

Abstract
In the normal brain, immune cell trafficking and immune responses are strictly controlled and limited. This unique homeostatic equilibrium, also called brain immune quiescence, is crucial to maintaining proper brain functions and is altered in various pathological processes, from chronic immunopathological disorders to cognitive and psychiatric impairments. To date, the precise nature of factors regulating the brain/immune system interrelationship is poorly understood. In the present study, we demonstrate that one of these regulating factors is Connexin 43 (Cx43), a gap junction protein highly expressed by astrocytes at the blood–brain barrier (BBB) interface. We show that, by setting the activated state of cerebral endothelium, astroglial Cx43 controls immune recruitment as well as antigen presentation mechanisms in the mouse brain. Consequently, in the absence of astroglial Cx43, recruited immune cells elaborate a specific humoral autoimmune response against the von Willebrand factor A domain-containing protein 5a, an extracellular matrix protein of the brain. Altogether, our results demonstrate that Cx43 is a new astroglial factor promoting the immune quiescence of the brain.
Keywords: astrocyte, autoimmunity, blood-brain barrier, connexin, immune quiescence
Introduction
In the brain, immune cell infiltration is normally kept at a very low level, and a unique microenvironment strictly restricts immune reactions (Rivest, 2009; Ransohoff and Engelhardt, 2012). Mechanisms regulating immune responses and interaction between the brain and the peripheral immune system are tightly controlled. Indeed, activated immune cells infiltrating the brain, by secreting inflammatory molecules and elaborating autoimmune mechanisms against brain proteins, promote neuronal death and lesions with obvious pathological consequences on neural processing. Thus, unraveling the molecular and cellular basis of mechanisms regulating the entry and activation of immune cells in the brain is a major challenge that could lead to the development of new biomarkers and treatments for neuroinflammatory diseases.
One of the key elements protecting the brain from immune-mediated harm is the blood–brain barrier (BBB), a highly specialized endothelium forming a tight barrier restricting entry of harmful molecules and immune cells into the brain (Ransohoff and Engelhardt, 2012). Barrier properties of brain endothelium also depend on surrounding specialized cells, pericytes (Sá-Pereira et al., 2012) and astrocytes, the major glial cells in the brain, which project their endfeed around vessels and elaborate the so-called neurovascular unit (Abbott et al., 2006). Astrocytes have been shown to perform a range of vascular regulatory activities, maintaining BBB integrity (Abbott et al., 2006; Alvarez et al., 2013), coordinating blood flow (Petzold and Murthy, 2011), and regulating metabolite transfer to neurons (Bélanger et al., 2011).
A typical feature of astrocytes is their high level of Connexin (Cx) expression, with two major Cxs, Cx43 and Cx30. Cxs assemble by six in hemichannels (Hc) permeable to ions and small signaling molecules up to 1–1.2 kDa that either dock together to form gap junction channels, allowing direct astrocyte-to-astrocyte communication and adhesive cell–cell contacts (Elias et al., 2007), or stay in an Hc conformation to mediate the direct exchange of molecules between the intracellular and extracellular milieu (Giaume et al., 2010; Chever et al., 2014a). To date, Cx30 and Cx43 have been shown to contribute to several aspects of brain physiology, jointly controlling energy metabolite trafficking (Rouach et al., 2008), neurogenesis (Kunze et al., 2009), myelin maintenance (Lutz et al., 2009; May et al., 2013), and BBB integrity (Ezan et al., 2012). They have also been shown to control synaptic strength together (Pannasch et al., 2011) and individually (Chever et al., 2014a; Pannasch et al., 2014). Interestingly, astroglial Cxs are highly concentrated in perivascular endfeed, in which they form large gap junction plaques, allowing for the elaboration of electrical and chemical astroglial networks around the brain endothelium (Rouach et al., 2008).
Here, we address the specific role of astroglial Cx43 in the brain vascular physiology. We show that inactivation of Cx43 in astrocytes induces endothelial activation, allowing leukocytes to infiltrate the brain and elaborate a specific humoral autoimmune response against Vwa5a, a von Willebrand factor A domain-containing extracellular matrix protein of the brain.
Materials and Methods
Mice.
Animals used in this study were kept in pathogen free conditions. Mice of either sex were used in this study except for the transcriptome analysis for which only males were used. The Cre-recombinase activity in the brain of Cx43fl/fl/hGFAP-Cre (Cx43KO) and Cx43fl/fl (Cx43FL) mice (Theis et al., 2001) was tested systematically before performing additional experiments by revealing the β-galactosidase activity (Roche). In addition, because a Cre germ-line activity occurs in the offspring of hGFAP-Cre transgenic females (Zhang et al., 2013), only hGFAP-Cre males were used for breedings.
Antibodies.
References for antibodies used in this study are provided in Table 1.
Table 1.
Antibody list
| Antigen | Reference | Dilution | Protocol |
|---|---|---|---|
| B220 | Rat anti-mouse CD45R RM2600 (Life Technologies) | 1:50 | Paraffin section |
| CD3 | Rabbit anti-human A0452 (Dako) | 1:50 | Paraffin section |
| Gr1 (LY6C/G) | Rat anti-mouse mAb (clone RB6-8C5) RM3030 (Life Technologies) | 1:100 | Paraffin section |
| Iba1 | Rabbit polyclonal 019-19741 (Wako) | 1:400 | Paraffin section |
| 1:400 | Fixed section | ||
| Gfap | Mouse anti-mouse (G3893) or rabbit anti-human (G9269) (Sigma) | 1:500 | Fixed or frozen section |
| MHC II | Anti-mouse MHC II (I-A/I-E) APC, clone MS 114.15.2 (Affymetrix eBioscience) | 1:1000 | Fixed section |
| IgG | Goat anti-mouse Alexa Fluor-conjugated A11029 or A21424 (Life Technologies) | 1:2000 | Frozen section |
| S100 | Rabbit S2644 (Sigma) | 1:1000 | Fixed section |
| Occludin | Rabbit anti-human 71-1500 (Life Technologies) | 1:500 | Western blot |
| ZO-1 | Mouse anti-human 339100 (Life Technologies) | 1:500 | Western blot |
| Claudin-5 | Rabbit anti-mouse 34-1600 (Invitrogen) | 1:100 | Western blot |
| GAPDH | Mouse anti-rabbit G9295 (Sigma) | 1:5000 | Western blot |
| CD3 | Armenian hamster anti-mouse PE-coupled (clone 1452c11) 100308 (Biolegend) | 1:200 | FACS |
| CD4 | Rat anti-mouse adenomatous polyposis coli–Cy7-coupled (clone GK1.5) 100404 (Biolegend) | 1:800 | FACS |
| CD11c | Rat anti-mouse adenomatous polyposis coli-coupled (clone H65) 561115 (BD Biosciences) | 1:200 | FACS |
| CD8 | FITC rat anti-mouse CD8a (clone 53-6.7) (BD Biosciences) | 1:200 | FACS |
| CD11b | Anti-mouse/human PE-Cy7-coupled (clone M1/70) 101216 (Biolegend) | 1:800 | FACS |
| C1q | Rabbit anti-C1qA (1160-1-AP) (Proteintech) | 1:500 | Western blot |
| C9Neo | Rabbit anti-rat C9 (provided by Prof. B. Paul Morgan, Institute for Infection and Immunity, University of Cardiff, UK) | 1:500 | Western blot |
| Vwa5a | Rabbit anti-human, mouse Vwa5a C-terminal region (Clinisicences) | 1:50 | Fixed cells |
Primers.
Primers sequences are provided in Table 2.
Table 2.
qPCR primer list
| Gene | Primer sequence 5′-3′ |
|---|---|
| Gbp1 | Forward, ACAACTCAGCTAACTTTGTGGG |
| Reverse, TGATACACAGGCGAGGCATATTA | |
| IgJ | Forward, TGACGACGAAGCGACCATTC |
| Reverse, TTCAAAGGGACAACAATTCGGA | |
| CCL5 | Forward, CTGCTGCTTTGCCTACCTCT |
| Reverse, gcaagcaatgacagggaagc | |
| CXCL10 | Forward, CGTGGTCACATCAGCTGCTA |
| Reverse, agctagggaggacaaggagg | |
| CXCL1 | Forward, GGCTTCCTTATGTTCAAACAGGG |
| Reverse, GCCGTTACTCGGGTAAATTACA | |
| CCL20 | Forward, TTTTGGGATGGAATTGGACAC |
| Reverse, TGCAGGTGAAGCCTTCAACC | |
| CXCL2 | Forward, CGCTGTCAATGCCTGAAG |
| Reverse, GGCGTCACACTCAAGCTCT | |
| CCL3 | Forward, TTCTCTGTACCATGACACTCTGC |
| Reverse, CGTGGAATCTTCCGGCTGTAG | |
| CCL2 | Forward, GCCCCACTCACCTGCTGCTA |
| Reverse, TTTACGGGTCAACTTCACATTCAA | |
| CXCL12 | Forward, TGCATCAGTGACGGTAAACCA |
| Reverse, TTCTTCAGCCGTGCAACAATC | |
| TNFα | Forward, GACCCTCACACTCAGATCATCTTCT |
| Reverse, CCTCCACTTGGTGGTTTGCT | |
| IL-1β | Forward, CTGGTGTGTGCAGTTCCCATTA |
| Reverse, CCGACAGCACGAGGCTTT | |
| MHC II | Forward, ACAGCTTAGGAATGGGGACT |
| Reverse, CACGGTGATGGGACTCTTCA | |
| IFNγ | Forward, GGCCATCAGCAACAACATAAGCGT |
| Reverse, TGGGTTGTTGACCTCAAACTTGGC | |
| Rorγt | Forward, CCGCTGAGAGGGCTTCA |
| Reverse, TGCAGGAGTAGGCCACATTACA | |
| t-Bet | Forward, AATCGACAACAACCCCTTTG |
| Reverse, AACTGTGTTCCCGAGGTGTC | |
| FoxP3 | Forward, AGGAGCCGCAAGCTAAAAGC |
| Reverse, TGCCTTCGTGCCCACTGT | |
| Gata-3 | Forward, GAACCGCCCCTTATCAAG |
| Reverse, CAGGATGTCCCTGCTCTCCTT | |
| IL-2 | Forward, ATGTACAGCATGCAGCTCGCATC |
| Reverse, GGCTTGTTGAGATGATGCTTTGACA | |
| IL-1ra | Forward, CTTTACCTTCATCCGCTCTGAGA |
| Reverse, TCTAGTGTTGTGCAGAGGAACCA | |
| IL-4 | Forward, AGCTAGTTGTCATCCTGCTCT |
| Reverse, GCATGGAGTTTTCCCATGTTT | |
| IL-10 | Forward, CCCTGGGTGAGAAGCTGAAG |
| Reverse, CACTGCCTTGCTCTTATTTTCACA | |
| Granzyme B | Forward, ATCCTGCTCTGATTACCCATCGT |
| Reverse, ATGGATATGAAGCCAGTCTTTGC | |
| Gfap | Forward, GGGGCAAAAGCACCAAAGAAG |
| Reverse, GGGACAACTTGTATTGTGAGCC | |
| Vimentin | Forward, CGGAAAGTGGAATCCTTGCA |
| Reverse, CACATCGATCTGGACATGCTGT | |
| Nestin | Forward, GTCTCAGGACAGTGCTGAGCCTTC |
| Reverse, TCCCCTGAGGACCAGGAGTCTC |
Study approval.
Experiments and techniques reported here complied with the ethical rules of the French agency for animal experimentation and with the Institute of Medicaments, Toxicology, Chemistry, and the Environment animal ethics committee (Paris Descartes University, Agreement 86-23).
RNA preparation, cDNA libraries, and RNA sequencing.
Brain vessels (endothelial, smooth muscle cells, and pericytes) from 3-month-old Cx43fl/fl/hGFAP-Cre (Cx43KO; Theis et al., 2001) and control wild-type (WT) mice were isolated from whole hemispheres (the cerebellum and the olfactory bulbs were dissected out) as described previously (Yousif et al., 2007), and mRNAs were subsequently purified and sequenced. Total RNA was extracted using the RNeasy Lipid tissue kit (Qiagen). Messenger [poly(A+)] RNAs were purified from 1 μg of total RNA using oligo-dT. Libraries were prepared using the strand nonspecific RNA Sequencing (RNA-Seq) library preparation TruSeq RNA Sample Prep Kits v2 (Illumina). Libraries were multiplexed on one single flow-cell lane and subjected to 50 bp single read sequencing on a HiSeq 2000 device. A mean of 56 ± 27 million passing illumina quality filter reads was obtained for each of the three samples.
The whole RNA-Seq data analysis was performed using the Eoulsan software version 1.1.6 (Jourdren et al., 2012) with the following parameters. Before mapping, poly N read tails were trimmed, reads ≤11 bases were removed, and reads with quality mean ≤12 were discarded. Reads were then aligned against the Mus musculus genome (mm10 genome assembly from the University of California, Santa Cruz) using the Bowtie mapper (version 0.12.7; Langmead et al., 2009) using the –best and –k 2 parameters. Alignments from reads matching more than once on the reference genome were removed. To compute gene expression, M. musculus GFF3 genome annotation from UCSC (mm10) was used. All overlapping regions between alignments and referenced exons were counted. Data normalization and differential analysis were performed using the DESeq package version 1.6.10 (Anders and Huber, 2010). We selected transcriptional changes observed in Cx43KO brain vessels compared with WT libraries, considering only transcripts read >50 times with a fold change >2. Selected events were further validated by qPCR on purified brain vessel RNAs. RNA sequencing data are available at http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=mfqjqmscvxqbtoh&acc=GSE54458.
Quantitative RT-PCR.
Reverse transcription was performed from RNA extracted from purified brain vessels, dissected cortex and hippocampus, or sorted cells using the RNeasy kit (Qiagen). The primer list is provided in Table 2. QuantiTect primer assays (Qiagen) were used for Ighg1, Igkc, Igkv4-50, Igkv6-15, P-Selectin, E-Selectin, VCAM-1, and ICAM-1. qPCR was conducted using SYBR Green PCR master kit (Applied Biosystems). PCR cycling conditions were 50°C for 2 min, 95°C for 10 min, and 40 cycles of 95°C for 15 s and 60°C for 1 min. All experiments were performed in triplicate on an LC480 Roche light cycler. The relative abundance of amplified cDNA was calculated as 2−ΔCt, where ΔCt (change in cycle threshold) equals Ct in Cx43KO minus Ct in Cx43FL. Results are expressed as the means of 2−ΔCt tested cDNA/2−ΔCt RNA 18S values.
Flow cytometry.
Mice were anesthetized with ketamine–xylazine (140 and 8 mg/kg, i.p., respectively), and 20 ml of PBS was administered by intracardiac perfusion to prevent contamination of the brain tissue with intravascular leukocytes. Brains were dissected and squeezed between two glass slides. Homogenates were suspended in DMEM containing 1% fetal calf serum, and the cells were isolated in a Percoll (GE Healthcare) gradient as follows: brain homogenates were suspended in 1 ml of 70% Percoll (GE Healthcare) and carefully added under 2 ml of 30% Percoll; 1 ml of PBS was added on the top. The tubes were centrifuged at room temperature for 20 min at 2700 rpm (Megafuge 1; Heraeus) with the brake off. The cellular phase between 30 and 70% was collected and strained through a 100 μm mesh (BD Biosciences) for additional analysis. Mouse spleen single-cell preparations were obtained from C57BL/6 mice. Cells were then stained (see antibody references in Table 1) and sorted on an FACSAria III cell sorter (BD Biosciences). Electronic compensation was performed with single-stained spleen cells. Electronic gates were applied to exclude cell debris and doublets with a forward scatter-gate. Propidium iodide was used to exclude dead cells. For CD3+/CD4+/CD8− and CD3+/CD4−/CD8+ T lymphocytes, we plotted only the cells in the CD11b− gate (7.4 and 16.3% of total brain lymphoid cells in Cx43FL and Cx43KO animals, respectively). For microglia, we plotted CD11b+ cells. Cells were collected in Buffer RLT (Qiagen) with 1% β-mercaptoethanol for RNA purification.
Vwa5a cloning, primary astrocyte cultures, transfection, and immunocytofluorescence.
Vwa5a cDNA was amplified by RT-PCR on adult C57BL/6 mouse cortex cDNAs using the Superscript II reverse transcriptase (Invitrogen) and the following primers: 5′-ATGGAGCATCACTGTGGTC-3′ and 5′-TTAGACGCCAAAGACAGCAGGATTC-3′, cloned into pcDNA3.1 Topo TA (Invitrogen) and sequenced on both strands. Primary cortical astrocytes were prepared at postnatal day 2 (P2) and grown on coverslips as described previously (Arama et al., 2012). Transient transfection of pcDNA–Vwa5a was performed with Lipofectamine 2000 (Invitrogen). At 24 h after transfection, cells were fixed in 4% phosphate-buffered (PBS) paraformaldehyde (PFA) for 10 min at room temperature and immunostained as described previously (Arama et al., 2012), with Cx43KO sera (1:100) and anti-Vwa5a (Table 1).
In situ hybridization.
Igkc cDNA was amplified by PCR on 6-week-old Cx43KO brain cDNAs (see above, Quantitative RT-PCR) using Taq polymerase (Qiagen) and the primers 5′-GATGCTGCACCAACTGTATCC-3′ and 5′- ACACTCATTCCTGTTGAAGCT-3′ and cloned into pCRII-TOPO (Life Technologies). Igkc sense and antisense probes were prepared using the Riboprobe in vitro transcription systems (Promega). Colorimetric in situ hybridization (ISH) was performed on frozen sections using digoxigenin (DIG)-labeled RNA probes. Color development was done by anti-DIG-AP Fab fragment from goat (Roche) using NBT/BCIP (Roche).
Immunohistochemistry and immunohistofluorescence.
Mice were anesthetized with ketamine–xylazine (140 and 8 mg/kg, i.p., respectively). For frozen sections, mice were sacrificed by intracardiac perfusion of PBS. Brains were dissected and frozen in isopentane at −25°C. Frozen sections, 20 μm thick, were fixed by immersion in PBS/4% PFA for 20 min at room temperature and immersed in the blocking solution (PBS/5% NGS/0.25% Triton-X-100) for 1 h at room temperature. For fixed sections, mice were killed by intracardiac perfusion of PBS/4% PFA. Brains were incubated in PBS/4% PFA overnight at 4°C and for 1–3 d in PBS/30% sucrose at 4°C. Sections, 40 μm thick, were immersed in the blocking solution (PBS/5% NGS/0.25% Triton X-100) for 1 h at room temperature. For paraffin sections, brains were removed and fixed in JB fixative (0.5% zinc acetate, 0.05% zinc chloride, and 0.05% calcium acetate in Tris buffer at pH 7) for 48 h and then embedded in low-melting point paraffin (Poly Ethylene Glycol Distearate; Sigma). Sections, 5 μm thick, were deparaffinated in absolute ethanol, rinsed in water, and immersed in the blocking solution (PBS/3% BSA) at room temperature.
Sections were incubated with primary antibodies (Table 1) diluted in the blocking solution 12 h at 4°C, rinsed three times in PBS, incubated 2 h at room temperature with secondary antibodies, rinsed three times in PBS, and embedded in Fluormount G for fluorescent revelation or first revealed for HRP activity using the DAB Peroxidase Substrate kit (Vector Laboratories). Secondary antibodies included the following: Alexa Fluor-conjugated goat anti-mouse IgG (A11029/A21424; Life Technologies), anti-rabbit IgG (1:2000; A11034/A21429; Life Technologies), N-histofine peroxidase anti-rabbit or rat (T414341F/T414311F; Microm Microtech), and HRP-conjugated goat anti-mouse or rabbit (1:2500; SC-2004/SC2005; Santa Cruz Biotechnology). Fluorescence was imaged on a SP5 confocal microscope (Leica). HRP stainings were scanned using the MIRAX SCAN (Zeiss).
Image analysis of Iba1, Gfap, and S100 stainings.
For each experiment, tissues were all prepared and analyzed together: dissection, fixation, sectioning, labeling, and imaging. Analysis was performed using the Fiji software. For Iba1 surface quantification, HRP signal was extracted (color deconvolution; vectors: hematoxylin and eosin, diaminobenzidine) from background (rolling, 10). After thresholding (maxEntropy), images were converted to masks, and particles were analyzed (size, 10 to infinity; circularity, 0.10–1). Two sections were analyzed per animal. For Gfap surface quantification, confocal projections (15 μm, z = 0.5 μm) obtained with the 63× objective were filtered (Huang or Li thresholding methods). Individual astrocytes were delimited, and the integrated density was measured (with limit to threshold). For cell counting (Iba1, S100), stitched confocal images (z = 3 μm) obtained with a 20× objective were blurred (radius, 4), and cells were counted with Find Maxima (noise tolerance, 10).
Western blot.
All procedures are described previously (Ezan et al., 2012).
In situ brain perfusion.
Mice were anesthetized with ketamine–xylazine (140 and 8 mg/kg, i.p., respectively), and a polyethylene catheter was inserted into the carotids. The heart was cut, and the perfusion started immediately (flow rate, 2.5 ml/min) to obtain a complete substitution of the blood by the artificial perfusion fluid, a Krebs' carbonate-buffered physiological saline (in mm: 128 NaCl, 24 NaHCO3, 4.2 KCl, 2.4 NaH2PO4, 1.5 CaCl2, 0.9 MgCl2, and 9 d-glucose) containing also [14C]sucrose (0.3 μCi/ml; PerkinElmer Life and Analytical Sciences) as a vascular and integrity marker, gassed with 95% O2/5% CO2 for pH control (7.4) and warmed to 37°C. Perfusion was terminated after 120 s by decapitating the mouse. The whole brain was removed from the skull and dissected out on a freezer pack. Brain hemispheres and two aliquots of perfusion fluid were placed in tared vials and weighed, digested with Solvable (PerkinElmer Life and Analytical Sciences), and mixed with Ultima gold XR (PerkinElmer Life and Analytical Sciences) for 14C dpm counting (Tri-Carb; PerkinElmer Life and Analytical Sciences). In some experiments, human serum albumin (40 g/L) (Vialebex) was added in the perfusion fluid to increase shear stress and the hydrostatic pressure (∼180 mmHg; Ezan et al., 2012). The brain vascular volume, Vv (microliters per gram), was calculated using the distribution of [14C]sucrose: Vv = Xv/Cv where Xv (disintegrations per minute/gram) is the [14C]sucrose measured in the hemispheres, and Cv (disintegrations per minute/microliter) is the concentration of [14C]sucrose in the perfusion fluid (Dagenais et al., 2000). Of note, sucrose is a low molecular weight (342 Da) and very hydrophilic disaccharide compound that does not bind to plasma proteins and has no dedicated transporter in mammals. Hence, it exhibits negligible passive diffusion, which allows its use as an integrity BBB marker (Takasato et al., 1984). In this context, variations in the level of sucrose brain distribution volume only reflect modified physical integrity of the BBB and never results from confounding effects related to transporter expression or changes in vascular flow.
Electron microscopy.
Mice were anesthetized with ketamine–xylazine (140 and 8 mg/kg, i.p., respectively) and transcardially perfused with the fixative (2% PFA, 3% glutaraldehyde, and 3 mm CaCl2 in 0.1 m cacodylate buffer, pH 7.4) for 12 min. Brains were removed and left overnight at 4°C in the same fixative. Brain fragments (0.3 × 1 × 1 mm3) were then postfixed first in 0.1 m cacodylate buffer, pH 7.4, plus 1% OsO4 for 1 h at 4°C and then in 1% aqueous uranyl acetate for 2 h at room temperature. After dehydration in graded ethanol, followed by propylene oxide, the fragments were embedded in Epon. Ultrathin (80 nm) sections were prepared, stained in lead citrate, and photographed in a Jeol 100S transmission electron microscope equipped with a 2000 × 2000 Orius 830 CCD camera (Roper Scientific).
Purification and analysis of Ig–protein complexes in Cx43KO.
Three-month-old Cx43KO and Cx43FL mice were anesthetized with ketamine–xylazine (140 and 8 mg/kg, i.p., respectively) and perfused with PBS. Brains were homogenized in binding buffer [PBS, 0.2% Triton X-100, and 1× Complete Protease Inhibitor (Roche)] with a Dounce tissue grinder at 4°C. Lysates were sonicated twice at 10 Hz (Vibra-Cell VCX130) and centrifuged 20 min at 10,000 × g at 4°C. Protein content of the cleared lysates was measured using the BCA protein assay (Thermo Fisher Scientific). G-Sepharose beads were pretreated by incubation with 500 μg of Cx43FL for 1 h at 4°C and washed five times with binding buffer. Five hundred micrograms of Cx43FL or Cx43KO proteins were incubated with the pretreated G-Sepharose beads overnight at 4°C. After five washes with binding buffer at 4°C, G-Sepharose beads were boiled in 5× Laemmli's loading buffer, and eluted proteins were separated by denaturing electrophoresis in two NuPAGE 4–12% SDS-polyacrylamide gradient gels (Invitrogen). One of the gels was transferred on nitrocellulose and probed with Cx43KO serum (1:200 dilution). The second gel was colored by colloidal blue staining (Bio-Rad), and gel slices between 100 and 64 kDa were analyzed by mass spectrometry.
Mass spectrometry.
Gel bands were reduced, alkylated, and subjected to digestion with trypsin (sequencing grade; Promega) overnight in 25 mm ammonium bicarbonate at 30°C. The extracted peptides were analyzed by nano-liquid chromatography-MS/MS using an Ultimate 3000 system (Dionex) coupled to an LTQ-Orbitrap XL mass spectrometer (Thermo Fisher Scientific). Samples are loaded on a C18 precolumn (300 μm inner diameter × 5 mm; Dionex) at 20 μl/min in 5% acetonitrile and 0.1% trifluoroacetic acid. After 3 min of desalting, the precolumn was switched online with the analytical C18 column (75 μm inner diameter × 50 cm; C18 PepMap; Dionex) equilibrated in solvent A (2% acetonitrile and 0.1% formic acid). Bound peptides were eluted using a 160 min linear gradient [from 0 to 30% (v/v)] of solvent B (80% acetonitrile and 0.085% formic acid) at a 150 nl/min flow rate and an oven temperature of 40°C. Data-dependent acquisition was performed on the LTQ-Orbitrap mass spectrometer in the positive ion mode. Survey MS scans were acquired in the Orbitrap on the 400–1200 mass-to-charge range with the resolution set to a value of 60,000. Each scan was recalibrated in real time by coinjecting an internal standard from ambient air into the C-trap (“lock mass option”). The five most intense ions per survey scan were selected for collision-induced dissociation fragmentation, and the resulting fragments were analyzed in the linear trap (LTQ). Target ions already selected for MS/MS were dynamically excluded for 180 s. Data were acquired using the Xcalibur software (version 2.2), and the resulting spectra were then analyzed via the Mascot Software (version 2.3) created with Proteome Discoverer (version 1.4; Thermo Fisher Scientific) using the SwissProt M. musculus database. Carbamidomethylation of cysteines, oxidation of methionine, and protein N-terminal acetylation were set as variable modifications for all Mascot searches. Specificity of trypsin digestion was set for cleavage after Lys or Arg except before Pro, and two missed trypsin cleavage sites were allowed. The mass tolerances in MS and MS/MS were set to 2 ppm and 0.5 Da, respectively. The resulting Mascot files were further processed using myProMS (Poullet et al., 2007). The estimated false discovery rate (nonparametric estimation of q values; Qvality) was set to 1% by automatically filtering the Mascot score of all peptide identifications.
Results
Transcriptome analysis of brain vessels purified from astroglial Cx43-deleted mice
To address the role of astroglial Cx43 in the regulation of cerebrovascular functions, we analyzed the molecular events occurring in the brain vascular system when astroglial Cx43 is absent. Brain vessels (endothelial, smooth muscle cells, and pericytes) were isolated from 3-month-old Cx43KO (Theis et al., 2001) and control WT mice by homogenization, density-gradient centrifugation, and filtration, as described previously (Yousif et al., 2007). RNAs were subsequently purified and sequenced. Because mutant mice were maintained on a mixed C57BL/6/BALBc genetic background and genetic background strongly influences gene transcription (Iacobas et al., 2012), two distinct WT RNAs libraries from C57BL/6 and C57BL/6/BALBc mice were prepared. We then compared the transcriptomes and selected changes observed in Cx43KO brain vessels regardless of the genetic background. Genes with <50 reads and with fewer than twofold changes were not considered. The selected transcriptional variations were further validated by qPCR on purified microvessels (Fig. 1A). In total, very few transcriptional variations fit our criteria. We observed a strong upregulation of guanylate binding protein-1 (Gbp1), an IFNγ-induced GTPase secreted by activated endothelial cells and monocytes (Hammon et al., 2011; Haudek-Prinz et al., 2012), as well as upregulation of several transcripts for Ig. Performing qPCR on P25, 6 week, and 3 month whole cortex and hippocampus RNAs, we next determined that these transcriptional variations started at ∼6 weeks of age in Cx43KO (Fig. 1B). These results suggested that leukocytes might be copurified with brain vessels in Cx43KO.
Figure 1.

Transcriptome analysis of brain vessels purified from astroglial Cx43-deleted mice. A, Transcriptional changes observed in 3-month-old Cx43KO purified brain vessels by RNA-Seq compared with C57BL/6/BALBc or C57BL/6 mice. baseMean, Number of reads; FC, fold changes. On the right, validation of the RNA-Seq results by qPCR on purified brain vessels of Cx43KO relative to C57BL6. “Infinite” means that the transcript was sequenced only in Cx43KO brain vessels. B, qPCR analysis on P25, 6 week, and 3 month Cx43KO and Cx43FL cortex and hippocampus. Only Ighg1 was tested for heavy chain transcripts. NE, Not expressed. Gbp1: at P25, not expressed, n = 4; at 6 weeks, 23.3 ± 9.3, p < 0.0001, n = 5; at 3 months, 16.3 ± 7, p = 0.05, n = 3; Igkc: at P25, 1.2 ± 0.1, p = 0.4, n = 4; at 6 weeks, 338.0 ± 111.2, p = 0.006, n = 4; at 3 months, 333.9 ± 57.9, p = 0.0001, n = 3; IgJ: at P25, 1.1 ± 0.0, p = 0.3, n = 4; at 6 weeks, 30.7 ± 10.9, p = 0.01, n = 3; at 3 months, 50.6 ± 7.9, p = 0.0002, n = 3; Ighg1: at P25, 0.7 ± 0.1, p = 0.6, n = 4; at 6 weeks, 348.2 ± 99.0, p = 0.0003, n = 4; at 3 months, 757.5 ± 105.5, p = 0.0001, n = 3; IgV50: at P25, not expressed, n = 4; at 6 weeks, 4.3 ± 0.9, p = 0.01, n = 4; at 3 months, 143.8 ± 62.0, p = 0.004 n = 3; IgV15: at P25, 1.2 ± 0.2, p = 0.7, n = 4; at 6 weeks, 626.6 ± 218.5, p = 0.05, n = 4; at 3 months, 51.8 ± 18.5, p = 0.0003, n = 3. Cx43FL values are set as 1. Data are presented as means ± SEMs. Mann–Whitney two-tailed test. nsp > 0.05, *p < 0.05, **p < 0.01, ***p < 0.001.
Immune cells migrate across the BBB in the absence of astroglial Cx43
These observations prompted us to search for the presence of immune cells infiltrated in the brain of Cx43KO. We performed immunolabeling, transmission electron microscopy (TEM), and flow cytometry (Fig. 2). At 6 weeks of age, lymphocytes comprising B220+ (Fig. 2Ai), IgG+ (B cells and plasma cells) (Fig. 2B), CD3+ cells (T cells) (Fig. 2Aii), Gr1+ macrophages (Fig. 2Ci), and neutrophils (Fig. 2Cii,Ciii) were detected in the Cx43KO brain parenchyma with no preferential area or inserted into the basal lamina (BL) surrounding brain vessels (Fig. 2Aiii,Bii,Ciii), whereas they were almost absent in Cx43fl/fl littermates (Cx43FL). Quantification of infiltrated cells at different postnatal stages revealed that immune recruitment started at ∼6 weeks of age in Cx43KO (Fig. 2D). We further characterized the brain-infiltrated CD3+ T lymphocytes by flow cytometry and qPCR and determined that they consisted mainly in two distinct populations of CD4+ and CD8+ (Fig. 2E). qPCR analysis of these cells demonstrated that they were composed of CD4+/Tbet+/Gata3+/Foxp3+/IL10+/IFNγ+/IL2−/IL4−/Rorγt− cells, thus mainly CD4+ activated T helper 1 (TH1), TH2, and regulatory T cells, as well as cytotoxic CD8+/IL10+/IFNγ+/GranzymeB+ T cells (Fig. 2F). Thus, the absence of astroglial Cx43 leads to the recruitment of various types of leukocytes in the brain, starting at ∼6 weeks of age.
Figure 2.

Immune cells migrate across BBB in the absence of astroglial Cx43. A–C, Representative images of intraparenchymal leukocytes (in red) in Cx43KO at 6 weeks. Nuclei (in gray) are counterstained with Hoechst. Ai, B220+ cell; Aii, CD3+ cells; Aiii, TEM image of a lymphocyte inserted into the perivascular BL. EC, Endothelial cell; A, astrocyte endfeed; L, lumen. Bi, IgG+ cell; Bii, Biii, TEM images of plasma cells located within the perivascular BL (Bii) or the parenchyma (Biii). Ci, Gr1+ macrophage (round nucleus); Cii, Gr1+ neutrophil (multi-lobed nucleus); Ciii, TEM image of a neutrophil inserted into the perivascular BL. D, Quantification of intraparenchymal leukocytes in Cx43KO and Cx43FL (Cx43KO/Cx43FL cell/mm2): Gr1+ cells at P25, 1.2 ± 0.1, p = 0.4, n = 3; at 6 weeks, 5.8 ± 1.1, p = 0.05, n = 3; at 3 months, 1.9 ± 0.5, p = 0.03, n = 6; at 12 months, 6.5 ± 4.3, p = 0.05, n = 3. B220+ cells at P25, 1.7 ± 0.3, p = 0.1, n = 3; at 6 weeks, 3.0 ± 0.5, p = 0.05, n = 3; at 3 months, 4.8 ± 1.1, p = 0.002, n = 6; at 12 months, 4.1 ± 1.8, p = 0.05, n = 3. CD3+ cells at P25, 0.8 ± 0.5, p = 0.9, n = 3; at 6 weeks, 16.9 ± 11.1, p = 0.05, n = 3; at 3 months, 4.5 ± 1.0, p = 0.05, n = 6; at 12 months, 10.1 ± 3.8, p = 0.05, n = 3. Data are presented as means ± SEMs. Mann–Whitney one-tailed test. nsp > 0.05, *p < 0.05, **p < 0.01. Cx43FL values are set as 1. E, FACS analysis of intraparenchymal CD3+ cells in 6-week-old Cx43KO and Cx43FL. Left plots show CD3 sorting in CD11b− cells isolated from the brain. Right plots show CD4 and CD8 sorting in CD3+ cells. SSC, Side scatter. F, qPCR analysis of Cx43KO brain-sorted CD3+/CD4+ and CD3+/CD8+ cells compared with CD3+ C57BL/6 spleen-sorted cells. NE, Not expressed. “Infinite” means that the transcript was amplified only in Cx43KO cells. Data are presented as means ± SEMs. Wilcoxon's signed-rank test, two tailed (n = 3). C57BL/6 spleen values are set as 1.
Immune cell recruitment in Cx43KO is not linked to BBB breakdown
Why are immune cells recruited into the Cx43KO brain? To address this question, we first assessed BBB integrity, because BBB breakdown can result in immune cell infiltration. Ultrastructure analysis of the gliovascular unit in 3-month-old Cx43KO compared with Cx43FL showed no irregularity in the BL and no accumulation of pinocytic vesicles, whereas astroglial perivascular endfeed were in close contact with the BL with a dense intracellular content showing no sign of edema (Fig. 3A). Moreover, tight junctions (TJs) extended from the vessel lumen to the BL with no apparent discontinuity (Fig. 3A). Accordingly, Western blot detection of TJ proteins ZO-1, Claudin 5, and Occludin in 6-week-old and 3-month-old Cx43KO and Cx43FL cortex and hippocampus revealed no difference (Fig. 3B). BBB integrity in Cx43KO and Cx43FL mice was further examined at P25, 6 weeks, and 3 months of age by performing in situ brain perfusion (Dagenais et al., 2000; Fig. 3C). Briefly, mice were anesthetized, and a catheter was inserted into the carotids. The heart was cut, and the perfusion started immediately at a constant and controlled flow rate to obtain a complete substitution of the blood by a plasma protein- and cell-free Krebs' carbonate-buffered physiological saline containing [14C]sucrose. Human serum albumin was added to create shear stress and increase hydrostatic pressure (180 mmHg; Ezan et al., 2012). [14C]Sucrose was used as a marker of the vascular space and integrity, because it does not cross the BBB significantly during short exposure. The level of [14C]sucrose (Vv) was then measured in the whole brain. Of note, given that our analysis involves the brain as a whole, we could not exclude small regional or cellular BBB integrity variations. Regardless of the age, [14C]sucrose Vv in Cx43FL (Fig. 3C, white bars) was in agreement with the physiological [14C]sucrose Vv described previously (Dagenais et al., 2000), indicating no BBB leakage in control animals. No difference could be noted in P25 and 6-week-old Cx43KO (Fig. 3C, black bars). In contrast, Vv in 3-month-old Cx43KO increased significantly compared with Cx43FL mice, indicating a loss of vascular integrity. However, this increase was not detected in the absence of shear stress and normal hydrostatic pressure (no albumin; Fig. 3C, gray bars). Hence, although BBB progressively weakens in Cx43KO, it remains tight when not submitted to shear and pressure stress, suggesting that immune cell recruitment does not originate from BBB breakdown in Cx43KO.
Figure 3.

BBB phenotype in the absence of astroglial Cx43. Ai, Representative images of the gliovascular unit ultrastructure in 3-month-old Cx43KO and Cx43FL (n = 6); Aii, details of TJs and BL. A, Astroglial perivascular endfeed; EC, endothelial cells. B, Western blot of the TJ proteins ZO-1, Claudin 5, and Occludin in 6-week-old and 3-month-old Cx43KO and Cx43FL cortex and hippocampus. GAPDH was used as the loading control (n = 3). C, BBB integrity in Cx43KO and Cx43FL mice at P25, 6 weeks, and 3 months of age was assessed by measuring the brain Vv (in microliters per gram) after in situ brain perfusion of [14C]sucrose under shear stress and increased hydrostatic vascular pressure [with (+) albumin, 180 mmHg, white and black bars] or under normal hydrostatic pressure and without shear stress [no (−) albumin, 120 mmHg, gray bars]. At P25 with albumin: Cx43FL, 21.6 ± 0.9 and Cx43KO, 19.0 ± 1.0, p = 0.7, n = 7; at 6 weeks with albumin: Cx43FL, 18.2 ± 0.2 and Cx43KO, 18.3 ± 0.8, p = 0.7, n = 7; at 3 months with albumin: Cx43FL, 18.1 ± 0.8 and Cx43KO, 39.3 ± 1.6, p = 0.005, n = 7; at 3 months without albumin: Cx43FL, 15.7 ± 0.2 and Cx43KO, 15.4 ± 0.3, p = 0.7, n = 7. Data are means ± SEMs. nsp > 0.05, **p < 0.001. Mann–Whitney two-tailed test.
Immune cell recruitment in Cx43KO is not linked to gliosis
We next assessed inflammatory mechanisms that are known to undermine the brain immune quiescence (Denes et al., 2010) by measuring the expression level of IL-1β and TNFα, two proinflammatory cytokines commonly associated with the breakdown of CNS immune control (Farina et al., 2007). qPCR on mutant and Cx43FL control littermates cortex and hippocampus showed no difference at P25. However, a strong burst of TNFα was observed at 6 weeks in Cx43KO, which moderated afterward (Fig. 4A). IL-1β expression followed the same profile, with a more modest upregulation (Fig. 4A). We wondered whether upregulation of inflammatory cytokines could originate from microglial cells, which are resident macrophages in the brain and play a pivotal role in the propagation of inflammatory signals (David and Kroner, 2011). qPCR analysis was performed on 6-week-old and 3-month-old Cx43KO and Cx43FL CD11b+ brain-sorted cells (mainly microglia and macrophages). Cx43KO cells did not express higher amounts of IL-1β and TNFα, but the anti-inflammatory IL-1ra was increased at 6 weeks of age (Fig. 4B). We further studied the microglial reactivity by measuring Iba1 immunostaining intensity. We observed stronger immunoreactivity in the Cx43KO cortex and hippocampus but only at 3 months of age (Fig. 4C,D). The total number of Iba1+ cells in Cx43FL and Cx43KO was not significantly different at 6 weeks and 3 months (Fig. 4E) but increased locally in 3-month-old Cx43KO areas of strong Iba1 staining (Fig. 4E). Interestingly, these areas were often centered on infiltrated leukocytes in the Cx43KO brain parenchyma (Fig. 4F). Astroglial reactivity was assessed by measuring the expression level of Gfap, Vimentin, and Nestin (Clarke et al., 1994; Duggal et al., 1997) by qPCR on cortex and hippocampus of P25, 6 week, and 3 month Cx43KO and Cx43FL (Fig. 5A). No difference could be noted at P25 and 6 weeks, although a slight increase of Gfap and Vimentin was detected at 3 months (Fig. 5A). Gfap immunoreactivity in the cortex and hippocampus remained comparable in both genotypes (Fig. 5B,C) but increased locally in areas of stronger Iba1 immunoreactivity in 3-month-old Cx43KO (Fig. 5D). The number of S100+ astrocytes remained constant (Fig. 5E). Thus, increases in inflammatory cytokines in 6-week-old Cx43KO occurred independently of glial reactivity. Microglia and astrocytes became reactive but only locally at ∼3 months of age and mainly around immune infiltrates. Moreover, microglia appeared to be rather polarized toward an anti-inflammatory phenotype, expressing IL-1ra (Rivest, 2009; David and Kroner, 2011). These results suggest that reactive gliosis does not trigger brain immune recruitment in Cx43KO.
Figure 4.

Microglial phenotype in the absence of astroglial Cx43. A, qPCR analysis of inflammatory cytokines on Cx43KO and Cx43FL cortex and hippocampus. IL-1β: at P25, 0.9 ± 0.2, p = 0.9; at 6 weeks, 2.8 ± 0.3, p = 0.0007, n = 4; at 3 months, 1.7 ± 0.2, p = 0.03, n = 3; TNFα: at P25, 0.7 ± 0.6, p = 0.8, n = 4; at 6 weeks, 7.9 ± 1.5, p = 0.0003. B, qPCR analysis of inflammatory and anti-inflammatory markers in CD11b+ brain-sorted cells of Cx43KO and Cx43FL mice. IL-1β: at 6 weeks, 2.0 ± 0.6, p = 0.4, n = 4; at 3 months, 0.9 ± 0.1, p = 0.7, n = 3; TNFα: at 6 weeks, 2.1 ± 0.2, p = 0.06, n = 4; at 3 months, 1.2 ± 0.5, p = 0.9, n = 3; IL-1ra: at 6 weeks, 31.6 ± 8.8, p = 0.05, n = 4; at 3 months, 1.6 ± 0.3, p = 0.4, n = 3. C, Representative images of Iba1 immunostaining in the cortex (Cx) and hippocampus (Hp; dashed lines) of 6-week-old and 3-month-old Cx43KO and Cx43FL. Squared area shows microglial cells strongly labeled by Iba1 and with processes mostly oriented centripetally. D, Evaluation of the Iba1 intensity in cortex and hippocampus in Cx43KO/Cx43FL: at 6 weeks, 1.4 ± 0.2, p = 0.2, n = 3; at 3 months, 2.9 ± 0.7, p = 0.004, n = 6. E, Evaluation of the number of Iba1+ cells of in the brain of 6-week-old and 3-month-old Cx43KO and Cx43FL and in 3-month-old Cx43KO strong Iba1-stained areas (Iba1 cells/mm2): at 6 weeks, 0.9 ± 0.07, p = 0.7, n = 3; at 3 months, 1.5 ± 0.2, p = 0.2, n = 3; in strong Iba1-stained areas, 2.55 ± 0.1, p = 0.007, n = 3. F, Coimmunofluorescent detection of Iba1 (green) and Gr1 or IgG (red) in 3-month-old Cx43KO. Cx43FL values are set as 1. Data are presented as means ± SEMs. Mann–Whitney two-tailed test. nsp > 0.05, *p < 0.05, **p < 0.01, ***p < 0.001. NE, Not expressed.
Figure 5.

Astroglial phenotype in the absence of astroglial Cx43. A, qPCR analysis of astroglial activation markers on Cx43KO and Cx43FL cortex and hippocampus. Gfap: at P25, 1.6 ± 0.2, p = 0.06, n = 4; at 6 weeks, 1.1 ± 0.1, p = 0.7, n = 4; at 3 months, 1.5 ± 0.1, p = 0.009, n = 3; Vimentin: at P25, 1.0 ± 0.1, p = 0.8, n = 4; at 6 weeks, 1.0 ± 0.2, p = 0.2, n = 4; at 3 months, 1.8 ± 0.2, p = 0.004, n = 3; Nestin: at P25, 1.2 ± 0.3, p = 0.6, n = 4; at 6 weeks, 0.6 ± 0.09, p = 0.1, n = 4; at 3 months, 1.3 ± 0.1, p = 0.2, n = 3. Data are presented as means ± SEMs. nsp > 0.05, *p < 0.05, **p < 0.01, ***p < 0.001. Mann–Whitney two-tailed test. Cx43FL values are set as 1. B, Representative images of Gfap immunohistostaining in the cortex and hippocampus (dashed lines) of 6-week-old and 3-month-old Cx43KO and Cx43FL. C, Evaluation of the Gfap staining in the cortex and hippocampus at 6 weeks, 1.5 ± 0.6, p = 0.5, n = 3; at 3 months, 1.3 ± 0.2, p = 0.2, n = 3. D, Coimmunostaining of Gfap and Iba1 in 3-month-old Cx43KO cortex. E, Evaluation of the number of S100-labeled cells in cortex and hippocampus (S100 cells/mm2) at 3 months, 0.9 ± 0.1, p = 0.9, n = 3. Data are presented as means ± SEMs. nsp > 0.05, **p < 0.01. Mann–Whitney two-tailed test. Cx43FL values are set as 1.
Absence of astroglial Cx43 leads to endothelial activation and chemoattraction
Endothelial activation and chemoattraction mechanisms are both mandatory for immune infiltration across the BBB (Ransohoff and Engelhardt, 2012). We first tested whether the increase in IL-1β and TNFα observed in the Cx43KO 6-week-old cortex and hippocampus (Fig. 5A) could originate from the vasculature by performing qPCR on purified brain vessels at P25 and 6 weeks of age (results for 3-month-old brain vessels correspond to the RNA-Seq study in Fig. 1). We found that TNFα strongly upregulated at 6 weeks in Cx43KO vessels but not at P25, whereas IL-1β levels stayed comparable in Cx43FL and Cx43KO at both stages (Fig. 6A). This result indicated that endothelial activation might occur at ∼6 weeks in Cx43KO. Therefore, we measured the expression of canonical endothelial activation markers, the adhesion molecules P-selectin, E-selectin, VCAM-1, and ICAM-1, in purified brain vessels (Ransohoff and Engelhardt, 2012). Interestingly, expression of ICAM-1 and VCAM-1 at P25, and ICAM-1 and P-selectin at 6 weeks increased in Cx43KO vessels (Fig. 6B). We finally addressed chemoattraction mechanisms, first testing the expression of several chemokines in dissected cortex and hippocampus at P25, 6 weeks, and 3 months. Among the tested chemokines (Table 2), CXCL10, CXCL12, and CCL5 were upregulated and as early as P25 for CCL5 and CXCL10 in Cx43KO (Fig. 6C). In contrast, in the purified vascular fraction, these three chemokines were upregulated only at 6 weeks but to a lesser extent, suggesting that other neural cells may account for the expression of these chemokines (Fig. 6D). These results suggest that the absence of astroglial Cx43 leads to endothelial activation and chemoattraction, which may likely contribute to immune recruitment in the brain.
Figure 6.

Endothelial activation and chemoattraction in the absence of astroglial Cx43. A, qPCR analysis of inflammatory cytokines on Cx43KO and Cx43FL purified brain vessels. IL-1β at P25, 1.1 ± 0.1, p = 0.2, n = 5; at 6 weeks, 1.03 ± 0.07, p = 0.5, n = 5; TNFα at P25, 0.8 ± 0.1, p = 0.4, n = 5; at 6 weeks, 3.7 ± 0.4, p < 0.0001, n = 5. B, qPCR analysis of endothelial activation markers on Cx43KO and Cx43FL purified brain vessels. P-Selectin at P25, 1.8 ± 0.3, p = 0.07, n = 5; at 6 weeks, 3.9 ± 1.3, p = 0.02, n = 3; E-selectin at P25, 0.9 ± 0.1, p = 0.8, n = 5; at 6 weeks, 2.3 ± 0.6, p = 0.06, n = 5; VCAM-1 at P25, 1.2 ± 0.1, p = 0.02, n = 5; at 6 weeks, 1.3 ± 0.1, p = 0.1, n = 5; ICAM-1 at P25, 1.2 ± 0.0, p = 0.02, n = 5; at 6 weeks, 1.8 ± 0.3, p = 0.01, n = 5. C, qPCR analysis of three of the tested chemokines on Cx43KO and Cx43FL cortex and hippocampus. CCL5 at P25, 8.6 ± 2.5, p = 0.004, n = 4; at 6 weeks, 76.3 ± 26.2, p = 0.0009, n = 4; at 3 months, 13.3 ± 4.0, p = 0.002, n = 6; CXCL10 at P25, 11.4 ± 2.1, p < 0.0001, n = 4; at 6 weeks, 27.4 ± 8.0, p < 0.0001, n = 4; at 3 months, 7.3 ± 0.9, p < 0.0001, n = 6; CXCL12 at P25, 0.9 ± 0.2, p = 0.3, n = 4; at 6 weeks, 7.0 ± 1.8, p = 0.0003, n = 4; at 3 months, 6.3 ± 0.7, p = 0.0002, n = 6. D, qPCR analysis of three of the tested chemokines on Cx43KO and Cx43FL purified brain vessels: CCL5 at P25, 2.0 ± 0.8, p = 0.9, n = 5; at 6 weeks, 1.8 ± 0.2, p = 0.005, n = 5; CXCL10 at P25, 1.6 ± 0.3, p = 0.16, n = 5; at 6 weeks, 9.8 ± 4.0, p < 0.0001, n = 5; CXCL12 at P25, 1.3 ± 0.08, p = 0.05, n = 5; at 6 weeks, 2.2 ± 0.2, p < 0.0001, n = 5. Cx43FL expression levels are set at 1. NE, Not expressed. Data are presented as means ± SEMs. nsp > 0.05, *p < 0.05, **p < 0.01, ***p < 0.001. Mann–Whitney two-tailed test. Cx43FL values are set as 1.
Infiltrated immune cells in Cx43KO elaborate humoral autoimmune responses
In addition to immune infiltration, the major histocompatibility complex class II molecule (MHC II) was upregulated strongly from 6 weeks in the Cx43KO cortex and hippocampus (Fig. 7A). By performing immunohistostaining, we detected MHC II in some intraparenchymal and perivascular Iba1+ cells (microglia and macrophages; Fig. 7B), suggesting that microglia became antigen-presenting cells in Cx43KO. Moreover, at 3 months of age, Cx43KO mice showed Ig deposits around most astrocytic cell bodies (Fig. 7C). These Igs, in the absence of BBB breaching and blood extravasation (Fig. 3), probably originated from B cells, which were the only ones labeled by ISH for Igkc transcript (Fig. 7D). Consistently, IgG was increased in brain protein extracts (Fig. 7E). In contrast, the level of C1q and C9 were not increased in Cx43KO compared with Cx43FL, suggesting that Ig deposition was not followed by activation of the complement cascade (Fig. 7E). Finally, compared with control mice, Cx43KO sera exhibited reactivity mainly to a protein of ∼90 kDa in whole brain extracts (Fig. 7G) and in G-Sepharose-purified immune complexes from Cx43KO brains (Fig. 7H). By performing mass spectrometry (Fig. 7I) and immunocytochemistry on transfected primary astrocytes (Fig. 7J), we further identified this protein as Vwa5a (also called LOH11CR2A or BCSC-1), a von Willebrand factor A domain-containing extracellular matrix protein (Whittaker and Hynes, 2002; Naba et al., 2012) whose function is yet unknown. Altogether, these results demonstrate that a specific humoral autoimmune response develops in Cx43KO.
Figure 7.
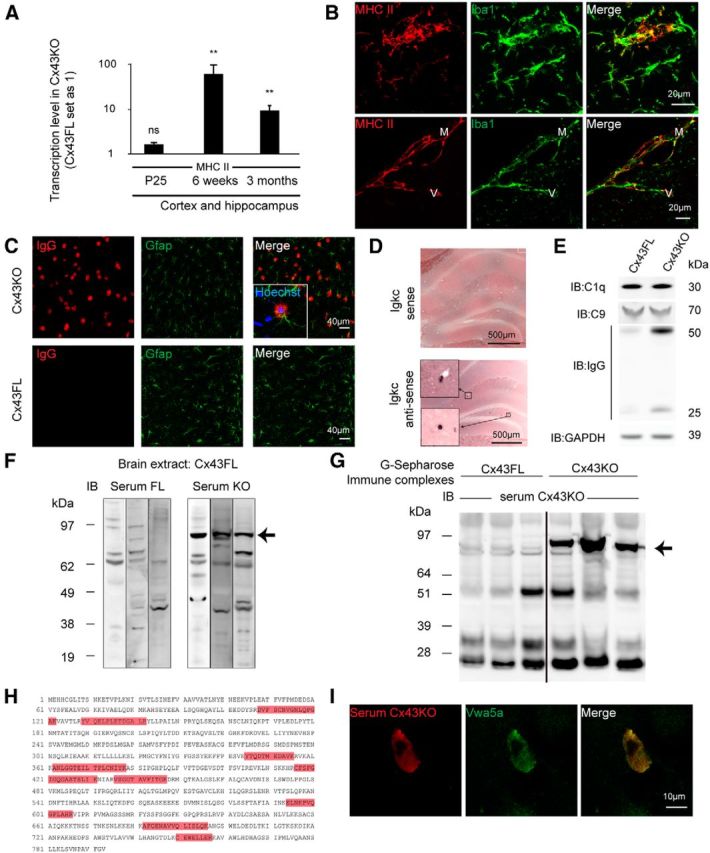
Humoral autoimmune response in the absence of astroglial Cx43. A, qPCR analysis of MHC II expression on Cx43KO and Cx43FL cortex and hippocampus at P25: 1.7 ± 0.2, p = 0.09, n = 4; at 6 weeks, 63.9 ± 38.0, p = 0.002, n = 4; at 3 months, 9.9 ± 2.7, p = 0.002, n = 5. Cx43FL values are set as 1. Data are presented as means ± SEMs. Mann–Whitney two-tailed test. nsp > 0.05, **p < 0.01. Cx43FL values are set as 1. B, Immunohistodetection of MHC II in the brain parenchyma and perivascular space of 6-week-old Cx43KO (n = 3). Microglia and macrophages are labeled with Iba1. MHC II labeling was absent in Cx43FL (our unpublished observation). C, Gfap (green) and IgG (red) double immunostaining in the cortex of 3-month-old Cx43KO and Cx43FL. Squared area shows a labeled astrocyte. D, Detection of Igkc transcript in Cx43KO hippocampus by ISH, with detail of labeled B cells (squared area). Sense probe is the negative control (n = 3). E, C1q, C9, and IgG heavy and light chain immunodetection on Western blots of 3-month-old Cx43KO and Cx43FL brain proteins (n = 3). F, Immunoblot (IB) of Cx43FL brain proteins using three different Cx43KO or Cx43FL sera. A band around 90 kDa is selectively revealed by Cx43KO sera (arrow; n = 7). G, G-Sepharose-purified immune complexes from Cx43KO and Cx43FL brain immunoblotted with a Cx43KO serum. A band around 90 kDa is selectively revealed in Cx43KO-purified proteins (arrow; n = 12). H, Matrix-assisted laser desorption/ionization time-of-flight analysis of these immune complexes revealed the presence of nine peptides (in red) belonging to Vwa5a in Cx43KO immune complexes only (n = 6). I, A Vwa5a-transfected primary astrocyte immunolabeled by both a Cx43KO serum (red) and a Vwa5a-specific antibody (green; n = 3).
Discussion
In this study, we address the role that astroglial Cx43, a gap junction protein highly expressed at the gliovascular interface (Simard et al., 2003; Ezan et al., 2012), plays in the brain vascular physiology. We show that the absence of astroglial Cx43 compromises the ability of the brain to maintain immune quiescence (Lampron et al., 2013), with recruitment of leukocytes, including B and T lymphocytes, macrophages and neutrophils, enhanced antigen presentation, and autoimmune reactions, in particular against Vwa5a, a yet uncharacterized extracellular matrix component of the brain (Whittaker and Hynes, 2002; Naba et al., 2012). This loss of immune quiescence does not originate from BBB breakdown, although BBB weakens progressively in Cx43KO. It is not linked to glial activation either, because this was only detected after the initiation of immune infiltration. In contrast, it correlates with an increase in TNFα, endothelial adhesion molecule, and chemokine expression in brain vessels, suggesting that endothelial activation and chemoattraction may be instrumental to the recruitment of immune cells. A small increase of IL-1β was also found in the brain of CX43KO, but its origin could not be clarified. The question then arises of how astroglial Cx43 participates in the regulation of these immune recruitment mechanisms. Cx43 mediates approximately half of the astroglial coupling (Rouach et al., 2008). It controls astrocyte cell volume, glutamatergic synaptic activity of hippocampal neurons (Chever et al., 2014b), and spreading depression velocity (Theis et al., 2003). Cx43 forms functional Hc in astrocytes in basal conditions, which tune the moment-to-moment glutamatergic synaptic transmission (Chever et al., 2014a). Cx43 channel-independent functions regulate astroglial morphology and polarity (Olk et al., 2009; Francis et al., 2011). Altogether, alteration of these functions could turn on the innate immune system and instigate an autoimmune response in the nervous system. In particular, glutamate levels have been shown to control MHC II and neuroinflammatory molecules expression (Jensen et al., 2013). Of note, although immune infiltration and endothelial activation started postnatally at ∼6 weeks in Cx43KO, we cannot exclude a link with developmental effects attributable to the early deletion of Cx43. Indeed, the human GFAP promoter used to drive Cx43 deletion in astrocytes is also active in neuroprogenitors, which express Cx43 (Casper and McCarthy, 2006). Along this line, knockdown of Cx43 nonchannel functions has been shown to impair the migration of neurons through adhesive interactions with radial glial fibers during embryogenesis (Elias et al., 2007, 2010). However, this phenotype was not seen even in the Cx43 full knock-out, probably because of compensation mechanisms (Elias et al., 2007), and their implication in effects described here is therefore unlikely.
Interestingly, endothelial activation and immune infiltration followed a bell-shaped progression, peaking at ∼6 weeks. This suggests that triggering events may either occur around this particular stage or that a threshold was reached in Cx43KO beyond which immune mechanisms were activated. Comparing astroglial functions in the absence of Cx43 at different postnatal stages may help to understand this point. Inflammation then resolved spontaneously with age, an outcome that might be related to the presence of infiltrated T lymphocytes in Cx43KO expressing high levels of anti-inflammatory IL-10 (O'Garra and Vieira, 2007). Moreover, upregulation of the anti-inflammatory protein IL-1ra by microglia, likely in response to IL-10 (Lang et al., 2002), suggests that they are polarized into a M2a immunomodulatory phenotype with a role in tissue reparation (Rivest, 2009; David and Kroner, 2011). Thus, self-limited inflammatory conditions were set in Cx43KO, probably restraining immunopathological effects.
The role of astrocytes in the brain immunity has been documented mainly in pathological conditions (Alvarez et al., 2013; Jensen et al., 2013). In particular, astrocytes have been shown to sense the presence of pathogens and to control the entry of immune cells during neuroinflammation through the secretion of factors such as VEGF, Sonic Hedgehog, or retinoic acid (Alvarez et al., 2011; Argaw et al., 2012; Mizee et al., 2014). Interestingly, we here show that astroglial Cx43 regulates the brain/immune system interrelationship in a nonpathological context. That being said, what could be the consequences of such modified brain/immune system homeostasis on brain functions? Loss of immune quiescence, with immune-competent cells abnormally crossing the BBB and developing autoimmune mechanisms, could be deleterious as suggested in pathological situations linked to a decreased expression of astroglial Cx43, i.e., multiple sclerosis and neuromyelytis optica (Masaki et al., 2013), depression (Ernst et al., 2011; Sun et al., 2012), and epilepsy (Kovacs et al., 2012). Moreover, the observed upregulation of chemokines, which are known neurotransmitters/neuromodulators (Rostene et al., 2011), may directly account for synaptic transmission defects reported in Cx43KO mice (Theis et al., 2003; Chever et al., 2014b).
Overall, our results reveal a novel and unexpected role for astroglial Cx43 in setting immune quiescence in the brain and designate Vwa5a as a new autoimmune target in the brain. These findings may open new avenues in the design of tools for the control of immune cell migration and antigen presentation in the normal brain, which are altered in several neurological disorders, including chronic immunopathological disorders such as multiple sclerosis (Wraith and Nicholson, 2012), epilepsy (Rodgers et al., 2009), and cognitive and psychiatric impairments (Jones and Thomsen, 2013; Najjar et al., 2013).
Footnotes
This work was supported by Fondation pour l'Aide à la Recherche sur la Sclérose en Plaques (ARSEP) and the MemoLife Laboratory of Excellence. We thank Vanessa Moreira, Nicole Quenech'du, and Jérémie Teillon for technical help and Sophie Hüe, Tarek Sharshar, Marika Sarfati, and Roberto Bruzzone for discussions and critical reading of this manuscript. We thank Prof. B. Paul Morgan for providing us with the anti-rat C9neo antibodies.
The authors declare no competing financial interests.
References
- Abbott NJ, Rönnbäck L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7:41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- Alvarez JI, Dodelet-Devillers A, Kebir H, Ifergan I, Fabre PJ, Terouz S, Sabbagh M, Wosik K, Bourbonnière L, Bernard M, van Horssen J, de Vries HE, Charron F, Prat A. The Hedgehog pathway promotes blood-brain barrier integrity and CNS immune quiescence. Science. 2011;334:1727–1731. doi: 10.1126/science.1206936. [DOI] [PubMed] [Google Scholar]
- Alvarez JI, Katayama T, Prat A. Glial influence on the blood brain barrier. Glia. 2013;61:1939–1958. doi: 10.1002/glia.22575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arama J, Boulay AC, Bosc C, Delphin C, Loew D, Rostaing P, Amigou E, Ezan P, Wingertsmann L, Guillaud L, Andrieux A, Giaume C, Cohen-Salmon M. Bmcc1s, a novel brain-isoform of Bmcc1, affects cell morphology by regulating MAP6/STOP functions. PLoS One. 2012;7:e35488. doi: 10.1371/journal.pone.0035488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argaw AT, Asp L, Zhang J, Navrazhina K, Pham T, Mariani JN, Mahase S, Dutta DJ, Seto J, Kramer EG, Ferrara N, Sofroniew MV, John GR. Astrocyte-derived VEGF-A drives blood-brain barrier disruption in CNS inflammatory disease. J Clin Invest. 2012;122:2454–2468. doi: 10.1172/JCI60842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bélanger M, Allaman I, Magistretti PJ. Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011;14:724–738. doi: 10.1016/j.cmet.2011.08.016. [DOI] [PubMed] [Google Scholar]
- Casper KB, McCarthy KD. GFAP-positive progenitor cells produce neurons and oligodendrocytes throughout the CNS. Mol Cell Neurosci. 2006;31:676–684. doi: 10.1016/j.mcn.2005.12.006. [DOI] [PubMed] [Google Scholar]
- Chever O, Lee CY, Rouach N. Astroglial connexin43 hemichannels tune Basal excitatory synaptic transmission. J Neurosci. 2014a;34:11228–11232. doi: 10.1523/JNEUROSCI.0015-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chever O, Pannasch U, Ezan P, Rouach N. Astroglial connexin 43 sustains glutamatergic synaptic efficacy. Philos Trans R Soc Lond B Biol Sci. 2014b;369:20130596. doi: 10.1098/rstb.2013.0596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke SR, Shetty AK, Bradley JL, Turner DA. Reactive astrocytes express the embryonic intermediate neurofilament nestin. Neuroreport. 1994;5:1885–1888. doi: 10.1097/00001756-199410000-00011. [DOI] [PubMed] [Google Scholar]
- Dagenais C, Rousselle C, Pollack GM, Scherrmann JM. Development of an in situ mouse brain perfusion model and its application to mdr1a P-glycoprotein-deficient mice. J Cereb Blood Flow Metab. 2000;20:381–386. doi: 10.1097/00004647-200002000-00020. [DOI] [PubMed] [Google Scholar]
- David S, Kroner A. Repertoire of microglial and macrophage responses after spinal cord injury. Nat Rev Neurosci. 2011;12:388–399. doi: 10.1038/nrn3053. [DOI] [PubMed] [Google Scholar]
- Denes A, Thornton P, Rothwell NJ, Allan SM. Inflammation and brain injury: acute cerebral ischaemia, peripheral and central inflammation. Brain Behav Immun. 2010;24:708–723. doi: 10.1016/j.bbi.2009.09.010. [DOI] [PubMed] [Google Scholar]
- Duggal N, Schmidt-Kastner R, Hakim AM. Nestin expression in reactive astrocytes following focal cerebral ischemia in rats. Brain Res. 1997;768:1–9. doi: 10.1016/S0006-8993(97)00588-X. [DOI] [PubMed] [Google Scholar]
- Elias LA, Wang DD, Kriegstein AR. Gap junction adhesion is necessary for radial migration in the neocortex. Nature. 2007;448:901–907. doi: 10.1038/nature06063. [DOI] [PubMed] [Google Scholar]
- Elias LA, Turmaine M, Parnavelas JG, Kriegstein AR. Connexin 43 mediates the tangential to radial migratory switch in ventrally derived cortical interneurons. J Neurosci. 2010;30:7072–7077. doi: 10.1523/JNEUROSCI.5728-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst C, Nagy C, Kim S, Yang JP, Deng X, Hellstrom IC, Choi KH, Gershenfeld H, Meaney MJ, Turecki G. Dysfunction of astrocyte connexins 30 and 43 in dorsal lateral prefrontal cortex of suicide completers. Biol Psychiatry. 2011;70:312–319. doi: 10.1016/j.biopsych.2011.03.038. [DOI] [PubMed] [Google Scholar]
- Ezan P, André P, Cisternino S, Saubaméa B, Boulay AC, Doutremer S, Thomas MA, Quenech'du N, Giaume C, Cohen-Salmon M. Deletion of astroglial connexins weakens the blood-brain barrier. J Cereb Blood Flow Metab. 2012;32:1457–1467. doi: 10.1038/jcbfm.2012.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farina C, Aloisi F, Meinl E. Astrocytes are active players in cerebral innate immunity. Trends Immunol. 2007;28:138–145. doi: 10.1016/j.it.2007.01.005. [DOI] [PubMed] [Google Scholar]
- Francis R, Xu X, Park H, Wei CJ, Chang S, Chatterjee B, Lo C. Connexin43 modulates cell polarity and directional cell migration by regulating microtubule dynamics. PLoS One. 2011;6:e26379. doi: 10.1371/journal.pone.0026379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaume C, Koulakoff A, Roux L, Holcman D, Rouach N. Astroglial networks: a step further in neuroglial and gliovascular interactions. Nat Rev Neurosci. 2010;11:87–99. doi: 10.1038/nrn2757. [DOI] [PubMed] [Google Scholar]
- Hammon M, Herrmann M, Bleiziffer O, Pryymachuk G, Andreoli L, Munoz LE, Amann KU, Mondini M, Gariglio M, Airó P, Schellerer VS, Hatzopoulos AK, Horch RE, Kneser U, Stürzl M, Naschberger E. Role of guanylate binding protein-1 in vascular defects associated with chronic inflammatory diseases. J Cell Mol Med. 2011;15:1582–1592. doi: 10.1111/j.1582-4934.2010.01146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haudek-Prinz VJ, Klepeisz P, Slany A, Griss J, Meshcheryakova A, Paulitschke V, Mitulovic G, Stockl J, Gerner C. Proteome signatures of inflammatory activated primary human peripheral blood mononuclear cells. J Proteomics. 2012 doi: 10.1016/j.jprot.2012.07.012. 76 Spec No.:150–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacobas S, Iacobas DA, Spray DC, Scemes E. The connexin43-dependent transcriptome during brain development: importance of genetic background. Brain Res. 2012;1487:131–139. doi: 10.1016/j.brainres.2012.05.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen CJ, Massie A, De Keyser J. Immune players in the CNS: the astrocyte. J Neuroimmune Pharmacol. 2013;8:824–839. doi: 10.1007/s11481-013-9480-6. [DOI] [PubMed] [Google Scholar]
- Jones KA, Thomsen C. The role of the innate immune system in psychiatric disorders. Mol Cell Neurosci. 2013;53:52–62. doi: 10.1016/j.mcn.2012.10.002. [DOI] [PubMed] [Google Scholar]
- Jourdren L, Bernard M, Dillies MA, Le Crom S. Eoulsan: a cloud computing-based framework facilitating high throughput sequencing analyses. Bioinformatics. 2012;28:1542–1543. doi: 10.1093/bioinformatics/bts165. [DOI] [PubMed] [Google Scholar]
- Kovacs R, Heinemann U, Steinhauser C. Mechanisms underlying blood-brain barrier dysfunction in brain pathology and epileptogenesis: role of astroglia. Epilepsia. 2012;53(Suppl 6):53–59. doi: 10.1111/j.1528-1167.2012.03703.x. [DOI] [PubMed] [Google Scholar]
- Kunze A, Congreso MR, Hartmann C, Wallraff-Beck A, Hüttmann K, Bedner P, Requardt R, Seifert G, Redecker C, Willecke K, Hofmann A, Pfeifer A, Theis M, Steinhäuser C. Connexin expression by radial glia-like cells is required for neurogenesis in the adult dentate gyrus. Proc Natl Acad Sci U S A. 2009;106:11336–11341. doi: 10.1073/pnas.0813160106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampron A, Elali A, Rivest S. Innate immunity in the CNS: redefining the relationship between the CNS and Its environment. Neuron. 2013;78:214–232. doi: 10.1016/j.neuron.2013.04.005. [DOI] [PubMed] [Google Scholar]
- Lang R, Patel D, Morris JJ, Rutschman RL, Murray PJ. Shaping gene expression in activated and resting primary macrophages by IL-10. J Immunol. 2002;169:2253–2263. doi: 10.4049/jimmunol.169.5.2253. [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz SE, Zhao Y, Gulinello M, Lee SC, Raine CS, Brosnan CF. Deletion of astrocyte connexins 43 and 30 leads to a dysmyelinating phenotype and hippocampal CA1 vacuolation. J Neurosci. 2009;29:7743–7752. doi: 10.1523/JNEUROSCI.0341-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masaki K, Suzuki SO, Matsushita T, Matsuoka T, Imamura S, Yamasaki R, Suzuki M, Suenaga T, Iwaki T, Kira J. Connexin 43 astrocytopathy linked to rapidly progressive multiple sclerosis and neuromyelitis optica. PLoS One. 2013;8:e72919. doi: 10.1371/journal.pone.0072919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May D, Tress O, Seifert G, Willecke K. Connexin47 protein phosphorylation and stability in oligodendrocytes depend on expression of Connexin43 protein in astrocytes. J Neurosci. 2013;33:7985–7996. doi: 10.1523/JNEUROSCI.5874-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizee MR, Nijland PG, van der Pol SM, Drexhage JA, van Het Hof B, Mebius R, van der Valk P, van Horssen J, Reijerkerk A, de Vries HE. Astrocyte-derived retinoic acid: a novel regulator of blood-brain barrier function in multiple sclerosis. Acta Neuropathol. 2014;128:691–703. doi: 10.1007/s00401-014-1335-6. [DOI] [PubMed] [Google Scholar]
- Naba A, Clauser KR, Hoersch S, Liu H, Carr SA, Hynes RO. The matrisome: in silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol Cell Proteomics. 2012;11:M111.014647. doi: 10.1074/mcp.M111.014647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najjar S, Pearlman DM, Alper K, Najjar A, Devinsky O. Neuroinflammation and psychiatric illness. J Neuroinflammation. 2013;10:43. doi: 10.1186/1742-2094-10-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Garra A, Vieira P. T(H)1 cells control themselves by producing interleukin-10. Nat Rev Immunol. 2007;7:425–428. doi: 10.1038/nri2097. [DOI] [PubMed] [Google Scholar]
- Olk S, Zoidl G, Dermietzel R. Connexins, cell motility, and the cytoskeleton. Cell Motil Cytoskeleton. 2009;66:1000–1016. doi: 10.1002/cm.20404. [DOI] [PubMed] [Google Scholar]
- Pannasch U, Vargová L, Reingruber J, Ezan P, Holcman D, Giaume C, Syková E, Rouach N. Astroglial networks scale synaptic activity and plasticity. Proc Natl Acad Sci U S A. 2011;108:8467–8472. doi: 10.1073/pnas.1016650108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannasch U, Freche D, Dallérac G, Ghézali G, Escartin C, Ezan P, Cohen-Salmon M, Benchenane K, Abudara V, Dufour A, Lübke JH, Déglon N, Knott G, Holcman D, Rouach N. Connexin 30 sets synaptic strength by controlling astroglial synapse invasion. Nat Neurosci. 2014;17:549–558. doi: 10.1038/nn.3662. [DOI] [PubMed] [Google Scholar]
- Petzold GC, Murthy VN. Role of astrocytes in neurovascular coupling. Neuron. 2011;71:782–797. doi: 10.1016/j.neuron.2011.08.009. [DOI] [PubMed] [Google Scholar]
- Poullet P, Carpentier S, Barillot E. myProMS, a web server for management and validation of mass spectrometry-based proteomic data. Proteomics. 2007;7:2553–2556. doi: 10.1002/pmic.200600784. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM, Engelhardt B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat Rev Immunol. 2012;12:623–635. doi: 10.1038/nri3265. [DOI] [PubMed] [Google Scholar]
- Rivest S. Regulation of innate immune responses in the brain. Nat Rev Immunol. 2009;9:429–439. doi: 10.1038/nri2565. [DOI] [PubMed] [Google Scholar]
- Rodgers KM, Hutchinson MR, Northcutt A, Maier SF, Watkins LR, Barth DS. The cortical innate immune response increases local neuronal excitability leading to seizures. Brain. 2009;132:2478–2486. doi: 10.1093/brain/awp177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostène W, Dansereau MA, Godefroy D, Van Steenwinckel J, Reaux-Le Goazigo A, Melik-Parsadaniantz S, Apartis E, Hunot S, Beaudet N, Sarret P. Neurochemokines: a menage a trois providing new insights on the functions of chemokines in the central nervous system. J Neurochem. 2011;118:680–694. doi: 10.1111/j.1471-4159.2011.07371.x. [DOI] [PubMed] [Google Scholar]
- Rouach N, Koulakoff A, Abudara V, Willecke K, Giaume C. Astroglial metabolic networks sustain hippocampal synaptic transmission. Science. 2008;322:1551–1555. doi: 10.1126/science.1164022. [DOI] [PubMed] [Google Scholar]
- Sá-Pereira I, Brites D, Brito MA. Neurovascular unit: a focus on pericytes. Mol Neurobiol. 2012;45:327–347. doi: 10.1007/s12035-012-8244-2. [DOI] [PubMed] [Google Scholar]
- Simard M, Arcuino G, Takano T, Liu QS, Nedergaard M. Signaling at the gliovascular interface. J Neurosci. 2003;23:9254–9262. doi: 10.1523/JNEUROSCI.23-27-09254.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun JD, Liu Y, Yuan YH, Li J, Chen NH. Gap junction dysfunction in the prefrontal cortex induces depressive-like behaviors in rats. Neuropsychopharmacology. 2012;37:1305–1320. doi: 10.1038/npp.2011.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takasato Y, Rapoport SI, Smith QR. An in situ brain perfusion technique to study cerebrovascular transport in the rat. Am J Physiol. 1984;247:H484–H493. doi: 10.1152/ajpheart.1984.247.3.H484. [DOI] [PubMed] [Google Scholar]
- Theis M, Mas C, Döring B, Krüger O, Herrera P, Meda P, Willecke K. General and conditional replacement of connexin43-coding DNA by a lacZ reporter gene for cell-autonomous analysis of expression. Cell Commun Adhes. 2001;8:383–386. doi: 10.3109/15419060109080758. [DOI] [PubMed] [Google Scholar]
- Theis M, Jauch R, Zhuo L, Speidel D, Wallraff A, Döring B, Frisch C, Söhl G, Teubner B, Euwens C, Huston J, Steinhäuser C, Messing A, Heinemann U, Willecke K. Accelerated hippocampal spreading depression and enhanced locomotory activity in mice with astrocyte-directed inactivation of connexin43. J Neurosci. 2003;23:766–776. doi: 10.1523/JNEUROSCI.23-03-00766.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittaker CA, Hynes RO. Distribution and evolution of von Willebrand/integrin A domains: widely dispersed domains with roles in cell adhesion and elsewhere. Mol Biol Cell. 2002;13:3369–3387. doi: 10.1091/mbc.E02-05-0259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wraith DC, Nicholson LB. The adaptive immune system in diseases of the central nervous system. J Clin Invest. 2012;122:1172–1179. doi: 10.1172/JCI58648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousif S, Marie-Claire C, Roux F, Scherrmann JM, Declèves X. Expression of drug transporters at the blood-brain barrier using an optimized isolated rat brain microvessel strategy. Brain Res. 2007;1134:1–11. doi: 10.1016/j.brainres.2006.11.089. [DOI] [PubMed] [Google Scholar]
- Zhang J, Dublin P, Griemsmann S, Klein A, Brehm R, Bedner P, Fleischmann BK, Steinhäuser C, Theis M. Germ-line recombination activity of the widely used hGFAP-Cre and nestin-Cre transgenes. PLoS One. 2013;8:e82818. doi: 10.1371/journal.pone.0082818. [DOI] [PMC free article] [PubMed] [Google Scholar]