

Abstract
Glutamate, a major neurotransmitter in the brain, activates ionotropic and metabotropic glutamate receptors (iGluRs and mGluRs, respectively). The two types of glutamate receptors interact with each other, as exemplified by the modulation of iGluRs by mGluRs. However, the other way of interaction (i.e., modulation of mGluRs by iGluRs) has not received much attention. In this study, we found that group I mGluR-specific agonist (RS)-3,5-dihydroxyphenylglycine (DHPG) alone is not sufficient to activate phospholipase C (PLC) in rat hippocampus, while glutamate robustly activates PLC. These results suggested that additional mechanisms provided by iGluRs are involved in group I mGluR-mediated PLC activation. A series of experiments demonstrated that glutamate-induced PLC activation is mediated by mGluR5 and is facilitated by local Ca2+ signals that are induced by AMPA-mediated depolarization and L-type Ca2+ channel activation. Finally, we found that PLC and L-type Ca2+ channels are involved in hippocampal mGluR-dependent long-term depression (mGluR-LTD) induced by paired-pulse low-frequency stimulation, but not in DHPG-induced chemical LTD. Together, we propose that AMPA receptors initiate Ca2+ influx via the L-type Ca2+ channels that facilitate mGluR5-PLC signaling cascades, which underlie mGluR-LTD in rat hippocampus.
Keywords: Cav1.2, Cav1.3, confocal imaging, electrophysiology, local calcium, mGluR-LTD
Introduction
Glutamate is a major excitatory neurotransmitter that activates ionotropic and metabotropic glutamate receptors (iGluRs and mGluRs, respectively). iGluRs are ligand-gated ion channels that mediate fast EPSCs, whereas mGluRs are G-protein-coupled receptors (GPCRs). Regulation of iGluRs by mGluRs was previously suggested to be important for integrative brain function (Doherty et al., 1997; Mannaioni et al., 2001). Surface expression of AMPA receptors are reduced by group I mGluR signaling to induce long-term depression (LTD) of synaptic currents (Lüscher and Huber, 2010), while NMDA currents are potentiated by group I mGluR agonists (Doherty et al., 1997). Importantly, impaired function of NMDA receptors in pathologic conditions could be reversed by the upregulation of group I mGluRs (Won et al., 2012), suggesting a possibility that this interaction can be applied to develop new therapeutic strategies. Likewise, the regulation of mGluR signaling by iGluRs may also have certain physiological significance, but this possibility has received relatively little attention.
While group I mGluRs (mGluR1 and mGluR5) link glutamatergic neurotransmission to a wide variety of signaling pathways and involve multiple partners (Conn and Pin, 1997), the Gq protein/phospholipase C (PLC)/inositol-3,4,5-triphosphate (IP3) signal cascades have been considered as the canonical pathway (Abe et al., 1992; Aramori and Nakanishi, 1992). However, independent studies have differed on how PLC is involved in group I mGluR effects. The stimulation of Schaffer collateral (SC)-CA1 neuron synapses activates group I mGluRs to mobilize intracellular Ca2+ by PLC and IP3 signal pathways (Nakamura et al., 1999, 2000; El-Hassar et al., 2011), but Ca2+ release by the group I mGluR agonist (RS)-3,5-dihydroxyphenylglycine (DHPG) was shown to be PLC independent (Sohn et al., 2011). In addition, mGluR-dependent LTD (mGluR-LTD) induced by DHPG occurs independently of PLC/IP3-dependent Ca2+ release or protein kinase C (PKC) activity at the SC-CA1 neuron synapse (Schnabel et al., 1999; Fitzjohn et al., 2001; Mockett et al., 2011), whereas mGluR-LTD at the same synapse induced by synaptic stimulation is dependent on intracellular Ca2+ rise and PKC activation (Bolshakov and Siegelbaum, 1994; Oliet et al., 1997; Otani and Connor, 1998). These results suggest that, unlike glutamate, application of DHPG is not sufficient for PLC activation. In agreement, when endoplasmic reticulum-containing spines of CA1 neurons were stimulated at a low frequency using glutamate uncaging, this induced group I mGluR-LTD specific to the stimulated synapse and this LTD was dependent on Ca2+ release (Holbro et al., 2009).
Together, we hypothesized that iGluRs may play a significant role in mGluR-mediated PLC activation. Considering that PLC activity per se is dependent on intracellular Ca2+ concentrations (Ryu et al., 1987), the activation of iGluRs may contribute to group I mGluR-mediated PLC activation possibly by inducing Ca2+ influx directly or indirectly. In this study, we found that AMPA receptors initiate Ca2+ influx through L-type Ca2+ channels that are specifically required for mGluR-mediated PLC activation. Furthermore, we found evidence that this mechanism is crucial for mGluR-LTD in the hippocampus induced by paired-pulse low-frequency synaptic stimulation (PP-LFS).
Materials and Methods
DNA constructs.
The PLCδ-pleckstrin homology domain (PHδ) cloned into pEGFP-N1 was obtained from Dr. P. Suh (Ulsan National Institute of Science and Technology, Korea). The Cav1.2 (mouse) cloned into vector pcDNA6/V5-His was purchased from Addgene (plasmid 26572). The vector pcDNA6/V5-His containing Cav1.3 (rat) sequence, which in turn contains exon 42a, was obtained from Dr. D. Lipscombe (Brown University, Providence, RI). Cav1.2-targeting short-hairpin RNA (shRNA) sequence (5′-GCCGAAATTACTTCAATATTTCAAGAGAATATTGAAGTAATTTCGGC-3′) was designed using the software tool available at the Whitehead Institute for Biomedical Research website (Yuan et al., 2004). The Cav1.3-targeting shRNA sequence (5′-GGAAACCATTTGACATATTTATTCAAGAGATAAATATGTCAAATGGTTTCC-3′) was purchased from Open Biosystems. The synthesized Cav1.2 and Cav1.3 targeting shRNA oligonucleotides (CosmoGenentech) were ligated into the lentiviral vector pLentiLox3.7 (pLL3.7; Rubinson et al., 2003), which coexpressed monomeric RFP. The pLL3.7 containing luciferase-targeting shRNA sequence (5′-TAAGGCTATGAAGAGATAC-3′) was used as nontargeting control (NT control; Dharmacon).
Cell culture and transfection.
All experiments were performed with the approval of the animal experiment ethics committee at the Seoul National University College of Medicine. Neuron-glia coculture protocol for low-density hippocampal primary culture was previously described (Kaech and Banker, 2006; Lee et al., 2012). Briefly, hippocampi were dissected from embryonic day 18 Sprague Dawley (SD) rats of either sex. Hippocampal neurons were dissociated by papain treatment and trituration, and were plated at a density of 1.1 × 104 cells/cm2 on poly-d-lysine (Sigma-Aldrich)-coated coverslips (Marienfeld) in serum-based cell culture media. The next day, coverslips were transferred onto glial cell feeder layers cultured for 14 d in vitro (DIV14) in B-27 (Invitrogen)-supplemented Neurobasal A (Invitrogen) media. To prevent proliferation of glial cells, 5 μm 1-β-d-cytosine-arabinofuranoside (AraC; Sigma-Aldrich) was added to the DIV4 cocultured neurons. For Western blotting experiments, hippocampal neurons were plated at a density of 4.4 × 104 cells/cm2 on a poly-d-lysine-coated culture dish. Primary cultured neurons (DIV4–DIV7) were transfected using a calcium phosphate method (Ryan et al., 2005). The culture media were removed and saved before transfection. Neurons were incubated with fresh Neurobasal A media (2 ml per 60 mm culture dish) containing 25 mm HEPES, pH 7.35. During this time, the DNA/calcium phosphate precipitate was prepared by mixing one volume of PHδ-GFP DNA construct (1 μg) alone or with NT control, shCav1.2, or shCav1.3 DNA (5 μg for confocal imaging and Ca2+ measurement; 10 μg for Western blotting) in 250 mm CaCl2 with an equal volume of 2 × HEPES-buffered saline (280 mm NaCl, 50 mm HEPES, 1.5 mm Na2HPO4, pH 7.1) using a Vortex mixer. The precipitate was allowed to form for 2 min at room temperature (RT) before being added to the culture. Two hundred microliters of DNA/calcium phosphate suspensions were added drop-wise to cultured hippocampal neurons. A layer of precipitate became obvious after a 15 min incubation period, when cells were quick-washed once; washed twice for 5 min, with fresh Neurobasal A media added between washings; and returned to the saved culture media.
HEK293 cells were plated at a density of 5 × 104 cells per 100 mm culture dish and maintained in DMEM (Invitrogen) supplemented with 10% FBS and 1% penicillin-streptomycin (PS). HEK293 cells were subcultured before reaching ∼80% confluence. HEK293 cells at ∼30% confluence were transfected as follows: (1) 5 μg of Cav1.2 or 5 μg of Cav1.3 alone; (2) 5 μg of Cav1.2 or 5 μg of Cav1.3 with 5 μg of NT control; (3) 5 μg of Cav1.2 or 5 μg of Cav1.3 with 5 μg of Cav1.2 shRNA or Cav1.3 shRNA, respectively. This transfection used the same calcium phosphate protocol for the primary culture except the medium was not changed before and after adding the DNA/calcium phosphate mixture to the culture. Cultures were maintained in a humidified incubator at 37°C in 5% CO2.
Organitypic slice culture and Sindbis viral transduction system.
For slice cultures, postnatal day P7–P9 SD rats of either sex were decapitated and their brains were obtained. The posterior part of the brain was cut into 350-μm-thick transverse slices using a vibratome (ZERO 1, Dosaka) in ice-cold Eagle's balanced salt solution (EBSS) supplemented with 12.5 mm HEPES. The entorhino-hippocampi were dissected out and cultured using membrane-interface techniques mostly according to a previously described procedure (De Simoni and Yu, 2006). Slices were placed on a porous (0.4 μm) membrane (Millicell-CM, Millipore) and fed with a mixture of 50% MEM, 25% horse serum, 24% EBSS, and 1% PS. Glucose was added to reach a final concentration of 36 mm. The media were changed to serum-free media (Neurobasal-A media with 2% B-27 supplement, 1% GlutaMAX-I, 1% PS, and 5 mm glucose) with 5 μm AraC the day after dissection. The media were changed every 2 d. We used a Sindbis virus system to overexpress PHδ-GFP in hippocampal organotypic slice cultures. PHδ-GFP was subcloned into SINrep (nsP2S726), a modified Sindbis viral vector that shows attenuated viral-induced cytotoxicity and higher expression levels of the protein of interest in neurons (Kim et al., 2004). The complementary RNAs (cRNAs) were synthesized from the linearized SINrep (nsP2S726)/PHδ-GFP and helper DH-BB (tRNA/TE12) plasmid DNA, using an in vitro transcription kit (mMessage mMachine, Ambion). BHK21 cells were electroporated with cRNAs of SINrep (nsP2S726)/PH-GFP and DH-BB (tRNA/TE12) according to the Sindbis Expression System manual (Invitrogen). The pseudovirions-containing media were collected after 48 h, and then cell debris was removed from the supernatant by centrifugation at 1400 × g for 10 min at 4°C and aliquots were stored at −80°C. Subsequently, cultured hippocampal slices were infected at 8–15 DIV with titer resulting in infection of <5% of neurons for 24 h.
Western blotting.
Primary cultured hippocampal neurons or HEK293 cells were harvested after 7 d or 24 h of DNA transfection, respectively. The cells were washed once with Dulbecco's PBS (Invitrogen) and solubilized in ice-cold lysis buffer containing 50 mm Tris-Cl, pH 7.4, 150 mm NaCl, 1 mm EDTA, 1% SDS, and 0.1% protease inhibitor mixture (Sigma-Aldrich). Cell lysates were then sonicated 20 times at 1 s intervals and denatured by 100°C boiling water for 5 min. Cell lysates were clarified by centrifugation at 8200 × g for 1 min at 4°C. Cell lysates were separated by SDS-PAGE and transferred onto a polyvinylidene difluoride membrane (Millipore). The resulting blots were blocked for 1 h in PBS plus 0.1% Triton X-100 (0.1% PBST) containing 5% skim milk (Difco). The blots were incubated overnight at 4°C with the following specific primary antibodies: mouse monoclonal anti-Cav1.2 (1:150; Neuromab), rabbit polyclonal anti-Cav1.3 (1:100; Alomone Labs), goat polyclonal anti-Cav1.3 (1:100; Santa Cruz Biotechnology), or goat polyclonal anti-GAPDH (1:1500; Santa Cruz Biotechnology) as loading controls. The blots were washed with PBS containing 1% NP-40 and 0.1% SDS (washing buffer) for 1 h and then twice with PBS for 30 min. After washing, the blots were incubated at RT for 1 h with the corresponding horseradish peroxidase-conjugated secondary antibodies: donkey anti-mouse IgG (1:2500; Jackson ImmunoResearch), donkey anti-rabbit IgG (1:2000; Abcam), or donkey anti-goat IgG (1:2000; Santa Cruz Biotechnology). The blots were then washed with washing buffer for 30 min and then twice with PBS for 15 min. Detection was performed using enhanced chemiluminescence reagent (GE Healthcare Bio-Sciences) and exposed to x-ray films (Agfa).
Imaging and analysis of PHδ-GFP translocation.
All images were obtained from DIV7–DIV14 cultured neurons. Transfected cells were imaged with a TCS-SP2 (Leica) confocal laser-scanning microscope using a 63× water-immersion objective (numerical aperture, 1.20; HCX PL APO 63×, Leica) or FV300 (Olympus) using a 63× water-immersion objective (numerical aperture, 0.9; LUMP-lanFl/IR, Olympus). Fluorochromes were excited with an argon laser at 488 and 543 nm. Appropriate emission filters were used for fluorescence detection. PHδ-GFP favors phosphatidylinositol 4,5-bisphosphate (PIP2) over phosphatidylinositol, phosphatidylinositol 3-phosphate, and phosphatidylinositol 3,4,5-triphosphate, but has ∼10 times higher affinity for IP3 than for PIP2 (Várnai and Balla, 1998). Because of this affinity difference, PIP2 hydrolysis by PLC causes PHδ-GFP to translocate from the plasma membrane to the cytosol. Dissociated hippocampal primary cultures were perfused with normal tyrode solution containing the following (in mm): 150 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 glucose, 10 HEPES, adjusted to pH 7.4 with Tris-OH while imaging. To make Ca2+-free normal tyrode solutions, CaCl2 was replaced by equimolar MgCl2 and 0.1 mm EGTA was added. Organotypic hippocampal slice cultures were perfused with artificial CSF (aCSF) containing the following (in mm): 124 NaCl, 26 NaHCO3, 3.2 KCl, 1.25 NaH2PO4, 2.5 CaCl2, 1.3 MgCl2, 10 glucose, 3 Na-pyruvate, 3 vitamin C, bubbled with mixture of 95% O2 and 5% CO2 to a final pH of 7.4. To statistically analyze PHδ-GFP translocation, regions of interest (ROIs) were defined in the cytosol areas of soma or dendrite. Differences in the fluorescence intensity (ΔF) inside ROIs were obtained and normalized to baseline values (F0) before first glutamate application (Figs. 1A–C, 2Ca,Cc; see 5B,Cb). The relative ΔF values were calculated by normalizing ΔF to the peak values of first glutamate application (ΔF1; Fig. 3A–D; see Fig. 4A,B). The ΔF2/ΔF1 values were obtained by calculating ratio between the ΔF induced by glutamate plus inhibitors (ΔF2) and the ΔF induced by glutamate only (ΔF1).
Figure 1.
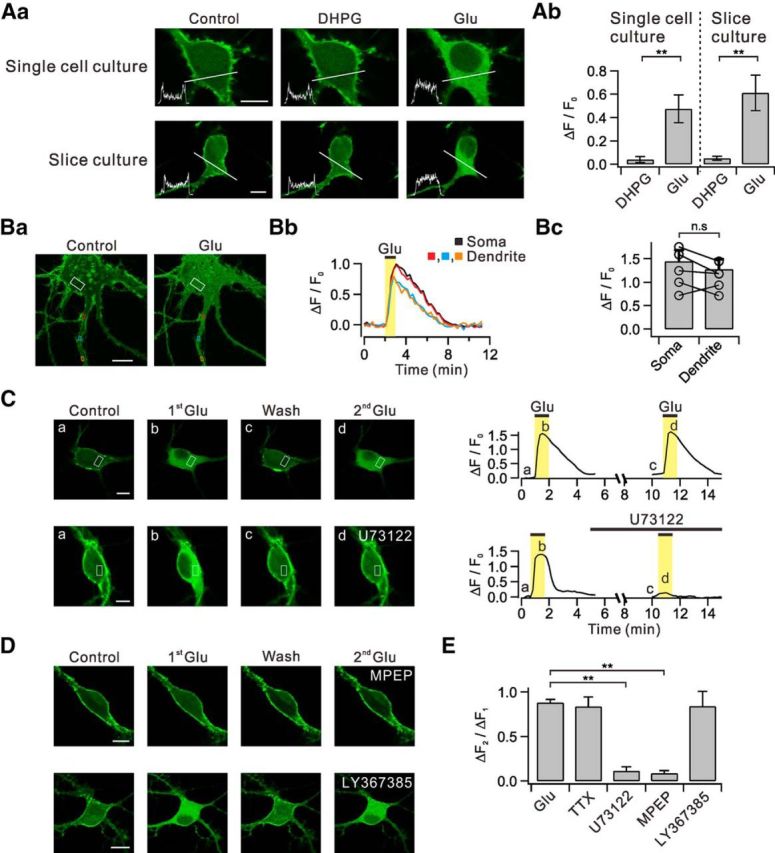
Glutamate, but not DHPG, induces translocation of PHδ-GFP via the mGluR5-PLC pathways. Aa, Dissociated hippocampal neurons in primary culture (single-cell culture) transfected with PHδ-GFP showed prominent green fluorescence signals in plasma membrane versus cytosol (Control). DHPG and glutamate (Glu) was applied to bath to see whether they induce PHδ-GFP translocation. Line profiles of fluorescence intensity (insets) were obtained across the white lines. Similar series of experiments were performed with hippocampal neurons in organotypic slice culture transfected with PHδ-GFP. Ab, Summary data showing the relative amplitudes of DHPG-induced and glutamate-induced PHδ-GFP translocation (ΔF/F0) in single-cell and slice culture conditions. Ba, Glutamate-induced PHδ-GFP translocation (ΔF/F0) was measured in ROIs of somatic cytosol (white) and multiple regions of dendritic cytosol (red, blue, and orange). Bb, Time courses of ΔF/F0 measured in designated ROIs. Bc, Bar graphs summarize the amplitudes of DHPG-induced or glutamate-induced PHδ-GFP translocation in soma and dendrites. C, Consecutive applications of 30 μm glutamate in control and in the presence of U73122. Images of PHδ-GFP-transfected neurons (left) and time courses of ΔF/F0 measured in designated ROIs (right). D, Effect of specific blockers for mGluR5 or mGluR1 on PHδ-GFP translocation. E, Bar graphs summarize ΔF2/ΔF1 in the presence of tetrodotoxin (TTX) and other experimental conditions described in C and D. n.s p > 0.05; **p < 0.01. Scale bar, 10 μm in all panels where indicated. Error bars represent SEM.
Figure 2.

Ca2+ influx triggered by AMPA receptor activation facilitates glutamate-induced PHδ-GFP translocation. Aa, Images demonstrate glutamate-induced translocation of PHδ-GFP in the presence of CNQX (AMPA receptor blocker) and APV (NMDA receptor blocker). Ab, Glutamate-induced [Ca2+]i in the presence of CNQX and APV. Error bars are shown in light colors. Ac, Bar graphs summarize mean ΔF2/ΔF1 (black) and [Ca2+]Glu2/[Ca2+]Glu1 (red). Ba, Images demonstrate glutamate-induced translocation of PHδ-GFP in Ca2+-free solutions or in the presence of thapsigargin (a sarco-endoplasmic reticulum Ca2+-ATPase blocker). Bb, Glutamate-induced [Ca2+]i in experimental conditions described in Ba. Bc, Bar graphs summarize mean ΔF2/ΔF1 (black) and [Ca2+]Glu2/[Ca2+]Glu1 (red). Ca, Images of PHδ-GFP translocation induced by 50 mm KCl only or 50 mm KCl plus DHPG (top) and time courses of ΔF/F0 measured in designated ROIs (bottom). Cb, Plot of [Ca2+]i versus different concentrations of KCl. Gray circles represent individual data. Cc, ΔF/F0 values are summarized in bar graph in experimental conditions described in Ca. **p < 0.01. Scale bar, 10 μm in all panels where indicated. Error bars represent SEM.
Figure 5.
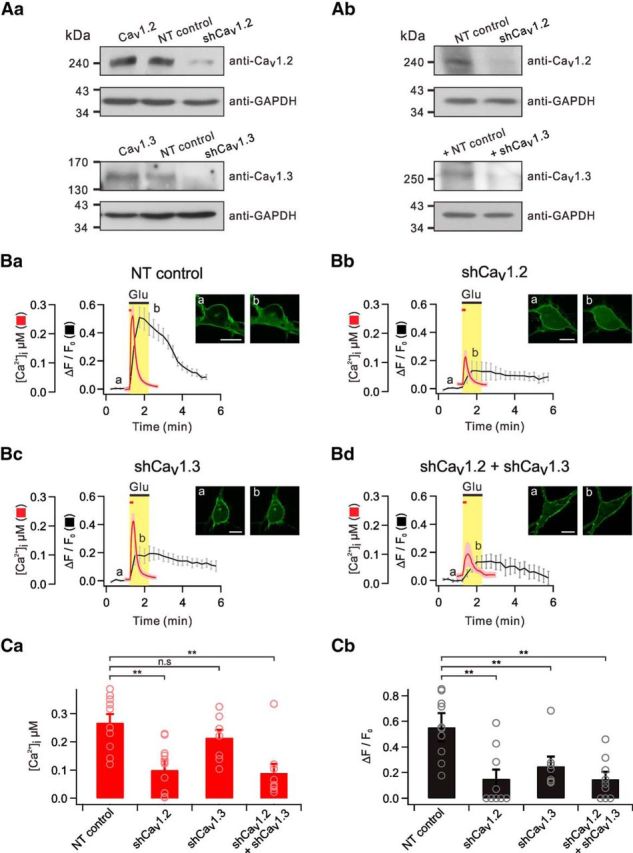
Both Cav1.2 and Cav1.3 contribute to glutamate-induced PHδ-GFP translocation. Aa, Western blotting of overexpressed Cav1.2 (top) and Cav1.3 (bottom) in HEK293 cells transfected with NT control, Cav1.2 shRNA (shCav1.2), or Cav1.3 shRNA (shCav1.3). Ab, Western blotting of endogenous Cav1.2 (top) and Cav1.3 (bottom) in primary cultured hippocampal neurons transfected with NT control, shCav1.2, or shCav1.3. GAPDH served as a loading control. Ba–Bc, Average of glutamate-induced [Ca2+]i and ΔF/F0 are plotted against time in cells transfected with NT control, shCav1.2, or shCav1.3. The insets show representative images of PHδ-GFP translocation at indicated time points (a, b). Scale bar, 10 μm. C, Bar graph summarizes [Ca2+]i (Ca) or ΔF/F0 (Cb) for each group. n.s p > 0.05; **p < 0.01. Error bars represent SEM.
Figure 3.

L-type Ca2+ channels provide Ca2+ for glutamate-induced PHδ-GFP translocation. A–D, Averages of glutamate-induced [Ca2+]i (red) and relative ΔF (black) are plotted against time for two successive applications of glutamate (first applications are glutamate only, and second applications are glutamate plus L-type, T-type, or N/P/Q-type Ca2+ channel blockers, or Ca2+-permeable AMPA receptor blocker). The time intervals of first and second glutamate applications are actual values (20 min) for ΔF, but for [Ca2+]i measurements the time intervals were 4 min, as described in Figure 2Ab: they were superimposed for comparison of time-dependent events. The insets show representative images of PHδ-GFP translocation at indicated time points (a–d). Scale bar, 10 μm. E, Bar graphs summarize mean ΔF2/ΔF1 (black) and [Ca2+]Glu2/[Ca2+]Glu1 (red). F, ΔF2/ΔF1 values are plotted versus [Ca2+]Glu2/[Ca2+]Glu1 values. Dashed line indicates linear relationship. **p < 0.01; *p < 0.05. Error bars represent SEM.
Figure 4.
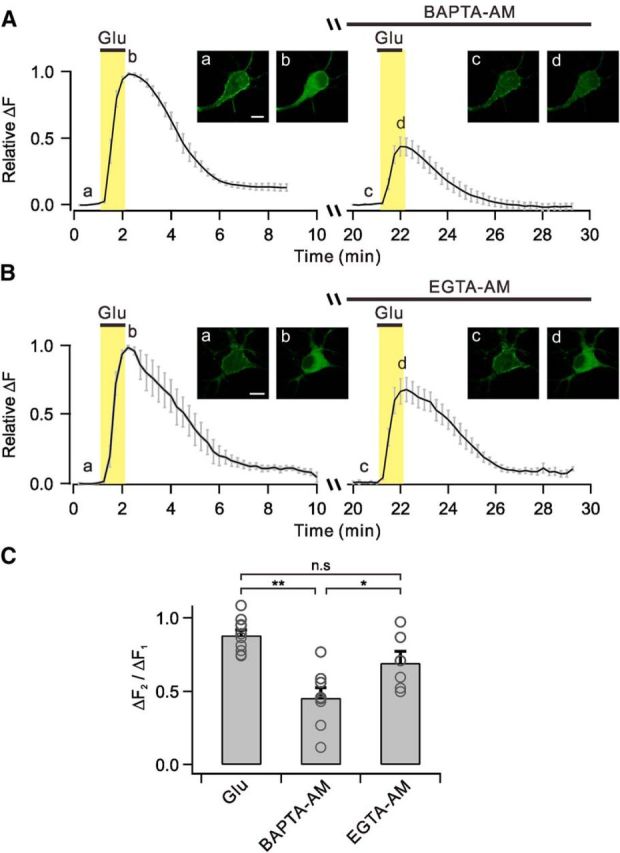
BAPTA, but not EGTA, inhibits glutamate-induced PHδ-GFP translocation. A–B, Averages of glutamate-induced relative ΔF are plotted against time with BAPTA-AM (A) or EGTA-AM (B) loadings before second glutamate applications. The insets show representative images of PHδ-GFP translocation at indicated time points (a–d). Scale bar, 10 μm. C, Bar graphs summarize mean ΔF2/ΔF1 in experimental conditions described in A and B. n.s p > 0.05; **p < 0.01; *p < 0.05. Error bars represent SEM.
Calcium measurements in hippocampal primary neurons.
DIV7–DIV14 primary cultured hippocampal neurons were loaded by incubation with 2 μm Fura-2-acetoxymethyl ester (Fura-2AM) plus 0.01% Pluronic F-127 in normal tyrode solution for 10 min at RT then washed for 10 min to remove excess calcium indicators. For fluorescence excitation, we used a polychromatic light source (xenon-lamp based, Polychrome-IV, TILL-Photonics), which was coupled to the epi-illumination port of an inverted microscope (IX70, Olympus) via a quartz light guide and a UV condenser. Microfluorometry was performed with a 40× water-immersion objective (numerical aperture, 1.15; UApo 40× W/340, Olympus) and a photodiode (TILL-Photonics). Standard two-wavelength protocol was used for fluorescence measurements of cells. Fluorescence intensity at an ROI including the soma was measured at 1 Hz with double-wavelength excitation at 340 nm (F340) and 380 nm (F380). The ratio r = F340/F380 was converted to [Ca2+]i values according to the following equation: [Ca2+]i = Keff (R − Rmin)/(Rmax − R), where Keff was estimated as 0.93 μm. Calibration parameters were determined by using in vivo calibration (Lee et al., 2000) and estimated Rmin and Rmax were typically 0.24 and 3.4, respectively.
Electrophysiological recordings.
For electrophysiological recordings, acute hippocampal brain slices were prepared from P16–P19 SD rats of either sex. Following decapitation, the whole brain was immediately removed and submerged in ice-cold aCSF. A vibratome (VT1200S, Leica) was used to prepare transverse hippocampal slices (350 μm thick). Slices were recovered at 32°C for 30 min and thereafter maintained at RT until used for recordings. Hippocampal CA1 pyramidal cells were visualized using an upright microscope equipped with differential interference contrast optics (BX51WI, Olympus). Electrophysiological recordings were made in somata with EPC-8 amplifier (HEKA Electronik) at a sampling rate of 10 kHz. All the recordings were performed at 32 ± 1°C and the rate of aCSF perfusion was maintained at 1–1.5 ml min−1. EPSCs at SC-CA1 synapses were recorded from CA1-PCs in a whole-cell configuration at a holding potential of −63 mV. Patch pipettes (2–4 MΩ) were filled with internal solutions containing the following (in mm): 130 potassium gluconate, 7 KCl, 2 NaCl, 1 MgCl2, 0.1 EGTA, 2 ATP-Mg, 0.3 Na-GTP, 10 HEPES, pH 7.3 with KOH, 295 mosmol l−1 with sucrose). Stimulator (Stimulus Isolator A360, WPI) connected to a monopolar electrode filled with aCSF was placed in stratum radiatum of the CA1 field to evoke synaptic responses. The intensity (100 μs duration; 5–25 μA) of extracellular stimulation was adjusted to evoke EPSC amplitudes in the range between 50 and 300 pA. After 5–10 min of stabilization from patch break-in, EPSCs were recorded at 0.1 Hz for 5 min baseline recordings, which was followed by DHPG application (8 min) or PP-LFS (1 Hz, 15 min) to induce a mGluR-LTD. The PP-LFS was performed in current-clamp mode. All recordings were performed in the presence of NMDAR antagonists (50 μm APV) to block NMDAR-dependent LTD.
Drugs.
DHPG, LY367385, MPEP, CNQX, APV, and ω-conotoxin MVIIC were purchased from Tocris Bioscience. U73122 was from Biomol. Fura-2AM, EGTA-AM, and BAPTA-AM were from Invitrogen. All other drugs were purchased from Sigma-Aldrich. Stock solutions of these drugs were made by dissolution in deionized water or DMSO and were stored at −20°C. On the day of the experiment one aliquot was thawed and used. The concentration of DMSO in solutions was maintained at <0.1%.
Statistical analysis.
Data were analyzed using IgorPro (version 4.1, WaveMetrics), OriginPro (version 8.0, Microcal) software, and Fiji-win 32 and are presented as mean ± SEM, where n represents the number of cells studied. The statistical significance of differences between the peaks was evaluated using a Student's t test with confidence levels of p < 0.01 (**), p < 0.05 (*) and p > 0.05 (not significant).
Results
Glutamate, but not DHPG, induces robust PLC activation in hippocampal neurons
The intracellular signaling cascades elicited by DHPG application were independent of PLC in previous studies (Schnabel et al., 1999; Fitzjohn et al., 2001; Ireland and Abraham, 2002; Sohn et al., 2007, 2011; Mockett et al., 2011). In addition, it is suspected that the apparent PLC independence of mGluR-LTD may be due to the use of DHPG to induce LTD instead of more physiological methods (Lüscher and Huber, 2010). Thus, we directly tested the idea that the ability to activate PLC may be different between DHPG and glutamate (the latter of which is released by fiber stimulation). To this end, we used PHδ-GFP constructs to visualize PLC activation, as described previously (Gamper et al., 2004; Horowitz et al., 2005).
We transfected primary cultured dissociated hippocampal neurons with PHδ-GFP construct (see Materials and Methods), and confirmed preferential localization of fluorescent signals at the plasma membrane (single-cell culture, Control; Fig. 1Aa). Interestingly, the application to the bath solutions of (RS)-3,5-DHPG (50 μm), a specific group I mGluR agonist, did not induce discernable PHδ-GFP translocation (DHPG: ΔF/F0 = 0.04 ± 0.03, n = 10), whereas glutamate (30 μm) caused a robust translocation of PHδ-GFP into the cytosol in the same population (Glu: ΔF/F0 = 0.47 ± 0.12, n = 10, p < 0.01; Fig. 1Aa,Ab). Line profiles of fluorescence intensity demonstrated that the cytosolic fluorescence, which reflects the generation of IP3, remained unchanged with DHPG treatment, but was significantly increased by glutamate treatment (Fig. 1Aa, insets). To rule out the possibility that these observations are limited to dissociated hippocampal neurons, we repeated similar series of experiments in hippocampal slices. In organotypic slice culture systems, we confirmed that glutamate, but not DHPG, induced PHδ-GFP translocation in the same population (DHPG: ΔF/F0 = 0.05 ± 0.02, n = 6; Glu: ΔF/F0 = 0.57 ± 0.13, n = 7, p < 0.01; Fig. 1Aa,Ab). Thus, our results suggest that stimulation of group I mGluR alone is not sufficient to activate PLC in hippocampal neurons.
Glutamate stimulates mGluR5 to activate PLC in hippocampal neurons
Glutamate-induced translocation of PHδ-GFP was observed in multiple regions of the proximal dendrites (Fig. 1Ba,Bb), and these were not significantly different from those observed in the soma (soma: ΔF/F0 = 1.44 ± 0.24; dendrite: ΔF/F0 = 1.28 ± 0.21, n = 6, p > 0.05; Fig. 1Bc). The increase in cytosolic fluorescence by glutamate was reliably repeated with similar magnitude when glutamate was reapplied after a ∼10 min interval (ΔF2/ΔF1 = 88.0 ± 3.6%, n = 10; Fig. 1C,E). For some experiments, we added tetrodotoxin (1 μm) in the bath solution before second glutamate application to block action potential-dependent events, and glutamate still induced reliable translocation of PHδ-GFP (ΔF2/ΔF1 = 83.6 ± 10.8%, n = 5, p > 0.05; Fig. 1E). The glutamate-induced translocation of PHδ-GFP was significantly inhibited by PLC inhibitor U73122 (1 μm; ΔF2/ΔF1 = 8.6 ± 3.1, n = 4, p < 0.01; Fig. 1C,E) and 25 μm MPEP, a specific mGluR5 blocker (ΔF2/ΔF1 = 11.2 ± 4.8%, n = 4, p < 0.01; Fig. 1D,E), but it was not affected by LY367385 (100 μm), a specific mGluR1 blocker (ΔF2/ΔF1 = 84.1 ± 16.7%, n = 5, p > 0.05; Fig. 1D,E). These results demonstrate that glutamate-induced translocation of PHδ-GFP is independent of action potential firing and occurs via the activation of mGluR5 and PLC.
Ca2+ influx via AMPA receptor activation is required for PLC activation by glutamate
Given the differential effects of DHPG and glutamate, we suspected that glutamate may provide additional signaling mechanisms required for PLC activation via iGluRs. To test this idea, we examined the contribution of iGluRs using 10 μm CNQX or 50 μm APV, specific antagonists for AMPA receptors and NMDA receptors, respectively. As shown in Figure 2Aa,Ac, glutamate-induced translocation of PHδ-GFP was blocked by CNQX (ΔF2/ΔF1 = 16.8 ± 4.2%, n = 5, p < 0.01), but it was not significantly affected by APV (ΔF2/ΔF1 = 69.3 ± 8.0%, n = 5, p > 0.05). We also found that glutamate-induced translocation of PHδ-GFP was completely suppressed in Ca2+-free bath solutions (ΔF2/ΔF1 = 9.0 ± 5.9%, n = 6, p < 0.01), but it was not significantly affected by 2 μm thapsigargin (ΔF2/ΔF1 = 89.6 ± 8.5%, n = 4, p > 0.05), which suggested a role of Ca2+ influx, but not Ca2+ mobilization (Fig. 2Ba,Bc). Thus, our results suggest that Ca2+ entry triggered by AMPA receptor activation may contribute to mGluR5-mediated PLC activation.
In a separate series of experiments, we characterized glutamate-induced Ca2+ transients ([Ca2+]i) to identify Ca2+ sources required for PLC activation by mGluR5 in primary cultured dissociated hippocampal neurons loaded with 2 μm Fura-2AM. Bath applications of glutamate (30 μm) caused a rapid increase in [Ca2+]i with an average amplitude of 438 ± 27 nm (n = 67). We found that glutamate-induced [Ca2+]i increase was completely suppressed in Ca2+-free bath solution ([Ca2+]2/[Ca2+]1 = 0.22 ± 0.1%, n = 4, p < 0.01), but it is not affected by 2 μm thapsigargin ([Ca2+]2/[Ca2+]1 = 89.5 ± 8.5%, n = 4, p > 0.05; Fig. 2Bb,Bc), indicating that the glutamate-induced [Ca2+]i increase is mostly mediated by Ca2+ influx, but not by Ca2+ release in cultured hippocampal neurons. We also confirmed that CNQX profoundly inhibits glutamate-induced [Ca2+]i ([Ca2+]2/[Ca2+]1 = 5.3 ± 3.4%, n = 5, p < 0.01), but APV has no effect ([Ca2+]2/[Ca2+]1 = 92.0 ± 11.1%, n = 5, p > 0.05; Fig. 2Ab,Ac). These results suggest that AMPA receptors, but not NMDA receptors, are largely responsible for the Ca2+ influx by glutamate. Given the calcium dependence of PLC activity (Ryu et al., 1987; Rebecchi and Pentyala, 2000), AMPA receptor-mediated depolarization and subsequent Ca2+ influx via voltage-gated Ca2+ channels (VGCCs) may facilitate PLC activation by mGluR5.
To test whether the opening of VGCCs independent of AMPA receptor stimulation can facilitate PLC activation by mGluR5, we elevated external K+ concentration from 5 to 50 mm. We added CNQX (10 μm) to the bath solution for these experiments so that we may exclude the contribution of AMPA receptor-mediated depolarization by glutamate that is released from presynaptic terminals in high-K+ conditions. Under this experimental condition, 50 mm KCl alone increased [Ca2+]i to 697 ± 86 nm (n = 12), but had no effects on PHδ-GFP translocation (ΔF/F0 = 0.08 ± 0.03, n = 5; Fig. 2Ca–Cc). When we added DHPG to 50 mm KCl, it robustly induced PHδ-GFP translocation (ΔF/F0 = 1.03 ± 0.2, n = 10, p < 0.01; Fig. 2Ca,Cc). These findings further support the idea that Ca2+ influx through VGCCs is required for PLC activation by mGluR5.
Both L-type and T-type Ca2+ channels mediate Ca2+ influx, but only L-type Ca2+ channels contribute to PLC activation
We used specific Ca2+ channel blockers to identify specific Ca2+ channel subtypes that mediate Ca2+ influx and contribute to PLC activation. We found that glutamate-induced [Ca2+]i is significantly inhibited when pretreated with nimodipine (10 μm), an L-type Ca2+ channel blocker ([Ca2+]2/[Ca2+]1 = 47.4 ± 10.1%, n = 7, p < 0.01; Fig. 3A,E). Consistent with the effects on Ca2+ influx, 10 μm nimodipine significantly blocked increase of cytosolic fluorescence (ΔF2/ΔF1 = 47.1 ± 7.9%, n = 5, p < 0.01; Fig. 3A,E). We confirmed that a lower concentration (1 μm) of nimodipine inhibited [Ca2+]i or PHδ-GFP translocation to a similar extent ([Ca2+]2/[Ca2+]1 = 44.1 ± 6.2%, n = 7, p < 0.01; ΔF2/ΔF1 = 52.9 ± 7.9%, n = 5, p < 0.01). We also confirmed that 10 μm nimodipine significantly inhibits increase of cytosolic fluorescence in the proximal dendrite as well (data not shown).
Mibefradil (5 μm), a T-type Ca2+ channel blocker, also significantly inhibited the glutamate-induced [Ca2+]i ([Ca2+]2/[Ca2+]1 = 53.8 ± 3.0%, n = 9, p < 0.01; Fig. 3B,E). In addition, simultaneous application of nimodipine (10 μm) and mibefradil (5 μm) had additive effect, inhibiting glutamate-induced [Ca2+]i by ∼75% ([Ca2+]2/[Ca2+]1 = 25.0 ± 5.3%, n = 5, p < 0.01). Interestingly, however, mibefradil did not inhibit glutamate-induced PHδ-GFP translocation (ΔF2/ΔF1 = 108.6 ± 24.8%, n = 6, p > 0.05; Fig. 3B,E). We obtained similar results with another T-type Ca2+ channel blocker, NiCl2 (100 μm; [Ca2+]2/[Ca2+]1 = 53.5 ± 4.4%, n = 5, p < 0.01; ΔF2/ΔF1 = 110.1 ± 22.4%, n = 5, p > 0.05; Fig. 3E).
The amplitude of glutamate-induced [Ca2+]i or PHδ-GFP translocation was not significantly affected by ω-conotoxin MVIIC (1 μm; [Ca2+]2/[Ca2+]1 = 97.9 ± 6.6%, n = 5, p > 0.05; ΔF2/ΔF1 = 94.4 ± 6.3%, n = 5, p > 0.05), indicating that N-type and P/Q type Ca2+ channels are not involved (Fig. 3C,E). It was noted that 1-naphthyl acetyl spermine (10 μm), a specific Ca2+-permeable AMPA blocker, slightly suppressed glutamate-induced [Ca2+]i ([Ca2+]2/[Ca2+]1 = 80.0 ± 1.7%, n = 4, p < 0.05), but had no effect on PHδ-GFP translocation (ΔF2/ΔF1 = 84.9 ± 11.7%, n = 6, p > 0.05; Fig. 3D,E).
We found that most glutamate-induced Ca2+ influx is mediated by L-type and T-type Ca2+ channels, but Ca2+ influx through the L-type Ca2+ channels exclusively contributes to PLC activation by mGluR5 stimulation. Thus, the contribution of each Ca2+ source (L-type and T-type Ca2+ channels) to global Ca2+ transients is not proportional to its contribution to PLC activation (Fig. 3F). These findings suggest that PLC activation by glutamate is not regulated by global Ca2+, but by local Ca2+. Therefore, it can be hypothesized that Ca2+ sensors involved in mGluR5-induced PLC activation is localized very closely to the L-type Ca2+ channels.
BAPTA, but not EGTA, inhibits PLC activation by glutamate significantly
The coupling distance between Ca2+ sources and Ca2+ sensors is a key determinant of signaling properties, and it can be probed by comparing the effects of BAPTA and EGTA. BAPTA, a fast Ca2+ chelator, effectively captures the Ca2+ on its way from the Ca2+ channels to the Ca2+ sensors in a short distance (<100 nm), whereas EGTA, a slow Ca2+ chelator, selectively spares nanodomain Ca2+ signals (Neher, 1998; Eggermann and Jonas, 2012). Therefore, signal transmission is impaired by only BAPTA, but not by EGTA, when the coupling distance is short (nanodomain coupling), whereas both BAPTA and EGTA are effective when the coupling distance is longer (microdomain coupling). We used the same experimental approach to probe the coupling distance between L-type Ca2+ channels and Ca2+ sensors involved in mGluR5-induced PLC activation. First, we confirmed that the glutamate-induced increase in global Ca2+ was almost completely abolished in cultured hippocampal neurons loaded with BAPTA-AM (100 μm) or EGTA-AM (100 μm) for 30 min (data not shown). Nonetheless, the glutamate-induced translocation of PHδ-GFP was significantly inhibited in BAPTA-loaded cells (ΔF2/ΔF1 = 45.4 ± 7.0%, n = 8, p < 0.01), but was not affected significantly in EGTA-loaded cells (ΔF2/ΔF1 = 69.3 ± 7.9%, n = 6, p > 0.05; p < 0.05 with BAPTA-loaded cells) supporting the nanodomain coupling between L-type Ca2+ channels and the Ca2+ sensors involved in PLC activation (Fig. 4A–C).
Both Cav1.2 and Cav1.3 contribute to PLC activation by glutamate
Cav1.2 and Cav1.3 are the most widely expressed L-type Ca2+ channels in neurons (Hell et al., 1993). We used an RNA interference technique to determine the contribution of Cav1.2 and Cav1.3 Ca2+ channels to the glutamate-induced PHδ-GFP translocation (see Materials and Methods). Immunoblot analyses of HEK293 cells or primary cultured hippocampal neurons transfected with respective DNA constructs revealed that the expression of both Cav1.2 and Cav1.3 was significantly decreased by targeted shRNAs compared with NT controls (Fig. 5Aa,Ab).
Glutamate-induced [Ca2+]i remained unchanged up to 7 d following transfection of primary cultured dissociated hippocampal neurons with NT control, and the average [Ca2+]i was 267.3 ± 30.4 nm (n = 10) at 7 d post-transfection (dpt; Fig. 5Ba,Ca). It was noted that glutamate-induced [Ca2+]i in neurons transfected with Cav1.2-targeting shRNA (shCav1.2) was significantly decreased at 7 dpt ([Ca2+]i = 99.7 ± 21.4, n = 14, p < 0.01; Fig. 5Bb,Ca). In contrast, glutamate-induced [Ca2+]i measured in neurons transfected with Cav1.3-targeting shRNA (shCav1.3) was slightly reduced, but was not significantly different from those transfected with NT control ([Ca2+]i = 215.0 ± 27.4, n = 8, p > 0.05; Fig. 5Bc,Ca). These data suggest that Cav1.2 is the major isoform that mediates glutamate-induced Ca2+ influx compared with Cav1.3.
Significant effects of Cav1.2 knock-down on glutamate-induced Ca2+ increase were observed after 5 dpt. Thus, the effects of Cav1.2 and Cav1.3 knock-down on glutamate-induced PHδ-GFP translocation were tested in neurons between 6 and 8 dpt. The normalized average amplitude (ΔF/F0) of the cytosolic fluorescence increment by glutamate in nontargeted cells was 0.55 ± 0.07 (n = 10), and it was significantly decreased to 0.15 ± 0.07 (n = 9, p < 0.01) in neurons transfected with shCav1.2 (Fig. 5Ba,Bb,Cb). The magnitude of inhibition by shCav1.2 of glutamate-induced PHδ-GFP translocation was comparable to that of glutamate-induced Ca2+ increase. Interestingly, glutamate-induced PHδ-GFP translocation was also significantly reduced by shCav1.3 (ΔF/F0 = 0.24 ± 0.06, n = 9, p < 0.01; Fig. 5Bc,Cb). Finally, the effects of Cav1.2 and Cav1.3 knock-down on Ca2+ transients ([Ca2+]i = 89.5 ± 32.4, n = 9, p < 0.01; Fig. 5Bd,Ca) and PHδ-GFP translocation (ΔF/F0 = 0.15 ± 0.05, n = 9, p < 0.01; Fig. 5Bd,Cb) were comparable to the effects of Cav1.2 knock-down alone. These data predict that the magnitude of Ca2+ influx is much larger for Cav1.2 than for Cav1.3, but both subtypes contribute to PLC activation by mGluR5.
mGluR-LTD is dependent on PLC and L-type Ca2+ channels at SC-CA1 pyramidal neuron excitatory synapse
We now have evidence that DHPG is not sufficient to activate PLC in hippocampal neurons, which may be a clue for answering the question why mGluR-LTD induced by DHPG (DHPG-LTD) is independent of PLC, PKC, IP3, and postsynaptic Ca2+ (Schnabel et al., 1999; Fitzjohn et al., 2001; Mockett et al., 2011). We confirmed that DHPG-LTD is not affected by the PLC inhibitor U73122 (Control: 59.9 ± 5.8% of baseline at 40 min after 100 μm DHPG, n = 5; U73122: 64.7 ± 3.7% of baseline at 40 min after 100 μm DHPG in the presence of 10 μm U73122, n = 5, p > 0.05; Fig. 6A).
Figure 6.

PLC is involved in mGluR-LTD induced by PP-LFS-LTD, but not in DHPG-LTD. EPSC amplitudes were normalized to the pre-DHPG or PP-LFS baseline values and were averaged. A, DHPG application induced a persistent depression of EPSC amplitude (black). Preincubation of slices in U73122 did not affect the magnitude of DHPG-LTD (red). The inset shows representative EPSCs during the baseline period (dark) and 40 min after (light) DHPG application in each condition. B, PP-LFS induced a similar magnitude of LTD compared with DHPG-LTD (black). PP-LFS-LTD was significantly blocked by U73122 (red). C, Pharmacological blockade of mGluR5 by MPEP almost completely blocked PP-LFS-LTD (red). D, PP-LFS-LTD was blocked by nimodipine (red). Insets (B–D) represent EPSCs during the baseline period (dark) and 35 min after (light) PP-LFS in each condition. Ea, Representative EPSCs elicited by paired-pulse stimulation (50 ms interval) during baseline (black, left) and after DHPG application or PP-LFS (gray, middle). Superimposed traces are shown in right panels. Scale bar, 50 ms (horizontal) and 100 pA (vertical). Eb, Bar graphs summarize paired-pulse ratio (PPR) changes in each group. n.s p > 0.05. Error bars represent SEM.
It should be noted that mGluR-LTD induced by PP-LFS (PP-LFS-LTD) was demonstrated to involve PLC and PLC-dependent signal pathways (e.g., PKC and postsynaptic Ca2+; Bolshakov and Siegelbaum, 1994; Oliet et al., 1997; Otani and Connor, 1998; Reyes-Harde and Stanton, 1998; Lee et al., 2005; Holbro et al., 2009). Possibly, PP-LFS to SC fibers induces Ca2+ channel activation associated with action potentials or dendritic Ca2+ spikes, thus contributing to PLC activation. To test this idea, we examined the effects of blocking PLC or Ca2+ channels on the changes of EPSC amplitude induced by PP-LFS (1 Hz for 15 min), which consistently induces mGluR-LTD (Lüscher and Huber, 2010). To inhibit NMDAR-dependent LTD, all recordings were performed in the presence of 50 μm APV. We confirmed that after PP-LFS to SC fibers, EPSC amplitude was significantly reduced and this reduction lasted as long as the recording was maintained (52.4 ± 3.7% of baseline at 35 min after PP-LFS, n = 8; Fig. 6B–D). The induction of LTD by PP-LFS was prevented by 10 μm U73122 (98.2 ± 2.3% of baseline at 35 min after PP-LFS, n = 6, p < 0.01; Fig. 6B). We also confirmed that mGluR-LTD induced by PP-LFS is almost completely suppressed by MPEP (86.3 ± 6.0% of baseline at 35 min after PP-LFS, n = 6, p < 0.01; Fig. 6C). We confirmed that the paired-pulse ratio remained unchanged by DHPG-LTD (Control: 150.4 ± 5.8%; DHPG-LTD: 157.8 ± 4.9%, n = 5, p > 0.05) and PP-LFS-LTD (Control: 148.8 ± 5.0%; PP-LFS-LTD: 160.7 ± 9.7%, n = 8, p > 0.05; Fig. 6Ea,Eb). These findings demonstrate that mGluR-LTD induced by PP-LFS of SC axons onto the CA1 pyramidal neurons requires PLC activation.
Finally, we tested whether L-type Ca2+ channels, which provide Ca2+ for PLC activation, are required for mGluR-LTD. We found that in the presence of 20 μm nimodipine, PP-LFS to SC pathway did not induce LTD (92.0 ± 5.8% of baseline at 35 min after PP-LFS, n = 7, p < 0.01; Fig. 6D). Together, these findings demonstrate that mGluR-LTD at SC-CA1 synapses induced by PP-LFS involves PLC activation, and Ca2+ influx via L-type Ca2+ channels serves as a prerequisite.
Discussion
In the present study, we demonstrated that mGluR5 stimulation is not sufficient for PLC activation in hippocampal neurons, but that Ca2+ influx via the L-type Ca2+ channels following AMPA receptor stimulation/membrane depolarization facilitates PLC activation. In addition, we found evidence that the coupling distance between L-type Ca2+ channels and Ca2+ sensors involved in PLC activation is very short (<100 nm). Finally, we have demonstrated the physiological significance of our findings by directly showing the involvement of PLC exclusively in PP-LFS-LTD at SC-CA1 synapses. Given that mGluR-LTD represents a decrease in AMPA receptor activity by group I mGluRs (Lüscher and Huber, 2010), it is interesting to note that AMPA receptors initiate Ca2+ influx, which facilitates mGluR5/PLC signaling, and this leads to a long-lasting decrease of AMPA receptor activity, which may be regarded as a negative feedback from mGluRs to AMPA receptors. Our data provide novel insight into the interaction between AMPA receptors and group I mGluRs as well as the Ca2+ dependence of mGluR5-PLC signaling cascades in hippocampal neurons.
Ca2+ dependence of PLC activation by group I mGluRs and muscarinic receptors was previously reported (Masgrau et al., 2000, 2001; Hashimotodani et al., 2005; Maejima et al., 2005). In cerebellar granule cells, activation of PLC by glutamate, which is mediated by mGluR1, was dependent on the external Ca2+ (Masgrau et al., 2001), whereas muscarinic receptor-mediated activation of PLC was modulated by changes in the loading state of intracellular Ca2+ stores (Masgrau et al., 2000). These results suggest that the Ca2+ source facilitating PLC activation is specific to each receptor. Notably, these results are consistent with the idea that receptor-mediated PLC activation is regulated by local Ca2+. However, the specific Ca2+ source to induce local Ca2+ increase remained unclear. In the present study, we excluded the involvement of Ca2+ stores in glutamate-induced PLC activation by showing that thapsigargin affected neither glutamate-induced [Ca2+]i increase nor PHδ-GFP translocation (Fig. 2). From the effects of Ca2+ removal, we confirmed the role of Ca2+ influx in glutamate-induced [Ca2+]i increase and PLC activation, and investigated the involvement of specific Ca2+ influx pathways. Both L-type and T-type Ca2+ currents contribute similarly to glutamate-induced Ca2+ influx (Fig. 3), which is consistent with previous studies showing abundant expression of L-type (Cav1.2 and Cav1.3) and T-type (Cav3.1, Cav3.2, and Cav3.3) Ca2+ channels in the soma and proximal dendrites of hippocampal neurons (McKay et al., 2006; Leitch et al., 2009). Interestingly, however, only L-type Ca2+ channels contribute to PLC activation (Figs. 3, 5). The discrepancy between the contribution of each Ca2+ channel to global Ca2+ and its contribution to PLC activation supports the local Ca2+ dependence of mGluR5-mediated PLC activation in hippocampal neurons. In addition, BAPTA was more effective than EGTA at inhibiting mGluR-mediated PLC activation (Fig. 4), which confirms the nanodomain coupling between L-type Ca2+ channels and Ca2+ sensors involved in PLC activation. It needs to be noted that both glutamate-induced Ca2+ increases and PHδ-GHP translocation were completely abolished in Ca2+-free solutions, but the inhibition of L-type Ca2+ channels did not result in complete inhibition. These results suggest the presence of another Ca2+ entry pathway that couples to PLC activation.
Among members of the L-type Ca2+ channel subfamily (Cav1.1 to Cav1.4), Cav1.2 and Cav1.3 channels are expressed in neurons. They are often present in the same cell, but accumulating evidence shows that they play differential roles (Berger and Bartsch, 2014). Using hippocampal neurons derived from genetically modified mice that either lack Cav1.3 or express dihydropyridine-insensitive Cav1.2, researchers found that Cav1.2 and Cav1.3 provide Ca2+ influx that is coupled to distinct Ca2+-dependent conductance (Hasreiter et al., 2014). Since there are no pharmacological drugs that are selective for either the Cav1.2 or the Cav1.3 isoform, shRNA for each isoform was used to investigate whether the specific L-type Ca2+ channel subunits provide local Ca2+ for PLC activation by mGluR5 (Fig. 5). Inhibition of Ca2+ response by shCav1.2 was large (63% reduction) and significant, while the effect of shCav1.3 was not significant. The effect of knock-down of both subunits was not different from the effect of shCav1.2 alone, suggesting that Cav1.2 is the major isoform that mediates L-type Ca2+ currents. Furthermore, inhibition of PHδ-GFP translocation by shCav1.2 (73% reduction) was comparable to its effect on Ca2+ response, supporting the idea that close coupling between Cav1.2 and PLC underlies the observed Ca2+ dependence of glutamate-induced PLC activation. This idea is consistent with results from a recent study showing that mGluR5 physically interacts with Cav1.2 subunits in hippocampal neurons (Kato et al., 2012). It was surprising, however, to find that shCav1.3, which showed little effect on Ca2+ response, significantly inhibited glutamate-induced PHδ-GFP translocation (55% reduction), suggesting a significant role of Cav1.3 in PLC activation. However, the effect of shCav1.3 on PHδ-GFP translocation was not added to the effect of shCav1.2 in double-knock-down experiments. These results may suggest that Cav1.3 is involved in glutamate-induced PLC activation, but not as a Ca2+ source. We know of no molecular mechanisms that might account for the Ca2+-independent role of Cav1.3, but such possibility needs to be explored in future studies.
We found that DHPG alone fails to induce PHδ-GFP translocation (Fig. 1A), but this finding does not necessarily mean that DHPG fails to activate PLC at all. When PLC activity was assessed by measuring the accumulation of 3H-labeled inositol monophosphate and polyphosphate (3H-InsPs), DHPG increased 3H-InsPs accumulation by 2–3-fold compared with the basal level (Alagarsamy et al., 2002, 2005). Admittedly, inositol phosphate (IP) formation assay using 3H-InsPs accumulation is a very sensitive measure of PLC activity: even the basal level of PLC activity is detected and the effects of agonists on PLC activation are expressed as fold increases in 3H-InsPs accumulation. In contrast, the change in basal level or DHPG-induced increase is hardly detected by the fluorescence-based assay using PHδ-GFP translocation, suggesting that the sensitivity of this method may not be as high as 3H-InsPs accumulation measurement. Thus, the lack of PHδ-GFP translocation by DHPG does not mean that DHPG has no effect, but that PHδ-GFP translocation assay is not sensitive enough to detect small changes. In fact, previous results obtained from the IP formation assay do not contradict but rather agree with our results: DHPG and glutamate increased 3H-InsPs accumulation to 362 and 1344% of the basal level, respectively (Masgrau et al., 2001). The merit of PHδ-GFP translocation assay is that we can monitor dynamic changes at the single-cell level to investigate underlying mechanisms of PLC activation. Another method for detecting PLC activation from single cells is to use the canonical transient receptor potential channel 6 (TRPC6) as a biosensor (Hashimotodani et al., 2005), since TRPC6 is activated by DAG (Delmas et al., 2002; Hashimotodani et al., 2005). With this method, activation of TRPC6 by DHPG application was detected in hippocampal neurons (Hashimotodani et al., 2005), suggesting that TRPC6 activation is a more sensitive method than PHδ-GFP translocation for detecting PLC activation. However, the limitation to TRPC6 channels is that they serve as Ca2+-permeating channels. In fact, overexpression of TRPC6 caused Ca2+ overload (Kuwahara et al., 2006). Therefore, the overexpression of TRPC6 in hippocampal neurons may have provided Ca2+ influx to promote mGluR-mediated PLC activation. In this respect, TRPC6 may not be suitable as a biosensor for investigating Ca2+ sources for PLC activation.
mGluR-LTD has been linked to the etiology of multiple neurological and/or psychological diseases (e.g., mental retardation, autism, Alzheimer's disease, Parkinson's disease, and drug addiction; Lüscher and Huber, 2010; Ménard and Quirion, 2012). It is well established that mGluR-LTD in the cerebellum is dependent on PLC activation (Kano et al., 2008). Cerebellar mGluR-LTD requires both parallel fiber stimulation and the depolarization of Purkinje cell (postsynaptic) membranes (Kano et al., 2008). Likewise, when hippocampal mGluR-LTD was induced by PP-LFS of SC (i.e., when released glutamate depolarizes the postsynaptic membrane via AMPA receptors), mGluR-LTD consistently required postsynaptic Ca2+ (either Ca2+ mobilization or Ca2+ influx), PKC, and PLC (Bolshakov and Siegelbaum, 1994; Oliet et al., 1997; Otani and Connor, 1998; Reyes-Harde and Stanton, 1998; Lee et al., 2005; Holbro et al., 2009). In contrast, DHPG-LTD was found to be independent of postsynaptic Ca2+, PKC, and PLC (Schnabel et al., 1999; Fitzjohn et al., 2001; Mockett et al., 2011). Therefore, we suggest that mGluR-LTD in physiological conditions requires PLC, whereas chemical LTD (DHPG-LTD) may bypass PLC activation and use alternative signaling pathways.
In summary, we have demonstrated in this study the importance of AMPA receptor-initiated Ca2+ influx via L-type Ca2+ channels in PLC activation by mGluR5 in hippocampal neurons. We also confirmed that hippocampal mGluR-LTD, induced by the stimulation of SC fibers, is dependent on PLC. We propose that in physiological conditions glutamate activates both AMPA and metabotropic glutamate receptors and their downstream events interact with each other to ensure reliable PLC activation.
Footnotes
This work was supported by a grant from the Korean Ministry of Health and Welfare (HI14C1846 to J.-W.S.), by a National Research Foundation grant from the Korean Ministry of Science and Future Planning (2014051826 to W.-K.H.), and by the Brain Korea 21 PLUS Program. We thank Dr. Joo Min Park (Jeju National University) for helpful discussions.
The authors declare no competing financial interests.
References
- Abe T, Sugihara H, Nawa H, Shigemoto R, Mizuno N, Nakanishi S. Molecular characterization of a novel metabotropic glutamate receptor mGluR5 coupled to inositol phosphate/Ca2+ signal transduction. J Biol Chem. 1992;267:13361–13368. [PubMed] [Google Scholar]
- Alagarsamy S, Rouse ST, Junge C, Hubert GW, Gutman D, Smith Y, Conn PJ. NMDA-induced phosphorylation and regulation of mGluR5. Pharmacol Biochem Behav. 2002;73:299–306. doi: 10.1016/S0091-3057(02)00826-2. [DOI] [PubMed] [Google Scholar]
- Alagarsamy S, Saugstad J, Warren L, Mansuy IM, Gereau RWt, Conn PJ. NMDA-induced potentiation of mGluR5 is mediated by activation of protein phosphatase 2B/calcineurin. Neuropharmacology. 2005;49(Suppl 1):135–145. doi: 10.1016/j.neuropharm.2005.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aramori I, Nakanishi S. Signal transduction and pharmacological characteristics of a metabotropic glutamate receptor, mGluR1, in transfected CHO cells. Neuron. 1992;8:757–765. doi: 10.1016/0896-6273(92)90096-V. [DOI] [PubMed] [Google Scholar]
- Berger SM, Bartsch D. The role of L-type voltage-gated calcium channels Cav1.2 and Cav1.3 in normal and pathological brain function. Cell Tissue Res. 2014;357:463–476. doi: 10.1007/s00441-014-1936-3. [DOI] [PubMed] [Google Scholar]
- Bolshakov VY, Siegelbaum SA. Postsynaptic induction and presynaptic expression of hippocampal long-term depression. Science. 1994;264:1148–1152. doi: 10.1126/science.7909958. [DOI] [PubMed] [Google Scholar]
- Conn PJ, Pin JP. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- Delmas P, Wanaverbecq N, Abogadie FC, Mistry M, Brown DA. Signaling microdomains define the specificity of receptor-mediated InsP(3) pathways in neurons. Neuron. 2002;34:209–220. doi: 10.1016/S0896-6273(02)00641-4. [DOI] [PubMed] [Google Scholar]
- De Simoni A, Yu LM. Preparation of organotypic hippocampal slice cultures: interface method. Nat Protoc. 2006;1:1439–1445. doi: 10.1038/nprot.2006.228. [DOI] [PubMed] [Google Scholar]
- Doherty AJ, Palmer MJ, Henley JM, Collingridge GL, Jane DE. (RS)-2-chloro-5-hydroxyphenylglycine (CHPG) activates mGlu5, but no mGlu1, receptors expressed in CHO cells and potentiates NMDA responses in the hippocampus. Neuropharmacology. 1997;36:265–267. doi: 10.1016/S0028-3908(97)00001-4. [DOI] [PubMed] [Google Scholar]
- Eggermann E, Jonas P. How the “slow” Ca(2+) buffer parvalbumin affects transmitter release in nanodomain-coupling regimes. Nat Neurosci. 2012;15:20–22. doi: 10.1038/nn.3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Hassar L, Hagenston AM, D'Angelo LB, Yeckel MF. Metabotropic glutamate receptors regulate hippocampal CA1 pyramidal neuron excitability via Ca(2)(+) wave-dependent activation of SK and TRPC channels. J Physiol. 2011;589:3211–3229. doi: 10.1113/jphysiol.2011.209783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzjohn SM, Palmer MJ, May JE, Neeson A, Morris SA, Collingridge GL. A characterisation of long-term depression induced by metabotropic glutamate receptor activation in the rat hippocampus in vitro. J Physiol. 2001;537:421–430. doi: 10.1111/j.1469-7793.2001.00421.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamper N, Reznikov V, Yamada Y, Yang J, Shapiro MS. Phosphatidylinositol 4,5-bisphosphate signals underlie receptor-specific Gq/11-mediated modulation of N-type Ca2+ channels. J Neurosci. 2004;24:10980–10992. doi: 10.1523/JNEUROSCI.3869-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimotodani Y, Ohno-Shosaku T, Tsubokawa H, Ogata H, Emoto K, Maejima T, Araishi K, Shin HS, Kano M. Phospholipase Cbeta serves as a coincidence detector through its Ca2+ dependency for triggering retrograde endocannabinoid signal. Neuron. 2005;45:257–268. doi: 10.1016/j.neuron.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Hasreiter J, Goldnagl L, Böhm S, Kubista H. Cav1.2 and Cav1.3 L-type calcium channels operate in a similar voltage range but show different coupling to Ca(2+)-dependent conductances in hippocampal neurons. Am J Physiol Cell Physiol. 2014;306:C1200–C1213. doi: 10.1152/ajpcell.00329.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hell JW, Westenbroek RE, Warner C, Ahlijanian MK, Prystay W, Gilbert MM, Snutch TP, Catterall WA. Identification and differential subcellular localization of the neuronal class C and class D L-type calcium channel alpha 1 subunits. J Cell Biol. 1993;123:949–962. doi: 10.1083/jcb.123.4.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holbro N, Grunditz A, Oertner TG. Differential distribution of endoplasmic reticulum controls metabotropic signaling and plasticity at hippocampal synapses. Proc Natl Acad Sci U S A. 2009;106:15055–15060. doi: 10.1073/pnas.0905110106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horowitz LF, Hirdes W, Suh BC, Hilgemann DW, Mackie K, Hille B. Phospholipase C in living cells: activation, inhibition, Ca2+ requirement, and regulation of M current. J Gen Physiol. 2005;126:243–262. doi: 10.1085/jgp.200509309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ireland DR, Abraham WC. Group I mGluRs increase excitability of hippocampal CA1 pyramidal neurons by a PLC-independent mechanism. J Neurophysiol. 2002;88:107–116. doi: 10.1152/jn.2002.88.1.107. [DOI] [PubMed] [Google Scholar]
- Kaech S, Banker G. Culturing hippocampal neurons. Nat Protoc. 2006;1:2406–2415. doi: 10.1038/nprot.2006.356. [DOI] [PubMed] [Google Scholar]
- Kano M, Hashimoto K, Tabata T. Type-1 metabotropic glutamate receptor in cerebellar Purkinje cells: a key molecule responsible for long-term depression, endocannabinoid signalling and synapse elimination. Philos Trans R Soc Lond B Biol Sci. 2008;363:2173–2186. doi: 10.1098/rstb.2008.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato HK, Kassai H, Watabe AM, Aiba A, Manabe T. Functional coupling of the metabotropic glutamate receptor, InsP3 receptor and L-type Ca2+ channel in mouse CA1 pyramidal cells. J Physiol. 2012;590:3019–3034. doi: 10.1113/jphysiol.2012.232942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Dittgen T, Nimmerjahn A, Waters J, Pawlak V, Helmchen F, Schlesinger S, Seeburg PH, Osten P. Sindbis vector SINrep(nsP2S726): a tool for rapid heterologous expression with attenuated cytotoxicity in neurons. J Neurosci Methods. 2004;133:81–90. doi: 10.1016/j.jneumeth.2003.09.029. [DOI] [PubMed] [Google Scholar]
- Kuwahara K, Wang Y, McAnally J, Richardson JA, Bassel-Duby R, Hill JA, Olson EN. TRPC6 fulfills a calcineurin signaling circuit during pathologic cardiac remodeling. J Clin Invest. 2006;116:3114–3126. doi: 10.1172/JCI27702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Min SS, Gallagher M, Kirkwood A. NMDA receptor-independent long-term depression correlates with successful aging in rats. Nat Neurosci. 2005;8:1657–1659. doi: 10.1038/nn1586. [DOI] [PubMed] [Google Scholar]
- Lee KH, Lee JS, Lee D, Seog DH, Lytton J, Ho WK, Lee SH. KIF21A-mediated axonal transport and selective endocytosis underlie the polarized targeting of NCKX2. J Neurosci. 2012;32:4102–4117. doi: 10.1523/JNEUROSCI.6331-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Rosenmund C, Schwaller B, Neher E. Differences in Ca2+ buffering properties between excitatory and inhibitory hippocampal neurons from the rat. J Physiol. 2000;525:405–418. doi: 10.1111/j.1469-7793.2000.t01-3-00405.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitch B, Szostek A, Lin R, Shevtsova O. Subcellular distribution of L-type calcium channel subtypes in rat hippocampal neurons. Neuroscience. 2009;164:641–657. doi: 10.1016/j.neuroscience.2009.08.006. [DOI] [PubMed] [Google Scholar]
- Lüscher C, Huber KM. Group 1 mGluR-dependent synaptic long-term depression: mechanisms and implications for circuitry and disease. Neuron. 2010;65:445–459. doi: 10.1016/j.neuron.2010.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maejima T, Oka S, Hashimotodani Y, Ohno-Shosaku T, Aiba A, Wu D, Waku K, Sugiura T, Kano M. Synaptically driven endocannabinoid release requires Ca2+-assisted metabotropic glutamate receptor subtype 1 to phospholipase C β4 signaling cascade in the cerebellum. J Neurosci. 2005;25:6826–6835. doi: 10.1523/JNEUROSCI.0945-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannaioni G, Marino MJ, Valenti O, Traynelis SF, Conn PJ. Metabotropic glutamate receptors 1 and 5 differentially regulate CA1 pyramidal cell function. J Neurosci. 2001;21:5925–5934. doi: 10.1523/JNEUROSCI.21-16-05925.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masgrau R, Servitja JM, Sarri E, Young KW, Nahorski SR, Picatoste F. Intracellular Ca2+ stores regulate muscarinic receptor stimulation of phospholipase C in cerebellar granule cells. J Neurochem. 2000;74:818–826. doi: 10.1046/j.1471-4159.2000.740818.x. [DOI] [PubMed] [Google Scholar]
- Masgrau R, Servitja JM, Young KW, Pardo R, Sarri E, Nahorski SR, Picatoste F. Characterization of the metabotropic glutamate receptors mediating phospholipase C activation and calcium release in cerebellar granule cells: calcium-dependence of the phospholipase C response. Eur J Neurosci. 2001;13:248–256. doi: 10.1046/j.0953-816X.2000.01384.x. [DOI] [PubMed] [Google Scholar]
- McKay BE, McRory JE, Molineux ML, Hamid J, Snutch TP, Zamponi GW, Turner RW. Ca(V)3 T-type calcium channel isoforms differentially distribute to somatic and dendritic compartments in rat central neurons. Eur J Neurosci. 2006;24:2581–2594. doi: 10.1111/j.1460-9568.2006.05136.x. [DOI] [PubMed] [Google Scholar]
- Ménard C, Quirion R. Group 1 metabotropic glutamate receptor function and its regulation of learning and memory in the aging brain. Front Pharmacol. 2012;3:182. doi: 10.3389/fphar.2012.00182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mockett BG, Guévremont D, Wutte M, Hulme SR, Williams JM, Abraham WC. Calcium/calmodulin-dependent protein kinase II mediates group I metabotropic glutamate receptor-dependent protein synthesis and long-term depression in rat hippocampus. J Neurosci. 2011;31:7380–7391. doi: 10.1523/JNEUROSCI.6656-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Barbara JG, Nakamura K, Ross WN. Synergistic release of Ca2+ from IP3-sensitive stores evoked by synaptic activation of mGluRs paired with backpropagating action potentials. Neuron. 1999;24:727–737. doi: 10.1016/S0896-6273(00)81125-3. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Nakamura K, Lasser-Ross N, Barbara JG, Sandler VM, Ross WN. Inositol 1,4,5-trisphosphate (IP3)-mediated Ca2+ release evoked by metabotropic agonists and backpropagating action potentials in hippocampal CA1 pyramidal neurons. J Neurosci. 2000;20:8365–8376. doi: 10.1523/JNEUROSCI.20-22-08365.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E. Vesicle pools and Ca2+ microdomains: new tools for understanding their roles in neurotransmitter release. Neuron. 1998;20:389–399. doi: 10.1016/S0896-6273(00)80983-6. [DOI] [PubMed] [Google Scholar]
- Oliet SH, Malenka RC, Nicoll RA. Two distinct forms of long-term depression coexist in CA1 hippocampal pyramidal cells. Neuron. 1997;18:969–982. doi: 10.1016/S0896-6273(00)80336-0. [DOI] [PubMed] [Google Scholar]
- Otani S, Connor JA. Requirement of rapid Ca2+ entry and synaptic activation of metabotropic glutamate receptors for the induction of long-term depression in adult rat hippocampus. J Physiol. 1998;511:761–770. doi: 10.1111/j.1469-7793.1998.761bg.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebecchi MJ, Pentyala SN. Structure, function, and control of phosphoinositide-specific phospholipase C. Physiol Rev. 2000;80:1291–1335. doi: 10.1152/physrev.2000.80.4.1291. [DOI] [PubMed] [Google Scholar]
- Reyes-Harde M, Stanton PK. Postsynaptic phospholipase C activity is required for the induction of homosynaptic long-term depression in rat hippocampus. Neurosci Lett. 1998;252:155–158. doi: 10.1016/S0304-3940(98)00496-0. [DOI] [PubMed] [Google Scholar]
- Rubinson DA, Dillon CP, Kwiatkowski AV, Sievers C, Yang L, Kopinja J, Rooney DL, Zhang M, Ihrig MM, McManus MT, Gertler FB, Scott ML, Van Parijs L. A lentivirus-based system to functionally silence genes in primary mammalian cells, stem cells and transgenic mice by RNA interference. Nat Genet. 2003;33:401–406. doi: 10.1038/ng1117. [DOI] [PubMed] [Google Scholar]
- Ryan XP, Alldritt J, Svenningsson P, Allen PB, Wu GY, Nairn AC, Greengard P. The Rho-specific GEF Lfc interacts with neurabin and spinophilin to regulate dendritic spine morphology. Neuron. 2005;47:85–100. doi: 10.1016/j.neuron.2005.05.013. [DOI] [PubMed] [Google Scholar]
- Ryu SH, Suh PG, Cho KS, Lee KY, Rhee SG. Bovine brain cytosol contains three immunologically distinct forms of inositolphospholipid-specific phospholipase C. Proc Natl Acad Sci U S A. 1987;84:6649–6653. doi: 10.1073/pnas.84.19.6649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnabel R, Kilpatrick IC, Collingridge GL. An investigation into signal transduction mechanisms involved in DHPG-induced LTD in the CA1 region of the hippocampus. Neuropharmacology. 1999;38:1585–1596. doi: 10.1016/S0028-3908(99)00062-3. [DOI] [PubMed] [Google Scholar]
- Sohn JW, Lee D, Cho H, Lim W, Shin HS, Lee SH, Ho WK. Receptor-specific inhibition of GABAB-activated K+ currents by muscarinic and metabotropic glutamate receptors in immature rat hippocampus. J Physiol. 2007;580:411–422. doi: 10.1113/jphysiol.2006.125914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn JW, Yu WJ, Lee D, Shin HS, Lee SH, Ho WK. Cyclic ADP ribose-dependent Ca2+ release by group I metabotropic glutamate receptors in acutely dissociated rat hippocampal neurons. PloS One. 2011;6:e26625. doi: 10.1371/journal.pone.0026625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Várnai P, Balla T. Visualization of phosphoinositides that bind pleckstrin homology domains: calcium- and agonist-induced dynamic changes and relationship to myo-[3H]inositol-labeled phosphoinositide pools. J Cell Biol. 1998;143:501–510. doi: 10.1083/jcb.143.2.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won H, Lee HR, Gee HY, Mah W, Kim JI, Lee J, Ha S, Chung C, Jung ES, Cho YS, Park SG, Lee JS, Lee K, Kim D, Bae YC, Kaang BK, Lee MG, Kim E. Autistic-like social behaviour in Shank2-mutant mice improved by restoring NMDA receptor function. Nature. 2012;486:261–265. doi: 10.1038/nature11208. [DOI] [PubMed] [Google Scholar]
- Yuan B, Latek R, Hossbach M, Tuschl T, Lewitter F. siRNA Selection Server: an automated siRNA oligonucleotide prediction server. Nucleic Acids Res. 2004;32:W130–W134. doi: 10.1093/nar/gkh366. [DOI] [PMC free article] [PubMed] [Google Scholar]