

Abstract
Multiple sclerosis (MS) is a progressive neurological disorder associated with myelin destruction and neurodegeneration. Oligodendrocyte precursor cells (OPCs) present in demyelinated lesions gradually fail to differentiate properly, so remyelination becomes incomplete. Protein tyrosine phosphatase receptor type Z (PTPRZ), one of the most abundant protein tyrosine phosphatases expressed in OPCs, is known to suppress oligodendrocyte differentiation and maintain their precursor cell stage. In the present study, we examined the in vivo mechanisms for remyelination using a cuprizone-induced demyelination model. Ptprz-deficient and wild-type mice both exhibited severe demyelination and axonal damage in the corpus callosum after cuprizone feeding. The similar accumulation of OPCs was observed in the lesioned area in both mice; however, remyelination was significantly accelerated in Ptprz-deficient mice after the removal of cuprizone. After demyelination, the expression of pleiotrophin (PTN), an inhibitory ligand for PTPRZ, was transiently increased in mouse brains, particularly in the neurons involved, suggesting its role in promoting remyelination by inactivating PTPRZ activity. In support of this view, oligodendrocyte differentiation was augmented in a primary culture of oligodendrocyte-lineage cells from wild-type mice in response to PTN. In contrast, these cells from Ptprz-deficient mice showed higher oligodendrocyte differentiation without PTN and differentiation was not enhanced by its addition. We further demonstrated that PTN treatment increased the tyrosine phosphorylation of p190 RhoGAP, a PTPRZ substrate, using an established line of OPCs. Therefore, PTPRZ inactivation in OPCs by PTN, which is secreted from demyelinated axons, may be the mechanism responsible for oligodendrocyte differentiation during reparative remyelination in the CNS.
SIGNIFICANCE STATEMENT Multiple sclerosis (MS) is an inflammatory disease of the CNS that destroys myelin, the insulation that surrounds axons. Associated damages to oligodendrocytes (the cells that produce myelin) and nerve fibers produce neurological disability. Most patients with MS have an initial relapsing-remitting course for 5–15 years. Remyelination during the early stages of the disease process has been documented; however, the molecular mechanism underlying remyelination has not been understood. Protein tyrosine phosphatase receptor type Z (PTPRZ) is a receptor-like protein tyrosine phosphatase preferentially expressed in the CNS. This study shows that pleiotrophin, an inhibitory ligand for PTPRZ, is transiently expressed and released from demyelinated neurons to inactivate PTPRZ in oligodendrocyte precursor cells present in the lesioned part, thereby allowing their differentiation for remyelination.
Keywords: multiple sclerosis, oligodendrocyte, pleiotrophin, PTPRZ, remyelination, tyrosine phosphorylation
Introduction
Myelination is an essential feature of the vertebrate nervous system that provides electrical insulation to axons, thereby facilitating the transmission of nerve impulses. Deficiencies in myelination in diseases such as multiple sclerosis (MS) lead to serious neurological disorders (Frohman et al., 2006). Most MS patients initially exhibit a relapsing-remitting disease course that eventually converts to a secondary progressive form of the disease with incomplete recovery. Oligodendrocyte precursor cells (OPCs), the principal source of myelinating oligodendrocytes (Goldman et al., 2012), differentiate to myelinate demyelinated axons again (remyelination) during remitting stages (Chang et al., 2002). Two animal disease models are widely accepted for studying the clinical and pathological features of MS lesions. Experimental autoimmune encephalomyelitis (EAE) is a T-cell-mediated inflammatory CNS demyelination model that is generated by immunization with the myelin/oligodendrocyte glycoprotein (MOG) (Høglund and Maghazachi, 2014). On the other hand, the cuprizone model of demyelination is induced, particularly in the corpus callosum in the CNS, by a T-cell-independent mechanism through feeding with the copper chelator cuprizone (Matsushima and Morell, 2001).
Previous studies identified protein tyrosine phosphorylation as a key event in oligodendrocyte differentiation and myelin formation (Krämer-Albers and White, 2011). The tyrosine phosphorylation of cellular proteins is controlled by the activities of protein tyrosine kinases (PTKs) and protein tyrosine phosphatases (PTPs). FYN tyrosine kinase signaling is a major pathway for oligodendrocyte differentiation, myelination, and remyelination (Umemori et al., 1994, 1999; Scarlato et al., 2000), whereas our knowledge regarding the functional role and regulatory mechanism of PTPs in these processes is limited.
PTPRZ [also called PTPζ or receptor-like PTPβ (RPTPβ)] is an RPTP that is predominantly expressed in the CNS (Maeda et al., 1994). Three isoforms have been generated by alternative splicing from a single PTPRZ gene: two transmembrane isoforms, PTPRZ-A and PTPRZ-B, and the secretory isoform PTPRZ-S (also known as phosphacan) (Maeda et al., 1994; Chow et al., 2008a). All three isoforms expressed in the CNS were shown to be highly glycosylated by chondroitin sulfate (Nishiwaki et al., 1998; Chow et al., 2008b; Kuboyama et al., 2012). The extracellular region of PTPRZ binds pleiotrophin (PTN)/heparin-binding growth-associated molecule (HB-GAM) (Maeda et al., 1996), midkine (Maeda et al., 1999), and IL-34 (Nandi et al., 2013) as its inhibitory ligands. PTN binding leads to inactivation of the intracellular catalytic activity of PTPRZ by inducing receptor dimerization or oligomerization (Meng et al., 2000; Kawachi et al., 2001; Fukada et al., 2006).
We recently demonstrated that PTPRZ functioned conversely to FYN tyrosine kinase in oligodendrocyte-lineage cells and negatively regulated the differentiation of oligodendrocytes and myelin formation in vivo (Kuboyama et al., 2012). The knock-out of Ptprz does not cause any gross anatomical abnormality in mice (Shintani et al., 1998), whereas adult Ptprz-deficient mice are markedly resistant to the MOG-mediated induction of EAE (Kuboyama et al., 2012). p190 RhoGAP, a GTPase-activating protein (GAP) for Rho GTPase, is one of the PTPRZ substrates in neurons and glial cells (Fukada et al., 2005; Tamura et al., 2006; Fujikawa et al., 2011; Kuboyama et al., 2012). The tyrosine phosphorylation of p190 RhoGAP by FYN activates its Rho GTPase activity (Wolf et al., 2001) and thereby induces oligodendrocyte differentiation and myelination (Wolf et al., 2001; Krämer-Albers and White, 2011). Therefore, PTPRZ functions as a negative regulator of oligodendrocyte differentiation, myelination, and remyelination (Kuboyama et al., 2012); however, the in vivo regulatory mechanism of PTPRZ activity in oligodendrocyte-lineage cells remains unclear.
In the present study, we found that Ptprz-deficient mice exhibited accelerated remyelination in the corpus callosum after cuprizone-induced demyelination. The expression of the inhibitory PTPRZ ligand PTN was strongly induced in demyelinated neurons. PTN-treated oligodendrocyte-lineage cells exhibited enhanced differentiation due to the inhibition of PTPRZ. The PTPRZ-mediated repression of oligodendrocyte differentiation appears to be relieved by PTN secreted from demyelinated nerve fibers for their remyelination.
Materials and Methods
Ethics statement.
All animal experimental protocols used in this study were approved by the Institutional Animal Care and Use Committee of National Institutes of Natural Sciences, Japan. All surgeries were performed under isoflurane anesthesia and all efforts were made to minimize suffering.
Animals.
Ptprz-deficient mice (Shintani et al., 1998) and p53-deficient mice (Gondo et al., 1994) backcrossed with the inbred C57BL/6 strain for more than 10 generations, respectively, were used with their wild-type counterparts as a control. All animals were maintained at 23°C under specific pathogen-free conditions.
Antibodies.
The purified rabbit polyclonal antibody against phosphorylated Tyr-1105 of p190 RhoGAP (anti-pY1105) was described previously (Tamura et al., 2006). We used commercially available antibodies against myelin basic protein (MBP, catalog #sc-13914; Santa Cruz Biotechnology), oligodendrocyte transcription factor 2 (Olig2; catalog #AB9610; Millipore), glial fibrillary acidic protein (GFAP, catalog #Z0334; DAKO), amyloid-β precursor protein (APP, catalog #ab15272; Abcam), neural/glial antigen 2 chondroitin sulfate proteoglycan (NG2 proteoglycan, catalog #AB5320; Millipore), pleiotrophin (PTN, catalog #ab79411; Abcam), midkine (catalog #sc-20715; Santa Cruz Biotechnology), interleukin-34 (IL-34, catalog #PRS4781; Sigma-Aldrich), RPTPβ (catalog #610179; BD Biosciences), β-tubulin III (catalog #T8660; Sigma-Aldrich), synapsin 1 (SYN1, catalog #574777; Millipore), p190 RhoGAP (catalog #610150; BD Biosciences and catalog #12164; Cell Signaling Technology), and phospho-Tyr (PY20, catalog #ab16389; Abcam).
Cuprizone-induced demyelination model.
Demyelination was induced in 2-month-old male mice by feeding a diet of powdered mouse chow (Rodent Diet CA-1; CLEA) containing 0.2% (w/w) cuprizone (bis-cyclohexanone oxaldihydrazone; Sigma-Aldrich) for 6 weeks, followed by a return to the normal pellet diet of CA-1 for remyelination.
Microcomputed tomography imaging.
For microcomputed tomography (micro-CT) imaging, mice anesthetized with isoflurane were transcardially perfused with 4% paraformaldehyde in PBS containing the following (in mm): 4.3 Na2HPO4, 1.4 KH2PO4, 137 NaCl, and 2.7 KCl, pH 7.4. The brains were removed and postfixed overnight at 4°C in the same fixative solution and then immersed in a graded series of Histodenz [5-(N-2,3-dihydroxypropylacetamido)-2,4,6-triiodo-N,N′-bis (2,3-dihydroxypropyl) isophthalamide; Sigma-Aldrich] solutions (7.5% and 15% for 1 d each and 22.5% for 3 d at 4°C) with mild rotation. The brains were gently wiped to remove surface solution and then scanned on a micro-CT system R_mCT2 (Rigaku) at 90 kV (160 μA) with a 20 mm field of view. Data were reconstructed by OsiriX software (Pixmeo) and the average CT numbers from two regions of interest (four adjacent pixels) in the dorsal corpus callosum and cerebral cortex were recorded for each image.
Luxol fast blue and immunohistochemical staining.
After CT scanning, the brains were immersed in 70% ethanol overnight and then subjected to conventional paraffin embedding. Paraffin-embedded brains were serially sectioned at 3 μm intervals in the coronal plane. Luxol fast blue (LFB) staining of deparaffinized sections was performed with LFB stain solution (catalog #4102; Muto Pure Chemicals). For immunohistochemistry, deparaffinized sections were pretreated with 3% hydrogen peroxide and 0.04% NP-40 in TBS (10 mm Tris-HCl, pH 7.4, 150 mm NaCl) for 30 min, blocked with 4% nonfat dry milk and 0.1% Triton X-100 in TBS, and then incubated with each specific primary antibody overnight at 4°C. Sections for anti-APP, anti-PTN, anti-β-tubulin III, or anti-SYN1 staining were microwaved in 10 mm citrate buffer, pH 6.0, at 98°C for 30 min as an antigen retrieval step. The primary antibodies were detected with horseradish peroxidase (HRP)-conjugated secondary antibodies and the Dako liquid diaminobenzidine chromogen system (DAKO), Alexa Fluor-conjugated secondary antibodies (Invitrogen), or DyLight Amine-Reactive Dyes (DyLight 488 NHS Ester; Thermo Scientific) according to the manufacturers' instructions. Digital photomicrographs of each specimen were taken with an Eclipse microscope Ci-L with a DS-Fi2 CCD (Nikon) or LSM 700 confocal microscope (Zeiss) and analyzed using Adobe Photoshop CS6 software.
Culture of primary mixed glial cells and isolation of oligodendrocyte lineage cells.
A primary mixed glial cell culture was prepared from wild-type and Ptprz-deficient mice as described previously (Kuboyama et al., 2012). Brain cortex tissues were obtained from mice on postnatal day 1, followed by papain digestion. Dissociated cells were cultured for 6 d on a poly-l-ornithine-coated 35 mm dish (2.0 × 104 cells per dish) with DMEM mixed 1:1 with Ham's F-12 (DMEM/F12; Life Technologies) supplemented with 1× GlutaMAX (Life Technologies), 1× N2 supplement (Life Technologies), 10 μg/ml of the AA homodimeric form of platelet-derived growth factor (PDGF-AA; Wako Pure Chemical), 0.5% fetal bovine serum (Nichirei Biosciences), and 100 μg/ml of bovine serum albumin (Sigma-Aldrich) in a humidified incubator at 37°C with 5% CO2. Cell differentiation was induced by the addition of 10 nm biotin (Sigma-Aldrich) and 30 ng/ml thyronine/thyroxine (Sigma-Aldrich). Recombinant human PTN produced in yeast was used as described previously (Murasugi et al., 2003; Fukada et al., 2005).
Mouse oligodendrocyte-lineage cells (OL1 cells) were established from the primary mixed glial cell culture from p53-deficient mice that have been used as a source of various cell lines (Tsukada et al., 1993). After a 10 d culture, cells were removed with 0.25% Trypsin-EDTA (Life Technologies) and replated on a poly-l-ornithine-coated 100 mm dish (4.0 × 105 cells per dish) with the following basal medium: knock-out DMEM/F12 (Life Technologies) supplemented with 1× GlutaMAX, 1× StemPro Neural Supplement (Life Technologies), 20 μg/ml of basic fibroblast growth factor (bFGF; Wako Pure Chemical), and 10 μg/ml of PDGF-AA. After a 14 d culture, the cell spheres that formed were collected with a sterilized micropipette (catalog #5242956.003; Eppendorf). Twenty cell spheres were added to each tube in 250 μl of CELLBANKER1 (Nippon Zenyaku Kogyo) and then stored in liquid nitrogen until later use. Before their use for assay purposes, the cell spheres were dissociated with 0.25% Trypsin-EDTA for 10 min and dissociated single cells were expanded as undifferentiated oligodendrocyte-lineage cells in a culture for 10 d with basal medium on a poly-l-ornithine-coated dish (20 spheres in a tube per 35 mm dish). The cells were then replated on a poly-l-ornithine-coated 35 mm dish (2.0 × 104 cells per dish) with basal medium and used.
Immunocytochemistry.
A fixative containing 4% paraformaldehyde and 20% sucrose in PBS was added to dishes carefully so as not to detach cells and fixation was allowed to proceed for 30 min. After permeabilizing and blocking with 0.1% Triton X-100 and 4% nonfat dry milk in TBS, the cells were incubated with the respective primary antibodies overnight at 4°C. The binding of specific antibodies was detected with Alexa Fluor-conjugated secondary antibodies. Digital photomicrographs of each specimen were taken with Biozero BZ-8000 (Keyence) or LSM 700 and analyzed using Adobe Photoshop CS6.
Immunoprecipitation and Western blotting.
OL1 cells were extracted with 1% NP-40 in TBS containing 1 mm vanadate, 10 mm NaF, and protease inhibitors (EDTA-free complete; Roche Molecular Biochemicals) with 250 μl of lysis buffer for a 35 mm dish. After precleaning the cell extracts with Protein G Sepharose (GE Healthcare), the immunocomplexes with an anti-p190 RhoGAP antibody (2 μl) were precipitated using Protein G Sepharose (20 μl beads volume) and eluted by boiling in a SDS-PAGE sample buffer. Samples were subjected to SDS-PAGE followed by semidry electroblotting onto a polyvinylidene difluoride membrane. The membrane was incubated with 4% nonfat dry milk and 0.1% Triton X-100 in TBS and then reacted overnight with the respective antibodies. The binding of these antibodies was detected with Luminata Forte Western HRP Substrate (Millipore). To detect phosphorylated proteins, the membranes were blocked with 1% BSA and 0.1% Triton X-100 in TBS and then incubated with HRP-conjugated anti-phospho-Tyr mAb or rabbit anti-pY1105 antibodies. Densitometry analyses were performed using ImageJ software.
Statistical analyses.
Statistical analyses were performed using IBM SPSS Statistics 20 software.
Results
To estimate cuprizone-induced demyelination in the mouse brain quantitatively, we optimized our contrast-enhanced CT imaging protocol (Almuriekhi et al., 2015) that detects differences in the penetration rate of Histodenz (Sigma-Aldrich) between the white matter (myelinated axon tracts), such as the corpus callosum, and the gray matter, such as the cortex. CT imaging of Histodenz-immersed normal brains showed a clear contrast between the corpus callosum and gray matter region of the cortex (Fig. 1A,B; left, normal). When mice were fed with a cuprizone-containing diet for 6 weeks, the CT number in the corpus callosum was markedly increased (turned from black to white in CT images; recovery, 0 week) due to the enhanced penetration of the contrast reagent by demyelination of axonal fibers. A scatterplot for the wild-type showed that the CT number difference between the corpus callosum and cortex significantly decreased after cuprizone feeding (Fig. 1C) and that this difference fully recovered 5 weeks after returning to the normal diet (Fig. 1C). The time course was consistent with previous findings obtained in C57BL/6 mice (Matsushima and Morell, 2001); therefore, the CT number difference in our analyses was considered to be a quantitative index of demyelination in the CNS. Ptprz-deficient mice also exhibited a similar degree of demyelination after cuprizone feeding (Fig. 1, 0 week). However, reparative remyelination was more evident in Ptprz-deficient mice 1–3 weeks after the removal of cuprizone than in wild-type mice (Fig. 1C), indicating accelerated remyelination in Ptprz-deficient mice.
Figure 1.

Micro-CT of cuprizone-treated mouse brains. A, B, Coronal plane reconstruction of micro-CT scans of Histodenz-immersed brains derived from wild-type (+/+) and Ptprz-deficient (−/−) mice. Images show the whole region (A) and dorsal corpus callosum corresponding to the square region in A (B). The CT number increases from black to white. Mice were fed a cuprizone-containing diet for 6 weeks (0) to induce demyelination, followed by spontaneous remyelination with the removal of cuprizone (1, 3, and 5 weeks). Normal mice were maintained on the regular diet. Scale bars, 1 mm. C, Scatterplot showing differences between the CT number of the dorsal corpus callosum and that of the cerebral cortex (each circle corresponds to an individual mouse). Open circles, Wild-type mouse; closed circles, Ptprz-deficient mouse. n = 10 for each group. *p < 0.05, **p < 0.01, two-way ANOVA with Bonferroni post hoc test.
We confirmed the validity of this CT assay by LFB myelin staining of tissue sections prepared from the same brains used for the CT imaging. The results of LFB staining (Fig. 2) were consistent with those of CT imaging. The immunohistochemical staining analysis indicated that the recovery of MBP expression (remyelination) occurred earlier in Ptprz-deficient mice than in wild-type mice (Fig. 3). The remyelination of lesioned nerve fibers requires the recruitment of OPCs to the lesion sites and their differentiation to myelin-carrying mature oligodendrocytes (Frohman et al., 2006). The number of oligodendrocyte-lineage cells (Olig2-positive) recruited to the lesioned area showed a similar pattern between the two genotypes (Fig. 4) in that Olig2-positive cells were decreased by cuprizone feeding, but increased 1–3 weeks after the removal of cuprizone and returned to normal levels by 5 weeks. These results suggested that accelerated remyelination in Ptprz-deficient mice was not induced by the enhanced recruitment of OPCs, but by their higher differentiation potential.
Figure 2.
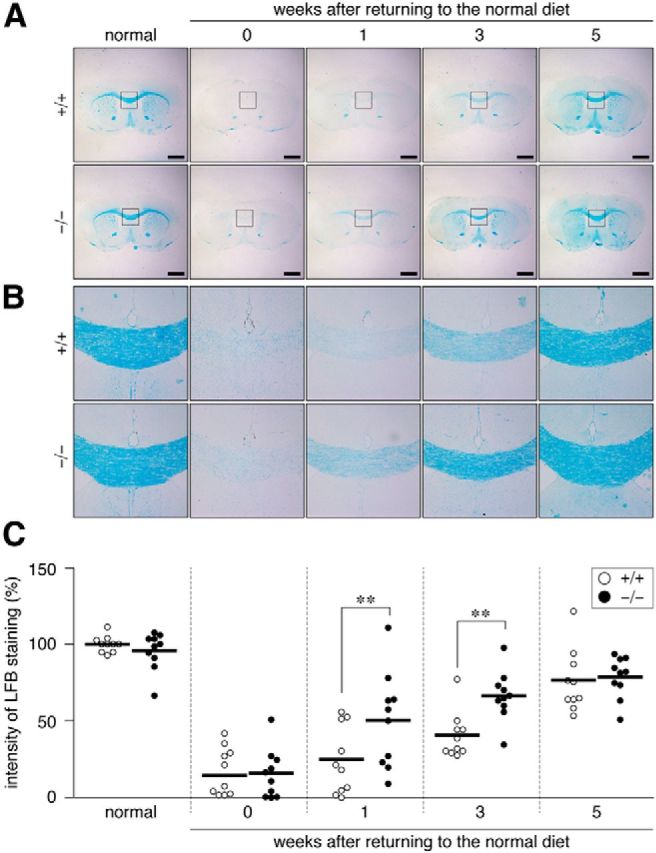
Accelerated remyelination in the corpus callosum of Ptprz-deficient mice after cuprizone-induced demyelination. A, B, LFB staining of wild-type (+/+) and Ptprz-deficient (−/−) mouse brains. Images show the whole region (A) and dorsal corpus callosum (B), respectively. After micro-CT scans, the Histodenz-immersed brains shown in Figure 1 were paraffin embedded and serially sectioned for histological examinations. Scale bars, 1 mm. C, Quantification of LFB staining of the dorsal corpus callosum. Open circles, Wild-type mouse; closed circles, Ptprz-deficient mouse. n = 10 for each group. **p < 0.01, two-way ANOVA with Bonferroni post hoc test.
Figure 3.

A, Accelerated reexpression of MBP in the corpus callosum of Ptprz-deficient mice after cuprizone-induced demyelination. Images show the dorsal corpus callosum. Coronal brain sections from wild-type (+/+) and Ptprz-deficient (−/−) mice were immunohistochemically analyzed using a specific polyclonal antibody against MBP (anti-MBP). Scale bars, 100 μm. B, Quantification of anti-MBP staining in the dorsal corpus callosum. Open circles, Wild-type mouse; closed circles, Ptprz-deficient mouse. n = 10 for each group. *p < 0.05, two-way ANOVA with Bonferroni post hoc test.
Figure 4.

A, Distribution of oligodendrocyte-lineage cells in the corpus callosum. Images show the dorsal corpus callosum. Coronal brain sections from wild-type (+/+) and Ptprz-deficient (−/−) mice were stained with anti-Olig2, a marker of oligodendrocyte-lineage cells. Scale bars, 50 μm. B, Scatterplot showing the number of anti-Olig2-stained nuclei in the dorsal corpus callosum. Open circles, Wild-type mouse; closed circles, Ptprz-deficient mouse. n = 10 for each group. No genotypic differences were detected by two-way ANOVA.
Astroglial cells and neurons also express PTPRZ (Shintani et al., 1998), so these cells may affect recovery from demyelinating lesions. However, no significant differences were observed in the accumulation of reactive astrocytes (GFAP-positive; Fig. 5) or the number of injured axons (APP-positive; Fig. 6) between the two genotypes. They gradually returned to normal levels in both genotypes by returning to the normal diet with a similar time course.
Figure 5.

A, Distribution of reactive astrocytes in the corpus callosum after the cuprizone treatment. Images show the dorsal corpus callosum. Brain sections from wild-type (+/+) and Ptprz-deficient (−/−) mice were stained with anti-GFAP, a marker of reactive astrocytes. Scale bars, 50 μm. B, Scatterplot showing the signal intensity of anti-GFAP staining. Relative values to the average value of the normal wild-type group. Open circles, Wild-type mouse; closed circles, Ptprz-deficient mouse. n = 10 for each group. No genotypic differences were detected by two-way ANOVA.
Figure 6.

A, Axonal damage induced in the dorsal corpus callosum by the cuprizone treatment. Stained images of damaged axons are shown. Brain sections from wild-type (+/+) and Ptprz-deficient (−/−) mice were stained with a specific antibody against APP (anti-APP) and then stained with hematoxylin. Scale bars, 50 μm. B, Scatterplot of the anti-APP-stained puncta in the dorsal corpus callosum. Open circles, Wild-type mouse; closed circles, Ptprz-deficient mouse. n = 10 for each group. No genotypic differences were detected by two-way ANOVA.
We then investigated by immunohistochemistry whether the expression of endogenous PTPRZ ligands such as PTN, midkine, and IL-34 were affected in lesions. The corpus callosum is a major cortical–cortical fiber tract connecting the two cerebral hemispheres (Jacobson and Trojanowski, 1974). PTN immunoreactivity was markedly increased both in the corpus callosum and the cortex after cuprizone-induced lesion (Fig. 7), but this gradually disappeared within 3 weeks after returning to the normal diet (Figs. 8, 9). On the other hand, we detected no changes in midkine or IL-34 expression (Fig. 7). Double-staining experiments revealed that PTN signals were present in the cortex neurons (β-tubulin III-positive; Fig. 10A) and the signals were colocalized with a synaptic vesicle marker synapsin 1 (SYN1), not only in the soma of cortex neurons (Fig. 10A), but also in axon fibers in the corpus callosum (Fig. 10B). The PTN signals also overlapped with APP signals in the corpus callosum (Fig. 10B), indicating that PTN was transiently expressed in damaged neurons and transported to demyelinated axons. Notably, the punctate signals of PTN were not detected in Olig2-positive oligodendrocyte-lineage cells or GFAP-positive astrocytes in the corpus callosum (Fig. 10B). PTN is likely released from demyelinated axonal fibers (see below).
Figure 7.
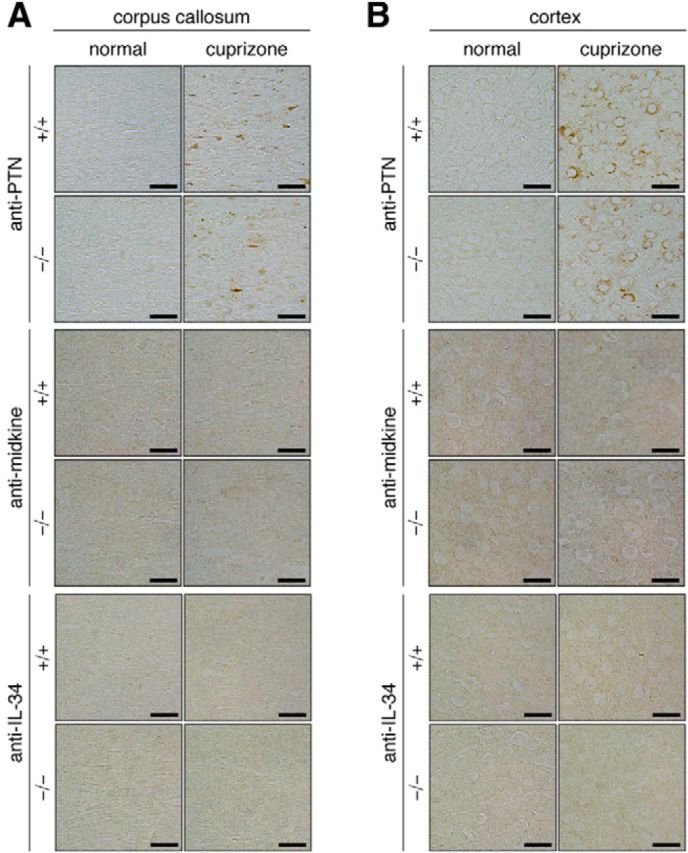
Increase in pleiotrophin, but not midkine or IL-34, expression in the brain by cuprizone-induced demyelination. A, B, Brain sections of normal mice and mice fed with a cuprizone-containing diet for 6 weeks were stained with a specific antibody against pleiotrophin (anti-PTN), midkine (anti-midkine), or IL-34 (anti-IL-34). Staining images of the dorsal corpus callosum (A) and cerebral cortex (B) of wild-type (+/+) and Ptprz-deficient (−/−) mice are shown. Scale bars, 25 μm.
Figure 8.

A, Pleiotrophin transiently expressed in the corpus callosum by the cuprizone treatment. Images show the dorsal corpus callosum. Brain sections from wild-type (+/+) and Ptprz-deficient (−/−) mice were stained with anti-PTN. Mice were fed a cuprizone-containing diet and then returned to a normal diet. Scale bars, 25 μm. B, Scatterplot showing the number of anti-PTN-stained puncta in the corpus callosum. Open circles, Wild-type mouse; closed circles, Ptprz-deficient mouse. n = 10 for each group. No genotypic differences were detected by two-way ANOVA.
Figure 9.

A, Pleiotrophin expression upregulated in the cerebral cortex by the cuprizone-induced demyelination. Anti-PTN staining of wild-type (+/+) and Ptprz-deficient (−/−) mouse brains. Scale bars, 25 μm. B, Scatterplot showing the percentage of anti-PTN positive neuron in the cortex. Open circles, Wild-type mouse; closed circles, Ptprz-deficient mouse. n = 10 for each group. No genotypic differences were detected by a two-way ANOVA.
Figure 10.

Transient increase in pleiotrophin expression in damaged neurons by cuprizone-induced demyelination. A, Double-immunofluorescence labeling of PTN (green) and β-tubulin III or SYN1 (red) in the cerebral cortex of normal mice and mice fed a cuprizone-containing diet for 6 weeks. Scale bars, 10 μm. B, Double immunostaining for PTN (green) along with APP, SYN1, Olig2, or GFAP (red) in the corpus callosum of cuprizone-treated mice. Scale bars, 10 μm.
We therefore investigated the potential role of PTN-PTPRZ signaling in oligodendrocyte differentiation using a primary culture of mixed mouse glial cells (Kuboyama et al., 2012). The PTN treatment alone did not induce oligodendrocyte maturation (i.e., the expression of MBP) in wild-type glial cells (Fig. 11A); however, PTN significantly enhanced the differentiation of oligodendrocyte induced by thyroid hormone (TH) (Fig. 11B). Ptprz-deficient glial cells showed higher differentiation activity than wild-type cells with the TH treatment (Fig. 11B), as described previously (Kuboyama et al., 2012); however, notably, the differentiation was not further potentiated by the addition of PTN (Fig. 11B).
Figure 11.

Accelerated oligodendrocyte differentiation through the PTN-PTPRZ interaction. A, B, Double-immunofluorescence labeling of a primary mixed glial culture obtained from Ptprz-deficient (−/−) and wild-type (+/+) brains using anti-NG2 proteoglycan (OPCs; red) and anti-MBP (oligodendrocytes, OLs; green) in conjunction with DAPI labeling of nuclei (blue). Glial cells were cultured in the absence or presence of 100 nm PTN without (A) or with (B) the oligodendrocyte differentiation factors biotin and TH for 6 d. Scale bars, 100 μm. The percentage of OPCs and OLs in all cells (DAPI-positive nuclei number) is shown in the bottom panel. Data are the mean ± SEM of four independent experiments. *p < 0.05, one-way ANOVA with Bonferroni post hoc test.
Because the mixed glial culture apparently contains a heterologous cell population, we established oligodendrocyte-lineage cells (OL1 cells) from the brains of p53-deficient mouse pups (Fig. 12A) and used them in biochemical analyses of PTN-PTPRZ signaling during differentiation. All OL1 cells were positive for an OPC marker NG2 and they differentiated into MBP-positive mature oligodendrocytes when cultured with TH (Fig. 12B,C), indicating that they exhibited normal differentiation properties. OL1 cells were strongly positive for the two receptor isoforms of PTPRZ-A and PTPRZ-B with chondroitin sulfate chains, whereas only PTPRZ-B was weakly detectable after differentiation (Fig. 12D,E).
Figure 12.
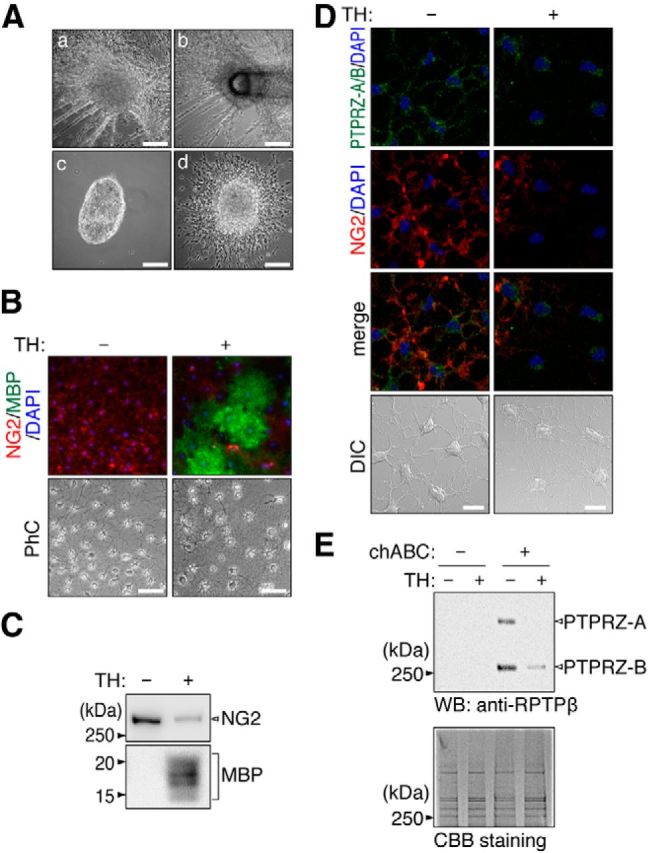
Establishment of OL1 cells. A, Isolation of OL1 cells. Dissociated brain cells from p53-deficient pups were cultured in basal medium (see the Materials and Methods section). Cell spheres (OL1 cells) formed after cultivation for 14 d (Aa). They were collected with a sterilized micropipette (Ab). A sphere was placed on a poly-l-ornithine-coated dish (Ac). Oligodendrocyte-lineage cells migrated out from the cell sphere after a 24 h culture (Ad). Scale bars, 200 μm. B, OL1 cells were cultured with or without the oligodendrocyte differentiation factors biotin and TH for 10 d. Cells were stained with anti-NG2 (red) and anti-MBP (green) antibodies. Scale bars, 100 μm. PhC, Phase contrast. C, Western blot analyses using anti-NG2 and anti-MBP antibodies. D, Double-immunofluorescence labeling of OL1 cells using anti-RPTPβ (anti-PTPRZ-A/B, green) and anti-NG2 proteoglycan (red) antibodies. Scale bars, 10 μm. DIC, Differential interference contrast. E, Western blot analysis using an anti-RPTPβ antibody in OL1 cells. The protein amounts applied were verified by Coomassie brilliant blue (CBB) staining. Because NG2 proteoglycan and PTPRZ receptors were expressed in OL1 cells as chondroitin-sulfate proteoglycans, the samples were treated with chABC before SDS-PAGE.
Immunocytochemistry showed that the PTN treatment expectedly enhanced the TH-induced differentiation of OL1 cells (Fig. 13A), as observed in the primary mixed glial culture (Fig. 11). Western blotting of OL1 cells also showed that PTN increased TH-induced MBP expression. The amount of NG2 protein was decreased by PTN independently of TH (Fig. 13B), suggesting that PTN has an activity to release PTPRZ-mediated blockage of OPC differentiation. It is known that p190 RhoGAP is primarily phosphorylated at Tyr-1105 by FYN tyrosine kinase in OPCs, thereby inducing differentiation into oligodendrocytes (Umemori et al., 1994; Wolf et al., 2001). However, phospho-Tyr-1105 in p190 RhoGAP is maintained at low levels by dephosphorylation by PTPRZ (Fukada et al., 2005; Tamura et al., 2006; Kuboyama et al., 2012). The phosphorylation of p190 RhoGAP at Tyr-1105 was enhanced in PTN-treated OL1 cells because of the inactivation of PTPRZ by PTN (Fig. 13C,D). It was concluded that the catalytic activity of PTPRZ receptor isoforms function to maintain OPCs in an undifferentiated state and PTN-induced inactivation of PTPRZ releases this blockage, thereby allowing for remyelination.
Figure 13.

PTN induced the oligodendrocyte differentiation of OL1 cells. A, B, OL1 cells were cultured with various combinations of TH and PTN for 10 d. Cells were analyzed by immunocytochemistry (A) and Western blotting (B) with anti-NG2 proteoglycan (OPCs; red) and anti-MBP (oligodendrocytes, OLs; green) antibodies. The percentage of OPCs and OLs in all cells (DAPI-positive nuclei, blue) are shown at the bottom in A. Scale bars, 100 μm. PhC, Phase contrast. Data are the mean ± SEM (n = 5). **p < 0.01, Student's t test. C, D, PTN-induced phosphorylation of p190 RhoGAP at Tyr-1105. OL1 cells were incubated for 30 min with or without PTN in the presence of TH. C, The overall tyrosine phosphorylation pattern and protein expression were analyzed with anti-phosphotyrosine PY20 and anti-p190 RhoGAP antibodies, respectively. D, Tyr-1105 phosphorylation of p190 RhoGAP. Extracts were immunoprecipitated with anti-p190 RhoGAP antibody-coated beads and binding proteins were analyzed by Western blotting with anti-pY1105 and anti-p190 RhoGAP antibodies. Tyr-1105 phosphorylation levels were determined by densitometric analyses. Data are the mean ± SEM (n = 7). **p < 0.01, Student's t test.
Discussion
MS is an inflammatory (immune)-mediated demyelinating disease of the human CNS. In the present study, we induced demyelination in the CNS with cuprizone and demonstrated that a Ptprz deficiency accelerated CNS remyelination after cuprizone-induced demyelination. In the lesioned area, transient expression of PTN was detected in demyelinated nerves. Extracellular PTN inhibited PTPRZ activity in OPCs and thus promoted their differentiation. The PTN-induced inactivation of PTPRZ was considered to be the adaptive mechanism underlying CNS remyelination in vivo.
Tyrosine phosphorylation of p190 RhoGAP by Fyn is a main signaling pathway involved in differentiation of OPCs to oligodendrocytes (Krämer-Albers and White, 2011). A 50% decrease in myelination has been reported in Fyn-knock-out mice (Umemori et al., 1994). p190 RhoGAP is a common crucial target for FYN (Wolf et al., 2001) and PTPRZ (Kuboyama et al., 2012) in oligodendrocyte-lineage cells. We recently reported that PTPRZ negatively regulates the differentiation of oligodendrocytes and myelin formation (Kuboyama et al., 2012) as a phosphatase that functions conversely to FYN tyrosine kinase. Ptprz-deficient mice showed the early onset of myelination in the developing brain, whereas the myelin sheath in the adult brain appears to be normal (Kuboyama et al., 2012). Moreover, adult Ptprz-deficient mice were less susceptible to EAE induced by active immunization with a MOG peptide than wild-type mice (Kuboyama et al., 2012). The enhanced tyrosine phosphorylation of p190 RhoGAP and the reduced loss of MBP in the EAE model of Ptprz-deficient mice (Kuboyama et al., 2012) suggested that a Ptprz deficiency was responsible for reduced demyelination, increased remyelination, or both. Although EAE is the most common animal model of MS, it is difficult to estimate the role of PTPRZ in remyelination itself because EAE is an immune-mediated demyelinating model (Høglund and Maghazachi, 2014). Therefore, we herein adopted a cuprizone model.
The feeding of toxic cuprizone has been shown to cause reproducible demyelination by a nonautoimmune mechanism in the corpus callosum (Matsushima and Morell, 2001). Ptprz-deficient mice, as well as wild-type mice, developed severe demyelination in the corpus callosum and axonal damage due to cuprizone (Figs. 1, 2, 3, 4, 5, 6), indicating that a Ptprz deficiency does not affect demyelination itself. Conversely, remyelination was accelerated in Ptprz-deficient mice after the removal of cuprizone (Figs. 1, 2, 3), which is consistent with our previous finding that PTPRZ is a negative regulator of oligodendrocyte differentiation (Kuboyama et al., 2012). No significant differences were observed in the accumulation of OPCs in the lesioned area (Fig. 4). These results indicate that the acceleration of remyelination in the Ptprz-deficient mouse was induced by a high differentiation ability of OPCs.
The functional role or regulatory mechanism of RPTPs has not been investigated as extensively as that of PTKs. PTPRZ is the first RPTP molecule in that its soluble extracellular ligands and intracellular substrate proteins have been identified as a set (Kawachi et al., 2001; Fukada et al., 2005, 2006). Although growth factors such as PTN, midkine, and IL-34 have been identified as endogenous inhibitory ligands for PTPRZ (Maeda et al., 1996, 1999; Kawachi et al., 2001; Fukada et al., 2006; Nandi et al., 2013), the in vivo significance of these ligand-receptor pairs has remained unclear. In the present study, we demonstrated accelerated remyelination in Ptprz-deficient mice (Figs. 1, 2, 3) and the transient induction of PTN in damaged neurons in the cuprizone-induced demyelination model (Figs. 7, 8, 9). PTN signals in axons in the corpus callosum were overlapped with the synaptic marker SYN1 and the damaged axon marker APP. APP is known to be packed into axonal vesicles that undergo kinesin 1-dependent anterograde movement along microtubules in axons (Amaratunga et al., 1995a, 1995b; Kaether et al., 2000).
Two groups previously reported the existence of vesicular glutamate release at axonal membranes in the corpus callosum and the optic nerve, suggesting that this process may be widespread in white matter (Kukley et al., 2007; Ziskin et al., 2007). The axonal release involves highly localized calcium microdomain signaling, similar to vesicle fusion at synapses (Kukley et al., 2007). Here, unmyelinated fibers are reportedly responsible for the signaling with NG2-positive glia (Ziskin et al., 2007). Although the mechanism of PTN secretion along the axon is not clear, our results suggest that PTN is thus released from demyelinated axons and inactivates the PTPRZ of OPCs in the lesioned area to achieve their differentiation and remyelination.
In a primary culture, PTN significantly enhanced TH-induced oligodendrocyte differentiation in wild-type cells, although PTN alone did not induce maturation (Fig. 11). In contrast, Ptprz-deficient glial cells showed higher TH-induced differentiation ability without PTN (Fig. 11); importantly, it was at similar levels to those of wild-type cells with PTN. These findings were essentially consistent with the in vivo results that the recovery of MBP expression was accelerated in Ptprz-deficient mice (Fig. 3). PTN-PTPRZ signaling is therefore considered to contribute to remyelination by enhancing OPC differentiation in vivo. Although we found no significant changes in the total number of oligodendrocyte-lineage cells (NG2-positive cells plus MBP-positive cells) after PTN treatment in the primary culture (Fig. 11), PTN was previously reported to contribute to the homeostatic self-renewal of human PDGFRα-sorted OPCs in culture (McClain et al., 2012).
We established a source of pure oligodendrocyte-lineage OL1 cells from the p53-deficient newborn mouse brain to determine the intracellular signaling pathways involved in OPC differentiation. The established OL1 cells maintained a normal differentiation ability and PTN sensitivity, similar to wild-type OPCs, in the primary culture (Figs. 12, 13). PTN increased the phosphorylation of the substrate molecules of PTPRZ, such as p190 RhoGAP, in OL1 cells (Fig. 13D), indicating the involvement of PTN-PTPRZ signaling mechanism in OPC differentiation. PTPRZ-A and PTPRZ-B isoforms were both expressed in OL1 cells in the immature state as chondroitin sulfate proteoglycans, whereas only PTPRZ-B proteins were weakly detectable after differentiation (Fig. 12D,E), which was very similar to the expression profiles of the receptor isoforms observed during the normal development of rodent brains (Nishiwaki et al., 1998). Together, these results demonstrated that PTPRZ receptors were required to maintain OPCs in the immature state and its ligand-induced inactivation released this blockage, thereby promoting oligodendrocyte differentiation and remyelination (Fig. 14). This PTN-PTPRZ signaling may be also involved in the oligodendrocyte differentiation during normal development.
Figure 14.

Proposed mechanism for remyelination after demyelinating lesions. OPCs, but not mature oligodendrocytes (OLs), abundantly express PTPRZ-A/B receptor proteins as chondroitin sulfate (CS) proteoglycans. The CS moiety of PTPRZ is important for achieving the high-affinity binding of PTN to the core protein (Maeda et al., 1996). PTPRZ activity is requisite to maintaining the immature state of OPCs. After cuprizone-induced oligodendrocyte death and demyelination, the expression of PTN is transiently upregulated in damaged neurons. PTN may be released from demyelinated axons and bind to PTPRZ at the cell surface of OPCs that are recruited to lesions, probably through PTPRZ-independent mechanisms. The binding of PTN results in receptor dimerization or oligomerization, thereby inhibiting its catalytic activity (Fukada et al., 2006). PTPRZ inactivation releases the block of differentiation in OPCs, so the remyelination of neighboring axons is initiated.
Despite the effectiveness of current therapies in controlling CNS inflammation in patients with MS, none prevents the chronic progressive process (Høglund and Maghazachi, 2014). Because OPCs are still present in the demyelinated regions of MS patients yet fail to differentiate, the development of therapeutic compounds is anticipated to enhance remyelination from this quiescent OPC population (Chang et al., 2002). The rationale for this concept has been already demonstrated by improving clinical outcomes in demyelination mouse models with several agents to promote OPC differentiation, such as: an antagonist antibody for LINGO-1 that is a negative regulator of FYN kinase in OPCs (Mi et al., 2005, 2007); benztropine, which depends on muscarinic receptor antagonism (Deshmukh et al., 2013); and miconazole via unknown targets (Najm et al., 2015). We detected no significant PTPRZ inhibition by miconazole in vitro (data not shown). Therefore, inhibitory ligands for PTPRZ may also be effective in the treatment of demyelinated lesions; however, midkine reportedly triggers relapses of EAE by increasing the number of autoreactive T-helper cells (Wang et al., 2008; Sonobe et al., 2012). Because the expression of PTPRZ is not detectable in T cells (Kuboyama et al., 2012), other midkine (and PTN) receptors, such as anaplastic lymphoma kinase, integrins, or low-density lipoprotein receptor-related proteins, may be involved in this event (Kadomatsu et al., 2013). We propose the development of selective inhibitors for the catalytic PTP domain of PTPRZ as a plausible and effective therapeutic strategy for demyelinating diseases.
Footnotes
This work was supported by Adaptable and Seamless Technology Transfer Program through Target-driven R&D (A-STEP), Japan Science and Technology Agency (M.N.); Imaging Science Project of the Center for Novel Science Initiatives; National Institutes of Natural Sciences (A.F.); and the Japan Society for the Promotion of Science (KAKENHI Grants 24700387 and 26830050 to K.K.). Micro-CT analyses were performed at the Model Animal Research Facility of the National Institute for Basic Biology (NIBB). Immunofluorescence photomicrograph images were acquired at the Spectrography and Bioimaging Facility, NIBB Core Research Facilities. We thank M. Katsuki for providing us with p53-deficient mice, Y. Isoshima and N. Nakanishi for technical assistance, and A. Kodama for secretarial assistance.
The authors declare no competing financial interests.
References
- Almuriekhi M, Shintani T, Fahiminiya S, Fujikawa A, Kuboyama K, Takeuchi Y, Nawaz Z, Nadaf J, Kamel H, Kitam AK, Samiha Z, Mahmoud L, Ben-Omran T, Majewski J, Noda M. Loss-of-function mutation in APC2 causes Sotos syndrome features. Cell Rep. 2015;10:1585–1598. doi: 10.1016/j.celrep.2015.02.011. [DOI] [PubMed] [Google Scholar]
- Amaratunga A, Fine RE. Generation of amyloidogenic C-terminal fragments during rapid axonal transport in vivo of beta-amyloid precursor protein in the optic nerve. J Biol Chem. 1995a;270:17268–17272. doi: 10.1074/jbc.270.29.17268. [DOI] [PubMed] [Google Scholar]
- Amaratunga A, Leeman SE, Kosik KS, Fine RE. Inhibition of kinesin synthesis in vivo inhibits the rapid transport of representative proteins for three transport vesicle classes into the axon. J Neurochem. 1995b;64:2374–2376. doi: 10.1046/j.1471-4159.1995.64052374.x. [DOI] [PubMed] [Google Scholar]
- Chang A, Tourtellotte WW, Rudick R, Trapp BD. Premyelinating oligodendrocytes in chronic lesions of multiple sclerosis. N Engl J Med. 2002;346:165–173. doi: 10.1056/NEJMoa010994. [DOI] [PubMed] [Google Scholar]
- Chow JP, Fujikawa A, Shimizu H, Noda M. Plasmin-mediated processing of protein tyrosine phosphatase receptor type Z in the mouse brain. Neurosci Lett. 2008a;442:208–212. doi: 10.1016/j.neulet.2008.07.028. [DOI] [PubMed] [Google Scholar]
- Chow JP, Fujikawa A, Shimizu H, Suzuki R, Noda M. Metalloproteinase- and gamma-secretase-mediated cleavage of protein-tyrosine phosphatase receptor type Z. J Biol Chem. 2008b;283:30879–30889. doi: 10.1074/jbc.M802976200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshmukh VA, Tardif V, Lyssiotis CA, Green CC, Kerman B, Kim HJ, Padmanabhan K, Swoboda JG, Ahmad I, Kondo T, Gage FH, Theofilopoulos AN, Lawson BR, Schultz PG, Lairson LL. A regenerative approach to the treatment of multiple sclerosis. Nature. 2013;502:327–332. doi: 10.1038/nature12647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frohman EM, Racke MK, Raine CS. Multiple sclerosis–the plaque and its pathogenesis. N Engl J Med. 2006;354:942–955. doi: 10.1056/NEJMra052130. [DOI] [PubMed] [Google Scholar]
- Fujikawa A, Fukada M, Makioka Y, Suzuki R, Chow JP, Matsumoto M, Noda M. Consensus substrate sequence for protein-tyrosine phosphatase receptor type Z. J Biol Chem. 2011;286:37137–37146. doi: 10.1074/jbc.M111.270140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukada M, Kawachi H, Fujikawa A, Noda M. Yeast substrate-trapping system for isolating substrates of protein tyrosine phosphatases: isolation of substrates for protein tyrosine phosphatase receptor type z. Methods. 2005;35:54–63. doi: 10.1016/j.ymeth.2004.07.008. [DOI] [PubMed] [Google Scholar]
- Fukada M, Fujikawa A, Chow JP, Ikematsu S, Sakuma S, Noda M. Protein tyrosine phosphatase receptor type Z is inactivated by ligand-induced oligomerization. FEBS Lett. 2006;580:4051–4056. doi: 10.1016/j.febslet.2006.06.041. [DOI] [PubMed] [Google Scholar]
- Goldman SA, Nedergaard M, Windrem MS. Glial progenitor cell-based treatment and modeling of neurological disease. Science. 2012;338:491–495. doi: 10.1126/science.1218071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gondo Y, Nakamura K, Nakao K, Sasaoka T, Ito K, Kimura M, Katsuki M. Gene replacement of the p53 gene with the lacZ gene in mouse embryonic stem cells and mice by using two steps of homologous recombination. Biochem Biophys Res Commun. 1994;202:830–837. doi: 10.1006/bbrc.1994.2005. [DOI] [PubMed] [Google Scholar]
- Høglund RA, Maghazachi AA. Multiple sclerosis and the role of immune cells. World J Exp Med. 2014;4:27–37. doi: 10.5493/wjem.v4.i3.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson S, Trojanowski JQ. The cells of origin of the corpus callosum in rat, cat and rhesus monkey. Brain Res. 1974;74:149–155. doi: 10.1016/0006-8993(74)90118-8. [DOI] [PubMed] [Google Scholar]
- Kadomatsu K, Kishida S, Tsubota S. The heparin-binding growth factor midkine: the biological activities and candidate receptors. J Biochem. 2013;153:511–521. doi: 10.1093/jb/mvt035. [DOI] [PubMed] [Google Scholar]
- Kaether C, Skehel P, Dotti CG. Axonal membrane proteins are transported in distinct carriers: a two-color video microscopy study in cultured hippocampal neurons. Mol Biol Cell. 2000;11:1213–1224. doi: 10.1091/mbc.11.4.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawachi H, Fujikawa A, Maeda N, Noda M. Identification of GIT1/Cat-1 as a substrate molecule of protein tyrosine phosphatase ζ/β by the yeast substrate-trapping system. Proc Natl Acad Sci U S A. 2001;98:6593–6598. doi: 10.1073/pnas.041608698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krämer-Albers EM, White R. From axon-glial signalling to myelination: the integrating role of oligodendroglial Fyn kinase. Cell Mol Life Sci. 2011;68:2003–2012. doi: 10.1007/s00018-010-0616-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuboyama K, Fujikawa A, Masumura M, Suzuki R, Matsumoto M, Noda M. Protein tyrosine phosphatase receptor type z negatively regulates oligodendrocyte differentiation and myelination. PLoS One. 2012;7:e48797. doi: 10.1371/journal.pone.0048797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukley M, Capetillo-Zarate E, Dietrich D. Vesicular glutamate release from axons in white matter. Nat Neurosci. 2007;10:311–320. doi: 10.1038/nn1850. [DOI] [PubMed] [Google Scholar]
- Maeda N, Hamanaka H, Shintani T, Nishiwaki T, Noda M. Multiple receptor-like protein tyrosine phosphatases in the form of chondroitin sulfate proteoglycan. FEBS Lett. 1994;354:67–70. doi: 10.1016/0014-5793(94)01093-5. [DOI] [PubMed] [Google Scholar]
- Maeda N, Nishiwaki T, Shintani T, Hamanaka H, Noda M. 6B4 proteoglycan/phosphacan, an extracellular variant of receptor-like protein-tyrosine phosphatase ζ/RPTPβ, binds pleiotrophin/heparin-binding growth-associated molecule (HB-GAM) J Biol Chem. 1996;271:21446–21452. doi: 10.1074/jbc.271.35.21446. [DOI] [PubMed] [Google Scholar]
- Maeda N, Ichihara-Tanaka K, Kimura T, Kadomatsu K, Muramatsu T, Noda M. A receptor-like protein-tyrosine phosphatase PTPzeta/RPTPbeta binds a heparin-binding growth factor midkine: involvement of arginine 78 of midkine in the high affinity binding to PTPzeta. J Biol Chem. 1999;274:12474–12479. doi: 10.1074/jbc.274.18.12474. [DOI] [PubMed] [Google Scholar]
- Matsushima GK, Morell P. The neurotoxicant, cuprizone, as a model to study demyelination and remyelination in the central nervous system. Brain Pathol. 2001;11:107–116. doi: 10.1111/j.1750-3639.2001.tb00385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClain CR, Sim FJ, Goldman SA. Pleiotrophin suppression of receptor protein tyrosine phosphatase-β/ζ maintains the self-renewal competence of fetal human oligodendrocyte progenitor cells. J Neurosci. 2012;32:15066–15075. doi: 10.1523/JNEUROSCI.1320-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng K, Rodriguez-Peña A, Dimitrov T, Chen W, Yamin M, Noda M, Deuel TF. Pleiotrophin signals increased tyrosine phosphorylation of beta beta-catenin through inactivation of the intrinsic catalytic activity of the receptor-type protein tyrosine phosphatase β/ζ. Proc Natl Acad Sci U S A. 2000;97:2603–2608. doi: 10.1073/pnas.020487997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi S, Miller RH, Lee X, Scott ML, Shulag-Morskaya S, Shao Z, Chang J, Thill G, Levesque M, Zhang M, Hession C, Sah D, Trapp B, He Z, Jung V, McCoy JM, Pepinsky RB. LINGO-1 negatively regulates myelination by oligodendrocytes. Nat Neurosci. 2005;8:745–751. doi: 10.1038/nn1460. [DOI] [PubMed] [Google Scholar]
- Mi S, Hu B, Hahm K, Luo Y, Kam Hui ES, Yuan Q, Wong WM, Wang L, Su H, Chu TH, Guo J, Zhang W, So KF, Pepinsky B, Shao Z, Graff C, Garber E, Jung V, Wu EX, Wu W. LINGO-1 antagonist promotes spinal cord remyelination and axonal integrity in MOG-induced experimental autoimmune encephalomyelitis. Nat Med. 2007;13:1228–1233. doi: 10.1038/nm1664. [DOI] [PubMed] [Google Scholar]
- Murasugi A, Kido I, Kumai H, Asami Y. Efficient production of recombinant human pleiotrophin in yeast, Pichia pastoris. Biosci Biotechnol Biochem. 2003;67:2288–2290. doi: 10.1271/bbb.67.2288. [DOI] [PubMed] [Google Scholar]
- Najm FJ, Madhavan M, Zaremba A, Shick E, Karl RT, Factor DC, Miller TE, Nevin ZS, Kantor C, Sargent A, Quick KL, Schlatzer DM, Tang H, Papoian R, Brimacombe KR, Shen M, Boxer MB, Jadhav A, Robinson AP, Podojil JR, Miller SD, Miller RH, Tesar PJ. Drug-based modulation of endogenous stem cells promotes functional remyelination in vivo. Nature. 2015;522:216–220. doi: 10.1038/nature14335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandi S, Cioce M, Yeung YG, Nieves E, Tesfa L, Lin H, Hsu AW, Halenbeck R, Cheng HY, Gokhan S, Mehler MF, Stanley ER. Receptor-type protein-tyrosine phosphatase ζ is a functional receptor for interleukin-34. J Biol Chem. 2013;288:21972–21986. doi: 10.1074/jbc.M112.442731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiwaki T, Maeda N, Noda M. Characterization and developmental regulation of proteoglycan-type protein tyrosine phosphatase ζ/RPTPβ isoforms. J Biochem (Tokyo) 1998;123:458–467. doi: 10.1093/oxfordjournals.jbchem.a021959. [DOI] [PubMed] [Google Scholar]
- Scarlato M, Beesley J, Pleasure D. Analysis of oligodendroglial differentiation using cDNA arrays. J Neurosci Res. 2000;59:430–435. doi: 10.1002/(SICI)1097-4547(20000201)59:3<430::AID-JNR18>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Shintani T, Watanabe E, Maeda N, Noda M. Neurons as well as astrocytes express proteoglycan-type protein tyrosine phosphatase ζ/RPTPβ: analysis of mice in which the PTPζ/RPTPβ gene was replaced with the LacZ gene. Neurosci Lett. 1998;247:135–138. doi: 10.1016/S0304-3940(98)00295-X. [DOI] [PubMed] [Google Scholar]
- Sonobe Y, Li H, Jin S, Kishida S, Kadomatsu K, Takeuchi H, Mizuno T, Suzumura A. Midkine inhibits inducible regulatory T cell differentiation by suppressing the development of tolerogenic dendritic cells. J Immunol. 2012;188:2602–2611. doi: 10.4049/jimmunol.1102346. [DOI] [PubMed] [Google Scholar]
- Tamura H, Fukada M, Fujikawa A, Noda M. Protein tyrosine phosphatase receptor type Z is involved in hippocampus-dependent memory formation through dephosphorylation at Y1105 on p190 RhoGAP. Neurosci Lett. 2006;399:33–38. doi: 10.1016/j.neulet.2006.01.045. [DOI] [PubMed] [Google Scholar]
- Tsukada T, Tomooka Y, Takai S, Ueda Y, Nishikawa S, Yagi T, Tokunaga T, Takeda N, Suda Y, Abe S. Enhanced proliferative potential in culture of cells from p53-deficient mice. Oncogene. 1993;8:3313–3322. [PubMed] [Google Scholar]
- Umemori H, Sato S, Yagi T, Aizawa S, Yamamoto T. Initial events of myelination involve Fyn tyrosine kinase signalling. Nature. 1994;367:572–576. doi: 10.1038/367572a0. [DOI] [PubMed] [Google Scholar]
- Umemori H, Kadowaki Y, Hirosawa K, Yoshida Y, Hironaka K, Okano H, Yamamoto T. Stimulation of myelin basic protein gene transcription by Fyn tyrosine kinase for myelination. J Neurosci. 1999;19:1393–1397. doi: 10.1523/JNEUROSCI.19-04-01393.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Takeuchi H, Sonobe Y, Jin S, Mizuno T, Miyakawa S, Fujiwara M, Nakamura Y, Kato T, Muramatsu H, Muramatsu T, Suzumura A. Inhibition of midkine alleviates experimental autoimmune encephalomyelitis through the expansion of regulatory T cell population. Proc Natl Acad Sci U S A. 2008;105:3915–3920. doi: 10.1073/pnas.0709592105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf RM, Wilkes JJ, Chao MV, Resh MD. Tyrosine phosphorylation of p190 RhoGAP by Fyn regulates oligodendrocyte differentiation. J Neurobiol. 2001;49:62–78. doi: 10.1002/neu.1066. [DOI] [PubMed] [Google Scholar]
- Ziskin JL, Nishiyama A, Rubio M, Fukaya M, Bergles DE. Vesicular release fo glutamate from unmyelinated axons in white matter. Nat Neurosci. 2007;10:321–330. doi: 10.1038/nn1854. [DOI] [PMC free article] [PubMed] [Google Scholar]