

Abstract
New memories are thought to be solidified (consolidated) by de novo synthesis of proteins in the period subsequent to learning. This view stems from the observation that protein synthesis inhibitors, such as anisomycin (ANI), administered during this consolidation period cause memory impairments. However, in addition to blocking protein synthesis, intrahippocampal infusions of ANI cause the suppression of evoked and spontaneous neural activity, suggesting that ANI could impair memory expression by simply preventing activity-dependent brain functions. Here, we evaluated the influence of intrahippocampal ANI infusions on allocentric spatial navigation using the Morris water maze, a task well known to require dorsal hippocampal integrity. Young, adult male Sprague Dawley rats were implanted with bilateral dorsal hippocampal cannulae, and their ability to learn the location of a hidden platform was assessed before and following infusions of ANI, TTX, or vehicle (PBS). Before infusion, all groups demonstrated normal spatial navigation (training on days 1 and 2), whereas 30 min following infusions (day 3) both the ANI and TTX groups showed significant impairments in allocentric navigation, but not visually cued navigation, when compared with PBS-treated animals. Spatial navigational deficits appeared to resolve on day 4 in the ANI and TTX groups, 24 h following infusion. These results show that ANI and TTX inhibit the on-line function of the dorsal hippocampus in a similar fashion and highlight the importance of neural activity as an intervening factor between molecular and behavioral processes.
SIGNIFICANCE STATEMENT The permanence of memories has long thought to be mediated by the production of new proteins, because protein synthesis inhibitors can block retrieval of recently learned information. However, protein synthesis inhibitors may have additional detrimental effects on neurobiological function. Here we show that anisomycin, a commonly used protein synthesis inhibitor in memory research, impairs on-line brain function in a way similar to an agent that eliminates electrical neural activity. Since disruption of neural activity can also lead to memory loss, it may be that memory permanence is mediated by neural rehearsal following learning.
Keywords: anisomycin, behavioral neuroscience, hippocampus, neural activity, protein synthesis inhibitor
Introduction
Memory consolidation is an active process that enables recently learned material to be stored in a stable state for long periods of time. Clear evidence for this process has been shown in experiments in which neuropharmacological manipulations following learning can significantly modulate subsequent retrieval (McGaugh, 2000). It is thought that the process that ultimately mediates memory stability is the production of new proteins (de novo protein synthesis), because translational inhibitors such as anisomycin (ANI) and cycloheximide administered during the consolidation period eliminate memory retrieval for recently learned events (Davis and Squire, 1984; Kandel, 2001; Nader, 2003). As a result, it is now almost an axiom that de novo protein synthesis is an essential component in memory consolidation.
The validity of the de novo protein synthesis hypothesis is directly dependent on the assumption that abolishing protein synthesis has no secondary effect on the neurons that support a memory trace. Indeed it has been previously argued that ANI, in particular, has no additional detrimental neurobiological effects other than its inhibition of protein synthesis (Davis and Squire, 1984). However, there is recent and mounting evidence that appears inconsistent with the idea of de novo protein synthesis as the substrate for memory consolidation. For example, spontaneous recovery of memories previously blocked by ANI has been repeatedly reported (Flexner and Flexner, 1967; Radyushkin and Anokhin, 1999). Furthermore, intracranial ANI has also been shown to catastrophically alter neuromodulatory tone, which by itself can interfere with memory consolidation (Canal et al., 2007; Qi and Gold, 2009; Sadowski et al., 2011). More recently, we have demonstrated that intrahippocampal microinfusions of ANI suppressed spontaneous and evoked local field potentials as well as hippocampal unit activity (Sharma et al., 2012). Importantly, the maximal doses of ANI used in these evaluative studies were equal to, or less than, the concentrations used in previous behavioral studies of memory. These collective results question the role of protein synthesis inhibition, per se, as the mediator of previously observed behavioral deficits using ANI.
By using behavior to assay for the on-line function of the brain, we previously showed that ventral (but not dorsal) hippocampal infusions of ANI impaired unconditioned anxiety-related responses in the elevated plus maze and shock-probe burying tests (Greenberg et al., 2014). These behavioral deficits were similar to those previously observed following direct suppression of the hippocampus using sodium channel blockers such as TTX and GABAA receptor agonists such as muscimol (Degroot and Treit, 2004; McEown and Treit, 2010). In the present study, we adopt this same behavioral approach and assess the influence of dorsal hippocampal applications of both ANI and TTX on spatial navigation in the Morris water maze. We demonstrate that both manipulations impair allocentric navigation in a similar fashion and thus provide further evidence that ANI acts to impair on-line brain function.
Materials and Methods
Subjects.
Data were obtained from 47 young, adult male Sprague Dawley rats weighing between 200 and 350 g. All rats were housed individually in 47 × 25 × 20.5 cm polycarbonate cages for the duration of the experiment with a 12 h light/dark cycle (lights on at 0600 h). Food and water were provided ad libitum. All experimental procedures followed guidelines published by the Canadian Council on Animal Care and received local ethical approval from the Biological Sciences Animal Policy and Welfare Committee of the University of Alberta.
Drugs.
Isoflurane (anesthetic; Halocarbon Product), Marcaine (preoperative analgesic; Hospira), Rimadyl (a postoperative anti-inflammatory; Pfizer), and Hibitane (postoperative antibiotic cream; Pfizer) were used during implantation surgery. During the testing protocol, anisomycin (Sigma-Aldrich; 100 μg/μl dissolved in 10 N HCl and brought to volume with PBS) and TTX (Abcam; 10 ng/μl dissolved in PBS) were used for dorsal hippocampus infusions.
Surgical implantation.
Animals were initially anesthetized in an enclosed 31 × 11 × 11 cm Plexiglas gas chamber filled with an isoflurane (4%), N2O (1.0 ml/min), and O2 (0.5 ml/min) vapor mix. Once the righting reflex was lost, animals were placed in a nose cone supplying a vapor mixture of isoflurane (1.5%), N2O (1.0 ml/min), and O2 (0.5 ml/min), which was maintained for the duration of the procedure. Before surgery animals were administered 0.9% saline (3 ml, i.p.) for hydration and Marcaine (0.3 ml, s.c., near the sagittal scalp line) as a local anesthetic. Animals were placed in a stereotaxic frame (Kopf Instruments) and implanted with stainless steel 22 gauge 5 mm guide cannulae (Plastics One) bilaterally into the dorsal hippocampus (AP, −3.3 mm; ML, ±2.2 mm; DV 3.0 mm) using a rat brain atlas (Paxinos and Watson, 1998). Guide cannulae were secured to the skull with dental acrylic anchored to a triad of jeweler's screws fixed in the skull. Dummy probes were inserted into the guide cannulae to prevent obstruction of the tracts. Postoperative Rimadyl (0.2 ml, i.p.) was administered to reduce inflammation during recovery. Hibitane was applied to the incision site postoperatively if there were any signs of infection. All animals were given at least 7 d of recovery time before any behavioral procedures and were handled for 10 min per day for 3 consecutive days before testing.
Training procedures.
A black circular maze (diameter 152 cm, depth 46 cm) was filled up to 4 cm below the top with tap water (21 ± 0.2°C SEM). The platform (diameter 17 cm, depth 46 cm) was located 3 cm below the surface of the water and painted to match the pool, concealing its location from the surface. Cued trials included placing a bright object (a small, inverted orange traffic cone) on the platform, which was easily discernible from the surface of the water. The platform was located in one of the four ordinal (NW, NE, SW, SE) positions, 38 cm away from the outside edge of the pool. Platform location was changed at the beginning of each day, but did not change across trials during a single day, similar to the matching-to-place task version of the water maze (Morris and Frey, 1997). Platform locations and starting points were randomized after each week of testing to control for systematic location bias and quadrant effects (Vorhees and Williams, 2006).
The animal was placed at a random cardinal starting position (N, W, S, E) at the beginning of each of the four trials during a single day. The animal was released in the pool facing the outer rim and was given 60 s to find the hidden platform. If the animal did not reach the platform in 60 s, it was guided there. The animal remained on the platform for 20 s before being returned to its cage for 2 min until the next trial. Before each cued trial, the platform was moved to a random ordinal location with the cue object placed upon it, but otherwise ran as previously described.
Measures of latency and path were assessed using an overhead contrast tracking device (VP200 advanced tracker) and further analyzed with HVS Water Tracking System software. The room was lit with soft diffuse lighting.
Each animal was run for four trials per day over the course of 4 consecutive days. Infusions of ANI, TTX, or PBS were made 30 min before testing on day 3. Two cued trials were also introduced on days 3 and 4, following the completion of the normal trials. Once the animal was released at the designated starting point, the experimenter returned to a set position in the room and remained there until the full trial had completed. Experimenters were blind to the experimental conditions of the animals. Defecates were removed from the water with a net after each trial and the pool was thoroughly cleaned after every session of testing.
Infusion procedures.
On day 3 of training, animals were randomly assigned to one of the three conditions: ANI (100 μg/μl dissolved in 10 N HCl and brought to volume with PBS), TTX (10 ng/μl dissolved in PBS), or a vehicle control group (PBS). It is important to note that the dosage of ANI was equal to or lower than that used in classic studies on protein synthesis inhibition and memory studies (Schafe et al., 1999; Nader et al., 2000). Solutions were infused into both dorsal hippocampal hemispheres through 26 gauge stainless steel internal cannulae attached to a 10 μl Hamilton syringe with polyethylene tubing at a rate of 0.5 μl/min for 2 min (total infusion volume of 1 μl per hemisphere) using a double infusion pump (Harvard Apparatus 22). Internal infusion cannulae were left in the guide cannulae for one additional minute to allow for diffusion of the drug out of the cannulae. Infusions were performed on awake animals that were lightly restrained by an experimenter. Behavioral testing occurred 30 min following the start of the infusion. This corresponds to the time period of maximal neural suppression for both TTX (Lorenzini et al., 1996) and ANI (Sharma et al., 2012).
Histology.
Following all experimentation, rats were deeply anesthetized using isoflurane inhalation and overdosed using intraperitoneal injections of urethane (1 ml of 0.67 g/ml). Following the loss of the righting reflex and lack of reflex withdrawal responses to toe pad pressure, animals were transcardially perfused with saline followed by formalin (4%). Brains were then extracted and stored in a sucrose (30%) formalin (4%) solution for a minimum of 24 h before slicing. Brains were flash frozen with compressed CO2 and sliced into 60-μm-thick coronal slices using a rotary microtome (1320 Microtome; Leica). The slices were mounted on gel-coated slides (Fisher Scientific), thionin stained (Sigma-Aldrich), and coverslipped using Permount (Fisher Scientific). Animals with cannulae placements above the pyramidal cell layer near CA1 were excluded from analysis. No lesions were found in any of the slices.
Data analysis.
Latency and path length were recorded using an overhead contrast camera (VP200 advance tracker), which relayed information to software (HVS Water Maze), producing an excel file output. Latency and path length averages of each trial per day and group were calculated using Microsoft Excel and were analyzed using SPSS (IBM Statistics 20). The path traveled by the animals was reconstructed using our overhead contrast tracking system and plotted using OriginPro 8.5 (OriginLab). Velocity was calculated by the tracking system as a function of distance over time of each trial.
A mixed-measures ANOVA was used to assess differences between trials within each day, between treatment groups, and to test for interactions. To reduce data noise and measurement error, the average of trials 2, 3, and 4 for each day and group were calculated. This measure was used to assess allocentric navigation performance after the first exposure to the platform location, which was further analyzed using another mixed-measures ANOVA. A one-way ANOVA was used to assess the effects of different treatment conditions on each day, as well as any differences in velocity between conditions.
Results
Consistent with previous research using the matching-to-place version of the Morris water maze (Vorhees and Williams, 2006), all groups demonstrated learning across trials on days 1 and 2 (Fig. 1). This is shown in terms of decreases in the latency and path length to find the hidden platform across the four trials. Using a mixed-measures 3 (Condition) × 4 (Trial) ANOVA design, we found significant decreases in performance measures across trials on day 1 (Latency: F(3,132) = 10.5, p < 0.001; Path: F(3,132) = 7.825, p < 0.001) and day 2 (Latency: F(2.76,121.62) = 30.99, p < 0.001; Path: F(2.614,115.014) = 34.117, p < 0.001), with no significant differences between groups on day 1 (Latency: F(2,44) = 0.375, p = 0.689; Path: F(2,44) = 0.51, p = 0.950) and day 2 (Latency: F(2,44) = 1.170, p = 0.320; Path: F(2,44) = 1.111, p = 0.338) or a significant interaction on day 1 (Latency: F(6,132) = 0.500, p = 0.807; Path: F(6,132) = 0.914, p = 0.487) or day 2 (Latency: F(4.968,109.301) = 0.474, p = 0.794; Path: F(4.717,103.773) = 0.200, p = 0.956). These data suggest that the initial performance of each randomized group was comparable before infusions.
Figure 1.
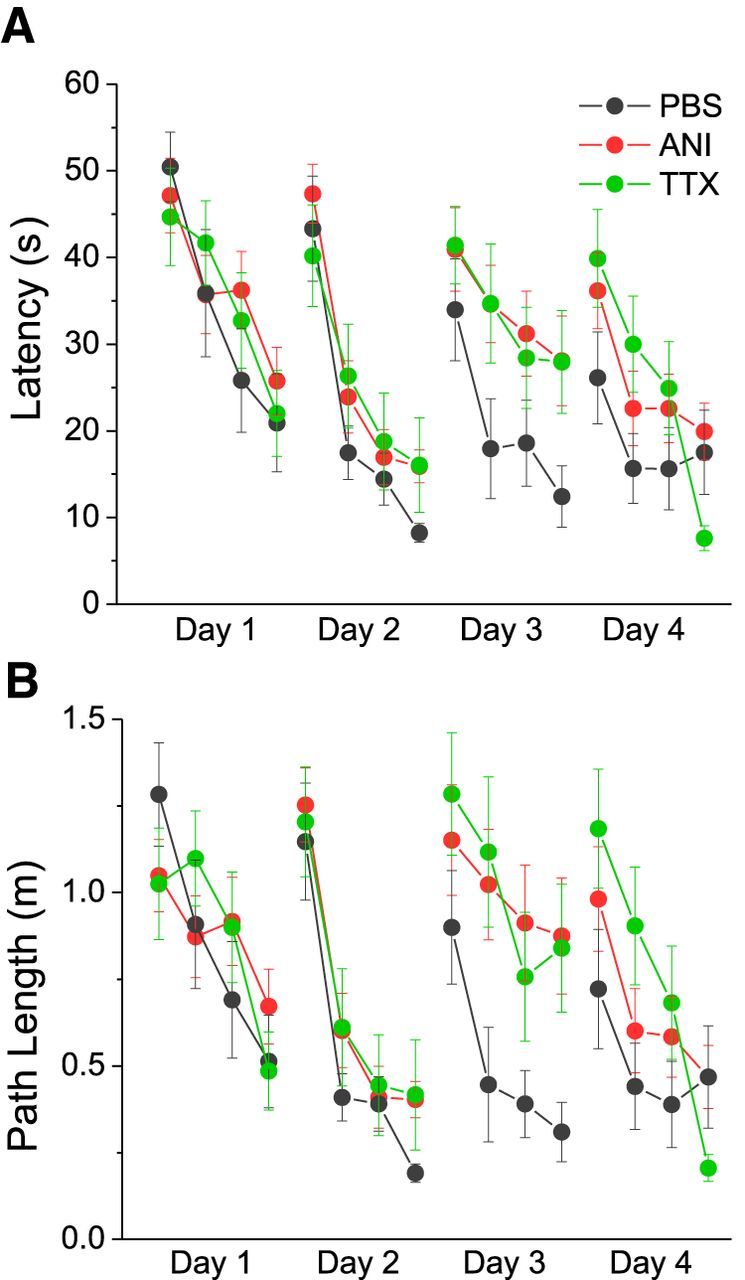
Averaged group performance (latency (A) and path length (B)) measures in the match-to-place version of the Morris water maze. Performance across trials shows learning across all groups (black symbols and lines represent the PBS group, red symbols and lines represent the ANI group, and green symbols and lines represent the TTX group) on days 1, 2, and 4, but not on day 3, for the ANI and TTX groups. Error bars reflect SEM.
As partially described, performance on day 2 across trials showed similar results to those on day 1, although all groups showed faster latencies overall. Indeed despite moving the platform to a novel location, performance measures appeared to optimize more rapidly across trials on day 2 compared with day 1, as shown in Figure 1. This suggests that all rats learned the overall contingencies of the task quite rapidly.
While the different groups showed similar performance on days 1 and 2, following infusions on day 3, there were marked differences apparent between the groups across trials. There were significant effects of both trial (Latency: F(3,132) = 5.897, p = 0.001; Path: F(3,132) = 5.298, p = 0.002) and group (Latency: F(2,44) = 3.464, p = 0.040; Path: F(2,44) = 4.445, p = 0.017), with no significant interaction (Latency: F(6,132) = 0.265, p = 0.952; Path: F(6,132) = 0.441, p = 0.850). These data (Fig. 1) show that TTX and ANI groups were impaired in terms of learning the new location of the platform since they did not show decreased latencies and path lengths (distance) on trials 2–4 as they previously did on day 2. In contrast, the PBS group did show marked improvement in latency and distance measures during trials 2–4, similar to their performance on day 2. Statistically, this was confirmed using post hoc comparisons (Tukey HSD). ANI and PBS groups were significantly different on both latency and distance measures (Latency: p = 0.043; Path length: p = 0.022) and TTX and PBS groups were significantly different for distance (p = 0.040) but not latency (p = 0.095) measures. ANI and TTX groups were not different from each other on either measure (Latency: p = 0.992; Path length p = 0.998). These results suggest that both ANI and TTX groups were equally impaired on the Morris water maze task following infusions.
The difference between groups appeared to resolve on day 4 because performance was comparable across groups and trials. Specifically, there were no significant effects for group (Latency: F(2,44) = 1.520, p = 0.230; Path length: F(2,44) = 1.343, p = 0.272), although there was a significant effect of trial (Latency: F(3,132) = 9.983, p < 0.001; Path: F(3,132) = 11.037, p < 0.001) without any significant interaction (Latency: F(6,132) = 1.702, p = 0.125; Path: F(6,132) = 1.755, p = 0.113). All groups performed similarly across trials on day 4, suggesting that the ANI and TTX groups recovered from the impairment observed on day 3.
Given that all groups appeared to show optimized performance on trials 2–4 on day 2 (and certainly in the control PBS group on day 3), we elected to compare performance by averaging measures for trials 2–4 and comparing across groups on each day (Vorhees and Williams, 2006). As shown in Figure 2, there were significant group differences (Latency: F(2,44) = 5.147, p = 0.010; Path: F(2,44) = 5.258, p = 0.009) as assessed using a 3 (Condition) × 4 (Day) mixed-measures ANOVA for these data. Using a Tukey HSD post hoc test, significant differences were found between the ANI and PBS groups (Latency: p = 0.012; Path length: p = 0.013) and the TTX and PBS groups (Latency: p = 0.030; Path length: p = 0.021), whereas the ANI and TTX groups did not differ (Latency: p = 0.997; Path length: p = 0.990).
Figure 2.
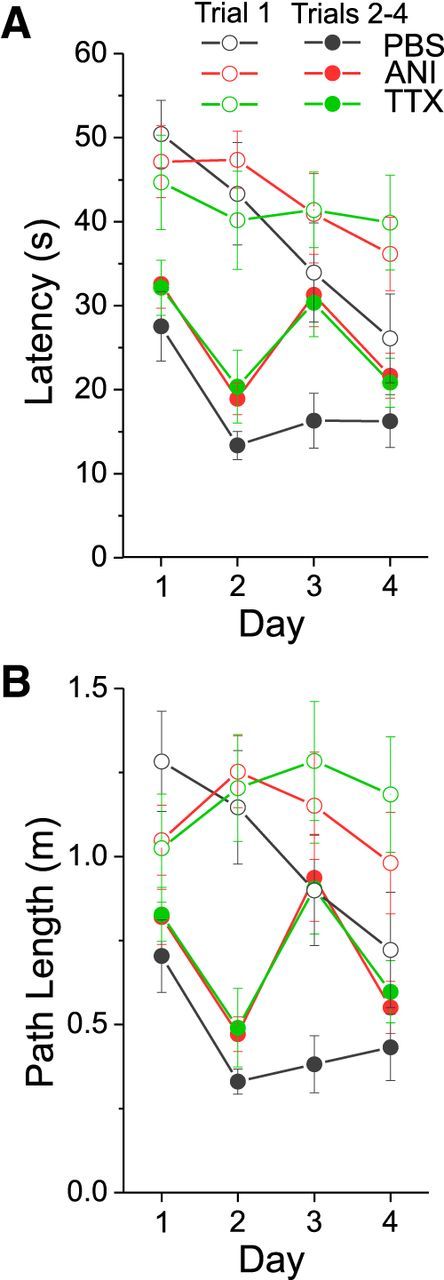
Averaged latency (A) and path length (B) measures across groups when grouping performance measures on trial 1 and trials 2–4 across all days. Black symbols and lines represent the PBS group, red symbols and lines represent the ANI group, and green symbols and lines represent the TTX group. Open symbols reflect average data for trial 1, and closed symbols reflect the average for trials 2–4. ANI and TTX groups demonstrate poor navigation on day 3 when comparing trial 1 to trials 2–4. Error bars reflect SEM.
Given these results, we conducted separate analyses (one-way ANOVAs) for each day for these same amalgamated measures of spatial navigation performance. All groups performed similarly on day 1 (Latency: F(2,44) = 0.570, p = 0.570; Path: F(2,44) = 0.465, p = 0.631), day 2 (Latency: F(2,44) = 1.452, p = 0.245; Path: F(2,44) = 1.156, p = 0.324), and day 4 (Latency: F(2,44) = 0.833, p = 0.442; Path: F(2,44) = 0.689, p = 0.507), but showed significant differences on day 3, i.e., post infusion (Latency: F(2,44) = 3.762, p = 0.031; Path: F(2,44) = 4.743, p = 0.014). As evaluated with a Tukey HSD post hoc test, ANI and TTX groups did not differ from each other on day 3 (Latency: p = 0.984; Path: p = 0.983), but there were significant group performance differences between ANI and PBS groups (Latency: p = 0.032; Path: p = 0.015) and between TTX and PBS groups (Path: p = 0.045). These results suggest that both ANI and TTX groups show similar impairments in the performance of the allocentric version of the Morris water maze, a task that has been repeatedly shown to depend upon the intact operation of the dorsal hippocampus.
We also reconstructed the swim paths across the different groups of animals following infusions on day 3. As exemplified in Figure 3, all groups tended to swim in a circular fashion around the outside circumference of the pool on the first trial. However, after finding or being placed on the platform during the first trial, the swim paths of control (PBS) animals on subsequent trials (i.e., on trials 2–4) showed a more direct navigational strategy. In contrast, and as shown in Figure 3, animals in the ANI or TTX infusion groups continued to demonstrate swim paths that were similar to those on the first trial. This pattern is suggestive of impaired spatial navigation, a behavior known to require on-line dorsal hippocampal function (Hostetter and Thomas, 1967; Good and Honey, 1997).
Figure 3.
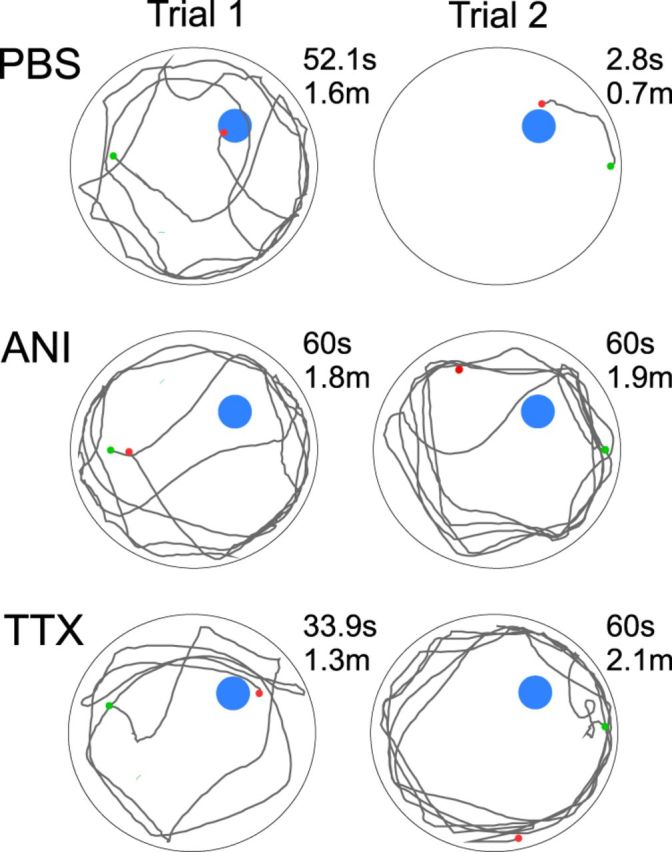
Swim paths of representative animals from each group on day 3. Start and end points are indicated as green and red dots, respectively. Trial 1 paths indicate a lack of knowledge of the location of the hidden platform that is uniform across groups. Trial 2 paths indicate a strong location preference for the PBS animal, which continues to be absent in ANI and TTX animals.
To assure that these deficits were not due to other factors, we assessed the average swim speed as well as the ability of all groups of animals to navigate to a visually cued platform (Fig. 4). There were no statistically significant differences in velocity between groups on any day (Day 1: F(2,213) = 2.413, p = 0.092; Day 2: F(2,213) = 1.524, p = 0.220; Day 3: F(2,213) = 0.254, p = 0.776; Day 4: (F(2,213) = 2.880, p = 0.058), as determined using a one-way ANOVA. There were also no significant differences between groups when measuring the average performance of the two cued trials on day 3 (Latency: F(2,44) = 0.646, p = 0.529; Path: F(2,44) = 1.884, p = 0.164) or on day 4 (Latency: F(2,44) = 1.648, p = 0.204; Path: F(2,44) = 2.050, p = 0.141). These results suggest that the impairment in ANI and TTX groups observed post infusion was not due to performance deficits or nonhippocampal-dependent learning.
Figure 4.
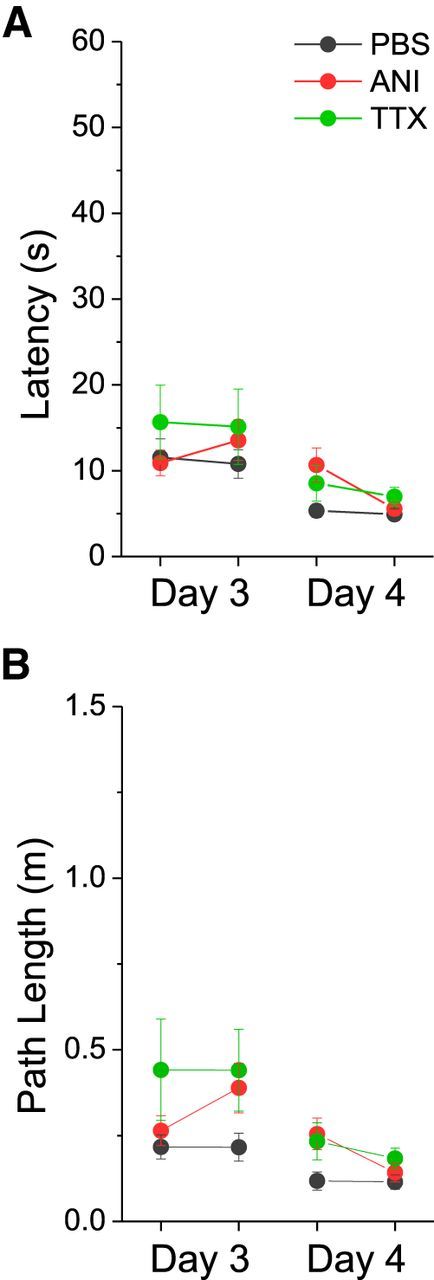
Averaged group performance (latency (A) and path length (B)) measures in the cued version of the water maze on days 3 and 4. Black symbols and lines represent the PBS group, red symbols and lines represent the ANI group, and green symbols and lines represent the TTX group. No differences were observed between groups. Error bars reflect SEM.
Discussion
We show here that dorsal hippocampal infusion of anisomycin disrupts the ability of rats to navigate in the allocentric (but not the cued) version of the Morris water maze. Given the similarity of these impairments to those observed with similar infusions of TTX (present study), together with our previous observation that similar doses of ANI disrupt electrical activity in the hippocampus (Sharma et al., 2012), we suggest that ANI impairs on-line brain function by inactivating neural tissue. This behavioral disruption dissipated 24 h following the infusions of both ANI and TTX, which suggests a similar transient and nonpermanent dysfunction of neural operations with both manipulations. Certainly, our results caution that the behavioral influences of translational inhibitors cannot be considered independently of their effects on neural activity. Indeed, they also imply that prior behavioral results using intracerebral applications of ANI [and other protein synthesis inhibitors (PSIs)] could very well be the result of neural silencing as opposed to suppression of protein synthesis per se. Moreover, they highlight the need to completely reconsider the use of PSIs in future behavioral paradigms that test for the importance of de novo protein synthesis in memory consolidation (or reconsolidation).
The importance of neural activity in memory consolidation
Coordinated neural activity has been previously shown to play a fundamental role in memory consolidation. The disruption or abolition of neural activity has been shown to produce profound memory deficits in studies using postlearning manipulations such as lesions (Morris et al., 1982; Morris, 1989; Zelikowsky et al., 2012); electroconvulsive shock (Duncan, 1949; Misanin et al., 1968); and neural inactivators such as TTX, lidocaine, or muscimol (Brioni et al., 1990; Packard and McGaugh, 1996; Ambrogi Lorenzini et al., 1999; Holt and Maren, 1999; Chang and Gold, 2003; Klement et al., 2005). More recent studies show that similarly timed disruptions of particular patterns of coordinated activity (such as sharp wave ripples and theta oscillations) can also result in memory deficits (Girardeau et al., 2009; Ego-Stengel and Wilson, 2010; Sauseng et al., 2010; Jadhav et al., 2012). These studies demonstrate that neural activity is an essential component in the consolidation period and may be paramount for the recall deficits previously ascribed to protein synthesis inhibition.
One way in which postlearning activity patterns may support long-term memory formation is through continuing plasticity among coactive local and even extended networks of neurons (Buzsáki, 1989). It is well known, for example, that neural stimulants and excitatory neuromodulators delivered in the postlearning period are effective in enhancing future recall and that these effects are likely activity dependent (McGaugh, 1999, 2000). Indeed boosting endogenous activity patterns, such as the slow oscillation through transcranial electrical stimulation, has also been shown to improve the consolidation of recently learned hippocampal-dependent memories (Marshall et al., 2006). Furthermore, impairing synaptic plasticity by the use of NMDA receptor blockers has been shown to have no effect on the learning of a new spatial location in a water maze, but did significantly alter the recall of its location after 24 h (McDonald et al., 2005). Moreover, when NMDA receptors were blocked immediately after learning, a similar recall deficit was found. These results, taken in tandem, suggest that an activity-dependent process requiring NMDA receptor-mediated neurotransmission is likely operational during the postlearning period and could be a causal element of memory consolidation.
De novo protein synthesis hypothesis of memory consolidation
Given that the suppressive effects of translational inhibitors, including ANI, on protein synthesis are likely inseparable from their detrimental effects on neurobiological processes (Radyushkin and Anokhin, 1999; Canal et al., 2007; Rudy, 2008; Qi and Gold, 2009; Sadowski et al., 2011; Sharma et al., 2012; Greenberg et al., 2014), what does this imply for the de novo protein theory of memory? That is, are new proteins necessary for supporting long-term memory changes? While this idea has good face validity based on the assumption that the neural instantiation of memory is mediated via maintained synaptic plasticity, which would include morphological changes, it is the evaluation of this hypothesis via the use of intracerebral PSIs that is problematic. In a commentary about the dependence of memory consolidation on protein synthesis, Rudy et al. (2006) concluded that the intracerebral application of ANI has many detrimental effects on neurobiological function, both in the immediate and long term (including the induction of apoptotic cell death) that would preclude a definitive answer in this respect. While permanent cell loss induced by ANI is certainly a concern for neurobiologists, our present findings suggest that the immediate effects are unlikely to involve this process since normal hippocampal function returned after 24 h. Regardless, it is surprising, given the numerous and varied demonstrations of neurobiological dysfunctions induced by translational inhibitors, that a re-evaluation of the de novo protein synthesis hypothesis has not taken place sooner.
To bypass the suppressive effects that direct intracerebral applications of ANI has on neural activity, systemic infusions of ANI might be considered. However, there is a great deal of evidence demonstrating that intraperitoneal infusions of ANI cause visceral malaise in rats, as measured through a lack of voluntary eating behavior (Davis and Squire, 1984; Hernandez and Kelley, 2004). Indeed the broad spectrum of adverse side effects produced from ANI must be overcome before attempting to resolve the true cause of the observed amnestic behaviors, which further complicates its use in memory research. In addition to malaise, systemic ANI can also affect the distribution of brain activity states during sleep (Rojas-Ramírez et al., 1977). Given the importance of off-line activity during sleep to memory consolidation (Buzsáki and Draguhn, 2004; Diekelmann and Born, 2010), any modulation of patterned neural activity, regardless of mechanism, is likely to impact behavioral measures.
While it is certainly the case that present-day molecular approaches to examine the longevity of memory have moved toward targeting individual proteins as opposed to broader disruption of all protein products, it is also clear that these specific manipulations can have ramifications on neural activity as well. For example, the antisense oligonucleotide knockdown of a specific tyrosine kinase receptor (MuSK) in the hippocampus that interfered with memory consolidation was also shown to disrupt hippocampal theta in vitro (Garcia-Osta et al., 2006). Further down the signaling pathway, the downregulation of CREB, an essential element in the long-term expression of synaptic potentiation, results in memory impairments, but may do so by simply reducing neuronal excitability (Han et al., 2006; Jancic et al., 2009). Conversely, the upregulation of CREB enhances memory at the same time that it increases neuronal excitability (Lopez de Armentia et al., 2007; Josselyn, 2010). Recently, we have shown that even the targeted post-translational interference of specific memory-related isoforms of PKC (PKMζ and/or PKCλ) also disrupt hippocampal activity (LeBlancq et al., 2014). These findings suggest that the ultimate mechanism of action when manipulating memory-related proteins may simply be mediated through effects on neural activity.
How do translational inhibitors disrupt neural activity?
In our previous work, we suggested that protein synthesis is essential for proper neural activity (Sharma et al., 2012). It is still unclear how translational inhibition might impact intrinsic and synaptic mechanisms of excitability. One potential mechanism is through the impairment of general cellular operations, particularly by blocking the function or production of protein classes necessary for cellular function. Protein classes such as cytoskeletal, metabolic, signaling, enzymatic, and membrane-associated peptides are all likely individually necessary for neuronal excitability, of which the impairment of any single class could prevent cellular activity. Indeed it seems implausible to suggest that a translational blocker, such as ANI, at the customary high doses found in past behavioral research studies, would leave these cellular operations intact. Another mechanism through which ANI could impair neural activity is through mitochondrial disruption. Bath applications of ANI to primary cortical cultures have been shown to decrease ATP production, suggesting that ANI specifically disrupts mitochondrial function (Zhou et al., 2008). Given that neural activity is richly dependent on ATP, it is likely that any manipulation that disrupts mitochondrial function would prominently affect neural signaling.
General conclusion
Our current findings demonstrate that ANI, similar to TTX, disrupts on-line brain function in a temporary manner. Given that both manipulations can impair the future expression of memory (i.e., retrieval), it is tempting to suggest that ANI may produce these effects via its suppression of activity, as opposed to its inhibition of protein synthesis. Certainly, our results support the idea that the divide between molecular biology and behavioral processes cannot be considered without the intervening factor of neural activity.
Footnotes
This work was supported by the Natural Science and Engineering Council of Canada grants 249861 to C.T.D. and 38726 to D.T., J.D.D. was supported by a Branch Out Neurological Foundation Student Summership. We acknowledge Wesley Vuong, Biruk Negash, and Lisa Rimstad for their contribution to data collection; Michelle Yeung for surgical training; and Shelbie LeBlancq, Lisa Rimstad, and Claire Scavuzzo for editorial comments. We would like to dedicate this article to the memory of Dr. Cornelius (Case) Vanderwolf.
The authors declare no competing financial interests.
References
- Ambrogi Lorenzini CG, Baldi E, Bucherelli C, Sacchetti B, Tassoni G. Neural topography and chronology of memory consolidation: a review of functional inactivation findings. Neurobiol Learn Mem. 1999;71:1–18. doi: 10.1006/nlme.1998.3865. [DOI] [PubMed] [Google Scholar]
- Brioni JD, Decker MW, Gamboa LP, Izquierdo I, McGaugh JL. Muscimol injections in the medial septum impair spatial learning. Brain Res. 1990;522:227–234. doi: 10.1016/0006-8993(90)91465-S. [DOI] [PubMed] [Google Scholar]
- Buzsáki G. Two-stage model of memory trace formation: a role for “noisy” brain states. Neuroscience. 1989;31:551–570. doi: 10.1016/0306-4522(89)90423-5. [DOI] [PubMed] [Google Scholar]
- Buzsáki G, Draguhn A. Neuronal oscillations in cortical networks. Science. 2004;304:1926–1929. doi: 10.1126/science.1099745. [DOI] [PubMed] [Google Scholar]
- Canal CE, Chang Q, Gold PE. Amnesia produced by altered release of neurotransmitters after intraamygdala injections of a protein synthesis inhibitor. Proc Natl Acad Sci U S A. 2007;104:12500–12505. doi: 10.1073/pnas.0705195104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Q, Gold PE. Intra-hippocampal lidocaine injections impair acquisition of a place task and facilitate acquisition of a response task in rats. Behav Brain Res. 2003;144:19–24. doi: 10.1016/S0166-4328(03)00063-9. [DOI] [PubMed] [Google Scholar]
- Davis HP, Squire LR. Protein synthesis and memory: a review. Psychol Bull. 1984;96:518–559. doi: 10.1037/0033-2909.96.3.518. [DOI] [PubMed] [Google Scholar]
- Degroot A, Treit D. Anxiety is functionally segregated within the septo-hippocampal system. Brain Res. 2004;1001:60–71. doi: 10.1016/j.brainres.2003.10.065. [DOI] [PubMed] [Google Scholar]
- Diekelmann S, Born J. The memory function of sleep. Nat Rev Neurosci. 2010;11:114–126. doi: 10.1038/nrn2762. [DOI] [PubMed] [Google Scholar]
- Duncan CP. The retroactive effect of electroshock on learning. J Comp Physiol Psychol. 1949;42:32–44. doi: 10.1037/h0058173. [DOI] [PubMed] [Google Scholar]
- Ego-Stengel V, Wilson MA. Disruption of ripple-associated hippocampal activity during rest impairs spatial learning in the rat. Hippocampus. 2010;20:1–10. doi: 10.1002/hipo.20707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flexner JB, Flexner LB. Restoration of expression of memory lost after treatment with puromycin. Proc Natl Acad Sci U S A. 1967;57:1651–1654. doi: 10.1073/pnas.57.6.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Osta A, Tsokas P, Pollonini G, Landau EM, Blitzer R, Alberini CM. MuSK expressed in the brain mediates cholinergic responses, synaptic plasticity, and memory formation. J Neurosci. 2006;26:7919–7932. doi: 10.1523/JNEUROSCI.1674-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girardeau G, Benchenane K, Wiener SI, Buzsáki G, Zugaro MB. Selective suppression of hippocampal ripples impairs spatial memory. Nat Neurosci. 2009;12:1222–1223. doi: 10.1038/nn.2384. [DOI] [PubMed] [Google Scholar]
- Good M, Honey RC. Dissociable effects of selective lesions to hippocampal subsystems on exploratory behavior, contextual learning, and spatial learning. Behav Neurosci. 1997;111:487–493. doi: 10.1037/0735-7044.111.3.487. [DOI] [PubMed] [Google Scholar]
- Greenberg A, Ward-Flanagan R, Dickson CT, Treit D. ANI inactivation: unconditioned anxiolytic effects of anisomycin in the ventral hippocampus. Hippocampus. 2014;24:1308–1316. doi: 10.1002/hipo.22312. [DOI] [PubMed] [Google Scholar]
- Han MH, Bolaños CA, Green TA, Olson VG, Neve RL, Liu RJ, Aghajanian GK, Nestler EJ. Role of cAMP response element-binding protein in the rat locus ceruleus: regulation of neuronal activity and opiate withdrawal behaviors. J Neurosci. 2006;26:4624–4629. doi: 10.1523/JNEUROSCI.4701-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez PJ, Kelley AE. Long-term memory for instrumental responses does not undergo protein synthesis-dependent reconsolidation upon retrieval. Learn Mem. 2004;11:748–754. doi: 10.1101/lm.84904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt W, Maren S. Muscimol inactivation of the dorsal hippocampus impairs contextual retrieval of fear memory. J Neurosci. 1999;19:9054–9062. doi: 10.1523/JNEUROSCI.19-20-09054.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hostetter G, Thomas GJ. Evaluation of enhanced thigmotaxis as a condition of impaired maze learning by rats with hippocampal lesions. J Comp Physiol Psychol. 1967;63:105–110. doi: 10.1037/h0024144. [DOI] [PubMed] [Google Scholar]
- Jadhav SP, Kemere C, German PW, Frank LM. Awake hippocampal sharp-wave ripples support spatial memory. Science. 2012;336:1454–1458. doi: 10.1126/science.1217230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jancic D, Lopez de Armentia M, Valor LM, Olivares R, Barco A. Inhibition of cAMP response element-binding protein reduces neuronal excitability and plasticity, and triggers neurodegeneration. Cereb Cortex. 2009;19:2535–2547. doi: 10.1093/cercor/bhp004. [DOI] [PubMed] [Google Scholar]
- Josselyn SA. Continuing the search for the engram: examining the mechanism of fear memories. J Psychiatry Neurosci. 2010;35:221–228. doi: 10.1503/jpn.100015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandel ER. Neuroscience—the molecular biology of memory storage: a dialogue between genes and synapses. Science. 2001;294:1030–1038. doi: 10.1126/science.1067020. [DOI] [PubMed] [Google Scholar]
- Klement D, Past'alková E, Fenton AA. Tetrodotoxin infusions into the dorsal hippocampus block non-locomotor place recognition. Hippocampus. 2005;15:460–471. doi: 10.1002/hipo.20072. [DOI] [PubMed] [Google Scholar]
- LeBlancq M, McKinney T, Dickson C. PKMζ unzipped: intrahippocampal infusion of zeta inhibitory peptide (ZIP) causes neural silencing. Soc Neurosci Abstr. 2014;40 303.16/C52. [Google Scholar]
- Lopez de Armentia M, Jancic D, Olivares R, Alarcon JM, Kandel ER, Barco A. cAMP response element-binding protein-mediated gene expression increases the intrinsic excitability of CA1 pyramidal neurons. J Neurosci. 2007;27:13909–13918. doi: 10.1523/JNEUROSCI.3850-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzini CA, Baldi E, Bucherelli C, Sacchetti B, Tassoni G. Role of dorsal hippocampus in acquisition, consolidation and retrieval of rat's passive avoidance response: a tetrodotoxin functional inactivation study. Brain Res. 1996;730:32–39. doi: 10.1016/S0006-8993(96)00427-1. [DOI] [PubMed] [Google Scholar]
- Marshall L, Helgadóttir H, Mölle M, Born J. Boosting slow oscillations during sleep potentiates memory. Nature. 2006;444:610–613. doi: 10.1038/nature05278. [DOI] [PubMed] [Google Scholar]
- McDonald RJ, Hong NS, Craig LA, Holahan MR, Louis M, Muller RU. NMDA-receptor blockade by CPP impairs post-training consolidation of a rapidly acquired spatial representation in rat hippocampus. Eur J Neurosci. 2005;22:1201–1213. doi: 10.1111/j.1460-9568.2005.04272.x. [DOI] [PubMed] [Google Scholar]
- McEown K, Treit D. Inactivation of the dorsal or ventral hippocampus with muscimol differentially affects fear and memory. Brain Res. 2010;1353:145–151. doi: 10.1016/j.brainres.2010.07.030. [DOI] [PubMed] [Google Scholar]
- McGaugh JL. The perseveration-consolidation hypothesis: Mueller and Pilzecker, 1900. Brain Res Bull. 1999;50:445–446. doi: 10.1016/S0361-9230(99)00126-4. [DOI] [PubMed] [Google Scholar]
- McGaugh JL. Memory—a century of consolidation. Science. 2000;287:248–251. doi: 10.1126/science.287.5451.248. [DOI] [PubMed] [Google Scholar]
- Misanin JR, Miller RR, Lewis DJ. Retrograde amnesia produced by electroconvulsive shock after reactivation of a consolidated memory trace. Science. 1968;160:554–555. doi: 10.1126/science.160.3827.554. [DOI] [PubMed] [Google Scholar]
- Morris RG. Synaptic plasticity and learning: selective impairment of learning rats and blockade of long-term potentiation in vivo by the N-methyl-D-aspartate receptor antagonist AP5. J Neurosci. 1989;9:3040–3057. doi: 10.1523/JNEUROSCI.09-09-03040.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RG, Frey U. Hippocampal synaptic plasticity: role in spatial learning or the automatic recording of attended experience? Philos Trans R Soc London B Biol Sci. 1997;352:1489–1503. doi: 10.1098/rstb.1997.0136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RG, Garrud P, Rawlins JN, O'Keefe J. Place navigation impaired in rats with hippocampal lesions. Nature. 1982;297:681–683. doi: 10.1038/297681a0. [DOI] [PubMed] [Google Scholar]
- Nader K. Memory traces unbound. Trends Neurosci. 2003;26:65–72. doi: 10.1016/S0166-2236(02)00042-5. [DOI] [PubMed] [Google Scholar]
- Nader K, Schafe GE, Le Doux JE. Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature. 2000;406:722–726. doi: 10.1038/35021052. [DOI] [PubMed] [Google Scholar]
- Packard MG, McGaugh JL. Inactivation of hippocampus or caudate nucleus with lidocaine differentially affects expression of place and response learning. Neurobiol Learn Mem. 1996;65:65–72. doi: 10.1006/nlme.1996.0007. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. San Diego, CA: Academic; 1998. [DOI] [PubMed] [Google Scholar]
- Qi Z, Gold PE. Intrahippocampal infusions of anisomycin produce amnesia: contribution of increased release of norepinephrine, dopamine, and acetylcholine. Learn Mem. 2009;16:308–314. doi: 10.1101/lm.1333409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radyushkin KA, Anokhin KV. Recovery of memory in chicks after disruption during learning: the reversibility of amnesia induced by protein synthesis inhibitors. Neurosci Behav Physiol. 1999;29:31–36. doi: 10.1007/BF02461355. [DOI] [PubMed] [Google Scholar]
- Rojas-Ramírez JA, Aguilar-Jiménez E, Posadas-Andrews A, Bernal-Pedraza JG, Drucker-Colín RR. The effects of various protein synthesis inhibitors on the sleep-wake cycle of rats. Psychopharmacology. 1977;53:147–150. doi: 10.1007/BF00426484. [DOI] [PubMed] [Google Scholar]
- Rudy JW. Is there a baby in the bathwater? Maybe: some methodological issues for the de novo protein synthesis hypothesis. Neurobiol Learn Mem. 2008;89:219–224. doi: 10.1016/j.nlm.2007.08.014. [DOI] [PubMed] [Google Scholar]
- Rudy JW, Biedenkapp JC, Moineau J, Bolding K. Anisomycin and the reconsolidation hypothesis. Learn Mem. 2006;13:1–3. doi: 10.1101/lm.157806. [DOI] [PubMed] [Google Scholar]
- Sadowski RN, Canal CE, Gold PE. Lidocaine attenuates anisomycin-induced amnesia and release of norepinephrine in the amygdala. Neurobiol Learn Mem. 2011;96:136–142. doi: 10.1016/j.nlm.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauseng P, Griesmayr B, Freunberger R, Klimesch W. Control mechanisms in working memory: a possible function of EEG theta oscillations. Neurosci Biobehav Rev. 2010;34:1015–1022. doi: 10.1016/j.neubiorev.2009.12.006. [DOI] [PubMed] [Google Scholar]
- Schafe GE, Nadel NV, Sullivan GM, Harris A, LeDoux JE. Memory consolidation for contextual and auditory fear conditioning is dependent on protein synthesis, PKA, and MAP kinase. Learn Mem. 1999;6:97–110. [PMC free article] [PubMed] [Google Scholar]
- Sharma AV, Nargang FE, Dickson CT. Neurosilence: profound suppression of neural activity following intracerebral administration of the protein synthesis inhibitor anisomycin. J Neurosci. 2012;32:2377–2387. doi: 10.1523/JNEUROSCI.3543-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorhees CV, Williams MT. Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat Protoc. 2006;1:848–858. doi: 10.1038/nprot.2006.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelikowsky M, Bissiere S, Fanselow MS. Contextual fear memories formed in the absence of the dorsal hippocampus decay across time. J Neurosci. 2012;32:3393–3397. doi: 10.1523/JNEUROSCI.4339-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Lam PY, Han D, Cadenas E. c-Jun N-terminal kinase regulates mitochondrial bioenergetics by modulating pyruvate dehydrogenase activity in primary cortical neurons. J Neurochem. 2008;104:325–335. doi: 10.1111/j.1471-4159.2007.04957.x. [DOI] [PubMed] [Google Scholar]