

Abstract
Alzheimer's disease (AD) is a frequent and irreversible age-related neurodegeneration without efficient treatment. Experimental AD in mice responds positively to decreased insulin-like growth factor I (IGF-I) signaling, a pathway also implicated in aging. Here we aimed to protect the aging brain from devastating amyloid pathology by making specifically adult neurons resistant to IGF signaling. To achieve that, we knocked out neuronal IGF-1R during adulthood in APP/PS1 mice. We found that mutants exhibited improved spatial memory and reduced anxiety. Mutant brains displayed fewer amyloid plaques, less amyloid-β (Aβ), and diminished neuroinflammation. Surprisingly, adult neurons undergoing IGF-1R knock-out reduced their apical soma and developed leaner dendrites, indicative of remarkable structural plasticity entailing condensed forebrain neuroarchitecture. Neurons lacking IGF-1R in AD showed less accumulation of Aβ-containing autophagic vacuoles. At the same time, plasma Aβ levels were increased. Our data indicate that neuronal IGF-1R ablation, via preserved autophagic compartment and enhanced systemic elimination, offers lifelong protection from AD pathology by clearing toxic Aβ. Neuronal IGF-1R, and possibly other cell size-controlling pathways are promising targets for AD treatment.
SIGNIFICANCE STATEMENT We found compelling evidence in vivo that Alzheimer's disease (AD) progression is significantly delayed when insulin-like growth factor (IGF) signaling is blocked in adult neurons. To show that, we built a novel mouse model, combining inducible neuron-specific IGF-1R knock-out with AD transgenics. Analysis of the experimental AD phenotype revealed less abundant amyloid-β (Aβ) peptides, fewer plaques, and diminished neuroinflammation in mutants with inactivated IGF signaling, together with clearly preserved behavioral and memory performances. We present for the first time evidence that IGF signaling has profound effects on neuronal proteostasis and maintenance of cell morphology in vivo. Our results indicate in a model highly pertinent to translational research that neuronal IGF resistance may represent a pathophysiologically relevant mechanism of the brain for preventing Aβ accumulation.
Keywords: Alzheimer's disease, amyloid-β, conditional mutagenesis, insulin-like growth factor, mouse model, neuron
Introduction
Alzheimer's disease (AD) is a progressive and irreversible neurodegenerative dementia, for which the main risk factor is age. Insulin-like growth factor I (IGF-I), through its cognate receptor IGF-1R and downstream effectors, controls stress resistance, aging, and lifespan (Holzenberger et al., 2003; Xu et al., 2014). Since aging is modulated by insulin-like signals in the brain (Taguchi et al., 2007; Kappeler et al., 2008; Zhang et al., 2013), this pathway could function as a molecular bridge between physiological aging and AD pathogenesis (Carro et al., 2002). In accord with that, constitutive heterozygous knock-out of IGF-1R in an APP/PS1 AD mouse model reduces astrogliosis, prevents neuronal death, and alleviates AD-associated cognitive deficits (Cohen et al., 2009). However, that model could not distinguish between benefits due to delayed aging in long-lived IGF-1R+/− mutants and brain-specific mechanisms. Knock-out of IGF-1R specifically in hippocampal neurons delays premature death of Tg2576 AD mice, strongly pointing to neurons as key players in IGF-dependent protection of the CNS against amyloid-β (Aβ) toxicity (Freude et al., 2009). Yet, high mortality in young Tg2576 mice, with half dying before 20 weeks of age, precludes elucidation of the important age-related pathophysiology and long-term cognitive outcome of this complex neurodegeneration. Meanwhile, evidence for a possible role of IGF in AD progression has also been obtained from clinical investigation. Indeed, in AD patients, hippocampus is less responsive to IGF-I stimulation ex vivo, as demonstrated by decreased activation of the downstream IRS-PI3K-Akt pathway (Talbot et al., 2012), yet it remains unknown whether this is a cause or consequence of the disease. Similarly, when hippocampal neurons are exposed to Aβ oligomers, they show increased serine inhibition of IRS-1 (Bomfim et al., 2012). Together, this suggests that regulation of IGF-I signaling in central neurons responds to local abundance of Aβ. Here, we investigated whether the aging brain can be protected from amyloid pathology by making adult neurons experimentally resistant to IGF signaling. To circumvent IGF developmental effects, we combined efficient inducible neuron-specific IGF-1R knock-out with the APP/PS1 murine AD model. We obtained consistent evidence that blocking neuronal IGF-I signaling during adulthood alleviates amyloid pathology and cognitive deficits. We found that Aβ accumulation in the brain is counteracted by a normalized autophagic compartment and enhanced plasma clearance. This study also revealed that IGF-1R regulates important structural plasticity of forebrain neurons, and we discuss how this may contribute to efficient protection against Aβ proteotoxicity.
Materials and Methods
Animal models.
APPswe/PS1dE9 transgenic mice (Jankowsky et al., 2004) were obtained from Jackson Laboratory (stock 5864), and CaMKCreERT2 transgenic mice (Erdmann et al., 2007) were obtained from European Mutant Mouse Archives (EMMA 02125). APPswe/PS1dE9 and CaMKCreERT2 transgenes were backcrossed to IGF-1Rflox/flox knock-in mice (Kappeler et al., 2008) with 129/SvPas genetic background to obtain APPswe/PS1dE9+/0;IGF-1Rflox/flox and CaMKCreERT2+/0;IGF-1Rflox/flox breeder mice. These double mutants were used to generate the final triple mutant APPswe/PS1dE9+/0;CaMKCreERT2+/0;IGF-1Rflox/flox. Littermates with genotype CaMKCreERT2+/0;IGF-1Rflox/flox were used as non-AD controls (abbreviated WT). Mice were maintained in a specific pathogen free barrier facility using individually ventilated cages (Tecniplast). Sentinel mice (8–12 per room and year) were tested at Harlan Laboratories following the recommendations of the Federation of European Laboratory Animal Science Associations; no infections or pathogens were detected. Mice were kept at 22–24°C air temperature, on a 14/10 h light/dark cycle and had ad libitum access to water and rodent chow (LASQCdiet Rod16, Genobios). Cages were equipped with mouse houses (Tecniplast) to prevent aggressive behavior. All experiments were conducted according to the EC Council Directive (86/609/EEC) for care and use of animals for experimental procedures, and complied with the regulations of Comité d'Ethique pour l'Expérimentation Animale “Charles Darwin” (specific approval No. Ce5/2012/043), registered at Comité National de Réflexion Ethique sur l'Expérimentation Animale (Ile-de-France, Paris, No. 5).
PCR genotyping.
WT, floxed, and knock-out IGF-1R alleles were simultaneously genotyped from skin biopsies using triplex PCR with forward primers 5′-ATCTTGGAGTGGTGGGTCTGTTTC-3′ and 5′-CCATGGGTGTTAAATGTTAATGGC-3′, and reverse primer 5′-ATGAATGCTGGTGAGGGTTGTCTT-3′. APPswe/PS1dE9 was detected using PS1dE9-specific forward 5′-AATAGAGAACGGCAGGAGCA-3′ and reverse primer 5′-GCCATGAGGGCACTAATCAT-3′. CaMKCreERT2 was detected using forward 5′-GCTTGCAGGTACAGGAGGTAGT-3′ and reverse primer 5′-GGTTCTCCGTTTGCACTCAGGA-3′.
Tamoxifen-induced neuronal IGF-1R knock-out.
All experimental animals had the same APPswe/PS1dE9+/0;CaMKCreERT2+/0;IGF-1Rflox/flox genotype. Both male and female mice were used. Mice matched by gender, age, genotype, and weight were randomly assigned to experimental groups. At 2 months, half of the mice received tamoxifen injections to generate ADINKO (AD and inducible neuronal IGF-1R knock-out) mutants (Fig. 1A). The other half was injected with vehicle alone to serve as littermate controls. Tamoxifen (T5648, Sigma-Aldrich), dissolved in sunflower seed oil/ethanol (10:1) at 10 mg ml−1, was administered intraperitoneally at 42 mg kg−1 body weight twice per day for 5 d. All genotypes and the individual Cre-loxP recombination efficiencies were checked after killing from brain biopsies by PCR. To confirm neuron specificity of CaMKCreERT2-inducible knock-out, CaMKCreERT2 mice were crossed with reporter mice (Jackson Laboratory, stock 7908) that express tdTomato upon Cre recombination (Madisen et al., 2010). Double transgenic CaMKCreERT2+/0;CAG-tdTomato+/0 mice were injected with tamoxifen at 2 months and analyzed 4 weeks later. To assess dendrite morphology, triple-mutant NestinCreERT2+/0;CAG-tdTomato+/0;IGF-1Rflox/flox mice (NestinCreERT2+/0 provided by Dr. A. J. Eisch) were obtained by crossing CAG-tdTomato+/0;IGF-1Rflox/flox and NestinCreERT2+/0;IGF-1Rflox/flox breeders. NestinCreERT2+/0;IGF-1RWT/WT mice were crossed to tdTomato reporter mice to generate controls. At 3 months, both groups received tamoxifen intraperitoneally and were analyzed at 16 months for tdTomato expression.
Figure 1.
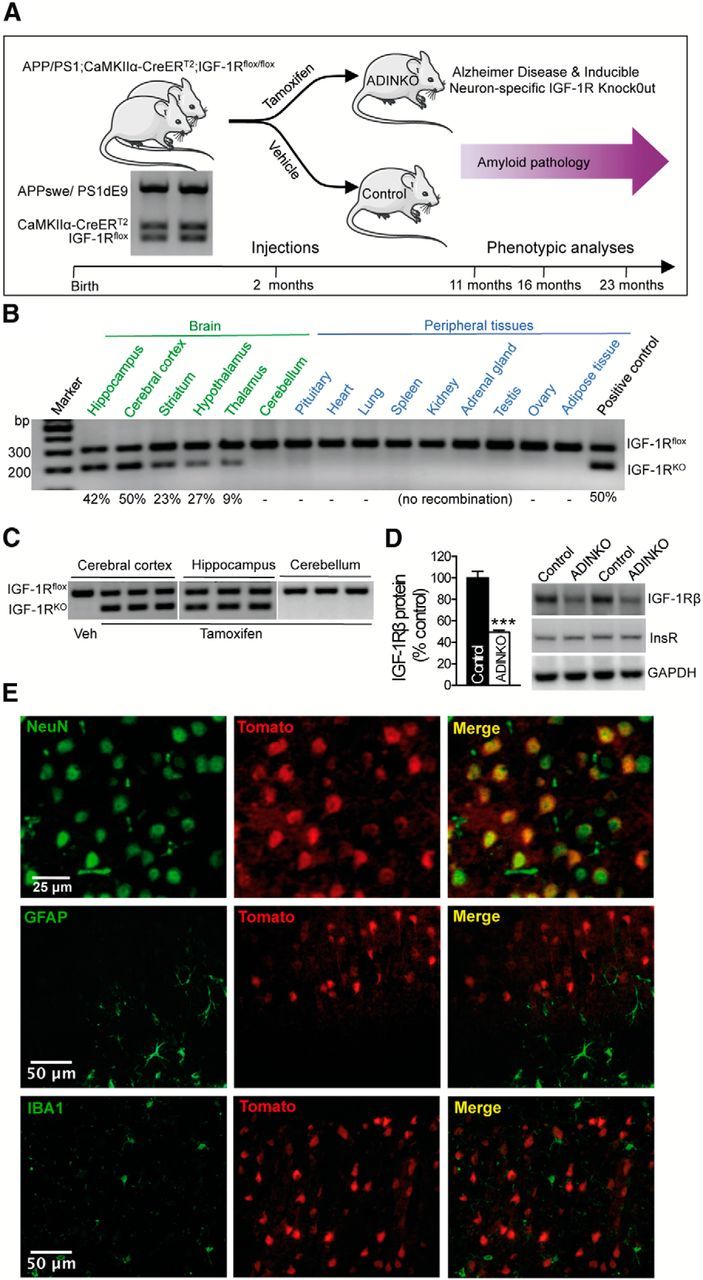
Study design and efficacy of inducible neuronal IGF-1R knock-out. A, Mutant strains were crossed to combine neuron-specific tamoxifen-inducible IGF-1R gene knock-out with transgenic APPswe/PS1dE9 AD model. All mice had Igf1r flox alleles (IGF-1Rflox) and harbored CaMKIIα-CreERT2 and APPswe/PS1dE9 (APP/PS1) transgenes (genotype by PCR). All mice developed plaques that increased in number and size throughout adult life. Tamoxifen (Tam) was administered at 2 months of age to induce IGF-1R knock-out (ADINKO), while controls received vehicle alone. Behavioral phenotyping comprised Barnes maze test at 11, 16, and 23 months, and Y-maze and open-field test at 23 months. Regarding postmortem studies, half the cohort was killed at 16 months for histopathology, biochemistry, and Aβ ELISA, the other half at 23 months, for Aβ ELISA. B, PCR revealed strong Cre-lox recombination (IGF-1RKO) in forebrain (cortex, hippocampus, and striatum), much less or no recombination elsewhere in the CNS, and none in peripheral tissues (n = 5). C, Efficiency and forebrain specificity of IGF-1R knock-out was highly reproducible, as shown for three brains 4 weeks after Tam administration. D, IGF-1R levels in ADINKO cortex were half the control values, while insulin receptor values (InsR) remained unchanged. Western blot, ***p < 0.001, Mann–Whitney U test, n = 10 per group, mean ± SEM. E, Cell type-specific markers in cortical sections of CaMKIIα-CreERT2+/0;CAG-tdTomato+/0 reporter mice (red fluorescence indicating Cre-lox excision) demonstrated that essentially all neurons underwent recombination, while astrocytes (GFAP) and microglia (Iba1) did not.
Behavioral and cognitive analyses.
Experiments were conducted by investigators blinded to treatment group in a room dedicated to behavioral analyses. Mice were allowed to acclimate for ≥30 min. All tests used automated video tracking (Viewer3, Biobserve).
Open-field test.
We used an open-field arena 50 by 30 cm wide with opaque 35 cm walls. Mice were allowed to explore freely for 10 min while activity was recorded. The arena was divided into 30 by 15 cm center area, 6-cm-wide corridors along the walls, and a 9 by 9 cm square in each corner, partly overlapping with corridors. The ratio of distance traveled in the center to total distance traveled, and the ratio of time spent in center to total test duration were used as measures of anxiety-related behavior. Mice returned to their home cage after each trial.
Barnes maze.
Spatial learning and memory were assessed using the Barnes maze, a circular dry-land maze (Barnes, 1979). We needed a test repeatedly applicable to mice of increasing age, including very old individuals. We chose the Barnes maze, which is similar to the Morris water maze, but less strenuous and less physically challenging, and which has been successfully used in AD mice (O'Leary and Brown, 2009; Stewart et al., 2011; Larson et al., 2012; Attar et al., 2013). This test works well with mice from 129/Sv genetic background. It consists of an elevated 92 cm platform, with 20 floor holes (diameter, 5 cm). Distant visual cues were placed on surrounding room walls and remained throughout the test. For training, the mouse was placed in the center of the maze under an opaque start box. After 10 s, the box was lifted, and the mouse allowed to explore the maze for 5 min. Indirect light (200 lx) and sound (10 kHz, 75 dB) were used to reinforce motivation to escape to a dark, recessed refuge box, hidden under one of the 20 holes. Mice were trained four times per day (≥15 min between trials) for 4 consecutive days. The maze was thoroughly cleaned after each trial. On day 5, mice were submitted to one test trial, with the refuge box removed, to evaluate long-term memory retention. Parameters calculated for training and test trials included distance traveled, average speed, and cumulative errors. Visiting a wrong hole was scored 1–10, depending on the distance between incorrect hole and true position of the refuge box during training. Cumulative error was the sum of scores.
Y-maze.
In the 23-month-old mice, a two-trial Y-maze task was performed 4 weeks after the Barnes paradigm to assess short-term, hippocampus-dependent spatial memory (Brouillette et al., 2012). Y-maze arms were 8 by 40 cm with transparent, 16-cm-high walls. The maze floor was thoroughly cleaned after each test and covered with fresh bedding. Distant cues were present on surrounding room walls. During exposure, mice could freely explore two arms (“start” arm and “other” arm) for 5 min, while the third (“new”) arm was blocked with an opaque door. The assignment of arms was counterbalanced within each group. Mice were then returned to their home cage for 3 min. During the following test period, they were placed at the end of their start arm and allowed to explore all three arms for 5 min. The percentage of visits to each arm was calculated. Mice were excluded from analysis based on the preset criterion: animal does not move during test. This corresponded to one 11-month-old ADINKO male and one 16-month-old control female in the probe test (day 5) of the Barnes maze procedure, out of 25 individuals in each group. In the Y-maze test, two control males, one ADINKO male, and one ADINKO female were excluded because they did not enter one of the three arms.
Antibodies used.
The following antibodies were used: NeuN (MAB377, 1:1000; Millipore), and doublecortin (DCX; sc-8066, 1:1000; Santa Cruz Biotechnology), GFAP (Z0334, 1:2000; Dako), Aβ (82E1, 1:1000; IBL International), Iba1 (019–19741, 1:1000; Wako), MBP (SMI-99P, 1:4000; Covance), IGF-1Rβ (3027, 1:1000; Cell Signaling Technology), Akt (9272, 1:1000; Cell Signaling Technology), phospho-Akt (Ser473; 4058, 1:1000; Cell Signaling Technology), IRβ (sc-711, 1:1000; Santa Cruz Biotechnology), APP and Aβ (6E10, 1:1000; Covance), C-terminal fragment (CTF; A8717, 1:1000; Sigma-Aldrich), drebrin (ab12350, 1:2000; Abcam), synapsin-I (AB1543P, 1:2000; Millipore Bioscience Research Reagents), LC3-I/II (ab58610, 1:1000; Abcam), p62 (NBP1-49956, 1:2000; Novus), beclin-1 (3738, 1:1000; Cell Signaling Technology), Atg5 (2630, 1:1000; Cell Signaling Technology), ADAM10 (sc-28358, 1:1000; Santa Cruz Biotechnology) and BACE1 (2253, 1:1000; ProSci), cathepsin D (AF1029, 1:1000; R&D Systems), secondary Alexa Fluor antibodies (Life Technologies), secondary IRDye 600LT or 800CW (1:15,000, LI-COR Biosciences), biotinylated secondary antibodies (BA-1000, 1:200; BA-2000, 1:200; Vector Laboratories), horseradish peroxidase-conjugated anti-mouse (626520, 1:3000; Life Technologies), anti-rabbit (656120, 1:3000; Life Technologies), or anti-goat (A5420, 1:3000; Sigma-Aldrich) secondary antibodies.
Immunofluorescence.
Three-month-old CaMKCreERT2+/0;CAG-tdTomato+/0 and 16-month-old NestinCreERT2+/0;CAG-tdTomato+/0 mice were anesthetized using 100 mg kg−1 body weight pentobarbital and transcardially perfused with cold PBS, followed by 4% paraformaldehyde. Brains were frozen in 30% sucrose, stored at −80°C, and later sectioned on a cryostat. Tissues were stained using specific antibodies. Immunostained sections were observed using a spinning disk confocal microscope DMI4000B (Leica).
Immunohistochemistry.
Sixteen-month-old ADINKO mice were weighed and anesthetized. Body length (nasoanal distance) was measured and brain removed, weighed, and split along the midline. One hemibrain was postfixed in 4% paraformaldehyde at 4°C for 24 h, dehydrated, paraffin embedded, and sectioned parasagittally (7 μm; microtome RM2125RT, Leica). Sections were stained with cresyl violet (C-5042, Sigma-Aldrich), or immunolabeled using specific antibodies and diamino-benzidine (Dako) reaction. Stained sections were observed using a DM5000B microscope (Leica), and micrographs were analyzed with ImageJ software (National Institutes of Health). Four sections, 0.5–1.0 mm lateral to midline (Paxinos and Franklin, 2001), were analyzed per animal to establish individual means. Results were expressed as the average number of cells or plaques per square millimeter.
Amyloid plaques and number of glial cells per plaque.
Sections were coimmunolabeled with anti-GFAP (Z0334, 1:5000; Dako), anti-Iba1 (1:1000; Wako), and anti-Aβ (82E1, 1:1000; IBL International), subsequently revealed by respective secondary Alexa Fluor antibodies (1:200; Life Technologies), and counterstained with DAPI. Immunostained sections were scanned using a spinning disk confocal microscope (DMI4000B, Leica). Images were merged and analyzed using ImageJ. Plaques were automatically delimited and those ≥50 μm2 considered. GFAP-positive astrocytes and Iba1-positive microglial cells with DAPI-stained nucleus located within 15 μm of the edge of a plaque were manually counted (Serrano-Pozo et al., 2010). Four sections were analyzed per animal to establish individual means. Results were expressed as average number of GFAP-positive astrocytes or microglial cells per plaque.
Neuronal soma size.
We measured neuronal soma size in 2D following Fricano et al. (2014). Per mouse, we used four cresyl violet-stained parasagittal (0.5–1.0 mm lateral to midline) paraffin-embedded tissue sections of 7 μm, 70 μm apart, and 6–8 mice per group. We used only neurons with visible nucleolus to ensure full-size cross section, and traced perimeters using ImageJ. Micrographs were taken using a DM5000B microscope (Leica). Neuronal cell surface was measured in motor cortex layer V using ImageJ algorithms based on dense blue cytoplasmic stain and filtering for particle size. Selections were confirmed manually by checking for neuronal morphology and clearly visible nucleus. On average, 110 neurons were measured per animal. Neuronal size was also measured in motor cortex layer II (20 neurons), hippocampal CA3 (40 neurons), and olfactory bulb (10 neurons).
Neuron and dendrite morphology.
Using 30 μm hippocampus sections from NestinCreERT2+/0;CAG-tdTomato+/0 mice and eight optical Z series acquired by spinning disk confocal microscopy (Roper/Leica; spinning head from Yokogawa), we analyzed five tdTomato-activated cells per mouse (n = 6 mice per group). Only NeuN-positive and DCX-negative cells showing an intact dendritic arbor within the same tissue section were considered as mature granular neurons of the dentate gyrus, and subsequently analyzed. Primary dendrite length, number of branch points, and their distances to neuronal soma were evaluated manually by following the branching path for each neuron. Neuronal volume and detailed branch thickness were measured on Z-stacks of 0.5 μm optical slices. We determined 3D cell soma volume using confocal microscopy. Briefly, single neurons were imaged into a set of uniform, 0.5-μm-thick layers, with the proximodistal axis in the plane of the layers. Each slice from this stack was analyzed using ImageJ. Total soma volume was defined as the sum of all soma volumes from the different layers (Chvátal et al., 2007). Distal dendrite morphology was assessed on 0.2 μm optical slices after deconvolution treatment. Three-dimensional reconstruction was performed using Fiji software.
Western immunoblot.
Animals were transcardially perfused with cold PBS, pH 7.4, to eliminate blood cells from capillaries. The brain was quickly removed and cortex dissected on ice, immediately frozen in isopentane at −70°C, and stored at −80°C. Tissues were homogenized in Tris (250 mm sucrose, 20 mm Tris base, 1 mm EDTA, 1 mm EGTA, 0.5% Triton X-100, 150 mm NaCl) or RIPA buffer (25 mm Tris-HCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 1 mm EDTA, 1 mm EGTA, 150 mm NaCl) for total protein extraction with protease and phosphatase inhibitors (Roche) using a Polytron (Kinematica), and centrifuged 14,000 rpm at 4°C for 20 min. For SDS-PAGE, 40–80 μg of protein per lane were loaded onto 4–20% Tris-glycine or 10–20% Tris-tricine gradient gels (Bio-Rad) and migrated at 100 V. Size-separated proteins were electrotransferred to PVDF membrane. Blocked membranes were incubated overnight at 4°C with primary antibodies, washed in TBS, and incubated for 60 min at room temperature with corresponding horseradish peroxidase or IRDye-conjugated secondary antibodies. Blots were developed with ECL (WP20005, Life Technologies) and bands analyzed using ChemiDoc and Quantity One 4.2.1 (Bio-Rad). Alternatively, fluorescence intensities were quantified using an Odyssey CLx (LI-COR). Sample loading and protein transfer were controlled using anti-GAPDH antibody (sc-32233, 1:3000; Santa Cruz Biotechnology).
ELISA.
To measure Aβ40 and Aβ42 levels in cerebral cortex, tissue samples were homogenized at 4°C in Tris buffer (50 mm Tris base, pH 7.4, 250 mm sucrose, 1 mm EDTA, 1 mm EGTA, 1 mm NaVO3, 10 mm NaF; 100 mg of tissue per milliliter) with protease inhibitors (Roche) using a Polytron. Soluble and insoluble Aβ were extracted in 0.4% diethylamine (DEA) and 70% formic acid (FA), respectively, as described previously (Minami et al., 2010). Crude 10% homogenate was mixed with an equal volume of 0.4% DEA, sonicated, and spun for 1 h at 4°C at 130,000 × g using a TLA 50.4 Ti rotor and Optima TL ultracentrifuge (Beckman Coulter). The DEA supernatant was neutralized with 0.5 m Tris base, pH 6.8 (10:1 v/v), and used for detection of soluble Aβ. For insoluble Aβ, the pellet was homogenized in 70% FA and again ultracentrifuged. FA-containing supernatants were neutralized with 1 m Tris base, pH 11 (1:20 v/v). Aβ40 and Aβ42 were detected using human Aβ ELISA (Life Technologies). For IGF-I detection, blood was collected under topical anesthesia from ocular sinus using EDTA, cooled on ice, and centrifuged (3000 rpm, 10 min, 4°C). Plasma was immediately frozen. IGF-I was measured using mouse IGF-I Quantikine ELISA (R&D Systems).
Statistical analysis.
Investigators were blinded to group allocation (sex, genotype) during experiments, data collection, and analysis. Mice were coded by numbers. For comparison between groups, normally distributed data were analyzed by paired or unpaired two-tailed Student's t test while non-normally distributed data were assessed by Mann–Whitney U test. Data from Barnes maze experiments were analyzed by repeated-measures two-way ANOVA followed by individual post hoc tests where applicable. We used SPSS (SPSS Institute) and Prism (GraphPad). Results are presented as means ± SEM.
Results
Ablating neuronal IGF-1R in adult APP/PS1 mice
We administered tamoxifen at 2 months of age to induce IGF-1R gene inactivation in APPswe/PS1dE9+/0;CaMK-CreERT2+/0;IGF-1Rflox/flox mice (ADINKO model; Fig. 1A). Efficient IGF-1R knock-out occurred swiftly in cortex and hippocampus, but not outside the forebrain or in peripheral tissues (Fig. 1B,C). In hippocampus and cortex, which strongly express CaMKIIa, we observed 50 and 42% Cre-lox excision, respectively, indicating that in these structures, virtually all neurons had lost the capacity to produce IGF-1R. Protein levels of IGF-1R in cortex were just 50% of controls (Fig. 1D), consistent with the proportions of neuronal versus other cell types and their respective IGF-1R expression (Herculano-Houzel et al., 2013). Insulin receptor protein expression remained unaffected (Fig. 1D). Cortical sections of CaMKIIα-CreERT2+/0;CAG-tdTomato+/0 reporter mice revealed that Cre recombination occurred exclusively in neurons, but not in astrocytes or microglia (Fig. 1E).
Upon knock-out induction, mutant mice did not show any overt phenotypic changes and remained outwardly normal. Endocrine phenotyping revealed elevated plasma IGF-I in adult ADINKO mice (males: control, 225 ± 18 ng ml−1; ADINKO, 310 ± 13 ng ml−1, p < 0.01; females: control, 215 ± 13 ng ml−1; ADINKO, 284 ± 15 ng ml−1, p < 0.01, n = 6–7), indicative of hormone upregulation in response to central IGF-I resistance. Final body length at 16 months was unchanged in ADINKO males, marginally increased in ADINKO females (males: control, 114 ± 1 mm; ADINKO, 115 ± 1 mm, not significant; females: control, 112 ± 1 mm; ADINKO, 116 ± 1 mm, p = 0.039, Student's t test, n = 7), and body weight did not differ between ADINKO and controls (males: control, 43.6 ± 2.7 g; ADINKO, 40.9 ± 2.3 g; females: control, 41.5 ± 2.0 g; ADINKO, 45.1 ± 3.2 g).
Blocking IGF-I signaling in adult forebrain neurons diminishes Aβ load
To measure the impact of neuronal IGF-1R knock-out on amyloid pathology, we performed anti-Aβ immunohistochemistry (IHC). Histomorphometry revealed a significant decrease of plaque density in cortex and hippocampus of female ADINKO mice compared with controls (Fig. 2A,B,D,E; female cortex, −28%, p < 0.01; female hippocampus, −19%, p < 0.05; Student's t test). Similarly, in ADINKO males, we observed a significant decrease in plaque density in cortex (Fig. 2A,C,D,F; male cortex, −36%, p < 0.05). Analysis of size distribution revealed that small plaques (cross section, <250 μm2) contributed most to the lower prevalence of plaques in ADINKO mice, in cerebral cortex as well as hippocampus (Fig. 2B,C,E,F). Of note, thioflavine-S staining confirmed the IHC results by revealing significant reduction in plaque density in ADINKO mice, in particular of small thioflavine-S-stained plaques (data not shown).
Figure 2.
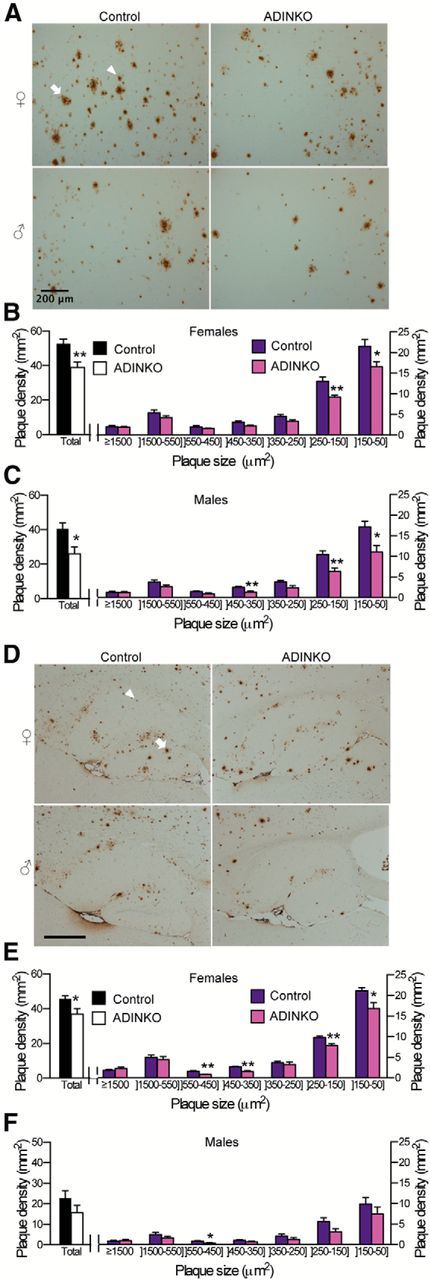
Amyloid plaque pathology is diminished in ADINKO mice. A, D, Representative micrographs of plaques (82E1 antibody) in cortex (A) and hippocampus (D). Arrowhead: plaque 50–150 μm2; arrow: plaque ≥1500 μm2. B, C, E, F, Prevalence of plaques, in particular small and middle-sized, was diminished in cortex and hippocampus from 16-month-old ADINKO males and females. Note that differences were not significant for male hippocampus. *p < 0.05; **p < 0.01, mean ± SEM; Student's t test, n = 7 per group, four sections used per animal.
To extend on amyloid plaque results, we measured cortical abundance of Aβ fractions by ELISA. ADINKO mice showed a marked decrease in Aβ40 and Aβ42 peptides in formic acid-extracted, insoluble fraction (Fig. 3A), which reflects amyloid plaque content. The extrapolated amounts of insoluble Aβ in the entire cortex were 8636 ± 1054 ng Aβ42 for female controls versus 5460 ± 445 ng for female ADINKO mice (p < 0.01). Concerning Aβ40, controls carried 1452 ± 236 ng versus 722 ± 122 ng for ADINKO mice (p < 0.01). Regarding DEA-extracted, soluble Aβ, a particularly neurotoxic fraction (Shankar et al., 2008; Mc Donald et al., 2010), female ADINKO mice exhibited significantly less Aβ40 (−50%, p = 0.028), and a trend to lower Aβ42 (−19%, p = 0.083; Fig. 3B). Regardless of genotype, males displayed 28–58% fewer plaques and half the levels of Aβ peptides found in females (Fig. 3A,B), consistent with studies reporting higher amyloid burden and plaque prevalence in female APP/PS1 mice (Wang et al., 2003; Hirata-Fukae et al., 2008; Minami et al., 2010). We next measured Aβ in cerebral cortex homogenates from 23-month-old mice. From 16 to 23 months, Aβ40 increased >10-fold and Aβ42 up to fivefold in this model (Fig. 3A–D). Brains from 23-month-old male and female ADINKO mice had again significantly less Aβ40 and Aβ42 in insoluble (Fig. 3C) and soluble fractions (Fig. 3D) compared with controls. Immunoblotting using antibody 6E10 confirmed these findings, showing that the Tris-soluble total Aβ pool was down by 65% in extracts from ADINKO cortex (Fig. 3E; p < 0.05). These results strongly suggested that reduced plaque load resulted from diminished Aβ abundance. Small oligomers may be particularly toxic (Shankar et al., 2008). Using anti-6E10 immunoblotting, we showed that ADINKO mice carried significantly less dimers, trimers, and high-molecular-weight oligomers (Fig. 3F). Thus, blocking IGF signaling in adult neurons diminished toxic Aβ concentrations in both sexes, and was particularly efficient when peptides accumulated massively with age. To finally check for possible hyperaggregation (Cohen et al., 2009), we established Aβ concentration within plaques. We computed the average volume fraction of plaques from anti-Aβ IHC data at 16 months and found 2.48% for control females and 1.75% for ADINKO females. We compared this with the total amount of insoluble, FA-extracted Aβ (Fig. 3A). The resulting average concentration of Aβ species within plaques was the same in controls and ADINKO brains (2.34 and 2.37 μg mg−1, respectively), suggesting no hyperaggregation in mutants. We conclude that hyperaggregation is unlikely to explain reduced number and smaller size of Aβ plaques in ADINKO mice.
Figure 3.
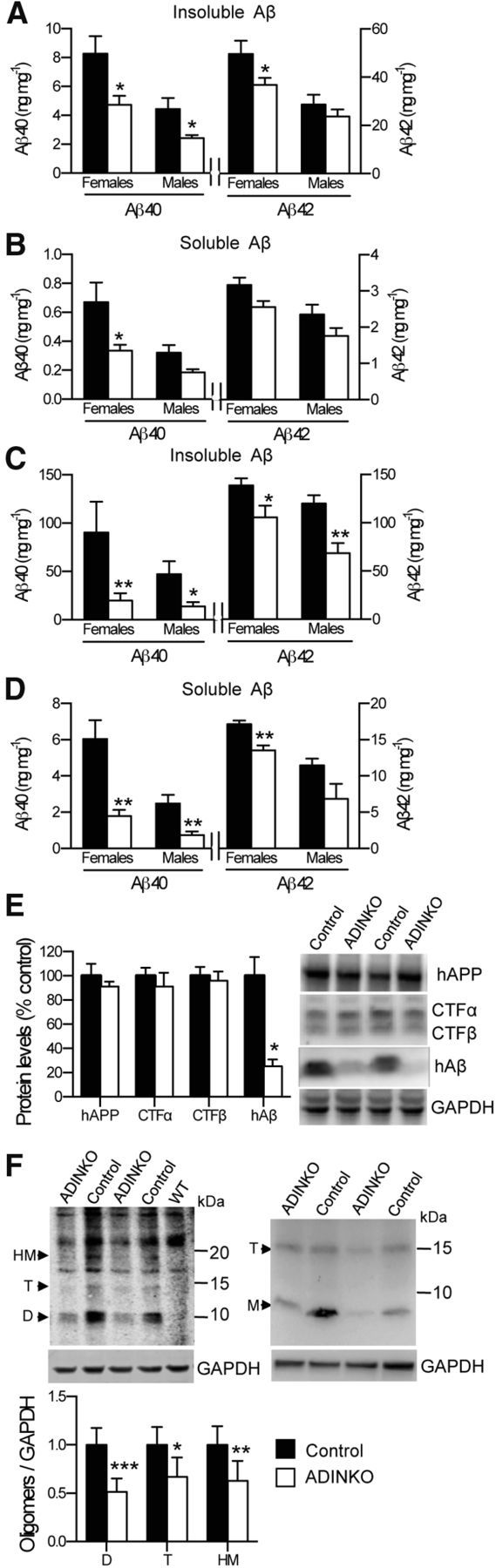
Levels of Aβ peptides are reduced in ADINKO cortex. A–D, Aβ40 and Aβ42 in cortical homogenates of 16-month-old (A, B) and 23-month-old (C, D) ADINKO mice and controls (n = 6–12). FA-treated insoluble and DEA-soluble fractions measured by human-specific ELISA. E, Western immunoblot (WIB) analysis of human APP (hAPP), CTF-α/β, human Aβ (hAβ) from cortical homogenates revealed diminished Aβ monomer levels with no change in APP processing. F, SDS-stable Aβ oligomers were extracted from cortex as detergent-soluble (left blot) and extracellular-enriched (right) fractions, and submitted to WIB using anti-Aβ antibody 6E10 and Odyssey detection (LI-COR). Specific bands migrated at 8 kDa (D, dimers), 12 kDa (T, trimers) and 18 kDa (4-mers or 5-mers; HM, high-molecular-weight oligomers; Mc Donald et al., 2010) and were diminished in ADINKO mice. M, Monomers. *p < 0.05, **p < 0.01, ***p < 0.001. Mean ± SEM, Mann–Whitney U test, n = 5 mice.
To determine whether neuronal inactivation of IGF-1R in APP/PS1 mice affected Aβ production, we examined APP metabolism. Protein expression of APP, of α and β secretases (ADAM10, BACE1), and of substrates of γ-secretase (CTF-α and CTF-β) was similar in cortical homogenates from control and ADINKO mice (Fig. 3E; data not shown), suggesting that diminished amyloid pathology observed in ADINKO mice did not result from reduced production of Aβ. Neither could diminished Aβ levels be ascribed to major Aβ metabolizing enzyme IDE (insulin degrading enzyme), which we found unchanged in the brain (controls, 100 ± 5.5%; ADINKO, 83 ± 6.9%, not significant). Collectively, these data indicated that IGF-1R inactivation in adult neurons provided protection from neurotoxicity of soluble Aβ and from amyloid pathology by counteracting Aβ accumulation and plaque formation, and pointed to neuronal and systemic Aβ clearance as possible mechanisms. This view is supported by the comparison of cortical with blood-borne pools of Aβ, which clearly indicated that systemic clearance is unfavorably low in control mice, especially in females, while ADINKO mice showed significantly higher peripheral Aβ elimination (Table 1).
Table 1.
Aβ40 and Aβ42 in blood plasma and cerebral cortex at 16 months
| Females |
Males |
|||
|---|---|---|---|---|
| Control | ADINKO | Control | ADINKO | |
| Aβ40 | ||||
| Plasma concentration (pg/ml) | 769 ± 74 | 953 ± 37** | 808 ± 42 | 798 ± 84 |
| Total amount in plasma (ng)a | 2.1 ± 0.3 | 3.0 ± 0.2* | 2.5 ± 0.1 | 3.0 ± 0.4 |
| Soluble amount in cortex (ng)b | 120 ± 27 | 51 ± 8* | 60 ± 10 | 28 ± 4* |
| Plasma/cortex ratio (%) | 3.0 ± 1.3 | 7.1 ± 0.9** | 6.3 ± 1.8 | 12.2 ± 2.2** |
| Aβ42 | ||||
| Plasma concentration (pg/ml) | 389 ± 45 | 541 ± 23* | 488 ± 39 | 485 ± 40 |
| Total amount in plasma (ng)a | 1.1 ± 0.1 | 1.7 ± 0.1** | 1.5 ± 0.1 | 1.8 ± 0.2 |
| Soluble amount in cortex (ng)b | 550 ± 47 | 382 ± 33* | 443 ± 54 | 267 ± 32* |
| Plasma/cortex ratio (%) | 0.21 ± 0.04 | 0.48 ± 0.04** | 0.44 ± 0.11 | 0.77 ± 0.11** |
aAβ plasma pool was determined from concentration and plasma volume (80 μl g−1 of body weight; (Gunji et al., 2002).
bAmount of soluble Aβ in cortex was calculated from concentration (Fig. 3B) and cortex weight.
*p < 0.05 and
**p < 0.01, ADINKO versus control.
Mean ± SEM, n = 7–12, Mann–Whitney U test.
Neuronal IGF-1R knock-out reduces brain inflammation
Neuroinflammation is an important component of AD progression (Choi et al., 2013). To determine whether fewer plaques and lower Aβ were correlated with glial activation, we counted astrocytic and microglial cells. IHC revealed that GFAP-positive astrocytes increased significantly by 14% in hippocampus of female ADINKO mice compared with controls, remaining unchanged in cortex (Fig. 4A,B). By contrast, Iba1-positive microglia were significantly reduced by 19–28% in male and female ADINKO mice, both in hippocampus and cortex (Fig. 4C,D), and male controls showed fewer microglia than females (Fig. 4D). These results suggested that microglial load was directly related to severity of amyloid pathology. In AD, microglial cells and astrocytes cluster around Aβ deposits, indicative of a role in plaque formation, maintenance, or clearance. We therefore selectively counted glial cells close to plaques. Strikingly, we found 81% more GFAP-positive astrocytes per plaque in female ADINKO mice (p < 0.01) and +37% astrocytes per plaque (p = 0.05) in males (Fig. 4E,F). Microglia concentration close to plaques was similar in ADINKO mice and controls (males: control, 0.46 ± 0.04 microglia per plaque; ADINKO, 0.43 ± 0.04 microglia per plaque; females: control, 0.56 ± 0.04 microglia per plaque; ADINKO, 0.56 ± 0.03 microglia per plaque). These data suggested that neuronal IGF-1R knock-out provoked redistribution of astrocytes toward plaques, possibly to limit amyloid deposition (Koistinaho et al., 2004). The observed decrease in microgliosis corroborates a potential beneficial role of astrocyte redistribution and is in agreement with the alleviated inflammatory status of ADINKO brains compared with those of controls.
Figure 4.
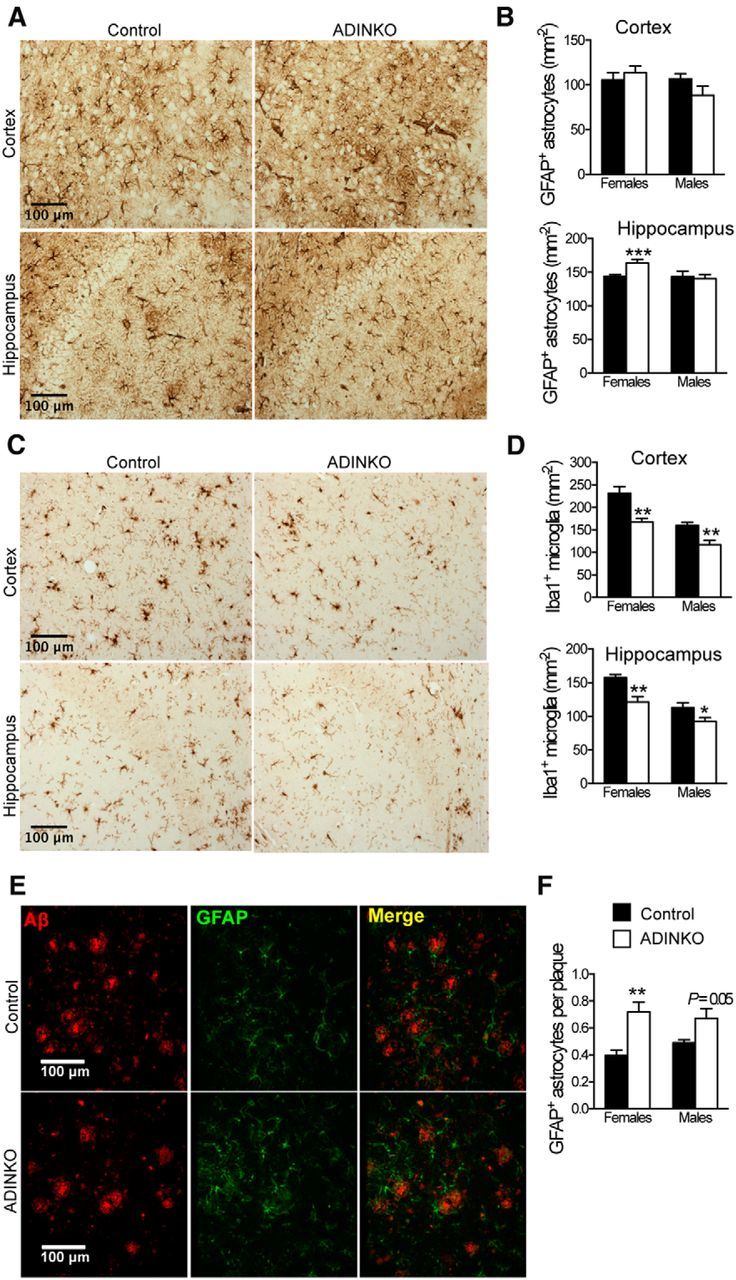
ADINKO brains show less microgliosis with astrocytes accumulating in amyloid plaque vicinity. A, Representative anti-GFAP IHC for astrocyte analysis. B, GFAP-positive astrocytes were more abundant in ADINKO female hippocampus, but not in motor cortex. C, Representative IHC of Iba1-positive microglia. D, Infiltration of Iba1-positive microglia was reduced in ADINKO motor cortex and hippocampus. Males had fewer microglia than females. E, Representative micrograph with plaques (red) and GFAP-positive astrocytes (green) accumulating close to plaques. F, More GFAP-positive astrocytes accumulated around plaques in ADINKO cortex. Student's t test, n = 7 per group, *p < 0.05, **p < 0.01, mean ± SEM.
Neuronal IGF-1R knock-out corrects AD memory and behavioral defects
To find out whether IGF-1R inactivation also ameliorated AD-related behavioral deficits, we analyzed spatial memory using the Barnes maze test. At 16 months, when APP/PS1 mice generally exhibit spatial memory deficits (Reiserer et al., 2007; Heneka et al., 2013), training in the Barnes maze improved performance in all groups as indicated by decreased distance traveled and fewer cumulative errors over days (Fig. 5A–E). Importantly, female ADINKO mice showed improved learning behavior. They traveled significantly less distance to find the refuge box and also tended to make fewer mistakes compared with controls (Fig. 5A,B). Twenty-four hours after the last acquisition trial, we measured long-term memory retention. Again, female ADINKO mice traveled less distance and made fewer mistakes searching the refuge box than controls (p < 0.05; Fig. 5D,E; age group, 16 months). At this age, no differences were seen in males (data not shown). All mice moved with similar speed regardless of genotype, both during acquisition and memory-retention test, indicating that differences in performance were not due to motor deficits (Fig. 5C). In fact, the better memory performance of female ADINKO mice was correlated to a strong trend in this group to use spatial search strategies over random or serial strategies [spatial search: female control, 50% (12 of 24); female ADINKO, 76% (19 of 25); Fisher's exact test, p = 0.079]. Progression of AD pathology is age dependent, and we therefore examined spatial memory performance also at 11 and 23 months. Temporal analysis revealed that IGF-1R inactivation delayed behavioral deficits in APP/PS1 females (Fig. 5D,E). Yet, at 23 months, spatial memory retention measured using the Barnes maze was similar in mutants and controls. At this age, and in contrast to the 16-month-old mice, all mice relied significantly more on nonspatial than on spatial strategies, suggesting that the test became too difficult for the 23-month-old mice (nonspatial strategy used by 37% of mice at 16 months, vs 68% at 23 months; Fisher's exact test, p = 0.010). Thus, we submitted the 23-month-old female mice to a less-demanding two-trial version of the Y-maze assessing short-term spatial memory. At this age, WT (non-APP/PS1) mice preferentially explored the novel arm compared with familiar and start arms, whereas control APP/PS1 mice were unable to discriminate between the novel and familiar arm (Fig. 5F). Female ADINKO mice displayed a significant preference for the novel arm compared with the previously exposed familiar and start arms, suggesting intact spatial memory (paired t test, p = 0.006; Fig. 5F).
Figure 5.

Neuron-specific IGF-1R inactivation in adult APP/PS1 mice improves spatial memory and reduces hyperactivity and anxiety-like behavior. A–E, Learning and memory assessed using Barnes maze. Sixteen-month-old females (n = 25 per group) were trained 4 times/d for 4 d. Training improved performance in all groups. Distance traveled to refuge box (A), cumulative error (B), and speed (C) are shown for each day. A, Repeated-measures ANOVA for distance over days revealed a significant main effect of neuronal IGF-1R knock-out in females (p = 0.042 for interaction, p < 0.001 for day; p = 0.035 for knock-out followed by multiple t test; *p < 0.05, mean ± SEM). D, E, Long-term retention was assessed on day 5. Distance traveled (D) and cumulative error (E) was evaluated as during acquisition. Number of mice is indicated in each bar. Mann–Whitney U test (*p < 0.05). F, Twenty-three-month-old females were tested in a two-trial Y-maze assay for short-term spatial memory, comparing visits to known (Other) versus unknown areas (New). Mean ± SEM and individual results of female control (n = 12) and female ADINKO mice (n = 11; paired Student's t test. G–J, Open-field test of 23-month-old female ADINKO (n = 12), controls (n = 13), and non-AD littermates (WT, n = 8). The WT group was included to validate tests. Total distance traveled (G) and time of active behavior (H) within a 10 min session. I, J, Ratios of the time spent and distance traveled in the center versus corners or corridor revealed decreased anxiety-like behavior of ADINKO mice. Student's t test, **p < 0.01, *p < 0.05; mean ± SEM.
Additional behavioral phenotyping using the open-field test revealed reduced hyperactivity in female ADINKO mice, suggesting that neuronal IGF-1R inactivation also improved neurobehavioral disturbances, such as AD-like psychomotor disinhibition (Fig. 5G,H). Moreover, female ADINKO mice explored the anxiogenic center of the arena more often than controls, indicative of reduced anxiety (Fig. 5I,J). Meanwhile, inactivation of IGF-1R did not cause any noticeable motor or balance deficits in rotarod test (data not shown). As regards the observed sex differences in cognitive amelioration, our data are in line with studies revealing more advanced AD phenotype in females (Wang et al., 2003; Hirata-Fukae et al., 2008; Minami et al., 2010), and not indicative of sex-specific effects of IGF-1R inactivation on disease progression. Thus, our findings clearly indicated that cognitive and behavioral defects in female APP/PS1 mice were significantly alleviated by IGF-1R inactivation induced at 2 months of age.
IGF-I signaling regulates soma size in differentiated neurons
To start unraveling mechanisms involved in the observed neurological benefits, we focused on ADINKO forebrain neurons. IGF-1R knock-out neurons looked smaller, and we found a conspicuous 19–23% reduction in cross section of neuronal soma in ADINKO cortex and hippocampus compared with controls (Fig. 6A–C; females, p < 0.01; males, p < 0.01, Student's t test). In these regions, all neurons were IGF-1R knock-out (Fig. 1B–E). By contrast, soma size of olfactory bulb periglomerular neurons, which do not lose IGF-1R in this model, remained unchanged (females: control, 95 ± 7 μm2; ADINKO, 102 ± 8 μm2; males: control, 101 ± 9 μm2; ADINKO, 98 ± 3 μm2, not significant). Smaller soma size of forebrain neurons contributed to 28–39% increased neuron density in male and female ADINKO cortex and hippocampus (Fig. 6D). At the same time, total volumes of ADINKO cortexes were diminished by 12% in females and 19% in males (data not shown), explaining lower overall brain weight in ADINKO mice (Fig. 6E). Yet, the macroanatomical structure of ADINKO brains always remained normal, as revealed by extensive observation of cresyl violet histology. Based on cerebral cortex volume and cell density at 16 months, an estimation of the overall number of neurons revealed that ADINKO males and females had 11.7 million cortical neurons, which is close to normal (Herculano-Houzel et al., 2013). Cortex of control females harbored on average 1.1 million fewer neurons (−9.7%), possibly due to the markedly more advanced amyloid pathology in this group. Average reduction in total neuron number was limited to 490,000 (−4.2%) in males. While neuronal soma was diminished in ADINKO mice, the levels of presynaptic (synapsin-I) and postsynaptic (drebrin) dendrite proteins remained unchanged and axonal integrity seemed also preserved, since myelin content in homogenates of hippocampus and cortex was increased compared with controls (Fig. 6F,G).
Figure 6.
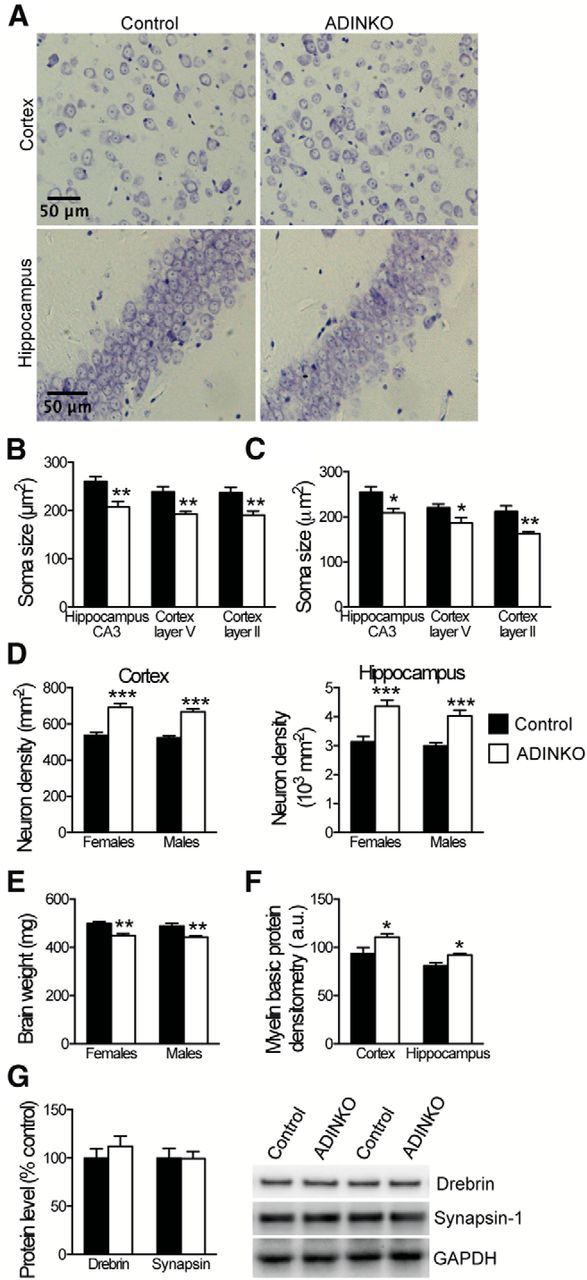
Altered neuronal morphology in ADINKO forebrain. A, Representative micrographs with cresyl violet staining for image analysis. B, C, Morphometry of neuronal soma in hippocampus CA3 and motor cortex layer V and II revealed reduced soma size in female (B) and male (C) ADINKO mice. D, E, Concomitantly, neuron density was increased in ADINKO cortex and hippocampus (D), and ADINKO mice presented with lower brain weight (E). F, Prevalence of MBP was increased in female ADINKO cortex and hippocampus. G, Western blot of drebrin and synapsin-I suggested preserved synaptic mass in ADINKO cortex. Student's t test, n = 6–7 per group; *p < 0.05; **p < 0.01; ***p < 0.001, mean ± SEM.
To check whether this compact neuromorphology was a cell-autonomous consequence of IGF-1R knock-out, we used an additional transgenic mouse model, where tdTomato staining and IGF-1R knock-out were induced selectively in adult-born neurons at 3 months. We analyzed dendritic arbors of tdTomato-activated granular neurons in dentate gyrus. Compared with IGF-1RWT/WT neurons, primary branch length was 47% shorter and primary branch diameter was 25% smaller in IGF-1RKO/KO neurons (p < 0.05; Fig. 7A–C). Yet, total number of branch points per neuron was identical (Fig. 7D) and no differences in spine density were detected on distal dendrites (Fig. 7A). Moreover, analysis of branch point distribution along the dendritic arbor revealed a significant shift, the first branch point in IGF-1RKO/KO neurons being closer to the soma than in IGF-1RWT/WT neurons (Fig. 7E). As in ADINKO mice, we found a significant 25% volume reduction in IGF-1RKO/KO neuronal soma compared with controls (Fig. 7F), while cell nucleus volume was unchanged (Table 2). Confocal microscopy suggested that reduced volume mainly resulted from smaller apical cytoplasm (Table 2). Thus, differences in neuromorphology between ADINKO and control mice appeared to be mainly the consequence of cell-intrinsic changes. Together, these results revealed that soma size and neuronal morphology of postmitotic forebrain neurons are controlled by IGF-I signaling.
Figure 7.
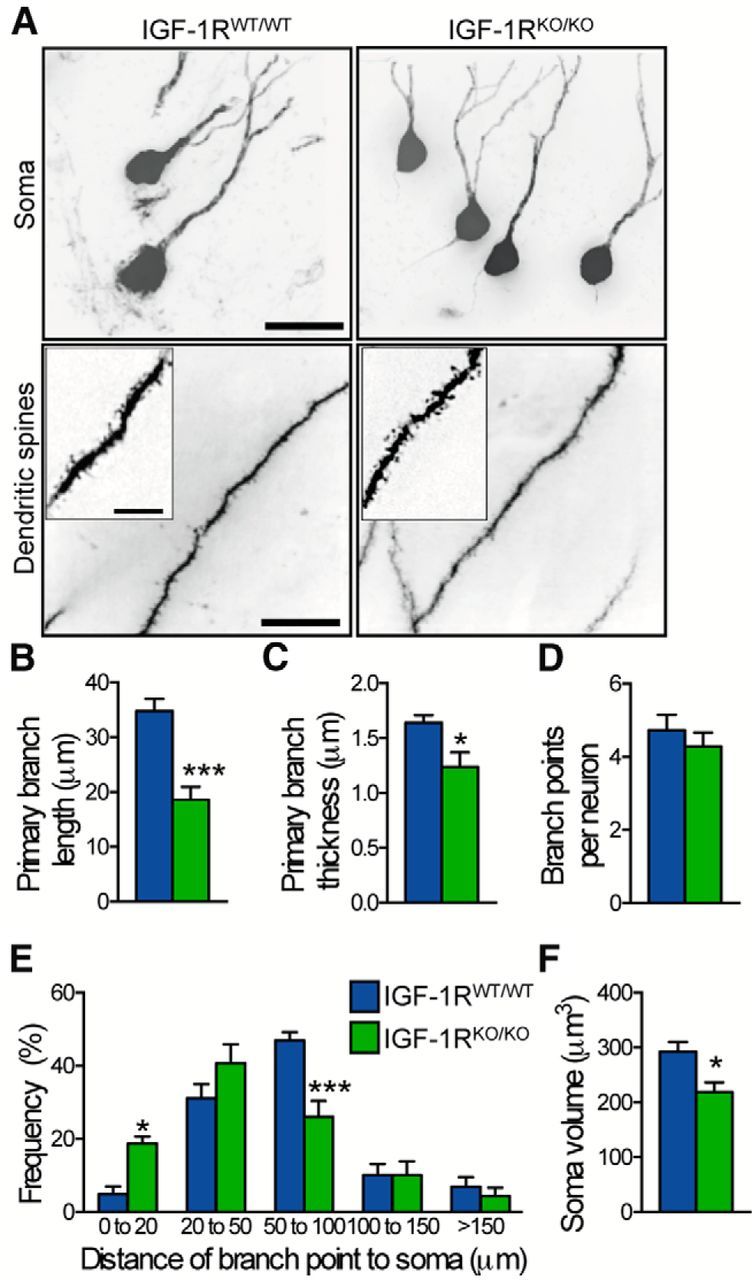
Cell-autonomous effect on neuronal morphology. A–F, Neuronal soma and dendrite morphology assessed on hippocampal sections from NestinCreERT2+/0;CAG-tdTomato+/0;IGF-1Rflox/flox mice (IGF-1RKO/KO) and NestinCreERT2+/0;CAG-tdTomato+/0;IGF-1RWT/WT controls (IGF-1RWT/WT). A, Representative confocal micrographs of tdTomato-activated hippocampal neurons. Insets show distal dendrites with synaptic spines. Scale bars, 10 μm. B–D, Primary branch length (B) and thickness (C) were reduced in IGF-1RKO/KO compared with IGF-1RWT/WT mice, while total number of branchings (D) was similar. E, Distribution of branchings along dendrites revealed increased frequency of proximal branchings. F, IGF-1RKO/KO neurons had smaller cell soma compared with IGF-1RWT/WT neurons. Student's t test, n = 6–7 per group; *p < 0.05, mean ± SEM.
Table 2.
Morphometry of IGF-1R knock-out neuronsa
| IGF-1RWT/WT | IGF-1RKO/KO | Percentage difference | P | |
|---|---|---|---|---|
| Neuronal soma length (μm) | 10.1 ± 0.3b | 9.2 ± 0.2b | −9% | 0.048 |
| Neuronal soma diameter (μm) | 6.0 ± 0.2 | 5.4 ± 0.3 | −10% | Not significant |
| Neuronal soma volume (μm3) | 293 ± 18 | 219 ± 18 | −25% | 0.016 |
| Nucleus volume (μm3) | 111 ± 11 | 117 ± 10 | −5% | Not significant |
| Cytoplasm volume (μm3) | 176 ± 10 | 108 ± 16 | −39% | 0.005 |
| Apical cytoplasm height (μm) | 1.69 ± 0.09 | 1.29 ± 0.08 | −24% | 0.009 |
| Basal cytoplasm height (μm) | 1.30 ± 0.13 | 1.38 ± 0.11 | +6% | Not significant |
| Primary dendrite length (μm) | 34.8 ± 2.2 | 18.6 ± 2.4 | −47% | < 0.001 |
| Primary dendrite thickness (μm) | 1.64 ± 0.07 | 1.24 ± 0.13 | −25% | 0.022 |
| Distal branch thickness (μm) | 0.62 ± 0.03 | 0.53 ± 0.02 | −15% | 0.021 |
aMeasurements were performed on dentate gyrus granular neurons from NestinCreERT2+/0;CAG-tdTomato+/0;IGF-1Rflox/flox mice (IGF-1RKO/KO) and NestinCreERT2+/0;CAG-tdTomato+/0;IGF-1RWT/WT controls (IGF-1RWT/WT).
bMean ± SEM, n = 6 mice per group, Student's t test.
Blocking IGF-I signaling in adult neurons rescues AD-related autophagy defects
Ablating neuronal IGF-1R led to 47% decrease in serine phosphorylation (Ser473) of Akt without changes in total Akt (Fig. 8A, top), indicating sustained downregulation of neuronal PI3K-Akt-mTOR signaling. As IGF-1R controls protein synthesis and autophagy through downstream PI3K-Akt-mTOR pathways, known to be defective in AD (Talbot et al., 2012; Nixon, 2013), we assessed selected autophagy markers. In macroautophagy, damaged organelles and proteins are sequestered in autophagosomes that fuse with lysosomes for hydrolysis. Accumulation of autophagosomes has been described in AD brain as a result of disturbed autophagy-lysosomal degradation (Nixon, 2013). Interestingly, we found a 26% decrease in autophagosome marker protein LC3-II in cortical homogenates of ADINKO mice compared with controls (Fig. 8A, bottom; p < 0.05), suggesting that IGF-1R inactivation diminished AD-related autophagosome accumulation. Other components of the autophagy pathway, namely p62/sequestosome-1, which is important for tau degradation, as well as beclin-1 and Atg5, which are involved in autophagosome formation, remained unchanged (Fig. 8A, bottom). The presence of beclin-1 and Atg5 at constant levels indicates that biogenesis of autophagic vacuoles (AVs) was the same in ADINKO mice and AD controls. In contrast, the protease cathepsin D, a marker of lysosomal-AVs, especially autophagolysosomes (Yang et al., 2011, 2014), was significantly increased in control AD cerebral cortex, while cathepsin D levels in ADINKO mice were as low as in non-AD WT mice (Fig. 8B). Similarly, IHC on hippocampus CA3 showed significantly less cathepsin D immunoreactivity in ADINKO mice (control, 100 ± 9.1%; ADINKO, 72.7 ± 1.7%, p < 0.01), confirming that the AV compartment was clearly diminished in ADINKO mice compared with control AD mice. We next coimmunolabeled cathepsin D and Aβ to quantify those AVs that contain Aβ. Neurons in control AD mice exhibited enlarged, perinuclear Aβ-positive AVs (Fig. 8C), characteristic of AD (Yang et al., 2011). Their number was significantly diminished in ADINKO mice compared with control AD mice (Fig. 8C, Mann–Whitney U test, p < 0.05). Analysis of size distribution revealed significantly fewer large and medium-sized Aβ-positive AVs in ADINKO mice, whereas the small vacuoles, typically lysosomes, remained unchanged. Together, these results indicate that ablation of IGF-1R contributed to reducing the Aβ-containing, enlarged AV compartment. We conclude that specific structural and molecular abnormalities of autophagy pathways observed in AD neurons were rescued in ADINKO mutants. Overall, these findings suggested that changes in neuronal size, morphology and autophagy pathway due to IGF-1R knock-out cooperate in preventing toxic Aβ accumulation in the brain.
Figure 8.
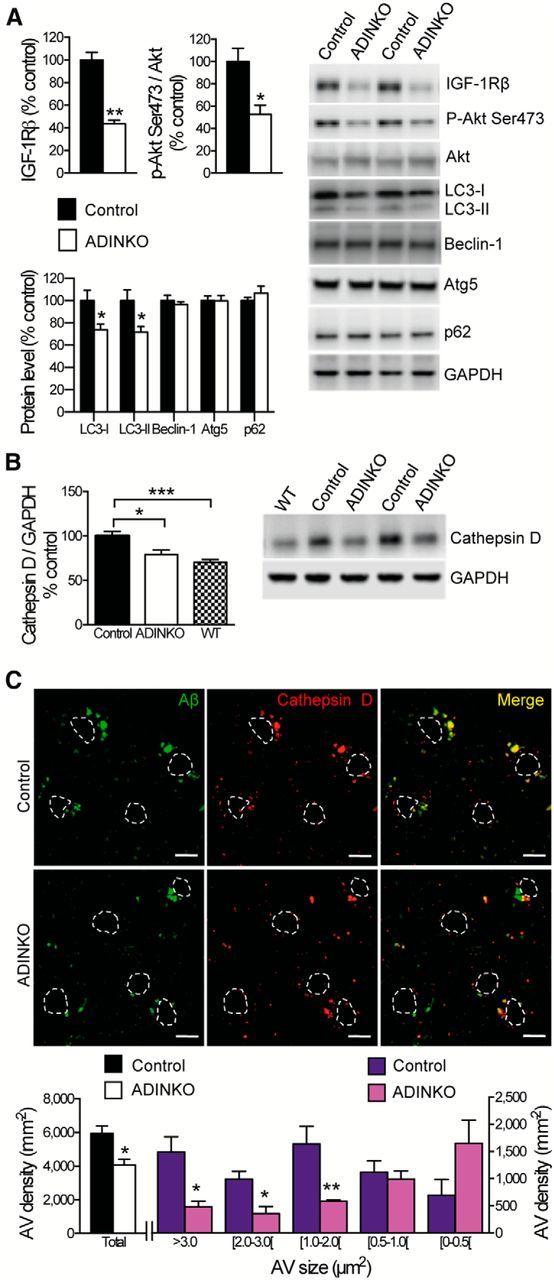
Diminished accumulation of Aβ-containing AVs in ADINKO forebrain. A, Diminished phospho-Akt (Ser473) and LC3 in ADINKO cortex compared with controls (representative Western blots). B, Cathepsin D levels in ADINKO cortex were reduced to WT levels (representative Western blot). C, Representative micrographs of double immunolabeling of Aβ (green) and cathepsin D-positive AVs (red). Nuclei of neurons are outlined in white. Scale bars, 10 μm. Size distribution revealed that prevalence of Aβ-positive AVs, in particular middle-sized and large, was diminished in hippocampus from ADINKO females. Mann–Whitney U test, n = 6 per group; *p < 0.05, **p < 0.01, mean ± SEM.
Discussion
We demonstrated that ablation of IGF-1R from adult neurons alleviated AD pathology and the associated cognitive deficits in transgenic mice. IGF-1R-deficient neurons had small soma, lean dendrites, and proximal shift of dendrite branching. In line with beneficial effects of partial IGF-1R inactivation on AD pathology (Cohen et al., 2009; Freude et al., 2009), we showed that blocking IGF signaling in adult neurons is neuroprotective. We found that IGF-1R ablation from postmitotic neurons in vivo diminished insoluble Aβ and amyloid plaques, and cleared soluble monomeric and oligomeric Aβ, thereby reducing proteotoxicity and limiting neuronal loss in the forebrain. This paralleled with preserved myelin content, as well as better neuroinflammatory and cognitive status in ADINKO brains. APP processing remained unaffected, but AD-related accumulation of Aβ-containing AVs was reduced in neurons. Figure 9 shows a synopsis of these findings.
Figure 9.

Neuronal IGF-1R signaling impacts AD progression in multiple ways. Blocking IGF-1R signaling in adult neurons affects cell maintenance and protein homeostasis. Neurons change to a more compact soma and leaner dendrites (1). Autophagy defects in AD, characterized by accumulation of Aβ containing autophagic vacuoles, normalize after IGF-1R inactivation (2). IGF-1R inactivation does not change APP production or processing. Significantly less insoluble Aβ (fewer plaques) and markedly diminished soluble Aβ point to facilitated clearance of toxic peptides from the brain (3). Consequently, neuronal microenvironment is less toxic, as reflected by preserved myelin content and diminished microglial infiltration, possibly preventing loss of neurons (4). Cytoarchitectural and functional changes observed after neuronal IGF-1R inactivation in the forebrain of AD improve behavioral and cognitive performances (5). Several different processes are improved in the absence of IGF signaling, suggesting that neuroprotective mechanisms are well adapted to low somatotropic tone.
PI3K-Akt is a well established kinase system upstream of mTOR that regulates protein synthesis and autophagy. In ADINKO mice, we revealed markedly reduced cortical Akt activation and low LC3-II. Moreover, while Aβ-containing autophagosome and autophagolysosome vesicles, known to accumulate specifically in AD neurons (Yang et al., 2011, 2014), were indeed enlarged in AD control hippocampus, we found this compartment reduced to almost normal size in ADINKO mice. These findings indicate that phenotypic defects of autophagy are at least partly rescued in ADINKO neurons and suggest that upon long-term IGF-1R inactivation, neuronal protein turnover and degradation are most likely handled more adequately with available cellular autophagic capacity. As judged from cellular morphology, ADINKO neurons present with improved health, possibly explaining enhanced neuroprotection and cell survival during AD progression. Recent data indicate that Aβ is both generated and degraded in autophagic vesicles, potentially contributing in a vicious circle to Aβ accumulation in AD (Nilsson et al., 2013). Therefore, a downsized autophagic compartment per se may also contribute to reducing Aβ efflux from ADINKO neurons, thereby potentially lowering the Aβ pool in the brain parenchyma. The relative contributions of smaller autophagic compartments in ADINKO brains versus possibly enhanced autophagic activity remain to be elucidated. Importantly, pertaining to effects on cell size and autophagy, ADINKO brains studied 9, 14, and 21 months after definitive ablation of IGF-1R are to be considered as a new steady state or endpoint. This situation is different from acute effects observed in vitro under short-term activation of autophagy, which may in turn explain why p62 levels and LC3-II/I ratios were not significantly altered in ADINKO brains, as could be expected at short term (Klionsky et al., 2012). Of note, some in vivo studies also report unchanged levels of soluble p62 upon induction of autophagy in AD brains (Steele et al., 2013) and in neurodegeneration (Ghazi-Noori et al., 2012).
Our data are consistent with results from other model systems. Inhibition of IGF-1R orthologue daf-2 in AD-like nematodes promotes detoxification via aggregation into high-molecular-weight Aβ deposits and increased autophagic clearance of Aβ oligomers (Cohen et al., 2006; Florez-McClure et al., 2007). Abnormalities in IGF-1R expression and activation, including IGF-I resistance, exist in AD brains (Rivera et al., 2005; Steen et al., 2005; Moloney et al., 2010; Bomfim et al., 2012; Talbot et al., 2012), although causality is not clear. Upon ligand binding, IGF-1R phosphorylates adaptor proteins IRS1 and IRS2 that subsequently activate PI3K-Akt and mTOR pathways. Brain-specific IRS2 knock-out increases lifespan, and AD mice lacking IRS2 accumulate less Aβ and are rescued from premature mortality (Taguchi et al., 2007; Freude et al., 2009; Killick et al., 2009). Our findings suggest that IGF-1R resistance observed in AD postmortem brains may represent a defense mechanism of aging neurons facing Aβ deposition. Furthermore, it is possible that the naturally prevailing endocrine status is characterized rather by low IGF signaling than by a full complement of IGF. This latter situation is predominating in ad libitum-fed laboratory mice (Kappeler et al., 2009) and may in fact accelerate AD-like progression. From that perspective, the IGF-1R knock-out mutation in ADINKO mice corrects an artificially high activated neuronal IGF-I pathway.
Interestingly, inhibition of insulin-like/insulin-signaling pathways in simple organisms postpones multiple age-related pathologies, extending health span (Cohen et al., 2006; Florez-McClure et al., 2007). This supports the idea that interventions slowing down aging also postpone age-related diseases, including AD (Kenyon, 2010; Iliadi et al., 2012). In humans, disease-free survival is high in families with exceptional longevity (Sebastiani and Perls, 2012), suggesting that health span and life span are linked. Recently, pharmacological inhibition of growth hormone and IGF-I was listed as a promising strategy to retard human aging (Longo et al., 2015). Here, we showed that blocking neuronal IGF signaling improves resistance to Aβ proteotoxicity and slows AD progression. Similarly, we demonstrated that damage induced by stroke-modeling ischemic-hypoxic insult is diminished when neuronal IGF-1R is knocked out (M. Holzenberger and S. Aïd, unpublished observations), and Biondi et al. (2015) reported that in a mouse model of spinal muscular atrophy, motoneurons are protected and neuromuscular behavior improved when IGF signaling is reduced. Thus, different IGF-1R loss-of-function mutations are beneficial in age-related neurological disorders, indicating that mechanisms of delayed aging can be traced to cell-autonomous changes in the brain. Genetic inactivation of IGF-1R in neurons also revealed that this pathway controls a high degree of cell-autonomous structural plasticity. This plasticity is all the more remarkable since diminution of cell soma occurred in fully differentiated neurons, and not during cell growth and development, as it is mostly the case when genes are implicated in structural plasticity. Moreover, ADINKO neurons all over the brain underwent size reduction, suggesting high phenotypic penetrance of this effect. Interestingly, IGF-dependent regulation of cell size has also been reported for chondrocytes (Wang et al., 1999; Cooper et al., 2013). We showed for the first time that adult neuron-specific IGF-1R knock-out markedly reduced apical cytoplasm and length of primary dendrite, and that reduction in cell size is linked to significant resistance to AD. These morphological changes did not affect dendrite complexity or axon integrity, important for normal neuronal function. Comparable size effects in CNS neurons have been reported in mice lacking phosphatase and tensin homolog (PTEN) and components of the mTOR pathway mTORC2 and mTORC1. The PI3K-mTOR pathway is a major signaling route activated by IGF-1R, and PTEN is counteracting PI3K signaling. The tuberous sclerosis protein complex TSC1/2 acts as a negative regulator of mTOR. Indeed, neurons lacking TSC1 or PTEN show a progressive enlargement of their soma during development (Backman et al., 2001; Kwon et al., 2001; Tavazoie et al., 2005; van Diepen et al., 2009), while neurons lacking mTORC1 or mTORC2 exhibited reduced soma size (Cloëtta et al., 2013; Thomanetz et al., 2013).
Our findings in adult brain are thus consistent with an important role of neuronal IGF-1R as upstream signal for cell size-controlling the PI3K-Akt-mTOR pathway. In agreement with our study, recent data demonstrated increased mTOR activity in AD mouse brain, while mTOR inhibition reduces Aβ accumulation and spatial memory deficits (Caccamo et al., 2010). Furthermore, increased plasma Aβ, as we observed here, points to more efficient Aβ clearance from the brain into the bloodstream. This finding fits well with the observation of reduced soluble Aβ in the brain. Slender neurons most likely cause other local changes in histoarchitecture, eventually affecting adjacent glial cells and endothelia that may all converge to alter interstitial fluid movements and local clearance. Xie et al. demonstrated elegantly that convective fluxes in the interstitial compartment of cerebral cortex affect Aβ clearance. Therefore, morphological changes of major tissue components in the CNS may influence Aβ steady state (Xie et al., 2013). Clearly, more work is needed to determine whether IGF-dependent changes in neuronal morphology readily impact the function of glymphatic compartments.
Our findings suggest that changes in neuronal size and morphology, and improvements in autophagy compartments due to IGF-1R knock-out cooperate to prevent accumulation of Aβ with aging, thereby efficiently diminishing neuronal toxicity. This study showed in vivo that IGF signaling is strongly involved in the control of proteostasis in aging neurons, and that suppression of neuronal IGF signaling efficiently slows down AD progression. IGF receptors, ligands, high-affinity binding proteins, and proteases constitute a gene family that can be targeted at multiple molecular levels. Therefore, this study opens new avenues of research, notably pointing to other cellular pathways controlling neuronal homeostasis during aging as potential targets for AD treatment.
Footnotes
This work was supported by Institut National de la Santé et de la Recherche Médicale, Université Pier et Marie Curie, France Alzheimer, and Ligue Européenne contre la Maladie d'Alzheimer. Fellowships were provided by the Fondation pour la Recherche Médicale (G.G.), the Ministère de l'Education Nationale, de l'Enseignement Supérieur et de la Recherche (G.G., C.G.), and the AXA Research Fund (Z.C.). France Alzheimer and Fondation Plan Alzheimer supported S.A. We thank P. Lacube, T. Ledent, and L. Dinard for valuable contributions.
The authors declare no competing financial interests.
References
- Attar A, Liu T, Chan WT, Hayes J, Nejad M, Lei K, Bitan G. A shortened Barnes maze protocol reveals memory deficits at 4-months of age in the triple-transgenic mouse model of Alzheimer's disease. PLoS One. 2013;8:e80355. doi: 10.1371/journal.pone.0080355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backman SA, Stambolic V, Suzuki A, Haight J, Elia A, Pretorius J, Tsao MS, Shannon P, Bolon B, Ivy GO, Mak TW. Deletion of Pten in mouse brain causes seizures, ataxia and defects in soma size resembling Lhermitte-Duclos disease. Nat Genet. 2001;29:396–403. doi: 10.1038/ng782. [DOI] [PubMed] [Google Scholar]
- Barnes CA. Memory deficits associated with senescence: a neurophysiological and behavioral study in the rat. J Comp Physiol Psychol. 1979;93:74–104. doi: 10.1037/h0077579. [DOI] [PubMed] [Google Scholar]
- Biondi O, Branchu J, Ben Salah A, Houdebine L, Bertin L, Chali F, Desseille C, Weill L, Sanchez G, Lancelin C, Aïd S, Lopes P, Pariset C, Lécolle S, Côté J, Holzenberger M, Chanoine C, Massaad C, Charbonnier F. IGF-1R reduction triggers neuroprotective signaling pathways in spinal-muscular-atrophy mice. J Neurosci. 2015 doi: 10.1523/JNEUROSCI.0608-15.2015. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bomfim TR, Forny-Germano L, Sathler LB, Brito-Moreira J, Houzel JC, Decker H, Silverman MA, Kazi H, Melo HM, McClean PL, Holscher C, Arnold SE, Talbot K, Klein WL, Munoz DP, Ferreira ST, De Felice FG. An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer's disease-associated Abeta oligomers. J Clin Invest. 2012;122:1339–1353. doi: 10.1172/JCI57256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouillette J, Caillierez R, Zommer N, Alves-Pires C, Benilova I, Blum D, De Strooper B, Buée L. Neurotoxicity and memory deficits induced by soluble low-molecular-weight amyloid-β1–42 oligomers are revealed in vivo by using a novel animal model. J Neurosci. 2012;32:7852–7861. doi: 10.1523/JNEUROSCI.5901-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caccamo A, Majumder S, Richardson A, Strong R, Oddo S. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: effects on cognitive impairments. J Biol Chem. 2010;285:13107–13120. doi: 10.1074/jbc.M110.100420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carro E, Trejo JL, Gomez-Isla T, LeRoith D, Torres-Aleman I. Serum insulin-like growth factor I regulates brain amyloid-beta levels. Nat Med. 2002;8:1390–1397. doi: 10.1038/nm1202-793. [DOI] [PubMed] [Google Scholar]
- Choi SH, Aid S, Caracciolo L, Minami SS, Niikura T, Matsuoka Y, Turner RS, Mattson MP, Bosetti F. Cyclooxygenase-1 inhibition reduces amyloid pathology and improves memory deficits in a mouse model of Alzheimer's disease. J Neurochem. 2013;124:59–68. doi: 10.1111/jnc.12059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chvátal A, Anderová M, Hock M, Prajerová I, Neprasová H, Chvátal V, Kirchhoff F, Syková E. Three-dimensional confocal morphometry reveals structural changes in astrocyte morphology in situ. J Neurosci Res. 2007;85:260–271. doi: 10.1002/jnr.21113. [DOI] [PubMed] [Google Scholar]
- Cloëtta D, Thomanetz V, Baranek C, Lustenberger RM, Lin S, Oliveri F, Atanasoski S, Rüegg MA. Inactivation of mTORC1 in the developing brain causes microcephaly and affects gliogenesis. J Neurosci. 2013;33:7799–7810. doi: 10.1523/JNEUROSCI.3294-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen E, Bieschke J, Perciavalle RM, Kelly JW, Dillin A. Opposing activities protect against age-onset proteotoxicity. Science. 2006;313:1604–1610. doi: 10.1126/science.1124646. [DOI] [PubMed] [Google Scholar]
- Cohen E, Paulsson JF, Blinder P, Burstyn-Cohen T, Du D, Estepa G, Adame A, Pham HM, Holzenberger M, Kelly JW, Masliah E, Dillin A. Reduced IGF-1 signaling delays age-associated proteotoxicity in mice. Cell. 2009;139:1157–1169. doi: 10.1016/j.cell.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper KL, Oh S, Sung Y, Dasari RR, Kirschner MW, Tabin CJ. Multiple phases of chondrocyte enlargement underlie differences in skeletal proportions. Nature. 2013;495:375–378. doi: 10.1038/nature11940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdmann G, Schütz G, Berger S. Inducible gene inactivation in neurons of the adult mouse forebrain. BMC Neurosci. 2007;8:63. doi: 10.1186/1471-2202-8-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florez-McClure ML, Hohsfield LA, Fonte G, Bealor MT, Link CD. Decreased insulin-receptor signaling promotes the autophagic degradation of beta-amyloid peptide in C. elegans. Autophagy. 2007;3:569–580. doi: 10.4161/auto.4776. [DOI] [PubMed] [Google Scholar]
- Freude S, Hettich MM, Schumann C, Stöhr O, Koch L, Köhler C, Udelhoven M, Leeser U, Müller M, Kubota N, Kadowaki T, Krone W, Schröder H, Brüning JC, Schubert M. Neuronal IGF-1 resistance reduces Abeta accumulation and protects against premature death in a model of Alzheimer's disease. FASEB J. 2009;23:3315–3324. doi: 10.1096/fj.09-132043. [DOI] [PubMed] [Google Scholar]
- Fricano CJ, Despenza T, Jr, Frazel PW, Li M, O'Malley AJ, Westbrook GL, Luikart BW. Fatty acids increase neuronal hypertrophy of Pten knockdown neurons. Front Mol Neurosci. 2014;7:30. doi: 10.3389/fnmol.2014.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghazi-Noori S, Froud KE, Mizielinska S, Powell C, Smidak M, Fernandez de Marco M, O'Malley C, Farmer M, Parkinson N, Fisher EM, Asante EA, Brandner S, Collinge J, Isaacs AM. Progressive neuronal inclusion formation and axonal degeneration in CHMP2B mutant transgenic mice. Brain. 2012;135:819–832. doi: 10.1093/brain/aws006. [DOI] [PubMed] [Google Scholar]
- Gunji H, Little RA, Hiraiwa K. Interleukin-6 deficiency increases blood volume without altering body composition in young mice. Cytokine. 2002;20:30–37. doi: 10.1006/cyto.2002.1977. [DOI] [PubMed] [Google Scholar]
- Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, Griep A, Axt D, Remus A, Tzeng TC, Gelpi E, Halle A, Korte M, Latz E, Golenbock DT. NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature. 2013;493:674–678. doi: 10.1038/nature11729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herculano-Houzel S, Watson C, Paxinos G. Distribution of neurons in functional areas of the mouse cerebral cortex reveals quantitatively different cortical zones. Front Neuroanat. 2013;7:35. doi: 10.3389/fnana.2013.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata-Fukae C, Li HF, Hoe HS, Gray AJ, Minami SS, Hamada K, Niikura T, Hua F, Tsukagoshi-Nagai H, Horikoshi-Sakuraba Y, Mughal M, Rebeck GW, LaFerla FM, Mattson MP, Iwata N, Saido TC, Klein WL, Duff KE, Aisen PS, Matsuoka Y. Females exhibit more extensive amyloid, but not tau, pathology in an Alzheimer transgenic model. Brain Res. 2008;1216:92–103. doi: 10.1016/j.brainres.2008.03.079. [DOI] [PubMed] [Google Scholar]
- Holzenberger M, Dupont J, Ducos B, Leneuve P, Géloën A, Even PC, Cervera P, Le Bouc Y. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature. 2003;421:182–187. doi: 10.1038/nature01298. [DOI] [PubMed] [Google Scholar]
- Iliadi KG, Knight D, Boulianne GL. Healthy aging—insights from Drosophila. Front Physiol. 2012;3:106. doi: 10.3389/fphys.2012.00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowsky JL, Fadale DJ, Anderson J, Xu GM, Gonzales V, Jenkins NA, Copeland NG, Lee MK, Younkin LH, Wagner SL, Younkin SG, Borchelt DR. Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum Mol Genet. 2004;13:159–170. doi: 10.1093/hmg/ddh019. [DOI] [PubMed] [Google Scholar]
- Kappeler L, De Magalhaes Filho C, Dupont J, Leneuve P, Cervera P, Périn L, Loudes C, Blaise A, Klein R, Epelbaum J, Le Bouc Y, Holzenberger M. Brain IGF-1 receptors control mammalian growth and lifespan through a neuroendocrine mechanism. PLoS Biol. 2008;6:e254. doi: 10.1371/journal.pbio.0060254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kappeler L, De Magalhaes Filho C, Leneuve P, Xu J, Brunel N, Chatziantoniou C, Le Bouc Y, Holzenberger M. Early postnatal nutrition determines somatotropic function in mice. Endocrinology. 2009;150:314–323. doi: 10.1210/en.2008-0981. [DOI] [PubMed] [Google Scholar]
- Kenyon CJ. The genetics of ageing. Nature. 2010;464:504–512. doi: 10.1038/nature08980. [DOI] [PubMed] [Google Scholar]
- Killick R, Scales G, Leroy K, Causevic M, Hooper C, Irvine EE, Choudhury AI, Drinkwater L, Kerr F, Al-Qassab H, Stephenson J, Yilmaz Z, Giese KP, Brion JP, Withers DJ, Lovestone S. Deletion of Irs2 reduces amyloid deposition and rescues behavioural deficits in APP transgenic mice. Biochem Biophys Res Commun. 2009;386:257–262. doi: 10.1016/j.bbrc.2009.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, Ahn HJ, Ait-Mohamed O, Ait-Si-Ali S, Akematsu T, Akira S, Al-Younes HM, Al-Zeer MA, Albert ML, Albin RL, Alegre-Abarrategui J, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koistinaho M, Lin S, Wu X, Esterman M, Koger D, Hanson J, Higgs R, Liu F, Malkani S, Bales KR, Paul SM. Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-β peptides. Nat Med. 2004;10:719–726. doi: 10.1038/nm1058. [DOI] [PubMed] [Google Scholar]
- Kwon CH, Zhu X, Zhang J, Knoop LL, Tharp R, Smeyne RJ, Eberhart CG, Burger PC, Baker SJ. Pten regulates neuronal soma size: a mouse model of Lhermitte-Duclos disease. Nat Genet. 2001;29:404–411. doi: 10.1038/ng781. [DOI] [PubMed] [Google Scholar]
- Larson ME, Sherman MA, Greimel S, Kuskowski M, Schneider JA, Bennett DA, Lesné SE. Soluble α-synuclein is a novel modulator of Alzheimer's disease pathophysiology. J Neurosci. 2012;32:10253–10266. doi: 10.1523/JNEUROSCI.0581-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo VD, Antebi A, Bartke A, Barzilai N, Brown-Borg HM, Caruso C, Curiel TJ, de Cabo R, Franceschi C, Gems D, Ingram DK, Johnson TE, Kennedy BK, Kenyon C, Klein S, Kopchick JJ, Lepperdinger G, Madeo F, Mirisola MG, Mitchell JR, et al. Interventions to slow aging in humans: are we ready? Aging Cell. 2015 doi: 10.1111/acel.12338. Advance online publication. Retrieved July 6, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, Lein ES, Zeng H. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010;13:133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mc Donald JM, Savva GM, Brayne C, Welzel AT, Forster G, Shankar GM, Selkoe DJ, Ince PG, Walsh DM, Walsh DM. The presence of sodium dodecyl sulphate-stable Abeta dimers is strongly associated with Alzheimer-type dementia. Brain. 2010;133:1328–1341. doi: 10.1093/brain/awq065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minami SS, Sidahmed E, Aid S, Shimoji M, Niikura T, Mocchetti I, Rebeck GW, Prendergast JS, Dealwis C, Wetzel R, Bosetti F, Matsuoka Y, Hoe HS, Turner RS. Therapeutic versus neuroinflammatory effects of passive immunization is dependent on Aβ/amyloid burden in a transgenic mouse model of Alzheimer's disease. J Neuroinflammation. 2010;7:57. doi: 10.1186/1742-2094-7-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moloney AM, Griffin RJ, Timmons S, O'Connor R, Ravid R, O'Neill C. Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer's disease indicate possible resistance to IGF-1 and insulin signalling. Neurobiol Aging. 2010;31:224–243. doi: 10.1016/j.neurobiolaging.2008.04.002. [DOI] [PubMed] [Google Scholar]
- Nilsson P, Loganathan K, Sekiguchi M, Matsuba Y, Hui K, Tsubuki S, Tanaka M, Iwata N, Saito T, Saido TC. Aβ secretion and plaque formation depend on autophagy. Cell Rep. 2013;5:61–69. doi: 10.1016/j.celrep.2013.08.042. [DOI] [PubMed] [Google Scholar]
- Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. 2013;19:983–997. doi: 10.1038/nm.3232. [DOI] [PubMed] [Google Scholar]
- O'Leary TP, Brown RE. Visuo-spatial learning and memory deficits on the Barnes maze in the 16-month-old APPswe/PS1dE9 mouse model of Alzheimer's disease. Behav Brain Res. 2009;201:120–127. doi: 10.1016/j.bbr.2009.01.039. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. San Diego: Academic; 2001. [Google Scholar]
- Reiserer RS, Harrison FE, Syverud DC, McDonald MP. Impaired spatial learning in the APPSwe + PSEN1DeltaE9 bigenic mouse model of Alzheimer's disease. Genes Brain Behav. 2007;6:54–65. doi: 10.1111/j.1601-183X.2006.00221.x. [DOI] [PubMed] [Google Scholar]
- Rivera EJ, Goldin A, Fulmer N, Tavares R, Wands JR, de la Monte SM. Insulin and insulin-like growth factor expression and function deteriorate with progression of Alzheimer's disease: link to brain reductions in acetylcholine. J Alzheimers Dis. 2005;8:247–268. doi: 10.3233/jad-2005-8304. [DOI] [PubMed] [Google Scholar]
- Sebastiani P, Perls TT. The genetics of extreme longevity: lessons from the New England Centenarian Study. Front Genet. 2012;3:277. doi: 10.3389/fgene.2012.00277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano-Pozo A, William CM, Ferrer I, Uro-Coste E, Delisle MB, Maurage CA, Hock C, Nitsch RM, Masliah E, Growdon JH, Frosch MP, Hyman BT. Beneficial effect of human anti-amyloid-beta active immunization on neurite morphology and tau pathology. Brain. 2010;133:1312–1327. doi: 10.1093/brain/awq056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele JW, Lachenmayer ML, Ju S, Stock A, Liken J, Kim SH, Delgado LM, Alfaro IE, Bernales S, Verdile G, Bharadwaj P, Gupta V, Barr R, Friss A, Dolios G, Wang R, Ringe D, Fraser P, Westaway D, St George-Hyslop PH, et al. Latrepirdine improves cognition and arrests progression of neuropathology in an Alzheimer's mouse model. Mol Psychiatry. 2013;18:889–897. doi: 10.1038/mp.2012.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steen E, Terry BM, Rivera EJ, Cannon JL, Neely TR, Tavares R, Xu XJ, Wands JR, de la Monte SM. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer's disease—is this type 3 diabetes? J Alzheimers Dis. 2005;7:63–80. doi: 10.3233/jad-2005-7107. [DOI] [PubMed] [Google Scholar]
- Stewart S, Cacucci F, Lever C. Which memory task for my mouse? A systematic review of spatial memory performance in the Tg2576 Alzheimer's mouse model. J Alzheimers Dis. 2011;26:105–126. doi: 10.3233/JAD-2011-101827. [DOI] [PubMed] [Google Scholar]
- Taguchi A, Wartschow LM, White MF. Brain IRS2 signaling coordinates life span and nutrient homeostasis. Science. 2007;317:369–372. doi: 10.1126/science.1142179. [DOI] [PubMed] [Google Scholar]
- Talbot K, Wang HY, Kazi H, Han LY, Bakshi KP, Stucky A, Fuino RL, Kawaguchi KR, Samoyedny AJ, Wilson RS, Arvanitakis Z, Schneider JA, Wolf BA, Bennett DA, Trojanowski JQ, Arnold SE. Demonstrated brain insulin resistance in Alzheimer's disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest. 2012;122:1316–1338. doi: 10.1172/JCI59903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavazoie SF, Alvarez VA, Ridenour DA, Kwiatkowski DJ, Sabatini BL. Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat Neurosci. 2005;8:1727–1734. doi: 10.1038/nn1566. [DOI] [PubMed] [Google Scholar]
- Thomanetz V, Angliker N, Cloëtta D, Lustenberger RM, Schweighauser M, Oliveri F, Suzuki N, Rüegg MA. Ablation of the mTORC2 component rictor in brain or Purkinje cells affects size and neuron morphology. J Cell Biol. 2013;201:293–308. doi: 10.1083/jcb.201205030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Diepen MT, Parsons M, Downes CP, Leslie NR, Hindges R, Eickholt BJ. MyosinV controls PTEN function and neuronal cell size. Nat Cell Biol. 2009;11:1191–1196. doi: 10.1038/ncb1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Zhou J, Bondy CA. Igf1 promotes longitudinal bone growth by insulin-like actions augmenting chondrocyte hypertrophy. FASEB J. 1999;13:1985–1990. doi: 10.1096/fasebj.13.14.1985. [DOI] [PubMed] [Google Scholar]
- Wang J, Tanila H, Puoliväli J, Kadish I, van Groen T. Gender differences in the amount and deposition of amyloidbeta in APPswe and PS1 double transgenic mice. Neurobiol Dis. 2003;14:318–327. doi: 10.1016/j.nbd.2003.08.009. [DOI] [PubMed] [Google Scholar]
- Xie L, Kang H, Xu Q, Chen MJ, Liao Y, Thiyagarajan M, O'Donnell J, Christensen DJ, Nicholson C, Iliff JJ, Takano T, Deane R, Nedergaard M. Sleep drives metabolite clearance from the adult brain. Science. 2013;342:373–377. doi: 10.1126/science.1241224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Gontier G, Chaker Z, Lacube P, Dupont J, Holzenberger M. Longevity effect of IGF-1R mutation depends on genetic background-specific receptor activation. Aging Cell. 2014;13:19–28. doi: 10.1111/acel.12145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang DS, Stavrides P, Mohan PS, Kaushik S, Kumar A, Ohno M, Schmidt SD, Wesson D, Bandyopadhyay U, Jiang Y, Pawlik M, Peterhoff CM, Yang AJ, Wilson DA, St George-Hyslop P, Westaway D, Mathews PM, Levy E, Cuervo AM, Nixon RA. Reversal of autophagy dysfunction in the TgCRND8 mouse model of Alzheimer's disease ameliorates amyloid pathologies and memory deficits. Brain. 2011;134:258–277. doi: 10.1093/brain/awq341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang DS, Stavrides P, Saito M, Kumar A, Rodriguez-Navarro JA, Pawlik M, Huo C, Walkley SU, Saito M, Cuervo AM, Nixon RA. Defective macroautophagic turnover of brain lipids in the TgCRND8 Alzheimer mouse model: prevention by correcting lysosomal proteolytic deficits. Brain. 2014;137:3300–3318. doi: 10.1093/brain/awu278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Li J, Purkayastha S, Tang Y, Zhang H, Yin Y, Li B, Liu G, Cai D. Hypothalamic programming of systemic ageing involving IKK-beta, NF-kappaB and GnRH. Nature. 2013;497:211–216. doi: 10.1038/nature12143. [DOI] [PMC free article] [PubMed] [Google Scholar]