

Abstract
The mutation F184C in Kv1.1 leads to development of episodic ataxia type I (EA1). Although the mutation has been said to alter activation kinetics and to lower expression, we show here that the underlying molecular mechanisms may be more complex. Although F184 is positioned in the “peripheral” S1 helix, it occupies a central position in the 3D fold. We show in cut-open oocyte voltage-clamp recordings of gating and ionic currents of the Shaker Kv channel expressed in Xenopus oocytes that F184 not only interacts directly with the gating charges of the S4, but also creates a functional link to the selectivity filter of the neighboring subunit. This link leads to impaired fast and slow inactivation. The effect on fast inactivation is of an allosteric nature considering that fast inactivation is caused by a linked cytosolic ball peptide. The extensive effects of F184C provide a new mechanism underlying EA.
SIGNIFICANCE STATEMENT Episodic ataxia (EA) is an inherited disease that leads to occasional loss of motor control in combination with variable other symptoms such as vertigo or migraine. EA type I (EA1), studied here, is caused by mutations in a voltage-gated potassium channel that contributes to the generation of electrical signals in the brain. The mechanism by which mutations in voltage-gated potassium channels lead to EA is still unknown and there is no consistent pharmacological treatment. By studying in detail one disease-causing mutation in Kv1.1, we describe a novel molecular mechanism distinct from mechanisms described previously. This mechanism contributes to the understanding of potassium channel function in general and might lead to a better understanding of how EA develops.
Keywords: episodic ataxia, gating currents, Kv channels, Xenopus oocytes
Introduction
Episodic ataxia (EA) consists of sporadic episodes of vertigo, migraine, and ataxia and is occasionally associated with epilepsy or hemiplegic migraine (for review, see Jen, 2008). The most frequent types of EA are caused by mutations in the genes kcna1 (type I) and cacna1a (type II) encoding for the KCNA (Kv1.1) and Cav2.1 channels, respectively. Although the genes responsible for EA have been identified, the underlying mechanism is not well understood and there is no distinct pharmacological treatment. In this study, we concentrate on a mutation found in the S1 of Kv1.1 (Browne et al., 1995). Kv1 channels are abundantly expressed in the CNS, where they are responsible for repolarization after an action potential and protect against too rapid reactivation. Therefore, they influence action potential duration and interspike intervals. Kv1.1 channels are highly expressed in hippocampal neurons, where they play an important role in the regulation of action potential firing. They are, however, also found in peripheral neurons. In the hippocampus, Kv1.1 coexpresses with the auxiliary subunit Kvβ1, which leads to rapid inactivation by a “ball peptide” blocking ion conduction (Pongs and Schwarz, 2010). Elsewhere, Kv1.1 leads to prolonged outward K+ currents.
Members of the Kv channel superfamily are tetrameric intrinsic membrane proteins, each monomer of which comprises six transmembrane helices (S1–S6; Fig. 1a). Helices S1–S4 of the four subunits with the highly cationic RxxRxxRxxR motif in the S4 form four peripheral voltage-sensing domains (VSDs), whereas the S5 and S6 of all four subunits form a single central ion-conducting pore (Long et al., 2005b). Upon depolarization, the charged S4 segment triggers a conformational change in the VSD, which is transferred to the pore and finally leads to pore opening and ion conduction (Bezanilla, 2008; Faure et al., 2012).
Figure 1.

a, Topology of Kv channels F184C (F244C in Shaker) is located at the C terminus of the S1. b, Position of F244 with respect to the arginines of S4 (PDB: 3LUT). c, Sequence alignment of the S1 of Kv1.1 and Shaker Kv channels.
We found previously that mutations in several positions that prove important for electromechanical coupling, i.e., the energetic coupling between the VSDs and the central pore (Blunck and Batulan, 2012), also led to EA1 (S. Barreto and R.B., unpublished data; Batulan et al., 2010). Alterations in the electromechanical coupling lead to modulation of channel function, most prominently a shift in the voltage dependence of activation and separation of gating charge movement from pore opening (Lu et al., 2002; Labro et al., 2008; Muroi et al., 2009; Batulan et al., 2010; Haddad and Blunck, 2011; Wall-Lacelle et al., 2011). These results suggest that modulation of channel function, rather than trafficking defects, may be the mechanisms underlying EA1.
Here, we investigated a specific mutation in the S1 of Kv1.1, F184C (F244C in Shaker), that leads to EA in heterozygous patients (Browne et al., 1995). Earlier electrophysiological investigations of this mutation found that activation kinetics were slightly slowed and the conductance voltage relation (GV) was shifted to more depolarized potentials (Adelman et al., 1995). These effects are explained by its position in the voltage sensor. Despite its location in the S1, the crystal structure of a Kv1.2/2.1 chimera (Long et al., 2007) suggests that it points toward the center of the VSD (Fig. 1b). In this work, we demonstrate the central role of this position as it not only influences S4 movement, but also affects directly the selectivity filter in the central pore as well as fast and slow inactivation.
We performed our experiments in the Shaker Kv channel because this Drosophila analog to the human Kv1 channels has been studied extensively and a plethora of information on its gating mechanism is available. Shaker shares a 49.4% sequence identity, which extends in particular to the transmembrane regions, investigated here, with 76% identity (Fig. 1c). The differences are largely restricted to the S2–S3 and S3–S4 loops. In addition, Kv1.1 does not contain the inactivation peptide in its N terminus, which is responsible for fast (N-type) inactivation. Fast inactivation in Kv1.1 channels is triggered by the inactivation peptide in the N terminus of the Kv β subunit. We used here both the Shaker wild-type and inactivation-removed (IR) mutant. The relation of Shaker to ataxia is evident in its name: “Shaker” was named after the phenotypic erratic motor control in Shaker knock-out flies.
Materials and Methods
Molecular biology.
Mutants were made in the background of the Shaker Kv channel clones zH4IR-W434F and zH4IR-A359C in the pBSta plasmid vector for gating current and ionic current measurements, respectively. In the IR mutant, positions 6–46 (inactivation ball peptide) have been deleted. W434F leads to nearly instantaneous inactivation (Perozo et al., 1993; Yang et al., 1997) and the mutation A359C has been introduced for eventual fluorescence measurements (data not shown here; Kalstrup and Blunck, 2013).
Point mutations were introduced using the Quikchange kit (Agilent) following standard protocols. For oocyte injection, plasmids were linearized using NotI and in vitro transcribed using the T7-mMachine kit (Ambion). Oocytes from Xenopus laevis, freed of follicular membrane, were injected with 4.6 ng of RNA and incubated in Barth solution at 18°C until experiments were performed 1–3 d after injection.
Tetramer constructs were made using the same approach as Gagnon et al. (2009) by linking each Shaker monomer, in frame, via unique restriction sites. First, the unique restriction site AatII (GACGTC) was engineered into monomer1 using PCR, eliminating the stop codon in the process. PCR was also used to insert the unique AatII site into the N terminus and to eliminate the start ATG codon of monomer 2. Dimer 1 was then obtained by introducing monomer1 into the new unique HindIII/AatII sites in the N terminus of monomer 2 using standard recombinant DNA techniques. Dimer 2 was created using the same method, by linking monomers 3 and 4 with the unique restriction site AvrII (CCTAGG). Finally, using the same approach, dimers 1 and 2 were linked with the restriction site Nde1 (CATATG) to create the tetramer. The six-base restriction sites introduce different residues between each monomer: aspartic acid and valine between the C terminus of monomer 1 and the N terminus of monomer 2, histidine and methionine between monomers 2 and 3, and proline and arginine between monomers 3 and 4.
Electrophysiology.
Gating and ionic currents were obtained from a cut-open oocyte voltage-clamp setup as described previously (Taglialatela et al., 1992; Batulan et al., 2010; Haddad and Blunck, 2011). Oocytes were placed in a cut-open chamber such that the lower cap, middle torus, and upper cap were accessed by the inner, guard, and top chamber, respectively. Perforation of the lower cap using saponin (0.2%) led to low resistance access to the inner oocyte and the guard and top chamber were clamped to the external potential. Gating currents were obtained by subtraction using online subtraction of linear components. External solutions contained 115 mm NMG (N-methyl-d-glucamine), 10 mm HEPES, and 2 mm Ca(OH)2 adjusted to pH 7.1 using methanesulfonic acid (MES). The internal solution contained the following (in mm): 10 HEPES, 2 EDTA, and 115 NMDG adjusted to pH 7.1 using MES for gating currents and 10 HEPES, 2 EDTA, and 115 KOH adjusted to pH 7.1 using MES for ionic currents. All measurements were done with an n ≥ 6 from at least 2 injections of independent oocyte batches.
Data analysis.
Gating-charge voltage (QV) and GV curves were normalized and fitted to simple Boltzmann relations. Due to the leak currents in the F244 mutants, the gating charge relations were obtained from the off-gating charges at the end of the depolarizing pulses. Leak currents were determined from the steady-state currents toward the end of a 50 ms pulse.
Results
Effect of F244C on voltage dependence
The mutation F244C (position numbering according to Shaker) has been described previously in Kv1.1 (Adelman et al., 1995; Zerr et al., 1998) and Shaker (Hong and Miller, 2000; Lacroix and Bezanilla, 2012; Lacroix et al., 2014). Adelman and coworkers (Adelman et al., 1995; Zerr et al., 1998) demonstrated that the GV was shifted to more depolarized potentials and that activation kinetics were slightly slowed. Replacing F244 with a bulky tryptophane in Shaker shifted the GV to +30 mV and slowed both activation and deactivation kinetics (Hong and Miller, 2000). Despite its “peripheral” position in the primary sequence, its central influence on the gating charges of the S4 voltage sensor becomes evident in the crystal structure of the Kv1.2–2.1 chimera (Fig. 1b; Long et al., 2007). In the open (inactivated) state, F244 points between the second (R2) and third (R3) arginine in the S4. We tested the influence of different F244 mutations on conductance and gating currents of the ShakerIR Kv channel (Fig. 2a,b). With the exception of F244Y, which was only slightly increased to −17 mV, all GVs were strongly shifted to more depolarized potentials with V1/2 between 0 and +50 mV (Fig. 2c). This was consistent with the value of +30 mV determined for F244W previously (Hong and Miller, 2000; Lacroix and Bezanilla, 2012). Any alteration at position F244 thus led to a shift of the GV to more depolarized potentials.
Figure 2.
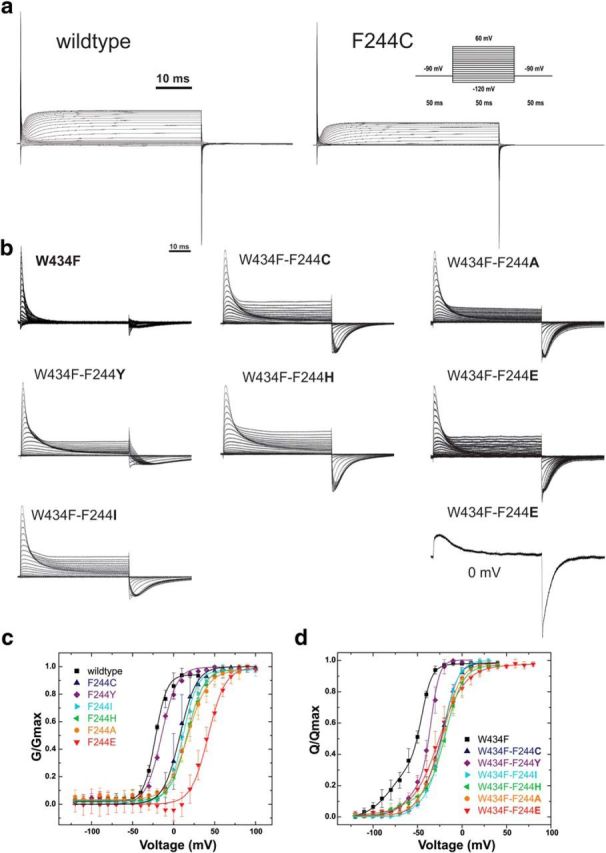
a, Current response of ShakerIR-WT and ShakerIR-F244C in response to depolarizing pulses to voltages between −120 and +60 mV from a holding potential of −90 mV. Activation is slowed in F244C. b, Gating currents in response to depolarizing pulses to voltages between −120 mV and +60 mV from a holding potential of −90 mV elicited from ShakerIR-W434F mutants and an additional mutation at position F244 as indicated. The response of F244E to a depolarizing pulse to 0 mV is shown independently. At low depolarizations, the slow onset of F244E leads to apparent larger QOFF than QON. c, GV relations of different F244 mutants elicited from protocols shown in a. d, QV relations of different F244 mutants in response to depolarizing pulses to voltages between −120 mV and +60 mV from a holding potential of −90 mV.
The same held true for the QV curves (Fig. 2d). We had to determine the QVs using the OFF-gating charge movement at the end of a depolarizing pulse because the ON-gating charge (during activation) was contaminated by development of leak current (Fig. 2b). The leak current will be addressed in detail below. Although all QVs were shifted to more depolarized potentials, there was little variation between the different mutants. F244Y, once again, was shifted only to −40 mV, whereas the other mutants had V1/2 ranging between −18 mV and −28 mV. For most mutants, the relation between V1/2 of QVs and GVs was linear, indicating that directly gating charge movement and not coupling to the pore was influenced. In F244E, however, the QV was shifted by only +25 mV, but the GV by +61 mV. The reason for this discrepancy might be in the very slow kinetics of the ON-gating currents, which led to an apparent shift of the QVON (Fig. 2b).
Influence on gating charge movement
The above results suggest that placing a negative charge at position F244(E) slows activation. However, an electrostatic interaction between 244E and the cationic arginines in S4 do not significantly alter the difference in free energy between the resting and the activated state. Accordingly, the QV curve of F244E is not more shifted than the uncharged F244C, F244-H, F244-I or F244-A. The reason is likely that the arginines pairing with F244E are replaced in the activated state with the ones positioned right below on the S4. We confirmed that the primary reason for the shift of the GV in the F244 mutants is not of an electrostatic nature by testing a double-mutant cycle with F244C and charge neutralizations of the first three cationic charges in S4 (Fig. 3). The difference in free energy, determined by generalized interaction-energy analysis (Chowdhury et al., 2014), per tetramer was <1.8 kcal/mol (ΔΔG = 1.23, 0.64 and −1.23 kcal/mol for R1, R2, and R3, respectively), which is considered nonspecific for homotetrameric channels (Horovitz, 1996; Chowdhury et al., 2014). We thereby assumed that F244C did not alter the charge per channel and neutralization of the arginines altered the charge per channel by the same amount in the presence and absence of F244C. The effect of the F244C replacement is thus independent of the charge of the arginines in S4. This is consistent with the findings of Lacroix et al. (2014), who suggested that it is the hydrophobic surface of the residue that decides the effect on gating charge movement.
Figure 3.

Normalized QV relations of ShakerIR-W434F (solid) and ShakerIR-F244C (open) in the absence (black) and presence (red) or neutralization of the first (R1, R362Q, left), second (R2, R365Q, center), and third (R3, R368Q, right) arginine of S4.
Development of leak currents in the C-type inactivated state
One remarkable feature of the F244C gating currents was the development of “leak currents” in the “instantly C-type-inactivated” W434F mutant (Fig. 2b). The W434F mutant should not allow any ionic current because the mutation W434F has been shown to accelerate C-type inactivation such that no ionic currents develop (Perozo et al., 1993; Yang et al., 1997). The leak was observed in all of the F244 mutants, with the strongest currents in F244E (normalized to gating currents; Fig. 4a). The ratio between leak current and gating charge remained constant independently of the expression level of the channels, indicating that the current originated from the channels and was not an unspecific leak induced by expression.
Figure 4.

a, Ratio between maximal leak current amplitude and maximal gating current amplitude. The ratio was constant for all mutants independently of expression level. b, Position of F244 with respect to the selectivity filter. F244 of one subunit (blue) is shown in red, as well as I429 and W434 of the neighboring subunit (light orange). Potassium in the selectivity filter is shown in green (PDB: 3LUT). c, Onset of leak current (solid black squares) compared with the gating currents (solid gray squares) and the ionic current (hollow black circles). d, Gating currents of ShakerIR-W434F-F244C with K+ as the dominant ion internally and NMG (left) or K (right) as dominant ion externally. e, Block of leak currents by externally applied 4-aminopyridine. f, Block of the leak current by fast (N-type) inactivation in Shaker-W434F-F244C without deletion of positions 6–46.
The question remained as to which path the current passes through the channel. With F244 on the S1 helix, two possibilities present themselves: (1) the current could pass through the voltage sensor or (2) it could pass through the “normal” ion conduction pore. Current passing through the voltage-sensing domain has been shown to occur in some mutants of the Shaker Kv channel at hyperpolarizing potentials (ω currents; Pathak et al., 2007; Tombola et al., 2007) or upon voltage sensor activation (proton currents; Starace and Bezanilla, 2001, 2004) in proton channels (Ramsey et al., 2006; Tombola et al., 2008; Berger and Isacoff, 2011) and mutants of voltage-gated sodium channels linked to arrhythmias (Sokolov et al., 2007; Moreau et al., 2015). Because F244 strongly influences voltage sensor gating and is located close to the arginines of the S4, it could induce such “gating pore” currents. Conversely, although a direct influence on the gating of the pore domain has never been demonstrated, the S1 has been shown to be in close proximity to the pore (Long et al., 2005b; Lee et al., 2009; Fig. 4b). The current could therefore also influence the ion-conducting pore and the selectivity filter.
Comparing the onset of the leak current for the F244C mutant with gating charge movement and ionic current revealed that it coincided with pore opening, suggesting that the current is carried by the pore domain (Fig. 4c). We further tested this by exchanging external NMG+ with K+ ions, which led to a significant reduction in current amplitude (Fig. 4d). We also blocked the current with 20 mm 4-aminopyridine, which does not influence charge movement but prevents the final transition to pore opening (Fig. 4e). Finally, we made the W434F mutation in the presence of the N-terminal ball peptide. Blocking of the pore by the N-terminal ball peptide (N-type inactivation) prevented leak currents (Fig. 4f). We could thus be confident that the leak current passed through the ion-conducting pore and that no “gating pore” developed. The mutations at position F244 in the S1 thus led to pore conduction.
Influence on C-type inactivation
As mentioned earlier, the absence of ion conduction in the W434F mutant is caused by extremely fast C-type inactivation. C-type inactivation of potassium channels has been shown to be caused by a rearrangement in the pore domain and, more specifically, in the selectivity filter, where a Tyr is thought to flip, thus disturbing some of the potassium-binding sites (Bernèche and Roux, 2005; Cordero-Morales et al., 2006; Cuello et al., 2010a). In the F244-W434F mutants, on the other hand, C-type inactivation seems to be impaired. The ratio of leak-to-gating currents suggests that C-type inactivation is not complete and the amplitude of the leak current lets us estimate that, in steady state, ∼1% of channels remain open. Alternatively, it is possible that, even in the inactivated state, a small leak conductance (∼1%) remained. How does this effect translate to the wild-type C-type inactivation (i.e., in the absence of the W434F mutation)? When comparing C-type inactivation of wild-type and F244 mutants, the time constants were altered (Fig. 5a); however, we found both accelerated (F244E) and slowed (F244A) time courses. The more important difference, however, seems to be the reduced number of channels entering C-type inactivation. Therefore, wild-type C-type inactivation is also affected by mutations at F244 in the S1. But how is the effect of mutating F244C transferred to the selectivity filter where C-type inactivation occurs? C-type inactivation has been shown previously to not only influence the selectivity filter and pore domain, but also to lead to conformational changes in the entire ion channel (Olcese et al., 2001; Claydon et al., 2007; Cuello et al., 2010b; Haddad and Blunck, 2011; Kalstrup and Blunck, 2013). Because mutations in F244 have a profound influence on voltage sensor gating, the influence on the pore region could be mediated indirectly through electromechanical coupling; that is, the S4–S5 linker and the C-terminal S6. Conversely, the close proximity of S1 and the pore domain would also be consistent with a direct interaction. In the crystal structure of the Kv1.2/2.1 chimera, F244 and W434 are only 16 Å apart from each other, separated by I429 at the end of the pore helix (Fig. 4b).
Figure 5.

a, Time course of slow (C-type) inactivation for ShakerIR-WT (black) and ShakerIR-F244X mutants (X = C, I, A, H, Y, E) in response to a depolarizing pulse to +60 mV from a holding potential of −90 mV. b, GV relation of ShakerIR-I429A elicited from depolarizing pulses from −90 mV to voltages between −100 mV and +50 mV. c, C-type inactivation elicited from pulses from −90 mV to +60 mV for Shaker-IR concatemers. Each concatemer contained four repeats corresponding to a Shaker-IR monomer. The W434F mutation leading to accelerated C-type inactivation is introduced in repeat II (red). Then the F244C mutation is introduced in the first (F1), second (F2), or third (F3) repeat (striped). d, Ratio of amplitudes of fast to slow component (left) and time constants (right) of exponential fits to data as shown in c. e, Illustration of the synthesis direction; based on our data, the tetramers are assembled counterclockwise when seen from the extracellular side.
To distinguish between both cases, we first set out to mutate the linking position I429. We found, however, that I429N and I429G did not yield functional channels. The mutant I429A yielded functional channels and showed a voltage dependence shifted to more depolarized potentials (V1/2 = −9 mV; Fig. 5b). The shifted voltage dependence of I429A indicates an influence on the voltage sensor movement, as we hypothesized based on its position relative to F244 and W434. However, the interaction does not have to be direct but could still be mediated via the S4–S5 linker because we were not able to obtain any functional double mutants with F244 to further characterize this interaction.
To prove a direct influence of F244 on the pore and selectivity filter, we rationalized that a direct effect should act exclusively onto the neighboring subunit (Fig. 4b), whereas an indirect effect should be cooperative because pore opening and the final opening steps occur cooperatively (Zagotta et al., 1994; Schoppa and Sigworth, 1998; Pathak et al., 2005; Kalstrup and Blunck, 2013). The major effect of the F244 mutants on the pore domain is the partial inhibition of C-type inactivation. We thus constructed a tetrameric ShakerIR Kv channel out of four repeats of the channel with a single W434F mutation in the second repeat (Tet; Fig. 5c). Consistent with previous constructs, C-type inactivation was accelerated compared with wild-type ShakerIR but slower than homotetrameric W434F channels (Yang et al., 1997).
We then introduced the mutation F244C in the first (F1), second (F2), or third (F3) repeat and determined the time course of C-type inactivation (Fig. 5c). The time courses were fitted to a double exponential decay (Fig. 5d, Table 1). C-type inactivation was altered for all three mutants. Whereas in the F1 and F2 concatemers, the slow component was reduced, it was the fast component in the F3 concatemer. Only the fast component originated from the second, W434F, repeat. Accordingly, the ratio of the amplitudes of fast and slow decay (inactivation) was reduced significantly in the F3 concatemer (Fig. 5d).
Table 1.
Exponential decay of concatemers (Fig. 5c)
| τ1 | τ2 | A(τ1)/A(τ2) | |
|---|---|---|---|
| Tet | 0.33 ± 0.03 | 6.91 ± 1.0 | 1.18 ± 0.38 |
| F1 | 0.30 ± 0.04 | 6.04 ± 0.57 | 1.57 ± 0.58 |
| F2 | 0.37 ± 0.09 | 6.05 ± 0.59 | 1.89 ± 1.18 |
| F3 | 0.30 ± 0.08 | 6.68 ± 1.30 | 0.55 ± 0.25 |
Inactivation was fitted with a two-component exponential with time constants τ1 and τ2. A(τ1) and A(τ2) are the amplitudes for the two components.
The effect that we observed here was the same as in F244C-W434F and F244C. In all cases, the steady-state value for entry into C-type inactivation was reduced. The F1 and F2 concatemers act upon the slow component, indicating that they influence exclusively a wild-type repeat. The F3 concatemer, on the other hand, influences exclusively the second (W434F) repeat. The mutation F244C thus only acts upon the neighboring subunit, as predicted for a direct interaction among F244, I429, and W434.
Our results obtained from the concatemers also indicate that the tetramer is always synthesized in the same direction—counterclockwise—when seen from the external side along the central axis. We only observed an effect on the W434F mutation if F244C was introduced in repeat III. According to the crystal structure, F244C of repeat III and W434F of repeat II are only in close proximity if the channel is assembled counterclockwise (Fig. 5e). It will be interesting to compare this result with the crystal structure of a voltage-gated sodium or calcium channel once they are available.
Effect of F244C on N-type inactivation and recovery from inactivation
The question remains in how far an effect on the conducting state of the selectivity filter (C-type inactivation) can have a physiological effect. In the hippocampus, Kv1.1 coexpresses with Kvβ1, leading to rapid N-type inactivation blocking ion conduction with the ball peptide (Pongs and Schwarz, 2010). In this case, the primary effect of C-type inactivation would be hidden because the channel would already be nonconducting. However, it has been shown that, in the presence of fast (N-type) inactivation, C-type inactivation is significantly accelerated and determines the rate of recovery from inactivation (Baukrowitz and Yellen, 1995). We therefore tested the recovery from inactivation after a 20 ms depolarizing pulse in the full-length Shaker Kv channel including the inactivating N terminus. As in the inactivation-removed ShakerIR Kv channel, F244C led to slowed activation kinetics (Fig. 6b). N-type inactivation was shifted parallel to activation to more depolarized potentials (−23.2 ± 0.6 mV and 5.5 ± 0.4 mV for WT and F244C, respectively; Fig. 6a). We also found that total inactivation was reduced even after prolonged depolarization (Fig. 6c). Impaired N-type inactivation has been described for Shaker Kv channels at high expression levels (Moran et al., 1992). However, expression levels were comparable to the control experiments because it has been shown that F184C in Kv1.1 does not alter single channel conductance or open probability (Bretschneider et al., 1999). Therefore, the induced conformational change in the pore domain related to C-type inactivation also influences the effectiveness of N-type inactivation, suggesting that the conformational changes by C-type inactivation also reach the lower pore region (Cuello et al., 2010b; Labro et al., 2012).
Figure 6.
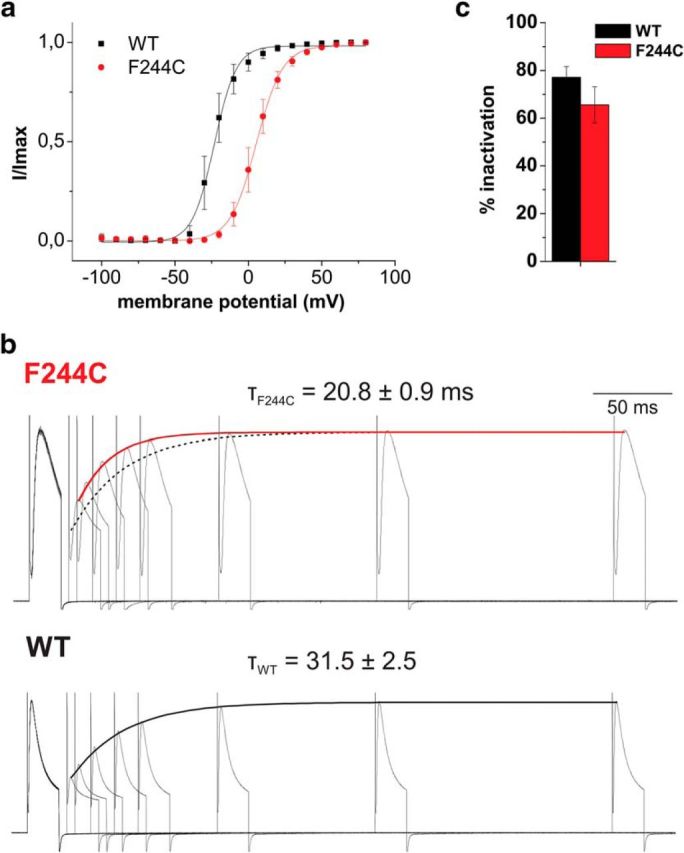
a, Voltage dependence of fast inactivation for Shaker-WT and Shaker-F244C. Inactivation was determined by a test pulse to +50 mV (wild-type, WT) or +80 mV (F244C) after a series of depolarizations. b, Time course of recovery from inactivation for Shaker-F244C and Shaker-WT elicited from two 20 ms pulses from −90 mV to +50 mV and +80 mV for WT and F244C, respectively, with varying interpulse intervals. The dotted line marks the recovery from inactivation of Shaker-WT. c, Percentage of fast inactivation for Shaker-WT and Shaker-F244C.
In addition to the already impaired N-type inactivation, recovery from inactivation was also accelerated, which is consistent with an effect of F244C on C-type inactivation governing recovery from inactivation (τWT = 31.5 ± 2.5 ms vs τF244C = 20.8 ± 0.9 ms; Fig. 6b).
Discussion
The disease mutation F244C in the outer S1 influences all three of the following processes: the gating charge movement (voltage dependence of activation), slow (C-type) inactivation (conductance state of the selectivity filter), and fast (N-type) inactivation. In the hippocampus, fast inactivation in Kv1.1 is induced by the N terminus of the coexpressed Kvβ1. The position F244 in the S1 thus plays a central role in the gating machinery of the Kv channels. It influences both the gating of the voltage-sensing domain and the ion conduction pathway. The gating of the voltage-sensing domain is shifted to more depolarized potentials with respect to wild-type. Proportionally, the conductance voltage relation is also shifted to more depolarized potentials. The position in the crystal structure suggests that F244 interacts directly with the arginines of the S4. More surprising was the fact that a negative charge at this position in F244E did not have a significantly different effect as, for example, an alanine or cysteine, suggesting that the interaction has no electrostatic component to it. One explanation would be that the additional cost of breaking the salt bridge would be compensated by the additional gain of forming another one with the next arginine in the S4. Alternatively, F244E replaces the aspartate at position E283 such that no additional energy is required. Consistent with the effect not being electrostatic, Lacroix et al. (2012, 2014) suggested that the van-der-Waals surface of F244 is responsible for this interaction. It had been proposed that F244 in the S1, together with F290 in the S2, form a “molecular clamp” that defines the movement of the S4.
In addition to its effect on the S4-gating energetics, F244 also influences C-type inactivation directly most likely via a link with I429 and W434 in the pore helix (Fig. 4b). Proximity between the S1 and the pore domain has been suggested previously by Lee et al. (2009). The investigators suggested that the interface between the S1 and the pore domain is evolutionary conserved. It had been shown previously that replacing residues in the pore domain with “bulky” tryptophanes leads to shifts in the voltage dependence of gating in Shaker, suggesting that they were facing the voltage-sensing domain. Interestingly, residue I429W did not yield functional channels (Li-Smerin et al., 2000). Here, we showed for the first time that the interface not only shows spatial proximity, but that there is a direct functional link between the S1 and the pore domain, influencing the selectivity filter and preventing it from undergoing complete C-type inactivation. Although I429A channels did not lead to leak currents in ShakerIR-W434F, they did shift the voltage dependence of gating charge movement to more depolarized potentials just like the F244 mutants. Furthermore, the concatemers show that the F244 mutation only acts upon the neighboring subunit as predicted if F244-I429 and W434 interact directly rather than through the electromechanical coupling region in the S4–S5 linker and the S6.
These results indicate that the intersubunit link between the voltage-sensing domains and the neighboring selectivity filter on the extracellular face influences the function of the channel. What does this mean for the electromechanical coupling between voltage sensing and pore opening? It has been shown that the region annealing the S4–S5 linker with the C-terminal S6 is crucial for coupling (Lu et al., 2001, 2002; Long et al., 2005a; Labro et al., 2008; Haddad and Blunck, 2011), including an intersubunit interaction on the cytosolic surface (Batulan et al., 2010). Mutating position I384 to asparagine led to uncoupling of VSD and pore, indicating that this region is the predominant mechanism. However, even in the uncoupled mutants, the small residual pore opening still showed a voltage dependence (Haddad and Blunck, 2011). This residual voltage dependence might be caused by the intersubunit coupling between the VSD and pore at the extracellular side, as described in this work. It might also be responsible for part of the voltage dependence of C-type inactivation. However, it would require more work to elucidate these possibilities.
The widespread influence of a single point mutation at a position that was thought previously to be “peripheral” emphasizes the complexity of the gating processes of voltage-gated ion channels. Although it is important to understand (and dissect) the different gating processes, they are closely interdependent. This “allosteric” influence becomes especially evident in the alteration of N-type inactivation by the F244C mutation in the S1. During N-type inactivation, the N terminus is thought to enter the inner pore, thus blocking conductance. Both the N terminus and the inner pore gate are far away from the mutated position F244.
The varied effects on the Kv channel also raise the question of which of them is most likely to be responsible for the EA1 phenotype. Given Kv1.1's role in repolarization, a shift in its voltage dependence of activation to more depolarized potentials and its slowed activation kinetics would lead to later onset of repolarization and earlier deactivation of Kv1.1. Both effects would cause action potentials to prolong.
Conversely, fast inactivation and recovery from slow inactivation of Kv1.1 has been suggested to be the mechanism underlying spike broadening upon repetitive firing (Geiger and Jonas, 2000). Impaired (incomplete) fast and slow inactivation of Kv1.1 would cause more channels to remain open until deactivation leading to faster repolarization. Taking into account the faster recovery from inactivation, even more Kv1.1 channels would be available for the next action potential, whereas sodium channel recovery from inactivation would remain normal, leading to prolonged interspike intervals for a given depolarization.
It is difficult to pinpoint the exact cell type responsible for the phenotype because Kv1.1 channels are widely expressed, being found in both excitatory and inhibitory neurons (Wang et al., 1994; Geiger and Jonas, 2000; Grosse et al., 2000). Both effects, the prolonged action potentials and the increased interspike intervals, would likely not play any significant role at the long interspike intervals found, for example, in pyramidal cells. However, the often short interspike intervals found in interneurons would probably lead to continuous problems, inconsistent with the sporadic nature of EA1.
The reason that small changes are well tolerated or compensated in the axons is the digital nature of the signals. Normally, presynaptic signals are transmitted by digital all-or-nothing responses. However, these spike-envoked responses may be modulated by subthreshold depolarization (analog; Alle and Geiger, 2006). Whereas the digital responses do not seem to be the prime suspects causing EA1 in both pyramidal cells and interneurons, the role that Kv1.1 plays for the analog modulation presents a possible mechanism. Kv1.1 has recently been related to analog digital (AD) facilitation (Kole et al., 2007; Shu et al., 2007; Bialowas et al., 2015). In CA3 pyramidal neurons, blocking of Kv1.1 prevents AD facilitation, which has been suggested to be linked to Kv1.1 inactivation. A shift of the voltage dependence and impairment of Kv1.1 inactivation would thus alter AD facilitation. The analog nature of the modulation also lets small modulations lead to alterations in the response as no all-or-nothing correction occurs.
At present, our conclusions are only speculative and it will require more specific investigations in situ to understand the molecular mechanisms leading to the EA1 phenotype. The sporadic or chaotic occurrence even hints at a combined effect of different cell types consistent with the widespread expression of Kv1.1 channels in the hippocampus. We have shown here that the molecular mechanisms underlying the EA1 mutation show a higher degree of complexity and allosteric components than previously thought and they give us a deep insight into the functioning of Kv channels.
Footnotes
This work was supported by the Canadian Institutes for Health Research (Grants MOP-102689 and MOP-136894 to R.B.). J.Z. holds a fellowship by the Natural Sciences and Engineering Research Council of Canada Collaborative Research and Training Experience (CREATE) program Cellular Dynamics of Macromolecular Complexes (CDMC). GÉPROM is a research group funded by the Fonds de recherche du Québec–Santé. We thank Yolaine Dodier and Mireille Marsolais for technical assistance and Dr. Roberto Araya for helpful discussion. The tetramer construct was based on a concatemer provided by Drs. Dominique Gagnon and Francisco Bezanilla.
The authors declare no competing financial interests.
References
- Adelman JP, Bond CT, Pessia M, Maylie J. Episodic ataxia results from voltage-dependent potassium channels with altered functions. Neuron. 1995;15:1449–1454. doi: 10.1016/0896-6273(95)90022-5. [DOI] [PubMed] [Google Scholar]
- Alle H, Geiger JR. Combined analog and action potential coding in hippocampal mossy fibers. Science. 2006;311:1290–1293. doi: 10.1126/science.1119055. [DOI] [PubMed] [Google Scholar]
- Batulan Z, Haddad GA, Blunck R. An intersubunit interaction between S4–S5 linker and S6 is responsible for the slow off-gating component in Shaker K+ channels. J Biol Chem. 2010;285:14005–14019. doi: 10.1074/jbc.M109.097717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baukrowitz T, Yellen G. Modulation of K+ current by frequency and external [K+]: a tale of two inactivation mechanisms. Neuron. 1995;15:951–960. doi: 10.1016/0896-6273(95)90185-X. [DOI] [PubMed] [Google Scholar]
- Berger TK, Isacoff EY. The pore of the voltage-gated proton channel. Neuron. 2011;72:991–1000. doi: 10.1016/j.neuron.2011.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernèche S, Roux B. A gate in the selectivity filter of potassium channels. Structure. 2005;13:591–600. doi: 10.1016/j.str.2004.12.019. [DOI] [PubMed] [Google Scholar]
- Bezanilla F. How membrane proteins sense voltage. Nat Rev Mol Cell Biol. 2008;9:323–332. doi: 10.1038/nrm2376. [DOI] [PubMed] [Google Scholar]
- Bialowas A, Rama S, Zbili M, Marra V, Fronzaroli-Molinieres L, Ankri N, Carlier E, Debanne D. Analog modulation of spike-evoked transmission in CA3 circuits is determined by axonal Kv1.1 channels in a time-dependent manner. Eur J Neurosci. 2015;41:293–304. doi: 10.1111/ejn.12787. [DOI] [PubMed] [Google Scholar]
- Blunck R, Batulan Z. Mechanism of electromechanical coupling in voltage-gated potassium channels. Front Pharmacol. 2012;3:166. doi: 10.3389/fphar.2012.00166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretschneider F, Wrisch A, Lehmann-Horn F, Grissmer S. Expression in mammalian cells and electrophysiological characterization of two mutant Kv1.1 channels causing episodic ataxia type 1 (EA-1) Eur J Neurosci. 1999;11:2403–2412. doi: 10.1046/j.1460-9568.1999.00659.x. [DOI] [PubMed] [Google Scholar]
- Browne DL, Brunt ER, Griggs RC, Nutt JG, Gancher ST, Smith EA, Litt M. Identification of two new KCNA1 mutations in episodic ataxia/myokymia families. Hum Mol Genet. 1995;4:1671–1672. doi: 10.1093/hmg/4.9.1671. [DOI] [PubMed] [Google Scholar]
- Chowdhury S, Haehnel BM, Chanda B. A self-consistent approach for determining pairwise interactions that underlie channel activation. J Gen Physiol. 2014;144:441–455. doi: 10.1085/jgp.201411184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claydon TW, Vaid M, Rezazadeh S, Kwan DC, Kehl SJ, Fedida D. A direct demonstration of closed-state inactivation of K+ channels at low pH. J Gen Physiol. 2007;129:437–455. doi: 10.1085/jgp.200709774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordero-Morales JF, Cuello LG, Zhao Y, Jogini V, Cortes DM, Roux B, Perozo E. Molecular determinants of gating at the potassium-channel selectivity filter. Nat Struct Mol Biol. 2006;13:311–318. doi: 10.1038/nsmb1069. [DOI] [PubMed] [Google Scholar]
- Cuello LG, Jogini V, Cortes DM, Perozo E. Structural mechanism of C-type inactivation in K(+) channels. Nature. 2010a;466:203–208. doi: 10.1038/nature09153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuello LG, Jogini V, Cortes DM, Pan AC, Gagnon DG, Dalmas O, Cordero-Morales JF, Chakrapani S, Roux B, Perozo E. Structural basis for the coupling between activation and inactivation gates in K(+) channels. Nature. 2010b;466:272–275. doi: 10.1038/nature09136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faure É, Starek G, McGuire H, Bernèche S, Blunck R. A limited 4 A radial displacement of the S4–S5 linker is sufficient for internal gate closing in Kv channels. J Biol Chem. 2012;287:40091–40098. doi: 10.1074/jbc.M112.415497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagnon DG, Bezanilla F. A single charged voltage sensor is capable of gating Shaker K + channel. J Gen Physiol. 2009;133:467–483. doi: 10.1085/jgp.200810082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger JR, Jonas P. Dynamic control of presynaptic Ca(2+) inflow by fast-inactivating K(+) channels in hippocampal mossy fiber boutons. Neuron. 2000;28:927–939. doi: 10.1016/S0896-6273(00)00164-1. [DOI] [PubMed] [Google Scholar]
- Grosse G, Draguhn A, Höhne L, Tapp R, Veh RW, Ahnert-Hilger G. Expression of Kv1 potassium channels in mouse hippocampal primary cultures: development and activity-dependent regulation. J Neurosci. 2000;20:1869–1882. doi: 10.1523/JNEUROSCI.20-05-01869.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddad GA, Blunck R. Mode shift of the voltage sensors in Shaker K+ channels is caused by energetic coupling to the pore domain. J Gen Physiol. 2011;137:455–472. doi: 10.1085/jgp.201010573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong KH, Miller C. The lipid-protein interface of a Shaker K(+) channel. J Gen Physiol. 2000;115:51–58. doi: 10.1085/jgp.115.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horovitz A. Double-mutant cycles: a powerful tool for analyzing protein structure and function. Folding and Design. 1996;1:R121–R126. doi: 10.1016/S1359-0278(96)00056-9. [DOI] [PubMed] [Google Scholar]
- Jen JC. Hereditary episodic ataxias. Ann NY Acad Sci. 2008;1142:250–253. doi: 10.1196/annals.1444.016. [DOI] [PubMed] [Google Scholar]
- Kalstrup T, Blunck R. Dynamics of internal pore opening in KV channels probed by a fluorescent unnatural amino acid. Proc Natl Acad Sci U S A. 2013;110:8272–8277. doi: 10.1073/pnas.1220398110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kole MH, Letzkus JJ, Stuart GJ. Axon initial segment Kv1 channels control axonal action potential waveform and synaptic efficacy. Neuron. 2007;55:633–647. doi: 10.1016/j.neuron.2007.07.031. [DOI] [PubMed] [Google Scholar]
- Labro AJ, Raes AL, Grottesi A, Van Hoorick D, Sansom MS, Snyders DJ. Kv channel gating requires a compatible S4–S5 linker and bottom part of S6, constrained by non-interacting residues. J Gen Physiol. 2008;132:667–680. doi: 10.1085/jgp.200810048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labro AJ, Lacroix JJ, Villalba-Galea CA, Snyders DJ, Bezanilla F. Molecular mechanism for depolarization-induced modulation of Kv channel closure. J Gen Physiol. 2012;140:481–493. doi: 10.1085/jgp.201210817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacroix JJ, Bezanilla F. Tuning the voltage-sensor motion with a single residue. Biophys J. 2012;103:L23–25. doi: 10.1016/j.bpj.2012.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacroix JJ, Hyde HC, Campos FV, Bezanilla F. Moving gating charges through the gating pore in a Kv channel voltage sensor. Proc Natl Acad Sci U S A. 2014;111:E1950–E1959. doi: 10.1073/pnas.1406161111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SY, Banerjee A, MacKinnon R. Two separate interfaces between the voltage sensor and pore are required for the function of voltage-dependent K(+) channels. PLoS Biol. 2009;7:e47. doi: 10.1371/journal.pbio.1000047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li-Smerin Y, Hackos DH, Swartz KJ. A localized interaction surface for voltage-sensing domains on the pore domain of a K+ channel. Neuron. 2000;25:411–423. doi: 10.1016/S0896-6273(00)80904-6. [DOI] [PubMed] [Google Scholar]
- Long SB, Campbell EB, Mackinnon R. Voltage sensor of Kv1.2: structural basis of electromechanical coupling. Science. 2005a;309:903–908. doi: 10.1126/science.1116270. [DOI] [PubMed] [Google Scholar]
- Long SB, Campbell EB, Mackinnon R. Crystal structure of a mammalian voltage-dependent shaker family K+ channel. Science. 2005b;309:897–903. doi: 10.1126/science.1116269. [DOI] [PubMed] [Google Scholar]
- Long SB, Tao X, Campbell EB, MacKinnon R. Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature. 2007;450:376–382. doi: 10.1038/nature06265. [DOI] [PubMed] [Google Scholar]
- Lu Z, Klem AM, Ramu Y. Ion conduction pore is conserved among potassium channels. Nature. 2001;413:809–813. doi: 10.1038/35101535. [DOI] [PubMed] [Google Scholar]
- Lu Z, Klem AM, Ramu Y. Coupling between voltage sensors and activation gate in voltage-gated K+ channels. J Gen Physiol. 2002;120:663–676. doi: 10.1085/jgp.20028696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran O, Schreibmayer W, Weigl L, Dascal N, Lotan I. Level of expression controls modes of gating of a K+ channel. FEBS Lett. 1992;302:21–25. doi: 10.1016/0014-5793(92)80275-L. [DOI] [PubMed] [Google Scholar]
- Moreau A, Gosselin-Badaroudine P, Delemotte L, Klein ML, Chahine M. Gating pore currents are defects in common with two Nav1.5 mutations in patients with mixed arrhythmias and dilated cardiomyopathy. J Gen Physiol. 2015;145:93–106. doi: 10.1085/jgp.201411304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muroi Y, Arcisio-Miranda M, Chanda B. Tryptophan scanning mutagenesis to identify the residues involved in coupling between the pore and DIII voltage-sensor of a sodium channel [abstract] Biophys J. 2009;96:248a. [Google Scholar]
- Olcese R, Sigg D, Latorre R, Bezanilla F, Stefani E. A conducting state with properties of a slow inactivated state in a shaker K(+) channel mutant. J Gen Physiol. 2001;117:149–163. doi: 10.1085/jgp.117.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak MM, Yarov-Yarovoy V, Agarwal G, Roux B, Barth P, Kohout S, Tombola F, Isacoff EY. Closing in on the resting state of the Shaker K(+) channel. Neuron. 2007;56:124–140. doi: 10.1016/j.neuron.2007.09.023. [DOI] [PubMed] [Google Scholar]
- Pathak M, Kurtz L, Tombola F, Isacoff E. The cooperative voltage sensor motion that gates a potassium channel. J Gen Physiol. 2005;125:57–69. doi: 10.1085/jgp.200409197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perozo E, MacKinnon R, Bezanilla F, Stefani E. Gating currents from a nonconducting mutant reveal open-closed conformations in Shaker K+ channels. Neuron. 1993;11:353–358. doi: 10.1016/0896-6273(93)90190-3. [DOI] [PubMed] [Google Scholar]
- Pongs O, Schwarz JR. Ancillary subunits associated with voltage-dependent K+ channels. Physiol Rev. 2010;90:755–796. doi: 10.1152/physrev.00020.2009. [DOI] [PubMed] [Google Scholar]
- Ramsey IS, Moran MM, Chong JA, Clapham DE. A voltage-gated proton-selective channel lacking the pore domain. Nature. 2006;440:1213–1216. doi: 10.1038/nature04700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoppa NE, Sigworth FJ. Activation of Shaker potassium channels. III. An activation gating model for wild-type and V2 mutant channels. J Gen Physiol. 1998;111:313–342. doi: 10.1085/jgp.111.2.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu Y, Yu Y, Yang J, McCormick DA. Selective control of cortical axonal spikes by a slowly inactivating K+ current. Proc Natl Acad Sci U S A. 2007;104:11453–11458. doi: 10.1073/pnas.0702041104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolov S, Scheuer T, Catterall WA. Gating pore current in an inherited ion channelopathy. Nature. 2007;446:76–78. doi: 10.1038/nature05598. [DOI] [PubMed] [Google Scholar]
- Starace DM, Bezanilla F. Histidine scanning mutagenesis of basic residues of the S4 segment of the shaker k+ channel. J Gen Physiol. 2001;117:469–490. doi: 10.1085/jgp.117.5.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starace DM, Bezanilla F. A proton pore in a potassium channel voltage sensor reveals a focused electric field. Nature. 2004;427:548–553. doi: 10.1038/nature02270. [DOI] [PubMed] [Google Scholar]
- Taglialatela M, Toro L, Stefani E. Novel voltage clamp to record small, fast currents from ion channels expressed in Xenopus oocytes. Biophys J. 1992;61:78–82. doi: 10.1016/S0006-3495(92)81817-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tombola F, Pathak MM, Gorostiza P, Isacoff EY. The twisted ion-permeation pathway of a resting voltage-sensing domain. Nature. 2007;445:546–549. doi: 10.1038/nature05396. [DOI] [PubMed] [Google Scholar]
- Tombola F, Ulbrich MH, Isacoff EY. The voltage-gated proton channel Hv1 has two pores, each controlled by one voltage sensor. Neuron. 2008;58:546–556. doi: 10.1016/j.neuron.2008.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall-Lacelle S, Hossain MI, Sauvé R, Blunck R, Parent L. Double mutant cycle analysis identified a critical leucine residue in the IIS4S5 linker for the activation of the Ca(V)2.3 calcium channel. J Biol Chem. 2011;286:27197–27205. doi: 10.1074/jbc.M111.237412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Kunkel DD, Schwartzkroin PA, Tempel BL. Localization of Kv1.1 and Kv1.2, two K channel proteins, to synaptic terminals, somata, and dendrites in the mouse brain. J Neurosci. 1994;14:4588–4599. doi: 10.1523/JNEUROSCI.14-08-04588.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Yan Y, Sigworth FJ. How does the W434F mutation block current in Shaker potassium channels? J Gen Physiol. 1997;109:779–789. doi: 10.1085/jgp.109.6.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagotta WN, Hoshi T, Aldrich RW. Shaker potassium channel gating. III: Evaluation of kinetic models for activation. J Gen Physiol. 1994;103:321–362. doi: 10.1085/jgp.103.2.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerr P, Adelman JP, Maylie J. Episodic ataxia mutations in Kv1.1 alter potassium channel function by dominant negative effects or haploinsufficiency. J Neurosci. 1998;18:2842–2848. doi: 10.1523/JNEUROSCI.18-08-02842.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]