

Abstract
Chronic pain is still a basic science and clinical challenge. Unraveling of the neurobiological mechanisms involved in chronic pain will offer novel targets for the development of therapeutic strategies. It is well known that central sensitization in the anterior cingulate cortex (ACC) plays a critical role in initiation, development, and maintenance of chronic pain. However, the underlying mechanisms still remain elusive. Here, we reported that caveolin-1 (Cav-1), a scaffolding protein in membrane rafts, was persistently upregulated and activated in the ACC neurons after chronic constriction injury (CCI) in mice. Knockdown or blocking of Cav-1 in the contralateral ACC to the injury side reversed CCI-induced pain behavioral and neuronal sensitization and overexpression of Cav-1 in the ipsilateral ACC-induced pain behavior in the unaffected hindpaw. Furthermore, we found that Cav-1 directly binding with NMDA receptor 2B subunit (NR2B) and promotion of NR2B surface levels in the ACC contributed to modulation of chronic neuropathic pain. Disrupting the interaction of Cav-1 and NR2B through microinjection of a short peptide derived from the C-terminal of NR2B into the ACC exhibited a significant anti-nociception effect associated with decrease of surface NR2B expression. Moreover, Cav-1 increased intracellular Ca2+ concentration and activated the ERK/CREB signaling pathway in an NR2B-dependent manner in the ACC. Our findings implicate that Cav-1 in the ACC neurons modulates chronic neuropathic pain via regulation of NR2B and subsequent activation of ERK/CREB signaling, suggesting a possible caveolin-mediated process would participate in neuronal transmission pathways implicated in pain modulation.
Keywords: anterior cingulate cortex, caveolin-1, NMDA receptor 2B subunit, neuropathic pain
Introduction
Preclinical and clinical brain imaging studies have shown that the ACC is a key brain region for pain perception and modulation underlying physiological and pathological pain status (Xu et al., 2008; Zhuo, 2008; Mohseni et al., 2012; Gasquoine, 2013; Meerwijk et al., 2013). Persistent changes of neuronal excitability associated with intrinsic plasticity are main cellular mechanisms for the modulatory effect of ACC on chronic pain (Zhao et al., 2006; Toyoda et al., 2009b; Li et al., 2010). The changes of neuronal excitability and intrinsic plasticity are dependent on functional regulation of NMDA receptors and their downstream signaling cascades (Mori et al., 2011; Sanchez et al., 2012). Emerging evidence suggests that NMDA receptor 2B subunit (NR2B)-mediated neuronal plasticity in the ACC contributes to inflammation-related or nerve injury-related persistent pain (Wu et al., 2005; Li et al., 2009; Zhuo, 2009, 2014). However, the regulatory mechanism of NR2B in ACC underlying chronic pain remains unknown.
Caveolae, a special type of membrane raft, are small invaginations of the plasma membrane in many vertebrate cells (Chidlow and Sessa, 2010; Ariotti and Parton, 2013). In the CNS, caveolae are implicated in the establishment of cell polarity (Ledesma et al., 1998), axon guidance (Guirland and Zheng, 2007), normal spine density and morphology (Hering et al., 2003), endocytosis, cell transport and metabolism, signaling transduction (Tsui-Pierchala et al., 2002; Allen et al., 2007), and receptor trafficking (Brusés et al., 2001; Francesconi et al., 2009). Formation and maintenance of caveolae are primarily due to the protein caveolins (Anderson, 1998). Caveolins may act as scaffolding proteins within caveolae membranes by compartmentalizing and concentrating signaling molecules including G-protein subunits, receptor and nonreceptor tyrosine kinases, and small GTPases through its caveolin-scaffolding domain (Mineo et al., 1996; Couet et al., 1997; Venema et al., 1997; Yamamoto et al., 1998). There are three homologous genes of caveolin expressed in mammalian cells: caveolin-1 (Cav-1), caveolin-2 (Cav-2), and caveolin-3 (Cav-3) (Krajewska and Maslowska, 2004; Williams and Lisanti, 2004). Cav-1 is expressed in neurons in several brain regions including cerebral cortex (Cameron et al., 1997), hippocampus (Petralia et al., 2003), and hypothalamus (Zschocke et al., 2002). A recent study revealed that Cav-1 is present at excitatory synapses and concentrates at the postsynaptic density during late-stage development (Petralia et al., 2003) and mediates regulation of group I metabotropic glutamate receptor trafficking and signaling (Francesconi et al., 2009), suggesting that Cav-1 might play an important role in synapse formation and plasticity. In addition to these physiological roles, the potential roles of dysregulation of Cav-1 expression and related impact on neuronal signaling in some pathological processes in the CNS, such as neuronal injury (Gaudreault et al., 2005), ischemia (Shen et al., 2006), and Alzheimer's disease (Gaudreault et al., 2004), have been reported. Since Cav-1 regulates neuronal plasticity and receptor trafficking and long-term plastic changes related to NR2B in ACC contribute to chronic pain modulation, we investigated the potential involvement of Cav-1 and its regulatory mechanism in a chronic constriction injury (CCI)-induced chronic neuropathic pain model.
Materials and Methods
Animals.
Adult male Kunming mice (20–22 g), provided by the Experimental Animal Center of Xuzhou Medical College, were used for these studies. Mice were housed under a 12 h light/dark cycle with food and water ad libitum. All experimental protocols were approved by the Animal Care and Use Committee of Xuzhou Medical College (Xuzhou, Jiangsu Province, China) and were in accordance with the Declaration of NIH Guide for Care and Use of Laboratory Animals (Publication No. 80-23, revised 1996).
CCI model.
CCI model was performed following a method described previously (Bennett and Xie, 1988). In brief, mice were anesthetized with 10% chloral hydrate (0.3 ml/100 g, i.p.). The right sciatic nerve was exposed at mid-thigh level through a small incision and a unilateral constriction injury just proximal to the trifurcation was performed with three loose ligatures using a 5-0 silk thread (spaced at 1 mm intervals). In sham-operated animals, the nerve was exposed but not ligated. The incision was closed in layers, and the wound was treated with antibiotics (penicillin).
Stereotaxic microinjection.
Mice were anesthetized under isoflurane and placed in a stereotaxic frame (Stoelting). Stereotaxic ACC microinjection was performed as described before. In brief, the skin over the scalp was incised and a hole was made in the skull to allow the insertion of a microinjection needle into the ACC. A Hamilton syringe (1 μl) driven by a motorized syringe pump (set at a rate of 0.05 μl/min) was used to perfuse the solution (0.5 μl each injection). The needle was withdrawn 5 min after completion of the injection. The coordinates of the injection were as follows: 1.0 mm anterior to bregma, 0.3 mm lateral to the midline, and 1.8 mm ventral to the surface of the skull. After completion of experiments, all animals were deeply anesthetized with 10% chloral hydrate (0.3 ml/100 g, i.p.) and perfused with 4% paraformaldehyde. Serial cryostat coronal sections (30 μm) of the ACC were stained with hematoxylin and eosin and the site of injection was confirmed under the microscope. All doses of drugs are based on the results of preliminary experiments. The dose of each drug and time points of treatment were presented in the related figures and legends.
Behavioral test.
Paw withdrawal latencies (PWLs) in response to thermal hyperalgesia were measured by the IITC Plantar Analgesia Meter (IITC Life Science) according to the method described previously (Hargreaves et al., 1988). Animals were habituated in the testing environments for 2 d and behavioral test was performed in a blinded manner. Mice were placed in transparent acrylic enclosures (7 × 9 × 11 cm) with a glass plate and allowed to acclimatize to test environment for 1 h in a temperature-controlled and noise-free room (23–26°C). The high-intensity, movable radiant heat source was placed underneath the glass and focused onto the plantar surface of each hindpaw. The nociceptive endpoint in the radiant heat test was characteristic lifting or licking of the hindpaw. The time from onset of radiant heat to endpoint was considered as the PWL. The radiant heat intensity was adjusted at the beginning of the experiment to obtain basal PWL of 12–15 s, and an automatic 25 s cutoff was used to prevent tissue damage. Each animal was tested three times on each hindpaw at intervals of 5 min.
Paw withdrawal threshold (PWT) in response to mechanical allodynia was assessed by using von Frey filaments (North Coast Medical), starting with 0.31 g and ending with 4.0 g filament as the cutoff value. Animals were placed in individual plastic boxes (20 × 25 × 15 cm) on a metal mesh floor and allowed to acclimate for 1 h. The filaments were presented, in ascending order of strength, perpendicular to the plantar surface with sufficient force to cause slight bending against the paw and held for 6–8 s. Brisk withdrawal or paw flinching was considered as a positive response. The PWT was determined by sequentially increasing and decreasing the stimulus strength (the “up-and-down” method), and the data were analyzed using the nonparametric method of Dixon, as described previously (Chaplan et al., 1994). The behavioral tests were performed by an investigator blinded to the treatment.
Evaluation of blood–brain barrier permeability.
Evan's Blue (EB) leakage, a well established protocol, was used to assess blood—brain barrier (BBB) permeability. In brief, mice were anesthetized with 10% chloral hydrate (0.3 ml/100 g, i.p.) and injected with EB (2%, 2 ml/kg; Sigma) from caudal vein 0.5 h before perfusion. Animals were transcardially perfused with 0.9% saline until the perfusate from the right atrium ran clear and were subsequently decapitated and brains rapidly removed. The ACC area was obtained and placed in the Eppendorf container.
For quantitative measurement of EB extravasation, the sample was weighed and then homogenized in 0.75 ml of PBS and 0.25 ml of 100% TCA solution (Sigma). Samples were cooled overnight at 4°C, and then centrifuged for 30 min at 1000 g. The supernatants of each sample were subsequently measured at 620 nm using an ELISA plate reader. All measurements were within the range of detection established by the standard curve. The dye concentration was calculated as the ratio of absorbance relative to the amount of tissue (Dickstein et al., 2006; Harford-Wright et al., 2014).
Immunohistofluorescence.
Mice were anesthetized with 10% chloral hydrate (0.3 ml/100 g, i.p.) and subjected to sternotomy followed by intracardial perfusion with 40 ml saline and 100 ml 4% ice-cold PFA in 0.1 mol/L phosphate buffer. The brain was removed, post fixed in 4% PFA overnight, and subsequently allowed to equilibrate in 30% sucrose in phosphate buffer overnight at 4°C. Thirty micrometer transverse series brain sections containing ACC were cut on a cryostat and stored in phosphate buffer. After washing in PBS, the sections were incubated in PBS containing 5% normal donkey serum and 0.3% Triton X-100 at room temperature for 2 h, and then incubated with primary antibodies—rabbit anti-p-Cav-1,rabbit anti-t-Cav-1, goat anti-NR2B, and mouse anti-CD34 (1:50; Santa Cruz Biotechnology); mouse anti-PSD-95 (1:100; Abcam); and mouse anti-NeuN (1:100; Millipore)—in 5% normal donkey serum for 24–48 h at 4°C. Excess antibody was removed by incubation with PBS for 15 min and incubated with Alexa Fluor 488- or 543-conjugated secondary antibody (1:200; Molecular Probe) for 2 h at room temperature. To remove excess secondary antibody, slices were washed three times at 5 min intervals with PBS. Images were captured using a high-resolution digital confocal microscope (Olympus) with a computer.
Immunocytofluorescence.
To assess the effects of Cav-1 on surface NR2B expression, the primary cultured ACC neurons were washed three times with PBS at 10 min intervals, fixed with 4% PFA for 30 min, and then blocked for 2 h at room temperature with PBS containing 5% donkey serum. Surface NR2B of neurons were labeled by overnight incubation (at 4°C) with rabbit anti-NR2B (extracellular epitope, 1:100; Santa Cruz Biotechnology) in blocking buffer and excess antibody was removed by incubation with PBS for 15 min. Then cells were incubated with Alexa Fluor 543-conjugated secondary antibody (1:200; Molecular Probe) for 2 h at room temperature. To remove excess secondary antibody, cells were washed six times at 5 min intervals with PBS. For labeling intracellular NR2B, all the neurons were incubated with primary antibodies (rabbit anti-NR2B, 1:100; Santa Cruz Biotechnology) for 24 h after permeabilization with 0.3% Triton X-100 for 15 min, and then neurons were washed and incubated with Alexa Fluor 405-conjugated secondary antibody (1:200; Molecular Probe) for 2 h at room temperature. Cells were washed and mounted onto microscope slides for confocal imaging. Identical acquisition parameters were used for a given set of labeled neurons without oversaturation or undersaturation of the acquired signals. NIH ImageJ software was used for image quantification and analysis. Both the average intensity and the area of each selected NR2B puncta were measured. Data are presented as the ratio of surface to total (surface + intracellular).
Immunohistochemistry.
Sections containing ACC obtained were the same for the immunohistofluorescence method. For the Fos protein assay, the sections were incubated in primary polyclonal rabbit-anti-Fos antibody (1:100; Santa Cruz Biotechnology) at 4°C for 48 h. The sections were then incubated in biotinylated goat anti-rabbit (1:200) at 37°C for 1 h and in avidin-biotin- peroxidase complex (1:100; Vector Laboratories) at 37°C for 2 h. Finally, the sections were treated with 0.05% diaminobenzidine for 5–10 min. Sections were rinsed in PBS to stop the reaction, mounted on gelatin-coated slides, air dried, dehydrated with 70–100% alcohol, cleared with xylene, and coverslipped for microscopic examination. All positive neurons were counted without considering the intensity of the staining.
Immunoprecipitation.
Immunoprecipitations (IPs) were performed using the Catch and Release Reversible Immunoprecipitation System kit (Millipore) according to the manufacturer's instructions. Catch and Release overcomes many of the limitations associated with traditional IP. Its unique Spin-Column format was designed to make IP faster, simpler, and more reproducible. Catch and Release enables the elution of the antigen: antibody complex without denaturation, while ensuring minimal contamination by nonspecific proteins in the eluate. Antibodies used for IPs were the following: caveolin-1 and NR2B (both from Santa Cruz Biotechnology).
Immunoblotting analysis.
Tissue samples were homogenized in lysis buffer containing the following (in mm): 20.0 Tris, 250.0 sucrose, 0.03 Na3VO4, 2.0 MgCl2, 2.0 EDTA, 2.0 EGTA, 2.0 phenylmethylsulfonyl fluoride, 0.1 dithiothreitol, and 0.02% (v/v) protease inhibitor cocktail, pH 7.4. The homogenates were centrifuged at 13,000 rpm for 30 min at 4°C. The supernatant was collected and protein concentration was performed according to the Bradford method using bovine serum albumin as a standard (Bradford, 1976). The protein samples were stored at −80°C.
Protein samples were separated using 10% SDS-PAGE (60 μg total protein per lane) and transferred onto polyvinylidene difluoride membrane (Millipore). In addition, the gels stained with Coomassie Blue were used to confirm the equal amounts of protein loaded on each lane. The membranes were incubated overnight at 4°C with the primary polyclonal rabbit anti-p-Cav-1,anti-t-Cav-1 antibody (1:500; Santa Cruz Biotechnology), anti-ERK1/2 or anti-CREB antibody (1:1000; Cell Signaling Technology), goat anti-NR2B (1:500; Santa Cruz Biotechnology), and mouse anti-β-actin (1:1000; Santa Cruz Biotechnology). The membranes were extensively applied with Tris-buffered saline Tween 20 and incubated for 2 h with the secondary antibody conjugated with alkaline phosphatase (1:500; Santa Cruz Biotechnology) or horseradish peroxidase-conjugated secondary antibody (1:2000; Cell Signaling Technology) at room temperature. The immune complexes were detected by using a nitro blue tetrazolium/5-bromo-4-chloro-3-indolyl phosphate assay kit (Sigma) or chemiluminescence (Pierce). β-Actin was used as a loading standard. Western blot densitometry analysis of signal intensity was performed using NIH ImageJ software.
PCR.
Total RNAs were extracted from mice ACC or cultured ACC neurons using High Pure RNA Tissue Kit (Roche) according to the manufacturer's instructions. The quantity and quality of the isolated RNAs were assessed using NanoDrop 2000 UV-Vis spectrophotometer (Thermo Scientific). qRT-PCR was performed to analyze transcript levels of Cav-1, Cav-2, and Cav-3 using EXPRESS One-Step SYBR Green ER SuperMix Kit (Invitrogen) for one-step qRT-PCR according to the manufacturer's instructions and a StepOne Real-Time PCR System (ABI). Data were analyzed using the ΔΔCt method (Livak and Schmittgen, 2001). The following real-time PCR protocol was used for all genes: activation of reverse transcriptase and cDNA synthesis (5 min, 50°C), PCR activation (30 s, 95°C), 40 cycles of denaturation (10 s, 95°C), annealing (20 s, 59°C), and extension (10 s, 72°C). At the end of the PCR, a melting curve analysis was performed by gradually increasing the temperature from 60 to 95°C with a heating rate of 0.3°C/s. Primers for the selected genes were designed using primer 5.0 (Premier) and their specificity was confirmed by BLAST (http://www.ncbi.nlm.nih.gov/blast/Blast.cgi). β-Actin was used as endogenous control gene. Primers were purchased from Invitrogen. The primers were used for qRT-PCR as follows: Cav-1: Forward 5′-ATTGCAGAACCAGAAGGGACAC-3′, Reverse 5′-CCATTGGGATGCCGAAGAT-3′, Amplified length, 121 bp; Cav-2: Forward 5′-TCATTGCGGGTATCCTGTTTG-3′, Reverse 5′-CACGGAAGGCAAGACCATT AG-3′, Amplified length, 95 bp; Cav-3: Forward 5′-CGACGGTGTATGGAAGGTGA G-3′, Reverse 5′-GGAGATACAGGCGAACAGGAAT-3′, Amplified length, 127 bp; β-actin: Forward 5′-CATTGTTACCAACTGGGACGACAT-3′, Reverse 5′-GCCTCGGTGAGCAGCACA-3′, Amplified length, 102 bp.
Mammalian two-hybrid assay.
For the mammalian two-hybrid assay, plasmid pBIND (encoding the yeast GAL4 DNA binding domain upstream of a multiple cloning region, MCR) and pACT (encoding the herpes simplex virus VP16 activation domain upstream of a MCR and expressing the Renilla reniformis luciferase) and the reporter plasmid encoding firefly luciferase (pG5Luc) were purchased from Promega (CheckMate Mammalian Two-Hybrid System). The expression plasmids were constructed according to the scheme shown in Fig. 7B. To generate pBIND-Cav1, a fusion protein of GAL4 DNA binding domain and coding sequence of Cav-1, the coding sequence of Cav-1 was amplified by PCR from cDNA of mouse brain and inserted into the MCR of pBIND vector. The C-terminal of NR2B was also amplified by PCR from cDNA of mouse brain and, respectively, fused to the VP16 domains of the expression plasmids pACT at the BamH1-EcoR V site to construct pACT-NR2B-C fusion activation expression vector. All constructs were verified by PCR and DNA sequencing.
Figure 7.

Cav-1 directly interacts with NR2B. A, The representative immunoblot demonstrates the interaction of Cav-1 and NR2B in a coimmunoprecipitation experiment. Equal amounts of total lysates (500 μg) were used for immunoprecipitation with a Cav-1 or NR2B-specific antibody and the membranes were probed with an NR2B or Cav-1-specific antibody. B, The CDS region of genes Cav1 was inserted into pBIND to produce Gal4-Cav-1 chimeric fusion expression vector (pBIND-Cav-1), and the C-terminal of NR2B was inserted into pACT to produce VP16-NR2B-C chimeric fusion expression vector (pACT-NR2B-C). C, pBIND-Cav1 and/or pACT-NR2B-C transfected into 293T cells with the reporter gene vector pG5luc or empty vector. Forty-eight hours after transfection, the interactions were measured by relative luciferase activity. In all groups, only pBIND-Cav1 and pACT-NR2B-C cotransfection with pG5luc indicated the strong luciferase activity. All experiments were repeated more than three times with triplication for each experiment. Data are shown as means ± SEM. D, Predicted Cav-1-binding motifs from NR2B. Peptide A and C is the sequence containing aromatic amino acid (red); peptide B is a peptide in which aromatic residues from A were replaced to alanine or glycine (blue). E, The mammalian two-hybrid assay was used to validate specific blockage of this direct interaction by peptide A, not by its control peptide B and C. All experiments were repeated more than three times with triplication for each experiment. ***p < 0.001, versus adjacent groups. Data are shown as means ± SEM. F, G, The representative immunoblot bands and the quantitative data demonstrated the increased interaction of Cav-1 and NR2B by CCI in a coimmunoprecipitation experiment. Intra-ACC injection of peptide A, not its control peptides, B or C (1.0 μg/0.5 μl each peptide), could effectively inhibit the formation of a signaling complex for Cav-1 and NR2B. ***p < 0.001, **p < 0.01, versus adjacent groups. n = 3. Data are shown as means ± SEM. IP, immunoprecipitation; WB, Western blot.
HEK293T cells were maintained in DMEM supplemented with 10% fetal calf serum and seeded in 24-well plates before transfection. Following the manufacturer's instructions, both or only one of the pGAL4 and pVP16 fusion constructs (pBIND-Cav1and pACT-NR2b-N or pACT-NR2b-C) are cotransfected with the pG5luc Vector into 293T cells using GenEscort I (Wisegen Biotechnology) with a molar ratio of 1:1:1 for pACT-NR2B-C:pBIND-Cav1:pG5luc Vector. After incubation for 48 h, luciferase activity was measured using Dual Luciferase Reporter Gene Assay Kit (Beyotime RG009) on the GloMax-Multi detection system (Promega). Results were expressed as firefly luciferase activity to Renilla luciferase activity ratio (F/R). The F/R ratio was calculated using the following formula: F/r = mean of firefly luciferase activity/mean of Renilla luciferase activity.
The ACC neuronal culture.
The primary ACC neuronal culture was prepared using a method described previously (Wu et al., 2011). In brief, the ACC was aseptically removed from 13- to 15-d-old mouse embryos, minced with sterile surgical blades, and incubated in 0.125% trypsin. The anterior cingulate cortical neurons were suspended in complete DMEM containing 15% fetal bovine serum and plated in poly-l-lysine-coated 24-well plates (Corning) at a density of 2.0 × 105cells · cm−2 or 0.3 × 105cells · cm−2. Cultures were kept in an incubator at 5% CO2 at 37°C. The medium was replaced by Neurobasal medium with supplemental B27 (Invitrogen) 4 h after cells were seeded. After 5 d in culture, purity of the neurons was assessed by staining with neuron-specific antibody against microtubule-associated protein 2 (Millipore). In our case, >98% of cells were positively stained. After culture for 7 d (for analysis of calcium signal) or 15 d (for analysis of surface NR2Bexpression), neurons were used for the follow-up experiments.
siRNA and lentivirus transfection.
The expression of caveolin-1 was suppressed in mice ACC neurons by using targeted siRNA (GenePharma). The specific caveolin-1 siRNA sequences were as follows: sense, GGAAAUUGAUCUGGUCAACtt; antisense, GUUGACCAGAUCAAU UUCCtt (Head et al., 2008). Cells were treated with 0.8 μg/ml siRNA with siRNA-mate for 6 h. The medium was replaced by Neurobasal medium with supplemental B27 (Invitrogen) and cultured for 72 h. Scrambled siRNA served as controls. Functional knockdown of protein expression was assessed by immunoblot. Lentivirus-Cav-1 (Lenti-Cav-1) and Lentivirus-empty (Lenti-empty; GenePharma) were directly added to neurons with a titer 6 × 107 TU/ml and tested after 72 h.
Determination of cytosolic-free calcium.
The primary ACC neuron cultured in 24-well plates was washed three times at 10 min intervals with PBS, then was incubated with 5 μmol/L Fluo-3 AM for 30 min at 37°C. Fluo-3 AM was removed from plates. Cells were washed three times at 10 min intervals with PBS. Fluorescence was imaged with a confocal laser scanning microscope (Olympus) and the fluorescence changes determined by Fluo-3 represent the cytoplasmic calcium concentration changes. Background was measured and subtracted from cell measurements. The fluorescence intensity variation was recorded from 20 to 40 neurons on average per experiment. All groups' densitometry analysis of signal intensity was performed using NIH ImageJ software.
Statistical analysis.
All data presented as mean ± SEM. P values <0.05 were considered to be statistically significant. Statistical analysis between two samples was performed using Student's t test. Statistical analysis of more than two groups was performed using one-way ANOVA followed by a Tukey's post hoc test. The significance of any differences in PWL in behavior test was assessed using two-way ANOVA followed by Tukey's post hoc tests. “Time” was treated as a “within-subjects” factor and “treatment” was treated as a “between-subjects” factor. Statistical analyses of data were generated using GraphPad Prism 5 (GraphPad Software).
Results
CCI increases the expression of Cav-1 in the ACC
To explore whether caveolins in the ACC are involved in the modulation of chronic neuropathic pain, we first determined the alteration of three subtypes of caveolin expression in mRNA level in CCI mice. Total RNA was obtained from the contralateral ACC 7 d after surgery in sham and CCI mice. The results from qRT-PCR indicated that the expression level of Cav-1 and Cav-2 mRNA was increased >30- and 3-fold in CCI group as compared with sham group, respectively (Fig. 1A). No significant difference in Cav-3 expression was seen (Fig. 1A). Based on the alteration of caveolin mRNA induced by CCI, we therefore chose Cav-1 to perform the following studies. We found that alteration of Cav-1 mRNA expression induced by CCI exhibited a significant brain region-specific expression pattern. No significant difference in Cav-1 expression was detected in contralateral hippocampus and motor cortex (Fig. 1B). We also could not see any changes of Cav-1 mRNA expression in ACC in a formalin-induced acute pain model (Fig. 1C). Next, time course analyses for total- and phospho-Cav-1 expression in contralateral ACC were performed in CCI mice. The results from Western blot showed that CCI produced a long-lasting ∼2- and 7-fold increase in the total- and phospho-Cav-1 expression (p-Cav-1) in contralateral ACC, respectively. Consistent with the finding in the acute pain model, no significant difference in total- and p-Cav-1 expression in ACC was detected in acute phase following CCI (at 4 h after injury; Fig. 1D–F). Then, we picked up a 7 d time point after CCI to assess the change of Cav-1 expression in bilateral ACC. The expression of t-Cav-1 was increased in bilateral ACC, and p-Cav-1 expression in ipsilateral ACC also showed an increasing trend (Fig. 1G–I). Immunofluorescence staining has further confirmed that the expression of both t-Cav-1 and p-Cav-1 was increased in bilateral ACC, and the expression in contralateral ACC to the injury side was higher than in ipsilateral ACC (Fig. 1J,K).
Figure 1.
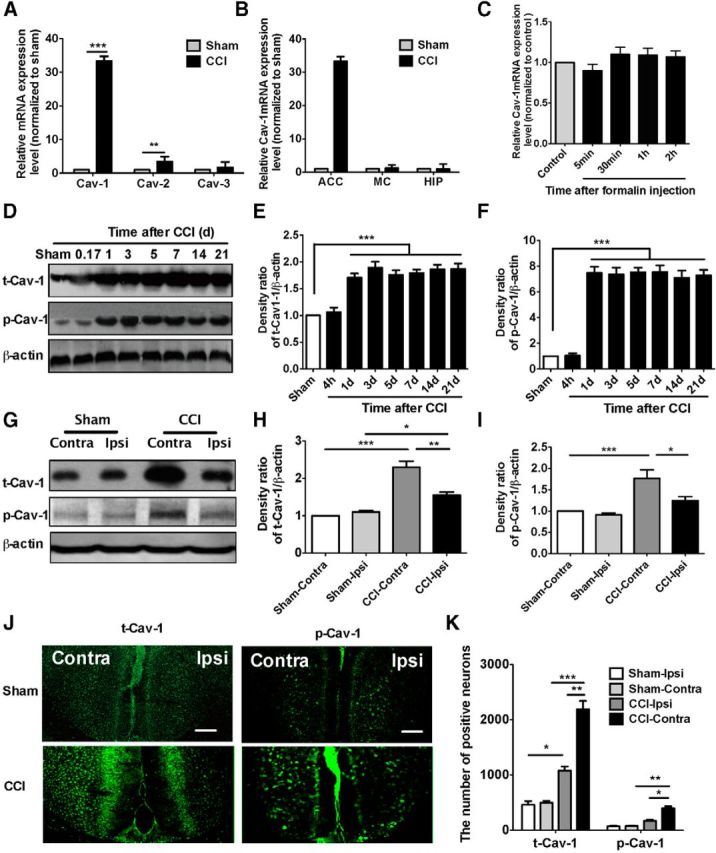
CCI increases the expression of Cav-1 in the ACC. A, RT-PCR showed that Cav-1 and Cav-2 mRNA, but not Cav-3 mRNA, were increased in the CCI group as compared with sham group (>30-fold increase for Cav-1 and threefold for Cav-2; n = 5). B, Alteration of Cav-1 mRNA expression by CCI exhibited a brain region-specific expression pattern. No significant difference in Cav-1 mRNA expression was detected in the hippocampus and motor cortex. Total RNA was obtained from the contralateral ACC, hippocampus, and motor cortex to the injury side 7 d after surgery in sham and CCI mice (n = 4). ***p < 0.001, **p < 0.01, compared with sham. C, No significant changes of Cav-1 mRNA expression in the ACC were detected in a formalin-induced acute pain. Total RNA was obtained from contralateral ACC on different time points after intraplantar injection of 4% formalin (40 μl; n = 4). D–F, The time course of the immunoblotting assay showed a long-lasting (at least up to 21 d after injury) increase of t-Cav-1 and p- Cav-1 protein in contralateral ACC (the representative bands shown in D and the quantitative data shown in E and F). The samples for immunoblotting were collected at different time points after injury and the immunoblot intensities of t-Cav-1 and p-Cav-1 expression were normalized to β-actin for semiquantitative analysis (n = 5). ***p < 0.001, compared with sham. G–I, The expression of t-Cav-1 was increased in bilateral ACC and p-Cav-1 expression in ipsilateral ACC also showed an increasing trend (the representative bands shown in G and the quantitative data shown in H and I). The samples for immunoblotting were collected at 7 d after injury and the immunoblot intensities of t-Cav-1 and p-Cav-1 expression were normalized to β-actin for semiquantitative analysis (n = 5). J, K, Immunofluorescence showed that t-Cav-1 and p-Cav-1 expressed in the ACC in CCI mice. The t-Cav-1 and p-Cav-1 expression in contralateral ACC was stronger than that in ipsilateral ACC. ***p < 0.001, **p < 0.01, *p < 0.05, versus adjacent groups (n = 5). Scale bar, 200 μm. Data are shown as means ± SEM. MC, motor cortex; HIP, hippocampus; Ipsi, ipsilateral; Contra, contralateral.
Furthermore, to determine the cell types expressing Cav-1 in the ACC, we performed double immunofluorescence studies using cell type-specific antibodies against NeuN (a mature neuron marker), GFAP (an astrocyte marker), and specific antibodies to t-Cav-1 and p-Cav-1. The present results show that the increased expression of t-Cav-1 and p-Cav-1 by CCI was mainly localized in mature neurons (Fig. 2A) and is not found in mature glial cells in ACC (data not shown). The increased t-Cav-1 and p-Cav-1 also colocalized with PSD-95, a postsynaptic marker (Fig. 2B), showing that Cav-1 were present in the PSD, which was a structural basis for its concentrating and organizing neurotransmitter receptors in the synaptic cleft and participating in the regulation of synaptic and neuronal function. These results suggest that brain region-specific and ongoing pain-dependent changes of Cav-1 expression in ACC neurons may be critical components in the development and maintenance of chronic pain behavioral phenotype.
Figure 2.

Cellular and subcellular localization of t-Cav-1 and p-Cav-1 expression in the ACC. A, Double immunofluorescence showed that t-Cav-1 and p-Cav-1 expression in contralateral ACC were increased and expressed in neurons in CCI mice (colocalization with NeuN; n = 4). Scale bar, 100 μm. B, The increased t-Cav-1 and p-Cav-1 colocalized with PSD-95, a postsynaptic marker (n = 5). Scale bar, 50 μm.
Manipulation of Cav-1 in the ACC modulates neuronal activity and pain behavior
Does the increase of Cav-1 in the ACC contribute to pain sensory modulation in CCI mice? To answer this question, we used Cav-1 siRNA to silence Cav-1 gene expression and the lentivirus system to upregulate Cav-1 gene expression. We first validated efficient delivery tools of siRNAs (Fig. 3A) and lentivirus (Fig. 3B) in vivo by intra-ACC injection. Microinjection of Cav-1 siRNA, but not scramble siRNA control, into contralateral ACC at 7 d after surgery significantly reversed CCI-induced thermal hyperalgesia and mechanical allodynia, and the analgesic effect of single-dose injection could last for at least 4 d (Fig. 3D). Consistent with the finding in siRNA-mediated knockdown of Cav-1 expression, blocking Cav-1 signaling by intracontralateral ACC injection of antibodies against t-Cav1 or p-Cav1, but not their control IgG, also significantly reversed CCI-induced thermal hyperalgesia and mechanical allodynia, and the analgesic effect of single-dose injection could last for at least 1 h (Fig. 3E).
Figure 3.
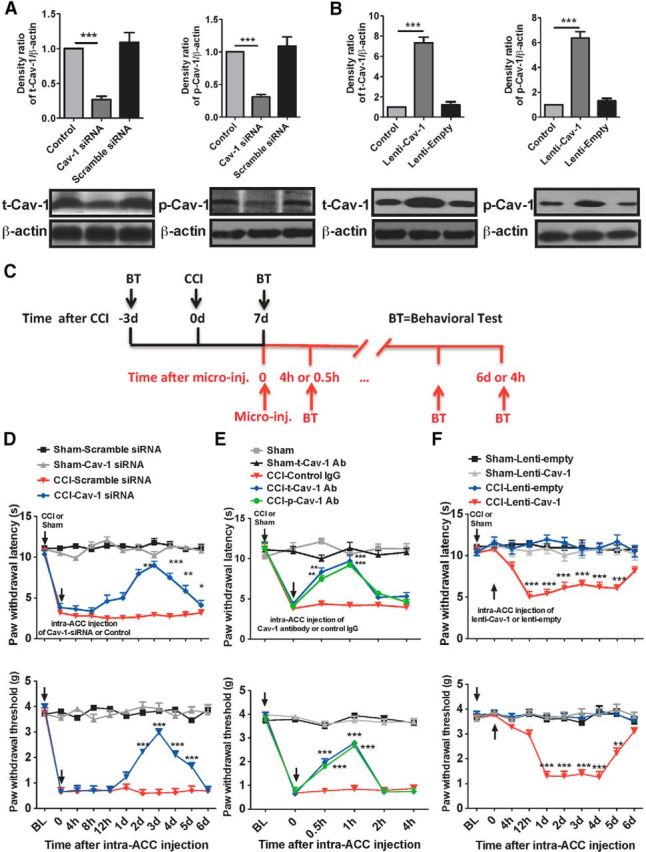
Manipulation of Cav-1 in the ACC modulates pain behavior. A, B, The expression of t-Cav-1 and p-Cav-1 by immunoblotting assay was used to validate efficient delivery tools of Cav-1 siRNAs (for knockdown of Cav-1) and lentivirus Cav-1 (Lenti-Cav-1; for overexpression of Cav-1) in vivo by intra-ACC injection. Microinjection of Cav-1 siRNAs (10 μg/0.5 μl) or Lenti-Cav-1 (0.5 × 106 TU/0.5 μl), not their controls, significantly inhibited or increased t-Cav-1 and p-Cav-1 expression in the ACC. The ACC samples were collected at 72 h after microinjection (n = 4). ***p < 0.001, compared with naive control or scramble siRNA or Lenti-empty control. C, Schematic illustration of experimental protocol. D, Microinjection of Cav-1 siRNAs (10 μg/0.5 μl), not their control scramble siRNAs, into contralateral ACC at 7 d after surgery significantly reversed CCI-induced thermal hyperalgesia and mechanical allodynia (n = 8). ***p < 0.001, **p < 0.01, and *p < 0.05, compared with CCI-scramble siRNA. E, Microinjection of anti-t-Cav-1 antibody (0.2 μg/0.5 μl) or anti-p-Cav-1 antibody (0.1 μg/0.5 μl), not its control IgG, into contralateral ACC at 7 d after surgery significantly reversed CCI-induced thermal hyperalgesia (top) and mechanical allodynia (bottom; n = 8). ***p < 0.001, **p < 0.01, compared with CCI-Control IgG. F, Microinjection of Lenti-Cav-1 (0.5 × 106 TU/0.5 μl), not its control Lenti-empty, into ipsilateral ACC at 7 d after surgery induced a significant decrease of thermal and mechanical pain threshold in the unaffected hindpaw (n = 8). ***p < 0.001, compared with CCI-Lenti-empty. BL, baseline.
Peripheral noxious stimuli or injury triggered c-Fos expression in the ACC, which can be used as a marker of neuronal sensitization and long-term plastic changes in ACC related to pain modulation (Hunt et al., 1987; Li et al., 2010). In agreement with the previous findings (Wei et al., 1999; Zhuo, 2008), although CCI produced a unilateral noxious response, it increased the c-Fos expression in the both sides of the ACC at 7 d after injury. However, the level of c-Fos expression in contralateral ACC to injury side was much higher than in ipsilateral ACC (data not shown). Knockdown of Cav-1 expression or blocking Cav-1 signaling could significantly inhibit CCI-induced c-Fos expression in contralateral ACC (Fig. 4A), suggesting that Cav-1 contributed to the neuronal sensitization and long-term plastic changes in ACC induced by chronic pain.
Figure 4.
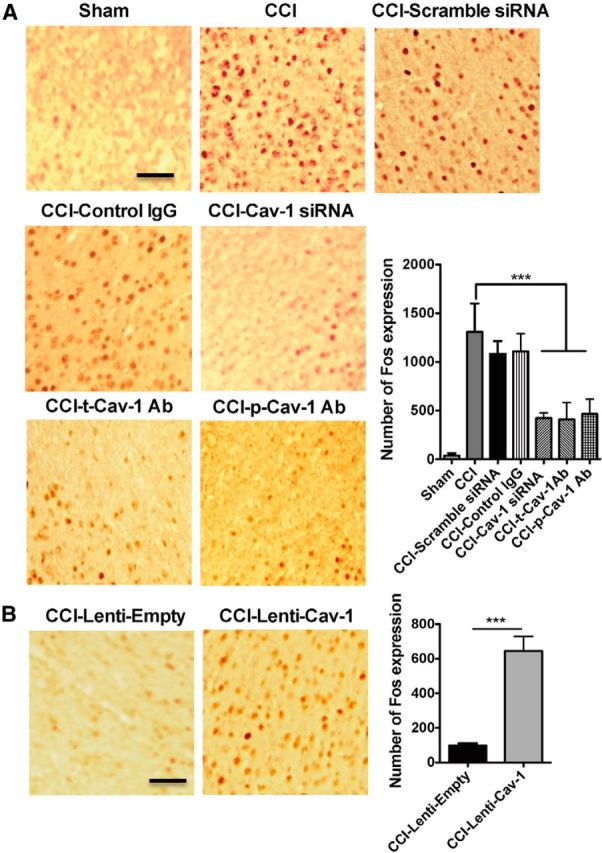
Manipulation of Cav-1 in the ACC regulates neuronal activity. A, The representative immunohistochemical staining and the quantitative data for the inhibiting CCI-induced Fos expression in contralateral ACC by microinjection of Cav-1 siRNAs (10 μg/0.5 μl) or anti-t-Cav-1 antibody (0.2 μg/0.5 μl) or anti-p-Cav-1 antibody (0.1 μg/0.5 μl), but not their controls (n = 6). ***p < 0.001, compared with CCI. B, The representative immunohistochemical staining and the quantitative data for the increasing CCI-induced Fos expression in ipsilateral ACC by microinjection of Lenti-Cav-1 (0.5 × 106 TU/0.5 μl), not its control Lenti-empty (n = 6). ***p < 0.001, compared with CCI-Lenti-empty. Scale bar, 100 μm. Data are shown as means ± SEM.
Since downregulation of Cav-1 in ACC could produce an analgesic effect, can upregulation of Cav-1 in ACC produce pain behavior? First, we microinjected Lenti-Cav-1 or its control Lenti-empty into ACC to upregulate Cav-1 expression in naive mice and then measure the thermal pain threshold at 2–6 d after injection. No significant change of thermal pain threshold was detected following this injection. Based on a bilateral pattern of c-Fos activation in ACC and a unilateral manner of pain behavioral sensitization induced by noxious stimuli, we supposed that a state of latent neuronal sensitization in ACC was developed underlying chronic pain and unmasking this latent sensitization by pronociceptive stimuli would trigger pain behavior in the unaffected hindpaw. To test whether upregulation of Cav-1 in ipsilateral ACC to the injury side could be a trigger to elicit pain behavior in the unaffected hindpaw, we microinjected the lenti-Cav-1 or its control Lenti-empty into ipsilateral ACC to upregulate Cav-1 expression in CCI or sham mice to see the change of thermal pain latency and mechanical pain threshold. As we expected, upregulation of Cav-1 expression in ipsilateral ACC produced a significant decrease of thermal pain latency and mechanical pain threshold in the unaffected hindpaw (Fig. 3F), which was accompanied with c-Fos activation in ipsilateral ACC (Fig. 4B). These findings suggested that Cav-1 in ACC contributes to the modulation of chronic pain processing.
The change of BBB permeability is not involved in pain modulation by Cav-1 in ACC
Dysfunction or overexpression of Cav-1 is associated with BBB-increased permeability and infiltration of inflammatory cells in the CNS (Nusrat et al., 2000; Song et al., 2007; Nag et al., 2009). Furthermore, some studies have demonstrated that increased permeability of the BBB could enhance pain processing (Beggs et al., 2010; Echeverry et al., 2011). Concerning the role of Cav-1 in the BBB homeostasis and the BBB in pain modulation, we wanted to know whether the change of the BBB permeability was involved in pain modulation by Cav-1 in ACC. We first found that the increased expression of t-Cav-1in ACC by CCI and Lenti-Cav-1 was not colocalized with CD34 as markers of endothelial cells (Fig. 5A). Next, we used the EB dye extravasation technique for determination of BBB permeability. The results showed that CCI had not increased extrinsic EB content in contralateral ACC to the injured side (Fig. 5B). Overexpression of Cav-1 in ipsilateral ACC to the injured side also could not increase extrinsic EB content (Fig. 5C). These results suggested that the change of ACC BBB permeability was not involved in pain modulation by Cav-1.
Figure 5.

BBB permeability is not involved in pain modulation by Cav-1 in ACC. A, The increased expression of t-Cav-1 in contralateral ACC by CCI and ipsilateral ACC by Lenti-Cav-1 was not colocalized with CD34 as markers of endothelial cells (n = 5). Scale bar, 50 μm. B, Quantification of EB leakage at day 7 after CCI showing no increased extrinsic EB content in contralateral ACC to the injured side (n = 5). C, Quantification of EB leakage at day 7 after CCI showed that overexpression of Cav-1 in ipsilateral ACC to the injured side also could not increase extrinsic EB content (n = 5). Data are shown as means ± SEM. EB, Evan's blue.
Cav-1 in ACC modulates neuropathic pain via regulation of NR2B
Cav-1 as an adaptor molecule or scaffolding protein needs to recruit the related signaling molecules and then integrate membrane signal transduction for exerting its effect. Next, we want to know what the signaling molecule is that interacts with Cav-1 in ACC for chronic neuropathic pain modulation. Previous studies have demonstrated that NR2B is localized predominantly in the forebrain including the ACC and plays critical roles in the neuronal plasticity and pain behavior sensitization induced by noxious stimuli (Monyer et al., 1994; Wu et al., 2005; Zhao et al., 2005). Cav-1 colocalizes NR2B on the cell body and along dendritic shafts and spines in cultured primary neurons (Head et al., 2008). Therefore, we test whether NR2B is a potential candidate for interacting with Cav-1 to mediate chronic neuropathic pain modulation in the ACC. First, we found that Cav-1 colocalized with NR2B in neuronal membrane in the ACC by double immunofluorescence staining and CCI increased in colocalization of Cav-1 and NR2B (Fig. 6A), which could be reversed by knockdown of Cav-1 mediated by Cav-1 siRNA, but not by its control scramble siRNA (Fig. 6A). Furthermore, immunoblot analysis also revealed that CCI increased the expression of NR2B protein in the ACC, which was reversed by knockdown of Cav-1 (Fig. 6B). Microinjection of NR2B inhibitor ifenprodil into contralateral ACC significantly inhibited CCI-induced thermal hyperalgesia and mechanical allodynia (Fig. 6C). We have already demonstrated that upregulation of Cav-1 expression in ipsilateral ACC induced pain behavior in the unaffected hindpaw in CCI mice. Furthermore, we found that upregulation of Cav-1 expression was accompanied with the increased expression of NR2B and increased colocalization of Cav-1 and NR2B in membrane in ipsilateral ACC (Fig. 6D). Treatment with ifenprodil significantly inhibited Cav-1 overexpression-induced thermal hyperalgesia and mechanical allodynia in the unaffected hindpaw in CCI mice (Fig. 6E,F). These results suggested that the regulation of expression and distribution of NR2B were required in Cav-1-related pain modulation in the ACC.
Figure 6.
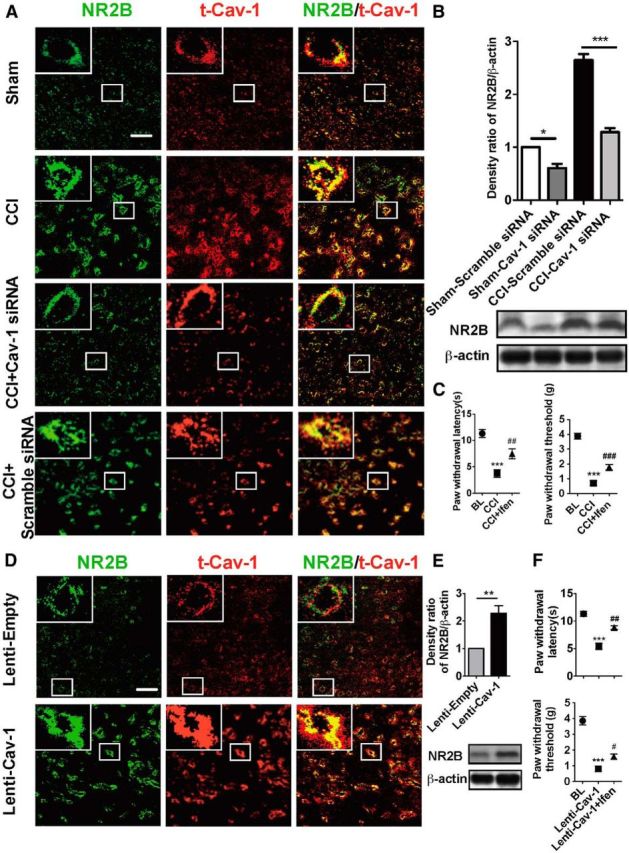
NR2B mediates pain modulation by Cav-1 in the ACC in CCI mice. A, Double immunofluorescence showed that Cav-1 colocalized with NMDA-NR2B in cell membrane in the ACC and CCI increased colocalization of Cav-1 and NMDA-NR2B. Knockdown of Cav-1 by intra-ACC injection of Cav-1 siRNA (10 μg/0.5 μl), but not by its control scramble siRNA, inhibited CCI-induced increase of colocalization of Cav-1 and NMDA-NR2B (n = 5). Scale bar, 50 μm. B, CCI increased the expression of NR2B in contralateral ACC, which could be reversed by knockdown of Cav-1 (n = 5). ***p < 0.001, *p < 0.05. C, Microinjection of NR2B inhibitor ifenprodil (0.5 μg/0.5 μl) into contralateral ACC significantly inhibited CCI-induced thermal hyperalgesia (left) and mechanical allodynia (right). Behavioral test was performed at 3 d before injury, 7 d after CCI, and 1 h after injection of ifenprodil (n = 8). ***p < 0.001, compared with baseline (BL), ##p < 0.01, ###p < 0.001, compared with CCI. D, Overexpression of Cav-1 with Lenti-Cav-1 (0.5 × 106 TU/0.5 μl), not its control Lenti-empty, increased colocalization of Cav-1 and NR2B in membrane in ipsilateral ACC (n = 5). Scale bar, 50 μm. E, Overexpression of Cav-1 with Lenti-Cav-1 (0.5 × 106 TU/0.5 μl), not its control Lenti-empty, increased expression of NR2B in ipsilateral ACC (n = 5). **p < 0.01. F, NR2B inhibitor ifenprodil (0.5 μg/0.5 μl) reversed thermal hyperalgesia (top) and mechanical allodynia (bottom) in the unaffected hindpaw induced by overexpression of Cav-1 in ipsilateral ACC in CCI mice (n = 8). Behavioral test was performed at 3 d before CCI, 72 h after Lenti-Cav-1 injection, and 1 h after injection of ifenprodil (n = 8). ***p < 0.001, compared with BL, #p < 0.05, ##p < 0.01, compared Lenti-Cav-1. Data are shown as means ± SEM. BL, baseline; Ifen, ifenprodil.
Cav-1 directly interacts with NR2B
Cav-1, as a scaffolding protein, can organize and concentrate certain caveolin-interacting proteins through caveolin-scaffolding domain to mediate its functional effects. In rat primary cortical neuron cultures, previous study has shown an interaction between Cav-1 and NR2B (Head et al., 2008). Consistent with this finding, we identified the formation of the signaling complex for Cav-1 and NR2B in the ACC by coimmunoprecipitation experiment and this formation of signaling complex was elevated by CCI (Fig. 7A,F).
Next, we performed the mammalian two-hybrid assay to identify a direct interaction between Cav-1 and NR2B. The CDS region of gene Cav-1 was inserted into pBIND to produce Gal4-Cav-1 chimeric fusion expression vector (pBIND-Cav1), and C-terminal of NR2B was inserted into pACT to produce VP16-NR2B-C chimeric fusion expression vectors (pACT-NR2B-C; Fig. 7B). The interaction between Cav-1 and NR2B was determined by cotransfection of the reporter gene vector pG5luc with pBIND-Cav1 and pACT-NR2B-C. Both pBIND-Cav1 and pACT-NR2B-C fusion expression vectors were expressed in 293T cells to examine their interaction as indicated by luciferase activity. pBIND and pACT were used as negative control plasmids. The results show that coexpression of pBIND-Cav-1 and pACT-NR2B-C significantly activated transcription of the reporter gene. No significant activation was observed in the cotransfection of reporter gene vector pG5luc and one of the chimeric fusion expression vectors, pBIND-Cav-1 or pACT-NR2B-C (Fig. 7C). This indicates a direct interaction between Cav-1 and NR2B.
It has been shown that a 20 aa membrane proximal region of the cytosolic N-terminal domain of caveolin (caveolin-scaffolding domain) is sufficient to mediate the interactions of caveolin and other signaling molecules. These interactions are strictly dependent on the presence of aromatic residues within these caveolin-selected peptide ligands. The short peptide sequence interacted directly with the caveolin-scaffolding domain and competitively inhibits the interaction of the caveolin-scaffolding domain with the appropriate region of the corresponding protein (Couet et al., 1997). Therefore, we selected and synthesized a short peptide that contains aromatic amino acids (the potential binding sites for interaction) derived from the C-terminal of NR2B (including two control peptides) according to a predicted protein active motif web (http://hits.isb-sib.ch/cgi-bin/motif_scan): peptide A, derived from C-terminal of NR2B; control peptide B, mutated phenylalanine of peptide A to Alanine, Tyrosine to Glycine; and control peptide C, randomly selected a peptide from the C-terminal of NR2B that contains aromatic residues (Fig. 7D). Furthermore, we used the mammalian two-hybrid assay to validate specific blockage of this direct interaction by peptide A, not by its control peptides B and C (Fig. 7E). Finally, we used coimmunoprecipitation assay to validate that peptide A, not its control peptides B and peptide C, could effectively inhibit the formation of signaling complex for Cav-1 and NR2B (Fig. 7F,G).
Cav-1 regulates NR2B surface levels in cultured ACC neurons
Given the importance of NR2B surface levels in mediating the potentiation of synaptic transmission during plasticity (Plattner et al., 2014), we assessed whether Cav-1 affects the subcellular location of NR2B subunits. To compare the change of NR2B surface levels in cultured ACC neurons derived from knockdown or overexpression of Cav-1, we measured the surface NR2B expression using an antibody against the N-terminal of NR2B and then measured intracellular NR2B expression after membrane permeabilization. As shown in Figure 8A, the levels of NR2B on the surface were markedly increased in KCl (50 mm)-treated ACC neurons, compared with that from the control group. Pretreatment with Cav-1 siRNA, not its control scramble siRNA, at 72 h before administration of KCl, could significantly inhibit KCl-induced increase of surface NR2B expression (Fig. 8A). Furthermore, overexpression of Cav-1 in the cultured ACC neurons with Lenti-Cav-1, but not its control Lenti-empty, increased the surface NR2B expression (Fig. 8B). These results demonstrate that NR2B surface levels are regulated by Cav-1.
Figure 8.
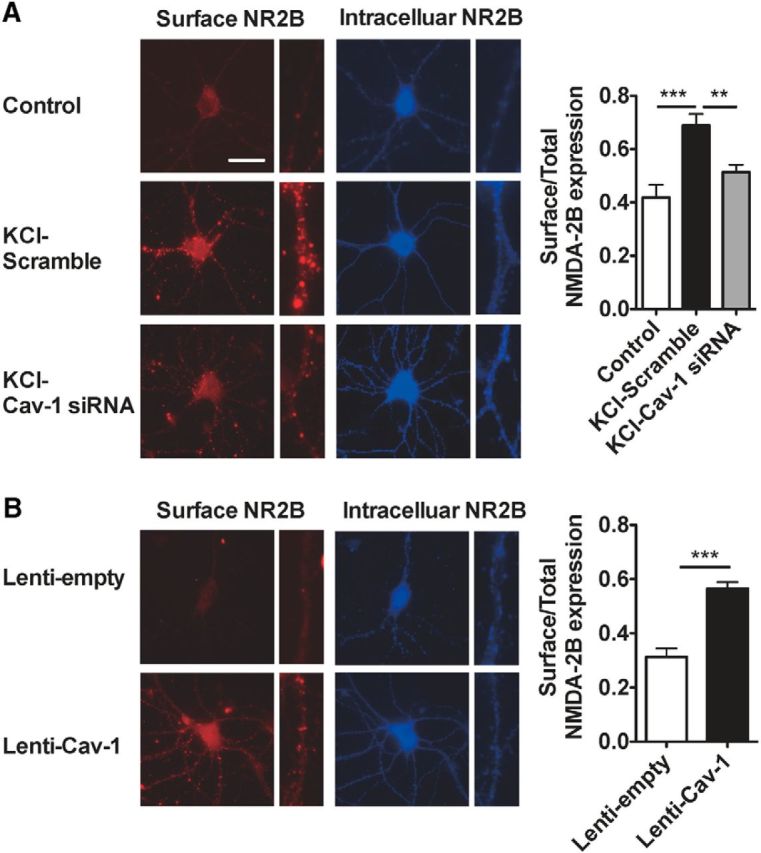
Cav-1 regulates NR2B surface levels in cultured ACC neurons. A, Representative images and quantification of the imaging experiments showing increased surface NR2B expression by treatment with KCl (50 mm) for 12 h in cultured ACC neurons and inhibition of this increase by pretreatment with Cav-1 siRNA, not by its control scramble siRNA. The cultured ACC neurons were treated with 0.8 μg/ml Cav-1 siRNA or its control scramble siRNA for 72 h before treatment with KCl. ***p < 0.001, **p < 0.01, versus adjacent groups(n = 25–30 cells in 3 independent experiments). B, Representative images and quantification of the imaging experiments show that overexpression of Cav-1 in the cultured ACC neurons with Lenti-Cav-1, but not its control Lenti-empty, increased the surface NR2B expression. The cultured ACC neurons were treated with 6 × 107 TU/ml Lenti-Cav-1 or its control Lenti-empty for 72 h. ***p < 0.001, versus adjacent groups(n = 25–30 cells in 3 independent experiments). The ratios of surface (red) to total (red + blue) fluorescence are presented. Data are shown as means ± SEM. Scale bars: 25 μm.
Disruption of the interaction of Cav-1 and NR2B in the ACC alleviates neuropathic pain
Due to Cav-1 directly interacting with NR2B and regulation of NR2B surface levels, we microinjected peptide A into contralateral ACC in CCI mice to disturb the interaction of Cav-1 and NR2B, and then investigated the effect of this intervention on neuropathic pain behavior and colocalization of t-Cav-1 and NR2B. We found that intra-ACC injection of peptide A, not B and C, could at least partly inhibit CCI-induced increase of NR2B expression and colocalization of t-Cav-1 and NR2B (Fig. 9A), which was accompanied with alleviation of CCI-induced thermal hyperalgesia and mechanical allodynia (Fig. 9B), suggesting that the interaction of Cav-1 and NR2B mediates the modulatory effect of Cav-1 on neuropathic pain in the ACC and Phenylalanine (1102) and Tyrosine (1109) of the NR2B C-terminal play an important role for mediating this interaction.
Figure 9.
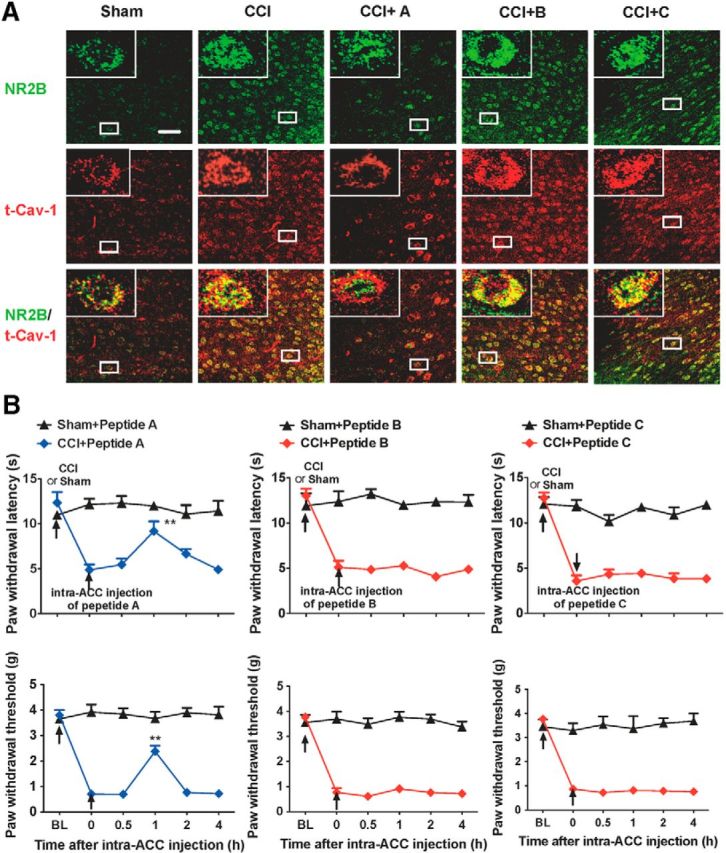
Disruption of the interaction of Cav-1 and NR2B in the ACC alleviates chronic neuropathic pain. A, Double immunofluorescence staining showed that intra-ACC injection of peptide A, not B and C, could inhibit CCI-induced increase of colocalization of t-Cav-1 and NR2B (n = 4). Scale bar, 50 μm. B, Intra-ACC injection of peptide A, not B or C, reversed CCI-induced thermal hyperalgesia (top) and mechanical allodynia (bottom). Behavioral test was performed at 3 d before CCI, 7 d after CCI, and 0.5–4 h after injection of peptides (n = 8). **p < 0.01, compared with 0 time point (before injection of peptides). Data are shown as means ± SEM. BL, baseline.
Cav-1 regulates intracellular Ca2+ concentration through NR2B
Previous studies have shown that the increased intracellular Ca2+ concentration mediated by activation of NMDAR contributed to plasticity related to chronic pain in the ACC (Zhuo, 2009, 2014). Therefore, we asked whether Cav-1 regulated intracellular Ca2+ concentration via NMDAR. To answer this question, the cultured ACC neurons were depolarized with 50 mm KCl to mimic sensitized in vivo neurons in the ACC by neuropathic pain. We found that Cav-1 mRNA expression was significantly increased in the cultured ACC neurons treated with 50 mm KCl for 12 h (Fig. 10A), which was accompanied with the increase of intracellular Ca2+ concentration (Fig. 10B). To explore the contribution of Cav-1 to the elevation of intracellular Ca2+ concentration, we pretreated the cultured ACC neurons with Cav-1 siRNA or its control scramble siRNA before administration of KCl. Results indicated that knockdown of Cav-1 with Cav-1 siRNA, but not control scramble siRNA, significantly inhibited KCl-induced increase of intracellular Ca2+ concentration (Fig. 10B). Furthermore, overexpression of Cav-1 in the cultured ACC neurons with Lenti-Cav-1, but not its control Lenti-empty, also increased the intracellular Ca2+ concentration (Fig. 10C), which could be inhibited by treatment with the NR2B inhibitor ifenprodil (Fig. 10D). The increase of intracellular Ca2+ concentration induced by KCl in the cultured ACC neurons was also inhibited by treatment with ifenprodil (Fig. 10E). These results suggest that Cav-1 regulates intracellular Ca2+ concentration through NR2B.
Figure 10.

Cav-1 regulates intracellular Ca2+ concentration through NR2B. A, RT-PCR assay showed that Cav-1 mRNA expression was significantly increased in the cultured ACC neurons treated with 50 mm KCl for 12 h. ***p < 0.001, compared with the KCl group. Total RNA was extracted from the cultured ACC neurons at 12 h after KCl treatment. The mRNA expression levels of Cav-1 were then measured in the total RNA by performing real-time RT-PCR, using β-actin as an internal control. These levels were expressed as a percentage of those in naive controls. RT-PCR assay was performed at three independent experiments.***p < 0.001, compared with the KCl group. B, The representative Fluo-3 fluorescence (left) and the quantitative data (right) showed that knockdown of Cav-1 with Cav-1 siRNA, but not control scramble siRNA, significantly inhibited KCl-induced increase of intracellular Ca2+ concentration in cultured ACC neurons. The cultured ACC neurons were treated with 0.8 μg/ml Cav-1 siRNA or its control scramble siRNA for 72 h before treatment with KCl. Then neurons in culture were loaded with Fluo-3 AM (5 μm for 30 min at 37°C) to measure changes in free intracellular Ca2+. The fluorescence changes determined by Fluo-3 represent the cytoplasmic calcium [Ca2+]i changes (n = 25–30 cells in 3 independent experiments). **p < 0.01, compared with control-scramble siRNA group or KCl-Cav-1 siRNA group. C, The representative Fluo-3 fluorescence (left) and the quantitative data (right) showed that overexpression of Cav-1 with Lenti-Cav-1, but not control Lenti-empty, significantly increased the intracellular Ca2+ concentration in cultured ACC neurons. The cultured ACC neurons were treated with 6 × 107 TU/ml Lenti-Cav-1 or its control Lenti-empty for 72 h before loading with Fluo-3 AM (n = 25–30 cells in 3 independent experiments). **p < 0.01. D, The representative Fluo-3 fluorescence (left) and the quantitative data (right) showed that NR2B inhibitor ifenprodil (6 μm) inhibited the increase of intracellular Ca2+ concentration induced by overexpression of Cav-1. The cultured ACC neurons were treated with 6 × 107 TU/ml Lenti-Cav-1 and ifenprodil (6 μm) or its vehicle for 72 h before loading with Fluo-3 AM (n = 25–30 cells in 3 independent experiments). **p < 0.01. E, The representative Fluo-3 fluorescence (left) and the quantitative data (right) showed that NR2B inhibitor ifenprodil (6 μm) inhibited the increase of intracellular Ca2+ concentration induced by KCl. The cultured ACC neurons at 7 d were pretreated in 6 μm ifenprodil 10 min before treatment by 50 mm KCl, then testing with Fluo-3 AM (n = 25–30 cells in 3 independent experiments), **p < 0.01. Data are shown as means ± SEM. Scale bar, 50 μm. Ifen, ifenprodil.
Activation of ERK/CREB signaling pathway contributes to the modulatory effect of Cav-1 on neuropathic pain in the ACC
Our studies have shown that CCI produced a long-lasting increase in the total- and phospho-Cav-1 expression in ACC and this high expression level was maintained for at least 21 d after surgery, suggesting that the increased Cav-1 expression in ACC played an important role in the development and maintenance of chronic neuropathic pain. The development and maintenance of chronic pain status is mainly dependent on maladaptive alterations in the expression, distribution, and activity of ion channels, receptors, and intracellular and intranuclear signal transduction pathways. Previous studies indicated that aberrant activation of ERK/CREB signaling pathway in the ACC is required for the central sensitization during the development and maintenance of hyperalgesia and allodynia induced by various pathological conditions (Wei et al., 2002; Wei and Zhuo, 2008; Toyoda, 2009a; Wang et al., 2012). To determine whether Cav-1 contributes to the activation of ERK/CREB in the ACC during chronic neuropathic pain, we performed experiments to knock down Cav-1 expression in contralateral ACC or to overexpress Cav-1 in ipsilateral ACC in CCI mice and then monitored activation of ERK/CREB by immunoblotting with an antibody that detects the activated form of ERK and CREB. Our results indicated that knockdown of Cav-1 with Cav-1 siRNA, but not its control scramble siRNA, inhibited the expression of p-ERK1/2 and p-CREB induced by CCI in contralateral ACC (Fig. 11A,B), and overexpression of Cav-1 with Lenti-Cav-1, but not its control Lenti-empty, increased expression of p-ERK1/2 and p-CREB in ipsilateral ACC in CCI mice (Fig. 11C,D). Furthermore, we found that thermal hyperalgesia and mechanical allodynia in the unaffected hindpaw induced by upregulation of Cav-1 expression in ipsilateral ACC could be reversed by microinjection of ERK inhibitor U0126 (Fig. 11E) or knockdown of CREB with CREB siRNA in ipsilateral ACC in CCI mice (Fig. 11F). These findings reveal that activation of ERK/CREB signaling pathway in the ACC is required for the modulatory effect of Cav-1 on neuropathic pain.
Figure 11.
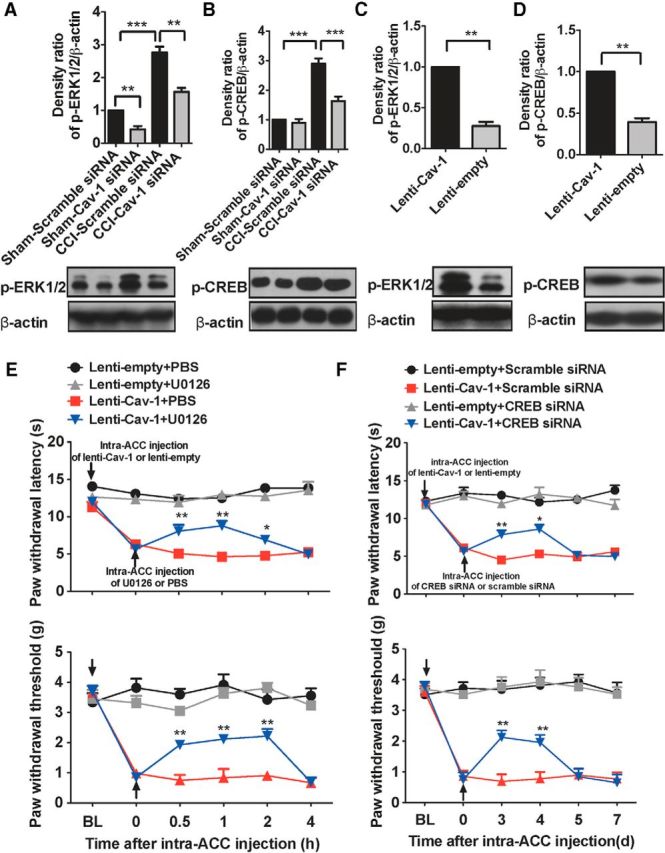
Cav-1 is involved in activation of ERK/CREB in the ACC in neuropathic pain mice. A, B, The representative immunoblotting bands (bottom) and the quantitative data (top) showed that knockdown of Cav-1 with Cav-1 siRNA, but not its control scramble siRNA, reversed the increased expression of p-ERK1/2 (A) and p-CREB (B) by CCI in contralateral ACC. Cav-1 siRNA (10 μg/0.5 μl), or its control, was injected into contralateral ACC at 7 d after CCI and contralateral ACC was collected for measuring expression of p-ERK1/2 and p-CREB at 72 h after microinjection (n = 5). **p < 0.01,***p < 0.001. C, D, The representative immunoblotting bands (bottom) and the quantitative data (top) showed that overexpression of Cav-1 with Lenti-Cav-1, but not its control Lenti-empty, increased expression of p-ERK1/2 (C) and p-CREB (D) in ipsilateral ACC in CCI mice. Lenti-Cav-1 (0.5 × 106 TU/0.5 μl), or its control, was injected into ipsilateral ACC at 7 d after CCI and ipsilateral ACC was collected for measuring expression of p-ERK1/2 and p-CREB at 72 h after microinjection (n = 5). **p < 0.01. E, F, The representative immunoblotting bands (bottom) and the quantitative data (top) showed that blockage of ERK/CREB signaling in ipsilateral ACC reversed thermal hyperalgesia (top) and mechanical allodynia (bottom) in the unaffected hindpaw induced by overexpression of Cav-1 expression in ipsilateral ACC in CCI mice. U0126 (75 ng/0.5 μl; E) or CREB siRNA (10 μg/0.5 μl; F), or their controls, was injected into ipsilateral ACC at 3 d after Lenti-Cav-1 or Lenti-empty injection and then pain behavior in the unaffected hindpaw was tested at different time points after the last microinjection (n = 8). *p < 0.05, **p < 0.01, compared with 0 time point (before last microinjection). Data are shown as means ± SEM. BL, baseline.
NR2B mediates activation of ERK/CREB signaling pathway by Cav-1 in the ACC
Several lines of evidence have demonstrated that the activation of NMDA receptors triggers postsynaptic calcium, leading to the activation of the calcium-stimulated ERK/CREB signaling pathway (Wei et al., 2002; Chen et al., 2007; Head et al., 2008). Expression of Cav-1 is essential for NMDA-mediated ERK1/2 phosphorylation in cultured neurons (Head et al., 2008). Therefore, we want to know whether NR2B mediates activation of ERK/CREB signaling pathway by Cav-1 in the ACC in CCI mice. Consistent with the finding by immunofluorescence staining, immunoblotting assay showed that NDMA-NR2B expression was significantly increased in contralateral ACC or ipsilateral ACC with overexpression of Cav-1 in CCI mice, which could be reversed by injection of the NR2B inhibitor ifenprodil, but not by its vehicle (Fig. 12A,D). We injected ifenprodil into contralateral ACC or into ipsilateral ACC after Lenti-Cav-1 injection and then monitored activation of ERK/CREB. The results show that inhibition of NR2B reversed the increased expression of p-ERK1/2 and p-CREB both induced by CCI in contralateral ACC (Fig. 12B,C) and by overexpression of Cav-1 in ipsilateral ACC in CCI mice (Fig. 12E,F), suggesting that activation of ERK/CREB by Cav-1 in the ACC is dependent on NR2B in CCI mice.
Figure 12.

NR2B mediates CCI-induced and overexpression of Cav-1-induced activation of ERK/CREB in ACC in chronic neuropathic pain mice. The representative immunoblotting bands (bottom) and the quantitative data (top) showed that ifenprodil inhibited CCI-induced (A–C) or overexpression of Cav-1-induced (D–F) increase of NR2B (A, D), p-ERK (B, E), and p-CREB (C, F) expression in ACC. Ifenprodil (0.5 μg/0.5 μl) was microinjected into contralateral ACC at 7 d after CCI or into ipsilateral ACC at 3 d after Lenti-Cav-1 injection for overexpression of Cav-1 on the same side in CCI mice. ACC was collected for measuring NR2B, p-ERK, and p-CREB expression at 1 h after ifenprodil injection (n = 5). *p < 0.05, **p < 0.01, ***p < 0.001. Data are shown as means ± SEM. Ifen, ifenprodil.
Discussion
The present study reveals that Cav-1 in the ACC contributes to the pain sensory modulation of chronic neuropathic pain and related neuronal sensitization through promotion of NR2B surface levels and NR2B-dependent activation of the ERK/CREB signaling pathway. The major findings are as follows: (1) a long-lasting increase of t-Cav-1 and p-Cav-1 expression in contralateral ACC neurons to the injury side is associated with development and maintenance of pain behavioral and neuronal sensitization, (2) Cav-1 directly binding with NR2B and promoting NR2B surface levels mediates modulation of chronic neuropathic pain by Cav-1 in the ACC, and (3) NR2B-dependent increase of intracellular Ca2+ and subsequent activation of ERK/CREB signaling is required for modulation of chronic neuropathic pain by Cav-1 in the ACC. These findings elucidate a novel mechanism for regulation of chronic neuropathic pain by Cav-1 in ACC.
Emerging evidence has shown that caveolins, as an adaptor protein that associates with lipid rafts and the main coat protein of caveolae/lipid rafts pathway, play an important role in integration of signal transduction and neuronal plasticity. For example, in hippocampal neurons, caveolin proteins are essential for the regulation of CREB phosphorylation after activation of estrogen-sensitive signaling pathways (Boulware et al., 2007). Overexpression of neuron-targeted Cav-1 in primary neurons enhances raft formation, neurotransmitter and neurotrophin receptor expression, NMDAR- and BDNF-mediated prosurvival kinase activation, agonist-stimulated cAMP formation, and dendritic growth (Head et al., 2011). Cav-1 regulates metabotropic glutamate receptor (mGluR) trafficking and mGluR-mediated calcium signaling, and Cav-1 knock-out mice exhibit impaired induction of mGluR-dependent long-term depression at CA3-CA1 synapses (Francesconi et al., 2009; Takayasu et al., 2010). Moreover, recent studies have also reported that caveolins may be involved in some CNS diseases including AD, MS (Jones et al., 2004; Zarif Yeganeh et al., 2009), and Huntington's disease (Trushina et al., 2006). The present study expands our knowledge about the functional role of Cav-1 in CNS. Using a CCI-induced neuropathic pain model, we found for the first time an upregulation of Cav-1 genes and a long-lasting increase of t-Cav-1 and p-Cav-1 expression in contralateral ACC to the injury side associated with pain behavioral and neuronal sensitization. Persistent Cav-1 upregulation and activation may be involved in the maintenance of chronic pain. Furthermore, we explored the relevance of altered Cav-1 expression and pain behavioral and neuronal sensitization. Our data demonstrate that knockdown or blocking of Cav-1 could reverse CCI-induced increase of Fos protein expression in contralateral ACC and pain behavior. Indeed, overexpression of Cav-1 in ipsilateral ACC could unmask latent neuronal sensitization and induce pain behavior in the unaffected hindpaw. A great number of studies have demonstrated that Cav-1-mediated change of BBB permeability plays an important role in CNS pathological conditions (Wang et al., 2011; Deng et al., 2012; Gu et al., 2012; Liu et al., 2012). Furthermore, we ruled out the role of BBB permeability in pain modulatory effect by Cav-1; present findings clearly demonstrate that Cav-1 expressed in ACC neurons contributes to the modulation of chronic pain processing.
The importance of central sensitization in the ACC for chronic pain modulation is well established. The initiation, development, and maintenance of central sensitization is dependent on maladaptive alterations in the expression, distribution and activity of ion channels, receptors and intracellular signal transduction pathways, and signaling integration.
Membrane rafts provide structural signaling platforms for receptor trafficking and for downstream signaling integration (Hering et al., 2003). Previous studies have demonstrated that Cav-1 coimmunoprecipitates and colocalizes with NR2B and that expression of caveolin is necessary for neuronal NMDA-mediated signaling in cultured primary cortical neurons (Head et al., 2008). Cav-1 is required for normal coupling of mGluR1/5 to downstream signaling cascades and induction of mGluR-LTD (Francesconi et al., 2009). Consistent with these findings, here we show that Cav-1 directly binding with NR2B and promoting surface NR2B expression mediates modulation of chronic neuropathic pain by Cav-1 in the ACC. We found that CCI increased colocalization of Cav-1 and NR2B in neuronal membrane, which could be reversed by knockdown of Cav-1. Overexpression of Cav-1 in ipsilateral ACC also promoted NR2B surface levels associated with pain sensitization in the unaffected hindpaw in CCI mice. Interaction of Cav-1 and NR2B in the ACC is further substantiated by our findings in coimmunoprecipitation and mammalian two-hybrid assay experiments that in the ACC extracts, Cav-1 (or NR2B) was present and increased by CCI in immunoprecipitates obtained with anti-NR2B (or Cav-1) antibody. Moreover, disruption of the interaction of Cav-1 and NR2B with a short peptide that contains aromatic amino acids derived from C-terminal of NR2B can reverse CCI-induced promotion of NR2B surface levels, which was accompanied with alleviation of chronic neuropathic pain. These findings suggested that the recruitment of NR2B to lipid rafts by Cav-1 could be an important mechanism for central sensitization in the ACC during chronic neuropathic pain process.
Phosphorylation of ion channels or receptors by protein kinases and new protein synthesis mediated by activation of transcription factor is an important mechanism in development and maintenance of chronic pain. Previous studies have shown that activation of the ERK/CREB signaling pathway may contribute to neuronal sensitization in the ACC and behavioral sensitization during development of chronic pain (Wei and Zhuo, 2008; Toyoda et al., 2009b; Wang et al., 2012). Activation of the ERK/CREB signaling pathway is dependent on NMDAR-mediated Ca2+ influx (Chen et al., 2007). A recent study reported that in primary rat cortical neurons, Cav-1 expression is essential for NMDAR-mediated ERK1/2 activation (Head et al., 2008). In the present study, we found that Cav-1 regulated intracellular Ca2+ in an NR2B-dependent manner in cultured ACC neurons. Knockdown of Cav-1 inhibited CCI-induced activation of the ERK/CREB signaling pathway in contralateral ACC. Overexpression of Cav-1 activated the ERK/CREB signaling pathway through NR2B in ipsilateral ACC in CCI mice. Pain behavior in the unaffected hindpaw induced by Cav-1 overexpression in ipsilateral ACC could be reversed by inhibition of the ERK/CREB signaling pathway. These findings reveal that subsequent activation of the NR2B/Ca2+/ ERK/CREB signaling pathway in the ACC is required for the modulation of chronic neuropathic pain by Cav-1.
In conclusion, this study provides insights into the role of Cav-1 in the ACC neurons in chronic neuropathic pain processing and elucidates the receptor and intracellular mechanisms underlying Cav-1-mediated pain modulation in the ACC. These findings may lead to novel targeted drug discovery for the treatment of chronic pain.
Footnotes
This study was supported by grants from the National Natural Science Foundation of China (81070888 and 81230025 to J.-L.C., 81200859 to H.-L.D.), Natural Science Foundation of Jiangsu Education Department (13KJB320024 to J.-X.Y.), Natural Science Foundation of Jiangsu Province (BK2011198 to J.-L.C.), The Scientific Innovation Group of “Qing Lan Project” of Jiangsu Province (to J.-L.C.), and Jiangsu Provincial Special Program of Medical Science (BL2014029). We thank Wen-Zhen Duan, Heng Cai, and Tian-Hui Gao for administrative assistance.
The authors declare no competing financial interests.
References
- Allen JA, Halverson-Tamboli RA, Rasenick MM. Lipid raft microdomains and neurotransmitter signaling. Nat Rev Neurosci. 2007;8:128–140. doi: 10.1038/nrn2059. [DOI] [PubMed] [Google Scholar]
- Anderson RG. The caveolae membrane system. Annu Rev Biochem. 1998;67:199–225. doi: 10.1146/annurev.biochem.67.1.199. [DOI] [PubMed] [Google Scholar]
- Ariotti N, Parton RG. SnapShot: caveolae, caveolins, and cavins. Cell. 2013;154:704–704.e1. doi: 10.1016/j.cell.2013.07.009. [DOI] [PubMed] [Google Scholar]
- Beggs S, Liu XJ, Kwan C, Salter MW. Peripheral nerve injury and TRPV1-expressing primary afferent C-fibers cause opening of the blood-brain barrier. Mol Pain. 2010;6:74. doi: 10.1186/1744-8069-6-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett GJ, Xie YK. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain. 1988;33:87–107. doi: 10.1016/0304-3959(88)90209-6. [DOI] [PubMed] [Google Scholar]
- Boulware MI, Kordasiewicz H, Mermelstein PG. Caveolin proteins are essential for distinct effects of membrane estrogen receptors in neurons. J Neurosci. 2007;27:9941–9950. doi: 10.1523/JNEUROSCI.1647-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein–dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Brusés JL, Chauvet N, Rutishauser U. Membrane lipid rafts are necessary for the maintenance of the (alpha)7 nicotinic acetylcholine receptor in somatic spines of ciliary neurons. J Neurosci. 2001;21:504–512. doi: 10.1523/JNEUROSCI.21-02-00504.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron PL, Ruffin JW, Bollag R, Rasmussen H, Cameron RS. Identification of caveolin and caveolin-related proteins in the brain. J Neurosci. 1997;17:9520–9535. doi: 10.1523/JNEUROSCI.17-24-09520.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Chen L, Miyamoto Y, Furuya K, Mori N, Sokabe M. PREGS induces LTP in the hippocampal dentate gyrus of adult rats via the tyrosine phosphorylation of NR2B coupled to ERK/CREB signaling. J Neurophysiol. 2007;98:1538–1548. doi: 10.1152/jn.01151.2006. [DOI] [PubMed] [Google Scholar]
- Chidlow JH, Jr, Sessa WC. Caveolae, caveolins, and cavins: complex control of cellular signalling and inflammation. Cardiovasc Res. 2010;86:219–225. doi: 10.1093/cvr/cvq075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couet J, Li S, Okamoto T, Ikezu T, Lisanti MP. Identification of peptide and protein ligands for the caveolin-scaffolding domain. Implications for the interaction of caveolin with caveolae-associated proteins. J Biol Chem. 1997;272:6525–6533. doi: 10.1074/jbc.272.10.6525. [DOI] [PubMed] [Google Scholar]
- Deng J, Huang Q, Wang F, Liu Y, Wang Z, Wang Z, Zhang Q, Lei B, Cheng Y. The role of caveolin-1 in blood-brain barrier disruption induced by focused ultrasound combined with microbubbles. J Mol Neurosci. 2012;46:677–687. doi: 10.1007/s12031-011-9629-9. [DOI] [PubMed] [Google Scholar]
- Dickstein DL, Biron KE, Ujiie M, Pfeifer CG, Jeffries AR, Jefferies WA. Abeta peptide immunization restores blood- brain barrier integrity in Alzheimer disease. FASEB J. 2006;20:426–433. doi: 10.1096/fj.05-3956com. [DOI] [PubMed] [Google Scholar]
- Echeverry S, Shi XQ, Rivest S, Zhang J. Peripheral nerve injury alters blood-spinal cord barrier functional and molecular integrity through a selective inflammatory pathway. J Neurosci. 2011;31:10819–10828. doi: 10.1523/JNEUROSCI.1642-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francesconi A, Kumari R, Zukin RS. Regulation of group I metabotropic glutamate receptor trafficking and signaling by the caveolar/lipid raft pathway. J Neurosci. 2009;29:3590–3602. doi: 10.1523/JNEUROSCI.5824-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasquoine PG. Localization of function in anterior cingulate cortex: from psychosurgery to functional neuroimaging. Neurosci Biobehav Rev. 2013;37:340–348. doi: 10.1016/j.neubiorev.2013.01.002. [DOI] [PubMed] [Google Scholar]
- Gaudreault SB, Dea D, Poirier J. Increased caveolin-1 expression in Alzheimer's disease brain. Neurobiol Aging. 2004;25:753–759. doi: 10.1016/j.neurobiolaging.2003.07.004. [DOI] [PubMed] [Google Scholar]
- Gaudreault SB, Blain JF, Gratton JP, Poirier J. A role for caveolin-1 in post-injury reactive neuronal plasticity. J Neurochem. 2005;92:831–839. doi: 10.1111/j.1471-4159.2004.02917.x. [DOI] [PubMed] [Google Scholar]
- Gu Y, Zheng G, Xu M, Li Y, Chen X, Zhu W, Tong Y, Chung SK, Liu KJ, Shen J. Caveolin-1 regulates nitric oxide-mediated matrix metalloproteinases activity and blood-brain barrier permeability in focal cerebral ischemia and reperfusion injury. J Neurochem. 2012;120:147–156. doi: 10.1111/j.1471-4159.2011.07542.x. [DOI] [PubMed] [Google Scholar]
- Guirland C, Zheng JQ. Membrane lipid rafts and their role in axon guidance. Adv Exp Med Biol. 2007;621:144–155. doi: 10.1007/978-0-387-76715-4_11. [DOI] [PubMed] [Google Scholar]
- Harford-Wright E, Lewis KM, Ghabriel MN, Vink R. Treatment with the NK1 antagonist emend reduces blood brain barrier dysfunction and edema formation in an experimental model of brain tumors. PLoS One. 2014;9:e97002. doi: 10.1371/journal.pone.0097002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Head BP, Patel HH, Tsutsumi YM, Hu Y, Mejia T, Mora RC, Insel PA, Roth DM, Drummond JC, Patel PM. Caveolin-1 expression is essential for N-methyl-D-aspartate receptor-mediated Src and extracellular signal-regulated kinase 1/2 activation and protection of primary neurons from ischemic cell death. FASEB J. 2008;22:828–840. doi: 10.1096/fj.07-9299com. [DOI] [PubMed] [Google Scholar]
- Head BP, Hu Y, Finley JC, Saldana MD, Bonds JA, Miyanohara A, Niesman IR, Ali SS, Murray F, Insel PA, Roth DM, Patel HH, Patel PM. Neuron-targeted caveolin-1 protein enhances signaling and promotes arborization of primary neurons. J Biol Chem. 2011;286:33310–33321. doi: 10.1074/jbc.M111.255976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hering H, Lin CC, Sheng M. Lipid rafts in the maintenance of synapses, dendritic spines, and surface AMPA receptor stability. J Neurosci. 2003;23:3262–3271. doi: 10.1523/JNEUROSCI.23-08-03262.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt SP, Pini A, Evan G. Induction of c-fos-like protein in spinal cord neurons following sensory stimulation. Nature. 1987;328:632–634. doi: 10.1038/328632a0. [DOI] [PubMed] [Google Scholar]
- Jones KA, Jiang X, Yamamoto Y, Yeung RS. Tuberin is a component of lipid rafts and mediates caveolin-1 localization: role of TSC2 in post-Golgi transport. Exp Cell Res. 2004;295:512–524. doi: 10.1016/j.yexcr.2004.01.022. [DOI] [PubMed] [Google Scholar]
- Krajewska WM, Maslowska I. Caveolins: structure and function in signal transduction. Cell Mol Biol Lett. 2004;9:195–220. [PubMed] [Google Scholar]
- Ledesma MD, Simons K, Dotti CG. Neuronal polarity: essential role of protein-lipid complexes in axonal sorting. Proc Natl Acad Sci U S A. 1998;95:3966–3971. doi: 10.1073/pnas.95.7.3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li TT, Ren WH, Xiao X, Nan J, Cheng LZ, Zhang XH, Zhao ZQ, Zhang YQ. NMDA NR2A and NR2B receptors in the rostral anterior cingulate cortex contribute to pain-related aversion in male rats. Pain. 2009;146:183–193. doi: 10.1016/j.pain.2009.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XY, Ko HG, Chen T, Descalzi G, Koga K, Wang H, Kim SS, Shang Y, Kwak C, Park SW, Shim J, Lee K, Collingridge GL, Kaang BK, Zhuo M. Alleviating neuropathic pain hypersensitivity by inhibiting PKMz in the anterior cingulate cortex. Science. 2010;330:1400–1444. doi: 10.1126/science.1191792. [DOI] [PubMed] [Google Scholar]
- Liu J, Jin X, Liu KJ, Liu W. Matrix metalloproteinase-2-mediated occludin degradation and caveolin-1-mediated claudin-5 redistribution contribute to blood-brain barrier damage in early ischemic stroke stage. J Neurosci. 2012;32:3044–3057. doi: 10.1523/JNEUROSCI.6409-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Meerwijk EL, Ford JM, Weiss SJ. Brain regions associated with psychological pain: implications for a neural network and its relationship to physical pain. Brain Imaging Behav. 2013;7:1–14. doi: 10.1007/s11682-012-9179-y. [DOI] [PubMed] [Google Scholar]
- Mineo C, James GL, Smart EJ, Anderson RG. Localization of epidermal growth factor-stimulated Ras/Raf-1 interaction to caveolae membrane. J Biol Chem. 1996;271:11930–11935. doi: 10.1074/jbc.271.20.11930. [DOI] [PubMed] [Google Scholar]
- Mohseni HR, Smith PP, Parsons CE, Young KS, Hyam JA, Stein A, Stein JF, Green AL, Aziz TZ, Kringelbach ML. MEG can map short and long-term changes in brain activity following deep brain stimulation for chronic pain. PLoS One. 2012;7:e37993. doi: 10.1371/journal.pone.0037993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994;12:529–540. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- Mori F, Ribolsi M, Kusayanagi H, Siracusano A, Mantovani V, Marasco E, Bernardi G, Centonze D. Genetic variants of the NMDA receptor influence cortical excitability and plasticity in humans. J Neurophysiol. 2011;106:1637–1643. doi: 10.1152/jn.00318.2011. [DOI] [PubMed] [Google Scholar]
- Nag S, Manias JL, Stewart DJ. Expression of endothelial phosphorylated caveolin-1 is increased in brain injury. Neuropathol Appl Neurobiol. 2009;35:417–426. doi: 10.1111/j.1365-2990.2008.01009.x. [DOI] [PubMed] [Google Scholar]
- Nusrat A, Parkos CA, Verkade P, Foley CS, Liang TW, Innis-Whitehouse W, Eastburn KK, Madara JL. Tight junctions are membrane microdomains. J Cell Sci. 2000;113:1771–1781. doi: 10.1242/jcs.113.10.1771. [DOI] [PubMed] [Google Scholar]
- Petralia RS, Wang YX, Wenthold RJ. Internalization at glutamatergic synapses during development. Eur J Neurosci. 2003;18:3207–3217. doi: 10.1111/j.1460-9568.2003.03074.x. [DOI] [PubMed] [Google Scholar]
- Plattner F, Hernández A, Kistler TM, Pozo K, Zhong P, Yuen EY, Tan C, Hawasli AH, Cooke SF, Nishi A, Guo A, Wiederhold T, Yan Z, Bibb JA. Memory enhancement by targeting Cdk5 regulation of NR2B. Neuron. 2014;81:1070–1083. doi: 10.1016/j.neuron.2014.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez JT, Seidl AH, Rubel EW, Barria A. Control of neuronal excitability by NMDA-type glutamate receptors in early developing binaural auditory neurons. J Physiol. 2012;590:4801–4818. doi: 10.1113/jphysiol.2012.228734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Ma S, Chan P, Lee W, Fung PC, Cheung RT, Tong Y, Liu KJ. Nitric oxide down-regulates caveolin-1 expression in rat brains during focal cerebral ischemia and reperfusion injury. J Neurochem. 2006;96:1078–1089. doi: 10.1111/j.1471-4159.2005.03589.x. [DOI] [PubMed] [Google Scholar]
- Song L, Ge S, Pachter JS. Caveolin-1 regulates expression of junction-associated proteins in brain microvascular endothelial cells. Blood. 2007;109:1515–1523. doi: 10.1182/blood-2006-07-034009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayasu Y, Takeuchi K, Kumari R, Bennett MV, Zukin RS, Francesconi A. Caveolin-1 knockout mice exhibit impaired induction of mGluR-dependent long-term depression at CA3-CA1 synapses. Proc Natl Acad Sci U S A. 2010;107:21778–21783. doi: 10.1073/pnas.1015553107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoda H, Zhao MG, Zhuo M. Enhanced quantal release of excitatory transmitter in anterior cingulate cortex of adult mice with chronic pain. Mol Pain. 2009a;5:4. doi: 10.1186/1744-8069-5-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoda H, Zhao MG, Ulzhöfer B, Wu LJ, Xu H, Seeburg PH, Sprengel R, Kuner R, Zhuo M. Roles of the AMPA receptor subunit GluA1 but not GluA2 in synaptic potentiation and activation of ERK in the anterior cingulate cortex. Mol Pain. 2009b;5:46. doi: 10.1186/1744-8069-5-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trushina E, Singh RD, Dyer RB, Cao S, Shah VH, Parton RG, Pagano RE, McMurray CT. Mutant huntingtin inhibits clathrin-independent endocytosis and causes accumulation of cholesterol in vitro and in vivo. Hum Mol Genet. 2006;15:3578–3591. doi: 10.1093/hmg/ddl434. [DOI] [PubMed] [Google Scholar]
- Tsui-Pierchala BA, Encinas M, Milbrandt J, Johnson EM., Jr Lipid rafts in neuronal signaling and function. Trends Neurosci. 2002;25:412–417. doi: 10.1016/S0166-2236(02)02215-4. [DOI] [PubMed] [Google Scholar]
- Venema VJ, Zou R, Ju H, Marrero MB, Venema RC. Caveolin-1 detergent solubility and association with endothelial nitric oxide synthase is modulated by tyrosine phosphorylation. Biochem Biophys Res Commun. 1997;236:155–161. doi: 10.1006/bbrc.1997.6921. [DOI] [PubMed] [Google Scholar]
- Wang H, Morishita Y, Miura D, Naranjo JR, Kida S, Zhuo M. Roles of CREB in the regulation of FMRP by group I metabotropic glutamate receptors in cingulate cortex. Mol Brain. 2012;5:27. doi: 10.1186/1756-6606-5-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Liu Y, Shang X, Xue Y. CRM197-induced blood-brain barrier permeability increase is mediated by upregulation of caveolin-1 protein. J Mol Neurosci. 2011;43:485–492. doi: 10.1007/s12031-010-9471-5. [DOI] [PubMed] [Google Scholar]
- Wei F, Zhuo M. Activation of Erk in the anterior cingulate cortex during the induction and expression of chronic pain. Mol Pain. 2008;4:28. doi: 10.1186/1744-8069-4-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei F, Li P, Zhuo M. Loss of synaptic depression in mammalian anterior cingulate cortex after amputation. J Neurosci. 1999;19:9346–9354. doi: 10.1523/JNEUROSCI.19-21-09346.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei F, Qiu CS, Kim SJ, Muglia L, Maas JW, Pineda VV, Xu HM, Chen ZF, Storm DR, Muglia LJ, Zhuo M. Genetic elimination of behavioral sensitization in mice lacking Calmodulin-stimulated adenylyl cyclases. Neuron. 2002;36:713–726. doi: 10.1016/S0896-6273(02)01019-X. [DOI] [PubMed] [Google Scholar]
- Williams TM, Lisanti MP. The Caveolin genes: from cell biology to medicine. Ann Med. 2004;36:584–595. doi: 10.1080/07853890410018899. [DOI] [PubMed] [Google Scholar]
- Wu LJ, Toyoda H, Zhao MG, Lee YS, Tang J, Ko SW, Jia YH, Shum FW, Zerbinatti CV, Bu G, Wei F, Xu TL, Muglia LJ, Chen ZF, Auberson YP, Kaang BK, Zhuo M. Upregulation of forebrain NMDANR2B receptors contributes to behavioral sensitization after inflammation. J Neurosci. 2005;25:11107–11116. doi: 10.1523/JNEUROSCI.1678-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu XM, Qian ZM, Zhu L, Du F, Yung WH, Gong Q, Ke Y. Neuroprotective effect of ligustilide against ischaemia-reperfusion injury via up-regulation of erythropoietin and down-regulation of RTP801. Br J Pharmacol. 2011;164:332–343. doi: 10.1111/j.1476-5381.2011.01337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Wu LJ, Wang H, Zhang X, Vadakkan KI, Kim SS, Steenland HW, Zhuo M. Presynaptic and postsynaptic amplifications of neuropathic pain in the anterior cingulate cortex. J Neurosci. 2008;28:7445–7453. doi: 10.1523/JNEUROSCI.1812-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Toya Y, Schwencke C, Lisanti MP, Myers MG, Jr, Ishikawa Y. Caveolin is an activator of insulin receptor signaling. J Biol Chem. 1998;273:26962–26968. doi: 10.1074/jbc.273.41.26962. [DOI] [PubMed] [Google Scholar]
- Zarif Yeganeh M, Ghaffarpour M, Farhud DD, Karimlou M, Ghabaee M, Haghighi Nazari A, Najmabadi H, Ohadi M. Skew in the human caveolin 1 gene upstream purine complex homozygote haplotype compartment in multiple sclerosis. J Neuroimmunol. 2009;216:103–107. doi: 10.1016/j.jneuroim.2009.09.007. [DOI] [PubMed] [Google Scholar]
- Zhao MG, Toyoda H, Lee YS, Wu LJ, Ko SW, Zhang XH, Jia Y, Shum F, Xu H, Li BM, Kaang BK, Zhuo M. Roles of NMDA NR2B subtype receptor in prefrontal long-term potentiation and contextual fear memory. Neuron. 2005;47:859–872. doi: 10.1016/j.neuron.2005.08.014. [DOI] [PubMed] [Google Scholar]
- Zhao MG, Ko SW, Wu LJ, Toyoda H, Xu H, Quan J, Li J, Jia Y, Ren M, Xu ZC, Zhuo M. Enhanced presynaptic neurotransmitter release in the anterior cingulate cortex of mice with chronic pain. J Neurosci. 2006;26:8923–8930. doi: 10.1523/JNEUROSCI.2103-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuo M. Cortical excitation and chronic pain. Trends Neurosci. 2008;31:199–207. doi: 10.1016/j.tins.2008.01.003. [DOI] [PubMed] [Google Scholar]
- Zhuo M. Plasticity of NMDA receptor NR2B subunit in memory and chronic pain. Mol Brain. 2009;2:4. doi: 10.1186/1756-6606-2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuo M. Long-term potentiation in the anterior cingulate cortex and chronic pain. Philos Trans R Soc Lond B Biol Sci. 2014;369:20130146. doi: 10.1098/rstb.2013.0146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zschocke J, Manthey D, Bayatti N, van der Burg B, Goodenough S, Behl C. Estrogen receptor alpha-mediated silencing of caveolin gene expression in neuronal cells. J Biol Chem. 2002;277:38772–38780. doi: 10.1074/jbc.M205664200. [DOI] [PubMed] [Google Scholar]