2152. doi: 10.1523/JNEUROSCI.22-06-02142.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Swanson RA. The glutamate transporters EAAT2 and EAAT3 mediate cysteine uptake in cortical neuron cultures. J Neurochem. 2003;84:1332–1339. doi: 10.1046/j.1471-4159.2003.01630.x. [DOI] [PubMed] [Google Scholar]
- Coco S, Verderio C, Trotti D, Rothstein JD, Volterra A, Matteoli M. Non-synaptic localization of the glutamate transporter EAAC1 in cultured hippocampal neurons. Eur J Neurosci. 1997;9:1902–1910. doi: 10.1111/j.1460-9568.1997.tb00757.x. [DOI] [PubMed] [Google Scholar]
- Codoñer-Franch P, Tavárez-Alonso S, Murria-Estal R, Megías-Vericat J, Tortajada-Girbés M, Alonso-Iglesias E. Nitric oxide production is increased in severely obese children and related to markers of oxidative stress and inflammation. Atherosclerosis. 2011;215:475–480. doi: 10.1016/j.atherosclerosis.2010.12.035. [DOI] [PubMed] [Google Scholar]
- Correia SS, Bassani S, Brown TC, Lisé MF, Backos DS, El-Husseini A, Maria Passafaro M, Esteban JA. Motor protein-dependent transport of AMPA receptors into spines during long-term potentiation. Nat Neurosci. 2008;11:457–466. doi: 10.1038/nn2063. [DOI] [PubMed] [Google Scholar]
- Deken SL, Beckman ML, Quick MW. PICKing on transporters. Trends Neurosci. 2001;24:623–625. doi: 10.1016/S0166-2236(00)01933-0. [DOI] [PubMed] [Google Scholar]
- De Stefani D, Bononi A, Romagnoli A, Messina A, De Pinto V, Pinton P, Rizzuto R. VDAC1 selectively transfers apoptotic Ca2+ signals to mitochondria. Cell Death Differ. 2012;19:267–273. doi: 10.1038/cdd.2011.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dringen R. Metabolism and functions of glutathione in brain. Prog Neurobiol. 2000;62:649–671. doi: 10.1016/S0301-0082(99)00060-X. [DOI] [PubMed] [Google Scholar]
- Fernández E, García-Moreno JM, Martín de Pablos A, Chacón J. May the evaluation of nitrosative stress through selective increase of 3-nitrotyrosine proteins other than nitroalbumin and dominant tyrosine-125/136 nitrosylation of serum α-synuclein serve for diagnosis of sporadic Parkinson's disease? Antioxid Redox Signal. 2013;19:912–918. doi: 10.1089/ars.2013.5250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Focant MC, Hermans E. Protein interacting with C kinase and neurological disorders. Synapse. 2013;67:532–540. doi: 10.1002/syn.21657. [DOI] [PubMed] [Google Scholar]
- Fournier KM, González MI, Robinson MB. Rapid trafficking of the neuronal glutamate transporter, EAAC1: evidence for distinct trafficking pathways differentially regulated by protein kinase C and platelet-derived growth factor. J Biol Chem. 2004;279:34505–34513. doi: 10.1074/jbc.M404032200. [DOI] [PubMed] [Google Scholar]
- Fujii K, Maeda K, Hikida T, Mustafa AK, Balkissoon R, Xia J, Yamada T, Ozeki Y, Kawahara R, Okawa M, Huganir RL, Ujike H, Snyder SH, Sawa A. Serine racemase binds to PICK1: potential relevance to schizophrenia. Mol Psychiatry. 2006;11:150–157. doi: 10.1038/sj.mp.4001776. [DOI] [PubMed] [Google Scholar]
- González MI, Susarla BT, Fournier KM, Sheldon AL, Robinson MB. Constitutive endocytosis and recycling of the neuronal glutamate transporter, excitatory amino acid carrier 1. J Neurochem. 2007;103:1917–1931. doi: 10.1111/j.1471-4159.2007.04881.x. [DOI] [PubMed] [Google Scholar]
- Griffith OW, Meister A. Potent and specific inhibition of glutathione synthesis by buthionine sulfoximine (S-n-butyl homocysteine sulfoximine) J Biol Chem. 1979;254:7558–7560. [PubMed] [Google Scholar]
- Hanley JG. PICK1: a multi-talented modulator of AMPA receptor trafficking. Pharmacol Ther. 2008;118:152–160. doi: 10.1016/j.pharmthera.2008.02.002. [DOI] [PubMed] [Google Scholar]
- Hanley JG, Henley JM. PICK1 is a calcium-sensor for NMDA-induced AMPA receptor trafficking. EMBO J. 2005;24:3266–3278. doi: 10.1038/sj.emboj.7600801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251–262. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Janssen WG, Rothstein JD, Morrison JH. Differential synaptic localization of the glutamate transporter EAAC1 and glutamate receptor subunit GluR2 in the rat hippocampus. J Comp Neurol. 2000;418:255–269. doi: 10.1002/(SICI)1096-9861(20000313)418:3<255::AID-CNE2>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Himi T, Ikeda M, Yasuhara T, Nishida M, Morita I. Role of neuronal glutamate transporter in the cysteine uptake and intracellular glutathione levels in cultured cortical neurons. J Neural Transm. 2003;110:1337–1348. doi: 10.1007/s00702-003-0049-z. [DOI] [PubMed] [Google Scholar]
- Jin W, Ge WP, Xu J, Cao M, Peng L, Yung W, Liao D, Duan S, Zhang M, Xia J. Lipid binding regulates synaptic targeting of PICK1, AMPA receptor trafficking, and synaptic plasticity. J Neurosci. 2006;26:2380–2390. doi: 10.1523/JNEUROSCI.3503-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin W, Shen C, Jing L, Zha XM, Xia J. PICK1 regulates the trafficking of ASIC1a and acidotoxicity in a BAR domain lipid binding-dependent manner. Mol Brain. 2010;3:39. doi: 10.1186/1756-6606-3-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joch M, Ase AR, Chen CX, MacDonald PA, Kontogiannea M, Corera AT, Brice A, Séguéla P, Fon EA. Parkin-mediated monoubiquitination of the PDZ protein PICK1 regulates the activity of acid-sensing ion channels. Mol Biol Cell. 2007;18:3105–3118. doi: 10.1091/mbc.E05-11-1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadar T, Dachir S, Shukitt-Hale B, Levy A. Sub-regional hippocampal vulnerability in various animal models leading to cognitive dysfunction. J Neural Transm. 1998;105:987–1004. doi: 10.1007/s007020050107. [DOI] [PubMed] [Google Scholar]
- Kirkland RA, Saavedra GM, Franklin JL. Rapid activation of antioxidant defenses by nerve growth factor suppresses reactive oxygen species during neuronal apoptosis: evidence for a role in cytochrome c redistribution. J Neurosci. 2007;27:11315–11326. doi: 10.1523/JNEUROSCI.3590-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korolainen MA, Pirttilä T. Cerebrospinal fluid, serum and plasma protein oxidation in Alzheimer's disease. Acta Neurol Scand. 2009;119:32–38. doi: 10.1111/j.1600-0404.2008.01057.x. [DOI] [PubMed] [Google Scholar]
- Li X, Sapp E, Chase K, Comer-Tierney LA, Masso N, Alexander J, Reeves P, Kegel KB, Valencia A, Esteves M, Aronin N, Difiglia M. Disruption of Rab11 activity in a knock-in mouse model of Huntington's disease. Neurobiol Dis. 2009;36:374–383. doi: 10.1016/j.nbd.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Valencia A, Sapp E, Masso N, Alexander J, Reeves P, Kegel KB, Aronin N, Difiglia M. Aberrant Rab11-dependent trafficking of the neuronal glutamate transporter EAAC1 causes oxidative stress and cell death in Huntington's disease. J Neurosci. 2010;30:4552–4561. doi: 10.1523/JNEUROSCI.5865-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim CS, Hoang ET, Viar KE, Stornetta RL, Scott MM, Zhu JJ. Pharmacological rescue of Ras signaling, GluA1-dependent synaptic plasticity, and learning deficits in a fragile X model. Genes Dev. 2014;28:273–289. doi: 10.1101/gad.232470.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisé MF, Wong TP, Trinh A, Hines RM, Liu L, Kang R, Hines DJ, Lu J, Goldenring JR, Wang YT, El-Husseini A. Involvement of myosin Vb in glutamate receptor trafficking. J Biol Chem. 2006;281:3669–3678. doi: 10.1074/jbc.M511725200. [DOI] [PubMed] [Google Scholar]
- Liu G, Shi QW, Lu GX. A newly discovered mutation in PICK1 in a human with globozoospermia. Asian J Androl. 2010;12:556–560. doi: 10.1038/aja.2010.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier S, Reiterer V, Ruggiero AM, Rothstein JD, Thomas S, Dahm R, Sitte HH, Farhan H. GTRAP3–18 serves as a negative regulator of Rab1 in protein transport and neuronal differentiation. J Cell Mol Med. 2009;13:114–124. doi: 10.1111/j.1582-4934.2008.00303.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao H, Elkin BS, Genthikatti VV, Morrison B, 3rd, Yang KH. Why is CA3 more vulnerable than CA1 in experimental models of controlled cortical impact-induced brain injury? J Neurotrauma. 2013;30:1521–1530. doi: 10.1089/neu.2012.2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazor D, Golan E, Philip V, Katz M, Jafe A, Ben-Zvi Z, Meyerstein N. Red blood cell permeability to thiol compounds following oxidative stress. Eur J Haematol. 1996;57:241–246. doi: 10.1111/j.1600-0609.1996.tb01370.x. [DOI] [PubMed] [Google Scholar]
- Moreira PI, Smith MA, Zhu X, Nunomura A, Castellani RJ, Perry G. Oxidative stress and neurodegeneration. Ann N Y Acad Sci. 2005;1043:545–552. doi: 10.1196/annals.1333.062. [DOI] [PubMed] [Google Scholar]
- Mosher KI, Wyss-Coray T. Microglial dysfunction in brain aging and Alzheimer's disease. Biochem Pharmacol. 2014;88:594–604. doi: 10.1016/j.bcp.2014.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noh H, Ha H. Reactive oxygen species and oxidative stress. Contrib Nephrol. 2011;170:102–112. doi: 10.1159/000324955. [DOI] [PubMed] [Google Scholar]
- Nunomura A, Castellani RJ, Zhu X, Moreira PI, Perry G, Smith MA. Involvement of oxidative stress in Alzheimer disease. J Neuropathol Exp Neurol. 2006;65:631–641. doi: 10.1097/01.jnen.0000228136.58062.bf. [DOI] [PubMed] [Google Scholar]
- Park M, Penick EC, Edwards JG, Kauer JA, Ehlers MD. Recycling endosomes supply AMPA receptors for LTP. Science. 2004;305:1972–1975. doi: 10.1126/science.1102026. [DOI] [PubMed] [Google Scholar]
- Parsons JL, Chipman JK. The role of glutathione in DNA damage by potassium bromate in vitro. Mutagenesis. 2000;15:311–316. doi: 10.1093/mutage/15.4.311. [DOI] [PubMed] [Google Scholar]
- Preiser JC. Oxidative stress. JPEN J Parenter Enteral Nutr. 2012;36:147–154. doi: 10.1177/0148607111434963. [DOI] [PubMed] [Google Scholar]
- Ren M, Xu G, Zeng J, De Lemos-Chiarandini C, Adesnik M, Sabatini DD. Hydrolysis of GTP on rab11 is required for the direct delivery of transferrin from the pericentriolar recycling compartment to the cell surface but not from sorting endosomes. Proc Natl Acad Sci U S A. 1998;95:6187–6192. doi: 10.1073/pnas.95.11.6187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein JD, Martin L, Levey AI, Dykes-Hoberg M, Jin L, Wu D, Nash N, Kuncl RW. Localization of neuronal and glial glutamate transporters. Neuron. 1994;13:713–725. doi: 10.1016/0896-6273(94)90038-8. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Van Kammen M, Levey AI, Martin LJ, Kuncl RW. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann Neurol. 1995;38:73–84. doi: 10.1002/ana.410380114. [DOI] [PubMed] [Google Scholar]
- Ruggiero AM, Liu Y, Vidensky S, Maier S, Jung E, Farhan H, Robinson MB, Sitte HH, Rothstein JD. The endoplasmic reticulum exit of glutamate transporter is regulated by the inducible mammalian Yip6b/GTRAP3–18 protein. J Biol Chem. 2008;283:6175–6183. doi: 10.1074/jbc.M701008200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasabe J, Chiba T, Yamada M, Okamoto K, Nishimoto I, Matsuoka M, Aiso S. D-serine is a key determinant of glutamate toxicity in amyotrophic lateral sclerosis. EMBO J. 2007;26:4149–4159. doi: 10.1038/sj.emboj.7601840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz JB, Lindenau J, Seyfried J, Dichgans J. Glutathione, oxidative stress and neurodegeneration. Eur J Biochem. 2000;267:4904–4911. doi: 10.1046/j.1432-1327.2000.01595.x. [DOI] [PubMed] [Google Scholar]
- Seet RC, Lee CY, Lim EC, Tan JJ, Quek AM, Chong WL, Looi WF, Huang SH, Wang H, Chan YH, Halliwell B. Oxidative damage in Parkinson disease: measurement using accurate biomarkers. Free Radic Biol Med. 2010;48:560–566. doi: 10.1016/j.freeradbiomed.2009.11.026. [DOI] [PubMed] [Google Scholar]
- Starkov AA. The role of mitochondria in reactive oxygen species metabolism and signaling. Ann N Y Acad Sci. 2008;1147:37–52. doi: 10.1196/annals.1427.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staudinger J, Zhou J, Burgess R, Elledge SJ, Olson EN. PICK1: a perinuclear binding protein and substrate for protein kinase C isolated by the yeast two-hybrid system. J Cell Biol. 1995;128:263–271. doi: 10.1083/jcb.128.3.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg JP, Takamiya K, Shen Y, Xia J, Rubio ME, Yu S, Jin W, Thomas GM, Linden DJ, Huganir RL. Targeted in vivo mutations of the AMPA receptor subunit GluR2 and its interacting protein PICK1 eliminate cerebellar long-term depression. Neuron. 2006;49:845–860. doi: 10.1016/j.neuron.2006.02.025. [DOI] [PubMed] [Google Scholar]
- Streit WJ, Walter SA, Pennell NA. Reactive microgliosis. Prog Neurobiol. 1999;57:563–581. doi: 10.1016/S0301-0082(98)00069-0. [DOI] [PubMed] [Google Scholar]
- Terashima A, Pelkey KA, Rah JC, Suh YH, Roche KW, Collingridge GL, McBain CJ, Isaac JT. An essential role for PICK1 in NMDA receptor-dependent bidirectional synaptic plasticity. Neuron. 2008;57:872–882. doi: 10.1016/j.neuron.2008.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trotti D, Danbolt NC, Volterra A. Glutamate transporters are oxidant-vulnerable: a molecular link between oxidative and excitotoxic neurodegeneration? Trends Pharmacol Sci. 1998;19:328–334. doi: 10.1016/S0165-6147(98)01230-9. [DOI] [PubMed] [Google Scholar]
- Valencia A, Morán J. Role of oxidative stress in the apoptotic cell death of cultured cerebellar granule neurons. J Neurosci Res. 2001;64:284–297. doi: 10.1002/jnr.1077. [DOI] [PubMed] [Google Scholar]
- Wang Z, Edwards JG, Riley N, Provance DW, Jr, Karcher R, Li XD, Davison IG, Ikebe M, Mercer JA, Kauer JA, Ehlers MD. Myosin Vb mobilizes recycling endosomes and AMPA receptors for postsynaptic plasticity. Cell. 2008;135:535–548. doi: 10.1016/j.cell.2008.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Wang YN, Sun CL, Yang D, Su LD, Xie YJ, Zhou L, Wang Y, Shen Y. C-terminal domain of ICA69 interacts with PICK1 and acts on trafficking of PICK1-PKCalpha complex and cerebellar plasticity. PLoS One. 2013;8:e83862. doi: 10.1371/journal.pone.0083862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilde GJ, Pringle AK, Wright P, Iannotti F. Differential vulnerability of the CA1 and CA3 subfields of the hippocampus to superoxide and hydroxyl radicals in vitro. J Neurochem. 1997;69:883–886. doi: 10.1046/j.1471-4159.1997.69020883.x. [DOI] [PubMed] [Google Scholar]
- Wilson JX. Antioxidant defense of the brain: a role for astrocytes. Can J Physiol Pharmacol. 1997;75:1149–1163. doi: 10.1139/y97-146. [DOI] [PubMed] [Google Scholar]
- Xia J, Chung HJ, Wihler C, Huganir RL, Linden DJ. Cerebellar long-term depression requires PKC-regulated interactions between GluR2/3 and PDZ domain-containing proteins. Neuron. 2000;28:499–510. doi: 10.1016/S0896-6273(00)00128-8. [DOI] [PubMed] [Google Scholar]
- Xie YJ, Zhou L, Jiang N, Zhang N, Zou N, Zhou L, Wang Y, Cowell JK, Shen Y. Essential roles of leucine-rich glioma inactivated 1 in the development of embryonic and postnatal cerebellum. Sci Rep. 2015;5:7827. doi: 10.1038/srep07827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Xia J. Structure and function of PICK1. NeuroSignals. 2006;15:190–201. doi: 10.1159/000098482. [DOI] [PubMed] [Google Scholar]
- Xu J, Kam C, Luo JH, Xia J. PICK1 mediates synaptic recruitment of AMPA receptors at neurexin-induced postsynaptic sites. J Neurosci. 2014;34:15415–15424. doi: 10.1523/JNEUROSCI.0296-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Xiao Y, Liu J, Ji Y, Liu H, Xu J, Jin X, Liu L, Guan MX, Jiang P. Loss of MED1 triggers mitochondrial biogenesis in C2C12 cells. Mitochondrion. 2014;14:18–25. doi: 10.1016/j.mito.2013.12.004. [DOI] [PubMed] [Google Scholar]
- Zerangue N, Kavanaugh MP. Interaction of L-cysteine with a human excitatory amino acid transporter. J Physiol. 1996;493:419–423. doi: 10.1113/jphysiol.1996.sp021393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Shao CY, Yang W, Zhang XM, Wu ZY, Zhou L, Wang XX, Li YH, Xia J, Luo JH, Shen Y. Chronic zinc exposure decreases the surface expression of NR2A-containing NMDA receptors in cultured hippocampal neurons. PLoS One. 2012;7:e46012. doi: 10.1371/journal.pone.0046012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zigdon H, Kogot-Levin A, Park JW, Goldschmidt R, Kelly S, Merrill AH, Jr, Scherz A, Pewzner-Jung Y, Saada A, Futerman AH. Ablation of ceramide synthase 2 causes chronic oxidative stress due to disruption of the mitochondrial respiratory chain. J Biol Chem. 2013;288:4947–4956. doi: 10.1074/jbc.M112.402719. [DOI] [PMC free article] [PubMed] [Google Scholar]