

Abstract
Parvalbumin (PV)-expressing perisoma-inhibiting interneurons (PIIs) of the dentate gyrus integrate rapidly correlated synaptic inputs and generate short-duration action potentials that propagate along the axon to their output synapses, supporting fast inhibitory signaling onto their target cells. Here we show that PV-PIIs in rat and mouse dentate gyrus (DG) integrate their intrinsic activity over time and can turn into a persistent firing mode characterized by the ability to generate long-lasting trains of action potentials at ∼50 Hz in the absence of additional inputs. Persistent firing emerges in the axons remote from the axon initial segment and markedly depends on hyperpolarization-activated cyclic nucleotide-gated channel (HCNC) activation. Persistent firing properties are modulated by intracellular Ca2+ levels and somatic membrane potential. Detailed computational single-cell PIIs models reveal that HCNC-mediated conductances can contribute to persistent firing during conditions of a shift in their voltage activation curve to more depolarized potentials. Paired recordings from PIIs and their target granule cells show that persistent firing supports strong inhibitory output signaling. Thus, persistent firing may emerge during conditions of intense activation of the network, thereby providing silencing to the circuitry and the maintenance of sparse activity in the dentate gyrus.
Keywords: basket cell, dentate gyrus, GABA, HCN, interneuron, persistent firing
Introduction
GABAergic inhibitory interneurons, in particular parvalbumin (PV)-expressing perisoma-inhibiting interneurons (PIIs), control fundamental functions of neuronal networks (Isaacson and Scanziani, 2011). They define the activity of large principal cell populations by determining the probability and timing of action potential discharges (Cobb et al., 1995; Pouille and Scanziani, 2001) and the generation of fast rhythmic activity patterns in neuronal networks (Buzsáki and Draguhn, 2004). These functional roles depend on the ability of PV-PIIs to discharge high-frequency action potentials that traverse reliably along their dense axon to their output synapses (Hu and Jonas, 2014).
In the classical view, PIIs generate action potentials at their axon initial segment during synaptic excitation (Hu and Jonas, 2014). However, recent investigations highlight the ability of neuronal subtypes to sustain long-lasting discharges in the absence of incoming excitatory synaptic inputs (Major and Tank, 2004; Sasaki, 2013). This activity mode was termed “persistent firing” and first identified in principal cells (Egorov et al., 2002; Knauer et al., 2013; Thuault et al., 2013). Recent studies demonstrated persistent firing in cortical interneuron subtypes, including CA1 perforant path-associated cells (Sheffield et al., 2011), CA1 and neocortical PV-expressing fast-spiking, ivy and neurogliaform cells (Krook-Magnuson et al., 2011; Suzuki et al., 2014). In these cells, persistent firing could be induced during integration of long-lasting trains of somatically evoked action potentials. In CA1 perforant path-associated cells, persistent firing emerged in the axon and required gap junction coupling and intracellular Ca2+ rise (Sheffield et al., 2013). However, the mechanism underlying persistent firing generation in other interneuron types remains essentially unknown. Here, we addressed this question by performing whole-cell recordings from PV-PIIs in slice preparations of the rodent dentate gyrus (DG) and show that persistent firing emerges in the remote axon during physiologically relevant synaptic activation. We provide experimental and theoretical evidence that persistent firing induction in PV-PIIs depends on hyperpolarization-activated cyclic nucleotide-gated channel (HCNC) activation and is reduced by intracellular Ca2+ rise.
Materials and Methods
Electrophysiology.
Acute slices were obtained from Wistar rats (P17–P23) or PV–Cre mice (P35–P70; The Jackson Laboratory; http://jaxmice.jax.org/strain/008069.html) of either sex in accordance with national and institutional legislations (licenses G-11/53 and X-12/20D). Slice preparation and whole-cell patch-clamp recordings (at 30–34°C) were performed as described previously (Bartos et al., 2001). Transverse hippocampal slices (300–350 μm) were incubated in ACSF containing 125 mm NaCl, 25 mm NaHCO3, 2.5 mm KCl, 1.25 mm NaH2PO4, 25 mm d-glucose, 2 mm CaCl2 and 1 mm MgCl2 (oxygenated with 95% O2/5% CO2) for 20–30 min (34°C) and stored at room temperature. Recording pipettes had resistances of 2–5 MΩ when filled with solution containing 120 mm K-gluconate, 20 mm KCl, 10 mm HEPES, 2 mm MgCl2, 2 mm Na2ATP, 10 mm EGTA, and 0.2% biocytin, pH 7.2, 290–310 mOsm. In some recordings, EGTA was replaced by BAPTA. For granule cell (GC) recordings, K-gluconate was replaced by KCl. Signals were amplified (Multiclamp 700B; Molecular Devices), filtered at 5 kHz, and digitized at 20–40 kHz (Power1401-2 interface; Cambridge Electronic Design). Series resistance (8–20 MΩ) was compensated in current clamp but not in voltage clamp [holding potential (Vhold) of approximately −70 mV]. For stimulus generation and data acquisition, we used custom-made Igor-based programs (FPulse). Paired recordings were performed in the presence of 4 mm kynurenic acid (KA). A subset of PIIs were recorded (n = 77) in the presence of 2 mm KA/5 μm gabazine. Persistent firing was induced by various protocols: (1) Protocol 1, somatic long-lasting suprathreshold current injections (3 s, 0.5–1.3 nA; every 15 s, 10–15 times); (2) Protocol 2, trains of short-duration suprathreshold somatic current injections (2 ms, 1–2 nA) at 30, 50, 75, or 100 Hz until persistent firing was induced; (3) Protocol 3, activation of the perforant path with a stimulation pipette filled with HEPES-buffered Na+-rich solution placed in the outer molecular layer (25 pulses at 30 Hz, repeated at 3 Hz); or (4) Protocol 4, trains of antidromic action potentials evoked with an extracellular electrode placed in the GC layer (GCL; 0.1 ms, 50 Hz pulses).
Data analysis.
Data were analyzed with IgorPro (Wavemetrics; Bartos et al., 2001). Action potential threshold was defined as the first voltage change exceeding 30 V/s. During paired recordings, single action potentials were induced in PIIs (2 ms, 1–2 nA), and unitary IPSCs (uIPSCs) were recorded in GCs. The functional properties of uIPSCs evoked by the last 30 action potentials (induced by somatic current injection) before persistent firing induction were compared with the last 30 uIPSCs during persistent firing trains. They showed comparable release probability derived from the failure rates (somatic, 26 ± 11% vs persistent firing, 28 ± 9%; eight pairs; p = 0.8). Synaptic latency was defined as the time from the steepest point in the rise of the presynaptic action potential to the onset of the uIPSC. Statistical analysis was performed using SigmaPlot 11 (Systat). Differences between two samples were assessed with a two-tailed unpaired or paired t test for independent and related samples, respectively. Nonparametric Mann–Whitney U tests were used if normality tests failed. Several datasets were compared with a one-way ANOVA on ranks and a post hoc Dunn's method for pairwise comparisons unless otherwise stated. Significance levels are indicated as p values. Data are presented as mean ± SEM.
Histology and immunohistochemistry.
Biocytin-filled cells were fixed in 4% PFA (1–24 h) and washed in 0.1 m phosphate buffer and subsequently in 0.025 m PBS, pH 7.3. Selected slices were incubated with primary antibodies against PV (polyclonal rabbit, 1:1000; Swant), HCN1 (rabbit polyclonal, 1:100; Alomone Labs), or Ankyrin-G (mouse, 1:100; Life Technologies) in PBS containing 10% goat serum and 0.2% Triton X-100 for 36 h (4°C). Cy3 anti-rabbit (1:500; Jackson ImmunoResearch) or Alexa Fluor-488 (1:500, anti-mouse) were incubated together with streptavidin-conjugated Alexa Fluor-647 (1:1000) for 24 h (4°C) and mounted in Mowiol. To test selectivity of the HCN1 antibody, we omitted the primary antibody. This resulted in the complete absence of specific staining (three of three PIIs; data not shown). Labeled neurons were examined with a confocal microscope (Zeiss LSM-710). Data presented in Figure 1 are based entirely on morphologically identified PIIs. A subset of these PIIs (24 cells) was reconstructed using NIH ImageJ Simple Neurite Tracer plug-in to determine their axonal length (Fig. 2C).
Figure 1.
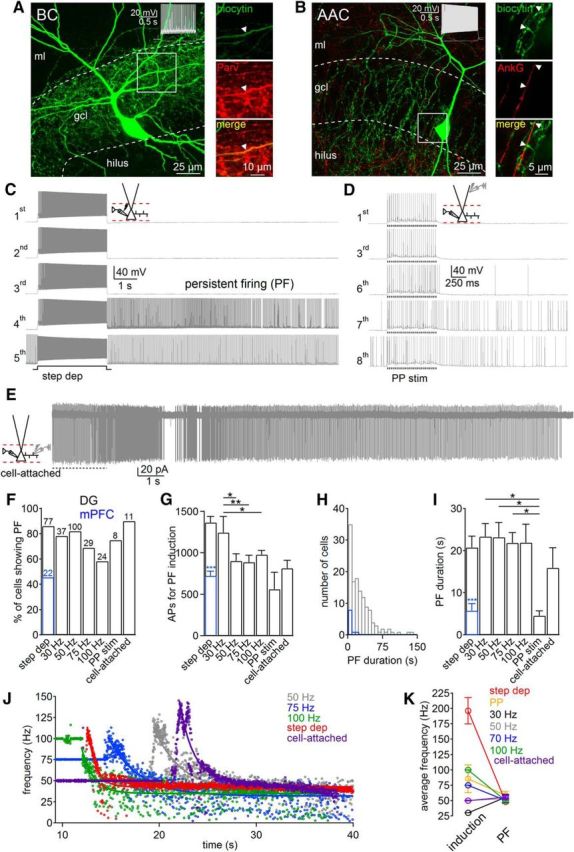
Induction conditions for persistent firing (PF) in PV-PIIs. A, Left, Confocal image of a DG basket cell (BC), filled with biocytin, visualized with streptavidin-conjugated Alexa Fluor-647. Right, Antibody-labeling against PV. B, Left, Confocal image of an axo-axonic cell (AAC). Right, Immunohistochemical labeling of Ankyrin-G (AnkG). Note putative AAC-mediated synapses at axon initial segments. Insets, Fast-spiking PII phenotype. C–E, Representative PII recordings show persistent firing induction with Protocol 1 (C), with perforant path (PP) stimulation (Protocol 3; D), and during cell-attached PII recordings (Protocol 4; E). F–I, Bars summarize the percentage of PIIs expressing persistent firing (F), the number of action potentials (APs) required for induction (G), and the duration of persistent firing (I) depending on the applied protocol. H, Histogram shows distribution of persistent firing duration (131 PIIs). J, Time course of persistent firing frequency induced by Protocol 1 (red), Protocol 2 (50–100 Hz), and Protocol 4 (cell attached; purple). Lines represent exponential fits to the data. K, Mean persistent firing discharge frequency is independent on activity during the induction protocol. Blue bars, Data obtained from PV-PIIs in the medial prefrontal cortex (mPFC). Step-dep, Stepwise depolarization for Protocol 1; somatically evoked AP trains at 30, 50, 75, or 100 Hz for Protocol 2; PP, perforant-path stimulation for Protocol 3; cell-attached recordings during extracellular PII axon stimulation (50 Hz) for Protocol 4; ml, molecular layer. Values in F represent the number of experiments. Circles and bars with lines represent mean ± SEM. *p ≤ 0.05; **p ≤ 0.01. G, Repeated-measures ANOVA (for Protocol 2, p = 0.007, 50 degrees of freedom) and post hoc Holm–Sidak pairwise comparison.
Figure 2.
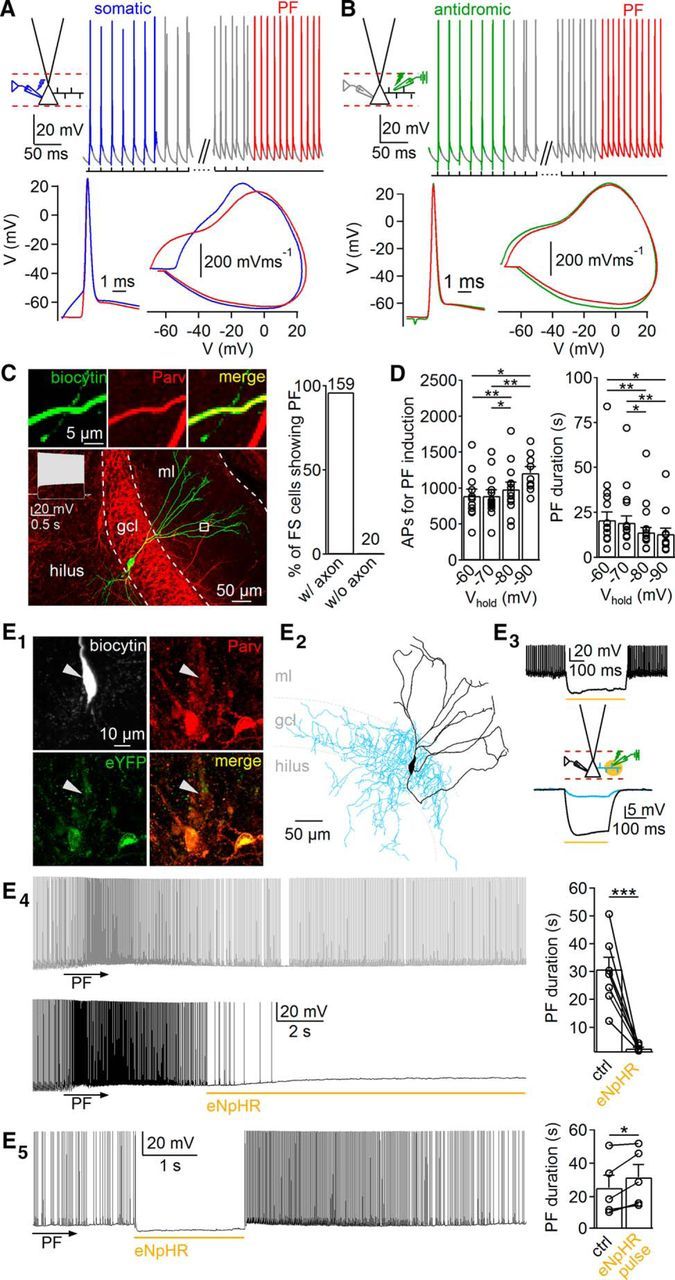
Persistent firing is induced in PII axons. A, B, Top, Representative traces from persistent firing (PF) induced by Protocol 2 (50 Hz; A) and by antidromic stimulation (50 Hz; B). Bottom, Superimposed individual action potentials (APs) and corresponding phase plots evoked by somatic current injection (blue), antidromic stimulation (green), or persistent firing induction (red). C, Left, Confocal image of a fast-spiking (FS) PV cell with cut axon. Insets, Colocalization of biocytin and PV in PII dendrites. Right, Graph summarizes the dependence of persistent firing in PV FS cells on the presence of the axon (without, mean axon length of 2.1 mm, 20 cells; with the axon, 143.4 ± 4.4 mm, 4 PIIs). ml, Molecular layer. D, Bar graphs summarize the effects of somatic Vhold on AP number required for persistent firing induction (left) and persistent firing duration (right). E1, Confocal images show expression of pAAV–eNpHR–eYFP in PV cells. Note the weak labeling of recorded PII attributable to PV wash out. E2, Reconstruction of the PV-PII shown in E1. E3, Top, FS discharge in the PV-PII shown in E1 and E2 is blocked by spot illumination of the soma (565 nm). Bottom, Effect of spot illumination of the PII axon (blue) and soma (black) on somatic voltage. E4, Left, Traces show persistent firing before (gray) and after (black) spot illumination of the PII axon. Right, Bar graph summarizes the effect of eNpHR activation on persistent firing duration. E5, Left, Short illumination (2 s) of the PII axon during persistent firing. Right, Bar graph summarizes the effect of short eNpHR activation on persistent firing duration. Yellow lines represent time of illumination. Circles connected by lines indicate individual experiments. Bars with lines represent means ± SEM. ctrl, Control. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001.
Optophysiology.
Recombinant adeno-associated viruses (AAVs) encoding halorhodopsin (eNpHR) were injected into ventral DGs (related to bregma: y, 2.55 mm; x, 2.9 mm; z, 2.3–2.9 mm) of PV–Cre mice as reported previously (Savanthrapadian et al., 2014). The expression cassette of the AAV contained enhanced yellow fluorescent protein (eYFP) and eNpHR between two inverted incompatible tandem loxP sites (pAAV–doublefloxed–eNpHR–EYFP–WPRE–pA; catalog #20949; Addgene). Slices were prepared >14 d after injection. A yellow light spot (565 nm; ∼50 μm diameter; pE-100; CoolLED) was applied for either 2 s (short illumination) or >5 s (long illumination) to activate eNpHR.
Single-cell modeling.
Single-cell simulations were performed with NEURON 7.3 using detailed passive models of PIIs (Nörenberg et al., 2010). Cable parameters were set uniformly with the specific axial resistance Ri of 170 Ω cm, the specific membrane resistance Rm of 10 kΩ cm2, and the specific membrane capacitance Cm of 0.9 μF/cm−2. The leak conductance (0.1 mS/cm2) had a reversal potential (Erev) of −79 mV. To reproduce the fast-spiking phenotype, we introduced modified Na+ and K+ conductances (Wang and Buzsáki, 1996) at experimentally defined densities (Hu et al., 2010; Hu and Jonas, 2014; ḠNa+, 100, 200, and 750 mS/cm−2 at dendrites, soma, and axon, respectively; uniform distribution of ḠK+, 200 mS/cm−2). The total Ih conductance (ḠIh) was inserted uniformly (Golding et al. 2005) with a density of 5 pS/μm−2 and channel properties defined previously in DG PIIs (Aponte et al., 2006). The default V½, which defines the half-activation potential of ḠIh, was −84 mV (Franz et al., 2000; Aponte et al., 2006). Erev was −27.4 mV, and the slope factor k was set to 13.1. The time step in simulations was 5 μs. Simulations were repeated in three different PII models (Nörenberg et al., 2010). Circles in Figure 4 represent the mean of three PII simulations.
Figure 4.

Single-cell PII modeling reveals the contribution of HCNCs in persistent firing during a positive shift in the membrane potential of their half-maximal activation. A, Morphology of one reconstructed PII used for single-cell modeling. Inset, Voltage trace in the presence (blue) and absence (gray) of the active Ih conductance (ḠIh) during negative current application to the soma (−100 pA, 1 s). Calibration: 250 ms, 1 mV. Right, Train of action potentials (APs) evoked at the soma (gray; 600 pA, 1 s) and propagating to the axon (red), recorded at a somatic distance of 531 μm. Recording sites correspond to pipettes shown schematically in A. GIh for both recording sites is shown below. B, Same as A with a midpoint potential of ḠIh activation (V½) set to −70 mV at times indicated by the arrow. Note the large GIh-induced axonal APs propagating to the soma. Right inset, Overshooting spikes result in inactivation of GIh and its recovery during the spike after hyperpolarization. C, AP frequency and GIh during persistent firing are plotted against V½. D, AP frequency increases with the density of ḠIh and ḠNa+. Circles represent means of simulations performed in PII models from three different reconstructed PIIs.
Results
Conditions for persistent firing induction in DG PV-PIIs
To examine the conditions of persistent firing induction in PIIs, we performed whole-cell recordings from fast-spiking cells with subsequent immunohistochemical and morphological identification (Fig. 1). Persistent firing analysis was based entirely on morphologically identified fast-spiking PIIs. The majority of PIIs expressed PV (59 of 65 PIIs; Fig. 1A). They deviated in two morphological subtypes: (1) basket cells with axon collaterals surrounding GC somata (97 of 159 PIIs; Figs. 1A, 3A); and (2) axo-axonic cells (AACs) forming chandelier-like axon collaterals targeting the axon initial segment of GCs (62 of 159 PIIs; see Figs. 1B, 5A; Soriano et al., 1990). In a subset of cells, we confirmed the identities of AACs by performing antibody labeling against Ankyrin-G, expressed selectively in the axon initial segment of GCs and targeted by putative AAC output synapses (three AACs tested; Fig. 1B; Gulyás et al., 2010).
Figure 3.
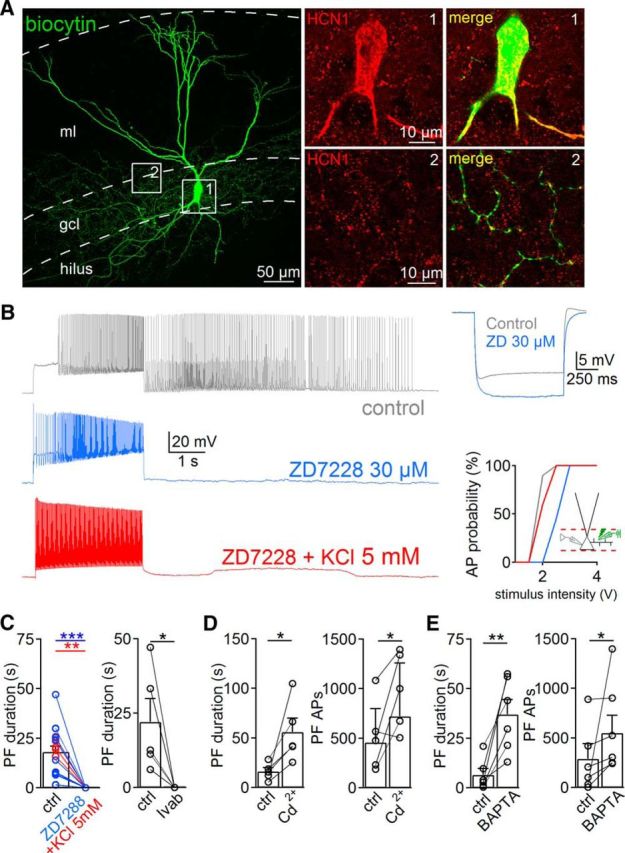
Persistent firing (PF) requires HCNC activation. A, Left, Confocal image from a biocytin-filled PII. Right, Antibodies against HCN1 label PII soma and axon collaterals. B, Left, Traces show persistent firing activity in the control condition (gray) and after bath application of 30 μm ZD7288 (blue). Coapplication of ZD7288 and 5 mm K+ does not recover persistent firing (red). Top right, Hyperpolarizing current injection in a PII (−200 pA, 1 s) in the control condition and in the presence of ZD7288. Bottom right, Probability of evoking antidromic action potentials (AP) as a function of extracellular stimulus intensity in control (ctrl; gray), in the presence of ZD7288 (blue), or in the presence of ZD7288 and 5 mm K+ (red). Note that elevated K+ compensates the effects of ZD7288 on axonal excitability. C–E, Bar graphs summarize the influence of ZD7288 (blue) or ZD7288 with K+ (C, red), 10–30 μm ivabradine (Ivab), 200 μm Cd2+, and intracellularly applied 10 mm BAPTA on PF duration or number of action potentials generated during persistent firing. Circles connected by lines represent individual experiments. Bars with lines represent mean ± SEM. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001.
Figure 5.

Perisomatic inhibition is maintained during persistent firing. A, Confocal image of an intracellularly labeled PII targeting a postsynaptic GC (depicted in purple). ml, Molecular layer. B, Trains of uIPSCs in a GC (top trace) induced by trains of action potentials (APs) in the presynaptic PII (bottom trace) in the pair shown in A. Numbers depict four activity phases: (1) somatically evoked APs; (2) simultaneous generation of somatic and persistent firing-mediated APs; (3) beginning of persistent firing (PF); and (4) end of PF. C, Magnified sections (1–4) from traces shown in B. D, Left, Superposition of single somatically induced (blue) and persistent firing-mediated (red) APs (bottom traces) and their corresponding averaged uIPSCs. Right, Synaptic latency during phases 1 and 4. E, Bar graphs summarize average 20–80% rise time (left) and half-duration (right) of uIPSCs. Circles connected by lines represent individual pairs and are means from 20–30 uIPSCs. Circles with error bars represent mean ± SEM. **p ≤ 0.01.
Persistent firing induction in PV-PIIs was tested with four protocols (Fig. 1C–K; see Material and Methods). In Protocol 1, intracellular suprathreshold current injection for 3 s (discharge frequency, 197 ± 21 Hz) interleaved by long-lasting resting periods reliably evoked persistent firing on average after trials 3–4 (1366 ± 73 action potentials; 66 PIIs; Fig. 1C). In Protocol 2, to mimic gamma activity patterns observed in the DG in vivo (Leutgeb et al., 2007), trains of somatically evoked action potentials were induced in PIIs at 30–100 Hz (see Figs. 2A,E, 5B). Persistent firing was evoked reliably at all induction frequencies tested (range, 58–85%; Fig. 1F). Interestingly, the required total action potential number was highest at the 30 Hz protocol and declined with increasing induction frequency (e.g., 30 Hz, 1279 ± 166 action potentials; 50 Hz, 945 ± 82 action potentials; 14 PIIs; p = 0.0033; Fig. 1G). In Protocol 3, to reproduce synaptic PII recruitment, we evoked perforant path-mediated 30 Hz bursts of suprathreshold EPSPs. Persistent firing was observed in 75% of recorded cells (six of eight PIIs, Fig. 1D,F). In Protocol 4, persistent firing induction was independent from changes in intracellular milieu and reproduced during somatic cell-attached recordings when persistent firing was evoked by extracellular PII axon stimulation in the GCL to generate antidromic action potentials (Fig. 1E,F). In summary, prolonged intrinsic gamma activity patterns reliably induce persistent firing in PIIs.
Properties of persistent firing in DG PV-PIIs
Several similarities and few differences in the properties of persistent firing were evident when the different induction protocols were applied. The mean duration of ∼20 s (Fig. 1I; Protocol 1, 20.7 ± 2.7 s; Protocol 2, induction frequency, 30 Hz, 23.3 ± 3 s; 50 Hz, 23.2 ± 3.5 s; 75 Hz, 21.8 ± 2.4 s; 100 Hz, 21.9 ± 4.4 s; Protocol 4, 15.9 ± 4.8 s) and discharge frequency of ∼50 Hz during persistent firing were similar among the applied protocols (Protocol 1, 47 ± 2 Hz; Protocol 2, induction frequency, 30 Hz, 55 ± 5 Hz; 50 Hz, 50 ± 4 Hz; 75 Hz, 50 ± 3 Hz; 100 Hz, 50 ± 4 Hz; Protocol 3, 58 ± 7 Hz; Protocol 4, 56 ± 3 Hz; Fig. 1K). One exception was formed by the duration of persistent firing induced during perforant-path stimulation (4.5 ± 1.1 s; Fig. 1I), which may be explained by the shorter induction period compared with the remaining protocols (∼800 ms vs >3 s). This difference resulted from the fact that high-frequency perforant-path stimulation induced subsequent EPSPs with declining amplitude during the trains (Sambandan et al., 2010) and thereby limited reliable PII recruitment. Moreover, persistent firing showed a biphasic time course consisting of an initial high-frequency discharging phase, followed by a long-lasting plateau phase at ∼50 Hz (Fig. 1J). Thus, persistent firing emerges consistently at gamma frequencies. Similar persistent firing characteristics were obtained from PV-PIIs in the medial prefrontal cortex (Fig. 1F–I). However, persistent firing duration was approximately fivefold shorter (5.7 ± 1.7 s; p = 10−6) and emerged less frequently than in the DG (45.5%; 10 of 22 PIIs). In summary, these data indicate that persistent firing is a general principle of cortical PIIs.
Persistent firing emerges in the distal PII axon
Action potentials evoked by somatic current injection appeared superimposed on depolarizing envelopes with a threshold (Vthres) of −48.1 ± 1.3 mV. In contrast, persistent firing-related action potentials emerged sharply from baseline with an apparent Vthres of −71.3 ± 1.3 mV (11 PIIs; p = 4 × 10−6), suggesting that persistent firing is generated at axon collaterals remote from the axon initial segment (Fig. 2A). Indeed, phase plots revealed similarities in the shape and Vthres of somatically recorded antidromic action potentials evoked by extracellular PII axon stimulation and persistent firing-related action potentials (Fig. 2B) but differences to the apparent Vthres of somatically evoked action potentials (Fig. 2A). Consistent with an axonal origin, persistent firing could not be induced by Protocols 1 and 2 (50 Hz) in reconstructed fast-spiking PV cells whose axons were short and very likely cut during slice preparation compared with morphologically identified PIIs with full axonal arborizations (without axon, 2.1 ± 0.2 mm axonal length, 20 fast-spiking PV cells vs with axon, 143.4 ± 4.4 mm, 4 reconstructed PIIs; p = 0.002; Fig. 2C).
To test whether the membrane potential of the somatodendritic compartment may influence the properties of persistent firing, we systematically altered the Vhold between −60 and −90 mV at the soma when persistent firing was induced by Protocol 2 at 50 Hz (Fig. 2D). Persistent firing could always be induced, but the number of required action potentials increased (e.g., −70 mV, 894 ± 82 action potentials; −90 mV, 1210 ± 87 action potentials; nine PIIs; p = 0.009) and the persistent firing duration declined with hyperpolarizing Vhold (e.g., 1.8 times longer at Vhold of −60 mV than Vhold of −90 mV; eight PIIs; p = 0.03; Fig. 2D). Thus, persistent firing is modulated by the steady-state somatodendritic membrane potential, consistent with the previously reported monotonic somatic influence on the axonal membrane potential (Nörenberg et al., 2010).
To further prove the axonal origin of persistent firing, we expressed pAAV–eNpHR–eYFP in PV–Cre mice (Fig. 2E1,E2). A yellow light spot applied to the soma of eYFP-expressing cells resulted in a marked hyperpolarization by −32.5 ± 5.3 mV (Vhold of −70 mV; five PIIs; Fig. 2E3; see Materials and Methods). In contrast, spot illumination of the PII axon in the GCL at a distance >150 μm from the cell body at the extracellular stimulation site evoked a weak voltage change at the soma of −2.3 ± 0.4 mV (four PIIs; Fig. 2E3). However, if the spotlight was applied ∼1.5 s after persistent firing induction, the duration of persistent discharges was reduced by 90.8 ± 1.7% (30.9 ± 4.3 vs 2.5 ± 0.4 s; eight PIIs; p < 0.001; Fig. 2E4). This effect depended on a long-lasting light-mediated silencing of PII axons of >5 s. Indeed, brief light-mediated PII silencing (≤2 s) reignited persistent firing and increased its total duration (25.3 ± 7.6 vs 31.6 ± 7.5 s; five PIIs; p = 0.040; Fig. 2E5). Thus, persistent firing seems to be induced in the distal PII axon.
HCNC activation induces persistent firing
The increased duration of persistent firing during short axonal hyperpolarization indicated the possible involvement of slow-gated HCNCs. Antibody labeling revealed HCN1 immunoreactivity in PV-PII soma and axons (four out of four PIIs; Fig. 3A). To test their contribution to the generation of persistent firing, we bath applied ZD7288 (4-ethylphenylamino-1,2-dimethyl-6-methylaminopyrimidinium chloride; 10–30 μm). It increased the apparent input resistance (79 ± 7.3 MΩ vs 100.7 ± 6 MΩ for ZD7288; 13 PIIs; p = 0.034) and reduced the somatic resting membrane potential (−71 ± 1.7 mV vs −74.4 ± 1.9 mV for ZD7288; nine PIIs; p = 0.02; Fig. 3B), consistent with a blockade of Ih conductances (Aponte et al., 2006). Under these conditions, persistent firing was fully abolished, regardless of the applied induction protocols (17 PIIs; Fig. 3B,C), an effect reproducible with the HCNC-selective blocker ivabradine (30 μm; five PIIs; Fig. 3C). ZD7288-mediated loss of persistent firing may have been caused by axonal hyperpolarization. To test this possibility, we measured the stimulus intensity required to evoke antidromic action potentials at 50% probability with an extracellular stimulation pipette positioned in the GCL. The required stimulus intensity was 133% higher in ZD7288 (Fig. 3B, right). To compensate for this apparent axonal hyperpolarization, we added 5 mm K+ to the extracellular medium and restored axonal excitability. Indeed, the spike probability-to-stimulus intensity relationship for antidromic action potential generation was similar to control conditions (Fig. 3B, right). However, persistent firing was still absent (five PIIs; Fig. 3C; but see Thuault et al., 2013), indicating its dependence on axonal HCNC activation.
Persistent firing in PIIs is modulated by intracellular Ca2+
Recent investigations in CA1 perforant path-associated cells proposed a marked facilitation of persistent firing by activation of voltage-gated Ca2+ channels (VGCCs; Sheffield et al., 2013). Therefore, we examined their role in persistent firing in DG PIIs by blocking VGCCs with bath-applied Cd2+ (200 μm). Interestingly, the duration and number of action potential trains during persistent firing were boosted instead of blocked (duration control, 16 ± 3.8 s vs 56 ± 8.6 s for Cd2+, five PIIs, p = 0.042; action potential number control, 508 ± 159 vs 973 ± 176 for Cd2+, five PIIs, p = 0.046; Fig. 3D). Similarly, loading PIIs with the Ca2+ chelator BAPTA (10 mm; for 90 ± 10 min) increased persistent firing duration and action potential number (6.5 ± 3.2 s for control vs 37 ± 7.5 s for BAPTA, six PIIs, p = 0.007; action potential number, 306 ± 128 vs 603 ± 189 for BAPTA, six PIIs, p = 0.031; Fig. 3E). Accordingly, during persistent firing, axonal Ca2+ may rise and reduce persistent firing in DG PIIs, suggesting interneuron type-specific or brain region-specific differences in the contribution of Ca2+-dependent mechanisms to persistent firing emergence.
Estimates on the role of HCNC-mediated conductances in persistent discharges
To infer the possible role of HCNC-mediated conductances in persistent firing, we used the previously developed detailed active single-cell model of DG PIIs that reproduce their fast-spiking phenotype (Hu et al., 2010; Hu and Jonas, 2014). We first introduced ḠIh with realistic gating properties (Fig. 4; Materials and Methods; Aponte et al., 2006), resulting in the emergence of a sag potential during negative current applications, similar to our experimental observations (Fig. 4A, inset). Next, we induced spike trains (∼100 Hz, 1 s) at the soma that propagated to the axon and obtained the amplitude and time course of the active Ih conductance (GIh) at both sites (Fig. 4A; 396 ± 68 μm from soma). Spike trains induced a monotonous decline of GIh in both compartments (Fig. 4A, right), suggesting that GIh may not be recruited during high-frequency activity patterns. However, V½, which defines the membrane potential at which half-maximal GIh emerges, has been shown to be tightly modulated by various intracellular signaling cascades and can be shifted to positive values by up to 25 mV (Chen et al., 2001; Biel et al., 2009; He et al., 2014). Therefore, we systematically changed V½ between −85 and −60 mV in PII axons. Figure 4B shows that a V½ shift to −70 mV in the axon causes a marked increase in GIh, followed by persistent discharges propagating to the PII soma. Quantitative analysis revealed that discharge frequency and GIh increased with positive values of V½, thereby reaching persistent firing analog activities at −70 to −75 mV (Fig. 4C). However, other factors, such as density of ḠIh and ḠNa+ (Fig. 4D) are also involved in setting the precise spike frequency. Thus, our computational analysis revealed that persistent firing in PII axons can be supported by HCNCs under conditions of a positive shift in their V½.
Persistent firing generates sustained perisomatic inhibition
To examine whether persistent firing permits inhibitory PII output signaling, we performed paired PII–GC recordings (Fig. 5). Persistent firing was induced by somatic action potential trains (Fig. 5A,B). During the induction, somatic action potentials evoked uIPSCs after short latencies (1.8 ± 0.3 ms; eight pairs; Fig. 5C,D; Bartos et al., 2001). However, during persistent firing induction, synaptic latency declined by 39% (1.1 ± 0.2 ms; p = 0.0078; Fig. 5D, right), indicating a shorter spatiotemporal distance between action potential generation and synaptic release site. The functional properties of uIPSCs were not significantly different between the somatically induced and the persistent firing-mediated events (amplitude, 81.7 ± 19.6 vs 84.4 ± 16.3 pA, p = 0.8; 20–80% rise time, 0.6 ± 0.03 vs 0.61 ± 0.05 ms, p = 0.57; half-duration, 8.0 ± 0.9 vs 8.2 ± 1.3 ms, p = 0.87, respectively; Fig. 5E). Thus, synaptic transmission at PII output synapses is unchanged during persistent firing, allowing reliable and strong inhibitory signaling in the DG.
Discussion
Here we showed that DG PIIs can be recruited efficiently into a persistent firing mode characterized by gamma frequency (∼50 Hz) discharges over prolonged time periods ranging from several seconds to minutes after sustained phases of enhanced activity. This finding fits to persistent firing identified recently in CA1 perforant path-associated, neurogliaform and neocortical layer 2/3 interneurons, as well as cortical principal cells, indicating that it is a general discharge mode independent of neuron type or cortical area (Egorov et al., 2002; Krook-Magnuson et al., 2011; Sheffield et al., 2011; Suzuki et al., 2014). Persistent firing induction depends on frequency and duration of intrinsic PII activity, which is controlled by the strength of excitatory synaptic inputs, suggesting that persistent firing may act as a balancing mechanism to provide powerful feedforward perisomatic inhibition in response to strong global network recruitment.
To the best of our knowledge, we provide the first evidence for HCNC activation as one major cellular mechanism underlying persistent firing induction in PV-PIIs. Indeed, HCNC block by ZD7288 abolished persistent firing. This effect could not be explained by the negative shift in resting membrane potential because elevation of PII membrane potential and axonal excitability to control values by extracellular KCl did not restore persistent firing (Fig. 3B,C). How are HCNCs activated? A midpoint potential of HCNC activation of approximately −84 mV (Aponte et al., 2006) points to membrane hyperpolarization being an important requirement for HCNC opening. Our single PII model data showed that, during times of overshooting spikes, the amount of active HCNCs declined as a result of inactivation and recovered during spike afterhyperpolarization (Fig. 4A). The slow activation time constant of HCNCs (∼180 ms at −70 mV; Aponte et al., 2006) did not allow a net activation of GIh during high frequent activity, which could be an important requirement for suprathreshold depolarization and persistent firing induction. Alternatively, gating kinetics of HCNCs stay under tight modulatory control by various compounds (Chen et al., 2001; He et al., 2014) that have been shown to accumulate during prolonged depolarization (e.g., cAMP; Mironov et al., 2009). They can accelerate open kinetics and facilitate HCNC activation at more depolarized membrane potentials, thereby shifting V½ to more positive values (Chen et al., 2001; Biel et al., 2009). Indeed, when this shift in the HCNC activation curve was reproduced in our PII model, strong Ih conductance emerged and induced persistent firing-analog discharges in PII axons (Fig. 4B). Thus, we propose that activity-dependent rising of HCNC-relevant modulatory signaling pathways (Biel et al., 2009; He et al., 2014) might be one possible mechanism underlying the induction of persistent firing.
How can HCNC activity be stopped? We propose that, once the modulatory effect is induced, it is long lasting and therefore could not be blocked by short-duration (≤2 s) silencing of PII axons using eNpHR activation (Fig. 2E5). On the contrary, persistent firing duration was prolonged, which may be explained by additional HCNC recruitment as a result of the transient axon hyperpolarization. We provide evidence that axonal Ca2+ accumulation (Bucurenciu et al., 2008) may contribute to the silencing of persistent firing. This observation contrasts with previous studies in CA1 perforant path-associated interneurons (Sheffield et al., 2013), suggesting neuron type-specific and brain area-specific differences in the mechanisms underlying persistent firing. Fast-spiking interneurons, including PIIs, express Ca2+-dependent K+ channels (Goldberg et al., 2005). The recruitment of Ca2+-dependent K+ conductances may hyperpolarize PIIs and thereby reduce (Fig. 2D) or even stop activity. In summary, different sets of intracellular signaling cascades may be involved in the initiation and silencing of persistent firing. The currently difficult access to distal PII axons using whole-cell recordings make investigations on the effect of the broad set of possible modulators and changes in axonal membrane potential on persistent firing highly challenging. Therefore, future investigations at the level of PII axons will be needed to fully understand the molecular and cellular mechanisms underlying start and stop of persistent discharges.
Persistent firing was induced under various conditions, including activation of the perforant-path transmitting multimodal information from the entorhinal cortex to the DG. Therefore, we propose that persistent firing may emerge under behaviorally relevant conditions when excitatory input strength to the DG is enhanced, such as during environmental exploration (Leutgeb et al., 2007). The high release probability of PII output synapses (Kraushaar and Jonas, 2000; Doischer et al., 2008) together with their broad axonal arborizations will allow PIIs to efficiently inhibit large GC populations throughout persistent firing. This may increase the signal-to-noise ratio in the network and its storage capacity of novel information. Alternatively, persistent firing may be important during high uncontrolled GC population activity, such as times preceding the emergence of local seizures (Bower and Buckmaster, 2008). Thus, synaptic inhibition during persistent firing may act as a homeostatic mechanism “resetting” the network to low baseline activity, thereby ensuring sparse coding during periods of strong circuit activation.
Footnotes
This work was funded by the Schram Foundation (M.B.), a Lichtenberg Professorship Award (Volkswagen Foundation; M.B.), German Research Foundation Grant BA1582/2-1 (M.B.) and the Research Unit; FOR2143, funded by the German Research Foundation; the Molecular and Translational Research in Freiburg (MOTI-VATE) Program of the Medical Faculty, University of Freiburg (Else Kröner-Fresenius Foundation; J.K.), and the BrainLinks–BrainTools, Cluster of Excellence funded by German Research Foundation Grant EXC 1086 (M.B.). We thank Dr. L. Alberi for providing pAAV–doublefloxed–eNpHR–EYFP–WPRE–pA and Drs. I. Vida and M. Strüber for reading previous versions of this manuscript. We thank K. Winterhalter and K. Semmler for technical assistance.
The authors declare no competing financial interests.
References
- Aponte Y, Lien CC, Reisinger E, Jonas P. Hyperpolarization-activated cation channels in fast-spiking interneurons of rat hippocampus. J Physiol. 2006;574:229–243. doi: 10.1113/jphysiol.2005.104042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartos M, Vida I, Frotscher M, Geiger JR, Jonas P. Rapid signaling at inhibitory synapses in a dentate gyrus interneuron network. J Neurosci. 2001;21:2687–2698. doi: 10.1523/JNEUROSCI.21-08-02687.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biel M, Wahl-Schott C, Michalakis S, Zong X. Hyperpolarization-activated cation channels: from genes to function. Physiol Rev. 2009;89:847–885. doi: 10.1152/physrev.00029.2008. [DOI] [PubMed] [Google Scholar]
- Bower MR, Buckmaster PS. Changes in granule cell firing rates precede locally recorded spontaneous seizures by minutes in an animal model of temporal lobe epilepsy. J Neurophysiol. 2008;99:2431–2442. doi: 10.1152/jn.01369.2007. [DOI] [PubMed] [Google Scholar]
- Bucurenciu I, Kulik A, Schwaller B, Frotscher M, Jonas P. Nanodomain coupling between Ca2+ channels and Ca2+ sensors promotes fast and efficient transmitter release at a cortical GABAergic synapse. Neuron. 2008;57:536–545. doi: 10.1016/j.neuron.2007.12.026. [DOI] [PubMed] [Google Scholar]
- Buzsáki G, Draguhn A. Neuronal oscillations in cortical networks. Science. 2004;304:1926–1929. doi: 10.1126/science.1099745. [DOI] [PubMed] [Google Scholar]
- Chen S, Wang J, Siegelbaum SA. Properties of hyperpolarization-activated pacemaker current defined by coassembly of Hcn1 and Hcn2 subunits and basal modulation by cyclic nucleotide. J Gen Physiol. 2001;117:491–504. doi: 10.1085/jgp.117.5.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb SR, Buhl EH, Halasy K, Paulsen O, Somogyi P. Synchronization of neuronal activity in hippocampus by individual GABAergic interneurons. Nature. 1995;378:75–78. doi: 10.1038/378075a0. [DOI] [PubMed] [Google Scholar]
- Doischer D, Hosp JA, Yanagawa Y, Obata K, Jonas P, Vida I, Bartos M. Postnatal differentiation of basket cells from slow to fast signaling devices. J Neurosci. 2008;28:12956–12968. doi: 10.1523/JNEUROSCI.2890-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egorov AV, Hamam BN, Fransén E, Hasselmo ME, Alonso AA. Graded persistent activity in entorhinal cortex neurons. Nature. 2002;420:173–178. doi: 10.1038/nature01171. [DOI] [PubMed] [Google Scholar]
- Franz O, Liss B, Neu A, Roeper J. Single-cell mRNA expression of HCN1 correlates with a fast gating phenotype of hyperpolarization-activated cyclic nucleotide-gated ion channels (Ih) in central neurons. Eur J Neurosci. 2000;12:2685–2693. doi: 10.1046/j.1460-9568.2000.00151.x. [DOI] [PubMed] [Google Scholar]
- Goldberg EM, Watanabe S, Chang SY, Joho RH, Huang ZJ, Leonard CS, Rudy B. Specific functions of synaptically localized potassium channels in synaptic transmission at the neocortical GABAergic fast-spiking cell synapse. J Neurosci. 2005;25:5230–5235. doi: 10.1523/JNEUROSCI.0722-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golding NL, Mickus TJ, Katz Y, Kath WL, Spruston N. Factors mediating powerful voltage attenuation along CA1 pyramidal neuron dendrites. J Physiol. 2005;568:69–82. doi: 10.1113/jphysiol.2005.086793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulyás AI, Szabó GG, Ulbert I, Holderith N, Monyer H, Erdélyi F, Szabó G, Freund TF, Hájos N. Parvalbumin-containing fast-spiking basket cells generate the field potential oscillations induced by cholinergic receptor activation in the hippocampus. J Neurosci. 2010;30:15134–15145. doi: 10.1523/JNEUROSCI.4104-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C, Chen F, Li B, Hu Z. Neurophysiology of HCN channels: from cellular functions to multiple regulations. Prog Neurobiol. 2014;112:1–23. doi: 10.1016/j.pneurobio.2013.10.001. [DOI] [PubMed] [Google Scholar]
- Hu H, Jonas P. A supercritical density of Na(+) channels ensures fast signaling in GABAergic interneuron axons. Nat Neurosci. 2014;17:686–693. doi: 10.1038/nn.3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Martina M, Jonas P. Dendritic mechanisms underlying rapid synaptic activation of fast-spiking hippocampal interneurons. Science. 2010;327:52–58. doi: 10.1126/science.1177876. [DOI] [PubMed] [Google Scholar]
- Isaacson JS, Scanziani M. How inhibition shapes cortical activity. Neuron. 2011;72:231–243. doi: 10.1016/j.neuron.2011.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knauer B, Jochems A, Valero-Aracama MJ, Yoshida M. Long-lasting intrinsic persistent firing in rat CA1 pyramidal cells: A possible mechanism for active maintenance of memory. Hippocampus. 2013;23:820–831. doi: 10.1002/hipo.22136. [DOI] [PubMed] [Google Scholar]
- Kraushaar U, Jonas P. Efficacy and stability of quantal GABA release at a hippocampal interneuron-principal neuron synapse. J Neurosci. 2000;20:5594–5607. doi: 10.1523/JNEUROSCI.20-15-05594.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krook-Magnuson E, Luu L, Lee SH, Varga C, Soltesz I. Ivy and neurogliaform interneurons are a major target of μ-opioid receptor modulation. J Neurosci. 2011;31:14861–14870. doi: 10.1523/JNEUROSCI.2269-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leutgeb JK, Leutgeb S, Moser MB, Moser EI. Pattern separation in the dentate gyrus and CA3 of the hippocampus. Science. 2007;315:961–966. doi: 10.1126/science.1135801. [DOI] [PubMed] [Google Scholar]
- Major G, Tank D. Persistent neural activity: prevalence and mechanisms. Curr Opin Neurobiol. 2004;14:675–684. doi: 10.1016/j.conb.2004.10.017. [DOI] [PubMed] [Google Scholar]
- Mironov SL, Skorova E, Taschenberger G, Hartelt N, Nikolaev VO, Lohse MJ, Kügler S. Imaging cytoplasmic cAMP in mouse brainstem neurons. BMC Neurosci. 2009;10:29. doi: 10.1186/1471-2202-10-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nörenberg A, Hu H, Vida I, Bartos M, Jonas P. Distinct nonuniform cable properties optimize rapid and efficient activation of fast-spiking GABAergic interneurons. Proc Natl Acad Sci U S A. 2010;107:894–899. doi: 10.1073/pnas.0910716107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouille F, Scanziani M. Enforcement of temporal fidelity in pyramidal cells by somatic feed-forward inhibition. Science. 2001;293:1159–1163. doi: 10.1126/science.1060342. [DOI] [PubMed] [Google Scholar]
- Sambandan S, Sauer JF, Vida I, Bartos M. Associative plasticity at excitatory synapses facilitates recruitment of fast-spiking interneurons in the dentate gyrus. J Neurosci. 2010;30:11826–11837. doi: 10.1523/JNEUROSCI.2012-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki T. The axon as a unique computational unit in neurons. Neurosci Res. 2013;75:83–88. doi: 10.1016/j.neures.2012.12.004. [DOI] [PubMed] [Google Scholar]
- Savanthrapadian S, Meyer T, Elgueta C, Booker SA, Vida I, Bartos M. Synaptic properties of SOM- and CCK-expressing cells in dentate gyrus interneuron networks. J Neurosci. 2014;34:8197–8209. doi: 10.1523/JNEUROSCI.5433-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffield ME, Best TK, Mensh BD, Kath WL, Spruston N. Slow integration leads to persistent action potential firing in distal axons of coupled interneurons. Nat Neurosci. 2011;14:200–207. doi: 10.1038/nn.2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffield ME, Edgerton GB, Heuermann RJ, Deemyad T, Mensh BD, Spruston N. Mechanisms of retroaxonal barrage firing in hippocampal interneurons. J Physiol. 2013;591:4793–4805. doi: 10.1113/jphysiol.2013.258418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano E, Nitsch R, Frotscher M. Axo-axonic chandelier cells in the rat fascia dentata: Golgi-electron microscopy and immunocytochemical studies. J Comp Neurol. 1990;293:1–25. doi: 10.1002/cne.902930102. [DOI] [PubMed] [Google Scholar]
- Suzuki N, Tang CSM, Bekkers JM. Persistent barrage firing in cortical interneurons can be induced in vivo and may be important for the suppression of epileptiform activity. Front Cell Neurosci. 2014;8:76. doi: 10.3389/fncel.2014.00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thuault SJ, Malleret G, Constantinople CM, Nicholls R, Chen I, Zhu J, Panteleyev A, Vronskaya S, Nolan MF, Bruno R, Siegelbaum SA, Kandel ER. Prefrontal cortex HCN1 channels enable intrinsic persistent neural firing and executive memory function. J Neurosci. 2013;33:13583–13599. doi: 10.1523/JNEUROSCI.2427-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XJ, Buzsáki G. Gamma oscillation by synaptic inhibition in a hippocampal interneuronal network model. J Neurosci. 1996;16:6402–6413. doi: 10.1523/JNEUROSCI.16-20-06402.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]