

Abstract
The development of the nervous system is a temporally and spatially coordinated process that relies on the proper regulation of the genes involved. Neurotrophins and their receptors are directly responsible for the survival and differentiation of sensory and sympathetic neurons; however, it is not fully understood how genes encoding Trk neurotrophin receptors are regulated. Here, we show that rat Bex3 protein specifically regulates TrkA expression by acting at the trkA gene promoter level. Bex3 dimerization and shuttling to the nucleus regulate the transcription of the trkA promoter under basal conditions and also enhance nerve growth factor (NGF)-mediated trkA promoter activation. Moreover, qChIP assays indicate that Bex3 associates with the trkA promoter within a 150 bp sequence, immediately upstream from the transcription start site, which is sufficient to mediate the effects of Bex3. Consequently, the downregulation of Bex3 using shRNA increases neuronal apoptosis in NGF-dependent sensory neurons deprived of NGF and compromises PC12 cell differentiation in response to NGF. Our results support an important role for Bex3 in the regulation of TrkA expression and in NGF-mediated functions through modulation of the trkA promoter.
Keywords: Bex3, differentiation, gene promoter, NGF, transcription, TrkA
Introduction
Nerve growth factor (NGF) was initially identified as a survival and differentiation factor for sensory and sympathetic neurons during development, but it has also been implicated in pathological conditions, including pain sensitivity and Alzheimer's disease (AD; Huang and Reichardt, 2001; Chao, 2003; Arévalo and Wu, 2006). NGF exerts its effects through two types of transmembrane receptors: tyrosine kinase receptor A (TrkA) and p75 neurotrophin receptor (Huang and Reichardt, 2003). TrkA is mainly expressed in the peripheral nervous system in sympathetic neurons, and in nociceptive neurons in dorsal root ganglia (DRGs) and in the trigeminal ganglia .(Martin-Zanca et al., 1990; Holtzman et al., 1992; Smeyne et al., 1994). During mouse development, trkA gene expression is regulated in DRG neurons, where expression begins at embryonic day 9.5 (E9.5), peaks at E13.5, and remains constant throughout development (Martin-Zanca et al., 1990).
The mouse and human trkA minimal promoters have been characterized by in vitro reporter assays using PC12 and neuroblastoma cell lines, and encompass 150 bp upstream from the transcription start site (Chang et al., 1998; Sacristán et al., 1999). In vivo, a 457 bp sequence, highly conserved in mouse, chick, and human genes, upstream from the transcription start site seems to be required to drive coordinated trkA expression in sensory and sympathetic neurons (Ma et al., 2000; Lei and Parada, 2007). Several transcription factors have been reported to influence trkA gene expression, including Sp1, Brn3a, Klf7, and Runx1 (Huang et al., 1999; Sacristán et al., 1999; Lei et al., 2001, 2005; Ma et al., 2003; Marmigère et al., 2006; Lanier et al., 2007). It has been established that NGF treatment increases trkA mRNA levels in PC12 cells and cholinergic neurons (Miller et al., 1991; Holtzman et al., 1992; Li et al., 1995; Zhou et al., 1995), and HAND transcription factors partially regulate trkA expression in response to NGF in cultured sympathetic neurons without affecting the basal expression of trkA (i.e., in the absence of NGF; Doxakis et al., 2008). However, at present it is unknown whether additional proteins modulate trkA gene expression in response to NGF.
Bex3 belongs to the bex (brain-expressed x-linked) gene family, composed of six members, bex1 to bex6 (Faria et al., 1998; Brown and Kay, 1999; Alvarez et al., 2005). Bex3 has also been referred to as a p75-associated death executor (NADE) and has been shown to interact with p75 in an NGF-dependent manner, leading to NGF-induced apoptosis (Mukai et al., 2000, 2002). However, the function of Bex3 remains unclear as subsequent studies did not find any link between Bex3 and p75-mediated cell death (Tong et al., 2003; Alvarez et al., 2005).
Here we describe an unanticipated role for Bex3 in regulating the trkA promoter. Depletion of Bex3 using shRNA decreased the survival of NGF-dependent neurons and impaired NGF-mediated PC12 cell differentiation as a consequence of the specific reduction of TrkA protein and trkA mRNA levels. Reporter assays indicated that Bex3 positively regulates basal trkA transcription and that dimerization of Bex3 is required for this function. Moreover, Bex3 overexpression enhanced the induction of trkA expression in response to NGF. Our results support a critical role for Bex3 in TrkA receptor expression through the regulation of the trkA promoter and, consequently, in NGF-mediated functions, such as survival and differentiation.
Materials and Methods
Materials.
The following antibodies were used: Trk (C-14), Bex3 (P-13), rabbit and goat IgGs (Santa Cruz Biotechnology), GFP (Clontech), β-tubulin III (Sigma-Aldrich), phosphor (p)-Trk (Y490), phospho-Akt and phospho-MAPK (Cell Signaling Technology), TrkA (RTA; a gift from L. Reichardt USCF, San Francisco, CA), and Trk (203); 705 is an antibody developed against Bex3 (see below). mNGF 2.5S was from Alomone Labs. Recombinant human BDNF was from Peprotech. Leptomycin B (LMB) and chloroquine were from Sigma-Aldrich. MG132 was from Calbiochem.
Plasmids.
The sequence 5′-caacaaccacaaccataac-3′ corresponding to nucleotides 78–96 of rat Bex3 cDNA and the sequence 5′-caacaacaacaaccacaac-3′ corresponding to nucleotides 108–126 and 78–96 of mouse Bex3 cDNA of isoforms A and B, respectively, were used to generate Bex3 shRNA using the pLVTHM lentiviral vector (Kim et al., 2004). Lentivirus expressing control shRNA was generated using the sequence 5′-gcgcgctttgtaggattcg-3′ from Euglena gracilis chloroplast DNA between s16 S and 16 S rRNA. The GFP-Bex3-expressing plasmid was constructed by inserting rat Bex3 cDNA in frame with GFP cDNA in the pEGFP–C3 vector. The HA-Bex3-expressing plasmid was constructed after PCR amplification of Bex3 from the cDNA library used to perform the yeast two-hybrid assay, sequenced, and cloned into pcDNA3. GFP-Bex3 L90,93A; GFP-Bex3 K89R; GFP-Bex3 C117S; and GFP-Bex3 C98S mutants were generated from the pEGFP-Bex3 plasmid by introducing the corresponding point mutations. GFP-Bex3 D1 and D2 were generated by introducing a TGA stop codon “in lieu” of nucleotides 288–290 and 178–180, respectively. A Bex3 cDNA resistant to the action of Bex3 shRNA was generated by site-directed mutagenesis of 5 nt in the third codon of the wild-type (WT) Bex3 sequence recognized by the shRNA. The sequences of the primers used for the mutagenesis were 5′-gctgcaaacaacaataatcataatcacaatcacaaccacaaccac-3′ and 5′-gtggttgtggttgtgattgtgattatgattattgttgtttgcagc-3′ (the bold nucleotides indicate the changes that were introduced in the cDNA). These changes did not affect the amino acid sequence of the protein, but the mRNA was resistant to Bex3 shRNA action. All constructs generated were confirmed by sequencing.
Yeast two-hybrid screening.
A yeast two-hybrid screening performed with the juxtamembrane region (ΔJM) of TrkB and a cDNA library from postnatal day 0 (P0) DRGs, as described previously (Kong et al., 2001; Yano et al., 2001). One of the proteins identified was Bex3.
Generation of anti-Bex3 antibodies.
Polyclonal antibodies against Bex3 (705) were generated in rabbits using a synthetic peptide present in rat Bex3 protein (NNNNHNHNHNHNHNHNHNHH) coupled to keyhole limpet hemocyanin. A first injection of 500 μg of coupled peptide was followed 3 weeks later by boosts of 200 μg every 2 weeks. Bleedings were obtained 7–10 d after the second boost. Antibodies were purified using a CNBr Sepharose column (Pharmacia) coupled with the synthetic peptide used to inject the rabbits. Antibody elution was performed with 0.1 m glycine, pH 2.5. The pH was neutralized with 1 m Tris-HCl, pH 9.0, and the solution was supplemented with 50 mm NaCl.
In vitro pull-down assays.
GST-TrkA juxtamembrane proteins were produced in Escherichia coli, as previously described (Yano et al., 2001). Bex3 protein was produced and labeled with 35S-Cys using the TnT Coupled Reticulocyte Lysate System (Promega) according to the manufacturer's instructions. Immobilized GST fusion proteins were incubated with labeled Bex3 at 4°C. The beads were then washed extensively, and the bound proteins were analyzed by 12% SDS-PAGE and detected by autoradiography.
Cell cultures.
HEK293 and HEK293FT cells were grown in DMEM supplemented with 10% bovine serum, 1% nonessential amino acids (NEAA), 2 mm l-glutamine and penicillin (pen; 100 U/ml), and streptomycin (strep; 100 μg/ml). Neuro2a (N2a) cells were grown in DMEM supplemented with 10% fetal bovine serum (FBS), 1% NEAA and pen/strep. PC12 and PC12-6/15 cells were grown in RPMI 1640 supplemented with 10% horse serum, 5% FBS, 2 mm l-glutamine, and pen/strep in collagen-coated plates.
DRG and hippocampal neuron cultures.
DRG neuron cultures were performed as previously described (Yu et al., 2011). Briefly, DRGs were dissected from E15.5 rat embryos of either sex and dissociated with 0.25% trypsin in L-15 media for 45 min at 37°C. Cells were centrifuged to eliminate trypsin and resuspended in plating medium (MEM, 10% FBS, 0.4% glucose, 2 mm glutamine, 25 U/ml pen/strep). A single-cell suspension was obtained by pipetting up and down 30 times with a 200 μl plastic pipette tip. After counting, cells were seeded using plating medium plus NGF (50 ng/ml) overnight on plastic plates coated with Growth Factor Reduced Matrigel (BD Biosciences) as a substrate. On the following day, the medium was changed to neurobasal (NB) medium (B-27, 0.4% glucose, 2 mm glutamine, 25 U/ml pen/strep), NGF, and 5-fluorodeoxyuridine (2.44 μg/ml) and uridine (2.44 μg/ml). Proliferating cells disappeared after 3–4 d, and >95% of cells at day in vitro 8 (DIV8) were neurons.
Hippocampi were dissected from E18.5 rat embryos of either sex and cultured as previously described (Arévalo et al., 2010). Briefly, primary hippocampal neurons were obtained from E18–E19 rat embryos. Cells were seeded on plating medium (MEM, 10% FBS, 0.37% glucose, 1 mm pyruvate, 2 mm glutamine, 25 U/ml penicillin, and 25 μg/ml streptomycin) overnight on poly-d-lysine-coated (PDL) plates. On the following day, the medium was changed to NB medium supplemented with B-27, 0.4% glucose, 2 mm glutamine, 25 U/ml penicillin, and 25 μg/ml streptomycin. Fluorodeoxyuridine (2.44 μg/ml) and uridine (2.44 μg/ml) were added to kill proliferating cells.
Transfection.
HEK293 cells were transfected using the calcium phosphate method. N2a cells, PC12 cells, and DRG neurons were transfected with Lipofectamine 2000 following the manufacturer's recommendations.
Lentivirus generation and infection.
The lentiviruses used in this study were generated as previously described (Arévalo et al., 2010). Briefly, HEK293FT cells were transfected with 9 μg of pLVTHM control shRNA, or pLVTHM rat or mouse Bex3 shRNA, together with 6 μg of psPAX2 and 5 μg of pMD.2G plasmids using the calcium phosphate method. The medium was changed after 12 h and collected 48–72 h later. The supernatant containing the lentiviruses was collected, centrifuged at 5000 rpm for 15 min, filtered (45 μm), and stored in aliquots at −80°C. The virus medium was used to infect cells, which were monitored by the expression of GFP. Following infection, Bex3 levels decreased by at least 70% within 4–5 d.
Western blot analyses.
Cells were lysed in lysis buffer (10 mm Tris, pH 7.4, 150 mm NaCl, 2 mm EDTA, 1% NP-40, 1 mm PMSF, 1 μg/ml aprotinin, 2 μg/ml leupeptin, 1 mm vanadate, 10 mm NaF, and 20 mm β-glycerophosphate) for 45 min at 4°C with gentle shaking and centrifuged at 14,000 × g for 15 min to eliminate cell debris. SDS buffer 5× was added to the lysates, and they were boiled for 7 min to denature the proteins. Proteins were resolved by SDS-PAGE, and Western blotting was performed with antibodies against the different proteins. To prevent interferences with the Ig chains, we used ProtA or ProtG conjugated with horseradish peroxidase when the same species of antibodies were used for both immunoprecipitation and Western blots. Quantification of the signal bands was accomplished using ImageJ software (NIH).
Coimmunoprecipitation assays.
Cells were lysed in 500 μl of lysis buffer (10 mm Tris pH 7.4, 150 mm NaCl, 2 mm EDTA, 1% NP-40, 1 mm PMSF, 1 μg/ml aprotinin, 2 μg/ml leupeptin, 1 mm vanadate, 10 mm NaF, and 20 mm β-glycerophosphate) for 45 min at 4°C with gentle shaking. Lysates were centrifuged at 13,000 rpm for 15 min at 4°C to eliminate debris, and the supernatant was incubated overnight with the corresponding antibody at 4°C. Protein–antibody complexes were precipitated with ProtA- or ProtG-agarose, washed four times with lysis buffer, and detected by Western blot assay.
Semiquantitative and quantitative RT-PCR.
TRIzol reagent was used to isolate total RNA from DRGs and hippocampal neurons, following the manufacturer's recommendations. cDNA was synthesized with 1 μg of RNA and reverse transcribed using the ImProm-II Reverse Transcription System and random primers. The cDNA concentration was determined by measuring absorbance with a Nanodrop 2000c (Thermo Scientific). For semiquantitative PCR, the relative amounts of cDNA and the range of PCR cycles that permit the linear amplification of bex3 (35 cycles) and β-actin (20 cycles) were determined. The primer sequences were as follows: 5′-agccatgtacgtagccatcc-3′ and 5-accctcatagatgggcacag-3′ for β-actin (used as a control); and 5′-cattcccaacaggcagatg-3′ and 5′-ggcataaggcagaattcatc-3′ for rat and mouse bex3. The PCR conditions for bex3 were as follows: 94°C for 30 s, 58°C for 30 s, followed by 72°C for 40 s. Using the NIH ImageJ software, each DNA band was tested for its relative band intensity. The relative expression of bex3 mRNA was evaluated by calculating the bex3/β-actin signal ratio. Quantitative PCRs (qPCRs) were performed in triplicate using SYBR-Green Power Master Mix (Applied Biosystems). The mRNA level of each gene was normalized using β-actin as a reference. The oligonucleotides used to amplify trkA (Ntrk1 reference NM_021589.1, with an efficiency of 106%) were 5′-tctccttctcgccagtggac-3′and 5′-acagccacagagaccccaaa-3′ for full-length trkB (Ntrk2 transcript variant 1 reference NM_012731.2, with an efficiency of 104.5%), were 5′-ccggacacatttgttcagca-3′and 5′-ttcccaaaggctccttctcc-3′ for β-actin (Actb reference NM_031144.3 with an efficiency of 107%), and were the same as those used in the semiquantitative RT-PCR. An ABI Prism 7300 detection system (Applied Biosystems) was used with the following conditions: 2 min at 50°C, then 10 min at 95°C followed by 40 cycles of 15 s at 95°C and 1 min at 61.5°C. Following amplification, the melting curves for the products were generated to ensure that a product represented a homogenous species. A comparative cycle of threshold fluorescence (Ct) was used, and the relative transcription level of the target gene was normalized to that of β-actin using the 2−ΔΔCt method.
DRG neuron survival.
NGF-dependent rat DRG neurons were transfected at DIV4–5 with the corresponding plasmids expressing GFP using Lipofectamine 2000 (Invitrogen). The following day, the medium was replaced, keeping the NGF for 24 h, after which it was removed. Transfected cells were recognized by GFP expression, and apoptotic neurons, identified by fragmented or condensed nuclei using Hoechst 33342 stain, were scored 48–72 h after NGF withdrawal. Images were taken in random selected fields using a Leica DMI3000B microscope equipped with a Leica DFC300FX camera.
In situ hybridization and immunocytochemistry.
Plasmids containing specific probes for mouse trkA (nucleotides 19–219 of Ntrk1 reference NM_001033124) and mouse bex3 (nucleotides 251–449 of bex3 ref NM_009750) were used to synthesize digoxigenin-labeled antisense riboprobes according to the supplier's instructions (Roche). bex3 and trkA riboprobes were used at final concentrations of 1 and 0.5 μg/ml, respectively.
Mouse embryos were obtained at different embryonic days considering the day the plug was observed as E0.5. Embryos were placed in 4% paraformaldehyde (PFA) in 0.1 m PB for 24 h. On the following day, they were placed in 4% PFA plus 20% sucrose in 0.1 m PB and then in 30% sucrose until they sank. Tissue sections (12 μm) were obtained. For in situ hybridization, sections were postfixed with 4% PFA for 10 min, treated with PBS containing 0.1% DEPC for 5 min, and then with PBS containing 20 μg/ml proteinase K for 6 min, followed by acetylation for 10 min. Sections were prehybridized for 30 min and then hybridized with the corresponding probe overnight at 55°C. On the next day, sections were rinsed with 5× SSC at 65°C, washed with 2× SSC/50% formamide for 30 min at 65°C, treated with 20 μg/ml RNase for 30 min at 37°C, washed with 2× SSC and 0.1× SSC, and subjected to anti-DIG-AP-conjugated antibody (Roche) staining and a BCIP/NBT (Roche) color reaction.
When in situ hybridization and immunocytochemistry were performed on the same section, the immunocytochemistry was done first. Sections were postfixed with 4% PFA for 10 min, washed three times with PBS, and preblocked in PBS with 5% NGS and 0.1% Triton X-100 for 30 min. Sections were incubated overnight at 4°C with RTA antibody that specifically recognizes TrkA. On the following day, sections were rinsed three times with PBS, incubated for 1 h at room temperature with Alexa Fluor 488 (Invitrogen), and rinsed three times with PBS. Then, in situ hybridization was performed as described above, using anti-DIG-rhodamine-conjugated antibody (Roche) to detect bex3. Images were collected with a Leica confocal microscope.
Reporter assays.
N2a or PC12 cells growing in 12-well plates were transfected using Lipofectamine 2000. The plasmids used were pEGFP, pEGFP-Bex3, pEGFP-Bex3 mutants, control shRNA, and Bex3 shRNA (1.5 μg), and the corresponding trkA promoter constructs (0.5 μg) were pMS8, pMS10.3, pMS16, and pMS22, which have been described previously (Sacristán et al., 1999). After 48 h, cell extracts from transfected cells were prepared, and CAT protein ELISA analyses were performed in duplicate using a CAT ELISA kit (Roche). CAT was measured using an LT-4000 microplate reader (LabTech) at 405 nm. Western blot analyses were performed to ensure that the expression levels were similar.
Immunofluorescence.
The HEK293 cells used to detect GFP-Bex3 localization were cultured on coverslips coated with 1 mg/ml PDL. After transfection with the corresponding expression vector, cells were treated or not with Leptomycin B for 6 h, fixed with 4% PFA in PBS for 5 min and washed three times for 5 min. Coverslips were mounted in Vectashield medium (Vector Laboratories).
Nuclear fractionation.
PC12-6/15 cells were stimulated or not with NGF (100 ng/ml) for 15 min, washed with cold PBS, and then lysed with cytoplasmic lysis buffer (10 mm HEPES, pH 7.5, 1.5 mm MgCl2, 10 mm KCl, 1 mm EDTA, 0.1% NP-40, 1 mm PMSF, 1 μg/ml aprotinin, 2 μg/ml leupeptin, 1 mm vanadate, 10 mm NaF, and 20 mm β-glycerophosphate) by pipetting up and down several times. Then, nuclei were pelleted by centrifugation at 500 × g for 10 min at 4°C. The nuclei were washed with cytoplasmic lysis buffer to remove any contaminating cytoplasm, and pelleted again. The nuclei were lysed with complete lysis buffer (50 mm HEPES pH 7.5, 1.5 mm MgCl2, 10 mm KCl, 10 mm NaCl, 1 mm EDTA, 1% NP-40, 1 mm PMSF, 1 μg/ml aprotinin, 2 μg/ml leupeptin, 1 mm vanadate, 10 mm NaF, and 20 mm β-glycerophosphate). Finally, proteins were quantified, and Western blots were performed. p-Histone3 was used as a nuclear marker.
Chromatin immunoprecipitation assays.
N2a cells were grown on 10 cm plates to confluence and then treated with 1% formaldehyde for 10 min at 37°C. Cells were washed with cold PBS twice, pelleted (3500 rpm, 4°C, 5 min), and lysed with SDS lysis buffer (1% SDS, 10 mm EDTA, 50 mm Tris-HCl, pH 8.0, and protease inhibitors) for 10 min at 4°C. Lysates were sonicated using an ultrasonic bath (USC100, VWR) and centrifuged at 13,000 rpm for 10 min at 4°C, and the supernatant was diluted 10-fold in chromatin immunoprecipitation (ChIP) dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mm EDTA, 16.7 mm Tris-HCl, pH 8.0, 167 mm NaCl, and protease inhibitors). For later input, 1% of the diluted supernatant was saved. The remaining supernatant was precleared with protein G-Plus Agarose for 1 h at 4°C with constant shaking. Chromatin was immunoprecipitated with 4 μg of normal goat IgG or Bex3 antibody (P-13) previously cross-linked with protein G-Plus Agarose overnight at 4°C. Complexes were washed sequentially with the following buffers and centrifuged at 2000 rpm for 3 min at 4°C: 2× low-salt buffer (0.1% SDS, 1% Triton X-100, 2 mm EDTA, 20 mm Tris-HCl, pH 8.0, 150 mm NaCl); 2× high-salt buffer (0.1% SDS, 1% Triton X-100, 2 mm EDTA, 20 mm Tris-HCl, pH 8.0, 500 mm NaCl); and 2× TE buffer. The immunoprecipitated complexes were eluted with 2× 250 μl elution buffer (1% SDS, 0.1 m NaHCO3), and cross-linking was reversed by the addition of 20 μl of 5 m NaCl and subsequent incubation for 5 h at 65°C. Then 10 μl of 0.5 m EDTA, 20 μl of 1 m Tris-HCl, pH 6.5, and 1 μl of 20 mg/ml proteinase K were added to the eluate, and incubated for 1 h at 45°C. The DNA was recovered by phenol/chloroform extraction. For N2a cell experiments, PCR was performed for 35 cycles at 94°C for 30 s, at 55°C for 30 s, and at 72°C for 30 s. The oligonucleotides used to amplify the trkA promoter were 5′-cgtgagggacatgaggaag-3′ and 5′-gggcggtgttaaagactagc-3′. Amplified products were detected by standard 2% agarose gel electrophoresis. For PC12 cell experiments, immunoprecipitations were performed as described above, but quantitative PCR was performed instead of PCR. Input and ChIP samples were analyzed by triplicate using SYBR Green fluorescence (ABI Prism 7300 detection System and Power SYBR Green Master Mix; Applied Biosystems). The detection system was used with the following conditions: 2 min at 50°C, then 10 min at 95°C, followed by 40 cycles of 15 s at 95°C and 1 min at 58°C. Following amplification, the melting curves for the products were generated to ensure that a product represented a homogenous species. The fragment amplified in both procedures is located in the promoter region described previously (Sacristán et al., 1999) between nucleotides −143 and −19, numbered with respect to the main trkA transcriptional start site. The relative fold difference of the amplified fragment was calculated relative to a control using the ΔΔCt method.
Statistics.
Results are expressed as the mean ± SEM. Statistical analyses were performed with GraphPad Prism version 4.
Results
Bex3 interacts with Trk neurotrophin receptors
To identify proteins that interact with the Trks, a yeast two-hybrid screen was performed using a P0 rat DRG library and the juxtamembrane domain of TrkB receptor as a bait (Kong et al., 2001; Lou et al., 2001; Yano et al., 2001). Bex3 was identified once among the 75 positive clones obtained. Considering that most rat DRG neurons at P0 are TrkA positive (Bennett et al., 1996; Benn et al., 2001; Guo et al., 2001), we assessed whether there was an association between Bex3 and TrkA. By performing coimmunoprecipitation assays in HEK293 cells transfected with plasmids expressing both proteins, we observed an interaction between them (Fig. 1A). In addition, endogenous Bex3 interacted with TrkA in PC12–6/15 cells both in the presence and in the absence of NGF (Fig. 1B,C). Furthermore, TrkA and Bex3 association was also observed in embryonic rat DRG neurons (Fig. 1D). These findings indicate that Bex3 interacts with the NGF receptor TrkA. To map the region of TrkA involved in this interaction, we performed in vitro pull-down assays using GST fusion proteins containing different fragments of the TrkA juxtamembrane region (Yano et al., 2001; Fig. 1E, left) and in vitro translated Bex3 protein. The results showed that all GST-TrkA protein fragments interacted with Bex3 (Fig. 1E, right). To determine whether Bex3 could bind to other domains of TrkA, a mutant TrkA lacking the ΔJM was used (Arévalo et al., 2004). Coimmunoprecipitation experiments showed that Bex3 did not interact with any other domains of TrkA aside from the juxtamembrane domain (Fig. 1F). Next, we examined which regions of Bex3 were involved in the interaction with TrkA by performing coimmunoprecipitation experiments in HEK293 cells transfected with TrkA and different Bex3 constructs. The deletion mutants Bex3-D1 and Bex3-D2, respectively lacking 33 and 73 aa of the C terminus of Bex3, failed to interact with TrkA, whereas the Bex3 proteins carrying L90,93A and C117S mutations, which disrupt the nuclear export sequence (NES) and CAAX motifs, respectively, showed reduced binding to TrkA (Fig. 1G). Notably, a point mutation in the C terminus of Bex3 that replaced lysine 89 by arginine, within a destruction box (D-box; Mukai et al., 2000), did not affect the interaction with TrkA (Fig. 1G). These data support the notion that the interaction between Bex3 and TrkA is mediated through the juxtamembrane region of TrkA and the C terminus of Bex3.
Figure 1.

Bex3 interacts with TrkA neurotrophin receptor. A, TrkA and Bex3 interact in HEK293-transfected cells. Lysates from HEK293 cells cotransfected with TrkA and HA-Bex3 were immunoprecipitated with Trk antibody (203) and Western blots were performed to detect the presence of Bex3 (HA) and Trk in the immunoprecipitates. The expression of TrkA and Bex3 in the lysates was detected with the corresponding antibodies. B, Endogenous Bex3 interacts with TrkA in PC12–6/15 cells. PC12–6/15 cell lysates were subjected to immunoprecipitation using Trk antibody (C-14) or rabbit-IgG as a control. Western blots were performed to detect the presence of Bex3 and Trk in the immunoprecipitates. C, Bex3 interacts with TrkA in PC12–6/15 cells in the presence and in the absence of NGF. PC12–6/15 cells were treated, or not, with NGF (100 ng/ml) for 15 min, and lysates were immunoprecipitated with Bex3 antibody (P-13). Western blots were performed to detect the presence of TrkA and Bex3 in the immunoprecipitates. The expression of TrkA and Bex3 in the lysates was detected with the corresponding antibodies. D, TrkA and Bex3 interact in rat DRGs. DRGs were dissected from E15.5 rat embryos and lysed for 2 h at 4°C. The lysates were immunoprecipitated with Trk antibody (203) or rabbit-IgG as a control, and Western blots were performed to detect the presence of Bex3 and Trk in the immunoprecipitates. E, The TrkA juxtamembrane domain participates in the interaction with Bex3. GST-TrkA recombinant proteins (left) were incubated with S35-labeled Bex3 and resolved in SDS-PAGE. Autoradiography was performed to detect the interaction, and Coomassie staining was used to check GST fusion protein levels (right). F, Bex3 interacts exclusively with the juxtamembrane region of TrkA. Lysates from HEK293 cells cotransfected with GFP-Bex3 and TrkA or TrkA-ΔJM were immunoprecipitated with Trk antibody (203), and Western blots were performed to detect the presence of Bex3 (GFP) and TrkA (RTA) in the immunoprecipitates and lysates. Arrow indicates GFP-Bex3, and asterisks indicate a nonspecific band. G, The C terminus of Bex3 mediates its interaction with TrkA. TrkA and GFP-fused Bex3 expression plasmids (left) were cotransfected in HEK293 cells, and lysates were immunoprecipitated with Trk antibody (203). TrkA-interacting Bex3 was detected using GFP antibodies. Immunoprecipitated TrkA and Bex3 levels in the lysates were detected with the corresponding antibodies (right). Actin was used as a loading control.
trkA and bex3 are coexpressed in most DRG neurons during development
To study the biological significance of Bex3 interaction with TrkA, we investigated the expression pattern of trkA and bex3 in mouse DRG neurons by in situ hybridization during development. bex3 and trkA mRNA expression were observed in DRGs at E11.5 and E12.5 (Fig. 2A). To address whether bex3 mRNA and TrkA protein were present in the same neurons, we simultaneously performed in situ hybridization for bex3 mRNA and immunohistochemistry for TrkA protein, using an antibody (RTA) that specifically recognizes TrkA, but not other Trk neurotrophin receptors (Clary and Reichardt, 1994). DRG neurons expressing bex3 mRNA also expressed TrkA protein (Fig. 2B). These data therefore demonstrate that bex3 mRNA and TrkA protein are coexpressed in most DRG neurons during development.
Figure 2.

Developmental expression pattern of bex3 and trkA. A, In situ hybridization with bex3 and trkA probes performed in consecutive sections of E11.5 and E12.5 mouse embryos. Scale bar, 100 μm. B, Coexpression of bex3 mRNA and TrkA protein during mouse DRG development. Immunofluorescence for TrkA and in situ hybridization for bex3 mRNA at E11.5 and E13.5. Note the colocalization of bex3 mRNA and TrkA in the same cells. Scale bar, 100 μm.
Bex3 protein levels modulate the survival of NGF-dependent DRG neurons and the differentiation of PC12 cells in response to NGF
To identify the potential roles for Bex3 in NGF/TrkA-dependent functions, we assessed the consequences of Bex3 depletion using shRNA in the survival of NGF-dependent DRG neurons and in the differentiation of PC12 cells. First, we tested bex3 mRNA levels in DRG neurons infected with lentivirus expressing either GFP and control shRNA or GFP and Bex3 shRNA (Fig. 3A, left). bex3 mRNA was efficiently downregulated in response to Bex3 shRNA (−67%; Fig. 3A, right). Consequently, Bex3 protein levels were decreased in DRG neurons infected with Bex3 shRNA lentivirus (Fig. 3B). We then assessed the survival of NGF-dependent DRG neurons in response to Bex3 depletion. Cultured sensory neurons were transfected with plasmids that expressed either GFP and control shRNA or GFP and Bex3 shRNA. The presence of GFP allowed transfected neurons to be identified unambiguously. The quantification of apoptotic cells was performed 48–72 h after NGF withdrawal (Fig. 3C, left), showing that Bex3 depletion significantly increased apoptosis in NGF-dependent neurons (+72%; Fig. 3C, right). These findings indicate that Bex3 protein reduction accelerates apoptosis of NGF-deprived sensory neurons.
Figure 3.
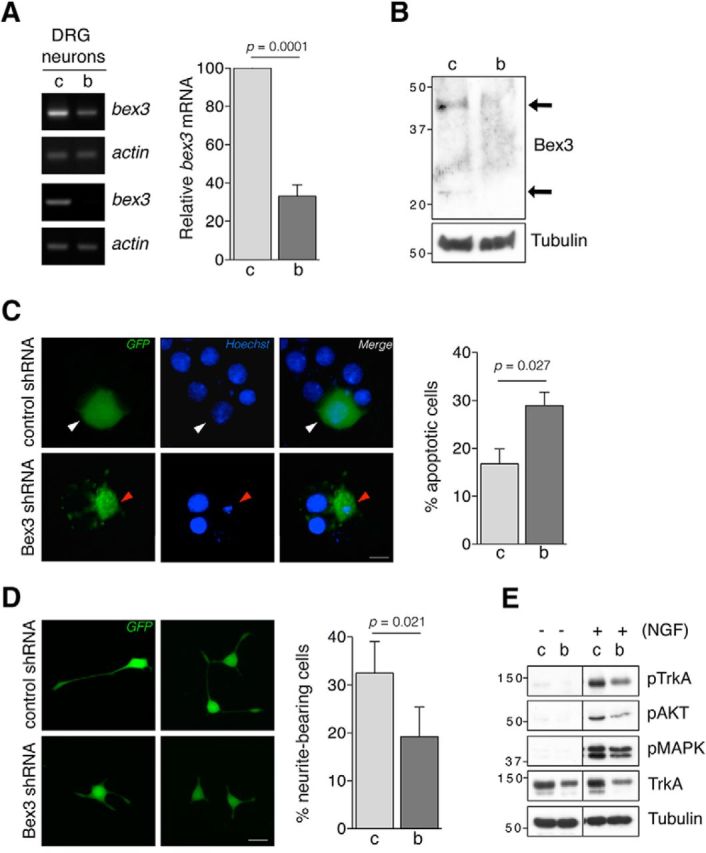
Bex3 protein levels modulate the survival of NGF-dependent neurons and the differentiation of PC12 cells. A, bex3 mRNA downregulation in DRG neurons by shRNA. Cultured DRG neurons were infected at DIV5 with lentivirus-expressing GFP and control shRNA (c) or GFP and Bex3 shRNA (b). RNA was obtained 5 d later, and semiquantitative RT-PCRs were performed as described in Materials and Methods. Actin was used as a control. Two representative RT-PCR experiments are shown (left). Quantification of bex3 and actin mRNA was performed using ImageJ software (right). The intensity of the DNA bands was quantified and normalized to that of actin in the control (100% vs 33.1 ± 5.9%, control vs Bex3 shRNA, respectively). Data are presented as the mean ± SEM (n = 7). The p value was calculated using a two-tailed Student's t test. B, Bex3 protein downregulation in DRG neurons. Cultured DRG neurons were infected as described in A, and extracts were obtained to analyze Bex3 protein by Western blot. A representative Western blot showing a decrease in Bex3 protein is depicted. Tubulin was used as a loading control. Arrows indicate Bex3 monomer and dimer. C, Bex3 protein levels modulate the survival of NGF-dependent neurons in vitro. Cultured DRG neurons were transfected with plasmids expressing either GFP and control shRNA (c) or GFP and Bex3 shRNA (b). Neurons were deprived of NGF for 48–72 h and stained with Hoechst 33342, indicating which ones were apoptotic (red arrowhead) or non-apoptotic (white arrowhead) within GFP-positive population. Left, Representative images of NGF-dependent DRG neurons transfected with control or Bex3 shRNA plasmids. Scale bar, 10 μm. Right, Quantification of apoptosis in GFP-positive cells. Data are shown as the mean ± SEM. The total number of quantified neurons was 249 and 464 in control and Bex3 shRNA-transfected groups respectively from four independent experiments (16.8 ± 3.1% vs 28.9 ± 2.8%, control vs Bex3 shRNA, respectively; right). The p value was calculated using a two-tailed Student's t test. D, Bex3 downregulation impairs neurite outgrowth of PC12. Cells were infected with lentivirus expressing GFP and control shRNA (c) or GFP and Bex3 shRNA (b) for 5 d, and then stimulated with NGF (100 ng/ml) for 72 h. Scale bar, 10 μm. Neurite outgrowth was quantified by assessing the percentage of GFP-positive cells bearing neurites at least twice the length of the cell body. Data are shown as the mean ± SEM. The total number of cells in the control and Bex3 shRNA-infected groups were respectively 204 and 210 from four independent experiments (32.5 ± 6.5% vs 19.3 ± 6.1%, control vs Bex3 shRNA, respectively). The p value was calculated using a two-tailed Student's t test. E, The downregulation of Bex3 impairs NGF-mediated signaling pathways in DRG neurons. DRG neurons were infected as indicated in A, and 5 d later they were treated with NGF (100 ng/ml) for 1 h. Cell extracts were obtained, and active TrkA, Akt, and MAPK were assessed using antibodies that recognize specific phosphorylated residues. Representative blots are shown (n = 6).
To address whether Bex3 also participates in neuronal differentiation in response to NGF, we infected PC12 cells with lentivirus expressing either GFP and control shRNA or GFP and Bex3 shRNA. Four days postinfection, cells were treated with NGF (100 ng/ml) for 72 h and images of GFP-positive cells were obtained (Fig. 3D, left). Quantification of neurite outgrowth showed that Bex3 depletion significantly decreased the percentage of differentiated cells in response to NGF (−59%; Fig. 3D, right). These results indicate that Bex3 levels modulate the differentiation of PC12 cells in response to NGF.
To explore how Bex3 regulates NGF-mediated survival and differentiation, we assessed the signaling pathways activated by NGF in sensory neurons infected with control or Bex3 shRNA lentivirus. We observed that the levels of phosphorylated TrkA, Akt, and MAPK proteins in response to NGF were reduced in neurons infected with Bex3 shRNA lentivirus (Fig. 3E). This was correlated with a clear reduction in total TrkA protein levels in Bex3-depleted neurons (Fig. 3E). Together, these data demonstrate that Bex3 regulates neuronal survival and differentiation, likely by altering NGF-dependent signaling pathways as a consequence of TrkA protein level modulation.
Bex3 downregulation reduces TrkA, but not TrkB, protein levels by decreasing trkA mRNA levels
To further confirm that Bex3 specifically modulated TrkA protein levels, we depleted Bex3 from DRG neurons and found that TrkA levels were significantly reduced (−76%; Fig. 4A). The reduction of TrkA protein levels in cells with Bex3 knocked down might have been caused by increased ubiquitin-mediated degradation of TrkA. Indeed, it has been shown that Bex3 inhibits Smac ubiquitination, affecting the stability of the protein (Yoon et al., 2004). To address whether Bex3 protein regulates TrkA ubiquitination, Bex3 and TrkA were expressed in HEK293 cells. No differences in TrkA ubiquitination were observed in the presence or absence of Bex3 (Fig. 4B). We also examined TrkA levels in DRG neurons infected with control or Bex3 shRNA in the presence of lysosome and/or proteasome inhibitors. TrkA levels were not rescued in the presence of any of the inhibitors when Bex3 was depleted (Fig. 4C). Therefore, the reduction of TrkA protein levels elicited by Bex3 downregulation is not the result of increased protein degradation.
Figure 4.

Bex3 levels regulates TrkA, but not TrkB, protein levels through modulation of trkA mRNA. A, TrkA protein levels are reduced in DRG neurons upon Bex3 downregulation. Cultured DRG neurons were infected as described in Figure 3A, and cell extracts were obtained to analyze TrkA expression by Western blot. Tubulin was used as a loading control. Two representative Western blots are shown (left). Quantification of TrkA levels in response to Bex3 downregulation (right). Western blots were scanned and quantified using ImageJ software. Data were normalized to the amount of tubulin and are presented as the mean ± SEM (100% vs 24.1 ± 5.1%, control vs Bex3 shRNA, respectively; n = 7). The p value was calculated using a two-tailed Student's t test. B, GFP-Bex3 expression does not affect TrkA ubiquitination. HEK293 cells were transfected with TrkA and different amounts of GFP-Bex3. TrkA was immunoprecipitated 24 h after transfection, and TrkA ubiquitination levels were assessed by Western blot. A representative experiment is shown (n = 3). Arrows indicate GFP or GFP-Bex3. C, Bex3 levels do not alter TrkA degradation. Cultured DRG neurons were infected at DIV5 with control shRNA (C) or Bex3 shRNA (B) lentivirus for 5 d, and then treated with chloroquine (10 μm), MG132 (50 μm), or both for 0, 3, or 6 h. Western blot analyses were performed to detect TrkA levels. A representative experiment is shown (n = 3). D, Bex3 downregulation decreases trkA mRNA levels. Cultured DRG neurons were infected as described in Figure 3A. RNA was obtained with TRIzol reagent, cDNA was synthesized, and quantitative PCR was performed as described in Materials and Methods. The relative transcription level of trkA was normalized to that of actin using the 2-ΔΔCt method. Data are presented as the mean ± SEM (1.10 ± 0.1 vs 0.57 ± 0.1, control vs Bex3 shRNA, respectively). Triplicate determinations from three independent experiments were quantified. The p value was calculated using a two-tailed Student's t test. E, bex3 mRNA downregulation in hippocampal neurons infected with lentivirus-expressing rat Bex3 shRNA. Experiments using cultured hippocampal neurons were performed and quantified in a similar way as described in Figure 3A. Data are presented as the mean ± SEM (100% vs 45.0 ± 11.4%, control and Bex3 shRNA, respectively; n = 4). The p value was calculated using a two-tailed Student's t test. F, TrkB protein levels are not altered upon Bex3 downregulation. Extracts from hippocampal neurons infected with control shRNA (c) or Bex3 shRNA (b) lentivirus were analyzed for TrkB expression. Tubulin was used as a loading control. Two representative Western blots are shown (left). TrkB levels (right) were quantified, as described in A. Data are shown as the mean ± SEM (100% vs 124.0 ± 23.9%, control and Bex3 shRNA, respectively; n = 4). The p value was calculated using a two-tailed Student's t test. G, Downregulation of Bex3 does not affect trkB mRNA. RNA was obtained from infected hippocampal neurons, and qPCR was performed as described in D. Data are shown as the mean ± SEM (1.0 ± 0.06 vs 0.98 ± 0.20, control and Bex3 shRNA, respectively). Triplicate determinations from three independent experiments were quantified.
To assess whether trkA mRNA levels were modulated by Bex3, total mRNA was obtained from infected DRG neurons, and its levels were measured by qPCR. A reduction in trkA mRNA levels was observed in Bex3-depleted DRG neurons (−52%; Fig. 4D). These data suggest that Bex3 regulates TrkA at the level of mRNA expression.
To determine whether the effects of Bex3 were specific for TrkA, we examined cultured hippocampal neurons, which express TrkB, but not TrkA, and Bex3 endogenously (Kendall et al., 2003). Neurons infected with lentivirus expressing Bex3 shRNA showed reduced levels of bex3 mRNA (−55%; Fig. 4E). TrkB protein levels were tested using Western blot (Fig. 4F, left). The quantification of several experiments did not reveal significant differences in TrkB protein levels in response to Bex3 depletion (Fig. 4F, right). Furthermore, trkB mRNA levels were not affected by Bex3 reduction in hippocampal neurons (Fig. 4G). Together, these data demonstrate that Bex3 levels specifically modulate TrkA protein through the regulation of trkA mRNA levels.
Bex3 shuttles between the cytoplasm and the nucleus
It has been described that Bex3 is located in the cytoplasm (Mukai et al., 2000; Alvarez et al., 2005). Since Bex3 protein has a NES, we tested whether Bex3 shuttles between the cytoplasm and the nucleus by performing immunofluorescence analyses. HEK293 cells transfected with GFP-Bex3 showed cytoplasmic staining (Fig. 5A, top panels), whereas the Bex3 mutant L90,93A, with a disrupted NES, exhibited mainly a nuclear localization (Fig. 5A, bottom panels). To address whether WT Bex3 was actively excluded from the nucleus, we treated transfected cells with leptomycin B, an inhibitor of nuclear export through CRM1 (Fornerod et al., 1997). Indeed, GFP-Bex3 was mainly located in the nucleus in the presence of leptomycin B (Fig. 5B, top panels). As a control of the specificity of the action of leptomycin B, we monitored the localization of GFP-ARMS, a protein without NES. GFP-ARMS was not accumulated in the nucleus in the presence of leptomycin B (Fig. 5B, bottom panels). Accordingly, Bex3 shuttles between the cytoplasm and the nucleus, and is actively excluded from the nucleus through its NES motif.
Figure 5.
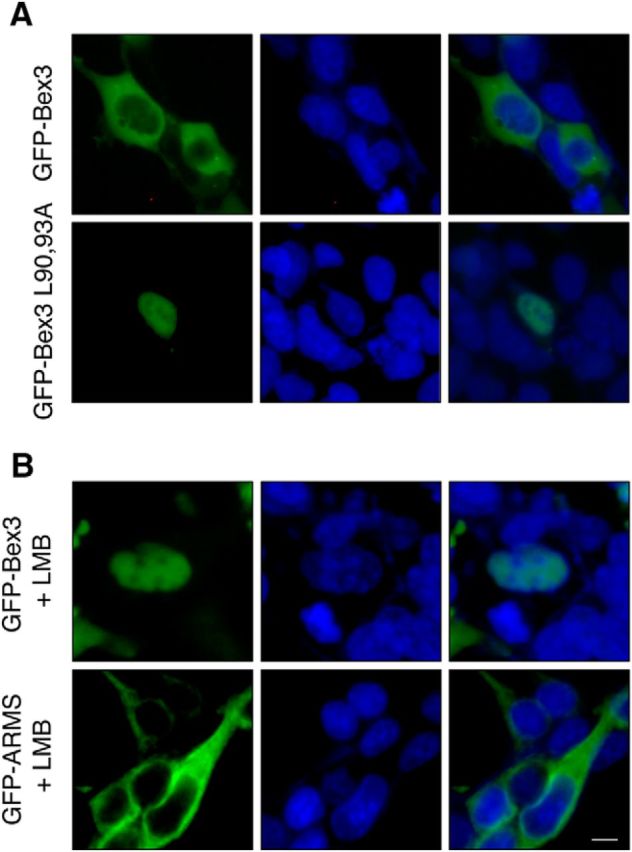
Bex3 shuttles between the cytoplasm and the nucleus. A, Subcellular localization of GFP-Bex3 protein in HEK293 cells. Cells were transfected with GFP-Bex3 or GFP-Bex3 L90,93A expression vectors. Nuclei were visualized with Hoechst 33342 stain. Note the accumulation of GFP-Bex3 L90,93A in the nucleus. B, Bex3 protein is accumulated in the nucleus in the presence of leptomycin B (LMB). HEK293 cells transfected with GFP-Bex3 or GFP-ARMS plasmids were treated with LMB (5 ng/ml) for 6 h. Nuclei were visualized with Hoechst 33342 stain. Note the nuclear accumulation of GFP-Bex3, but not of GFP-ARMS, upon LMB treatment. Scale bar, 20 μm.
Bex3 dimerization enhances transcriptional activity of the mouse trkA gene promoter
Our results demonstrated that Bex3 protein regulated trkA mRNA levels (Fig. 4B). To assess whether Bex3 controlled the transcriptional activity of the trkA promoter, we performed reporter assays using N2a cells, which express Bex3 and have been used previously to map the mouse trkA promoter (Sacristán et al., 1999). First, we transfected N2a cells with the reporter construct pMS8 (see Materials and Methods; Sacristán et al., 1999) and a plasmid that expresses control or Bex3 shRNA (Fig. 6A, top). Bex3 depletion reduced trkA promoter activity significantly (−36%; Fig. 6A, bottom). To determine whether the Bex3 shRNA-induced phenotype was directly attributable to the specific knockdown of Bex3 and not to the off-target effects of the shRNA, a rescue experiment was performed. We designed a GFP-fused Bex3 coding sequence harboring multiple silent mutations that rendered the resulting bex3 mRNA resistant to shRNA knockdown (GFP-Bex3-resistant; see Materials and Methods). The expression of this mRNA not only rescued the effect of endogenous Bex3 depletion but also induced a significant increase in the transcriptional activity of the trkA promoter above basal levels (+103%; Fig. 6A, bottom). These findings indicate that the observed decrease in the transcriptional activity of the trkA promoter in response to Bex3 shRNA is a result of the specific knockdown of Bex3 and suggest that the overexpression of Bex3 enhances trkA promoter activity.
Figure 6.

Bex3 dimerization potentiates the transcriptional activity of the mouse trkA promoter. A, Bex3 depletion reduces mouse trkA promoter activity. Top, Experiments using N2a cells were performed as described in Figure 3A upon infection with lentivirus-expressing control shRNA (c) and mouse Bex3 shRNA (b). Bottom, N2a cells were transfected with the reporter construct pMS8 together with control shRNA (c), Bex3 shRNA (b), or Bex3 shRNA + GFP-Bex3-resistant (R) expression vectors. Data are shown as the mean ± SEM (100% vs 64.3 ± 5.6%, control shRNA vs Bex3 shRNA, respectively); 100% vs 203 ± 12.5%, control shRNA vs Bex3 shRNA + GFP-Bex3-resistant expression vectors, respectively; n = 3–5 using duplicates). The p values were calculated using a paired two-tailed Student's t test. B, GFP-Bex3 expression increases the transcriptional activity of the mouse trkA promoter in N2a cells. Data are shown as the mean ± SEM (100% vs 188.7 ± 16.9%, GFP vs GFP-Bex3, respectively; 188.7 ± 16.9% vs 366 ± 20.5%, GFP-Bex3 vs GFP-Bex3 C117S, respectively; n = 4–9 using duplicates). The p values were calculated using a paired two-tailed Student's t test. Note the increased trkA promoter activity induced by GFP-Bex3, GFP-Bex3 K89R, and GFP-Bex3 C117S protein, and the lack of effect on the trkA promoter upon GFP-Bex3 mutants L90,93A, D1, and D2 expression. C, Representative Western blot showing GFP-Bex3 protein expression in N2a transfected cells, in duplicate. Note the presence of monomers, dimers, and tetramers of GFP-Bex3 proteins, indicated by arrows. D, Bex3 dimerization is required to enhance the transcriptional activity of the trkA promoter. Western blot showing the expression of WT Bex3 and Bex3 C98S, which does not dimerize in N2a cells (left). Reporter assays were performed as described in A (right). Data are shown as the mean ± SEM (205 ± 15.25% vs 123.6 ± 10.34%, GFP-Bex3 vs GFP-Bex3 C98S, respectively; n = 5 using duplicates). The p values were calculated using a paired two-tailed Student's t test. Arrows indicate GFP-Bex3 monomers, dimers, and tetramers. E, Bex3 overexpression increases neurite outgrowth of PC12 cells in response to NGF. Cells were transfected with plasmids expressing GFP, GFP-Bex3, GFP-Bex3 L90,93A, and GFP-Bex3 C98S, and the next day were stimulated with NGF (10 ng/ml) for 96 h. Neurite outgrowth was quantified as described in Figure 3D. Data are shown as the mean ± SEM. The percentage of neurite-bearing cells versus the total number of cells scored in each case was 31.1% and 376 cells for GFP, 43.9% and 275 cells for GFP-Bex3, 35.7% and 281 cells for GFP-Bex3 L90,93A and 33.2% and 230 cells for GFP-Bex3 C98S. The p value was calculated using a paired two-tailed Student's t test. F, Overexpression of Bex3 protein induces apoptosis of NGF-dependent sensory neurons in vitro. Cultured DRG neurons were transfected with plasmids expressing GFP, GFP-Bex3, GFP-Bex3 L90,93A, and GFP-Bex3 C98S. Apoptotic neurons were quantified as described in Figure 3C. Data are shown as the mean ± SEM. The percentage of apoptotic cells versus the total number of cells scored in each case was 36.0% and 257 cells for GFP, 56.0% and 127 cells for GFP-Bex3, 35.6% and 142 cells for GFP-Bex3 L90,93A, and 58.1% and 120 cells for GFP-Bex3 C98S. The p value was calculated using a paired two-tailed Student's t test.
To further address the involvement of Bex3 in the transcriptional activation of the trkA promoter, we performed reporter assays using different Bex3 expression constructs. Transfection of a plasmid expressing WT Bex3 fused to GFP increased trkA promoter activity (+89%; Fig. 6B). Disruption of the nuclear export sequence (Bex3 L90,93A) or deletion of the C terminus (Bex3 D1 and Bex3 D2 mutants) of Bex3 abolished the enhancing effect caused by WT Bex3 (Fig. 6B). A Bex3 K89R mutant, which disrupts the D-box, showed the same activity as WT Bex3; surprisingly, Bex3 C117S, which disrupts the CAAX motif, displayed enhanced activity compared with WT Bex3 (+195%; Fig. 6B). Expression levels of each protein were similar, as determined by Western blot analyses. In addition, we observed the presence of oligomers (dimers and tetramers) of GFP-Bex3, GFP-Bex3 K89R, and GFP-Bex3 C117S (Fig. 6C). The presence of oligomers was directly correlated with the increased trkA promoter activity elicited by the expression of these Bex3 proteins (Fig. 6B,C). Furthermore, the GFP-Bex3 D1, D2, and L90,93A mutants, which failed to increase trkA promoter activity (Fig. 6B), did not show or had greatly reduced numbers of oligomers, despite being expressed at similar or even higher levels than WT Bex3 (Fig. 6C). Together, these data indicate that Bex3 C terminus is necessary to enhance trkA promoter transcription and that oligomerization of Bex3 might be required for this function.
To determine the requirement of Bex3 oligomerization, we took advantage of a previously described Bex3 mutant (C98S) that does not dimerize due to the lack of a disulfide bond (Kimura et al., 2001). First, we confirmed that Bex3 C98S did not dimerize (Fig. 6D, left). Reporter assays showed reduced capability of Bex3 C98S to induce trkA promoter transcription compared with WT Bex3 (−60%; Fig. 6D, right). These results support a role of Bex3 dimerization in enhancing trkA promoter activity.
To assess the physiological consequences of overexpressing different Bex3 proteins, we performed functional assays. First, we analyzed the effect of ectopic expression of Bex3 proteins on PC12 cell differentiation in response to NGF. An increase in neurite outgrowth was observed with GFP-Bex3-expressing cells compared with GFP cells (+41%), whereas the expression of GFP-Bex3 L90,93A, which is mainly nuclear, or GFP-Bex3 C98S, which does not dimerize, had no effect (Fig. 6E). In the absence of NGF, the expression of none of the Bex3 proteins evoked a response. These results suggest that the overexpression of WT Bex3 potentiates PC12 differentiation and that nuclear–cytoplasmic shuttling and dimerization of Bex3 play an important role in neurite outgrowth. Additionally, we assessed survival in NGF-dependent sensory neurons. Interestingly, GFP-Bex3 and GFP-Bex3 C98S overexpression enhanced the percentage of apoptotic neurons deprived of NGF compared with GFP overexpression (+56% and +61%, respectively; Fig. 6F). On the contrary, the overexpression of GFP-Bex3 L90,93A did not promote apoptosis further than GFP overexpression. These data suggest that overexpression and nuclear–cytoplasmic shuttling of Bex3 evokes an apoptotic response in the absence of NGF.
Bex3 binds to the mouse trkA minimal promoter
To identify the region of the trkA promoter where Bex3 exerts its function, we used several trkA reporter constructs (Fig. 7A, top). Previously it has been reported that plasmids pMS10.3 and pMS16 containing 934 and 150 bp, respectively, upstream from the transcription start site, display transcriptional activity similar to pMS8, whereas pMS22, which only includes 43 bp upstream from the start site, has no transcriptional activity (Sacristán et al., 1999). Bex3 overexpression enhanced the transcriptional activity of pMS8, pMS10.3, and pMS16 to a similar extent, whereas pMS22 did not show any activity in response to Bex3 expression (Fig. 7A, bottom). Thus, Bex3 modulation of the trkA promoter activity requires the 150 bp sequence upstream from the trkA transcription start site. To assess whether Bex3 was associated with this sequence, we performed ChIP assays in N2a cells. Upon endogenous Bex3 immunoprecipitation, PCR assays yielded a DNA fragment extending from nucleotide −143 to −19 that was not observed when control IgGs were used (Fig. 7B). Furthermore, these data were confirmed in PC12 cells using qChIP assays. A sevenfold increase in trkA promoter sequence amount was observed when endogenous Bex3 protein was immunoprecipitated compared with control conditions (Fig. 7C). Thus, Bex3 binds to trkA promoter sequences within a 150 bp region upstream from the transcription start site.
Figure 7.

Bex3 increases the mouse trkA gene promoter activity and binds close to the transcriptional start site. A, Mapping the region of the mouse trkA promoter modulated by GFP-Bex3. The transcriptional activities of different reporter plasmids containing different sequences from the 5′ region of the mouse trkA promoter (top) are shown. Data are presented as the mean ± SEM. Data are reported as GFP versus GFP-Bex3, respectively (100% vs 188.7 ± 16.9%, for pMS8; 99.95 ± 28.75% vs 214.8 ± 19.01%, for pMS10.3; 113.7 ± 28.75% vs 196.2 ± 20.49%, for pMS16; 14.05 ± 2.2% vs 16.54 ± 3.17%, for pMS22; n = 4–9 using duplicates). The p values were calculated using a paired two-tailed Student's t test. B, Endogenous Bex3 protein binds to the mouse trkA promoter in N2a cells. ChIP analysis of N2a cell lysates using anti-Bex3-ProtG-agarose. Goat IgG-ProtG-agarose was used as a control. A representative experiment is shown (n = 5). C, Quantitative ChIP assays in PC12 cells. Cell lysates were immunoprecipitated using the anti-Bex3-ProtG-agarose or goat IgG-ProtG-agarose as a control. qPCRs were performed using the oligonucleotides described in Materials and Methods. The p value was calculated using a two-tailed Student's t test (n = 3 by triplicate).
Bex3 enhances the transcriptional activation of the trkA promoter in response to NGF
It has previously been reported that NGF increases trkA expression in PC12 cells and in cholinergic basal forebrain neurons (Miller et al., 1991; Holtzman et al., 1992; Li et al., 1995; Zhou et al., 1995). To address whether Bex3 potentiates NGF-mediated trkA transcriptional activity, we performed reporter assays in PC12 cells. GFP or GFP-Bex3 plasmids were cotransfected with pMS8 in PC12 cells to assess trkA promoter activity in response to NGF. The basal trkA promoter activity (i.e., in the absence of NGF) was enhanced in PC12 cells overexpressing GFP-Bex3 (+60%; Fig. 8A), an effect similar to that observed in N2a cells (Fig. 6A,B). No increase in promoter activation above basal levels was detected after 30 min of NGF treatment in GFP- or GFP-Bex3-expressing PC12 cells (Fig. 8A). NGF treatment for 18 h in GFP-transfected cells led to an increase in the activation of the trkA promoter compared with nonstimulated cells (+22%; Fig. 8A), which is in agreement with previous reports (Miller et al., 1991; Zhou et al., 1995). Furthermore, Bex3 overexpression in PC12 cells stimulated with NGF for 18 h elicited an increase in the activation of the trkA promoter compared with nonstimulated cells (+39%; Fig. 8A). Therefore, Bex3 protein overexpression enhanced the effect of NGF on the trkA promoter.
Figure 8.

Bex3 enhances NGF-mediated trkA promoter activity. A, Enhanced NGF-mediated trkA promoter activity by Bex3 overexpression. PC12 cells were transfected with the following plasmids: reporter construct pMS8 and GFP or GFP-Bex3. On the following day, cells were treated, or not, with NGF (100 ng/ml) for 0.5 and 18 h, and reporter assays were performed with all samples in parallel. Data are shown as the mean ± SEM (100% vs 160 ± 13.58%, GFP vs GFP-Bex3, respectively; 100% vs 121.7 ± 9.94%, no NGF vs 18 h NGF, respectively; 160 ± 13.58% vs 222.3 ± 27.91%, no NGF vs 18 h NGF, respectively; n = 3 using duplicates). The p values were calculated using a paired two-tailed Student's t test. Note the significant increase in trkA promoter activity in samples expressing GFP-Bex3 treated with NGF for 18 h. B, Bex3 dimers accumulate in the nucleus in response to NGF. Endogenous Bex3 was monitored by Western blot in nuclear fractions of PC12–6/15 cells stimulated, or not, with NGF (100 ng/ml) for 15 min. Phospho-Histone3 (pH3) was used as a nuclear marker. Arrows indicate Bex3 monomers and dimers.
It has been described that NGF increases Bex1 export from the nucleus (Vilar et al., 2006). Bex3 also shuttles between cytoplasm and nucleus (Mukai et al., 2000; Alvarez et al., 2005). To address whether NGF alters Bex3 levels in the nucleus, we obtained nuclear fractions from PC12-6/15 cells expressing endogenous Bex3 stimulated or not with NGF. Bex3 dimers were increased in the nucleus of NGF-stimulated cells (Fig. 8B). Therefore, these data suggest that NGF induces accumulation of Bex3 in the nucleus as a dimer.
Discussion
Here we show that Bex3 protein specifically regulates TrkA protein expression by enhancing trkA promoter activity. Consequently, Bex3 regulates the survival of NGF-dependent DRG neurons and the differentiation of PC12 cells. These conclusions are supported by several lines of experimental evidence, as follows: (1) a reduction in Bex3 levels using shRNA leads to a specific decrease in TrkA, but not TrkB, protein levels in neurons; (2) the reduction in TrkA protein levels is paralleled by a decrease in trkA mRNA; (3) the activity of the trkA promoter is directly regulated by Bex3 protein levels; (4) ChIP assays indicate that Bex3 associates with the trkA promoter; (5) NGF-induced trkA transcription is enhanced and accompanied by Bex3 accumulation and dimerization in the nucleus; and (6) Bex3 protein levels modulate NGF-mediated signaling, neurite outgrowth, and NGF-dependent survival.
The results presented here demonstrate that Bex3 protein specifically regulates TrkA protein levels, an effect that is mediated via the modulation of trkA mRNA transcription. These data are consistent with the coincident temporal expression patterns of TrkA and Bex3 in developing sensory neurons (Fig. 2), which is comparable to that observed for several transcription factors known to influence trkA mRNA expression (Huang et al., 1999; Lei et al., 2001; Ma et al., 2003). A puzzling result of Bex3 depletion was that the decrease in trkA mRNA was 50% (Fig. 4B), whereas TrkA protein levels decreased by 75% (Fig. 4A). This result may be due to Bex3 having additional functions aside from regulating trkA promoter activity. It has been reported that Bex3 associates with eIF3 protein (Tong et al., 2003), which plays an important role in translation initiation; it is therefore possible that Bex3 regulates trkA mRNA translation and that this may contribute to the strong reduction in TrkA protein levels observed when Bex3 is depleted.
The regulation of trkA promoter activity by the effect of Bex3 was addressed using gain-of-function and loss-of-function experiments. The overexpression of WT Bex3 resulted in an increase in trkA promoter activity, whereas a reduction of endogenous Bex3 impaired it. Other members of the Bex family, such as Bex2 and Bex4, have also been implicated in the transcriptional regulation of different genes (Han et al., 2005; Chien et al., 2008). We have identified the C terminus of Bex3 as a key region for the regulation of the trkA promoter, as well as the trkA promoter sequences close to the transcription start site through which Bex3 acts. An unexpected result was that WT Bex3 and mutants capable of enhancing the activation of the trkA promoter formed oligomers, whereas Bex3 mutant proteins, unable to increase trkA promoter activity, behaved as monomers. Bex3 is the only member of Bex family that shows oligomerization (Mukai et al., 2000; Alvarez et al., 2005). The direct correlation observed between the ability of Bex3 mutant proteins to form oligomers and to activate trkA promoter suggests that Bex3 oligomerization is important for Bex3 functions. The increased activation of the trkA promoter elicited by the overexpression of Bex3 C117S was particularly remarkable. This mutation in Bex3 prevents lipid modification, thus limiting anchoring of this mutant protein to membranes (Kim et al., 2004). In addition, the Bex3 C117S-impaired interaction with TrkA may facilitate its access to the nucleus, promoting enhanced trkA transcription compared with WT Bex3. In contrast, Bex3 C98S, which has been previously shown not to dimerize (Kimura et al., 2001), lacked any increase in the transcriptional activity of trkA promoter (Fig. 6D). Thus, Bex3 dimerization is crucial to its function on the trkA promoter.
The basal expression of trkA mRNA is regulated by several different factors (Lei and Parada, 2007). To date, only HAND transcription factors have been implicated in the expression of trkA mRNA in response to NGF (Doxakis et al., 2008). The effect mediated by HAND proteins is modest, suggesting the participation of additional proteins. Our results indicate for the first time that Bex3 enhances both the basal transcriptional activation of the trkA promoter as well as the NGF-mediated upregulation of trkA mRNA levels. Furthermore, Bex3 expression is enhanced by NGF treatment in oligodendrocytes (Mukai et al., 2000), and NGF modulates the expression of different genes, including several transcription factors (Yang et al., 2013). It is tempting to speculate that NGF would first increase Bex3 protein levels and dimerization, and that Bex3 would subsequently act on the trkA promoter.
How does Bex3 shuttle between the cytoplasm and the nucleus? It has been reported that Bex1 is mainly accumulated in the nucleus (Alvarez et al., 2005; Vilar et al., 2006), but in cells expressing p75, and in response to NGF, Bex1 rapidly relocates from the nucleus to the cytoplasm (Vilar et al., 2006). By contrast, Bex3 is mainly located in the cytoplasm (Fig. 5; Mukai et al., 2000; Alvarez et al., 2005). Bex3 trafficking may be controlled by the following different mechanisms: (1) Bex3 is efficiently excluded from the nucleus via the NES motif (Mukai et al., 2000; Alvarez et al., 2005; Fig. 5); (2) Bex3 interacts with 14-3-3ε (Kimura et al., 2001), and hence it is possible that 14-3-3 binding might exclude Bex3 from the nucleus, whereas in response to different stimuli Bex3 is released and enters the nucleus (it has been reported that 14-3-3 proteins sequester c-Abl in the cytosol, and in response to genotoxic agents 14-3-3 is phosphorylated and releases c-Abl, which translocates to the nucleus; Yoshida et al., 2005); (3) our results indicating that Bex3 C117S elicited a higher transcriptional activity of the trkA promoter than WT Bex3 (Fig. 6B) suggest that WT Bex3 may be kept outside of the nucleus because it is attached to membranes, where it can easily interact with Trk receptors; therefore, the binding of TrkA to Bex3 might regulate Bex3 function on the trkA promoter; and (4) a NGF-dependent interaction of Bex3 with p75 has been described in oligodendrocytes (Mukai et al., 2000), and it is known that, upon cleavage, the intracellular domain (ICD) of p75 enters the nucleus in response to NGF (Kanning et al., 2003; Frade, 2005). Accordingly, it is possible that Bex3 translocates into the nucleus associated with p75 ICD. Additional experiments will be required to address these questions.
A consequence of Bex3 increasing trkA mRNA expression is the regulation of NGF-mediated signaling, and hence neuronal survival and differentiation, which are dependent on the NGF/TrkA system during development. In agreement with this, the overexpression of WT Bex3, but not a mutant that is mainly nuclear (Bex3 L90,93A) or a mutant that does not dimerize (Bex3 C98S), enhances neurite outgrowth (Fig. 6E). This supports the idea that nuclear–cytoplasmic shuttling and dimerization of Bex3 are crucial events in promoting neurite outgrowth. Interestingly, the overexpression of WT Bex3 or Bex3 C98S, but not Bex3 L90,93A, enhances the apoptosis of sensory neurons that occurs in response to NGF deprivation (Fig. 6F). This might be explained by the effects of Bex3 expression inducing apoptosis through p75 neurotrophin receptor that have been previously reported (Mukai et al., 2000, 2002; Kimura et al., 2001). Under pathological conditions, TrkA expression has been proposed to be a predictive marker of favorable outcome in low-stage neuroblastomas, leading to enhanced patient survival and terminal neuronal differentiation in the presence of NGF or apoptosis in the absence of NGF, whereas TrkA expression is low or absent in most advanced-stage tumors, which do not undergo differentiation or regression (Brodeur et al., 2009). On the other hand, it has been reported that trkA mRNA levels are strongly reduced in different areas of postmortem brains of patients with AD (Boissiere et al., 1997; Hock et al., 1998). The reduced expression of TrkA contributes to impaired NGF signaling and a decreased retrograde transport of NGF in cholinergic neurons in the basal forebrain and striatum. Future studies should determine the levels of Bex3 in different neuroblastoma cell lines and in samples from AD patients to assess the possible roles of Bex3 in these disease states.
Footnotes
This work was supported by Ministerio de Ciencia e Innovación Grant BFU2008-00162, by Ministerio de Economía y Competitividad Grant (BFU2011-22898 to J.C.A. and BFU2013-39394-P to P.P.), by a Marie Curie International Reintegration Grant within the European Community 7th Framework Programme, by the Conserjería de Educación Grant SA074A08, and by a Conserjería de Sanidad de Junta Castilla y León grant to J.C.A. This work was in part supported by the FP7-PAINCAGE integrative project. D.M.-Z. is funded by a grant from the Spanish “Instituto de Saludo Carlos III” PI1202847. L.C. has been the recipient of a Predoctoral Fellowship from the University of Salamanca. J.C.A. was a “Ramón y Cajal” Investigator from the University of Salamanca and a NARSAD 2009 Young Investigator Awardee. We thank Louis Reichardt for the RTA antibody and Javier Herrero for technical support.
References
- Alvarez E, Zhou W, Witta SE, Freed CR. Characterization of the Bex gene family in humans, mice, and rats. Gene. 2005;357:18–28. doi: 10.1016/j.gene.2005.05.012. [DOI] [PubMed] [Google Scholar]
- Arévalo JC, Wu SH. Neurotrophin signaling: many exciting surprises! Cell Mol Life Sci. 2006;63:1523–1537. doi: 10.1007/s00018-006-6010-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arévalo JC, Yano H, Teng KK, Chao MV. A unique pathway for sustained neurotrophin signaling through an ankyrin-rich membrane spanning protein. EMBO J. 2004;23:2358–2368. doi: 10.1038/sj.emboj.7600253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arévalo JC, Wu SH, Takahashi T, Zhang H, Yu T, Yano H, Milner TA, Tessarollo L, Ninan I, Arancio O, Chao MV. The ARMS/Kidins220 scaffold protein modulates synaptic transmission. Mol Cell Neurosci. 2010;45:92–100. doi: 10.1016/j.mcn.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benn SC, Costigan M, Tate S, Fitzgerald M, Woolf CJ. Developmental expression of the TTX-resistant voltage-gated sodium channels Nav1.8 (SNS) and Nav1.9 (SNS2) in primary sensory neurons. J Neurosci. 2001;21:6077–6085. doi: 10.1523/JNEUROSCI.21-16-06077.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett DL, Averill S, Clary DO, Priestley JV, McMahon SB. Postnatal changes in the expression of the trkA high-affinity NGF receptor in primary sensory neurons. Eur J Neurosci. 1996;8:2204–2208. doi: 10.1111/j.1460-9568.1996.tb00742.x. [DOI] [PubMed] [Google Scholar]
- Boissiere F, Faucheux B, Ruberg M, Agid Y, Hirsch EC. Decreased TrkA gene expression in cholinergic neurons of the striatum and basal forebrain of patients with Alzheimer's disease. Exp Neurol. 1997;145:245–252. doi: 10.1006/exnr.1997.6443. [DOI] [PubMed] [Google Scholar]
- Brodeur GM, Minturn JE, Ho R, Simpson AM, Iyer R, Varela CR, Light JE, Kolla V, Evans AE. Trk receptor expression and inhibition in neuroblastomas. Clin Cancer Res. 2009;15:3244–3250. doi: 10.1158/1078-0432.CCR-08-1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AL, Kay GF. Bex1, a gene with increased expression in parthenogenetic embryos, is a member of a novel gene family on the mouse X chromosome. Hum Mol Genet. 1999;8:611–619. doi: 10.1093/hmg/8.4.611. [DOI] [PubMed] [Google Scholar]
- Chang BB, Persengiev SP, de Diego JG, Sacristan MP, Martin-Zanca D, Kilpatrick DL. Proximal promoter sequences mediate cell-specific and elevated expression of the favorable prognosis marker TrkA in human neuroblastoma cells. J Biol Chem. 1998;273:39–44. doi: 10.1074/jbc.273.1.39. [DOI] [PubMed] [Google Scholar]
- Chao MV. Neurotrophins and their receptors: a convergence point for many signalling pathways. Nat Rev Neurosci. 2003;4:299–309. doi: 10.1038/nrn1078. [DOI] [PubMed] [Google Scholar]
- Chien J, Narita K, Rattan R, Giri S, Shridhar R, Staub J, Beleford D, Lai J, Roberts LR, Molina J, Kaufmann SH, Prendergast GC, Shridhar V. A role for candidate tumor-suppressor gene TCEAL7 in the regulation of c-Myc activity, cyclin D1 levels and cellular transformation. Oncogene. 2008;27:7223–7234. doi: 10.1038/onc.2008.360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clary DO, Reichardt LF. An alternatively spliced form of the nerve growth factor receptor TrkA confers an enhanced response to neurotrophin 3. Proc Natl Acad Sci U S A. 1994;91:11133–11137. doi: 10.1073/pnas.91.23.11133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doxakis E, Howard L, Rohrer H, Davies AM. HAND transcription factors are required for neonatal sympathetic neuron survival. EMBO Rep. 2008;9:1041–1047. doi: 10.1038/embor.2008.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faria TN, LaRosa GJ, Wilen E, Liao J, Gudas LJ. Characterization of genes which exhibit reduced expression during the retinoic acid-induced differentiation of F9 teratocarcinoma cells: involvement of cyclin D3 in RA-mediated growth arrest. Mol Cell Endocrinol. 1998;143:155–166. doi: 10.1016/S0303-7207(98)00127-0. [DOI] [PubMed] [Google Scholar]
- Fornerod M, Ohno M, Yoshida M, Mattaj IW. CRM1 is an export receptor for leucine-rich nuclear export signals. Cell. 1997;90:1051–1060. doi: 10.1016/S0092-8674(00)80371-2. [DOI] [PubMed] [Google Scholar]
- Frade JM. Nuclear translocation of the p75 neurotrophin receptor cytoplasmic domain in response to neurotrophin binding. J Neurosci. 2005;25:1407–1411. doi: 10.1523/JNEUROSCI.3798-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo A, Simone DA, Stone LS, Fairbanks CA, Wang J, Elde R. Developmental shift of vanilloid receptor 1 (VR1) terminals into deeper regions of the superficial dorsal horn: correlation with a shift from TrkA to Ret expression by dorsal root ganglion neurons. Eur J Neurosci. 2001;14:293–304. doi: 10.1046/j.0953-816x.2001.01665.x. [DOI] [PubMed] [Google Scholar]
- Han C, Liu H, Liu J, Yin K, Xie Y, Shen X, Wang Y, Yuan J, Qiang B, Liu YJ, Peng X. Human Bex2 interacts with LMO2 and regulates the transcriptional activity of a novel DNA-binding complex. Nucleic Acids Res. 2005;33:6555–6565. doi: 10.1093/nar/gki964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hock C, Heese K, Müller-Spahn F, Hulette C, Rosenberg C, Otten U. Decreased trkA neurotrophin receptor expression in the parietal cortex of patients with Alzheimer's disease. Neurosci Lett. 1998;241:151–154. doi: 10.1016/S0304-3940(98)00019-6. [DOI] [PubMed] [Google Scholar]
- Holtzman DM, Li Y, Parada LF, Kinsman S, Chen CK, Valletta JS, Zhou J, Long JB, Mobley WC. p140trk mRNA marks NGF-responsive forebrain neurons: evidence that trk gene expression is induced by NGF. Neuron. 1992;9:465–478. doi: 10.1016/0896-6273(92)90184-F. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci. 2001;24:677–736. doi: 10.1146/annurev.neuro.24.1.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Zang K, Schmidt A, Saulys A, Xiang M, Reichardt LF. POU domain factor Brn-3a controls the differentiation and survival of trigeminal neurons by regulating Trk receptor expression. Development. 1999;126:2869–2882. doi: 10.1242/dev.126.13.2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanning KC, Hudson M, Amieux PS, Wiley JC, Bothwell M, Schecterson LC. Proteolytic processing of the p75 neurotrophin receptor and two homologs generates C-terminal fragments with signaling capability. J Neurosci. 2003;23:5425–5436. doi: 10.1523/JNEUROSCI.23-13-05425.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendall SE, Ryczko MC, Mehan M, Verdi JM. Characterization of NADE, NRIF and SC-1 gene expression during mouse neurogenesis. Brain Res Dev Brain Res. 2003;144:151–158. doi: 10.1016/S0165-3806(03)00166-4. [DOI] [PubMed] [Google Scholar]
- Kim AJ, Lee CS, Schlessinger D. Bex3 associates with replicating mitochondria and is involved in possible growth control of F9 teratocarcinoma cells. Gene. 2004;343:79–89. doi: 10.1016/j.gene.2004.08.031. [DOI] [PubMed] [Google Scholar]
- Kimura MT, Irie S, Shoji-Hoshino S, Mukai J, Nadano D, Oshimura M, Sato TA. 14-3-3 is involved in p75 neurotrophin receptor-mediated signal transduction. J Biol Chem. 2001;276:17291–17300. doi: 10.1074/jbc.M005453200. [DOI] [PubMed] [Google Scholar]
- Kong H, Boulter J, Weber JL, Lai C, Chao MV. An evolutionarily conserved transmembrane protein that is a novel downstream target of neurotrophin and ephrin receptors. J Neurosci. 2001;21:176–185. doi: 10.1523/JNEUROSCI.21-01-00176.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanier J, Quina LA, Eng SR, Cox E, Turner EE. Brn3a target gene recognition in embryonic sensory neurons. Dev Biol. 2007;302:703–716. doi: 10.1016/j.ydbio.2006.10.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei L, Parada LF. Transcriptional regulation of Trk family neurotrophin receptors. Cell Mol Life Sci. 2007;64:522–532. doi: 10.1007/s00018-006-6328-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei L, Ma L, Nef S, Thai T, Parada LF. mKlf7, a potential transcriptional regulator of TrkA nerve growth factor receptor expression in sensory and sympathetic neurons. Development. 2001;128:1147–1158. doi: 10.1242/dev.128.7.1147. [DOI] [PubMed] [Google Scholar]
- Lei L, Laub F, Lush M, Romero M, Zhou J, Luikart B, Klesse L, Ramirez F, Parada LF. The zinc finger transcription factor Klf7 is required for TrkA gene expression and development of nociceptive sensory neurons. Genes Dev. 2005;19:1354–1364. doi: 10.1101/gad.1227705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Holtzman DM, Kromer LF, Kaplan DR, Chua-Couzens J, Clary DO, Knüsel B, Mobley WC. Regulation of TrkA and ChAT expression in developing rat basal forebrain: evidence that both exogenous and endogenous NGF regulate differentiation of cholinergic neurons. J Neurosci. 1995;15:2888–2905. doi: 10.1523/JNEUROSCI.15-04-02888.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou X, Yano H, Lee F, Chao MV, Farquhar MG. GIPC and GAIP form a complex with TrkA: a putative link between G protein and receptor tyrosine kinase pathways. Mol Biol Cell. 2001;12:615–627. doi: 10.1091/mbc.12.3.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Merenmies J, Parada LF. Molecular characterization of the TrkA/NGF receptor minimal enhancer reveals regulation by multiple cis elements to drive embryonic neuron expression. Development. 2000;127:3777–3788. doi: 10.1242/dev.127.17.3777. [DOI] [PubMed] [Google Scholar]
- Ma L, Lei L, Eng SR, Turner E, Parada LF. Brn3a regulation of TrkA/NGF receptor expression in developing sensory neurons. Development. 2003;130:3525–3534. doi: 10.1242/dev.00582. [DOI] [PubMed] [Google Scholar]
- Marmigère F, Montelius A, Wegner M, Groner Y, Reichardt LF, Ernfors P. The Runx1/AML1 transcription factor selectively regulates development and survival of TrkA nociceptive sensory neurons. Nat Neurosci. 2006;9:180–187. doi: 10.1038/nn1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Zanca D, Barbacid M, Parada LF. Expression of the trk proto-oncogene is restricted to the sensory cranial and spinal ganglia of neural crest origin in mouse development. Genes Dev. 1990;4:683–694. doi: 10.1101/gad.4.5.683. [DOI] [PubMed] [Google Scholar]
- Miller FD, Mathew TC, Toma JG. Regulation of nerve growth factor receptor gene expression by nerve growth factor in the developing peripheral nervous system. J Cell Biol. 1991;112:303–312. doi: 10.1083/jcb.112.2.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukai J, Hachiya T, Shoji-Hoshino S, Kimura MT, Nadano D, Suvanto P, Hanaoka T, Li Y, Irie S, Greene LA, Sato TA. NADE, a p75NTR-associated cell death executor, is involved in signal transduction mediated by the common neurotrophin receptor p75NTR. J Biol Chem. 2000;275:17566–17570. doi: 10.1074/jbc.C000140200. [DOI] [PubMed] [Google Scholar]
- Mukai J, Shoji S, Kimura MT, Okubo S, Sano H, Suvanto P, Li Y, Irie S, Sato TA. Structure-function analysis of NADE: identification of regions that mediate nerve growth factor-induced apoptosis. J Biol Chem. 2002;277:13973–13982. doi: 10.1074/jbc.M106342200. [DOI] [PubMed] [Google Scholar]
- Sacristán MP, de Diego JG, Bonilla M, Martín-Zanca D. Molecular cloning and characterization of the 5′ region of the mouse trkA proto-oncogene. Oncogene. 1999;18:5836–5842. doi: 10.1038/sj.onc.1202963. [DOI] [PubMed] [Google Scholar]
- Smeyne RJ, Klein R, Schnapp A, Long LK, Bryant S, Lewin A, Lira SA, Barbacid M. Severe sensory and sympathetic neuropathies in mice carrying a disrupted Trk/NGF receptor gene. Nature. 1994;368:246–249. doi: 10.1038/368246a0. [DOI] [PubMed] [Google Scholar]
- Tong X, Xie D, Roth W, Reed J, Koeffler HP. NADE (p75NTR-associated cell death executor) suppresses cellular growth in vivo. Int J Oncol. 2003;22:1357–1362. [PubMed] [Google Scholar]
- Vilar M, Murillo-Carretero M, Mira H, Magnusson K, Besset V, Ibáñez CF. Bex1, a novel interactor of the p75 neurotrophin receptor, links neurotrophin signaling to the cell cycle. EMBO J. 2006;25:1219–1230. doi: 10.1038/sj.emboj.7601017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang FC, Tan T, Huang T, Christianson J, Samad OA, Liu Y, Roberson D, Davis BM, Ma Q. Genetic control of the segregation of pain-related sensory neurons innervating the cutaneous versus deep tissues. Cell Rep. 2013;5:1353–1364. doi: 10.1016/j.celrep.2013.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano H, Lee FS, Kong H, Chuang J, Arevalo J, Perez P, Sung C, Chao MV. Association of Trk neurotrophin receptors with components of the cytoplasmic dynein motor. J Neurosci. 2001;21:RC125. doi: 10.1523/JNEUROSCI.21-03-j0003.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon K, Jang HD, Lee SY. Direct interaction of Smac with NADE promotes TRAIL-induced apoptosis. Biochem Biophys Res Commun. 2004;319:649–654. doi: 10.1016/j.bbrc.2004.05.043. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Yamaguchi T, Natsume T, Kufe D, Miki Y. JNK phosphorylation of 14-3-3 proteins regulates nuclear targeting of c-Abl in the apoptotic response to DNA damage. Nat Cell Biol. 2005;7:278–285. doi: 10.1038/ncb1228. [DOI] [PubMed] [Google Scholar]
- Yu T, Calvo L, Anta B, López-Benito S, Southon E, Chao MV, Tessarollo L, Arévalo JC. Regulation of trafficking of activated TrkA is critical for NGF-mediated functions. Traffic. 2011;12:521–534. doi: 10.1111/j.1600-0854.2010.01156.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Valletta JS, Grimes ML, Mobley WC. Multiple levels for regulation of TrkA in PC12 cells by nerve growth factor. J Neurochem. 1995;65:1146–1156. doi: 10.1046/j.1471-4159.1995.65031146.x. [DOI] [PubMed] [Google Scholar]