

Abstract
In Alzheimer's disease, accumulation of soluble oligomers of β-amyloid peptide is known to be highly toxic, causing disturbances in synaptic activity and neuronal death. Multiple studies relate these effects to increased oxidative stress and aberrant activity of calcium-permeable cation channels leading to calcium imbalance. The transient receptor potential melastatin 2 (TRPM2) channel, a Ca2+-permeable nonselective cation channel activated by oxidative stress, has been implicated in neurodegenerative diseases, and more recently in amyloid-induced toxicity. Here we show that the function of TRPM2 is augmented by treatment of cultured neurons with β-amyloid oligomers. Aged APP/PS1 Alzheimer's mouse model showed increased levels of endoplasmic reticulum stress markers, protein disulfide isomerase and phosphorylated eukaryotic initiation factor 2α, as well as decreased levels of the presynaptic marker synaptophysin. Elimination of TRPM2 in APP/PS1 mice corrected these abnormal responses without affecting plaque burden. These effects of TRPM2 seem to be selective for β-amyloid toxicity, as ER stress responses to thapsigargin or tunicamycin in TRPM2−/− neurons was identical to that of wild-type neurons. Moreover, reduced microglial activation was observed in TRPM2−/−/APP/PS1 hippocampus compared with APP/PS1 mice. In addition, age-dependent spatial memory deficits in APP/PS1 mice were reversed in TRPM2−/−/APP/PS1 mice. These results reveal the importance of TRPM2 for β-amyloid neuronal toxicity, suggesting that TRPM2 activity could be potentially targeted to improve outcomes in Alzheimer's disease.
SIGNIFICANCE STATEMENT Transient receptor potential melastatin 2 (TRPM2) is an oxidative stress sensing calcium-permeable channel that is thought to contribute to calcium dysregulation associated with neurodegenerative diseases, including Alzheimer's disease. Here we show that oligomeric β-amyloid, the toxic peptide in Alzheimer's disease, facilitates TRPM2 channel activation. In mice designed to model Alzheimer's disease, genetic elimination of TRPM2 normalized deficits in synaptic markers in aged mice. Moreover, the absence of TRPM2 improved age-dependent spatial memory deficits observed in Alzheimer's mice. Our results reveal the importance of TRPM2 for neuronal toxicity and memory impairments in an Alzheimer's mouse model and suggest that TRPM2 could be targeted for the development of therapeutic agents effective in the treatment of dementia.
Keywords: Alzheimer's disease, β-amyloid, cognitive impairment, mouse model, neurotoxicity, TRPM2
Introduction
In Alzheimer's disease (AD), a progressive biochemical cascade leading to neuronal dysfunction takes place many years before the appearance of cognitive deficits (Selkoe, 2011). These alterations derive at least in part from the accumulation of β-amyloid peptides (Aβ) whose toxicity depends on its interactions with distinct cell surface proteins. These include, but are not limited to, the prion protein (Laurén et al., 2009; Caetano et al., 2011; Ostapchenko et al., 2013b), NMDAR (Snyder et al., 2005; Decker et al., 2010; Malinow, 2012; You et al., 2012), mGluR5 (Renner et al., 2010), and α7 nicotinic acetylcholine receptors (α7nAChRs) (Wang et al., 2000). Although precise mechanisms of toxicity remain unknown, dysregulation of Ca2+ homeostasis is widely accepted as being intimately associated with Aβ toxicity (LaFerla, 2002; Mattson, 2004). In light of the marked toxicity of Aβ toward glutamate synapses (Lambert et al., 1998; Walsh et al., 2002; Lacor et al., 2007; Laurén et al., 2009; Ma et al., 2009; Ferreira and Klein, 2011), one particularly attractive candidate mechanism contributing to Ca2+ dysregulation in AD is via NMDARs (De Felice et al., 2007). However, block of NMDARs as a therapeutic avenue is problematic as this can impair learning and memory (Newcomer et al., 1999; Rammsayer, 2001), cause psychotic-like symptoms in patients (Lahti et al., 1995; Breier et al., 1997; Newcomer et al., 1999), and exacerbate neurodegeneration (Ikonomidou et al., 2000). Identifying secondary sources of Ca2+ entry, recruited downstream of aberrant NMDAR activation, may provide attractive alternative targets for novel AD therapeutics.
Transient receptor potential melastatin 2 (TRPM2) is a Ca2+-permeable nonselective cation channel most highly expressed in the CNS (Fonfria et al., 2006a; Olah et al., 2009; Bai and Lipski, 2010). Notably, TRPM2 is reported to modulate neuronal vulnerability to ischemia (Bai and Lipski, 2010; Alim et al., 2013), and a variant of TRPM2 has been linked to neurodegenerative diseases (Hermosura et al., 2008). Functionally, TRPM2 channel opening is coupled to oxidative stress via the production of ADP-ribose (ADPR), an intracellular TRPM2 agonist (Knowles et al., 2013). However, ADPR alone is insufficient to gate TRPM2 channels rather channel activation is contingent on the binding of Ca2+ to an intracellular channel domain (Tong et al., 2006). In this regard, we recently showed that TRPM2 channels are intimately coupled to NMDARs via Ca2+ influx (Olah et al., 2009) and are necessary for the induction of NMDAR-dependent long-term depression (Xie et al., 2011).
Here we show that Aβ oligomers (AβOs) augment TRPM2 currents in cultured neurons. Moreover, the absence of TRPM2 decreases endoplasmic reticulum (ER) stress and abnormal microglial activation in TRPM2−/−/APP/PS1 mice, without affecting plaque burden. Critically, elimination of TRPM2 rescues cognitive deficits in aged TRPM2−/−/APP/PS1 mice. Our results reveal an important contribution of abnormal TRPM2 activation to the cognitive deficits observed in aged AD model mice and suggest that this channel represents a new therapeutic target in AD.
Materials and Methods
Animals.
TRPM2-null mice (TRPM2−/−) were generated in a C57BL/6J background as described previously (Yamamoto et al., 2008). Wild-type and TRPM2−/− mice were derived from heterozygous mating. Genotyping was performed as previously described (Yamamoto et al., 2008). The APP/PS1 mouse model of AD, carrying the genes of human APP-Swedish mutation and the ΔE9 mutation of the human presenilin 1 (Jankowsky et al., 2003), in a C57BL/6J background was obtained from The Jackson Laboratory (stock #005864). TRPM2−/− mice were crossed to APP/PS1 and the offspring intercrossed to generate the desired genotypes. Mouse cohorts of desired genotypes were generated and used longitudinally for behavioral analysis. Randomized littermates were used for behavioral analysis. Only male mice were used; and in all experiments, the experimenter was blind to the genotypes. All animal experiments were conducted with approval from the University of Western Ontario Animal Use Subcommittee (2008-127). Once the mouse cohorts reached 20–24 months, they were killed and used for biochemical and pathological analysis.
Electrophysiology.
Primary cultured hippocampal neurons were derived from time-pregnant CD1 or C57BL/6J or TRPM2−/− mice at E18 as previously described (Xie et al., 2011). Whole-cell voltage-clamp recordings were performed from neurons (14–21 DIV) treated for 24 h with control medium or medium containing AβOs (1 μm), which is neurotoxic (Ostapchenko et al., 2013b). AβOs were prepared and routinely checked by Western blotting as described previously (Ostapchenko et al., 2013b). In short, synthetic Aβ(1–42) peptide (rPeptide) was monomerized in hexafluoroisopropanol, reconstituted in DMSO, and diluted in PBS to a final concentration of 100 μm. Oligomers were allowed to form for 24 h at 4°C and were then used for experiments. For recording, the intracellular solution contained the following (in mm): 150 cesium gluconate, 10 HEPES, 2 MgCl2, and 1 ADPR. Standard extracellular solution contained the following (in mm): 140 NaCl, 5.4 KCl, 25 HEPES, 33 glucose, 2 CaCl2, 0.003 d-serine, and 0.0002 TTX. ADPR-dependent TRPM2 currents were evoked in response to repeated applications of 100 μm NMDA (10 s, repeated every 60 s) using a multibarrelled rapid perfusion system (SF-77B; Warner Instruments). Once TRPM2 currents stabilized, a calcium free solution was applied to inactivate TRPM2 channels (total divalent concentration was maintained by replacing CaCl2 with equimolar BaCl2). Data were filtered at 2 kHz, digitized, and acquired using pCLAMP and Axoscope software (Molecular Devices).
Real-time quantitative PCR.
RNAs from WT and TRPM2−/− hippocampi (three for each genotype) were purified as described previously (Ostapchenko et al., 2013a). To quantify expression levels of genes of interest, RNA samples were amplified in real-time quantitative PCR using the following primers: Trpm2 (forward, CTGTCTCCGTCTCATGCACA; reverse, AGACGTCCTTCATCATCCGC); Trpm7 (forward, TCTACTTCTCTTCACTCGGTGC; reverse, TGTTCCTCCTTCAGATGGCATT); GluN1 (forward, ACACAAGGATGCCCGTAGGA; reverse, TTTTAGGGTCGGGCTCTGCT); GluN2A (forward, CATTGGGAGCGGGTACATCTTT; reverse, TGGCTGCTCATCACCTCATTC); GluN2B (forward, GAGCGCCAATCCGTGATGAA; reverse, TAGACAGAGGACTCACGGCG); GluA2 (forward, GTGAGTCCTGGCATGGGAATG; reverse, TATCGGATGCCTCTCACCACTT); Cav1.2 (forward, ACGTCAGCAACCTGTAGTCC; reverse, AGCTCTGAGGCTTATCCCGA); Chrna7 (forward, TGGAGGACGAGAATTGGGGT; reverse, TGCCGGTGATGGGTGTAAGA). As the reference gene, Pgk1 (forward, GTCGTGATGAGGGTGGACTT; reverse, TTGGCTCCATTGTCCAAGCA) was used as it is consistently expressed in mouse hippocampus (Boda et al., 2009).
Western blotting, ELISA, and immunohistochemistry.
Mice were anesthetized using ketamine (100 mg/kg) and transcardially perfused with 0.9% saline. For each mouse, the cortex and hippocampus from one hemibrain were used for ELISA and Western blotting analyses; the other hemibrain was postfixed as described below and used for immunostaining.
For hippocampus fractionation, tissues were homogenized in 50 mm Tris-HCl, pH 7.2, 200 mm NaCl, 2 mm sodium EDTA, with protease inhibitor cocktail III (Millipore), centrifuged at 100,000 × g, and the supernatants were used as the Tris-soluble fraction of the proteins. The sediments were resuspended in radioimmunoprecipitation assay buffer (50 mm Tris-HCl, pH 8, 150 mm NaCl, 1% NP-40, 1% sodium deoxycholate, and 1% sodium dodecyl sulfate) containing protease inhibitor cocktail III (Millipore) and phosphatase inhibitors 10 mm NaF and 0.5 mm Na3VO4, followed by 3 cycles (power 3) of sonication for 10 s with 1 min intervals between the cycles using Sonic Dismembrator M100 (Fisher Scientific) on ice. Samples were centrifuged at 16,000 × g for 30 min, and the supernatants were used as the cellular fraction of the proteins. Remaining sediments were solubilized in 5 m guanidine-HCl, 50 mm HEPES, pH 7.3, 5 mm EDTA, followed by rotation at room temperature for 3 h, and used as insoluble protein fraction. Protein concentration was measured using the Bradford assay.
For Western blotting, a total of 30 μg of each sample was resolved by electrophoresis in SDS-polyacrylamide gels using 13.5% Tris-tricine gels or 4%–12% PAGEr Gold precast gels (Lonza), followed by a standard blotting procedure using anti-protein disulfide isomerase (PDI) (1:1000, catalog #3501), anti-synaptophysin (1:2000, catalog #5467, both Cell Signaling Technology), or anti β-amyloid (6E10, 1:2000, Covance) antibodies.
ELISA was performed using the kit for human Aβ(1–42) (catalog #KHB3544, Invitrogen). Tris-soluble and insoluble fractions were diluted 30- and 500-fold in dilution buffer and the Aβ(1–42) concentration was measured using synthetic peptide as a standard.
For immunohistochemistry, hemibrains were postfixed for 48 h with 4% PFA in phosphate-buffered saline. Coronal sections (50 μm) were cut using a vibratome. Polyclonal anti-human Aβ antibody (catalog #44344, Invitrogen), anti-peIF2α (Ser51) antibody (catalog #9721, Cell Signaling Technology), anti-Synaptophysin antibody (catalog #sc-9116, Santa Cruz Biotechnology), and anti-Iba1 antibody (catalog #PA5-27436, Thermo Fisher Scientific) were used on free-floating sections. Aβ was retrieved by boiling the sections in 10 mm sodium citrate buffer. Nonspecific binding was prevented by incubation with 2% horse serum (Invitrogen) and 2% BSA in TBS with 0.3% Triton X-100. Sections were then processed for immunostaining by overnight incubation at 4°C in the primary antibody (1:200) diluted in TBS. After rinsing twice in TBS, sections were incubated for 2.5 h at 4°C with the secondary antibody diluted in TBS containing 1% horse serum and 1% BSA, rinsed three times and mounted.
For thioflavin S staining, sections were incubated in 0.25% KMnO4 for 5 min, rinsed with water, and treated with bleaching solution (1% Na2S2O5 + 1% axalic acid) for 2 min. After rinsing with water, sections were placed in blocking solution (1% NaOH + 0.9% H2O2) for 30 s, washed, and incubated in 0.25% acetic acid for 5 s. Sections were then washed, placed on silanized slides, and stained with 1.25% thioflavin S solution in 50% ethanol for 5 min. Slices were rinsed once with 50% ethanol, twice with water, and mounted.
Mounted slices were imaged using a Zeiss LSM-510 confocal microscope equipped with either 10×/0.4 or 20×/0.75 objectives. Image analysis was done using ImageJ software (National Institutes of Health).
ER stress in primary neuronal cultures and surface sensing of translation (SUnSET) protein synthesis assay.
ER stress in cultured hippocampal wild-type and TRPM2−/− neurons at 14–18 DIV was induced by treating them with either 1 μm thapsigargin or 3 μg/ml tunicamycin and incubation for 18 h at 37°C in CO2 atmosphere. Cells were then washed two times with ECF containing the following (in mm): 140 NaCl, 25 HEPES, 5.4 KCl, 33 glucose, 2 CaCl2, 1 MgCl2, and proteins, extracted with 2 × Laemmli buffer added to each well (4% SDS, 20% glycerol, 10% 2-mercaptoethanol, 0.125 mm Tris HCl, pH 6.8, 0.004% bromophenol blue). Lysates were collected and incubated at 95°C for 5 min before loading on a 12% SDS-PAGE gel at 20 μl per lane. Proteins were transferred to BioTrace NT membrane (Pall Life Science), followed by standard Western blotting with antibodies against p-eIF2α (1:1,000), eIF2α (1:1000) (Cell Signaling Technology, catalog #3398 and 2103, respectively), and GRP78/BiP (1:300) (Abcam, catalog #ab21685). All secondary antibodies were from Jackson ImmunoResearch Laboratories.
Newly synthesized protein was measured following ER stress with thapsigargin by using a modified SUnSET assay (Schmidt et al., 2009). Cultured neurons from wild-type or TRPM2−/− mice (14 DIV) were treated with 1 μm thapsigargin or were left untreated, and placed in the incubator for 18 h; 2 μg/ml puromycin was added to all culture wells and incubated for a further 2 h to allow for incorporation of puromycin into new proteins. Cells were then washed in ECF and lysed in 2 × Laemmli buffer. SDS-PAGE, transfer, and Western blotting were done as described above. Anti-puromycin (clone 12D10, Millipore) was used at 1 μg/ml.
Elevated plus maze and locomotor activity.
Anxiety-like behavior was assessed using the elevated plus maze test, performed as described previously (Martins-Silva et al., 2011). Sessions were recorded and analyzed using the ANY-Maze software (Stoelting) to determine total time spent in the open and closed arms. Spontaneous locomotor activity in a new environment, a measure of exploratory behavior, was recorded using automated locomotor boxes as described previously (Guzman et al., 2011).
Barnes maze.
The Barnes maze consists of a white, circular platform (92 cm in diameter) with 20 equally spaced holes (5 cm in diameter; 7.5 cm between holes), elevated 105 cm above the floor (San Diego Instruments). Mice were trained at 3, 6, 9, 12, and 15 months of age, with all tests performed between 09:00 and 16:00 h as previously described (Martyn et al., 2012). For each new acquisition phase, the location of the target hole was changed. To test reference memory, a probe trial was conducted on the fifth day in which the target hole was barred and the number of nose-pokes to each maze hole was recorded. Path length, the number of hole-pokes before reaching the target (errors), and latency to reach the barred target hole were also measured. Sessions were recorded and analyzed using the ANY-Maze software. Results were expressed as number of target hole visits and preference of target hole over others (target hole visits /average nontarget visits) during the probe trial.
Morris water maze (MWM).
The MWM was performed as previously described (Martyn et al., 2012). Briefly, mice received 4 consecutive training days, with four trials per day separated by 15 min inter test intervals. A hidden platform was kept in a constant location in the center of one quadrant of a 2-m-wide round pool. For each trial, mice were randomly placed at one of the four equally spaced starting locations within the pool. Each trial consisted of a swim followed by a 10 s platform sit. To assess spatial memory, a probe trial was performed on the fifth day. For this trial, the platform was removed from the pool and the mice were allowed to swim freely for 60 s. Experiments were recorded using a video camera and analyzed using ANY-Maze video tracking software (Stoelting).
Statistical analyses.
Western blots and immunohistochemistry data were quantified using ImageJ software (National Institutes of Health) and analyzed using one-way ANOVA with Tukey's post hoc or by Student's t test using GraphPad Prism software. For behavioral studies, ANOVA followed by an appropriate post hoc test was used to compare multiple groups, using GraphPad Prism or SigmaStat software.
Results
Aβ oligomer treatment sensitizes ADPR-dependent TRPM2 currents generated by repeated NMDAR stimulation
We previously reported that repeated applications of NMDA to cultured hippocampal neurons initiate the development of large ADPR-dependent TRPM2 currents (Olah et al., 2009; Belrose et al., 2012). To investigate whether TRPM2 channel function can be modulated by AβOs, we treated cultured hippocampal neurons for 24 h with AβOs, a procedure known to increase neuronal death (Kudo et al., 2012; Ostapchenko et al., 2013b). As shown in Figure 1A, B, NMDA-induced TRPM2 currents were augmented by AβO treatment.
Figure 1.

TRPM2 is responsive to AβOs and contributes to increased ER stress in APP/PS1 mice. A, Representative traces show that NMDA facilitated ADPR-induced TRPM2 currents evoked in cultured hippocampal neurons from CD1 mice in the absence or presence of AβOs. These could be inhibited by omitting Ca2+ from the extracellular recording solution. TRPM2 current amplitude was determined as indicated by bidirectional arrows positioned between the stippled lines. B, Summary bar graphs show that treatment with 1 μm AβOs for 24 h increases TRPM2 current amplitude and density. Data from at least five cells for each treatment were analyzed with Student's t test. **p < 0.01. C, D, Immunoblot analysis of PDI levels in hippocampal extracts. At least four brain extracts obtained from each genotype were analyzed. E, F, Immunofluorescence analysis of phospho-eIF2α(Ser51) (green) levels in CA1, CA2, and CA3 areas of mouse hippocampus. βIII-tubulin (red) and Hoechst dye (blue) were used to label neurons and nuclei, respectively. At least four coronal slices from each mouse brain and at least three brains of each genotype were used for immunostaining. *p < 0.05 (one-way ANOVA followed by Tukey's post hoc test). **p < 0.01 (one-way ANOVA followed by Tukey's post hoc test). ***p < 0.001 (one-way ANOVA followed by Tukey's post hoc test). #p > 0.05 when comparing pSER51 WT levels to TRPM2−/−APP/PS1 levels. Scale bar, 90 μm.
Decreased levels of ER stress in brains of APP/PS1 mice lacking TRPM2
To study the influence of TRPM2 channels on Aβ neuronal toxicity, we crossed APP/PS1 mice (Jankowsky et al., 2003) to TRPM2−/− mice (Yamamoto et al., 2008). The offspring were then intercrossed to generate the four genotypes used in this study (WT, TRPM2−/−, APP/PS1, and TRPM2−/−/APP/PS1).
In AD patients and AD mouse models, increased protein misfolding leads to ER stress and the activation of the unfolded protein response (Matus et al., 2011; Scheper et al., 2011; Hetz and Mollereau, 2014). Among ER stress proteins, PDI has been found increased in the brain of AD patients (Kim et al., 2000; Andreu et al., 2012). Using immunoblots, we tested whether expression of PDI is changed in the hippocampus of 20- to 24-month-old APP/PS1 and TRPM2−/−/APP/PS1 mice compared with WT controls. Similar to previous reports in AD patients, APP/PS1 mice showed increased levels of PDI (Fig. 1C,D). In contrast, the levels of PDI in TRPM2−/−/APP/PS1 mice were no different from that of WT controls or TRPM2−/− mice (Fig. 1C,D).
Increased phosphorylation of the eukaryotic initiation factor 2α (eIF2α) at Ser51 in response to ER stress inhibits global protein translation and contributes to synaptic pathophysiology in the APP/PS1 mouse model of AD (Yasuda et al., 2002; Ma et al., 2013). We therefore tested whether the absence of TRPM2 could prevent changes in eIF2α phosphorylation in neurons of APP/PS1 mice. To specifically compare the response in neurons, we used immunofluorescence and quantified the staining of neuron-rich hippocampal areas. In areas CA1, CA2, and CA3 of the hippocampus of APP/PS1 mice, pSer51-eIF2α immunoreactivity was higher compared with wild-type controls (Fig. 1E,F). Conversely, in TRPM2−/−/APP/PS1 mice, hippocampal pSer51-eIF2α immunoreactivity was similar to the levels found in the hippocampus of TRPM2−/− mice (Fig. 1E,F).
TRPM2−/− neurons do not have altered ER stress responses or major compensatory responses
To examine the possibility that ER stress responses in general are altered in TRPM2−/− neurons, the response to ER stressors, thapsigargin (Tg) or tunicamycin (Tn), was determined by monitoring the changes in the expression levels of pSer51-eIF2α and Grp78/BiP, an ER chaperone and master regulator of ER stress. In cultured hippocampal neurons from both WT and TRPM2−/−, Tg or Tn treatment resulted in increased levels of pSer51-eIF2α and Grp78/BiP. No difference in the ER stress response between genotypes was observed (Fig. 2A–D). Consistent with the increased levels of pSer51-eIF2α in response to ER stress, general protein translation, determined using nonradioactive SUnSET, was reduced by treatment with Tg. Again, no difference in this response was observed in the absence of TRPM2 compared with control neurons (Fig. 2E,F).
Figure 2.
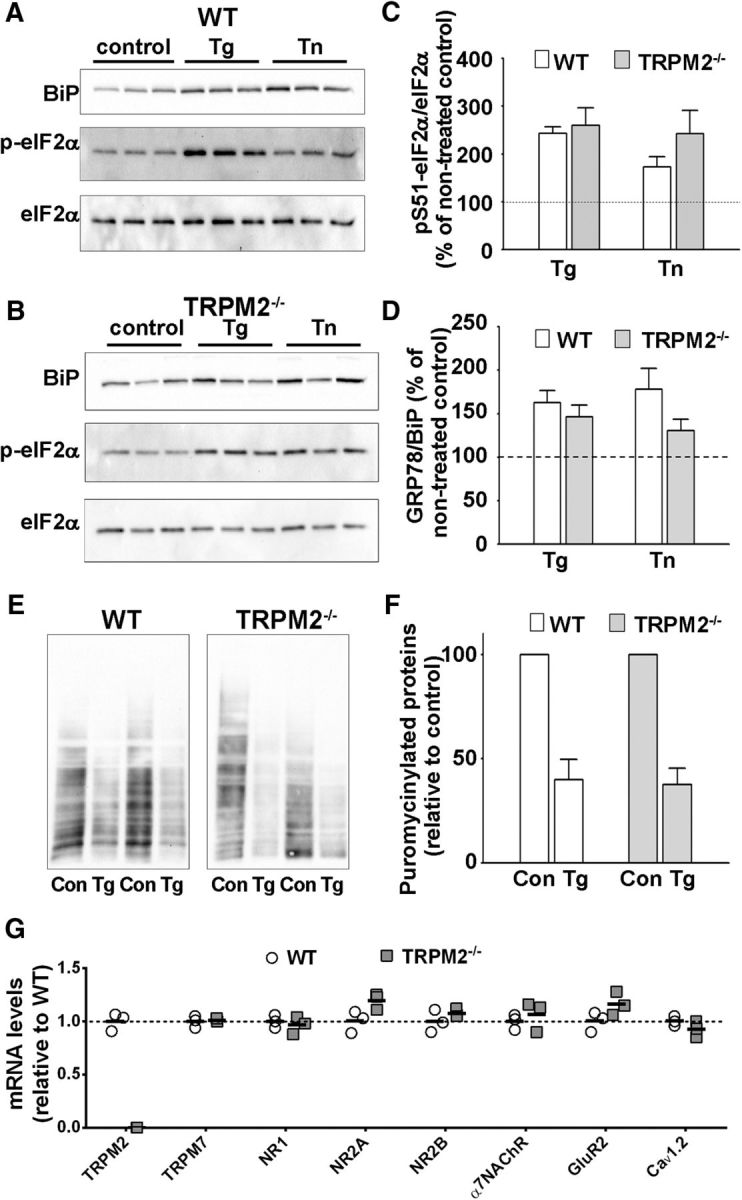
TRPM2−/− neurons do not have altered ER stress responses or major compensatory responses. A–D, Representative immunoblots of wild-type (A) and TRPM2−/− (B) primary hippocampal neuronal cultures, nontreated or treated with thapsigargin (Tg) or tunicamysin (Tn) as described in Materials and Methods. Analysis of p(S51)-eIF2α (C) and GRP78/BiP (D) levels. At least six samples were analyzed for each condition. For each treatment condition (Tg or Tn), data were analyzed with Student's t test. E, F, SUnSET assay of protein synthesis in control and thapsigargin-treated cultured hippocampal neurons. E, Representative blots. F, Analysis of puromycin immunostaining was done for three samples for each condition. Data were analyzed with Student's t test. G, qRT-PCR comparison of hippocampal mRNA levels of TRPM2, TRPM7, GluN1, GluN2A, GluN2B, GluA2, Cav 1.2, and α7NAChR in WT and TRPM2−/− mice. Data were collected from three samples for each genotype, normalized by Pgk1 mRNA levels, and analyzed with Student's t test.
To further examine whether the absence of TRPM2 can elicit compensatory responses in transcripts that regulate pathways related to Ca2+-induced toxicity, we used qRT-PCR. We tested the expression of genes previously linked to NMDAR/AβO excitotoxicity in the hippocampus of controls and TRPM2−/− mice. For NMDAR subunits, we limited our analysis to GluN1, GluN2A, and GluN2B, the most highly expressed subtypes in the adult hippocampus and cortex (Monyer et al., 1994). We also measured transcript levels of the AMPAR subunit GluA2, inclusion of which renders AMPARs Ca2+-impermeable, and of genes implicated in Ca2+ dysregulation in AD (Demuro et al., 2010), including the α7 nicotinic acetylcholine receptor and L-type voltage-gated Ca2+ channels (CaV 1.2). Last, as TRPM7 and TRPM2 expression has previously been suggested to be interdependent (Aarts et al., 2003), we examine whether TRPM7 expression was altered in TRPM2−/− mice. qRT-PCR analysis of hippocampal extracts from WT and TRPM2−/− mice confirmed the absence of TRPM2 mRNA in TRPM2−/− mice (Fig. 2G). There were no changes in the hippocampal mRNA levels of NMDA receptor subunits (i.e., GluN1, GluN2A, and GluN2B), GluA2, α7 nicotinic acetylcholine receptor CaV 1.2, or TRPM7 (Fig. 2G).
Prevention of synapse loss in APP/PS1 mice lacking TRPM2
Given that synapse loss and decreased levels of synaptic proteins are early correlates of AD severity (Pozueta et al., 2013), we compared the expression levels of the synaptic marker synaptophysin in the hippocampus of APP/PS1 mice with that of TRPM2−/−/APP/PS1 mutants. Immunofluorescence analysis showed that synaptophysin levels were decreased in the hippocampus of APP/PS1 mice compared with WT controls. On the other hand, synaptophysin staining in the hippocampus of TRPM2−/−/APP/PS1 mutants was similar to that observed for WT controls (Fig. 3A–D). Likewise, labeling of synaptophysin in the hippocampus of TRPM2−/− was not different from WT controls (Fig. 3A–D). These results suggest that elimination of TRPM2 prevents the decrease in synaptophysin levels observed in the brains of 20- to 24-month-old APP/PS1 mice.
Figure 3.
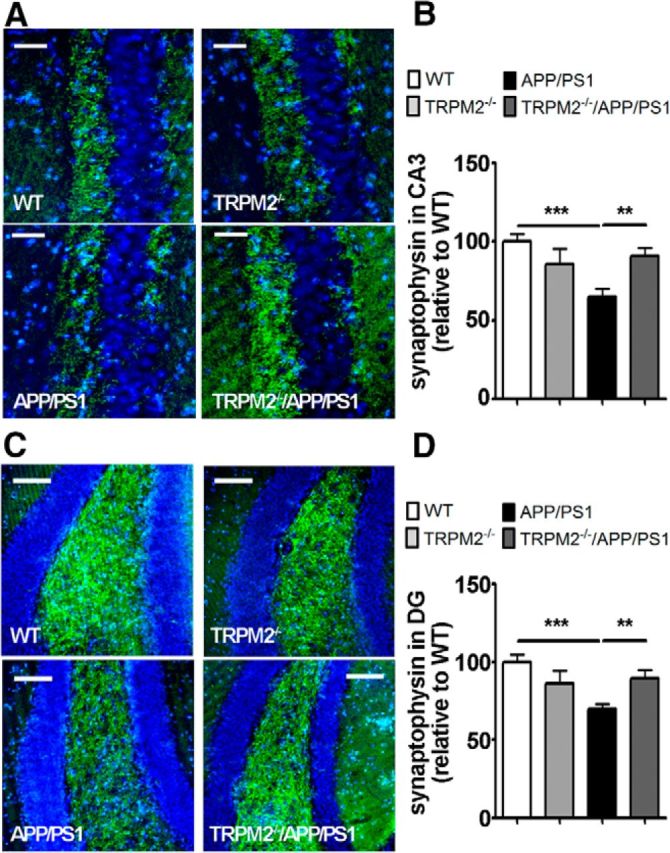
Levels of synaptophysin are rescued in brains of TRPM2−/−/APP/PS1 mice. A–D, Immunofluorescence analysis of synaptophysin levels (green) in CA3 (A, B) and DG (C, D) areas of mouse hippocampus. Hoechst dye (blue) was used to label nuclei. At least four coronal slices from each mouse brain and at least 3 brains of each genotype were used for immunostaining. **p < 0.01 (one-way ANOVA followed by Tukey's post hoc test). ***p < 0.001 (one-way ANOVA followed by Tukey's post hoc test). Scale bars: A, 45 μm; C, 90 μm.
Decreased microglial activation in APP/PS1 mice lacking TRPM2
We tested whether the absence of TRPM2 affects microglial activation in APP/PS1 mice by measuring Iba1 immunostaining, a marker of microglia. Our analysis shows that, while Iba1 immunoreactivity was increased in the hippocampus of APP/PS1 mice compared with WT controls, there was no difference in Iba1 staining when comparing TRPM2−/−/APP/PS1 mutants with WT controls (Fig. 4A,B). These results suggest that, while brains of APP/PS1 mice show increased microglial response, this response is absent in the brains of TRPM2−/−/APP/PS1 mutants. Together, these data provide multiple lines of evidence demonstrating that elimination of TRPM2 expression improves endpoints related to pathological hallmarks in APP/PS1 mice.
Figure 4.
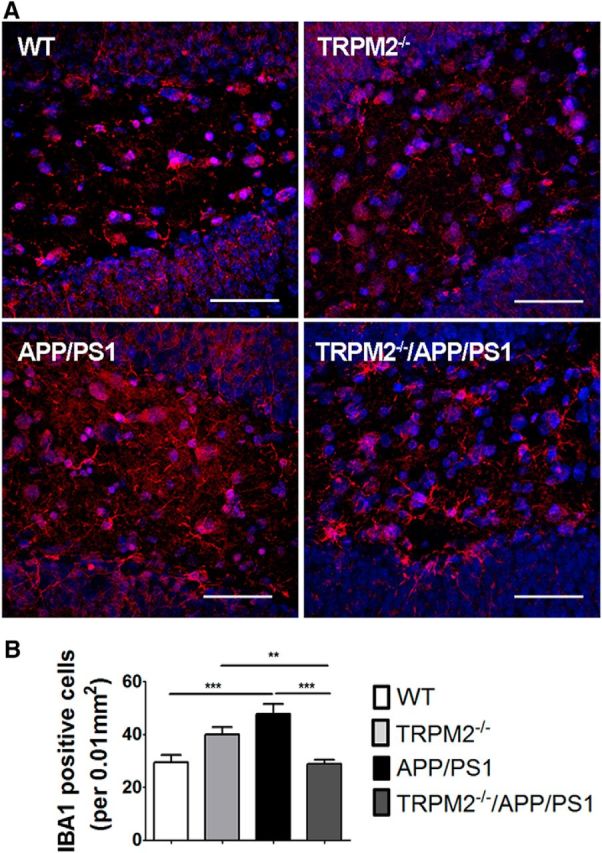
Decreased microglial activation in brains of TRPM2−/−/APP/PS1 mice. Levels of activated microglia in DG (A, B) area of mouse hippocampus were analyzed by Iba1 immunostaining (red). Hoechst dye (blue) was used to label nuclei. At least four coronal slices from each mouse brain and at least 3 brains of each genotype were used for immunostaining. **p < 0.01 (one-way ANOVA followed by Tukey's post hoc test). ***p < 0.001 (one-way ANOVA followed by Tukey's post hoc test). Scale bar, 90 μm.
TRPM2 elimination does not change plaques or soluble Aβ peptides but slightly decreases the levels of small molecular weight oligomers
It is possible that the absence of TRPM2 could influence amyloid processing and accumulation, which may contribute to altered levels of ER stress markers. Hence, we analyzed Aβ(1–42) levels in soluble, membrane-bound and insoluble amyloid plaque-associated fractions of hippocampal proteins. We did not detect any difference between APP/PS1 or TRPM2−/−/APP/PS1 mutants when comparing them for Aβ(1–42) immunofluorescence staining or thioflavin S-positive plaques (Fig. 5A,B). Similarly, ELISA determination of Aβ(1–42) content in insoluble and Tris-soluble fractions from APP/PS1 and TRPM2−/−/APP/PS1 hippocampal samples showed no change (Fig. 5C,D). To further investigate whether different oligomeric fractions of Aβ peptides may be affected by lack of TRPM2, we resolved low molecular weight oligomers by Tris-tricine gel electrophoresis of hippocampal lysates. These experiments revealed a decreased amount of Aβ trimers and tetramers in RIPA-soluble fractions of TRPM2−/−/APP/PS1 hippocampi compared with APP/PS1 mice (Fig. 5E). As overall Aβ peptides and amyloid plaques are not affected by lack of TRPM2, the decreased levels of cell-associated low molecular weight AβOs detected in TRPM2−/−/APP/PS1 mutants may reflect decreased cellular stress level compared with APP/PS1 mice. This may improve the turnover of misfolded proteins and, therefore, cell-bound AβOs.
Figure 5.

Normal plaque load in brains of TRPM2−/−/APP/PS1 mice. Aβ levels in mouse hippocampus. A, B, Brain slices were stained for plaques with anti-Aβ(1–42) IgG (A) and thioflavin S (B). At least four coronal slices from each mouse brain and at least three brains of APP/PS1 and TRPM2−/−/APP/PS1 mice were used for each immunostaining experiment. C, D, Levels of Aβ(1–42) of insoluble (C) and Tris-soluble (D) fractions of at least 6 hippocampi of APP/PS1 and TRPM2−/−/APP/PS1 mice were analyzed by ELISA. E, Western blot analysis of intracellular and cell membrane-bound fractions of hippocampus. The lower band corresponds to the trimers, and the upper band to the tetramers of Aβ(1–42). At least five extracts obtained from each genotype were analyzed. *p < 0.05 (Student's t test).
Elimination of TRPM2 in APP/PS1 mice does not change age-related hypoactivity
To determine whether improved AD pathological hallmarks observed in APP/PS1 mice lacking TRPM2 channels translates into improved behavioral performance, we compared the performance of TRPM2−/−/APP/PS1 mutants with that of APP/PS1, WT, and TRPM2−/− controls. Importantly, the tests were performed longitudinally in the same cohort of mice at different time points, so that we could evaluate age-related progression of behavior deficits.
As hypoactivity is frequently observed both in human AD patients and transgenic mouse models of AD (Ferguson et al., 2013; Iqbal et al., 2013), we investigated locomotor activity using automated open field chambers. Locomotor activity was recorded at 3, 6, 9, 12, and 15 months of age. We detected no difference in activity in the four genotypes until 12 months of age (Fig. 6A). At 12–15 months of age, both APP/PS1 and TRPM2−/−/APP/PS1 mutants showed decreased locomotor activity compared with WT controls and TRPM2−/− mice [Fig. 6A; two-way ANOVA, significant main effect of APP/PS1 carriers: F(1,33) = 11.12, p = 0.0021; no differences between noncarriers (WT and TRPM2−/−): F(1,33) = 0.9240, p = 0.3438; and no interaction]. These results suggest that the hypoactivity phenotype was not affected by ablation of TRPM2 channels.
Figure 6.
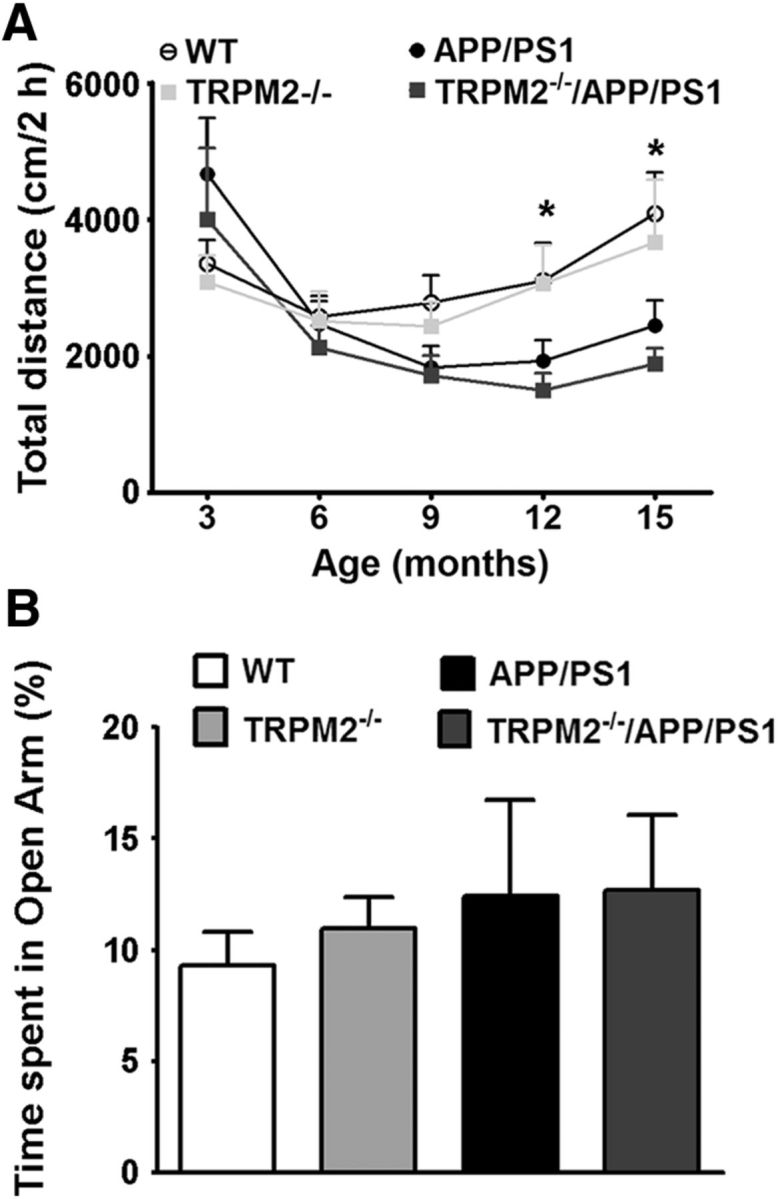
Elimination of TRPM2 in APP/PS1 mice does not change age-related hypoactivity or anxiety. A, Exploratory activity in the open field test of 3- to 15-month-old mice. At 15 months, carriers of APP/PS1 transgenes show decreased locomotion compared with noncarriers. B, Representative elevated plus maze exploration of 15-month-old mice. Bars represent the percentage of time spent by the four genotypes in the Open Arm of the maze. *p < 0.05. N for WT = 8–12, N for TRPM2−/− = 7–10, N for APP/PS1 = 10 or 11, N for TRPM2−/−/APP/PS1 = 10 or 11.
To test for anxiety behaviors, we performed the elevated plus-maze task (Fig. 6B). There were no differences in the time spent in the open arms among the four mouse genotypes (representative result for 15-month-old mice, ANOVA, no effect of APP/PS1 F(1,38) = 0.7546, p = 0.3905; no differences between noncarriers: F(1,38) = 0.1139, p = 0.7376; and no interaction). Hence, the APP/PS1 transgenes did not affect anxiety-like behavior. Moreover, TRPM2 elimination did not affect anxiety-like behavior.
Elimination of TRPM2 in APP/PS1 mice rescues spatial memory deficits
To test whether loss of TRPM2 had any effect on cognitive deficits in APP/PS1 mice, we used two spatial memory tests, the Barnes maze and the MWM. We tested mice at 3, 6, 9, 12, and 15 months of age in the Barnes maze and at 12 and 15 months in the MWM. Similar to previous reports (Savonenko et al., 2005), our Barnes maze analysis did not detect consistent spatial memory deficits when APP/PS1 mice were compared with controls until 12 months of age (Fig. 7A–D). However, APP/PS1 mice showed clear spatial deficits in the Barnes maze at 15 months of age (Fig. 7E,F). It is important to note that, because of the longitudinal nature of the study, the same cohort of mice was tested in the Barnes maze at 5 different time points. Even though the location of the target hole was changed between tests, analysis of latency time to reach the target role suggests that mice from all genotypes seem to remember the “task rules” as they performed well already on the first day of training from 6 months up to 12 months (Fig. 7A,B). Probe trials at 12 months showed no differences among genotypes for either the number of times mice visited the target hole or the target role preference index (Fig. 7C,D). At 15 months of age, no differences were observed among genotypes in the number of errors made before reaching the target hole (Fig. 7A), but mice carrying the APP/PS1 transgenes presented longer latencies to first visit the target hole than control mice [two-way repeated-measures ANOVA, significant main effect of genotype: F(3,119) = 4.883, p = 0.006, post hoc analysis showed significant differences, p < 0.05, when noncarriers (WT and TRPM2−/−) were compared against carriers (TRPM2−/−/APP/PS1 and APP/PS1); Fig. 7B]. Considering that both APP/PS1 and TRPM2−/−/APP/PS1 mice showed decreased locomotor activity at 15 months (Fig. 6A), the difference observed in latency to visit the target hole is more likely due to hypoactivity than to a learning deficiency. Strikingly, 15-month-old APP/PS1 mice made fewer target hole visits than the other genotypes during the probe trial (F(3,37) = 18.49, p = 0.0006, post hoc analysis showed significant differences, p < 0.05, when APP/PS1 mice were compared vs WT, TRPM2−/−, and TRPM2−/−/APP/PS1). In contrast, TRPM2−/−/APP/PS1 mice made as many visits to the target hole and preference index as WT or TRPM2−/− mice (Fig. 7E,F). These results suggest that elimination of TRPM2 alleviates the spatial memory deficits in the Barnes maze observed at 15 months in the AD mouse model.
Figure 7.

Elimination of TRPM2 in APP/PS1 mice rescues spatial memory deficits in the Barnes maze. Age-dependent impairment of APP/PS1 mice performance in Barnes maze task is rescued with TRPM2 removal. A, Errors to reach the target role for 3- to 15-month-old mice. B, Escape latency for 3- to 15-month old mice. C, D, On the fifth day, mice were subjected to a 90 s probe trial, with their spatial memory recall expressed as number of visits to the target hole (C) and the preference for the target hole over other holes during the probe trial (D) for 12-month-old mice. E, F, Similar to C, D, but for 15-month-old mice. Results are expressed as mean ± SEM. *p < 0.05. **p < 0.01. ***p < 0.001. N for WT = 8, N for TRPM2−/− = 8, N for APP/PS1 = 10, N for TRPM2−/−/APP/PS1 = 13.
Similar results were observed in MWM task (Fig. 8). Analysis of escape latency to the platform showed a decrease in the time to find the platform over training day for all genotypes at 12 months (Fig. 8A; two-way repeated-measures ANOVA, significant effect of training day: F(3,126) = 37.95, p < 0.001) and 15 months (Fig. 7E; two-way repeated-measures ANOVA, significant effect of training day: F(3,117) = 11.55, p < 0.001). This suggests that all genotypes were able to learn the task. Interestingly, mice carrying the APP/PS1 transgenes showed longer latencies to find the platform at both 12 months (two-way repeated-measures ANOVA, significant effect of genotype: F(3,126) = 3.632, p = 0.0203, post hoc analysis showed significant differences, p < 0.05, between WT and TRPM2−/− compared with TRPM2−/−/APP/PS1 and APP/PS1) and 15 months (two-way repeated-measures ANOVA, significant effect of genotype: F(3,117) = 4.167, p = 0.0118, post hoc analysis showed significant differences, p < 0.05, between WT and TRPM2−/− compared with TRPM2−/−/APP/PS1 and APP/PS1). Analysis of the distance traveled to find the target at 12 months (Fig. 7B) or 15 months (Fig. 7F) indicated significant main effects of genotype (12 months: two-way repeated-measures ANOVA, F(3,126) = 4.531, p = 0.0077; 15 months: two-way repeated-measures ANOVA, F(3,117) = 4.167, p = 0.0118) and day (12 months: two-way repeated-measures ANOVA, F(3,126) = 25.90, p < 0.0001; 15 months: two-way repeated-measures ANOVA, F(3,117) = 11.55, p < 0.0001). Overall, mice carrying the APP/PS1 transgenes, independent of the presence or absence of TRPM2, exhibited increased path lengths. However, at day 4, the performance of all genotypes was identical both at 12- and 15-month old mice. Additionally, swim speed analysis showed no differences in performance among genotypes at 12 months (Fig. 8C; two-way repeated-measures ANOVA, no differences in genotype: F(3,123) = 0.5156, p = 0.6738) or 15 months (Fig. 8G; two-way repeated-measures ANOVA, no differences in genotype: F(3,117) = 0.9244, p = 0.4380), indicating that differences observed in the MWM between mice expressing the APP/PS1 transgenes compared with those without it are not due to the decreased physical activity in the pool. Together, these results suggest that 12- and 15-month-old mice carrying the APP/PS1 transgenes showed difficulties learning the MWM spatial task as they took longer than WT and TRPM2−/− to learn the location of the platform; however, the deficit did not seem to be extensive, as it disappeared after repetitive training.
Figure 8.

Elimination of TRPM2 in APP/PS1 mice rescues age-dependent spatial memory deficits in the MWM. MWM testing of 12-month-old mice (A–D) and 15-month-old mice (E–H) was performed as described in Materials and Methods. A, E, Learning curves showing the average latency. B, F, Distance traveled to reach the platform. C, G, Mean swimming speed during the acquisition of the MWM paradigm. D, H, The percentage of time spent by each genotype swimming on the quadrants during the probe trial. Data are mean ± SEM. *p < 0.05 (vs T). **p < 0.01 (vs T). ***p < 0.001 (vs T). ****p < 0.0001 (vs T). T, Target hole; O, opposite quadrant; L, left quadrant; R, right quadrant. N values for WT = 12, TRPM2−/− = 10, APP/PS1 = 11, TRPM2−/−/APP/PS1 = 13.
The MWM probe trial in 12-month-old mice showed no significant differences among genotypes (Fig. 8D) and a significant main effect of quadrant (F(3,140) = 52.178, p < 0.001). Further analysis indicated that all genotypes spent more time in the target quadrant than in any other quadrant (p < 0.001, for Target vs opposite, Target vs right, and Target vs left). Analysis of the percentage of time searching in the target quadrant showed no differences in memory retention among the four genotypes (F(3,35) = 0.3544, p = 0.7863). These results indicate that there are no memory impairments in any of the mouse groups at 12 months. At 15 months (Fig. 8H), the results showed that APP/PS1 mice spent significantly less time in the target area than the other genotypes (F(3,39) = 5.887, p = 0.0020, post hoc analysis: p < 0.05 vs TRPM2−/−/APP/PS1, WT, and TRPM2−/−), whereas TRPM2−/−/APP/PS1 mice performed similarly to the WT animals (p > 0.05). These results show that 15-month-old APP/PS1 mice were unable to recall where the platform was located, and this spatial memory deficit was rescued in TRPM2−/−/APP/PS1 mice.
In conclusion, Barnes maze and MWM results indicate that APP/PS1 mice present clear age-dependent spatial memory deficits and that elimination of TRPM2 channels ameliorates these deficits.
Discussion
The importance of TRPM2 to CNS function is underscored by studies implicating TRPM2 in diseases, including stroke/ischemia (Jia et al., 2011; Verma et al., 2012; Alim et al., 2013), neurodegenerative diseases (Fonfria et al., 2005; Hermosura et al., 2008), bipolar disorder (Xu et al., 2006; Jang et al., 2015), and epilepsy (Katano et al., 2012). These studies suggest that abnormal TRPM2 activity can profoundly influence CNS function and pathology. We now show that Aβ oligomers can facilitate TRPM2 channel function and that elimination of TRPM2 can reverse pathological and behavioral deficits observed in the AD mouse model APP/PS1.
APP/PS1 mice are considered a less aggressive model of Aβ toxicity, probably reflecting a prodromal phase of AD (Ashe and Zahs, 2010; Ferretti et al., 2012). Starting at 6 months of age, APP/PS1 present significant amounts of soluble and plaque-associated Aβ (Savonenko et al., 2005; Ostapchenko et al., 2013a). Most transgenic AD mouse models present spatial learning and memory deficits, as assessed by MWM performance (Webster et al., 2014) or by Barnes maze (O'Leary and Brown, 2009). However, there are discrepancies for when these deficits first manifest (Ding et al., 2008; Timmer et al., 2010). Our experiments suggest that the performance of APP/PS1 mice on a battery of behavioral tasks cannot be distinguished from that of control mice until reaching 12–15 months of age. At this time, APP/PS1 mice exhibit spatial memory deficits in both MWM and Barnes maze tasks compared with wild-type counterparts. Importantly, elimination of TRPM2 channels ameliorated these deficits. The improved performance in both the Barnes maze and MWM tests with loss of TRPM2 expression in APP/PS1 is not related to changes in physical activity as both mouse lines showed similar levels of activity at all ages tested. Nonetheless, APP/PS1 mice, with or without TRPM2 expressed, showed decreased activity by 12–15 months of age, which corroborate previous findings (Ferguson et al., 2013). Although ablation of TRPM2 channels improved many AD pathology hallmarks in APP/PS1 mice, it did not improve the hypoactivity phenotype. This hypoactivity could be the result of Aβ-derived disturbances in muscular function (Mukhamedyarov et al., 2014). Indeed, Aβ peptides are elevated in skeletal muscles of AD patients (Kuo et al., 2000), where they have been suggested to cause membrane depolarization in fibers by inhibiting the Na+/K+-ATPase and by forming membrane pores (Mukhamedyarov et al., 2011, 2014). Moreover, Aβ oligomers have been detected in the muscle fibers of sporadic inclusion body myositis patients (Nogalska et al., 2010). TRPM2 expression in skeletal muscles is low (Fonfria et al., 2006a), which might explain the lack of improvement in AD-related hypoactivity.
Although the precise mechanisms underlying the improvement of spatial memory deficits by removal of TRPM2 are unknown, they are likely related to the reduced pathological markers noted in aged TRPM2−/−/APP/PS1 compared with APP/PS1 mice. The longitudinal design of our study precluded biochemical and histological analyses at time points corresponding with behavioral assays. Nevertheless, similar to AD patients (Kim et al., 2000; Andreu et al., 2012), the brains of 20- to 24-month-old APP/PS1 mice present upregulated PDI and increased phosphorylation of eIF2α, two molecular markers of ER stress (Matus et al., 2011; Scheper et al., 2011; Hetz and Mollereau, 2014). Interestingly, levels of pSer51-eIF2α immunoreactivity in TRPM2−/− mice were slightly higher than in controls, but not due to increased ER stress, as PDI levels between these two genotypes were not different. Additionally, 20- to 24-month-old APP/PS1 mice presented decreased levels of synaptophysin and increased levels of Iba1, suggesting that their brains presented synaptic loss and increased microglial activation. Brains of 20- to 24-month-old APP/PS1 mice lacking TRPM2 channels (TRPM2−/−/APP/PS1 mice) did not show any of these deficits, indicating that removal of the Ca2+-permeable oxidative stress sensor TRPM2 can help to normalize the unfolded protein response and thereby moderate ER stress, synaptic loss, and inflammation in AD mice. In agreement with this possibility, in the absence of TRPM2, we did not detect abnormal expression of other genes that could regulate Ca2+ dynamics. Interestingly, the fact that elimination of TRPM2 does not affect ER stress responses due to thapsigargin and tunicamycin treatment suggests that TRPM2 elimination does not affect ER stress responses indiscriminately but rather is selective for Aβ-mediated effects.
Other mechanisms may also be at play after TRPM2 removal. For example, genetic ablation of TRPM2 is associated with increased phosphorylation of Akt and inactivation of glycogen synthase kinase GSK3β (Xie et al., 2011; Jang et al., 2015), which was recently found to be beneficial in AD mouse models (Kazim et al., 2014). This effect could balance GSK3β activation by PP2B, a phosphatase whose activity has been directly linked to NMDAR dysregulation by Aβ (Snyder et al., 2005). Therefore, loss of TRPM2 may alter the balance between prosurvival and prodeath signaling in neurons. Also, TRPM2 is present in microglia, where it can activate microglia through ROS- and LPS-mediated signaling, promoting cytokine release and inflammation (Kraft et al., 2004; Fonfria et al., 2005, 2006b; Wehrhahn et al., 2010). AD is also characterized by microglia activation (Kalaria et al., 1996; Dhawan et al., 2012; Ferretti et al., 2012), which we confirm in the present study. Genetic deletion of TRPM2 mitigates this response providing an additional beneficial outcome via reduced neuroinflammation. Last, increasing evidence supports the correlation between cognitive deficits with the levels of soluble AβOs, both in AD patients and AD models (Lesné et al., 2006, 2013; DaRocha-Souto et al., 2011). Although TRPM2 deletion did not change the production of Aβ in APP/PS1 mice, it decreased the amount of small cell membrane-associated AβOs, particularly the trimer and tetramer species. This may consequently reduce Aβ synaptotoxicity (Figueiredo et al., 2013; Ostapchenko et al., 2013b).
Several mechanisms through which Aβ peptides augment TRPM2 function can be suggested, including oxidative stress, elevated Ca2+ and aberrant NMDAR stimulation (Mattson et al., 1999; De Felice et al., 2007; Malinow, 2012; Um et al., 2012; You et al., 2012). TRPM2 gating by oxidative/nitrosative stress occurs via the production of its agonist, NAD+-derived ADPR (Perraud et al., 2001). Mitochondria represent a major cellular store of NAD+ and ADPR derived from this source contributes to TRPM2 activation during oxidative stress (Perraud et al., 2005). Aβ-induced oxidative/nitrosative stress, associated with mitochondrial damage (Mattson et al., 1999), may represent one source of ADPR. Alternatively, ADPR production may be stimulated by DNA repair pathways downstream of oxidative/nitrosative stress-induced DNA damage (Park et al., 2014). In addition to ADPR, gating of TRPM2 requires binding of Ca2+/calmodulin to the N terminus of TRPM2 (Tong et al., 2006; Du et al., 2009). We previously reported that ADPR-promoted TRPM2 activation requires Ca2+ influx through voltage-gated Ca2+ or NMDAR channels (Olah et al., 2009). Coincidentally, Aβ can rapidly associate with excitatory synapses in close proximity, if not directly to NMDARs themselves (Lacor et al., 2004), and provoke Ca2+ dysregulation via aberrant NMDAR stimulation (De Felice et al., 2007; Shankar et al., 2008; Li et al., 2011; You et al., 2012). Of note, the action of Aβ peptides upon NMDARs is multifaceted with either increased (Li et al., 2011; You et al., 2012) or decreased function (Snyder et al., 2005; Shankar et al., 2007) having been reported. Reconciling these two opposing functional endpoints, recent evidence suggests that an initial increase in NMDAR function by Aβ treatment is followed by a loss of surface expression and receptor function with extended treatment (Um et al., 2012). Importantly, TRPM2 function can be augmented regardless of the direction of change in NMDAR function as a consequence of intracellular Ca2+ elevations and ROS/RNS production associated with Aβ treatment.
It is important to mention that several distinct genetic manipulations in mice have shown promising effects on the toxicity of Aβ peptides or levels of APP metabolites (Lambert et al., 1998; Ohno et al., 2006; Matus et al., 2011; Ma et al., 2013; Jiang et al., 2014). Not all manipulations are expected to lead to therapeutic strategies in humans due to potential experimental limitations. Of note, independent experiments, published during review of our manuscript, have also shown that TRPM2 in endothelial cells in the brain vasculature is a target for Aβ peptides (Park et al., 2014). Particularly, Aβ-induced oxidative stress and DNA damage resulted in overproduction of ADPR, leading to TRPM2 activation and calcium imbalance in cerebrovascular cells and eventually impaired cerebral blood flow. Together with our findings, these results argue that TRPM2 could be a connecting point between oxidative stress and cytotoxic effects in AD. Moreover, the improvement in molecular markers of AD pathology and spatial memory deficits in APP/PS1 mice lacking TRPM2 suggests that development of drugs selectively targeting this critical channel could find use for treating AD. Importantly, we did not detect any major behavior deficits in mice lacking TRPM2. In stark contrast to the limited therapeutic index observed for drugs targeting the NMDAR (Farber et al., 2002), this portends the possibility of an improved therapeutic index for drugs aimed at reducing aberrant TRPM2 activation. This is especially important given the paucity of available drugs for treating AD patients.
Footnotes
This work was supported by PrioNet-Canada and the Canadian Institutes of Health Research CIHR, MOP 93651, MAMP, MOP89919, VFP, MOP-97771 to J.F.M. and M.F.J.
The corresponding authors dedicate this manuscript to the memory of our dear friend and mentor, Dr. John F. MacDonald. Dr. MacDonald was a key contributor to the development of this project, having proposed the initial hypothesis and experimental design. He also supervised M.C. during her MSc thesis work. His keen intellect and scientific insight, his dedication and loyalty to his trainees, collaborators and colleagues as well as his sincere friendship will be greatly missed.
The authors declare no competing financial interests.
References
- Aarts M, Iihara K, Wei WL, Xiong ZG, Arundine M, Cerwinski W, MacDonald JF, Tymianski M. A key role for TRPM7 channels in anoxic neuronal death. Cell. 2003;115:863–877. doi: 10.1016/S0092-8674(03)01017-1. [DOI] [PubMed] [Google Scholar]
- Alim I, Teves L, Li R, Mori Y, Tymianski M. Modulation of NMDAR subunit expression by TRPM2 channels regulates neuronal vulnerability to ischemic cell death. J Neurosci. 2013;33:17264–17277. doi: 10.1523/JNEUROSCI.1729-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreu CI, Woehlbier U, Torres M, Hetz C. Protein disulfide isomerases in neurodegeneration: from disease mechanisms to biomedical applications. FEBS Lett. 2012;586:2826–2834. doi: 10.1016/j.febslet.2012.07.023. [DOI] [PubMed] [Google Scholar]
- Ashe KH, Zahs KR. Probing the biology of Alzheimer's disease in mice. Neuron. 2010;66:631–645. doi: 10.1016/j.neuron.2010.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai JZ, Lipski J. Differential expression of TRPM2 and TRPV4 channels and their potential role in oxidative stress-induced cell death in organotypic hippocampal culture. Neurotoxicology. 2010;31:204–214. doi: 10.1016/j.neuro.2010.01.001. [DOI] [PubMed] [Google Scholar]
- Belrose JC, Xie YF, Gierszewski LJ, MacDonald JF, Jackson MF. Loss of glutathione homeostasis associated with neuronal senescence facilitates TRPM2 channel activation in cultured hippocampal pyramidal neurons. Mol Brain. 2012;5:11. doi: 10.1186/1756-6606-5-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boda E, Pini A, Hoxha E, Parolisi R, Tempia F. Selection of reference genes for quantitative real-time RT-PCR studies in mouse brain. J Mol Neurosci. 2009;37:238–253. doi: 10.1007/s12031-008-9128-9. [DOI] [PubMed] [Google Scholar]
- Breier A, Malhotra AK, Pinals DA, Weisenfeld NI, Pickar D. Association of ketamine-induced psychosis with focal activation of the prefrontal cortex in healthy volunteers. Am J Psychiatry. 1997;154:805–811. doi: 10.1176/ajp.154.6.805. [DOI] [PubMed] [Google Scholar]
- Caetano FA, Beraldo FH, Hajj GN, Guimaraes AL, Jürgensen S, Wasilewska-Sampaio AP, Hirata PH, Souza I, Machado CF, Wong DY, De Felice FG, Ferreira ST, Prado VF, Rylett RJ, Martins VR, Prado MA. Amyloid-beta oligomers increase the localization of prion protein at the cell surface. J Neurochem. 2011;117:538–553. doi: 10.1111/j.1471-4159.2011.07225.x. [DOI] [PubMed] [Google Scholar]
- DaRocha-Souto B, Scotton TC, Coma M, Serrano-Pozo A, Hashimoto T, Serenó L, Rodríguez M, Sánchez B, Hyman BT, Gómez-Isla T. Brain oligomeric beta-amyloid but not total amyloid plaque burden correlates with neuronal loss and astrocyte inflammatory response in amyloid precursor protein/tau transgenic mice. J Neuropathol Exp Neurol. 2011;70:360–376. doi: 10.1097/NEN.0b013e318217a118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker H, Jürgensen S, Adrover MF, Brito-Moreira J, Bomfim TR, Klein WL, Epstein AL, De Felice FG, Jerusalinsky D, Ferreira ST. N-methyl-d-aspartate receptors are required for synaptic targeting of Alzheimer's toxic amyloid-beta peptide oligomers. J Neurochem. 2010;115:1520–1529. doi: 10.1111/j.1471-4159.2010.07058.x. [DOI] [PubMed] [Google Scholar]
- De Felice FG, Velasco PT, Lambert MP, Viola K, Fernandez SJ, Ferreira ST, Klein WL. Abeta oligomers induce neuronal oxidative stress through an N-methyl-d-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J Biol Chem. 2007;282:11590–11601. doi: 10.1074/jbc.M607483200. [DOI] [PubMed] [Google Scholar]
- Demuro A, Parker I, Stutzmann GE. Calcium signaling and amyloid toxicity in Alzheimer disease. J Biol Chem. 2010;285:12463–12468. doi: 10.1074/jbc.R109.080895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhawan G, Floden AM, Combs CK. Amyloid-beta oligomers stimulate microglia through a tyrosine kinase dependent mechanism. Neurobiol Aging. 2012;33:2247–2261. doi: 10.1016/j.neurobiolaging.2011.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y, Qiao A, Wang Z, Goodwin JS, Lee ES, Block ML, Allsbrook M, McDonald MP, Fan GH. Retinoic acid attenuates beta-amyloid deposition and rescues memory deficits in an Alzheimer's disease transgenic mouse model. J Neurosci. 2008;28:11622–11634. doi: 10.1523/JNEUROSCI.3153-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Xie J, Yue L. Intracellular calcium activates TRPM2 and its alternative spliced isoforms. Proc Natl Acad Sci U S A. 2009;106:7239–7244. doi: 10.1073/pnas.0811725106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farber NB, Kim SH, Dikranian K, Jiang XP, Heinkel C. Receptor mechanisms and circuitry underlying NMDA antagonist neurotoxicity. Mol Psychiatry. 2002;7:32–43. doi: 10.1038/sj.mp.4000912. [DOI] [PubMed] [Google Scholar]
- Ferguson SA, Sarkar S, Schmued LC. Longitudinal behavioral changes in the APP/PS1 transgenic Alzheimer's disease model. Behav Brain Res. 2013;242:125–134. doi: 10.1016/j.bbr.2012.12.055. [DOI] [PubMed] [Google Scholar]
- Ferreira ST, Klein WL. The Abeta oligomer hypothesis for synapse failure and memory loss in Alzheimer's disease. Neurobiol Learn Mem. 2011;96:529–543. doi: 10.1016/j.nlm.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferretti MT, Bruno MA, Ducatenzeiler A, Klein WL, Cuello AC. Intracellular Abeta-oligomers and early inflammation in a model of Alzheimer's disease. Neurobiol Aging. 2012;33:1329–1342. doi: 10.1016/j.neurobiolaging.2011.01.007. [DOI] [PubMed] [Google Scholar]
- Figueiredo CP, Clarke JR, Ledo JH, Ribeiro FC, Costa CV, Melo HM, Mota-Sales AP, Saraiva LM, Klein WL, Sebollela A, De Felice FG, Ferreira ST. Memantine rescues transient cognitive impairment caused by high-molecular-weight abeta oligomers but not the persistent impairment induced by low-molecular-weight oligomers. J Neurosci. 2013;33:9626–9634. doi: 10.1523/JNEUROSCI.0482-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonfria E, Marshall IC, Boyfield I, Skaper SD, Hughes JP, Owen DE, Zhang W, Miller BA, Benham CD, McNulty S. Amyloid beta-peptide(1–42) and hydrogen peroxide-induced toxicity are mediated by TRPM2 in rat primary striatal cultures. J Neurochem. 2005;95:715–723. doi: 10.1111/j.1471-4159.2005.03396.x. [DOI] [PubMed] [Google Scholar]
- Fonfria E, Murdock PR, Cusdin FS, Benham CD, Kelsell RE, McNulty S. Tissue distribution profiles of the human TRPM cation channel family. J Recept Signal Transduct Res. 2006a;26:159–178. doi: 10.1080/10799890600637506. [DOI] [PubMed] [Google Scholar]
- Fonfria E, Mattei C, Hill K, Brown JT, Randall A, Benham CD, Skaper SD, Campbell CA, Crook B, Murdock PR, Wilson JM, Maurio FP, Owen DE, Tilling PL, McNulty S. TRPM2 is elevated in the tMCAO stroke model, transcriptionally regulated, and functionally expressed in C13 microglia. J Recept Signal Transduct Res. 2006b;26:179–198. doi: 10.1080/10799890600637522. [DOI] [PubMed] [Google Scholar]
- Guzman MS, De Jaeger X, Raulic S, Souza IA, Li AX, Schmid S, Menon RS, Gainetdinov RR, Caron MG, Bartha R, Prado VF, Prado MA. Elimination of the vesicular acetylcholine transporter in the striatum reveals regulation of behaviour by cholinergic-glutamatergic co-transmission. PLoS Biol. 2011;9:e1001194. doi: 10.1371/journal.pbio.1001194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermosura MC, Cui AM, Go RC, Davenport B, Shetler CM, Heizer JW, Schmitz C, Mocz G, Garruto RM, Perraud AL. Altered functional properties of a TRPM2 variant in Guamanian ALS and PD. Proc Natl Acad Sci U S A. 2008;105:18029–18034. doi: 10.1073/pnas.0808218105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C, Mollereau B. Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat Rev Neurosci. 2014;15:233–249. doi: 10.1038/nrn3689. [DOI] [PubMed] [Google Scholar]
- Ikonomidou C, Stefovska V, Turski L. Neuronal death enhanced by N-methyl-d-aspartate antagonists. Proc Natl Acad Sci U S A. 2000;97:12885–12890. doi: 10.1073/pnas.220412197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal K, Flory M, Soininen H. Clinical symptoms and symptom signatures of Alzheimer's disease subgroups. J Alzheimers Dis. 2013;37:475–481. doi: 10.3233/JAD-130899. [DOI] [PubMed] [Google Scholar]
- Jang Y, Lee SH, Lee B, Jung S, Khalid A, Uchida K, Tominaga M, Jeon D, Oh U. TRPM2, a susceptibility gene for bipolar disorder, regulates glycogen synthase kinase-3 activity in the brain. J Neurosci. 2015;35:11811–11823. doi: 10.1523/JNEUROSCI.5251-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowsky JL, Xu G, Fromholt D, Gonzales V, Borchelt DR. Environmental enrichment exacerbates amyloid plaque formation in a transgenic mouse model of Alzheimer disease. J Neuropathol Exp Neurol. 2003;62:1220–1227. doi: 10.1093/jnen/62.12.1220. [DOI] [PubMed] [Google Scholar]
- Jia J, Verma S, Nakayama S, Quillinan N, Grafe MR, Hurn PD, Herson PS. Sex differences in neuroprotection provided by inhibition of TRPM2 channels following experimental stroke. J Cereb Blood Flow Metab. 2011;31:2160–2168. doi: 10.1038/jcbfm.2011.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang T, Tan L, Zhu XC, Zhang QQ, Cao L, Tan MS, Gu LZ, Wang HF, Ding ZZ, Zhang YD, Yu JT. Upregulation of TREM2 ameliorates neuropathology and rescues spatial cognitive impairment in a transgenic mouse model of Alzheimer's disease. Neuropsychopharmacology. 2014;39:2949–2962. doi: 10.1038/npp.2014.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalaria RN, Cohen DL, Premkumar DR. Cellular aspects of the inflammatory response in Alzheimer's disease. Neurodegeneration. 1996;5:497–503. doi: 10.1006/neur.1996.0069. [DOI] [PubMed] [Google Scholar]
- Katano M, Numata T, Aguan K, Hara Y, Kiyonaka S, Yamamoto S, Miki T, Sawamura S, Suzuki T, Yamakawa K, Mori Y. The juvenile myoclonic epilepsy-related protein EFHC1 interacts with the redox-sensitive TRPM2 channel linked to cell death. Cell Calcium. 2012;51:179–185. doi: 10.1016/j.ceca.2011.12.011. [DOI] [PubMed] [Google Scholar]
- Kazim SF, Blanchard J, Dai CL, Tung YC, LaFerla FM, Iqbal IG, Iqbal K. Disease modifying effect of chronic oral treatment with a neurotrophic peptidergic compound in a triple transgenic mouse model of Alzheimer's disease. Neurobiol Dis. 2014;71C:110–130. doi: 10.1016/j.nbd.2014.07.001. [DOI] [PubMed] [Google Scholar]
- Kim HT, Russell RL, Raina AK, Harris PL, Siedlak SL, Zhu X, Petersen RB, Shimohama S, Smith MA, Perry G. Protein disulfide isomerase in Alzheimer disease. Antiox Redox Signal. 2000;2:485–489. doi: 10.1089/15230860050192260. [DOI] [PubMed] [Google Scholar]
- Knowles H, Li Y, Perraud AL. The TRPM2 ion channel, an oxidative stress and metabolic sensor regulating innate immunity and inflammation. Immunol Res. 2013;55:241–248. doi: 10.1007/s12026-012-8373-8. [DOI] [PubMed] [Google Scholar]
- Kraft R, Grimm C, Grosse K, Hoffmann A, Sauerbruch S, Kettenmann H, Schultz G, Harteneck C. Hydrogen peroxide and ADP-ribose induce TRPM2-mediated calcium influx and cation currents in microglia. Am J Physiol Cell Physiol. 2004;286:C129–C137. doi: 10.1152/ajpcell.00331.2003. [DOI] [PubMed] [Google Scholar]
- Kudo W, Lee HP, Zou WQ, Wang X, Perry G, Zhu X, Smith MA, Petersen RB, Lee HG. Cellular prion protein is essential for oligomeric amyloid-beta-induced neuronal cell death. Human Mol Genet. 2012;21:1138–1144. doi: 10.1093/hmg/ddr542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo YM, Kokjohn TA, Watson MD, Woods AS, Cotter RJ, Sue LI, Kalback WM, Emmerling MR, Beach TG, Roher AE. Elevated abeta42 in skeletal muscle of Alzheimer disease patients suggests peripheral alterations of AbetaPP metabolism. Am J Pathol. 2000;156:797–805. doi: 10.1016/S0002-9440(10)64947-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Chang L, Fernandez SJ, Gong Y, Viola KL, Lambert MP, Velasco PT, Bigio EH, Finch CE, Krafft GA, Klein WL. Synaptic targeting by Alzheimer's-related amyloid beta oligomers. J Neurosci. 2004;24:10191–10200. doi: 10.1523/JNEUROSCI.3432-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, Viola KL, Klein WL. Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer's disease. J Neurosci. 2007;27:796–807. doi: 10.1523/JNEUROSCI.3501-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFerla FM. Calcium dyshomeostasis and intracellular signalling in Alzheimer's disease. Nat Rev Neurosci. 2002;3:862–872. doi: 10.1038/nrn960. [DOI] [PubMed] [Google Scholar]
- Lahti AC, Koffel B, LaPorte D, Tamminga CA. Subanesthetic doses of ketamine stimulate psychosis in schizophrenia. Neuropsychopharmacology. 1995;13:9–19. doi: 10.1016/0893-133X(94)00131-I. [DOI] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Abeta1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurén J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature. 2009;457:1128–1132. doi: 10.1038/nature07761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesné SE, Sherman MA, Grant M, Kuskowski M, Schneider JA, Bennett DA, Ashe KH. Brain amyloid-beta oligomers in ageing and Alzheimer's disease. Brain. 2013;136:1383–1398. doi: 10.1093/brain/awt062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesné S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- Li S, Jin M, Koeglsperger T, Shepardson NE, Shankar GM, Selkoe DJ. Soluble Abeta oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J Neurosci. 2011;31:6627–6638. doi: 10.1523/JNEUROSCI.0203-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma QL, Yang F, Rosario ER, Ubeda OJ, Beech W, Gant DJ, Chen PP, Hudspeth B, Chen C, Zhao Y, Vinters HV, Frautschy SA, Cole GM. Beta-amyloid oligomers induce phosphorylation of tau and inactivation of insulin receptor substrate via c-Jun N-terminal kinase signaling: suppression by omega-3 fatty acids and curcumin. J Neurosci. 2009;29:9078–9089. doi: 10.1523/JNEUROSCI.1071-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma T, Trinh MA, Wexler AJ, Bourbon C, Gatti E, Pierre P, Cavener DR, Klann E. Suppression of eIF2alpha kinases alleviates Alzheimer's disease-related plasticity and memory deficits. Nat Neurosci. 2013;16:1299–1305. doi: 10.1038/nn.3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinow R. New developments on the role of NMDA receptors in Alzheimer's disease. Curr Opin Neurobiol. 2012;22:559–563. doi: 10.1016/j.conb.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins-Silva C, De Jaeger X, Guzman MS, Lima RD, Santos MS, Kushmerick C, Gomez MV, Caron MG, Prado MA, Prado VF. Novel strains of mice deficient for the vesicular acetylcholine transporter: insights on transcriptional regulation and control of locomotor behavior. PLoS One. 2011;6:e17611. doi: 10.1371/journal.pone.0017611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martyn AC, De Jaeger X, Magalhães AC, Kesarwani R, Gonçalves DF, Raulic S, Guzman MS, Jackson MF, Izquierdo I, Macdonald JF, Prado MA, Prado VF. Elimination of the vesicular acetylcholine transporter in the forebrain causes hyperactivity and deficits in spatial memory and long-term potentiation. Proc Natl Acad Sci U S A. 2012;109:17651–17656. doi: 10.1073/pnas.1215381109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP. Pathways towards and away from Alzheimer's disease. Nature. 2004;430:631–639. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Pedersen WA, Duan W, Culmsee C, Camandola S. Cellular and molecular mechanisms underlying perturbed energy metabolism and neuronal degeneration in Alzheimer's and Parkinson's diseases. Ann N Y Acad Sci. 1999;893:154–175. doi: 10.1111/j.1749-6632.1999.tb07824.x. [DOI] [PubMed] [Google Scholar]
- Matus S, Glimcher LH, Hetz C. Protein folding stress in neurodegenerative diseases: a glimpse into the ER. Curr Opin Cell Biol. 2011;23:239–252. doi: 10.1016/j.ceb.2011.01.003. [DOI] [PubMed] [Google Scholar]
- Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994;12:529–540. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- Mukhamedyarov MA, Teplov AY, Grishin SN, Leushina AV, Zefirov AL, Palotás A. Extraneuronal toxicity of Alzheimer's beta-amyloid peptide: comparative study on vertebrate skeletal muscles. Muscle Nerve. 2011;43:872–877. doi: 10.1002/mus.22000. [DOI] [PubMed] [Google Scholar]
- Mukhamedyarov MA, Volkov EM, Khaliullina DF, Grigoryev PN, Zefirov AL, Palotás A. Impaired electro-genesis in skeletal muscle fibers of transgenic Alzheimer mice. Neurochem Int. 2014;64:24–28. doi: 10.1016/j.neuint.2013.10.014. [DOI] [PubMed] [Google Scholar]
- Newcomer JW, Farber NB, Jevtovic-Todorovic V, Selke G, Melson AK, Hershey T, Craft S, Olney JW. Ketamine-induced NMDA receptor hypofunction as a model of memory impairment and psychosis. Neuropsychopharmacology. 1999;20:106–118. doi: 10.1016/S0893-133X(98)00067-0. [DOI] [PubMed] [Google Scholar]
- Nogalska A, D'Agostino C, Engel WK, Klein WL, Askanas V. Novel demonstration of amyloid-beta oligomers in sporadic inclusion-body myositis muscle fibers. Acta Neuropathol. 2010;120:661–666. doi: 10.1007/s00401-010-0737-3. [DOI] [PubMed] [Google Scholar]
- Ohno M, Chang L, Tseng W, Oakley H, Citron M, Klein WL, Vassar R, Disterhoft JF. Temporal memory deficits in Alzheimer's mouse models: rescue by genetic deletion of BACE1. Eur J Neurosci. 2006;23:251–260. doi: 10.1111/j.1460-9568.2005.04551.x. [DOI] [PubMed] [Google Scholar]
- Olah ME, Jackson MF, Li H, Perez Y, Sun HS, Kiyonaka S, Mori Y, Tymianski M, MacDonald JF. Ca2+-dependent induction of TRPM2 currents in hippocampal neurons. J Physiol. 2009;587:965–979. doi: 10.1113/jphysiol.2008.162289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Leary TP, Brown RE. Visuo-spatial learning and memory deficits on the Barnes maze in the 16-month-old APPswe/PS1dE9 mouse model of Alzheimer's disease. Behav Brain Res. 2009;201:120–127. doi: 10.1016/j.bbr.2009.01.039. [DOI] [PubMed] [Google Scholar]
- Ostapchenko VG, Beraldo FH, Mohammad AH, Xie YF, Hirata PH, Magalhaes AC, Lamour G, Li H, Maciejewski A, Belrose JC, Teixeira BL, Fahnestock M, Ferreira ST, Cashman NR, Hajj GN, Jackson MF, Choy WY, MacDonald JF, Martins VR, Prado VF, Prado MA. The prion protein ligand, stress-inducible phosphoprotein 1, regulates amyloid-beta oligomer toxicity. J Neurosci. 2013b;33:16552–16564. doi: 10.1523/JNEUROSCI.3214-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostapchenko VG, Beraldo FH, Guimarães AL, Mishra S, Guzman M, Fan J, Martins VR, Prado VF, Prado MA. Increased prion protein processing and expression of metabotropic glutamate receptor 1 in a mouse model of Alzheimer's disease. J Neurochem. 2013a;127:415–425. doi: 10.1111/jnc.12296. [DOI] [PubMed] [Google Scholar]
- Park L, Wang G, Moore J, Girouard H, Zhou P, Anrather J, Iadecola C. The key role of transient receptor potential melastatin-2 channels in amyloid-beta-induced neurovascular dysfunction. Nat Commun. 2014;5:5318. doi: 10.1038/ncomms6318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perraud AL, Fleig A, Dunn CA, Bagley LA, Launay P, Schmitz C, Stokes AJ, Zhu Q, Bessman MJ, Penner R, Kinet JP, Scharenberg AM. ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature. 2001;411:595–599. doi: 10.1038/35079100. [DOI] [PubMed] [Google Scholar]
- Perraud AL, Takanishi CL, Shen B, Kang S, Smith MK, Schmitz C, Knowles HM, Ferraris D, Li W, Zhang J, Stoddard BL, Scharenberg AM. Accumulation of free ADP-ribose from mitochondria mediates oxidative stress-induced gating of TRPM2 cation channels. J Biol Chem. 2005;280:6138–6148. doi: 10.1074/jbc.M411446200. [DOI] [PubMed] [Google Scholar]
- Pozueta J, Lefort R, Shelanski ML. Synaptic changes in Alzheimer's disease and its models. Neuroscience. 2013;251:51–65. doi: 10.1016/j.neuroscience.2012.05.050. [DOI] [PubMed] [Google Scholar]
- Rammsayer TH. Effects of pharmacologically induced changes in NMDA-receptor activity on long-term memory in humans. Learn Mem. 2001;8:20–25. doi: 10.1101/lm.33701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renner M, Lacor PN, Velasco PT, Xu J, Contractor A, Klein WL, Triller A. Deleterious effects of amyloid beta oligomers acting as an extracellular scaffold for mGluR5. Neuron. 2010;66:739–754. doi: 10.1016/j.neuron.2010.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savonenko A, Xu GM, Melnikova T, Morton JL, Gonzales V, Wong MP, Price DL, Tang F, Markowska AL, Borchelt DR. Episodic-like memory deficits in the APPswe/PS1dE9 mouse model of Alzheimer's disease: relationships to beta-amyloid deposition and neurotransmitter abnormalities. Neurobiol Dis. 2005;18:602–617. doi: 10.1016/j.nbd.2004.10.022. [DOI] [PubMed] [Google Scholar]
- Scheper W, Nijholt DA, Hoozemans JJ. The unfolded protein response and proteostasis in Alzheimer disease: preferential activation of autophagy by endoplasmic reticulum stress. Autophagy. 2011;7:910–911. doi: 10.4161/auto.7.8.15761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt EK, Clavarino G, Ceppi M, Pierre P. SUnSET, a nonradioactive method to monitor protein synthesis. Nat Methods. 2009;6:275–277. doi: 10.1038/nmeth.1314. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease. Cold Spring Harbor Perspect Biol. 2011;3 doi: 10.1101/cshperspect.a004457. piia004457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27:2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, Greengard P. Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci. 2005;8:1051–1058. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- Timmer NM, van Dijk L, van der Zee CE, Kiliaan A, de Waal RM, Verbeek MM. Enoxaparin treatment administered at both early and late stages of amyloid beta deposition improves cognition of APPswe/PS1dE9 mice with differential effects on brain Abeta levels. Neurobiol Dis. 2010;40:340–347. doi: 10.1016/j.nbd.2010.06.008. [DOI] [PubMed] [Google Scholar]
- Tong Q, Zhang W, Conrad K, Mostoller K, Cheung JY, Peterson BZ, Miller BA. Regulation of the transient receptor potential channel TRPM2 by the Ca2+ sensor calmodulin. J Biol Chem. 2006;281:9076–9085. doi: 10.1074/jbc.M510422200. [DOI] [PubMed] [Google Scholar]
- Um JW, Nygaard HB, Heiss JK, Kostylev MA, Stagi M, Vortmeyer A, Wisniewski T, Gunther EC, Strittmatter SM. Alzheimer amyloid-beta oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nature Neurosci. 2012;15:1227–1235. doi: 10.1038/nn.3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma S, Quillinan N, Yang YF, Nakayama S, Cheng J, Kelley MH, Herson PS. TRPM2 channel activation following in vitro ischemia contributes to male hippocampal cell death. Neurosci Lett. 2012;530:41–46. doi: 10.1016/j.neulet.2012.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Wang HY, Lee DH, D'Andrea MR, Peterson PA, Shank RP, Reitz AB. beta-Amyloid(1–42) binds to alpha7 nicotinic acetylcholine receptor with high affinity: implications for Alzheimer's disease pathology. J Biol Chem. 2000;275:5626–5632. doi: 10.1074/jbc.275.8.5626. [DOI] [PubMed] [Google Scholar]
- Webster SJ, Bachstetter AD, Nelson PT, Schmitt FA, Van Eldik LJ. Using mice to model Alzheimer's dementia: an overview of the clinical disease and the preclinical behavioral changes in 10 mouse models. Front Genet. 2014;5:88. doi: 10.3389/fgene.2014.00088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehrhahn J, Kraft R, Harteneck C, Hauschildt S. Transient receptor potential melastatin 2 is required for lipopolysaccharide-induced cytokine production in human monocytes. J Immunol. 2010;184:2386–2393. doi: 10.4049/jimmunol.0902474. [DOI] [PubMed] [Google Scholar]
- Xie YF, Belrose JC, Lei G, Tymianski M, Mori Y, Macdonald JF, Jackson MF. Dependence of NMDA/GSK-3beta mediated metaplasticity on TRPM2 channels at hippocampal CA3-CA1 synapses. Mol Brain. 2011;4:44. doi: 10.1186/1756-6606-4-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Macciardi F, Li PP, Yoon IS, Cooke RG, Hughes B, Parikh SV, McIntyre RS, Kennedy JL, Warsh JJ. Association of the putative susceptibility gene, transient receptor potential protein melastatin type 2, with bipolar disorder. Am J Med Genet. 2006;141B:36–43. doi: 10.1002/ajmg.b.30239. [DOI] [PubMed] [Google Scholar]
- Yamamoto S, Shimizu S, Kiyonaka S, Takahashi N, Wajima T, Hara Y, Negoro T, Hiroi T, Kiuchi Y, Okada T, Kaneko S, Lange I, Fleig A, Penner R, Nishi M, Takeshima H, Mori Y. TRPM2-mediated Ca2+ influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nat Med. 2008;14:738–747. doi: 10.1038/nm1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda Y, Kudo T, Katayama T, Imaizumi K, Yatera M, Okochi M, Yamamori H, Matsumoto N, Kida T, Fukumori A, Okumura M, Tohyama M, Takeda M. FAD-linked presenilin-1 mutants impede translation regulation under ER stress. Biochem Biophys Res Commun. 2002;296:313–318. doi: 10.1016/S0006-291X(02)00859-8. [DOI] [PubMed] [Google Scholar]
- You H, Tsutsui S, Hameed S, Kannanayakal TJ, Chen L, Xia P, Engbers JD, Lipton SA, Stys PK, Zamponi GW. Abeta neurotoxicity depends on interactions between copper ions, prion protein, and N-methyl-d-aspartate receptors. Proc Natl Acad Sci U S A. 2012;109:1737–1742. doi: 10.1073/pnas.1110789109. [DOI] [PMC free article] [PubMed] [Google Scholar]