

Abstract
Complexins (Cplxs) are small synaptic proteins that cooperate with SNARE-complexes in the control of synaptic vesicle (SV) fusion. Studies involving genetic mutation, knock-down, or knock-out indicated two key functions of Cplx that are not mutually exclusive but cannot easily be reconciled, one in facilitating SV fusion, and one in “clamping” SVs to prevent premature fusion. Most studies on the role of Cplxs in mammalian synapse function have relied on cultured neurons, heterologous expression systems, or membrane fusion assays in vitro, whereas little is known about the function of Cplxs in native synapses. We therefore studied consequences of genetic ablation of Cplx1 in the mouse calyx of Held synapse, and discovered a developmentally exacerbating phenotype of reduced spontaneous and evoked transmission but excessive asynchronous release after stimulation, compatible with combined facilitating and clamping functions of Cplx1. Because action potential waveforms, Ca2+ influx, readily releasable SV pool size, and quantal size were unaltered, the reduced synaptic strength in the absence of Cplx1 is most likely a consequence of a decreased release probability, which is caused, in part, by less tight coupling between Ca2+ channels and docked SV. We found further that the excessive asynchronous release in Cplx1-deficient calyces triggered aberrant action potentials in their target neurons, and slowed-down the recovery of EPSCs after depleting stimuli. The augmented asynchronous release had a delayed onset and lasted hundreds of milliseconds, indicating that it predominantly represents fusion of newly recruited SVs, which remain unstable and prone to premature fusion in the absence of Cplx1.
Keywords: calyx of Held, complexin, MNTB, presynaptic mechanisms, synaptic transmission, synaptic vesicle fusion
Introduction
The fusion of synaptic vesicles (SVs) at chemical synapses is an exquisitely fast and precisely controlled process. Triggered by local Ca2+ domains that build-up near open Ca2+ channels after arrival of a presynaptic action potential (AP), the probability of SV fusion increases transiently more than 100,000-fold for less than 1 ms. This requires the transmitter release machinery to be in a very responsive state to be called into action with minimum delay when [Ca2+]i is elevated, but also to be very reluctant to operate at resting [Ca2+]i to minimize uncoordinated fusion events in the absence of presynaptic APs.
To achieve such speed and temporal precision, presynaptic terminals maintain a pool of readily releasable SVs (RRP) which are docked and primed in close proximity to voltage-gated Ca2+ channels (VGCCs). A brief Ca2+ transient together with the highly nonlinear Ca2+ sensitivity of the machinery for SV fusion guarantees a rapid and short-lived release transient. Generating the RRP and stabilizing primed SVs in a highly fusogenic state critically depends on numerous regulatory proteins (for review, see Wojcik and Brose, 2007; Südhof and Rothman, 2009; Südhof and Rizo, 2011). Among those are complexins (Cplxs), small hydrophilic proteins that bind to assembled SNARE complexes (McMahon et al., 1995; Reim et al., 2001; Chen et al., 2002). Mammalian Cplxs, of which four paralogues are known (Cplx1–Cplx4; McMahon et al., 1995; Reim et al., 2005), operate at a postpriming step in SV fusion and their loss causes profound deficits in release probability (Reim et al., 2001; Xue et al., 2007, 2008). Cplxs also regulate SV priming in certain synapses (Yang et al., 2013), and they can exert an inhibitory “clamp-like” effect on transmitter release, depending on the species, Cplx variant, or synapse type studied (Huntwork and Littleton, 2007; Xue et al., 2009).
So far, most studies on the role of Cplxs in mammalian synapse function have relied on cultured neurons, heterologous expression systems, or membrane fusion assays in vitro, whereas little is known about their role in native synapses. We therefore investigated the function of Cplx1 at the mouse calyx of Held, a glutamatergic synapse of the mammalian brainstem, during synapse maturation from postnatal day (P)8–P21. Because of the large size of its presynaptic terminal, this synapse allows one to directly study functional presynaptic parameters, such as Ca2+ influx, AP waveform, RRP size, and Ca2+ influx-release coupling. We show that genetic deletion of Cplx1 results in a developmentally exacerbating phenotype of reduced spontaneous and evoked synaptic transmission at resting synapses, but excessive asynchronous release following synaptic stimulation. Our findings indicate that: (1) Cplx1 enhances spontaneous and evoked transmitter release at the calyx by increasing the fusogenicity of SVs, (2) Cplx1 stabilizes newly primed SVs and thereby prevents their premature release, and (3) Cplx1 is required to achieve the tight colocalization between VGCCs and docked SVs that is observed in mature calyces, as SV fusion remains highly sensitive to the slow Ca2+ chelator EGTA in the absence of Cplx1.
Materials and Methods
Animals and genotyping.
The generation of Cplx1 knock-out mice was described previously (Reim et al., 2001). Cplx1 deficient mice were backcrossed for at least eight generations into the C57BL/6N genetic background. PCR genotyping was performed with primers 1118/1119 for wild-type (wt) mice and 1111/1112 for Cplx1−/− mice sequences (1118, AGT ACT TTT GAA TCC CCT GGT GA; 1119, TAG CTA TCC CTT CTT GTC CTT GTG;1111, CGC GGC GGA GTT GTT GAC CTC G;1112, CTG GCT TGT CCC TGA ATC CTG TCC). Homozygous Cplx1+/+, Cplx1−/−, and heterozygous Cplx1+/− littermates, as well as a limited number of C57BL/6N mice at the age of P8–P30 were included in this study. All experiments complied with national animal care guidelines (LAVES permit 33.9-42502-04-13/1359).
Slice preparation.
Cplx1+/+ and Cplx1−/− littermates of either sex were used for most of the experiments. For comparison, some recordings were also obtained from wt C57BL/6N mice of either sex. Brainstem slices were prepared as described previously (Taschenberger and von Gersdorff, 2000). After decapitation, the whole brain was immediately immersed into ice-cold low-Ca2+ artificial CSF (aCSF) containing the following (in mm): 125 NaCl, 2.5 KCl, 3 MgCl2, 0.1 CaCl2, 10 glucose, 25 NaHCO3, 1.25 NaH2PO4, 0.4 ascorbic acid, 3 myo-inositol, and 2 Na-pyruvate at pH 7.3 when bubbled with carbogen (95% O2, 5% CO2). The brainstem was glued onto the stage of a VT1000S vibratome (Leica), and 200-μm-thick coronal slices containing the medial nucleus of the trapezoid body (MNTB) were cut. Slices were incubated for 30–40 min at 35°C in a chamber containing normal aCSF and kept at room temperature (RT; 21–24°C) for up to 5 h thereafter. The composition of normal aCSF was identical to that of low-Ca2+ aCSF, except that 3 mm MgCl2 and 0.1 mm CaCl2 were replaced with 1 mm MgCl2 and 2 mm CaCl2.
Electrophysiology.
Whole-cell patch-clamp recordings were made from calyx of Held terminals and principal neurons (PNs) of the MNTB using an EPC-10 amplifier controlled by “Pulse” or “PatchMaster” software (HEKA Elektronik). Sampling intervals and filter settings were 20 μs and 5.0 kHz, respectively. Cells were visualized by infrared-differential interference contrast microscopy through 40× or 60× water-immersion objectives using an upright BX51WI microscope (Olympus). During experiments, slices were continuously perfused with normal aCSF solution. All experiments were performed at RT.
Presynaptic recordings.
Presynaptic patch pipettes were pulled from borosilicate glass (Science Products) on a P-87 micropipette puller (Sutter Instrument) and coated with dental wax to reduce stray capacitance. Open tip resistance was 4–5 MΩ. Access resistance (Rs) was ≤20 MΩ and routinely compensated by 50–60% during presynaptic voltage-clamp experiments. The holding potential (Vh) was −80 mV. For measuring presynaptic ICa(V) and changes in membrane capacitance (ΔCm), pipettes were filled with a solution containing the following (in mm): 140 Cs-gluconate, 20 TEA-Cl, 10 HEPES, 5 Na2-phosphocreatine, 5 ATP-Mg, 0.3 GTP, pH 7.3, with CsOH. The pipette solution was supplemented with varying concentrations of the Ca2+ buffer EGTA (0.5 or 5 mm). During experiments, slices were continuously perfused with 1 μm TTX, 1 mm 4-AP, and 40 mm TEA-Cl to suppress voltage-gated Na+ and K+ currents. Liquid junction potentials were not corrected. Changes in membrane capacitance (ΔCm) were monitored using the Sine+DC technique (Lindau and Neher, 1988) with a software lock-in amplifier (implemented in HEKA Pulse/PatchMaster) by adding a 1 kHz sinewave voltage command (peak-to-peak amplitude ±35 mV) to Vh. To avoid a contamination of ΔCm estimates after long-lasting presynaptic depolarizations with small Cm transients unrelated to SV exocytosis (Yamashita et al., 2005), ΔCm was estimated from the averaged Cm values during 50–100 ms after the end of the depolarizations. The number of SVs was estimated by the following equation: N = ΔCm/(Cms × πd2), where Cms × πd2 is the average capacitance contribution from a single SV given the values of typical membrane capacitance (Cms) with 10 fF/μm2 and an averaged SV diameter (d) with ∼50 nm, yielding an estimation of single SV capacitance contribution of ∼80 aF (Taschenberger et al., 2002; He et al., 2006, 2009; Sakaba, 2006). Presynaptic recordings with a leak current >200 pA were excluded from the analysis. Calyceal APs were elicited by afferent fiber stimulation via a bipolar stimulation electrode placed halfway between the brainstem midline and the MNTB. Stimulation pulses (100 μs duration) were applied using a stimulus isolator unit (AMPI), with the output voltage set to 1–2 V above threshold for AP generation (≤25 V) to exclude stimulation failures at higher frequencies. Calyceal APs were measured in the current-clamp mode of the EPC-10 after adjusting the fast-capacitance cancellation while in cell-attached mode using pipettes that were filled with a solution consisting of the following (in mm): 100 K-gluconate, 60 KCl, 10 HEPES, 0.5 EGTA, 5 Na2-phosphocreatine, 4 ATP-Mg, 0.3 Na2-GTP, pH 7.3 with KOH.
Postsynaptic recordings.
For postsynaptic recordings, patch pipettes were pulled from thin-walled glass (World Precision Instruments) on a PIP-5 puller (HEKA Elektronik). Open-tip resistance was 2.5–3.5 MΩ. Rs ranged from 4 to 7 MΩ. Rs compensation was set to ≥84% (2 μs delay). The holding potential was −70 mV. For measuring postsynaptic APs, pipettes were filled with a solution consisting of the following (in mm): 100 K-gluconate, 60 KCl, 5 Na2-phosphocreatine, 10 HEPES, 5 EGTA, 0.3 Na2-GTP, and 4 ATP-Mg, pH 7.3, with KOH. For EPSC recordings, pipettes were filled with a solution consisting of the following (in mm): 140 Cs-gluconate, 20 TEA-Cl, 10 HEPES, 5 EGTA, 5 Na2-phosphocreatine, 5 ATP-Mg, 0.3 GTP, pH 7.3, with CsOH. No corrections were made for liquid junction potentials.
EPSCs were elicited by afferent fiber stimulation as described above. For each AP-evoked EPSC (eEPSC) the series resistance (Rs) value was updated and stored with the data using the automated Rs compensation routine implemented in Pulse.
Immunostaining.
Freshly dissected brains from P8, P16, and P21 Cplx1+/+ and Cplx1−/− littermates were gently removed and fixed at room temperature for 2 h in 4% paraformaldehyde (PFA) PBS. Thereafter, the brains were immersed overnight at 4°C in 4% PFA PBS. Fifty-micrometer-thick coronal sections of the MNTB were cut at 0°C and placed onto SuperFrost microscope slides (Fisher Scientific). After cutting, MNTB slices were washed three times for 10 min each time in PBS and incubated for 1 h in goat serum dilution buffer (GSDB; 16% normal goat serum, 450 mm NaCl, 0.3% Triton X-100, 20 mm phosphate buffer, pH 7.4) in a wet chamber at room temperature. Primary antibodies were dissolved in GSDB buffer and applied overnight at 4°C in a wet chamber. After washing three times for 10 min each time (wash buffer: 450 mm NaCl, 20 mm phosphate buffer, 0.3% Triton X-100), the tissue was incubated with secondary antibodies in GSDB in a wet, light-protected chamber for 1 h at room temperature. Then, the preparations were washed three times for 10 min each time in wash buffer and one time for 10 min in 5 mm phosphate buffer, placed onto the glass microscope slides with a drop of fluorescence mounting medium (Dako), and covered with thin glass coverslips. The following antibodies were used: rabbit anti-Cplx1/2 (1:300–700; Synaptic Systems), guinea pig anti-vesicular glutamate transporter 1 (VGLUT1; 1:4000; Synaptic Systems), and secondary AlexaFluor 488- and AlexaFluor 568-labeled antibodies (1:200; Invitrogen). All immunostaining experiments were repeated at least three times.
Confocal microscopy and image analysis.
Confocal images were acquired using a laser-scanning confocal microscope (Leica TCS SP5, Leica Microsystems) with 488 nm (Ar) and 561 nm (He-Ne) lasers for excitation and 10× air or 40×/63× oil-immersion objectives. To produce three-dimensional reconstructions of the specimen, a z-axis stack of two-dimensional images was taken with a step size of 0.2 μm at a pixel size of 0.09 × 0.09 μm2. Images were processed using ImageJ and assembled for display in Adobe Photoshop software.
Immunoblotting.
Brain tissue extracts of the MNTB from P8, P16, P21 wt C57BL/6N mice and P17 Cplx1+/+, and Cplx1−/− littermates (5–8 animals per age group) were analyzed by SDS-PAGE and Western blotting using standard procedures. Immunoreactive proteins were visualized with ECL (GE Healthcare). The following antibodies were used: polyclonal rabbit antibodies to Cplx1/2 (1:2500, Synaptic Systems), Cplx3 (1:500), and Cplx4 (1:500; Reim et al., 2005), as well as monoclonal antibodies to Syntaxin 1 (1:20,000), SNAP25 (1:106), and Synaptobrevin 2 (1:7500) which were obtained from Synaptic Systems. Expression levels were normalized to those of tubulin (1: 20,000, Sigma-Aldrich), which served as loading control. ECL films were scanned and signal intensities were estimated with Igor Pro (Wavemetrics). All Western blot experiments were repeated three times.
Reverse transcription and real-time PCR.
Brain tissue of the cochlear nucleus region was dissected from P16–P18 C57BL/6 mice (5–8 animals per experiment), followed by total RNA extraction with TRIzol reagent (Invitrogen). Reverse transcription (1 h at 42°C and 10 min at 70°C) of the total RNA (800 ng, 1 μg per sample) was performed in first-strand cDNA synthesis mix containing the following (after the final dilution; in mm): 50 Tris-HCl, 75 KCl, 5 MgCl2, and 5 DTT adjusted to pH 8.3 and 100 U of SuperScript II reverse transcriptase (Invitrogen), 40 U of RNaseOUT ribonuclease inhibitor (Invitrogen), as well as 12.5 ng/μl oligo-dT primers (Invitrogen). Real-time PCRs were performed in triplicate for each gene transcript using the Qiagen QuantiTect master mix. Quantitative PCR (qPCR) was done using a Mastercycler ep realplex (Eppendorf) system, with Cplx1–4 probes (QT02332687; QT00494662; QT00123025; QT00133805) and the house keeping gene Hprt1 (F: CTG GTG AAA AGG ACC TCT CGA AG; R, CCA GTT TCA CTA ATG ACA CAA ACG) with temperature cycles of 95°C for 10 min initially, followed by 40 cycles at 95°C (15 s each) and 60°C (1 min each). In the fluorescence reporter plot, the cycle threshold (CT) was defined in the exponential phase of the amplification curve, at a level of 0.2 fluorescence units. A plot of CT value versus the logarithm of the input amount of total RNA was fitted with a line to estimate the amplification efficiency of each probe. All quantitative PCR experiments were repeated three times.
Off-line analysis.
Off-line analysis was performed using Igor Pro (Wavemetrics) and “R” (R Core Team, 2014). After correction for remaining series-resistance errors (Traynelis, 1998) using the Rs values stored in the data files (assuming a linear I–V relationship with a reversal potential of 0 mV), EPSCs were offset corrected and low-pass filtered (fcutoff = 5 kHz) using a 10-pole software Bessel filter. Miniature EPSCs (mEPSCs) were detected using a sliding template algorithm (Jonas et al., 1993; Clements and Bekkers, 1997). All data are presented as mean ± SEM. Statistical significance of difference was evaluated with the two-tailed unpaired Student's t test and p < 0.05 was taken as the level of statistical significance.
Results
Cplx1 is the predominant Cplx isoform at calyx of Held terminals
Four Cplx genes (Cplx1–4) encode four Cplx isoforms in the mammalian CNS (McMahon et al., 1995; Reim et al., 2001, 2005). Cplx1 and Cplx2 share 86% protein sequence identity and are ubiquitously expressed in most brain regions with overlapping expression patterns. During development, Cplxs are first detectable at P6 in the rat brain and their expression increases to reach a plateau at ∼20 d after birth, when most synapses have been formed (Ishizuka et al., 1997; Reim et al., 2005). To clarify which Cplx isoforms are expressed at the calyx of Held synapse, we performed Western blot analyses (Fig. 1A). Protein extracts were obtained from small tissue samples containing the MNTB region, which were cut out from acute brainstem slices of P17 Cplx1−/− and wt littermates. For comparison, protein extracts from whole-brain (P2 fraction) and retina were included in the analysis. These experiments revealed that among all Cplxs, Cplx1 is the predominantly expressed paralog in the MNTB region, and only minor coexpression of Cplx2 was detected. Although the antibody against Cplx1 is not isoform-specific and cross-reacts with Cplx2, both proteins can be discriminated by their different molecular weight (Fig. 1A). Cplx3 or Cplx4 expression was not detectable in the MNTB region, but the two proteins were readily detected in retina samples (Reim et al., 2005). Notably, the expression of Cplx1 was completely abolished in Cplx1−/− mice and no compensatory upregulation of Cplx2 was observed. We also examined the expression of several key synaptic proteins, including Syntaxin 1, SNAP 25, and Synaptobrevin 2, to assess possible changes in protein composition in Cplx1−/− MNTB synapses. As shown in Figure 1A, the analyzed neuronal SNARE complex components were expressed in the MNTB and their levels of expression were found unaltered in Cplx1−/− samples.
Figure 1.
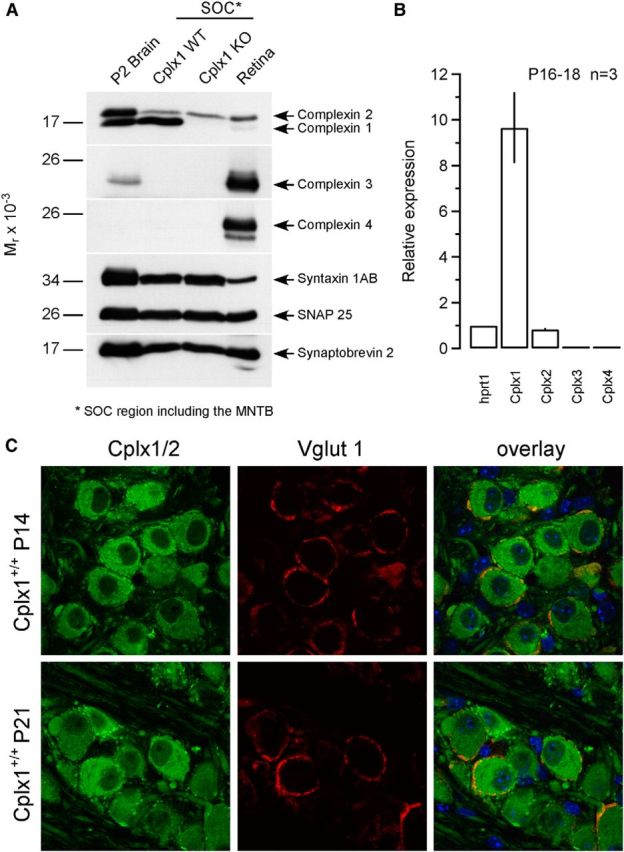
Analysis of Cplx expression in the mouse MNTB. A, P2 fraction from adult brain and tissue homogenates from adult retina and the MNTB region of the superior olivary complex (SOC) cut out from acute brainstem slices of P17 Cplx1+/+ and Cplx1−/− littermates (10 μg per lane) were analyzed by SDS-PAGE using antibodies against Cplx1/2, Cplx3, and Cplx4, as well as antibodies against the presynaptic marker proteins Syntaxin 1AB, Synaptobrevin 2, and SNAP 25. B, Validation of mRNA expression level of different Cplx isoforms in the AVCN region cut out from acute brainstem slices of P16–P18 Cplx1+/+ mice by quantitative real-time PCR. The expression level was calculated according to 2−(Ctgene − Cthprt1), where Ctgene and Cthprt1 represent the threshold levels of detection for the genes tested and for the housekeeping gene, respectively. The relative expression was then obtained by normalizing the expression levels to that of the housekeeping gene hprt1 (n = 3; technical replicates). C, Cplx1/2 localization patterns in the developing mouse calyx of Held. Immunofluorescence images representing projections of confocal sections of MNTB Cplx1+/+ mice costained with an anti-Cplx1/2 antibody (green, left), and an anti-Vglut1 antibody (red, middle) at P14 and P21. Cplx1/2 immunofluorescent MNTB PNs are surrounded by ring-like Cplx1/2- and Vglut1-positive structures representing calyx terminals. Right, The corresponding overlay images.
Because the protein extract of the tissue samples subjected to Western blot analysis contained presynaptically as well as postsynaptically expressed proteins of a nonhomogenous cell population, the described expression pattern may not necessarily reflect the protein expression levels in presynaptic calyces. Therefore, we used quantitative real-time PCR to examine the Cplx expression pattern at the mRNA level. Because calyx terminals originate from the globular bushy cells in the respective contralateral anteroventral cochlear nucleus (AVCN; Harrison and Irving, 1966; Kuwabara et al., 1991), the ACVN region was dissected out from P16–P18 wt mice to collect mRNA from bushy cell somata. After mRNA extraction and reverse transcription, qPCR analysis was performed. Cplx1, Cplx2, Cplx3, and Cplx4 probes were included in the reactions to validate their relative expression levels. Figure 1B illustrates that the expression level of Cplx1 was 12 times higher than that of Cplx2 (p < 0.01), whereas Cplx3/4 expression was undetectable. These data agree with previous studies showing strong expression of Cplx1 in the cochlear nucleus by in situ hybridization (Freeman and Morton, 2004) or immunocytochemistry (Strenzke et al., 2009). Together with the Western blot analysis described above, these results show that Cplx1 is the dominant Cplx isoform present at calyx of Held terminals.
To characterize the cellular and subcellular distribution of Cplxs in the MNTB, we performed immunocytochemical analyses of P14 and P21 MNTB slices (Fig. 1C). Colabeling with an antibody against the presynaptic marker protein Vglut1 (vesicular glutamate transporter 1) revealed strong Cplx1/2 immunoreactivity in P14 and P21 calyx terminals. Interestingly, immunolabeling was present not only in presynaptic calyces but also in MNTB PNs. Using the same antibody, strong Cplx1/2 immunoreactivity of somata has been reported before for spiral ganglion neurons and PNs of the cochlear nucleus (Strenzke et al., 2009), and for retinal ganglion cells (Reim et al., 2005). We therefore conclude that Cplx1/2 are expressed presynaptically as well as postsynaptically within the MNTB. The abundant expression of Cplx1/2 in the MNTB PNs raises the possibility that genetic ablation of Cplx1 may not only affect presynaptic but also postsynaptic function.
Synaptic strength is reduced in calyx synapses of Cplx1−/− mice
Because Cplx1 is by far the most highly expressed Cplx isoform in calyx of Held synapses and because genetic ablation of Cplx1 expression did not result in compensatory upregulation of other Cplxs in the MNTB (Fig. 1A), we used Cplx1−/− mice as a model to examine the role of Cplx1 in synaptic transmission at this synapse. We first recorded AP-evoked EPSCs elicited by afferent fiber stimulation in wt and Cplx1−/− synapses during postnatal development from P8 to P30 to test for changes in synaptic strength. Synaptic transmission was detectable at all ages tested as indicated by the presence of eEPSCs. Representative examples of eEPSCs recorded at three different ages in wt and Cplx1−/− mice are illustrated in Figure 2A. Synaptic strength was essentially unchanged in young (≤P12) Cplx1−/− synapses compared with wt synapses (Fig. 2A, left). However, eEPSC amplitudes of Cplx1−/− synapses started to decline strongly at the age of P14 and were severely reduced compared with those of wt mice at the age of P20–P30 (Fig. 2A, middle and right). Figure 2B illustrates AP-evoked EPSC amplitudes as a function of postnatal age for a developmental range from P14 to P21. Linear regression analysis of the scatter plot revealed a gradual decline of the mean eEPSC amplitude in Cplx1−/− calyx synapses from −9.8 nA (P14) to −2.1 nA (P21) (Pearson's r = 0.55, p < 0.001), whereas the mean eEPSC amplitudes of wt mice increased slightly [from −11.8 nA (P14) to −12.1 nA (P21); Joshi and Wang, 2002]. Despite considerable synapse-to-synapse variability, eEPSC amplitudes were on average reduced to ∼37% in P16–P21 Cplx1−/− calyx synapses compared with wt calyx synapses (Table 1). Because synaptic strength did not appear to be altered in Cplx1−/− calyx synapses at P14 and younger ages, we subsequently focused primarily on the characterization of synaptic transmission in posthearing-onset Cplx1−/− calyx synapses (P16–P21).
Figure 2.
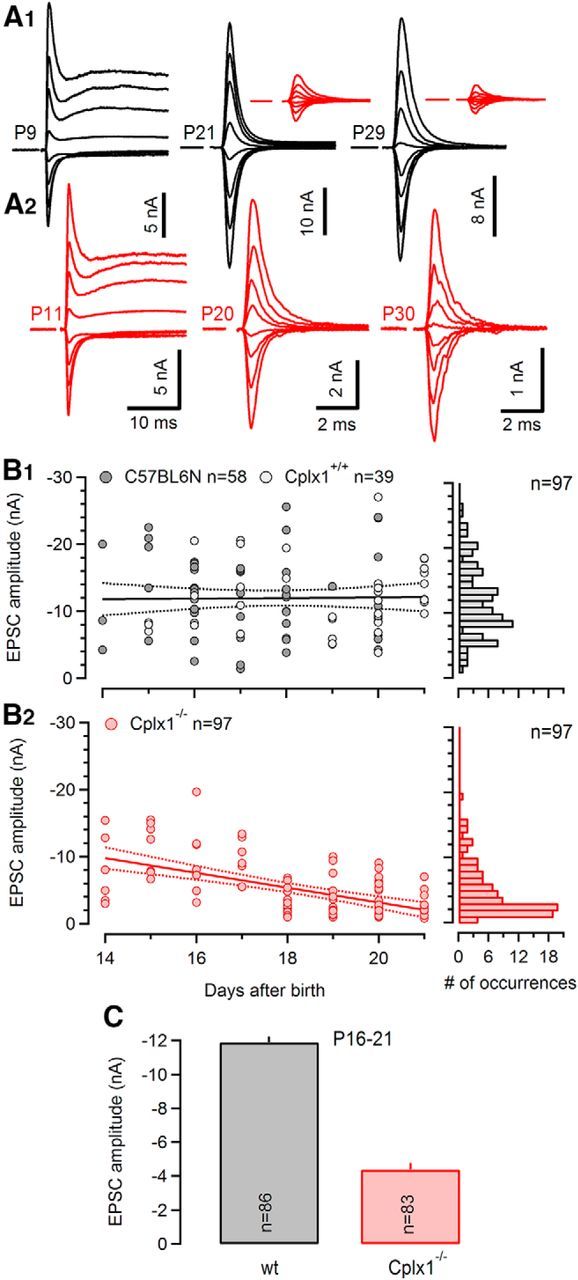
Pronounced developmental decline of synaptic strength in calyx synapses of Cplx1−/− mice. A, Families of representative EPSCs evoked by afferent fiber stimulation and recorded at various Vh ranging from −70 to +70 mV (20 mV increments) in calyx synapses of Cplx1+/+ mice at P9, P21, and P29 (A1) and Cplx1−/− mice at P11, P20, and P30 (A2). Insets, Top right and top middle show the same traces from the corresponding Cplx1−/− synapses drawn at scale to facilitate comparison. In both, wt and Cplx1−/− calyx synapses, NMDAR-mediated EPSC components are strongly reduced after P10 and virtually undetectable at ∼P30. B, Left, Scatter plots of AMPA EPSC peak amplitudes versus age for wt (B1) and Cplx−/− (B2) mice. Right, Corresponding frequency distribution of EPSC peak amplitudes. Because EPSC amplitudes recorded in Cplx1+/+ mice (light gray circles) were statistically not different from those obtained in wt C57BL6N mice (dark gray circles) the results were pooled. Solid and dotted lines represent linear regressions and 95% confident intervals, respectively. On average, amplitudes increased slightly (∼50 pA/d) in wt (B1) but showed a pronounced decrease (∼1 nA/d) in Cplx1−/− mice (B2) during postnatal week 3. C, Mean EPSC amplitudes. Bar graphs represent summary data for P16–P21 Cplx1−/− (red) and wt (gray) mice. Number of synapses tested as indicated. For P16–21 synapses, the average EPSC amplitudes were reduced by ∼63% in Cplx1−/− synapses.
Table 1.
Functional parameters of synaptic transmission in P16–21 Cplx1−/− and Cplx1+/+ mouse calyx of Held synapses
| Parameter | Cplx1+/+ | Cplx1−/− | Statistical significance of differences in mean |
|---|---|---|---|
| eEPSC peak amplitude | −11.88 ± 0.06 nA (n = 86) | −4.37 ± 0.04 nA (n = 83) | p < 0.001 |
| eEPSC rise time (20–80%) | 148 ± 2 μs (n = 86) | 154 ± 3 μs (n = 83) | n.s. |
| eEPSC weighted τdecay | 419 ± 11 μs (n = 85) | 440 ± 14 μs (n = 83) | n.s. |
| eEPSC half-width | 517 ± 8 μs (n = 86) | 552 ± 12 μs (n = 83) | p = 0.015 |
| mEPSC peak amplitude | −62.1 ± 2.9 pA (n = 39) | −63.6 ± 2.4 pA (n = 55) | n.s. |
| mEPSC weighted τdecay | 240 ± 8 μs (n = 39) | 238 ± 6 μs (n = 55) | n.s. |
| mEPSC frequency | 4.79 ± 0.43 s−1 (n = 39) | 2.14 ± 0.19 s−1 (n = 55) | p < 0.001 |
| Peak release ratea | 823 ± 62 SV/ms (n = 24) | 309 ± 36 SV/ms (n = 36) | p < 0.001 |
| Quantal content mb | 287 ± 21 SV (n = 38) | 113 ± 13 (n = 55) | p < 0.001 |
| Presynaptic ICa(V)c | −1.49 ± 0.08 nA (n = 20) | −1.44 ± 0.09 nA (n = 28) | n.s. |
| RRP sized | 351 ± 51 fF = 4388 ± 638 SV (n = 13) | 368 ± 39 fF = 4600 ± 488 SV (n = 18) | n.s. |
| Release probability pre | 13.1 ± 1.0% (n = 38) | 4.9 ± 0.6% (n = 55) | p < 0.001 |
| Asynchronous release ratef after high-frequency trains of: 100 Hz | 93 ± 17 SV/s (n = 28) | 1188 ± 92 SV/s (n = 44) | p < 0.001 |
| 200 Hz | 147 ± 26 SV/s (n = 32) | 1832 ± 143 SV/s (n = 47) | p < 0.001 |
| 300 Hz | 125 ± 37 SV/s (n = 15) | 2132 ± 172 SV/s (n = 43) | p < 0.001 |
n.s., Not significant.
aPeak release rates were obtained by deconvolving eEPSCs with the respective mEPSC waveforms (see Materials and Methods).
bm was calculated as the peak amplitude ratio eEPSCs/mEPSC and corrected with a factor of 1.4 to account for dispersion of release (Taschenberger et al., 2005).
cPeak ICa(V) values were obtained from currents evoked by step depolarizations of ≥10 ms.
dRRP size represents the sum of fast and slowly releasing vesicles as described by Sakaba and Neher (2001a) and was estimated from ΔCm measured in response to 50 ms step depolarizations.
epr was calculated as the ratio of m/(0.5 × RRP), assuming that those vesicles that contribute to synchronous AP-evoked release account for ∼50% of the total RRP (“fast releasing vesicles”; Sakaba and Neher, 2001a).
fAsynchronous release rates were obtained from line fits to the cumulative release measured after high-frequency trains (Fig. 7B) during a 100 ms interval starting 25 ms after the end of stimulation.
When comparing eEPSC waveforms, we noticed that the time course of eEPSC of Cplx1−/− calyx synapses was slightly slower than that of wt calyx synapses. This observation prompted us to analyze the eEPSC kinetics of Cplx1−/− synapses in more detail. During early postnatal development of calyx synapses, the decay time course of their AMPA receptor (AMPAR)-mediated eEPSCs accelerates markedly. This acceleration of the eEPSC kinetics, which is mainly caused by changes in the expression pattern of AMPARs during development, plays an indispensable role in the refinement of high-fidelity transmission at mature calyx of Held synapses (Taschenberger and von Gersdorff, 2000; Iwasaki and Takahashi, 2001; Koike-Tani et al., 2005). We found the kinetics of eEPSCs in Cplx1−/− calyx synapses to be slightly but consistently slower compared with that of wt synapses, as indicated by the slower rise times, slower weighted decay time constants and larger half-width values in Cplx1−/− synapses (Table 1). Because the time course of eEPSCs is in part determined by the synchrony of SV fusion (Diamond and Jahr, 1995; Isaacson and Walmsley, 1995; Taschenberger et al., 2005) in addition to the postsynaptic AMPAR kinetics, it is possible that genetic ablation of Cplx1 affects the synchrony of the vesicular release process in Cplx1−/− synapses. We therefore analyzed the time course of transmitter release in P16–P21 calyx synapses by deconvolving the waveforms of eEPSCs with those of the corresponding mEPSCs obtained from the same synapses. Deconvolution of eEPSCs was performed in the frequency domain using fast Fourier transform routines yielding the time course of quantal release. As shown in Table 1, the peak release rates of wt synapses were >2.5 times higher than those of Cplx1−/− synapses. In addition, the late component of synchronous release decayed slightly more slowly in Cplx1−/− synapses (data not shown), consistent with a stronger contribution of delayed release to the AP-evoked release transient in these synapses.
Mechanisms underlying the reduced synaptic strength in Cplx1−/− calyces
Several mechanisms upstream or downstream of SV fusion contribute to the regulation of synaptic strength, including changes in presynaptic Ca2+ influx and AP waveform, which both can result in altered presynaptic release probability (p), alterations in the size of the RRP (N), or changes in quantal size (q). Previous work indicates that Cplx deletion primarily affects presynaptic release probability (Reim et al., 2001). However, in none of the previously studied preparations, functional presynaptic parameters, such as Ca2+ influx, AP waveform, or RRP size could be measured directly. Taking advantage of the large size of the calyx terminal, we studied these properties by direct presynaptic recordings from calyx terminals to clarify the mechanisms underlying the reduced synaptic strength in Cplx1−/− calyx synapses.
Quantal size
To exclude the possibility that a decline in synaptic strength and/or the slowing of eEPSCs was in part mediated by an altered size and/or changed kinetics of quantal events, we first compared spontaneous miniature EPSCs (mEPSCs) in Cplx1−/− and wt calyx synapses. Amplitudes and decay time constants of the mEPSCs were found unaltered (Table 1; Fig. 3A,D,E), indicating that removal of Cplx1 is unlikely to affect SV filling and/or the composition or density of postsynaptic AMPARs at the calyx of Held synapse. However, we noticed that the mEPSC frequency was significantly reduced in Cplx1−/− compared with wt calyx synapses (Table 1; Fig. 3B,C), which may be indicative of a reduced presynaptic release probability in calyx synapses of Cplx1−/− mice. This result of reduced mEPSC frequency in Cplx1−/− calyx synapses is in sharp contrast to studies on Drosophila neuromuscular synapses, where Cplx deletion causes a 20-fold increase in spontaneous release rate (Huntwork and Littleton, 2007). Interestingly, the differences in mEPSC frequency between Cplx1−/− and wt calyx synapses diminished when raising external Ca2+ from 2 to 6 mm (Fig. 3E, right; p = 0.6).
Figure 3.
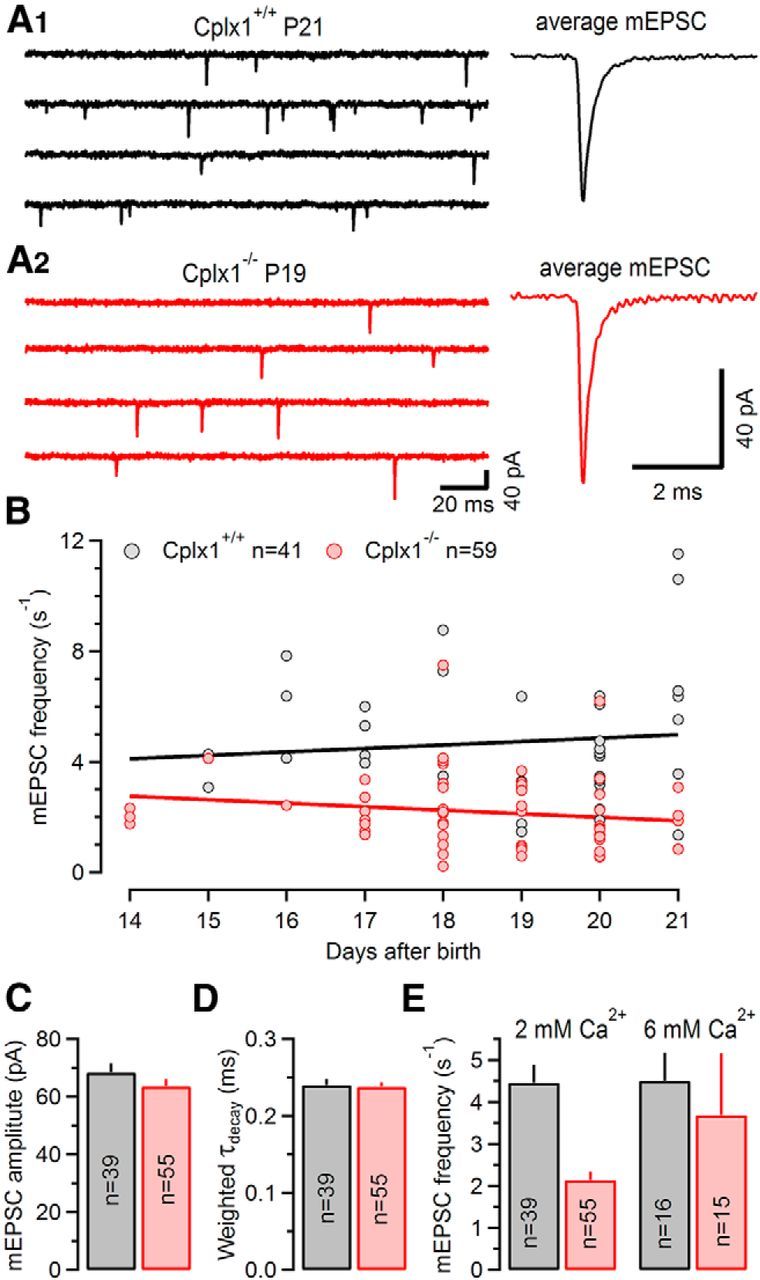
Unaltered mEPSC amplitudes and kinetics but strongly reduced frequency of spontaneous mEPSC in Cplx1−/− calyx synapses. A, Consecutive sweeps of representative mEPSC recordings (left) and corresponding average mEPSC waveforms (right) from a P21 Cplx1+/+ (A1, black) and a P19 Cplx1−/− mouse (A2, red). B, Scatter plot of average mEPSCs frequencies versus age from P14–P21 Cplx1+/+ (n = 41; gray circles) and Cplx1−/− (n = 59; red circles) mice. Solid lines represent linear regressions. Note the contrasting developmental trends in Cplx1+/+ versus Cplx1−/− calyx synapses. C–E, Summary data showing average values for mEPSC amplitude (C), mEPSC decay time constants (D), and for mEPSC frequency (E, left, normal Ca2+; right, elevated external Ca2+) in P16–P21 calyx synapses. Number of synapses analyzed as indicated. The average mEPSCs frequency of Cplx1−/− mice (red) was ∼48% of that in Cplx1+/+ (gray) mice, whereas mEPSC amplitudes and mEPSC decay kinetics were unchanged.
When plotting the rate of spontaneous release as a function of age, we found opposite developmental trends for the mEPSC frequency in wt compared with Cplx1−/− synapses (Fig. 3B). Although the mEPSC frequency increased slightly during postnatal maturation in wt synapses, a decrease with age was found in Cplx1−/− synapses (Spearman's rank correlation ρ = −0.267, p = 0.041). Together, these findings indicate that the reduction of synaptic strength in Cplx1−/− mice is mainly caused by changes in transmitter release rather than in quantal size.
Presynaptic Ca2+ influx and size of the readily releasable pool
To test whether genetic deletion of Cplx1 expression can alter VGCC function and/or the number of releasable SVs, we recorded presynaptic voltage-gated Ca2+ currents (ICa(V)) and changes in membrane capacitance (ΔCm) by directly voltage-clamping wt and Cplx1−/− calyx terminals. As shown in Figure 4A,C,E, peak amplitudes of ICa(V) in Cplx1−/− calyces were essentially unchanged compared with wt calyces. In both, Cplx1−/− and wt mice, the calyceal ICa(V) increased during development, such that mean amplitudes of ICa(V) amounted to −0.96 ± 0.05 nA at P8–P12 (n = 27) and −1.49 ± 0.08 nA at P16–P20 (n = 20) for wt terminals and −1.06 ± 0.07 nA at P8–P12 (n = 30) and −1.44 ± 0.09 nA at P16–P20 (n = 28) for Cplx1−/− terminals. This result indicates that Cplx1 is not involved in regulating the number of presynaptically expressed VGCCs.
Figure 4.
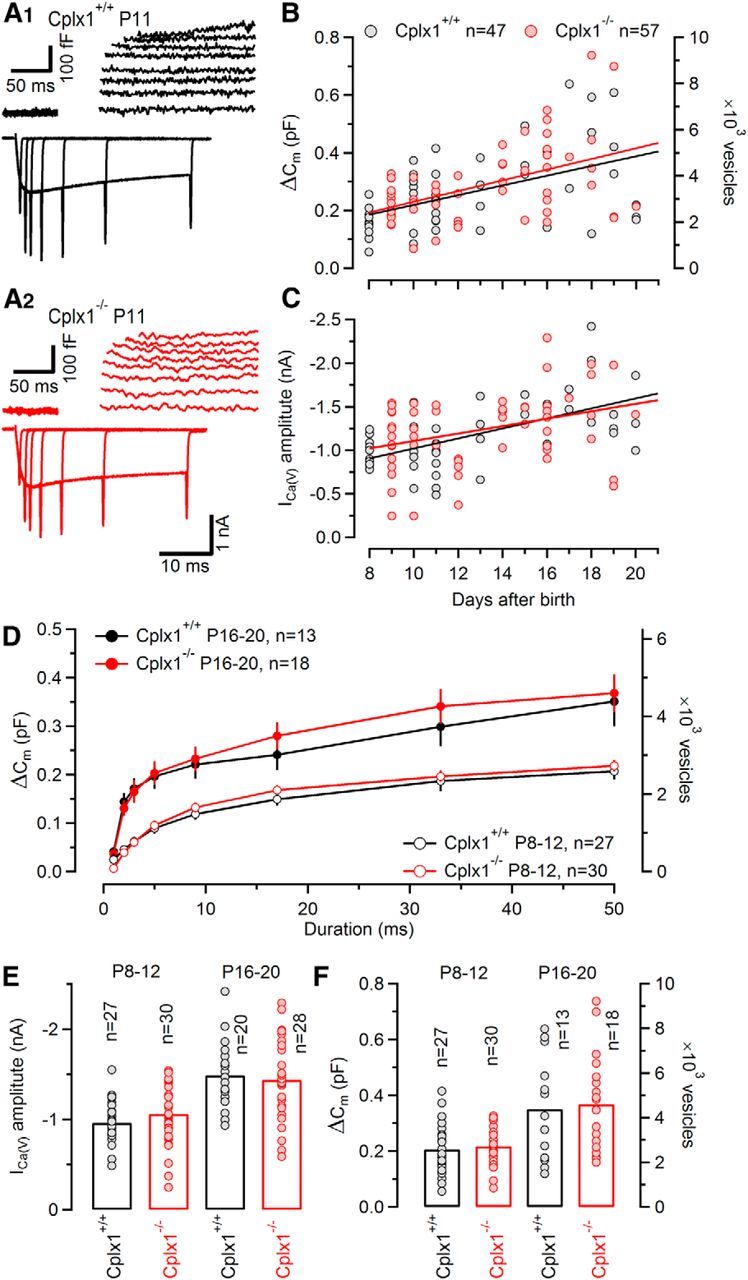
Unaltered voltage-gated Ca2+ currents and similar RRP size in Cplx1−/− calyx terminals. A, SV exocytosis triggered by presynaptic Ca2+ influx and recorded by capacitance measurements. Representative presynaptic ICa(V) and the corresponding changes in membrane capacitance (ΔCm) evoked by step depolarizations of 1, 2, 3, 5, 9, 17, 33, and 50 ms duration from Vh = −80 to 0 mV were recorded with 0.5 mm EGTA in the pipette solution in a Cplx1+/+ (A1) and a Cplx1−/− (A2) terminal. B,C, Scatter plots of peak amplitudes of ICa(V) (B) and corresponding maximum ΔCm values (obtained from 50 ms step depolarizations) as a function of postnatal age. Solid lines represent linear regressions. Both ICa(V) as well as exocytosis increased similarly in Cplx1+/+ (gray circles) and Cplx1−/− (red circles) terminals with maturation. Number of terminals tested as indicated. D, Average ΔCm values plotted as a function of step depolarization length for Cplx1+/+ (black) and Cplx1−/− (red) terminals. Data were grouped into two developmental stages: P8–P10 (open symbols) and P16–P20 (filled symbols). ΔCm values were similar for wt and Cplx1−/− terminals throughout development. E, F, Averaged peak amplitudes of ICa(V) (E) and ΔCm values (50 ms step depolarizations; F) for Cplx1+/+ (black) and Cplx1−/− (red) terminals. Number of terminals tested as indicated.
Next, we tested whether the size of the RRP is altered in Cplx1−/− synapses. We used presynaptic ΔCm measurements in voltage-clamped calyx terminals of wt and Cplx1−/− mice and evoked SV fusion by step depolarizations of 1–50 ms duration (from Vh= −80 to 0 mV) to gradually deplete the RRP. Example traces obtained during such experiments are shown in Figure 4A. ΔCm values were converted into SV numbers by assuming a single SV capacitance of 80 aF (see Materials and Methods). Considering that the reduction of synaptic strength in Cplx1−/− calyx synapses appeared relatively late during development, we categorized the experiments into two age groups (P8–P12 and P16–P20). In Figure 4D, ΔCm values are plotted against the duration of the presynaptic depolarization. ΔCm increased steeply as the duration of depolarization was extended up to ∼10 ms. Longer step depolarizations resulted in further but smaller increases in ΔCm. The relationship between exocytosis and the duration of depolarization was well approximated by a bi-exponential function, indicating heterogeneity among readily releasable SVs, reminiscent of the fast and slowly releasable pools described by Sakaba and Neher (2001a). For simplicity, we approximated the total RRP (sum of fast and slowly releasable SVs) by the ΔCm value measured after a 50 ms depolarization. This value may be an overestimation if significant SV pool replenishment occurs within the 50 ms step. On the other hand, ongoing endocytosis may lead to an underestimation of the RRP by ΔCm measurements. After converting the ΔCm values into SV numbers, we estimated that Cplx1−/− terminals harbor a RRP of ∼2720 SVs (218 ± 12 fF, n = 30) and 4600 SVs (368 ± 39 fF, n = 18) at P8–P12 and P16–P21, respectively. These results are similar to those obtained from wt mice with an average ∼2560 SVs (207 ± 17 fF, n = 27) and 4388 SVs (351 ± 51 fF, n = 13) at P8–P12 and P16–P21, respectively (Fig. 4D,F; Taschenberger et al., 2002; Lin et al., 2011). Thus, presynaptic capacitance recordings yielded no evidence in favor of an altered RRP size in Cplx1−/− calyces, indicating that the decrease in synaptic strength is mainly caused by a decrease in release probability.
Calyceal AP waveform and Ca2+ influx-secretion coupling
The presynaptic AP waveform is a strong regulator of release probability. AP broadening can increase the number of VGCCs that open during the AP and prolong their open time, and thereby increase presynaptic Ca2+ influx (Sabatini and Regehr, 1997; Borst and Sakmann, 1999). Therefore, we tested whether the calyceal AP waveform is changed in Cplx1−/− synapses. Presynaptic APs were evoked by afferent fiber stimulation in P16–P21 Cplx1−/− or wt calyces, and recorded in current-clamp mode. As illustrated in Figure 5A, AP waveforms were similar in Cplx1−/− (n = 7) and wt (n = 8) calyces (AP amplitude: 112.8 ± 0.3 mV in Cplx1−/− vs 115.2 ± 0.2 mV in wt, AP half-width: 262 ± 13 μs in Cplx1−/− vs 260 ± 14 μs in wt; Fig. 5B,C). Thus, changes in AP waveform are unlikely to contribute to the reduced synaptic strength in Cplx1−/− synapses.
Figure 5.

Unaltered calyceal AP waveform but higher sensitivity of SV exocytosis to intracellular EGTA in P16–P21 Cplx1−/− calyx terminals. A, Representative presynaptic calyceal APs elicited by afferent fiber stimulation and recorded in current-clamp configuration in a P17 Cplx1+/+ (black) and a P18 Cplx1−/− (red) terminal. B, Average AP amplitudes (left) and AP half-widths (right) were similar in Cplx1+/+ (black) and Cplx1−/− (red) terminals. Number of terminals tested as indicated. C, AP waveform is stable during repetitive presynaptic high-frequency firing. AP trains consisting of 35 APs elicited at a frequency of 100 Hz in a Cplx1+/+ (black) and a Cplx1−/− (red) terminal. Left, The first three and last three APs in the trains. Right, The first and last AP are shown superimposed for comparison. D–F, Presynaptic Ca2+ influx (D1, E1, F1) and SV exocytosis (D2, E2, F2) in response to AP-like depolarizations (1 ms steps to 0 mV). D, Representative recordings from a Cplx1+/+ (black) and a Cplx1−/− (red) terminal with 5 mm EGTA in the pipette solution. Note the similar ICa(V) but strongly attenuated ΔCm in the Cplx1−/− calyx terminal. E,F, Average traces for ICa(V) and ΔCm recorded in Cplx1+/+ (E) and Cplx1−/− (F) terminals with either 0.5 mm EGTA (black traces) or 5 mm EGTA (red traces) in the pipette solution. Light gray and light red areas in (E2, F2) represent ±SEM. Although exocytosis in Cplx1+/+ terminals was relatively insensitive to the slow Ca2+ chelator EGTA (E), the latter attenuated the average ΔCm value by ∼50% in Cplx1−/− terminals (F).
During postnatal maturation of calyx synapses, a tighter spatial coupling between VGCCs and docked SVs seems to compensate for the briefer mature AP waveform that leads to reduced Ca2+ influx (Taschenberger and von Gersdorff, 2000; Nakamura and Takahashi, 2007). The tighter colocalization between VGCCs and docked SVs exposes the latter to higher local-domain Ca2+ concentrations (Wang et al., 2008; Kochubey et al., 2009; Chen et al., 2015; Nakamura et al., 2015). We therefore considered that genetic ablation of Cplx1 expression could interfere with this developmental refinement and result in less tight spatial coupling between VGCCs and docked SVs in Cplx1−/− synapses, which in turn, may lower release probability (Fedchyshyn and Wang, 2005; Leão and von Gersdorff, 2009). To test this possibility, we performed experiments to compare the effect of different concentrations of the slow Ca2+ chelator EGTA (0.5 mm vs 5 mm) on SV exocytosis elicited by AP-like presynaptic depolarizations (1 ms steps from −80 to 0 mV). If VGCCs are tightly coupled to SVs, even high concentrations of EGTA (5 mm) cannot intercept Ca2+ ions before they reach the Ca2+ sensor of the release machinery. Thus, if addition of 5 mm EGTA to the pipette solution effectively attenuates release, the VGCCs are likely to be located more distantly from the SVs, providing sufficient time for EGTA to bind Ca2+ ions in transit (Naraghi and Neher, 1997). With patch pipettes containing either 5 mm or 0.5 mm EGTA, ΔCm following AP-like step depolarizations was measured in P16–P21 Cplx1−/− and wt terminals. Figure 5D illustrates two representative recordings obtained with 5 mm EGTA. Both calyces exhibit very different amount of exocytosis while having nearly identical ICa(V) amplitudes. The experiments are summarized in Figure 5E,F. SV exocytosis was strongly attenuated by 5 mm EGTA in Cplx1−/− mice (n = 12, p < 0.01) with an average capacitance jump ΔCm of 17.3 ± 4.8 fF (∼216 SVs, n = 15) but not in wt terminals (ΔCm of 44.7 ± 2.3 fF, ∼559 SVs, n = 15) of the same age. This result indicates that the spatial coupling between VGCCs and docked SVs is less tight in Cplx1−/− calyces. Given the similar ICa(V) amplitudes, this may, in part, account for a lower release probability during AP-evoked release in Cplx1−/− calyx synapses. In addition, if genetic ablation of Cplx1 interferes with the developmental tightening of the Ca2+ influx-release coupling in calyx terminals, it is actually expected to cause stronger functional deficits in more mature terminals (Fig. 2) because compensation for reduced Ca2+ influx due to the briefer mature presynaptic AP waveform can no longer occur.
Comparison of short-term plasticity in calyx synapses of Cplx1−/− and wt mice
Before hearing onset (P12–P13), pronounced short-term depression is generally observed in calyx of Held synapses. During maturation, the magnitude of depression decreases (Taschenberger and von Gersdorff, 2000; Iwasaki and Takahashi, 2001; Joshi and Wang, 2002). To study the effect of Cplx1 deletion on synaptic plasticity, eEPSC trains evoked by afferent-fiber stimulation at different frequencies (50, 100, and 200 Hz) were recorded in Cplx1−/−, Cplx1+/− and wt calyx synapses. Sample eEPSC trains are shown in Figure 6A. Consistent with previous reports, eEPSC trains of wt calyx synapses showed mostly depression (Erazo-Fischer et al., 2007; Lipstein et al., 2013). In contrast, eEPSCs of Cplx1−/− calyx synapses (n = 50, P16–P21) initially strongly facilitated. The maximum facilitation, which usually occurred following the second or third stimulus of the train, was 107 ± 3%, 120 ± 4%, and 140 ± 6% relative to the first eEPSC amplitude (eEPSC1) for frequencies of 50, 100 and 200 Hz, respectively. During trains of 35 stimuli, eEPSCs showed less depression in Cplx1−/− (72 ± 6%, 52 ± 5%, and 37 ± 5% of EPSC1 for 50, 100, and 200 Hz, respectively) compared with wt synapses (25 ± 2%, 17 ± 1%, and 10 ± 1% of eEPSC1 for 50, 100, and 200 Hz, respectively; Fig. 6B,C). Heterozygous Cplx1+/− mice showed a moderately reduced eEPSC amplitude with −8.45 ± 1.42 nA (n = 11, P16–P21) but virtually unchanged synaptic plasticity with 28 ± 3%, 23 ± 2%, and 14 ± 1% depression at steady-state relative to eEPSC1 for 50, 100, and 200 Hz trains, respectively. The reduced depression and enhanced synaptic facilitation of eEPSCs during train stimulation in Cplx1−/− synapses is consistent with a lower release probability in these synapses.
Figure 6.

Reduced short-term depression during EPSC trains in P16–P21 calyx synapses of Cplx1−/− mice. A, Representative 100 Hz trains consisting of 35 EPSCs evoked by afferent-fiber stimulation in a Cplx1+/+ (black), a Cplx1± (blue), and a Cplx1−/− (red) calyx synapse. B, C, Pooled data of eEPSC train amplitudes before (B) and after (C) normalizing to the peak of the eEPSC1 plotted against stimulus number. Stimulus trains consisted of 35 APs elicited at a frequency of 50, 100, or 200 Hz. Same color code as in A. Cplx1−/− mice had reduced average eEPSC amplitudes and, in contrast to Cplx1+/+ mice, showed synaptic facilitation for all stimulus frequencies tested. Slightly reduced synaptic depression is observed in heterozygous Cplx1+/− mice. eEPSC amplitudes were obtained from averages of at least five repetitions per synapse and stimulus frequency. D–F, Short-term plasticity in Cplx1−/− synapses is largely restored to wt-like pattern by raising external [Ca2+]. D, Representative 100 Hz eEPSC trains recorded in a Cplx1−/− synapse in normal (D1; 2 mm), as well as elevated (D2; 4 mm) external [Ca2+]. Changes in amplitude of EPSC1 after switching from 2 to 4 mm external [Ca2+] are plotted for nine Cplx1−/− synapses in the inset. On average, initial EPSC peak amplitudes increased approximately fivefold. E, F, Summary data obtained from 9 Cplx1−/− synapses showing average EPSC train amplitudes before (E) and after (F) normalizing to EPSC1 for recordings in normal (2 mm; light red) and elevated (4 mm; dark red) external [Ca2+].
To obtain a mean vesicular release probability (pr), we estimated the quantal content (m) of single AP-evoked EPSCs in wt and Cplx1−/− synapses. Pr can then be obtained by dividing this quantity by the total number of release-ready SVs. Because fast, synchronous AP-evoked release predominantly recruits SVs from the fast-releasing SV pool (∼50% of the total RRP) (Sakaba and Neher, 2001a; Sakaba, 2006), we divided our estimates for m by 50% of the total RRP (as estimated by the average ΔCm in response to a 50 ms depolarization, see above). Consistent with the reduction of eEPSC amplitudes, pr was ∼2.5-fold reduced in Cplx1−/− mice, with an averaged value of pr = 3.5 ± 0.4% compared with pr = 9.3 ± 0.6% in wt mice. These pr values can be regarded as a lower estimate because deriving m from eEPSC amplitudes does not take into account the temporal dispersion of release but rather assumes that quanta are released in perfect synchrony. Neglecting any late quanta consequently underestimates m (Borst and Sakmann, 1996). Using the correction that was derived in a previous study (Taschenberger et al., 2005), we therefore multiplied the amplitude ratio eEPSC/mEPSC by a factor of ∼1.4 to estimate m (Table 1).
To test whether eEPSCs and synaptic plasticity of Cplx1−/− calyx synapses can be rescued to a wt-like pattern (Reim et al., 2001), we raised pr by increasing the external Ca2+ concentration ([Ca2+]e) during the recordings. As summarized in Figure 6D–F, eEPSC amplitudes were strongly augmented from an average value of −1.3 ± 0.2 nA to −5.1 ± 1.0 nA (n = 9, p < 0.01) and synaptic facilitation in Cplx1−/− calyx synapses was converted to wt-like synaptic depression when we compared AP-evoked 100 Hz eEPSCs trains in normal external Ca2+ (2 mm) with those recorded after elevating [Ca2+]e to 4 mm.
Excessive asynchronous release causes aberrant AP firing in Cplx1−/− calyx synapses
Following high-frequency EPSC trains, we observed excessive asynchronous release in mature Cplx1−/− calyx synapses (P16–P21; Fig. 7A), that was absent from wt synapses of the same age, and was also never observed in younger homozygous Cplx1−/− (P8–P10), heterozygous Cplx1+/− or wt calyx synapses. The delayed release persisted for ≥500 ms after the end of the eEPSC trains (Fig. 7A). Although observed at a variety of synapses (Goda and Stevens, 1994; Vincent and Marty, 1996; Lu and Trussell, 2000; Otsu et al., 2004; Hefft and Jonas, 2005), asynchronous release is generally scarce at the calyx of Held (Erazo-Fischer et al., 2007; Scheuss et al., 2007), consistent with its role as a fast, faithful, and temporally precise relay. To estimate the total number of asynchronously released quanta after stimulus trains in Cplx1−/− synapses, we integrated the postsynaptic current after offset correction by subtracting a single or double exponential fit to the baseline. The baseline current, possibly generated by residual glutamate in the synaptic cleft, was determined by dividing the current into time segments of 10 ms duration, determining the minimum absolute current in such segments, and interconnecting these local minima. Cumulative charges versus time are plotted in Figure 7B. Current integrals were converted to SV numbers by division by the average mEPSC charge (Table 1). We estimated total numbers of 305, 492, and 562 asynchronously released SVs after 100, 200, and 300 Hz trains, respectively.
Figure 7.
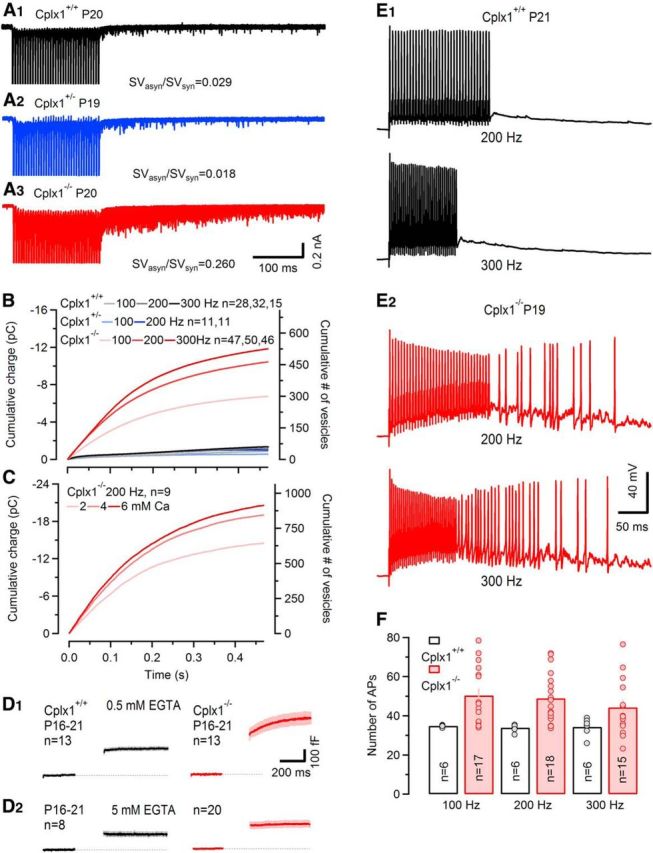
Strongly enhanced asynchronous release in P16–P21 Cplx1−/− synapses promotes aberrant postsynaptic AP firing. A, Ten consecutive traces of 200 Hz trains consisting of 35 EPSCs evoked by afferent-fiber stimulation and recorded in a P20 Cplx1+/+ (black), a P19 Cplx1+/− (blue), and a P20 Cplx1−/− (red) calyx synapse are shown superimposed. Amplitudes of AP-evoked synchronous EPSCs are truncated. Note the strongly enhanced asynchronous release during the first 500 ms following the stimulus trains in the Cplx1−/− synapse. B, Average cumulative charge of asynchronously released quanta plotted versus time after the end of stimulation for different stimulus frequencies (100, 200, and 300 Hz) for Cplx1+/+ (gray and black), heterozygous Cplx1+/− (blue), and Cplx1−/− (red) synapses. Cumulative charge of asynchronous release was obtained by integration after subtracting an exponential fit to the baseline to take into account a small current component activated by residual glutamate. C, Average cumulative charge of asynchronous release after 200 Hz trains plotted versus time for recordings in normal (2 mm) and elevated (4 and 6 mm) external [Ca2+]. Elevating external [Ca2+] further augmented asynchronous release in Cplx1−/− synapses. D, SV exocytosis after stimulus trains assayed by presynaptic membrane capacitance measurements. Stimulus trains consisted of 35 steps (1 ms from Vh = −80 to 0 mV) at a frequency of 200 Hz. Average ΔCm traces are shown. Number of terminals tested as indicated. D1, In the presence of 0.5 mm EGTA, Cplx1−/− synapses (red) showed ongoing delayed SV fusion after train stimulation. Delayed SV fusion was nearly undetectable in Cplx1+/+ terminals (black). D2, With 5 mm EGTA in the pipette solution, delayed release in Cplx1−/− mice was suppressed. E, 200 Hz (top) and 300 Hz (bottom) postsynaptic AP trains elicited by afferent fiber stimulation and recorded in a Cplx1+/+ (E1) and a Cplx1−/− (E2) MNTB PN in current-clamp mode. Whereas wt synapses generally followed presynaptic stimulation faithfully, Cplx1−/− neurons generated numerous aberrant spikes during and/or following the stimulus trains. F, Average numbers of APs elicited by high-frequency stimulus trains (100, 200, and 300 Hz, 35 stimuli) obtained from wt (black) and Cplx1−/− (red) synapses. Number of cells recorded as indicated. Average numbers of APs in Cplx1+/+ MNTB PNs were 35, 34, and 34 for 100, 200, and 300 Hz, respectively, whereas Cplx1−/− PNs fired on average >44 postsynaptic APs for any of these frequencies.
For comparison, we calculated the ratio of total asynchronous release during the first 500 ms following 200 Hz stimulation versus total synchronous release during the stimulus train (SVasyn/SVsyn). The total synchronous release was estimated by dividing cumulative eEPSC amplitudes by the average mEPSC amplitude obtained for the same synapse, and multiplying this value with 1.4 to correct for temporal dispersion of release (see above; Taschenberger et al., 2005).
The average ratio SVasyn/SVsyn for 200 Hz stimulation was dramatically higher in Cplx1−/− compared with wt synapses (26.0 ± 1.0%, n = 50 vs 2.9 ± 0.4%, n = 32, p < 0.001) or Cplx1+/− synapses (1.8 ± 0.5%, n = 11; Fig. 7A). As expected, asynchronous release increased with higher stimulus frequency, most likely due to the increase in residual [Ca2+]i (Fig. 7B). In Cplx1−/− synapses, the rates of asynchronous release measured immediately after high-frequency trains were ≥12 times higher compared with wt synapses for each frequency tested (Table 1). Nevertheless, during the final phase of 200 Hz stimulus trains, the average rates of synchronous release (number of synchronously released SVs divided by stimulus interval; 4423 ± 328 SV/s, n = 40, Cplx1−/−; 6095 ± 535 SV/s, n = 28, Cplx1+/+) were still significantly higher than the average rates of asynchronous release measured shortly after stimulation (Table 1). Thus, even in Cplx1−/− calyx synapses only a minority of SVs was released asynchronously during presynaptic AP firing.
Pronounced delayed and asynchronous SV fusion was also observed in presynaptic ΔCm measurements after repetitive stimulation (Fig. 7D). Presynaptic terminals were stimulated using trains of 35 1 ms steps to 0 mV (from Vh = −80 mV) at 200 Hz. In P16–P21 Cplx1−/− calyces, there was a continuous increase in membrane capacitance even after cessation of stimulation in addition to the ΔCm jump seen immediately after the stimulus train, indicating ongoing asynchronous release. Such a delayed release component was absent from presynaptic ΔCm recordings in wt mice of the same age (Fig. 7D1). Asynchronous release is thought to require accumulation of residual Ca2+ after intense stimulation (Katz and Miledi, 1968; Barrett and Stevens, 1972). Ca2+ chelators, such as EGTA preferentially inhibit asynchronous release during high-frequency action-potential trains but are less effective in suppressing synchronous release (Otsu et al., 2004). To determine whether asynchronous release in Cplx1−/− mice is triggered by elevated intracellular Ca2+, we included a high concentration of the Ca2+ buffer EGTA (5 mm) into the presynaptic patch pipette to chelate residual Ca2+ in the terminals. Under such conditions, no delayed asynchronous SV fusion was observed in Cplx1−/− calyces (Fig. 7D2).
As a fast and reliable relay synapse, the calyx of Held needs to faithfully transmit presynaptic activity over a wide range of input frequencies to provide well timed inhibition to other brainstem nuclei (for review, see Borst and Soria van Hoeve, 2012). High levels of asynchronous release after high-frequency trains in Cplx1−/− synapses may impede faithful and temporarily precise transmission. We therefore studied the impact of enhanced asynchronous release during physiologically relevant stimulus trains on postsynaptic AP generation in MNTB PNs (Fig. 7E,F). Under current-clamp conditions, stimulation of afferent fibers with 100–300 Hz trains consisting of 35 stimuli reliably elicited an average number of ∼35 spikes in MNTB PNs of P16–P21 wt mice. In contrast, numerous aberrant APs were observed during and following such stimulus trains in all recordings from PNs of Cplx1−/− MNTB (Fig. 7E). The average number of APs generated in response to 35 stimuli in Cplx1−/− mice varied depending on stimulation frequency (100 Hz, 50 ± 4, n = 17; 200 Hz, 49 ± 3, n = 18; 300 Hz, 44 ± 4, n = 15; Fig. 7F). These observations indicate that Cplx1 expression is essential for faithful and precise high-frequency transmission at the calyx of Held synapse.
Excessive asynchronous release was a robust phenomenon seen in all Cplx1−/− calyx synapses regardless of their initial eEPSC amplitudes. A scatter plot of total asynchronous release after 200 Hz trains versus initial amplitudes of the eEPSCs trains shows that peak amplitudes of AP-evoked eEPSC of wt and Cplx1−/− synapses overlapped to a large extend (P16–P21; Fig. 8A). However, all Cplx1−/− synapses that had initial eEPSC amplitudes within the range of wt synapses showed clearly higher values of cumulative asynchronous release. The latter parameter was above −5 pC for most Cplx1−/− synapses, although it rarely exceeded that number in wt synapses. It thus appears that the consequences of genetic ablation of Cplx1 for synaptic strength on the one hand and for asynchronous release on the other hand are not strongly correlated in individual Cplx1−/− synapses (Fig. 8A). Figure 8B exemplifies two recordings in Cplx1−/− calyx synapses showing either strongly reduced or wt-like synaptic strength. Despite their very different initial eEPSC amplitudes, both synapses showed similarly enhanced asynchronous release following the 200 Hz EPSC trains.
Figure 8.
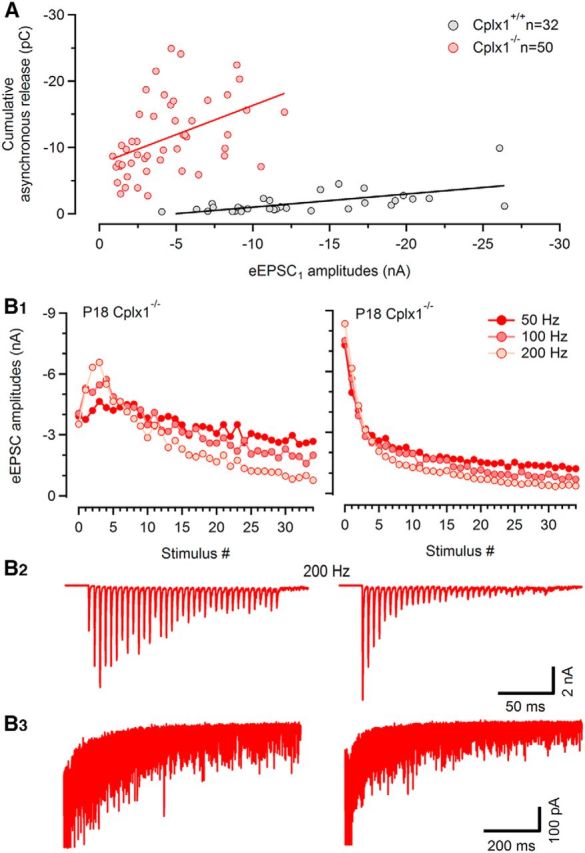
Effects of Cplx1 ablation on initial eEPSC amplitudes and amount of asynchronous release after train stimuli are weakly correlated in individual Cplx1−/− synapses. A, Scatter plot of cumulative asynchronous release following 200 Hz stimulus trains versus initial EPSC amplitude for 32 Cplx1+/+ (black) and 50 Cplx1−/− (red) synapses. Note the large overlap of the two datasets with respect to initial EPSC amplitudes. In contrast, significantly higher rates of asynchronous release were consistently observed after train stimulation in Cplx1−/− synapses, even if initial, synchronous EPSCs were wt-like. B, Examples for two P18 Cplx1−/− synapses showing either low (left column) or much higher, wt-like synaptic strength (right column) to exemplify consistency among Cplx1−/− synapses regarding augmented asynchronous release despite their heterogeneous EPSC amplitudes and short-term plasticity pattern. B1, Average EPSC amplitudes obtained from three repetitions for 50, 100, and 200 Hz trains were plotted as a function of stimulus number. B2, Representative 200 Hz EPSC train sample traces for the same cells as shown in (B1). B3, Delayed release following 200 Hz EPSC trains shown expanded and at a faster time scale. Four consecutive traces are shown superimposed.
Cplx1 stabilizes newly recruited SVs to prevent their premature fusion
As shown above, synchronous and asynchronous release components were differentially affected by Cplx1 deletion: asynchronous release was strongly augmented in all Cplx1−/− calyx synapses, whereas synchronous eEPSC amplitudes were severely reduced in a majority of them. We therefore considered the possibility that such divergent effects are observed because distinct SV pools generate asynchronous and synchronous release components. If, on the other hand, both asynchronously and synchronously released SVs originate from the same pool, competition between both modes of transmitter release may be observed (Otsu et al., 2004). During high-frequency trains, the amount of steady-state release is limited by the SV replenishment rate, which is significantly accelerated in a Ca2+-dependent manner at the end of such stimulus trains and shortly thereafter (Wang and Kaczmarek, 1998; Sakaba and Neher, 2001b; Lipstein et al., 2013). After cessation of stimulation, the global [Ca2+]i quickly returns to basal levels (Müller et al., 2007), such that ∼0.5 s later, the SV replenishment rate is expected to correspond to the much lower, basal rate of SV replenishment (Hosoi et al., 2007). Assuming a replenishment rate constant of ∼¼ s−1 (von Gersdorff et al., 1997), and a total number of sites contributing to AP-evoked release of approximately one-half of the RRP as estimated by ΔCm measurements (0.5 × 4600 = 2300; Fig. 4), we obtain a basal rate of SV replenishment of ∼575 SV/s for a fully depleted RRP. Because immediately after 200 Hz trains the average rate of asynchronous release exceeds the replenishment rate (Table 1), recovery from synaptic depression should be delayed in Cplx1−/− synapses if asynchronously and synchronously released SVs originate from the same pool. Figure 9A,B shows that this is indeed the case. Using 200 Hz trains consisting of 25 stimuli, recovery from depression was monitored by eliciting test eEPSCs at different post-train recovery intervals (250 ms to 16 s) in P14–P21 Cplx1−/− and wt synapses. The 250 ms time interval was chosen as the shortest interval because at that time remaining Ca2+-dependent facilitation of presynaptic ICa(V) and/or postsynaptic AMPA-receptor desensitization are expected to be small. To calculate the recovery ratio, we normalized the test eEPSC amplitudes to that of the first eEPSC elicited by the conditioning trains. Figure 9B plots the recovery ratio versus time. For Cplx1−/− synapses, the curve exhibits a drop in the early phase of the recovery time course with a minimum at 1 s recovery interval, indicating that indeed more SVs were consumed than replenished. For intervals ≥2 s, the recovery time course appeared to be similar in Cplx1−/− and wt synapses. We compared the time course of recovery for larger intervals (from 2 to 16 s, excluding the initial recovery from 0.25 to 1 s) by fitting single exponentials to the curves. The average time constant for Cplx1−/− synapses (τ = 4.8 ± 0.3 s, n = 12) was similar to that in wt synapses (τ = 4.0 ± 0.6 s, n = 17). Together, these observations indicate that loss of Cplx1 does not affect the replenishment rate, but excessive asynchronous release in Cplx1−/− calyx synapses delays pool replenishment after depletion, indicating that asynchronously and synchronously released SVs originate from the same reservoir.
Figure 9.
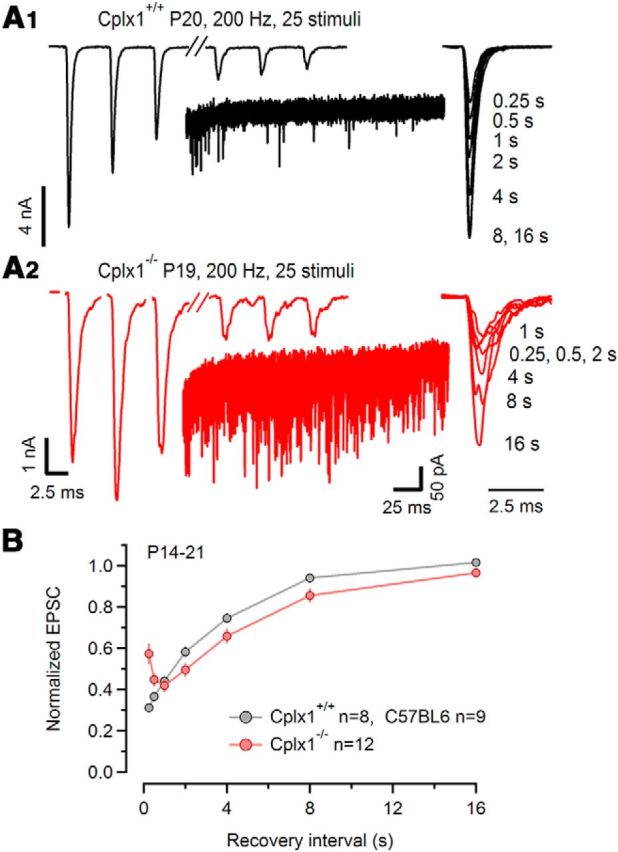
Asynchronously released SVs are recruited from the same pool that is contributing to synchronous EPSCs. Delayed recovery of synchronous EPSCs from depression during stimulus trains in Cplx1−/− calyx synapses. A, Sample traces of conditioning 200 Hz EPSC trains evoked by afferent-fiber stimulation (25 stimuli) that largely depleted the readily releasable SV pool (left) recorded in a P20 Cplx1+/+ synapse (A1) and a P19 Cplx1−/− synapse (A2). Only the first three and last three EPSCs of the trains are shown for clarity. Recovery of the RRP was tested by eliciting single EPSCs after the indicated recovery intervals (250 ms to 16 s; right). Insets, Comparison of asynchronous release during the first 240 ms immediately following the stimulus trains. Four sweeps are shown superimposed. B, Average time course of recovery from depression induced by 200 Hz trains. Peak amplitudes of the test EPSC were normalized by division by that of the first EPSCs of the conditioning trains. Note that maximum depression of EPSC amplitudes is observed after a recovery interval of 1 s in Cplx1−/− synapses, because strong asynchronous release after conditioning trains further augments synaptic depression and delays recovery of EPSC amplitudes in these synapses. Pooled data from 17 Cplx1+/+ and 12 Cplx1−/− synapses.
Because (1) the rate of spontaneous SV fusion was reduced in Cplx1−/− calyces, (2) enhanced asynchronous release was seen in Cplx1−/− calyces after stimuli leading to strong SV consumption, and because (3) augmented asynchronous release was long-lasting in Cplx1−/− calyces, possibly outlasting the residual presynaptic [Ca2+]i, we hypothesized that the asynchronously released quanta predominantly represent uncoordinated release of SVs newly recruited to the RRP and that Cplx1 is required to stabilize these SVs and prevent their premature fusion (Dhara et al., 2014). To test this hypothesis, we quantified changes in spontaneous SV fusion after eliciting a single presynaptic AP (Fig. 10). In wt calyx synapses, we noticed a small but sizeable drop of the mEPSC rate (from 3.69 ± 0.5 to 3.31 ± 0.59 Hz, n = 11) after a single presynaptic AP consistent with a small reduction of the RRP (Fig. 10A). In contrast, the mEPSC release rate remained elevated for several hundreds of milliseconds in Cplx1−/− calyx synapses. On average, the mEPSC frequency increased from 1.06 ± 0.14 to 2.55 ± 0.31 Hz after stimulating with a single AP in Cplx1−/− calyx synapses (n = 15; Fig. 10A). To study onset and decay kinetics of the delayed asynchronous release in more detail, we repeated these experiments but used either elevated [Ca2+]e (Fig. 10B) or delivered a short burst of three APs instead of a single AP (Fig. 10C). After such stimuli, the time course of changes in mEPSC rate could be resolved more clearly. On average, the delayed increase in mEPSC frequency peaked at ∼230–250 ms after stimulation. Its time course could be well approximated with a double-exponential function having a τrise of ∼150–190 ms and τdecay of 420–430 ms. These results indicate that newly recruited SVs remain unstable and are more prone to being released prematurely in calyx synapses of Cplx1−/− mice.
Figure 10.
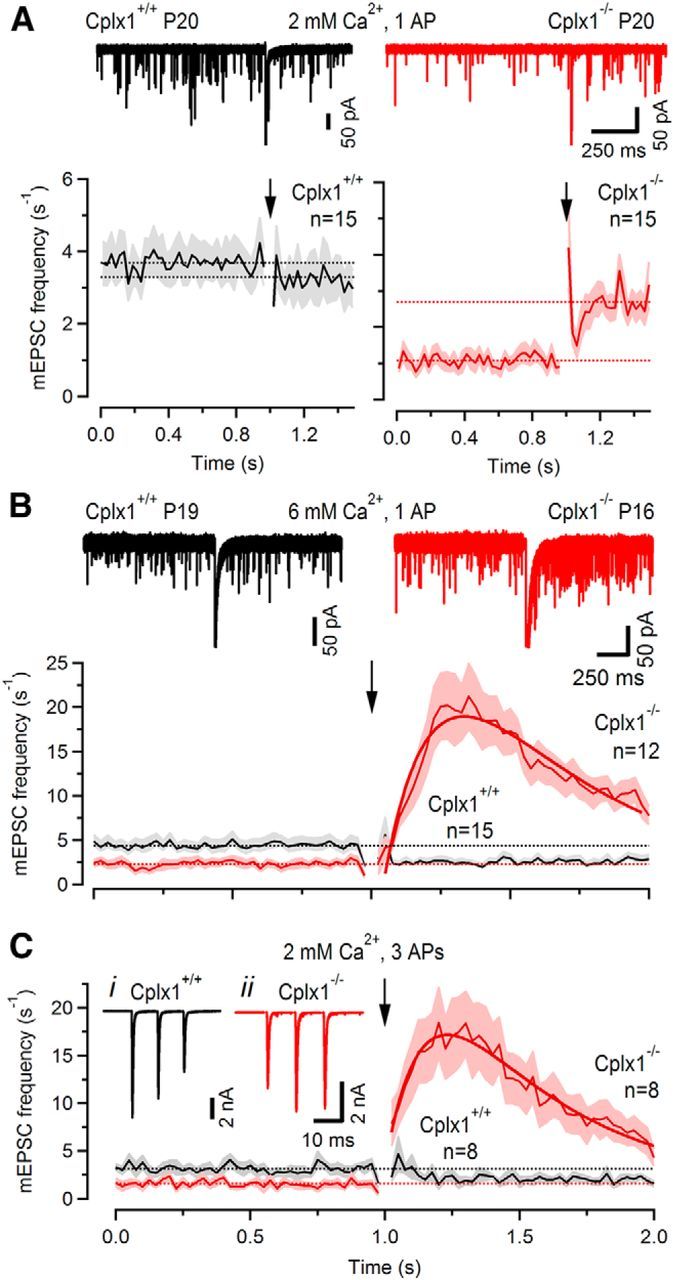
Asynchronously released quanta represent predominantly newly primed SVs. A, Changes in mEPSCs frequency observed after eliciting a single AP-evoked eEPSCs in Cplx1+/+ (black traces) Cplx1−/− (red traces) synapses. Top, Twenty consecutive traces are plotted superimposed. eEPSCs are peak truncated. mEPSCs frequencies were monitored 1 s before and 0.5 s after stimulation. Arrows mark the time point of stimulation. Solid lines represent average mEPSC frequencies calculated for 25 ms bins and the light gray and light red areas represent ±SEM (bottom). The mEPSC frequency was slightly reduced after stimulation in Cplx1+/+ (black) but strongly enhanced in Cplx1−/− (red) mice. B, Similar experiment as in A but with external Ca2+ raised from 2 to 6 mm to more clearly resolve the onset and decay of delayed asynchronous release. Traces from two representative experiments are shown in the top, average data are shown in the bottom. The smooth, solid red curve in B (bottom) represents a double-exponential fit to the asynchronous release rate yielding τrise ∼190 ms and τdecay ∼420 ms. C, Similar experiment as in B but applying a short train of three APs instead of a single AP. Rise and decay time constants of the asynchronous release rate were ∼150 ms and ∼430 ms, respectively. Insets in C illustrate AP-evoked synchronous eEPSCs from two representative experiments. Note the delayed onset of the increase in mEPSC frequency in B, C, which peaked at >200 ms after stimulation.
Expression of Cplx2 protects immature Cplx1−/− calyx of Held synapses from functional deficits
The absence of Cplx1 from calyx synapses led to functional deficits only relatively late in development (≥P14), whereas synaptic transmission in young synapses (≤P12) appeared normal. Because several Cplx isoforms can be coexpressed in individual neurons (Reim et al., 2001), other Cplxs may compensate for the loss of Cplx1 early in development when the mature pattern of protein expression is not yet fully established. To test such a possibility, we studied changes in Cplx2 expression during postnatal development of the MNTB by Western blot analysis of wt tissue and by anti-Cplx1/2 immunofluorescence staining of Cplx1−/− MNTB in which the remaining immunofluorescence represent anti-Cplx2 immunoreactivity (Fig. 11). Because our antibody and all other antibodies available detect both, Cplx1 and Cplx2, a specific Cplx2 staining in wt MNTB is impossible. Figure 11A,B illustrates that the Cplx2/Cplx1 expression ratio in the MNTB is reduced from P8 to P16 and remains low thereafter. The lack of somatic anti-Cplx2 immunoreactivity in MNTB PNs of P7 and P21 Cplx1−/− mice indicates that these neurons solely express Cplx1 (Fig. 11C, compare Fig. 1C). Although absent from PN somata, anti-Cplx2 staining that shows the typical morphology of calyx terminals at this developmental stage was abundant in the P7 Cplx1−/− MNTB in many continuous regions surrounding PNs (Fig. 11C, top row). Colocalization with anti-Vglut1 staining verified expression of Cplx2 in calyx terminals at this developmental stage. In contrast, anti-Cplx2 immunoreactivity was much weaker in the MNTB of Cplx1−/− mice at P21, when it was restricted to a few small patches and did not overlap with anti-Vglut1 staining (Fig. 11C, bottom row). Possibly, this remaining staining reflects Cplx2 expression in small inhibitory, glycinergic boutons at this developmental stage. Together, these results indicate that immature calyx terminals coexpress Cplx1 and Cplx2, and the developmental loss of Cplx2 from calyx terminals accounts, at least in part, for more severe functional deficits of the Cplx1 deletion in older (≥P14) calyx synapses.
Figure 11.
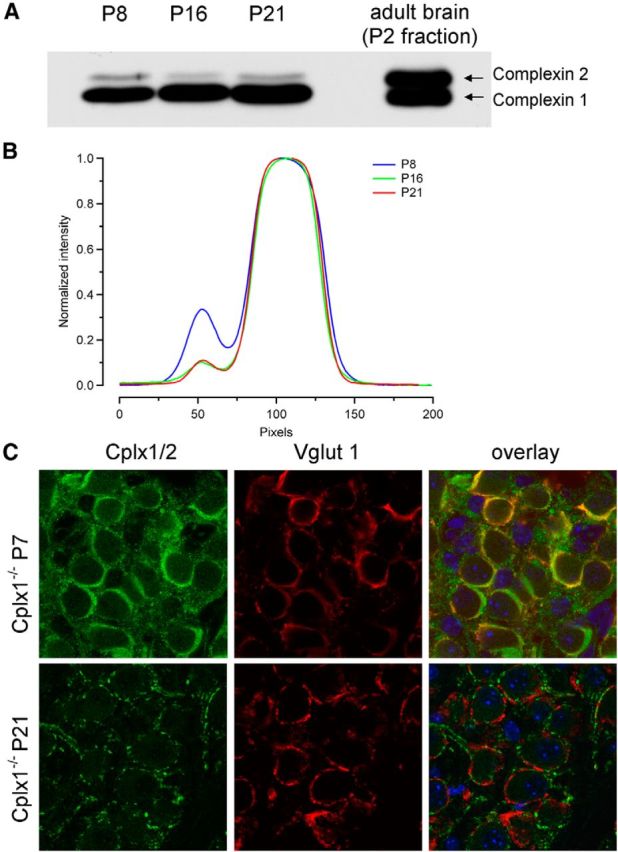
Developmental downregulation of Cplx2 expression in calyx terminals. A, Westernblot analysis of developmental changes in Cplx1/2 expression. Brain homogenates obtained from dissected MNTB regions of P8, P16, and P21 wt mice (10 μg protein per lane) were analyzed by SDS-PAGE and immunoblotting using an anti-Cplx1/2 antibody. Positions of Cplx1 and Cplx2 protein are indicated. B, Intensity profiles obtained from the blot shown in A. Note the decrease in relative contribution of the Cplx2 isoform to the total complexin expression. Different exposure times were used for the analysis to account for the changes in absolute Cplx expression levels from P8 to P21. C, Immunofluorescence images representing confocal sections of MNTB regions of Cplx1−/− mice costained with an anti-Cplx1/2 (green, left) and anti-Vglut1 (red, middle) antibody at P7 (top row) and P21 (bottom row). Right columns show the corresponding overlays. Note the overlap of the Cplx2 fluorescence with that of Vglut1 at P7 (orange) but not at P21, indicating undetectable expression of Cplx2 in calyx terminals of the latter age. The Cplx2-positive puncta that were not costained with Vglut1 presumably represent Cplx2-expressing inhibitory synapses.
A hypothetical model of Cplx functions at the calyx of Held synapse
Based on the experimentally observed functional deficits in Cplx1−/− mice, we propose the following mechanistic model for the functions of Cplx1 at the calyx synapse (Fig. 12): Following transmitter release, the RRP is replenished by newly recruited SVs, which dock in the proximity of VGCCs. After docking, SVs undergo a two-step priming process. Before transition into the stable ‘fully primed’ state, they first enter an unstable “semiprimed” state from which premature fusion events are likely to occur at low [Ca2+]i (Figs. 7, 8). This intrinsically unstable semiprimed state may require the presence of Cplx1 to “clamp” SVs to prevent premature fusion (Dhara et al., 2014). Alternatively, Cplx1 may serve as a “specificity factor” that promotes the association of SVs and SNARE complexes with the “correct” Ca2+ sensor (Neher, 2010), i.e., Syt2 at the calyx of Held. SV may be in the semiprimed state as long as they, or their SNARE complexes, are (transiently) bound to a Ca2+ sensor different from Syt2 (Walter et al., 2011). After transition into the fully primed state, SVs fuse primarily after AP-evoked Ca2+ increase and premature fusion events are unlikely to occur. Because their RRP is intact and spontaneous fusion rates are reduced in Cplx1−/− calyx synapses, we postulate that the majority of SVs eventually reside in the fully primed state at resting conditions, even in the absence of Cplxs. After presynaptic AP firing, the RRP is partially depleted and semiprimed vesicles transiently accumulate that may prematurely fuse in the absence of Cplx1.
Figure 12.
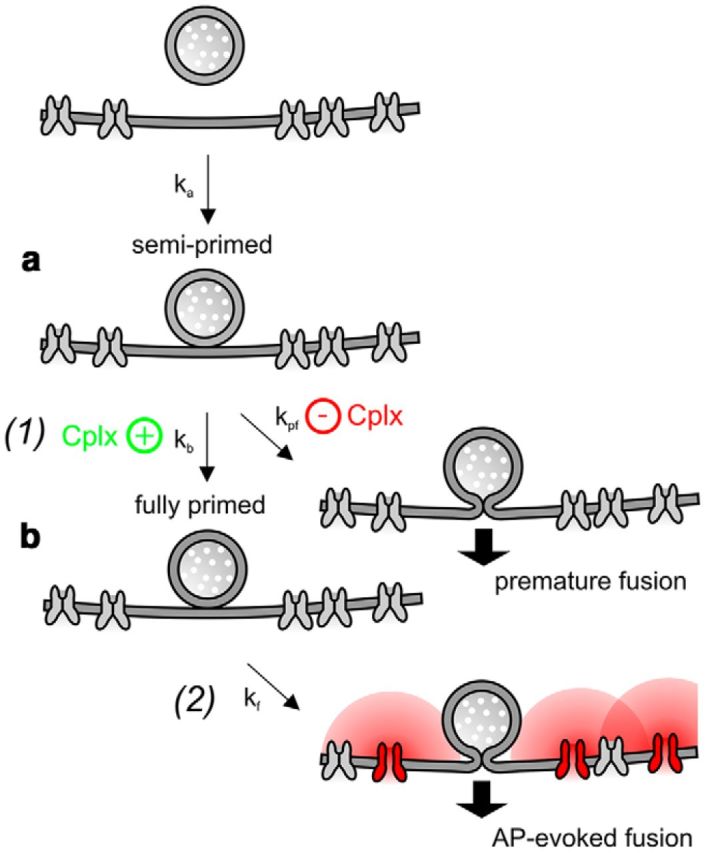
Hypothetical model of Cplx function at the calyx of Held synapse. After pool depletion by AP-evoked SV release, newly recruited SVs need to dock and prime near VGCCs. We postulated that priming is a two-step process. Initially, a semiprimed state (a) is achieved from which SVs can easily undergo premature fusion at low or even resting [Ca2+]i. This semiprimed SV state may represent an intrinsically unstable state or correspond to SVs that are initially bound to a Ca2+ sensor other than Syt2. After transition into the fully primed state (b), SVs fuse primarily after AP-evoked Ca2+ increase and premature fusion events are unlikely to occur. We hypothesize that the presence of Cplx1 in calyx terminals (right column) hast two consequences: (1) it accelerates the transition into the fully primed state making the semiprimed a very short-lived state and/or prevents premature fusion of semiprimed vesicles. (2) Cplx1 increases the secretory rate of fully primed vesicles and thereby enhances spontaneous as well as evoked release.
Within the scope of this model, the presence of Cplx1 in wt calyx terminals (Fig. 12) has thus two consequences: (1) Cplx1 accelerates the transition into the fully primed state, making the semiprimed state a very short-lived transition state, and/or prevents premature fusion of semiprimed vesicles. (2) Cplx1 increases the secretory rate and thereby enhances spontaneous and AP-evoked synchronous release.
Discussion
We studied the functional consequences of Cplx1 removal from calyx of Held synapses of juvenile mice. Our results can be summarized by the following eight key observations: (1) Mature calyces solely express Cplx1, whereas younger ones also coexpress a smaller amount of Cplx2. Cplx1 expression is not limited to presynaptic terminals but also abundant in postsynaptic MNTB PNs, which do not express Cplx2. (2) Genetic deletion of Cplx1 neither affects developmental profiles nor the number of synaptic GluRs. Quantal size, as well as the developmental pruning of NMDARs from postsynaptic densities, remain unaltered in Cplx1−/− mice. (3) Synaptic strength is strongly reduced in the absence of Cplx1. This functional defect appears relatively late during postnatal maturation (≥P16). (4) The presynaptic AP waveform, presynaptic Ca2+ influx, RRP size, and quantal size are unaltered in Cplx1−/− calyces, indicated that the reduced synaptic strength upon Cplx1 deletion is mediated by a decrease in release probability, which in turn is caused by mechanisms downstream of presynaptic Ca2+ influx, such as a changed Ca2+ sensitivity of the release machinery and/or altered spatial coupling between docked/primed SVs and VGCCs. (5) Ca2+-triggered SV fusion in Cplx1−/− calyces shows higher sensitivity to EGTA, indicating a defect in the developmental tightening of the spatial coupling between VGCCs and docked/primed SVs in mature Cplx1−/− terminals. (6) Excessive asynchronous release follows presynaptic AP burst firing in Cplx1−/− synapses. EPSPs generated by asynchronously released quanta trigger aberrant APs in MNTB PNs during and after stimulus trains, thus greatly reducing the temporal fidelity of transmission. (7) Recovery of synchronous eEPSCs after pool depletion is delayed by asynchronously released quanta in Cplx1−/− synapses, indicating that asynchronous and synchronous release components compete for the same pool of readily releasable SVs. (8) Even though the average rate of spontaneous release is lower in resting Cplx1−/− synapses, the mEPSC frequency is strongly and long-lastingly enhanced even after single APs or short AP bursts, indicating that newly primed SVs remain unstable and are prone to being released prematurely in the absence of Cplx1.
Collectively, our data indicate that Cplx1 serves three functions at the calyx of Held synapse. (1) Cplx1 acts as a positive modulator of SV exocytosis and enhances AP-evoked synchronous release. (2) Cplx1 stabilizes newly primed SVs that replenish the RRP after SV consumption. (3) Cplx1 function is required for the developmental tightening of the spatial coupling between SVs and VGCCs that usually occurs during calyx maturation and seems to be abolished in the absence of Cplx1. All of these functions strongly promote synchronous over asynchronous SV exocytosis.
An unresolved role of postsynaptically expressed Cplx1 at the calyx of Held
Although somatic anti-Cplx1/2 immunofluorescence has been reported before (Yamada et al., 1999; Reim et al., 2005; Strenzke et al., 2009) and Cplx is present in dendritic spines and shafts (McMahon et al., 1995; Yamada et al., 1999), its role in basal postsynaptic function remains obscure. Ahmad et al. (2012) reported a lack of postsynaptic effects following Cplx1 knock-down in hippocampal CA1 pyramidal cells where neither the AMPA/NMDA receptor ratio nor the composition of AMPARs or NMDARs were changed. We found strong anti-Cplx1/2 immunostaining in MNTB PNs which was abolished in Cplx1−/− mice. However, neither quantal size nor mEPSC kinetics were altered in the absence of Cplx1, indicating similar subunit composition of synaptic AMPARs in Cplx1−/− and wt synapses. In addition, we found the ratio of NMDAR-mediated to AMPAR-mediated EPSC components unaltered in young Cplx1−/− synapses (data not shown), and the developmental pruning of NMDARs from postsynaptic densities occurred similarly in mature Cplx1−/− and wt calyx synapses, so that NMDARs contributed ≤3% to the eEPSC amplitude in both genotypes. Thus, a lack of Cplx1 does not seem to affect the developmental expression pattern of postsynaptic GluRs in MNTB PNs.
Multiple roles of Cplx1 in regulating SV exocytosis
Depending on the experimental conditions and species tested, Cplxs were found to facilitate or inhibit SNARE-complex-mediated SV fusion. In most Cplx-deficient synapses tested so far, fast synchronous Ca2+-triggered release is decreased, indicating that Cplxs are positive regulators of SV fusion (Reim et al., 2001; Huntwork and Littleton, 2007; Xue et al., 2007; Yang et al., 2010a; Hobson et al., 2011; Martin et al., 2011). A number of studies further reported a decreased rate of spontaneous release in the absence of Cplxs (Reim et al., 2001; Xue et al., 2007, 2008; Strenzke et al., 2009; Lin et al., 2013). Other experimental results indicate an inhibitory clamp-like function of Cplxs, because: (1) genetic deletion of Cplx increases spontaneous release at Drosophila and Caenorhabditis elegans neuromuscular synapses (Huntwork and Littleton, 2007; Hobson et al., 2011; Martin et al., 2011), (2) Cplx knock-down causes increased spontaneous release rates in mouse cortical neurons (Maximov et al., 2009) and olfactory bulb neurons (Cao et al., 2013) in culture, and (3) Cplxs inhibit SNARE-mediated fusion in vitro (Giraudo et al., 2006).
At the calyx of Held synapses, synaptic strength and spontaneous release are reduced in Cplx1−/− mice, indicating that Cplx1 is a positive regulator of SV fusion at this synapse. On the other hand, asynchronous release was augmented in Cplx1−/− calyx synapses for several hundreds of milliseconds after single presynaptic APs and short AP bursts, and strongly enhanced after high-frequency stimulus trains. Both of these observations are consistent with an additional inhibitory role of Cplx1, albeit not as a fusion clamp of spontaneous release, i.e., at resting [Ca2+]i, because the increased fusion events uncovered by the Cplx1 loss seem to be triggered by elevations of [Ca2+]i (Fig. 7C,D).
Cplxs are not the only presynaptic proteins assigned with positive and negative regulatory functions. Synaptotagmin (Syt) is indispensable for synchronous SV fusion but might additionally be required to suppress asynchronous and spontaneous release (Maximov and Südhof, 2005; Pang et al., 2006; Kochubey and Schneggenburger, 2011). Tang et al. (2006) suggested that Cplxs bind to partially assembled SNARE complexes during priming, forcing completion of SNARE-complex assembly from a loose to a tightly assembled state that serves as a substrate for Ca2+-bound Syt. If Cplx acts as a fusion clamp that is later removed by Syt, then the loss of Syt should arrest SVs in a clamped state. The opposite was however observed in Syt-deficient synapses, which show dramatically enhanced spontaneous release (Pang et al., 2006; Kochubey and Schneggenburger, 2011), indicating that Cplx action is not simply reverted by Syt.
Cplx1 loss causes a defect in the developmental tightening of the coupling between VGCCs and docked SVs
To elucidate the mechanisms accounting for the attenuated synchronous release in Cplx1−/− mice, we measured calyceal AP waveform, presynaptic Ca2+ influx, RRP size and quantal size, and found all of these parameters unaltered compared with wt synapses. However, presynaptic capacitance measurements revealed a higher sensitivity to the slow Ca2+ chelator EGTA in Cplx1−/− terminals. This result is consistent with a less tight coupling between docked/primed SVs and VGCCs. Rapid AP-triggered neurotransmitter release requires an exquisite active zone (AZ) organization allowing interactions between VGCCs and readily releasable SVs at the shortest possible distances. Properly positioning SVs to VGCCs becomes an important requirement especially in mature calyx terminals that are characterized by very brief APs (Young Jr and Neher, 2009). The distance between VGCCs and docked SVs determines the level of the local [Ca2+]i sensed by the release machinery and thereby regulates release probability (for review, see Neher and Sakaba, 2008). The presynaptic Rab-3 interacting molecule (RIM) plays an important role in maintaining a high VGCCs density at calyx of Held synapses (Han et al., 2011). Furthermore, septin, a filamentous protein, regulates the distance between VGCCs and docked/primed SVs during maturation of the calyx terminal, and thus participates in the developmental switch from Ca2+ microdomain to nanodomain signaling (Yang et al., 2010b). A larger distance between docked SVs and VGCCs is expected to lower the peak release rates and broaden the release transients. Indeed, deconvolution of eEPSCs recorded in Cplx1−/− calyx synapses established a strongly reduced peak release rate and additionally a slightly slower rise and decay of the release time course. Although Cplxs are not known to directly interact with VGCCs, it is possible that they assist in the process of SV positioning via interaction with other AZ proteins. Thereby, the absence of Cplx1 and the consequent functional defects may indirectly affect the developmental maturation of calyx synapses.
Cplx1 expression in calyx terminals is required for high-fidelity transmission at calyx of Held synapses
MNTB PNs provide precisely timed inhibition to their target neurons. To preserve the timing of APs along the auditory pathway, transmission at calyx of Held synapses is optimized for minimum temporal jitter and maximum reliability (for review, see Borst and Soria van Hoeve, 2012). After genetic ablation of Cplx1 expression, which strongly shifts the balance from synchronous toward asynchronous release at calyx synapses, the temporal fidelity of transmission is severely compromised. Desynchronization of presynaptic and postsynaptic AP firing at the level of the MNTB may contribute to delayed peaks and decremented amplitudes of late components of auditory brainstem responses observed in young adult mice in vivo (Strenzke et al., 2009).
Footnotes
This work was supported by the Cluster of Excellence and DFG Research Center Nanoscale Microscopy and Molecular Physiology of the Brain (E.N., N.B., H.T.), the European Commission (EUROSPIN, FP7-HEALTH-F2-2009-241498; E.N., N.B.) and the Max Planck Society (E.N., N.B.). S.C. was a student of the Göttingen Graduate School for Neurosciences and Molecular Biosciences. We thank Kun-Han Lin and Noa Lipstein for valuable discussions and comments on the paper, F. Würriehausen for expert advice on programming, and I. Herfort and T. Hellmann for excellent technical assistance.
The authors declare no competing financial interests.
References
- Ahmad M, Polepalli JS, Goswami D, Yang X, Kaeser-Woo YJ, Südhof TC, Malenka RC. Postsynaptic complexin controls AMPA receptor exocytosis during LTP. Neuron. 2012;73:260–267. doi: 10.1016/j.neuron.2011.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett EF, Stevens CF. The kinetics of transmitter release at the frog neuromuscular junction. J Physiol. 1972;227:691–708. doi: 10.1113/jphysiol.1972.sp010054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borst JG, Sakmann B. Calcium influx and transmitter release in a fast CNS synapse. Nature. 1996;383:431–434. doi: 10.1038/383431a0. [DOI] [PubMed] [Google Scholar]
- Borst JG, Sakmann B. Effect of changes in action potential shape on calcium currents and transmitter release in a calyx-type synapse of the rat auditory brainstem. Philos Trans R Soc Lond B Biol Sci. 1999;354:347–355. doi: 10.1098/rstb.1999.0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borst JG, Soria van Hoeve J. The calyx of held synapse: from model synapse to auditory relay. Annu Rev Physiol. 2012;74:199–224. doi: 10.1146/annurev-physiol-020911-153236. [DOI] [PubMed] [Google Scholar]
- Cao P, Yang X, Südhof TC. Complexin activates exocytosis of distinct secretory vesicles controlled by different synaptotagmins. J Neurosci. 2013;33:1714–1727. doi: 10.1523/JNEUROSCI.4087-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Tomchick DR, Kovrigin E, Araç D, Machius M, Südhof TC, Rizo J. Three-dimensional structure of the complexin/SNARE complex. Neuron. 2002;33:397–409. doi: 10.1016/S0896-6273(02)00583-4. [DOI] [PubMed] [Google Scholar]
- Chen Z, Das B, Nakamura Y, DiGregorio DA, Young SM., Jr Ca2+ channel to synaptic vesicle distance accounts for the readily releasable pool kinetics at a functionally mature auditory synapse. J Neurosci. 2015;35:2083–2100. doi: 10.1523/JNEUROSCI.2753-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements JD, Bekkers JM. Detection of spontaneous synaptic events with an optimally scaled template. Biophys J. 1997;73:220–229. doi: 10.1016/S0006-3495(97)78062-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhara M, Yarzagaray A, Schwarz Y, Dutta S, Grabner C, Moghadam PK, Bost A, Schirra C, Rettig J, Reim K, Brose N, Mohrmann R, Bruns D. Complexin synchronizes primed vesicle exocytosis and regulates fusion pore dynamics. J Cell Biol. 2014;204:112–1140. doi: 10.1083/jcb.201311085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond JS, Jahr CE. Asynchronous release of synaptic vesicles determines the time course of the AMPA receptor-mediated EPSC. Neuron. 1995;15:1097–1107. doi: 10.1016/0896-6273(95)90098-5. [DOI] [PubMed] [Google Scholar]
- Erazo-Fischer E, Striessnig J, Taschenberger H. The role of physiological afferent nerve activity during in vivo maturation of the calyx of Held synapse. J Neurosci. 2007;27:1725–1737. doi: 10.1523/JNEUROSCI.4116-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedchyshyn MJ, Wang LY. Developmental transformation of the release modality at the calyx of held synapse. J Neurosci. 2005;25:4131–4140. doi: 10.1523/JNEUROSCI.0350-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman W, Morton AJ. Differential messenger RNA expression of complexins in mouse brain. Brain Res Bull. 2004;63:33–44. doi: 10.1016/j.brainresbull.2003.12.003. [DOI] [PubMed] [Google Scholar]
- Giraudo CG, Eng WS, Melia TJ, Rothman JE. A clamping mechanism involved in SNARE-dependent exocytosis. Science. 2006;313:676–680. doi: 10.1126/science.1129450. [DOI] [PubMed] [Google Scholar]
- Goda Y, Stevens CF. Two components of transmitter release at a central synapse. Proc Natl Acad Sci U S A. 1994;91:12942–12946. doi: 10.1073/pnas.91.26.12942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Kaeser PS, Südhof TC, Schneggenburger R. RIM determines Ca2+ channel density and vesicle docking at the presynaptic active zone. Neuron. 2011;69:304–316. doi: 10.1016/j.neuron.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison JM, Irving R. Ascending connections of the anterior ventral cochlear nucleus in the rat. J Comp Neurol. 1966;126:51–63. doi: 10.1002/cne.901260105. [DOI] [PubMed] [Google Scholar]
- He L, Wu XS, Mohan R, Wu LG. Two modes of fusion pore opening revealed by cell-attached recordings at a synapse. Nature. 2006;444:102–105. doi: 10.1038/nature05250. [DOI] [PubMed] [Google Scholar]
- He L, Xue L, Xu J, McNeil BD, Bai L, Melicoff E, Adachi R, Wu LG. Compound vesicle fusion increases quantal size and potentiates synaptic transmission. Nature. 2009;459:93–97. doi: 10.1038/nature07860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hefft S, Jonas P. Asynchronous GABA release generates long-lasting inhibition at a hippocampal interneuron-principal neuron synapse. Nat Neurosci. 2005;8:1319–1328. doi: 10.1038/nn1542. [DOI] [PubMed] [Google Scholar]
- Hobson RJ, Liu Q, Watanabe S, Jorgensen EM. Complexin maintains vesicles in the primed state in C. elegans. Curr Biol. 2011;21:106–113. doi: 10.1016/j.cub.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoi N, Sakaba T, Neher E. Quantitative analysis of calcium-dependent vesicle recruitment and its functional role at the calyx of Held synapse. J Neurosci. 2007;27:14286–14298. doi: 10.1523/JNEUROSCI.4122-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntwork S, Littleton JT. A complexin fusion clamp regulates spontaneous neurotransmitter release and synaptic growth. Nat Neurosci. 2007;10:1235–1237. doi: 10.1038/nn1980. [DOI] [PubMed] [Google Scholar]
- Isaacson JS, Walmsley B. Counting quanta: direct measurements of transmitter release at a central synapse. Neuron. 1995;15:875–884. doi: 10.1016/0896-6273(95)90178-7. [DOI] [PubMed] [Google Scholar]
- Ishizuka T, Saisu H, Suzuki T, Kirino Y, Abe T. Molecular cloning of synaphins/complexins, cytosolic proteins involved in transmitter release, in the electric organ of an electric ray (Narke japonica) Neurosci Lett. 1997;232:107–110. doi: 10.1016/S0304-3940(97)00586-7. [DOI] [PubMed] [Google Scholar]
- Iwasaki S, Takahashi T. Developmental regulation of transmitter release at the calyx of Held in rat auditory brainstem. J Physiol. 2001;534:861–871. doi: 10.1111/j.1469-7793.2001.00861.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas P, Major G, Sakmann B. Quantal components of unitary EPSCs at the mossy fibre synapse on CA3 pyramidal cells of rat hippocampus. J Physiol. 1993;472:615–663. doi: 10.1113/jphysiol.1993.sp019965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi I, Wang LY. Developmental profiles of glutamate receptors and synaptic transmission at a single synapse in the mouse auditory brainstem. J Physiol. 2002;540:861–873. doi: 10.1113/jphysiol.2001.013506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz B, Miledi R. The role of calcium in neuromuscular facilitation. J Physiol. 1968;195:481–492. doi: 10.1113/jphysiol.1968.sp008469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochubey O, Schneggenburger R. Synaptotagmin increases the dynamic range of synapses by driving Ca2+-evoked release and by clamping a near-linear remaining Ca2+ sensor. Neuron. 2011;69:736–748. doi: 10.1016/j.neuron.2011.01.013. [DOI] [PubMed] [Google Scholar]
- Kochubey O, Han Y, Schneggenburger R. Developmental regulation of the intracellular Ca2+ sensitivity of vesicle fusion and Ca2+-secretion coupling at the rat calyx of Held. J Physiol. 2009;587:3009–3023. doi: 10.1113/jphysiol.2009.172387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike-Tani M, Saitoh N, Takahashi T. Mechanisms underlying developmental speeding in AMPA-EPSC decay time at the calyx of Held. J Neurosci. 2005;25:199–207. doi: 10.1523/JNEUROSCI.3861-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwabara N, DiCaprio RA, Zook JM. Afferents to the medial nucleus of the trapezoid body and their collateral projections. J Comp Neurol. 1991;314:684–706. doi: 10.1002/cne.903140405. [DOI] [PubMed] [Google Scholar]
- Leão RM, von Gersdorff H. Synaptic vesicle pool size, release probability and synaptic depression are sensitive to Ca2+ buffering capacity in the developing rat calyx of Held. Braz J Med Biol Res. 2009;42:94–104. doi: 10.1590/S0100-879X2009000100014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin KH, Oleskevich S, Taschenberger H. Presynaptic Ca2+ influx and vesicle exocytosis at the mouse endbulb of held: a comparison of two auditory nerve terminals. J Physiol. 2011;589:4301–4320. doi: 10.1113/jphysiol.2011.209189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MY, Rohan JG, Cai H, Reim K, Ko CP, Chow RH. Complexin facilitates exocytosis and synchronizes vesicle release in two secretory model systems. J Physiol. 2013;591:2463–2473. doi: 10.1113/jphysiol.2012.244517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindau M, Neher E. Patch-clamp techniques for time-resolved capacitance measurements in single cells. Pflugers Arch. 1988;411:137–146. doi: 10.1007/BF00582306. [DOI] [PubMed] [Google Scholar]
- Lipstein N, Sakaba T, Cooper BH, Lin KH, Strenzke N, Ashery U, Rhee JS, Taschenberger H, Neher E, Brose N. Dynamic control of synaptic vesicle replenishment and short-term plasticity by Ca2+-calmodulin-Munc13–1 signaling. Neuron. 2013;79:82–96. doi: 10.1016/j.neuron.2013.05.011. [DOI] [PubMed] [Google Scholar]
- Lu T, Trussell LO. Inhibitory transmission mediated by asynchronous transmitter release. Neuron. 2000;26:683–694. doi: 10.1016/S0896-6273(00)81204-0. [DOI] [PubMed] [Google Scholar]
- Martin JA, Hu Z, Fenz KM, Fernandez J, Dittman JS. Complexin has opposite effects on two modes of synaptic vesicle fusion. Curr Biol. 2011;21:97–105. doi: 10.1016/j.cub.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maximov A, Südhof TC. Autonomous function of synaptotagmin 1 in triggering synchronous release independent of asynchronous release. Neuron. 2005;48:547–554. doi: 10.1016/j.neuron.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Maximov A, Tang J, Yang X, Pang ZP, Südhof TC. Complexin controls the force transfer from SNARE complexes to membranes in fusion. Science. 2009;323:516–521. doi: 10.1126/science.1166505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon HT, Missler M, Li C, Südhof TC. Complexins: cytosolic proteins that regulate SNAP receptor function. Cell. 1995;83:111–119. doi: 10.1016/0092-8674(95)90239-2. [DOI] [PubMed] [Google Scholar]
- Müller M, Felmy F, Schwaller B, Schneggenburger R. Parvalbumin is a mobile presynaptic Ca2+ buffer in the calyx of held that accelerates the decay of Ca2+ and short-term facilitation. J Neurosci. 2007;27:2261–2271. doi: 10.1523/JNEUROSCI.5582-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y, Takahashi T. Developmental changes in potassium currents at the rat calyx of Held presynaptic terminal. J Physiol. 2007;581:1101–1112. doi: 10.1113/jphysiol.2007.128702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y, Harada H, Kamasawa N, Matsui K, Rothman JS, Shigemoto R, Silver RA, DiGregorio DA, Takahashi T. Nanoscale distribution of presynaptic Ca2+ channels and its impact on vesicular release during development. Neuron. 2015;85:145–158. doi: 10.1016/j.neuron.2014.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naraghi M, Neher E. Linearized buffered Ca2+ diffusion in microdomains and its implications for calculation of [Ca2+] at the mouth of a calcium channel. J Neurosci. 1997;17:6961–6973. doi: 10.1523/JNEUROSCI.17-18-06961.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E. Complexin: does it deserve its name? Neuron. 2010;68:803–806. doi: 10.1016/j.neuron.2010.11.038. [DOI] [PubMed] [Google Scholar]
- Neher E, Sakaba T. Multiple roles of calcium ions in the regulation of neurotransmitter release. Neuron. 2008;59:861–872. doi: 10.1016/j.neuron.2008.08.019. [DOI] [PubMed] [Google Scholar]
- Otsu Y, Shahrezaei V, Li B, Raymond LA, Delaney KR, Murphy TH. Competition between phasic and asynchronous release for recovered synaptic vesicles at developing hippocampal autaptic synapses. J Neurosci. 2004;24:420–433. doi: 10.1523/JNEUROSCI.4452-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang ZP, Melicoff E, Padgett D, Liu Y, Teich AF, Dickey BF, Lin W, Adachi R, Südhof TC. Synaptotagmin-2 is essential for survival and contributes to Ca2+ triggering of neurotransmitter release in central and neuromuscular synapses. J Neurosci. 2006;26:13493–13504. doi: 10.1523/JNEUROSCI.3519-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. R: a language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2014. [Google Scholar]
- Reim K, Mansour M, Varoqueaux F, McMahon HT, Südhof TC, Brose N, Rosenmund C. Complexins regulate a late step in Ca2+-dependent neurotransmitter release. Cell. 2001;104:71–81. doi: 10.1016/S0092-8674(01)00192-1. [DOI] [PubMed] [Google Scholar]
- Reim K, Wegmeyer H, Brandstätter JH, Xue M, Rosenmund C, Dresbach T, Hofmann K, Brose N. Structurally and functionally unique complexins at retinal ribbon synapses. J Cell Biol. 2005;169:669–680. doi: 10.1083/jcb.200502115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini BL, Regehr WG. Control of neurotransmitter release by presynaptic waveform at the granule cell to Purkinje cell synapse. J Neurosci. 1997;17:3425–3435. doi: 10.1523/JNEUROSCI.17-10-03425.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaba T. Roles of the fast-releasing and the slowly releasing vesicles in synaptic transmission at the calyx of held. J Neurosci. 2006;26:5863–5871. doi: 10.1523/JNEUROSCI.0182-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaba T, Neher E. Quantitative relationship between transmitter release and calcium current at the calyx of Held synapse. J Neurosci. 2001a;21:462–476. doi: 10.1523/JNEUROSCI.21-02-00462.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaba T, Neher E. Calmodulin mediates rapid recruitment of fast-releasing synaptic vesicles at a calyx-type synapse. Neuron. 2001b;32:1119–1131. doi: 10.1016/S0896-6273(01)00543-8. [DOI] [PubMed] [Google Scholar]
- Scheuss V, Taschenberger H, Neher E. Kinetics of both synchronous and asynchronous quantal release during trains of action potential-evoked EPSCs at the rat calyx of Held. J Physiol. 2007;585:361–381. doi: 10.1113/jphysiol.2007.140988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strenzke N, Chanda S, Kopp-Scheinpflug C, Khimich D, Reim K, Bulankina AV, Neef A, Wolf F, Brose N, Xu-Friedman MA, Moser T. Complexin-I is required for high-fidelity transmission at the endbulb of Held auditory synapse. J Neurosci. 2009;29:7991–8004. doi: 10.1523/JNEUROSCI.0632-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC, Rizo J. Synaptic vesicle exocytosis. Cold Spring Harb Perspect Biol. 2011;3:a005637. doi: 10.1101/cshperspect.a005637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC, Rothman JE. Membrane fusion: grappling with SNARE and SM proteins. Science. 2009;323:474–477. doi: 10.1126/science.1161748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J, Maximov A, Shin OH, Dai H, Rizo J, Südhof TC. A Complexin/synaptotagmin 1 switch controls fast synaptic vesicle exocytosis. Cell. 2006;126:1175–1187. doi: 10.1016/j.cell.2006.08.030. [DOI] [PubMed] [Google Scholar]
- Taschenberger H, von Gersdorff H. Fine-tuning an auditory synapse for speed and fidelity: developmental changes in presynaptic waveform, EPSC kinetics, and synaptic plasticity. J Neurosci. 2000;20:9162–9173. doi: 10.1523/JNEUROSCI.20-24-09162.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taschenberger H, Leão RM, Rowland KC, Spirou GA, von Gersdorff H. Optimizing synaptic architecture and efficiency for high-frequency transmission. Neuron. 2002;36:1127–1143. doi: 10.1016/S0896-6273(02)01137-6. [DOI] [PubMed] [Google Scholar]
- Taschenberger H, Scheuss V, Neher E. Release kinetics, quantal parameters and their modulation during short-term depression at a developing synapse in the rat CNS. J Physiol. 2005;568:513–537. doi: 10.1113/jphysiol.2005.093468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis SF. Software-based correction of single compartment series resistance errors. J Neurosci Methods. 1998;86:25–34. doi: 10.1016/S0165-0270(98)00140-X. [DOI] [PubMed] [Google Scholar]
- Vincent P, Marty A. Fluctuations of inhibitory postsynaptic currents in Purkinje cells from rat cerebellar slices. J Physiol. 1996;494:183–199. doi: 10.1113/jphysiol.1996.sp021484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Gersdorff H, Schneggenburger R, Weis S, Neher E. Presynaptic depression at a calyx synapse: the small contribution of metabotropic glutamate receptors. J Neurosci. 1997;17:8137–8146. doi: 10.1523/JNEUROSCI.17-21-08137.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter AM, Groffen AJ, Sørensen JB, Verhage M. Multiple Ca2+ sensors in secretion: teammates, competitors or autocrats? Trends Neurosci. 2011;34:487–497. doi: 10.1016/j.tins.2011.07.003. [DOI] [PubMed] [Google Scholar]
- Wang LY, Kaczmarek LK. High-frequency firing helps replenish the readily releasable pool of synaptic vesicles. Nature. 1998;394:384–388. doi: 10.1038/28645. [DOI] [PubMed] [Google Scholar]
- Wang LY, Neher E, Taschenberger H. Synaptic vesicles in mature calyx of Held synapses sense higher nanodomain calcium concentrations during action potential-evoked glutamate release. J Neurosci. 2008;28:14450–14458. doi: 10.1523/JNEUROSCI.4245-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcik SM, Brose N. Regulation of membrane fusion in synaptic excitation-secretion coupling: speed and accuracy matter. Neuron. 2007;55:11–24. doi: 10.1016/j.neuron.2007.06.013. [DOI] [PubMed] [Google Scholar]
- Xue M, Reim K, Chen X, Chao HT, Deng H, Rizo J, Brose N, Rosenmund C. Distinct domains of complexin I differentially regulate neurotransmitter release. Nat Struct Mol Biol. 2007;14:949–958. doi: 10.1038/nsmb1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue M, Stradomska A, Chen H, Brose N, Zhang W, Rosenmund C, Reim K. Complexins facilitate neurotransmitter release at excitatory and inhibitory synapses in mammalian central nervous system. Proc Natl Acad Sci U S A. 2008;105:7875–7880. doi: 10.1073/pnas.0803012105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue M, Lin YQ, Pan H, Reim K, Deng H, Bellen HJ, Rosenmund C. Tilting the balance between facilitatory and inhibitory functions of mammalian and Drosophila Complexins orchestrates synaptic vesicle exocytosis. Neuron. 2009;64:367–380. doi: 10.1016/j.neuron.2009.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada M, Saisu H, Ishizuka T, Takahashi H, Abe T. Immunohistochemical distribution of the two isoforms of synaphin/complexin involved in neurotransmitter release: localization at the distinct central nervous system regions and synaptic types. Neuroscience. 1999;93:7–18. doi: 10.1016/S0306-4522(99)00104-9. [DOI] [PubMed] [Google Scholar]
- Yamashita T, Hige T, Takahashi T. Vesicle endocytosis requires dynamin-dependent GTP hydrolysis at a fast CNS synapse. Science. 2005;307:124–127. doi: 10.1126/science.1103631. [DOI] [PubMed] [Google Scholar]
- Yang X, Kaeser-Woo YJ, Pang ZP, Xu W, Südhof TC. Complexin clamps asynchronous release by blocking a secondary Ca2+ sensor via its accessory alpha helix. Neuron. 2010a;68:907–920. doi: 10.1016/j.neuron.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Cao P, Südhof TC. Deconstructing complexin function in activating and clamping Ca2+-triggered exocytosis by comparing knockout and knockdown phenotypes. Proc Natl Acad Sci U S A. 2013;110:20777–20782. doi: 10.1073/pnas.1321367110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YM, Fedchyshyn MJ, Grande G, Aitoubah J, Tsang CW, Xie H, Ackerley CA, Trimble WS, Wang LY. Septins regulate developmental switching from microdomain to nanodomain coupling of Ca2+ influx to neurotransmitter release at a central synapse. Neuron. 2010b;67:100–115. doi: 10.1016/j.neuron.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young SM, Jr, Neher E. Synaptotagmin has an essential function in synaptic vesicle positioning for synchronous release in addition to its role as a calcium sensor. Neuron. 2009;63:482–496. doi: 10.1016/j.neuron.2009.07.028. [DOI] [PubMed] [Google Scholar]