

Abstract
Sleep-active neurons located in the ventrolateral preoptic nucleus (VLPO) play a crucial role in the induction and maintenance of slow-wave sleep (SWS). However, the cellular and molecular mechanisms responsible for their activation at sleep onset remain poorly understood. Here, we test the hypothesis that a rise in extracellular glucose concentration in the VLPO can promote sleep by increasing the activity of sleep-promoting VLPO neurons. We find that infusion of a glucose concentration into the VLPO of mice promotes SWS and increases the density of c-Fos-labeled neurons selectively in the VLPO. Moreover, we show in patch-clamp recordings from brain slices that VLPO neurons exhibiting properties of sleep-promoting neurons are selectively excited by glucose within physiological range. This glucose-induced excitation implies the catabolism of glucose, leading to a closure of ATP-sensitive potassium (KATP) channels. The extracellular glucose concentration monitors the gating of KATP channels of sleep-promoting neurons, highlighting that these neurons can adapt their excitability according to the extracellular energy status. Together, these results provide evidence that glucose may participate in the mechanisms of SWS promotion and/or consolidation.
SIGNIFICANCE STATEMENT Although the brain circuitry underlying vigilance states is well described, the molecular mechanisms responsible for sleep onset remain largely unknown. Combining in vitro and in vivo experiments, we demonstrate that glucose likely contributes to sleep onset facilitation by increasing the excitability of sleep-promoting neurons in the ventrolateral preoptic nucleus (VLPO). We find here that these neurons integrate energetic signals such as ambient glucose directly to regulate vigilance states accordingly. Glucose-induced excitation of sleep-promoting VLPO neurons should therefore be involved in the drowsiness that one feels after a high-sugar meal. This novel mechanism regulating the activity of VLPO neurons reinforces the fundamental and intimate link between sleep and metabolism.
Keywords: glucose, homeostasis, hypothalamus, polysomnography, preoptic nucleus, sleep
Introduction
An organism's energy status has a significant impact on the regulation of vigilance states, as evidenced by several reports (Benington and Heller, 1995; Scharf et al., 2008; Siegel, 2009). Specifically, wakefulness is enhanced during fasting, whereas resting (i.e., sleeping/napping) is favored after food intake (Roky et al., 1999; Minet-Ringuet et al., 2004; Jenkins et al., 2006). Orexinergic neurons involved in arousal maintenance (Adamantidis et al., 2007; Tsujino and Sakurai, 2009) are inhibited by a rise in extracellular glucose concentration (Yamanaka et al., 2003; Burdakov and Alexopoulos, 2005; Burdakov et al., 2005), suggesting that ingesting a sweet meal may lead to an inhibition of these arousal-related neurons and therefore a sensation of sleepiness. Despite the strong support for these observations, it has never been investigated whether neurons responsible for sleep induction and maintenance are influenced directly by the availability of energy and how they can adjust the animal's behavior to accommodate the energy state.
Numerous studies have shown that sleep-active neurons located within the ventrolateral preoptic nucleus (VLPO) play a crucial role in promoting and maintaining slow-wave sleep (SWS) (Sherin et al., 1996; Szymusiak et al., 1998; Saper et al., 2001; Takahashi et al., 2009). These neurons contain GABA and project to the wake-promoting systems (Sherin et al., 1998; Steininger et al., 2001). Neurons within the VLPO are therefore in a suitable position to favor SWS onset through an overall repression of the wake-promoting neurons. However, the mechanisms responsible for VLPO activation at sleep onset and deactivation during arousal remain poorly understood. To address this issue, we hypothesize that sleep-promoting VLPO neurons may integrate metabolic signals such as ambient glucose to modulate sleep propensity.
Here, we demonstrate in mice that bilateral microinjections of glucose into the VLPO significantly increase sleep and the density of c-Fos-expressing neurons specifically within the VLPO. In acute VLPO brain slices, we found that glucose specifically increases the excitability of neurons exhibiting properties of sleep-promoting neurons. This effect requires the intraneuronal metabolization of glucose to ATP, which in turn inhibits ATP-sensitive potassium (KATP) channels. These novel results provide strong evidence that VLPO neurons may be considered as a central glucose-sensing structure that contributes directly to sleep control in response to the fine tuning of the organism's metabolic status.
Materials and Methods
Animal experiments.
Animal experiments were performed in accordance with the guidelines of the Ecole Supérieure de Physique et de Chimie Industrielles ParisTech/University Claude Bernard, Lyon I/Centre National de la Recherche Scientifique Animal Care and Use Committee.
In vivo glucose microinjection and sleep monitoring.
To monitor the effects of glucose microinjections into the VLPO on vigilance states, 8 male C57BL/6J mice (10 weeks of age; Charles River) were anesthetized intraperitoneally with a ketamine/xylazine mix (100 and 10 mg/kg, respectively), mounted on a stereotaxic frame (David Kopf Instruments), and implanted with a bilateral guide cannula (26G; Plastics One) placed 1 mm above the targeted bilateral VLPO (AP: −0.1 mm; ML: 0.7 mm; DV: 4.5 mm) according to the Franklin and Paxinos atlas (2007). Mice were then prepared for polysomnographic recordings. Two stainless-steel screws (Plastics One) were implanted over the parietal cortices and one screw was implanted over the cerebellar cortex for EEG recordings. Two gold-coated electrodes were inserted into neck muscles to record EMG activity. The cannula guide and EEG/EMG electrodes soldered to a miniature plug (Plastics One) were fixed to the skull using Super-Bond (Sun Medical) and finally secured with acrylic dental cement (Paladur). For sleep recording and glucose infusion, mice were allowed to recover from surgery for 7 d and then housed in individual Plexiglas barrels with woodchip bedding and connected to a cable allowing free movements during continuous polysomnographic recording. Barrels were placed in an insulated soundproof box maintained under the following standard laboratory conditions: a 12 h/12 h light-dark cycle (lights on at 8:00 A.M.), 22 ± 1°C temperature, and access to water and food pellets ad libitum. After habituation, mice were treated with 0.3 μl of artificial CSF (ACSF) vehicle or 5 mm glucose (in ACSF vehicle) or 10 mm glucose (in ACSF vehicle) by pressure infusion in each hemisphere. ACSF vehicle contained the following (in mm): 137 NaCl, 2.7 KCl, 1.3 CaCl2, 1 MgCl2, 10 Na2HPO4, and 1.76 KH2PO4, pH 7.4. In all solutions, osmolarity was equilibrated by adjusting sucrose concentration. All solutions were stored at −20°C at their final concentration. All animals underwent 3 bilateral injections of vehicle, glucose 5 mm, and glucose 10 mm in a randomized order with treatments spaced by 2 d. For this, a 5 μl Hamilton syringe placed in a syringe pump (0.1 μl/min; New Era Pump Systems) was connected to the internal cannula (33G; Plastics One), which protruded out 1 mm from the guide cannula into VLPOs. Injections started at 9:00 A.M. Under isoflurane sedation (induction 2%, maintenance 1.5%) to minimize contention stress, injection cannulas were lowered into the brain and left in place for 5 min before and after infusion. Animals regained consciousness immediately after the removal of isoflurane. Isoflurane exposure never exceeded 18 min. During the injection procedure, animals were unplugged from the recording cable. Immediately after the end of the procedure, animals were returned to their barrels, reconnected to the cable, and polysomnographic recordings were begun. Acquired EEG and EMG signals were amplified (MCP+; Alpha-Omega Engineering), digitalized, and collected via a CED interface with Spike2 software (Cambridge Electronic Design) at a sampling frequency of 520.8 Hz. Three vigilance states, wake (W), SWS, and paradoxical sleep (PS), were scored and quantified offline in 5 s epochs. During W, desynchronized low-amplitude EEG was accompanied by a sustained EMG activity with phasic bursts; SWS was distinguished by high-voltage slow waves and the disappearance of phasic muscle activity. PS onset was signaled by a muscle atonia associated with decreased EEG amplitude including a regular and pronounced theta rhythm. Hypnograms were then drawn directly using a custom script in Spike2 (Cambridge Electronic Design). The values were finally exported to calculate standard parameters for each vigilance state (durations, percentage, number, and episode duration expressed as mean ± SEM). In addition, normalized EEG spectral power was calculated during W, SWS, and PS with a Spike2 script computing a fast Fourier transform on 5 s EEG epochs (0.1 Hz resolution). The mean (±SEM) power for each EEG spectral band was then calculated as follows: δ, 0.5–4.5 Hz; θ, 5–9 Hz; σ, 10–14 Hz; β, 15–30 Hz; γ, 31–48 Hz). To detect statistical significance, data were analyzed as follows: for each vigilance state (W, SWS, PS), effect of glucose concentrations (vs vehicle) was tested using nonparametric Friedman tests. To detect pairwise differences, post hoc Wilcoxon tests were applied and corrected for multiple comparisons (Bonferroni correction). Each animal served as its own control. Significance was set at p < 0.05.
c-Fos immunohistochemistry.
For c-Fos immunohistochemistry experiments, the same mice used for bilateral injections (described above) were injected under isoflurane sedation a fourth time into the VLPO with glucose (10 mm) in one hemisphere (randomly chosen) and vehicle in the contralateral hemisphere. Killing and perfusion of animals were performed 90 min after treatment. Under deep pentobarbital anesthesia (150 mg/kg, i.p.; Ceva Santé Animale), mice were transcardially perfused with Ringer's lactate solution containing 0.1% heparin, followed by cold 4% paraformaldehyde solution in 0.1 m phosphate buffer, pH 7.4. Brains were removed, postfixed overnight in 4% paraformaldehyde at 4°C and cryoprotected in 30% sucrose solution for two d at 4°C. Brains were then frozen in dry-ice-cooled methylbutane and cut into 30-μm-thick coronal sections on a cryostat (Microm). Collected free-floating sections were successively incubated in the following: (1) rabbit anti-c-Fos antiserum (1:8000; Merck) in PBS containing 0.3% Triton X-100 and 0.1% sodium azide for 3 d at 4°C; (2) a biotinylated goat anti-rabbit IgG (1:1000; Vector Laboratories) for 90 min at room temperature; and (3) an ABC-HRP complex (1:1000; Elite kit; Vector Lab) for 90 min at room temperature. The sections were then immersed in 0.05 m Tris-HCl buffer, pH 7.6, containing 0.025% 3,3′-diaminobenzidine-4HCl (DAB; Sigma-Aldrich), 0.003% H2O2, and 0.6% nickel ammonium sulfate for 20 min at room temperature. Three 10 min washes were performed between each incubation step. The staining appeared as a dense black nuclear coloration of c-Fos-immunoreactive (Fos+) neurons. The stained sections were finally mounted on glass gelatin-coated slides, counterstained with neutral red to delineate forebrain areas, dried, dehydrated, and coverslipped with DEPEX (VWR). The anti-c-Fos antiserum was made against a synthetic peptide corresponding to the N-terminal part (residues 4–17) of human Fos. This part of the protein displays 100% homology among human, rat, and mouse and shares no homology with Fos-related antigens such as Fos B, Jun B, Fra-1 and Fra-2 (Blast 2 sequences; National Center for Biotechnology Information).
For each mouse used (n = 8), Fos-labeled sections evenly spaced throughout the rostrocaudal extent of the VLPO (n = 4 sections) were analyzed with an Axioskop 2 plus microscope (Zeiss) equipped with a motorized X–Y-sensitive stage and a video camera connected to a computerized image analysis system. Fos+ neurons were plotted using the Mercator version 2 software (ExploraNova). Mean amounts of Fos+ cells (± SEM) were calculated in both hemispheres (glucose vs vehicle) for the VLPO and three neighboring basal forebrain nuclei, the magnocellular nucleus (MCPO), the lateral preoptic area (LPOA) and the medial preoptic area (MPOA). The counting of Fos+ cell bodies within the MCPO, LPOA, and MPOA was computed according to their respective boundaries and surface areas; the latter two were determined by the changes in cellular densities and morphologies as revealed by neutral red counterstaining. In the VLPO, the quantification of Fos+ cells was performed by considering a half-ellipsoid area in the most ventral part of the basal forebrain (200–250 μm wide, 300–400 μm deep) drawn laterally to the optic chiasma (rostral sections) or to the supraoptic nucleus (caudal sections) (Takahashi et al., 2009). Fos+ neuronal densities were computed using surfaces of areas determined with neutral red staining. Photomicrographs were taken with a CCD Color 10-bit QiCam camera, imported into Adobe Photoshop version 7.0, digitally adjusted for brightness and contrast, and assembled into plates at a resolution of 300 dpi.
Slice preparation for electrophysiology and single-cell RT-PCR.
In preparation for electrophysiology and single-cell RT-PCR (scRT-PCR), brain slices containing the VLPO were obtained from 14- to 18-d old male C57BL/6J mice (Charles River) maintained on a 12 h/12 h light-dark cycle (lights on at 8:00 A.M.). Food and water were continuously available. All experiments were performed during the light phase. The animals were decapitated and brains were quickly removed. Coronal slices (300 μm thick) were cut with a vibrating microtome (VT2000S; Leica) in cold slicing ACSF at 4°C. Slices were left to recover for at least 30 min before transfer to the recording chamber, where they were superfused with ACSF saturated with 95% O2 and 5% CO2 (at 32°C).
Electrophysiological experiments.
ACSF contained the following (in mm): 130 NaCl, 5 KCl, 2.4 CaCl2, 20 NaHCO3, 1.25 KH2PO4, 1.3 MgSO4, 10 d-glucose, and 15 sucrose, pH 7.35, and was constantly oxygenated (95% O2/5% CO2). During slicing, 1 mm kynurenate was added to the ACSF. Individual slices were then transferred to a thermoregulated (32.5°C) chamber (Badcontroller V; Luigs & Neumann), placed under a microscope (Axioscop2FS; Zeiss) equipped with an infrared CCD camera (CoolSNAP HQ2; Roper Scientific,). Slices were maintained immersed and continuously surperfused at 3–5 ml/min with oxygenated kynurenate-free ACSF. All electrophysiological experiments were performed with a MultiClamp700B (Molecular Devices) amplifier connected to an acquisition board (Digidata 1440; Molecular Devices) attached to a computer running pCLAMP software (Molecular Devices).
Extracellular recordings in loose-cell-attached configuration.
Loose-cell-attached patch-clamp recordings were performed to monitor stable spontaneous firing activity of VLPO neurons for long periods of time, which is necessary for completing pharmacological experiments. Infrared videomicroscopy was used to choose VLPO neurons visually. Loose-cell-attached recordings were performed from the soma with patch micropipettes (3–6 MΩ) pulled from borosilicate glass capillary tubes (1.5 mm outer diameter, 0.86 mm inner diameter; Harvard Apparatus) on a horizontal puller (model P-1000; Sutter Instruments). Micropipettes were filled with oxygenated ACSF and fixed to an electric microdrive (Luigs & Neumann). The micropipette was placed in contact with the soma of a selected neuron under visual control. During recordings, a seal resistance of 10–15 MΩ was maintained to avoid damage or mechanical stimulation to the cell. Data are expressed as mean ± SEM. Values were compared using nonparametric Wilcoxon tests or permutation tests according to sample sizes. A p value ≤ 0.05 was considered statistically significant.
Patch-clamp recordings in whole-cell configuration.
In whole-cell patch-clamp configuration, recordings were performed with patch-clamp pipettes (3–6 MΩ) filled with 8 μl of internal solution containing the following (in mm): 144 K-gluconate, 3 MgCl2, 0.2 EGTA, 10 HEPES, 1.25 ATP, pH 7.2, 285–295 mOsm. The pipette was slowly brought to on the selected neuron to be recorded in whole-cell configuration using infrared videomicroscopy guidance and clamped at −60 mV. During whole-cell patch-clamp recordings, the intrinsic membrane properties of neurons were assessed by applying current steps (800 ms) from −100 pA to firing saturation in 10 pA increments.
Pharmacological studies.
The following drugs were used: noradrenaline (NA, 100 μm; Sigma-Aldrich); α-cyano-4-hydroxycinnamate (4-CIN, 500 μm; Sigma-Aldrich); pinacidil (500 μm; Sigma-Aldrich); diazoxide (300 μm; Sigma-Aldrich); and tolbutamide (500 μm; Sigma-Aldrich). Stock solutions of NA (100 mm; water), 4-CIN (500 mm; DMSO), pinacidil (500 mm; DMSO), diazoxide (300 mm; NaOH 0.1 m), and tolbutamide (500 mm; NaOH 0.1 m) were stored as frozen aliquots at −20°C. Before application, drugs were dissolved in ACSF to their working concentrations. Various concentrations of d-glucose (1, 2.5, 5, 10, 25 mm), 2-deoxy-d-glucose (2-DG, 7.5 mm; Sigma-Aldrich), and alloxan (10 mm; Sigma-Aldrich) in the ACSF were tested on the activity of VLPO neurons. For these experiments, ACSF osmolarity was equilibrated systematically by adjusting the sucrose concentration. Note that the addition of sucrose to compensate medium osmolarity has no effect on energetic metabolism. Indeed, our RT-PCR control experiments confirmed the absence in the VLPO of mRNAs that encode the enzyme sucrose isomaltase, which is involved in breaking down sucrose and maltose, by comparing RT-PCR products of cDNA originating from 2.8 ng of RNA prepared from microdissected VLPO (homemade) and small intestine (Biochain). Sucrase isomaltase was detected in the small intestine and not in the VLPO (data not shown). Eventually, the postsynaptic action of glucose was tested in the synaptic uncoupling condition on neurons recorded in the loose-cell-attached configuration. For these experiments, slices were first submitted to a pretreatment (∼10 min) with a modified ACSF containing low Ca2+ (0.1 mm) and high Mg2+ (10 mm) concentrations. This condition is known to completely block the release of calcium-dependent neurotransmitters. The effects of glucose in the synaptic uncoupling condition were evaluated under various concentrations in this low Ca2+/high Mg2+ medium.
Studies of the KATP-dependent current were performed in voltage-clamp mode. The membrane potential was maintained at −75 mV and membrane currents were measured with two 100 ms potential steps at −65 mV and −85 mV separated by 100 ms. Steps were followed after 100 ms by a potential ramp from −115 mV to −45 mV within 400 ms. This stimulation protocol was repeated every 10 s. All membrane potentials were corrected for liquid junction potential (−15 mV). We applied a hyperpolarizing voltage step (10 mV) before each stimulation to monitor passive electrical properties of the recorded cell, as well as access resistance, which did not vary >20% during the analyzed recording period. Access resistance was not compensated.
scRT-PCR.
The intrinsic membrane properties were determined in whole-cell current-clamp mode in a period not exceeding 10 min to minimize potential mRNA degradation. The cytoplasm was then aspirated into the patch pipette under visual control. The quality of the seal was monitored during harvesting to prevent extracellular contamination. The content of the pipette was expelled into an RNase-free PCR tube and the RT-PCR protocol was performed as described previously (Lambolez et al., 1992; Gallopin et al., 2006). The scRT-PCR protocol was designed to detect simultaneously the expression of the two isoforms of glutamic acid decarboxylase (GAD65 and GAD67), the four subunits composing KATP channels (Kir6.1, Kir6.2, SUR1, SUR2), one glucose transporter (GLUT3), and glucokinase (GK), a requirement for the entry of glucose into glycolysis. To amplify GK, GAD65, and GAD67 mRNAs, two primers sets, GK(1) and GK(2), GAD65(1), GAD65(2), GAD67(1), and GAD67(2), respectively (Table 1), were designed. After the RT reaction, the cDNAs present in 10 μl of solution were first amplified simultaneously using the primers listed in Table 1. Taq polymerase (2.5 U; QIAGEN) and 20 pmol concentrations of each primer (Table 1) were added to the buffer supplied by the manufacturer (final volume, 100 μl) and 21 cycles of PCR were run (94°C for 30 s; 60°C for 30 s; 72°C for 35 s). The second amplification was performed using 1 μl of the first PCR product as a template. Each cDNA was amplified individually with a second primer pair set internal to the pair used in the first PCR (nested primers; Table 1). Next, 35 cycles of PCR were performed and 10 μl of each individual PCR product were run on a 2% agarose gel with a 100 bp ladder (Promega) as molecular weight markers and stained with ethidium bromide. The sizes of PCR-generated fragments were as predicted by the mRNA sequences (Table 1). The RT-PCR protocol was tested on 500 pg of total RNA purified from microdissected mouse VLPO and all transcripts were detected.
Table 1.
PCR primers
| GenBank accession no. | First PCR primers | Size (bp) | Second PCR nested primers | Size (bp) |
|---|---|---|---|---|
| GAD65 (1) | PPM25176G-200 | 84 | PPM25176G-200 | 84 |
| NM 008078 | ||||
| GAD65 (2) | CCAAAAGTTCACGGGCGG, TCCTCCAGATTTTGCGGTTG | 375 | CACCTGCGACCAAAAACCCT, GATTTTGCGGTTGGTCTGCC | 248 |
| NM 008078 | ||||
| GAD67 (1) | PPM36500F-200 | 121 | PPM36500F-200 | 121 |
| NM 008077 | ||||
| GAD67 (2) | ATGATACTTGGTGTGGCGTAGC, GTTTGCTCCTCCCCGTTCTTAG | 253 | CAATAGCCTGGAAGAGAAGAGTCG, GTTTGCTCCTCCCCGTTCTTAG | 177 |
| NM 008077 | ||||
| SUR1 | CACCGCTGTTCCTGCTGTCT, CGCTCTGGGTTGCTTGTCCT | 523 | TCATCGGAGGCTTCTTCACC, TCTTCTCCCTCGCTGTCTGG | 208 |
| NM_011510.3 | ||||
| SUR2 | GCAGTCAAAGCCATCATCAGTGT, CAAGGATGGCAAGGAGAAGAG | 373 | CTTGAGCGATGAGATTGGTGAGG, AGATGATTTTCCACAACCCACTT | 310 |
| NM_001044720.1 | ||||
| Kir6.1 | GCTGGCTGCTCTTCGCTATC, CATACAGGGGGCTACGCTTG | 588 | TGGGTTTGGAGGGAGAATGA, TGGCGTCGTGGTTTTCTTGA | 286 |
| NM_008428.4 | ||||
| Kir6.2 | GGGTGGTAACGGCATCTTCC, ATGCTAAACTTGGGCTTGGC | 420 | CGCTCTACGACCTGGCTCCT, GGGTCAGGGCATCCAGCAG | 285 |
| NM_010602.2 | ||||
| GK (1) | GGCTGCAACGCCTGCTACAT, GCTCCTTGAAGCTCGGGTGC | 580 | GGGGCGCATGTGTGTCAACA, CACAATGTCGCAGTCGGCGA | 358 |
| NM_010292.4 | ||||
| GK (2) | PPM05095A-200 | 115 | PPM05095A-200 | 115 |
| NM 010292 | ||||
| GLUT3 | ATGGGGACAACGAAGGTGAC, GCCACAGAAAATGCCAATGA | 377 | CATCTCTGGTGTTCGCCGTG, CAGCATTTCAACAGACTCCGC | 338 |
| NM_011401.4 |
Results
Somnogenic properties of glucose when infused into the VLPO
Here, we investigated whether glucose (5 or 10 mm vs vehicle) can modify the sleep-waking cycle when bilaterally infused into the VLPO of mice chronically prepared for polysomnographic recordings. These animals received a randomized sequence of vehicle, glucose 5 mm, and glucose 10 mm, each spaced by 2 d. Seven of 8 mice treated had injection sites centered on both VLPO (Fig. 1A). In these animals, a significant effect of glucose injections was observed on both SWS durations (Friedman test, p < 0.01) and PS durations (Friedman test, p < 0.05) during the first 2 h after glucose or vehicle injections. Post hoc tests confirmed that SWS durations significantly increased between vehicle and both 5 and 10 mm glucose (Wilcoxon tests, p < 0.05; Fig. 1C, Table 2), whereas no significant pairwise difference was detected between groups for PS durations (Fig. 1C, Table 2). This somnogenic glucose-dependent effect is due to a severe reduction in waking amounts compared with the vehicle (Friedman test, p < 0.01; Table 2) and significantly shortened latencies to SWS (Friedman test, p < 0.001; Fig. 1B) and PS (Friedman test, p < 0.01). The post hoc analysis revealed a dose-dependent decrease in SWS latencies with significant differences between all groups (Wilcoxon tests, p < 0.05 for all comparisons; Fig. 1B, Table 2). Together, these results indicate that glucose would be able to modulate the activity of VLPO neurons and promote sleep.
Figure 1.
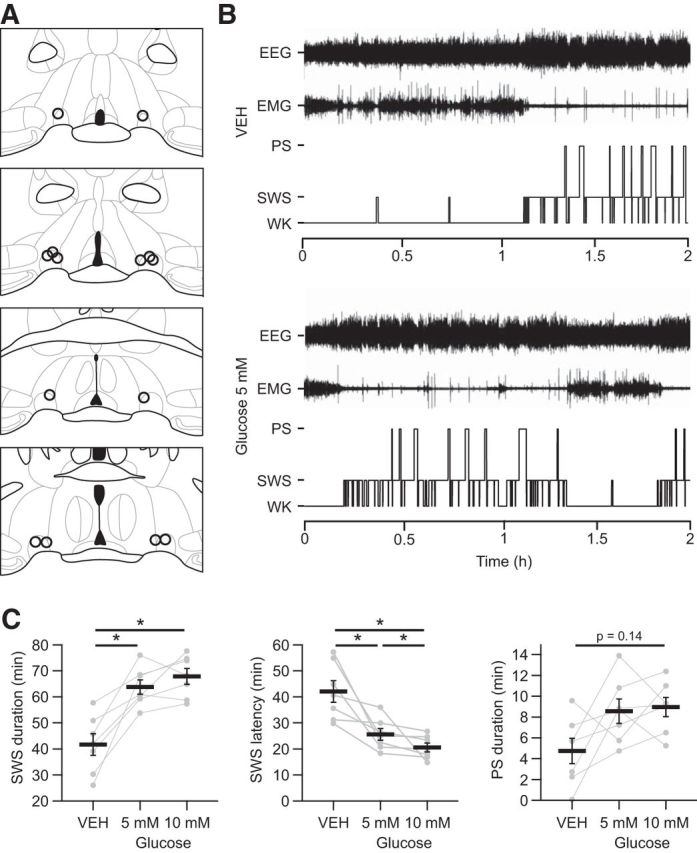
Microinjections of glucose into the VLPO increase sleep. A, Camera lucida drawings of frontal sections illustrating all injection sites (one open circle per animal). The four different sections are evenly spaced (120 μm intervals) throughout the rostrocaudal extent of the VLPO. Bregma coordinates range approximately from 0.26 to −0.22 mm according to the Franklin and Paxinos atlas (2007). B, Hypnograms of a representative animal illustrating the organization of the sleep-waking cycle during the 2 h after a bilateral injection of vehicle (VEH; top) or glucose 5 mm (bottom) into the VLPO. Recordings began immediately after the injection procedure once the animals were returned to their home barrels and reconnected to the recording setup. C, Quantification of SWS durations (left), SWS latencies (middle), and PS (right) durations during the 2 h after bilateral glucose or VEH injection (n = 7). Individual data for each mouse injected bilaterally with glucose or VEH (gray dots and lines) are displayed and data are presented as mean ± SEM (black bars). *p < 0.05, Wilcoxon test.
Table 2.
Sleep architecture after bilateral injections of glucose versus vehicle
| 0–2 h after injection |
|||
|---|---|---|---|
| Vehicle | Glucose 5 mm | Glucose 10 mm | |
| SWS | |||
| Duration (%) | 34.7 ± 3.5 | 53.2 ± 2.3* | 56.6 ± 2.5* |
| No. of episodes | 43.0 ± 4.6 | 66.0 ± 3.4 | 62.1 ± 2.4 |
| Episode duration (s) | 59.0 ± 4.1 | 58.5 ± 2.5 | 66.0 ± 3.3 |
| Latency (min) | 42.1 ± 4.1 | 25.6 ± 2.3* | 20.6 ± 1.7*# |
| PS | |||
| Duration (%) | 4.0 ± 1.0 | 7.1 ± 1.0 | 7.5 ± 0.8 |
| No. of episodes | 7.3 ± 1.9 | 14.0 ± 2.5 | 12.9 ± 1.8 |
| Episode duration (s) | 39.1 ± 8.6 | 38.6 ± 4.3 | 44.7 ± 4.5 |
| Latency (min) | 88.4 ± 7.3 | 47.2 ± 5.1* | 37.3 ± 2.2* |
| W | |||
| Duration (%) | 61.3 ± 4.5 | 39.6 ± 3.1* | 35.9 ± 3.0* |
For each experimental condition, the percentage of time spent in SWS, PS, and W was scored per 5 s epoch during the first 2 h (0–2 h) after injections (n = 7). The number of SWS and PS episodes, the mean duration of PS and SWS episodes, and the latency to the first SWS and PS episodes are also reported. Significance levels are given for post hoc Bonferroni-corrected Wilcoxon tests between conditions.
*p < 0.05 versus vehicle;
#p < 0.05 versus glucose 5 mm.
Moreover, to test whether glucose could activate VLPO neurons, we performed on the same mice a final microinjection of glucose (10 mm) in one hemisphere and vehicle in the contralateral VLPO. The mice were then killed 90 min after this injection to perform c-Fos immunohistochemistry. Sleep analysis during the period after injection and before killing (90 min) revealed that unilateral glucose injection led to a significant increase in the duration of SWS compared with that obtained after bilateral VEH infusions (40.6 ± 2.9% vs 26.7 ± 3.5%, respectively; p < 0.05). Notwithstanding, c-Fos immunostaining (Fig. 2A,B) revealed a density of c-Fos-labeled neurons that was significantly increased in the VLPO when infused with glucose compared with the contralateral VLPO infused with vehicle (Wilcoxon test p < 0.05; Fig. 2B,C, Table 3). In sharp contrast, the density of cells labeled for c-Fos remained unchanged in three adjacent forebrain areas (Fig. 2A): the MCPO (Fig. 2D, Table 3), the LPOA (Fig. 2E, Table 3), and the MPOA (Fig. 2F, Table 3). Together, these physiological and functional data strongly suggest that sleep-promoting VLPO neurons could be activated by glucose treatment. In addition, these results support the view that glucose may participate in the mechanisms of SWS promotion and/or consolidation.
Figure 2.

Density of c-Fos-labeled neurons in anterior hypothalamic regions after glucose infusion into the VLPO. A, Schema illustrating the VLPO and surrounding brain areas examined for c-Fos counting, including the VLPO (red), MCPO (blue), LPOA (orange), and MPOA (green). B, Photomicrographs of sections stained against c-Fos after unilateral infusions of glucose (left) and VEH (right) and counterstained with neutral red in a representative animal (c-Fos-labeled cells are shown in the bottom left inset at a higher magnification Scale bar, 10 μm. C–F, Quantification of the density of c-Fos-labeled neurons in the VLPO (C), MCPO (D), LPOA (E), and MPOA (F) of animals that received glucose in one hemisphere and VEH in the other hemisphere (n = 7). Individual data for each hemisphere injected with glucose or VEH (gray dots and lines) are displayed and data are presented as mean ± SEM (black bars). *p < 0.05, Wilcoxon test: ns.
Table 3.
Expression of c-Fos-labeled cells in the VLPO and surrounding regions after glucose versus vehicle treatment
| c-Fos+ neuronal density (cells/mm2) |
|||
|---|---|---|---|
| Glucose (10 mm) | Vehicle | p value, Wilcoxon test | |
| VLPO | 213.3 ± 25.3 | 72.0 ± 8.8 | 0.016* |
| MPOA | 361.7 ± 37.7 | 295.8 ± 51.0 | 0.219 (ns) |
| LPOA | 320.6 ± 32.5 | 292.3 ± 70.9 | 0.578 (ns) |
| MCPO | 60.2 ± 10.2 | 60.9 ± 6.2 | 0.938 (ns) |
*p ≤ 0.05.
To determine how glucose can modulate the activity of VLPO neurons, we performed ex vivo electrophysiological experiments.
Glucose responsiveness of VLPO neurons
To assess the glucose sensitivity of VLPO neurons, 49 spontaneously active neurons were recorded in a loose-cell-attached patch configuration. We observed that a rise in extracellular glucose concentrations led to a reversible increase in the firing rate in ∼73% (36/49) of recorded neurons (Fig. 3), whereas the remaining cells (13/49) were insensitive (Fig. 4). Figure 3A illustrates a typical response of a glucose-excited VLPO neuron. A decrease in the glucose concentration from 5 to 1 mm induced a significant reduction in the firing frequency of these cells (n = 8, Wilcoxon test p < 0.01; Fig. 3A,B). This effect was reversible because a subsequent rise in glucose concentration to 5 mm induced a significant increase in the firing rate of the same cells (n = 8, Wilcoxon test p < 0.01; Fig. 3A,B). We noted that the inhibitory effect associated with lowering glucose appears faster (2.55 ± 0.50 min) than the excitatory effect (7.39 ± 1.75 min) induced by increasing glucose (n = 8, Wilcoxon test p < 0.05). Glucose (1–25 mm) excited VLPO neurons in a dose-dependent manner across all concentrations tested, with an EC50 of 4.06 mm (Fig. 3C); this is compatible with the physiological glucose concentration range in the hypothalamus (Silver and Erecińska, 1994; Routh, 2002; de Vries et al., 2003).
Figure 3.
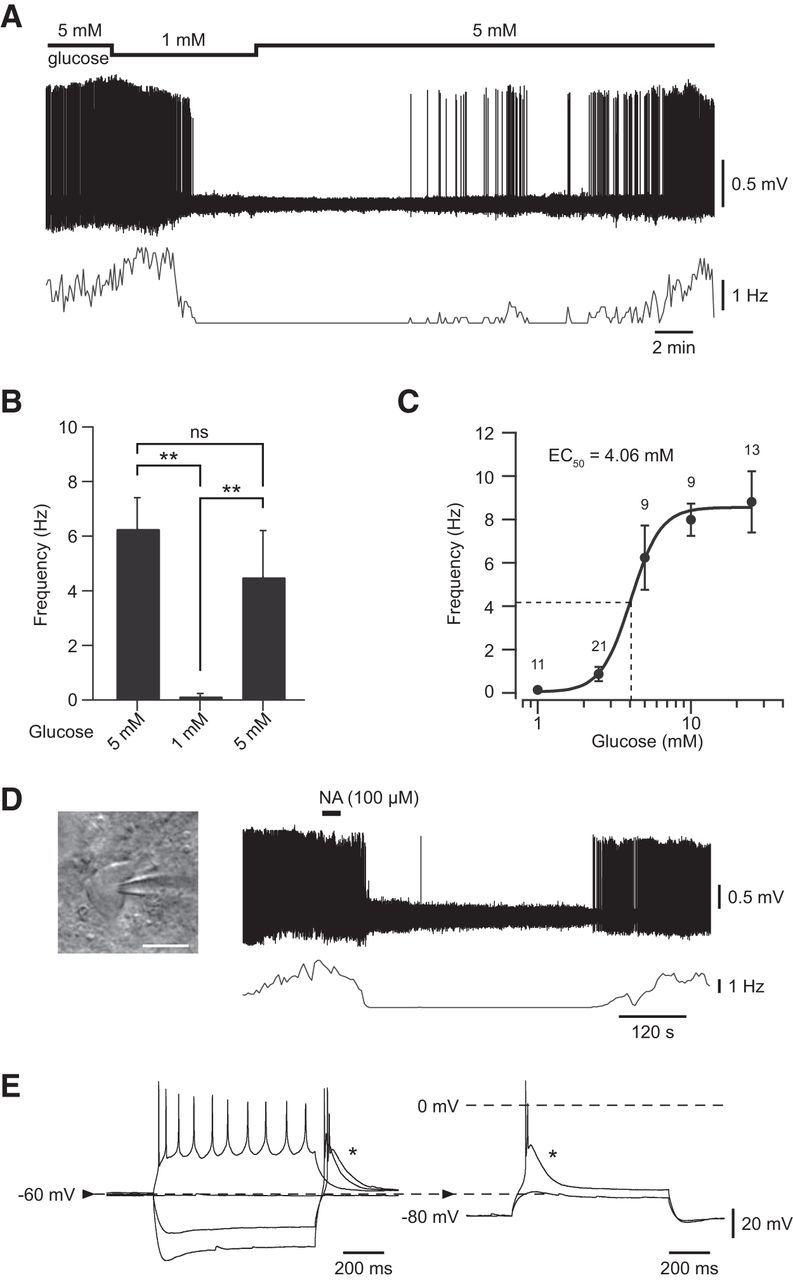
Effect of glucose applications on putative sleep-promoting VLPO neurons. A, Reversible inhibitory effect of decreasing extracellular glucose concentration from 5 to 1 mm on the spontaneous firing activity of a VLPO neuron recorded in loose-cell-attached configuration. B, Quantification of the excitatory effect of glucose (from 1 to 5 mm) on the spontaneous firing activity of glucose-responsive VLPO cells (n = 8). Data are presented as mean ± SEM. **p < 0.01, Wilcoxon test: ns. C, Dose–response relationship of the excitatory effect of glucose on glucose-sensitive VLPO cells. Data fit a Hill curve with an EC50 of 4.06 mm and are presented as mean ± SEM with group sizes for each glucose concentration (1, 2.5, 5, 10, 25 mm). D, Reversible inhibitory effect of bath-applied NA (100 μm; right) on the spontaneous firing activity of the same cell shown in A characterized by a multipolar shape (left). Scale bar, 20 μm. E, Current-clamp recording of the same cell shown in A characterized by the presence of a potent LTS (*).
Figure 4.

Absence of glucose effect on identified non-sleep-promoting neurons. A, Absence of effect of acute increase in extracellular glucose concentration on the spontaneous firing activity of a VLPO neuron. B, Spontaneous firing activity of VLPO neurons showing insensitivity to changes in extracellular glucose concentration for 1 mm (n = 6), 2.5 mm (n = 5), 10 mm (n = 8), and 25 mm (n = 5) glucose. Data are presented as mean ± SEM. Mann–Whitney test: ns. C, Reversible excitatory effect of bath-applied NA (100 μm) to the same cell shown in A. D, Current-clamp recording of VLPO cell devoid of LTS characterized by a bipolar shape (insert) Scale bar, 20 μm.
Previous studies led to the identification of the electrophysiological, biochemical, morphological, and pharmacological properties of neurons active during SWS in the VLPO (Gallopin et al., 2000; Matsuo et al., 2003; Gallopin et al., 2004; Saint-Mleux et al., 2004; Gallopin et al., 2005; Saint-Mleux et al., 2007; Liu et al., 2010; Moore et al., 2012). Based on these studies, putative sleep-active neurons in the VLPO can be characterized ex vivo by the presence of a low threshold calcium spike (LTS) and by their inhibition after bath application of NA. Therefore, to establish that glucose-excited VLPO cells exhibit properties of sleep-promoting neurons, we investigated whether they displayed LTS and/or if they were inhibited by NA. Bath application of NA (100 μm) to glucose-excited VLPO neurons recorded in a loose-cell-attached configuration induced a decrease in their firing rate (n = 7/7; Fig. 3D). Moreover, 17 glucose-excited neurons repatched in a whole-cell configuration systematically exhibited LTS (n = 17/17; Fig. 3E). This NA-induced inhibition and the presence of LTS were both identified in 4 glucose-excited cells (n = 4/4).
In addition, 13 VLPO neurons remained insensitive to changes in the extracellular glucose concentration, even in the case of an acute change from 2.5 to 25 mm (n = 6, Wilcoxon test p = 0.7; Fig. 4A,B). In this sample of cells, 10 of the 13 neurons were used for further characterization, revealing that they were excited by NA (100 μm, n = 7/7) and devoid of any LTS (n = 5/5; Fig. 4C,D). This indicates that these cells should not correspond to sleep-active cells (Gallopin et al., 2000; Matsuo et al., 2003; Liu et al., 2010; Wang et al., 2013). Together, these results suggest that neurons with properties of putative sleep-promoting VLPO neurons are specifically excited by glucose and could be considered as glucose-sensing neurons.
Characterization of glucose-sensing mechanisms in VLPO neurons
Next, we addressed the mechanisms underlying this glucose sensitivity in VLPO neurons. We first demonstrated that the excitatory effect of glucose identified in NA-inhibited neurons persisted in synaptic uncoupling conditions using a modified ACSF with low Ca2+/high Mg2+ (n = 4; Fig. 5A). This result indicates that glucose does have a direct postsynaptic action on the neurons. To further investigate the role of glucose metabolism in glucose-sensing VLPO neurons, we tested the effect of 2-DG, a nonmetabolizable analog of glucose. The firing rate of NA-inhibited neurons did not change when 2-DG (7.5 mm) was added to the control ACSF (2.5 mm glucose; n = 4, exact permutation test, p = 1; Fig. 5B). In contrast, replacement of 2-DG by an equimolar concentration of glucose (7.5 mm for a total concentration of 10 mm glucose) in the same cells led to a significant increase in the firing frequency of 4.83 ± 0.87 Hz (n = 4, exact permutation test, p < 0.05; Fig. 5B). This suggests that glucose is metabolized into cells to induce its excitatory effect.
Figure 5.

Glucose-induced excitation implies the catabolism of glucose in neurons. A, Excitatory effect of glucose on the spontaneous firing activity of identified sleep-active cells under synaptic uncoupling conditions (low Ca2+/high Mg2+) (n = 4). Data are presented as mean ± SEM. *p < 0.05, permutation test. B, Effect of the application of a nonmetabolizable glucose analog (2-DG) on the spontaneous firing activity of identified putative sleep-promoting and glucose-excited neurons (n = 4). Data are presented as mean ± SEM. *p < 0.05, permutation test: ns. C, Effect of the blockade of lactate transporters (4-CIN) and glucokinase inhibition (alloxan) on the glucose-induced increase in resting membrane potential of identified sleep-active neurons. Data are presented as mean ± SEM with group sizes for each condition. **p < 0.01, Mann–Whitney test: ns. D, Gel electrophoresis of scRT-PCR products of three characterized sleep-promoting cells expressing GK mRNAs, with the third neuron expressing GK and GLUT3 mRNAs. MW, Molecular weight (100 bp ladder).
Glucose is not the only energy supplier in the brain. Indeed, lactate derived from astrocytes is regarded as the privileged substrate for neurons (Pellerin et al., 1998; Magistretti et al., 2003; Levin et al., 2004). It is thus important to determine whether extracellular glucose is metabolized directly by glucose-excited VLPO neurons and/or if it is converted earlier to lactate by astrocytes. Therefore, we tested the excitatory effect of glucose in the presence of an inhibitor of monocarboxylate transporters, 4-CIN (500 μm), or in the presence of the GK inhibitor alloxan (10 mm). The effects of drugs were tested independently on the resting membrane potential (RMP) of VLPO LTS neurons recorded in a whole-cell configuration. To avoid any voltage variability related to dialysis of the cells, RMP was measured immediately after passing to whole-cell configuration.
Consistent with our previous findings (Fig. 3), the mean RMP of LTS neurons was significantly more depolarized when the extracellular glucose concentration was 10 mm compared with 2.5 mm (n = 10 per group, Mann–Whitney test p < 0.01; Fig. 5C). Under 4-CIN treatment, the same increase in extracellular glucose concentration produced a similar and significant increase in RMP (n = 12 per condition; Mann–Whitney test p < 0.01; Fig. 5C). These results imply that the excitatory effect of glucose would not rely on the entry of astrocyte-derived lactate into neurons. In contrast, the RMP of identified LTS neurons remained unaffected by changes in extracellular glucose concentration under alloxan treatment (n = 12 per condition; Mann–Whitney test p = 0.8; Fig. 5C). This latter result indicates that glucose must be metabolized by these VLPO neurons to obtain sensitivity toward glucose. Using the scRT-PCR technique after patch-clamp recordings, we found that LTS VLPO neurons expressed mRNAs encoding glucose transporter type 3 (GLUT3, n = 10/13; Fig. 5D). GLUT1 and GLUT2 mRNAs were never detected in our sample of VLPO neurons. We also detected GK mRNAs in three neurons of our sample (Fig. 5D). Combined, the presence of GK mRNA in subset of VLPO neurons and the pharmacological blockage of glucose-induced excitation by GK inhibition indicate that putative VLPO sleep-active neurons are able to sense variations in glucose concentration directly without glial contribution.
VLPO neurons express functional KATP channels
Glucose-sensing neurons in the ventromedial hypothalamus (VMH) (Miki et al., 2001), the arcuate nucleus (Wang et al., 2004), and the lateral hypothalamus (Miki et al., 2001; Burdakov et al., 2006; Kong et al., 2010) engage KATP channels for coupling the cellular metabolic status to the membrane potential (Seino, 1999). These results motivated us to consider the involvement of these KATP channels in the glucose-induced excitation of VLPO neurons.
The KATP channel is composed of four pore-forming Kir6.1 or Kir6.2 subunits and four regulatory sulfonylurea receptor (SUR) SUR1 or SUR2 subunits (Seino, 1999; Moreau et al., 2005). We detected in the VLPO functional Kir6.2/SUR1-containing KATP channels in LTS neurons inhibited by NA (Fig. 6). Indeed, bath application of diazoxide (300 μm), a SUR activator, induced an outward current in neurons at −65 mV recorded in whole-cell configuration (50.5 ± 12.2 pA, n = 12/12; Fig. 6B). These effects were completely reversed after application of the KATP channel inhibitor tolbutamide (500 μm; Fig. 6B). Under the same conditions, application of pinacidil (500 μm), a selective SUR2 activator, did not induce any significant effect on VLPO neurons (n = 3/3; Fig. 6B). Depolarizing voltage ramps (from −115 mV to −45 mV in 400 ms) revealed that the diazoxide-induced current reversed at −81.5 ± 2.7 mV (n = 12; Fig. 6C), which is close to the theoretical potassium equilibrium potential in our conditions (EK+ = −89 mV). Using the scRT-PCR technique, we detected mRNAs encoding the Kir6.2, but not Kir6.1, subunit in VLPO LTS neurons (n = 10/24; Fig. 6D). Although pharmacological experiments confirmed the presence of SUR1 in VLPO neurons, we could not detect mRNAs encoding SUR subunits by scRT-PCR. Together, these results indicate that VLPO neurons displaying intrinsic properties of putative sleep-promoting neurons express functional KATP channels that are composed of Kir6.2 and SUR1 subunits.
Figure 6.

Characterization of KATP channels in VLPO neurons. A, Current-clamp recording of a sleep-promoting neuron identified by the presence of a potent LTS (*) and a multipolar shape (insert) Scale bar, 20 μm. B, Effect of pinacidil (500 μm), diazoxide (300 μm), and tolbutamide (500 μm) on the stationary currents recorded at −65 mV for the same cell shown in A. C, Effect of diazoxide (blue) and tolbutamide (red) on the I–V relationship of the same neuron shown in A and B. I–V plot of the net KATP-related current (black), which reverses near the K+ equilibrium potential. D, Gel electrophoresis of scRT-PCR products of a sleep-promoting cell revealing the expression of Kir6.2 but not SUR1 or SUR2 mRNAs. MW, Molecular weight (100 bp ladder).
Extracellular glucose concentration monitors the gating of KATP channels
To investigate whether glucose-induced excitation of VLPO neurons is related to a change in the opening state of KATP channels, we evaluated the change in holding current of VLPO LTS cells <10 mm and 2.5 mm glucose concentrations. For this purpose, the baseline holding current of LTS neurons, held at −65 mV, was measured immediately after passing into whole-cell configuration and then during diazoxide and tolbutamide applications (Fig. 7). We found that the holding current measured just after passing in whole-cell (cell opening) configuration was significantly higher under an extracellular glucose concentration of 2.5 mm than that of cells at <10 mm glucose (−4.0 ± 4.0 pA; −19.3 ± 2.7 pA, respectively; n = 8 per conditions; Mann–Whitney test p < 0.05; Fig. 7A,B). This result is in agreement with our finding that the extracellular glucose concentration can modulate the excitability of VLPO cells.
Figure 7.

Extracellular glucose level monitors KATP channel opening state in sleep-promoting cells. A–B, Top, Representative traces of stationary currents recorded at −65 mV in 2.5 mm (A) and 10 mm (B) glucose and during diazoxide (300 μm) and tolbutamide (500 μm) application. Note the difference in currents induced by diazoxide and tolbutamide compared with the initial current. Bottom, Currents recorded during 100 ms voltage steps from −65 to −75 mV at times indicated by a–d in recordings illustrated above. C, Effect of diazoxide and tolbutamide applications on holding current of LTS neurons held at −65 mV. Data are presented as changes induced by pharmacological treatments from the initial value of the holding current at cell opening. Data are presented as mean ± SEM; n = 8 per condition. *p < 0.05, **p < 0.01, Wilcoxon test vs initial current at cell opening; #p < 0.05, Mann–Whitney test 2.5 mm vs 10 mm glucose.
Finally, we assessed the effects of diazoxide and tolbutamide on the holding current of LTS neurons at 2.5 and 10 mm extracellular glucose concentrations. The current values presented in Figure 7C correspond to the difference between the steady-state holding current during diazoxide and tolbutamide treatments and the initial holding current at cell opening. At an extracellular glucose concentration of 2.5 mm, diazoxide (300 μm) induced an outward potassium current (31.2 ± 10.4 pA, n = 8; Fig. 7A,C) at −65 mV, which was reversed after tolbutamide application (500 μm). During tolbutamide treatment, the stabilized holding current was significantly lower than the initial current evaluated before dialysis (−14.1 ± 3.3 pA, n = 8, Wilcoxon test p < 0.01; Fig. 7A,C).
Diazoxide (300 μm) provoked a significantly larger outward current at 10 mm glucose (60.5 ± 16.7 pA, n = 8, Mann–Whitney test p = 0.040; Fig. 7B,C), which was reversed by tolbutamide (500 μm). However, tolbutamide did not change the steady-state holding current of LTS neurons significantly compared with the holding current at cell opening (−4.3 ± 2.6 pA, n = 8, Wilcoxon test p = 0.1; Fig. 7B,C).
Our results indicate that, at 10 mm glucose, KATP channels would be in a more closed state than at 2.5 mm glucose, as seen by the absence of tolbutamide effect. Conversely, at 2.5 mm glucose, KATP channels would be likely in a more opened state compared with 10 mm glucose, as seen by the significant tolbutamide effect and the weaker diazoxide effect. Therefore, extracellular glucose may modulate the gating of KATP channels within VLPO LTS neurons, leading to their previously described glucose sensitivity.
Together, our results support the view that glucose may enhance the neuronal excitability of sleep-promoting neurons located in the VLPO and therefore may participate in the mechanisms of SWS promotion and/or consolidation.
Discussion
Using an integrated approach, we found here that infusion of glucose into the VLPO of mice promotes SWS and increases the density of c-Fos-labeled neurons selectively in the VLPO. Furthermore, we have demonstrated ex vivo that a rise in extracellular glucose increases the firing rate of putative sleep-promoting VLPO neurons. ATP synthesis in these neurons would proceed through the metabolism of glucose, which closes the KATP channels and leads to cellular depolarization. Functionally, the present results highlight that glucose likely contributes to sleep onset facilitation by increasing the excitability of sleep-promoting VLPO neurons and thus sleep propensity.
Glucose supply in the VLPO promotes SWS
We hypothesized that sleep-promoting VLPO neurons integrate the metabolic signal conveyed by glucose, thus enabling a metabolic drive of vigilance states. We tested this hypothesis here by investigating the influence of glucose on sleep regulation in vivo after direct bilateral glucose microinjection into the VLPO. We found that an increase in glucose concentration induces a significant increase in the time spent in SWS associated with a decrease in the latency to the first SWS episode. This is the first demonstration that local injections of glucose can affect vigilance states. Furthermore, this result illustrates a glucose effect on both the induction and maintenance of sleep because glucose appeared to increase sleep duration and sleep propensity. This finding is consistent with previous studies demonstrating a role for the VLPO in promotion and maintenance of SWS (Szymusiak and McGinty, 2008; Saper et al., 2010).
We performed c-Fos immunostaining after infusion of glucose to investigate whether glucose can activate VLPO neurons and if the effect that we observed on vigilance states could be restricted to the VLPO. In the VLPO, levels of c-Fos expression have been correlated with sleep durations, thus constituting a marker of sleep rebound (Lu et al., 2000; Gvilia et al., 2006; Dentico et al., 2009). In our experiments, no significant variation in c-Fos-labeled cells was observed in neighboring regions such as the MPOA, LPOA, and MCPO, whereas we observed a selective increase in c-Fos expression in the VLPO. These results strongly suggest that the increase in SWS elicited by glucose injection is a consequence of sleep-promoting VLPO neuronal activation.
Ex vivo identification of putative sleep-promoting VLPO neurons
Previous studies characterized the intrinsic and pharmacological properties of sleep-promoting VLPO neurons ex vivo. Indeed, a large majority of VLPO GABAergic neurons generate a potent LTS and are inhibited by wake-promoting neurotransmitters such as NA (Gallopin et al., 2000; Matsuo et al., 2003; Gallopin et al., 2004; Liu et al., 2010; Moore et al., 2012; McCarren et al., 2014). The inhibition of these VLPO GABAergic neurons by wake-promoting neurotransmitters is in agreement with their inactivity during wakefulness, strongly supporting the idea that they correspond to the sleep-active cells identified in vivo and are involved in the inhibition of arousal systems (Gallopin et al., 2000; Saper et al., 2001).
Glucose induces excitation of VLPO neurons through a metabolic pathway
Different neuronal populations in the brain that regulate glucose homeostasis or feeding behavior increase their firing according to a rise in ambient glucose (Levin et al., 2004; Burdakov et al., 2005). Using a nonmetabolizable glucose analog (2-DG), we have shown that glucose metabolization is required to induce excitation of VLPO neurons. This metabolization of glucose could occur in neurons and/or in astrocytes. Indeed, astrocyte-derived lactate is considered to be the major source of metabolic energy for neurons (Pellerin et al., 1998; Magistretti et al., 2003). We have demonstrated that the inhibition of the enzyme GK selectively expressed in neurons (Dunn-Meynell et al., 2002) abolished the glucose-induced excitation of putative sleep-promoting neurons, whereas blockade of lactate transport by 4-CIN did not affect the magnitude of the excitatory effect of glucose. Although we cannot exclude the possibility that astrocyte-derived glucose metabolites could influence VLPO neuronal activity, our results indicate that glucose by itself is sufficient to excite putative sleep-promoting VLPO neurons. Furthermore, single-cell mRNA detection revealed that VLPO LTS neurons express mRNAs that encode the glucose transporter GLUT3 and the enzyme GK. The single-cell mRNA detection of GK in a subset of cells is reinforced by the pharmacological effect of a GK inhibitor on the glucose-induced excitation. The expression of GK in neurons is reported to play an important role in the neuronal mechanisms of glucose sensing by triggering a shift in metabolism in response to glucose supply changes (Kang et al., 2006; Dunn-Meynell et al., 2009).
Together, our results reveal that glucose affects VLPO neuronal excitability in a manner similar to what occurs in subsets of hypothalamic glucose-excited neurons and in insulin-secreting pancreatic β-cells (Matschinsky, 1990). In these mechanisms, glucose enters the cell through specific glucose transporters, where it is phosphorylated by GK and converted to energy by ATP production (Gonzàlez et al., 2009). This increase in the cytosolic ATP concentration closes the KATP channels, leading to the excitation of the cell.
KATP channels as a link between cellular energetics and excitability of VLPO neurons
The cooperative action of GK and KATP channels in some glucose-sensing neurons plays a crucial role in glucose-induced excitation by coupling metabolism with membrane excitability (Routh, 2002; Kang et al., 2006). KATP channels could differ in their combinations of Kir6.1 or Kir6.2 and SUR1 or SUR2 subunits. On the basis of our pharmacological and scRT-PCR experiments, we favor the view that the KATP channels of sleep-promoting VLPO neurons are composed of Kir6.2 and SUR1 subunits. We showed that application of pinacidil, a selective SUR2 activator, did not induce any significant effect on VLPO neurons, indicating the absence of the SUR2 isoform. This result is in agreement with several studies indicating that pinacidil activates Kir6.2/SUR2, but not Kir6.2/SUR1 channels (Shindo et al., 1998; Ashcroft and Gribble, 2000; Wheeler et al., 2008). Moreover, we showed that diazoxide (a KATP channel opener) induces an outward current that was systematically reversed by tolbutamide (a KATP channel inhibitor), confirming the presence of functional KATP channels in VLPO neurons. However, we could not detect mRNAs encoding SUR subunits in individual cells. This is a common problem seen in scRT-PCR when mRNAs are present at low levels (Gallopin et al., 2006, Tsuzuki et al., 2001). Indeed, SUR mRNAs are present in very low abundance in neurons, even in the canonical glucose-sensing neurons of the VMH (Kang et al., 2004).
Moreover, the low proportion of putative sleep-active cells that are positive for GK at the cellular level could also be explained due to the very low amount of these mRNA species in cells even in glucose-responsive neurons (Lynch et al., 2000; Dunn-Meynell et al., 2002; Kang et al., 2004).
Concerning the expression of the Kir6 isoform, we detected the presence of Kir6.2 mRNAs in 41% of the harvested VLPO neurons (which might be underestimate by our detection method). However, this proportion is totally consistent with previous observations in glucose-excited neurons in the VMH (42%; Kang et al., 2004). In contrast to the widespread distribution of the Kir6.2 subunit in the brain, immunoreactivity of Kir6.1 in neurons has only been reported in specific brain regions that do not include the VLPO (Thomzig et al., 2005).
Energy metabolism and sleep homeostasis
Various pathways can regulate sleep pressure associated with drowsiness, including circadian and homeostatic drives, which can both modulate the activity of VLPO neurons (Saper et al., 2005). Here, we demonstrate for the first time that putative sleep-promoting VLPO neurons are also able to integrate energetic signals such as ambient glucose directly to modulate behavioral states. Importantly, these results complement previous reports showing that glucose modulates the excitability of orexin-expressing neurons located in the lateral hypothalamus, which are involved in arousal maintenance (Tsujino and Sakurai, 2009). Indeed, ex vivo experiments have revealed that glucose inhibits orexinergic neurons (Yamanaka et al., 2003; Burdakov et al., 2006).
Physiological and physiopathological studies have illustrated that sleep maintains a close and cooperative relationship with metabolism. Metabolism-related compounds such as glucose, glycogen, lactate, and/or ATP have been shown to fluctuate over vigilance states (Netchiporouk et al., 2001; Shram et al., 2002; Dworak et al., 2010). Food deprivation in rodent models increases wakefulness, which is compatible with the development of food-seeking behaviors (Jacobs and McGinty, 1971; Dewasmes et al., 1989; Minet-Ringuet et al., 2004). In contrast, carbohydrate intake appears to enhance sleep (Danguir and Nicolaidis, 1979; Nicolaidis, 2006). Similar effects have been observed in humans, where it was shown that even if glucose intake leads to short-term cognitive improvement, it could eventually lead to an enhanced sleepiness and facilitated sleep onset (Landström et al., 2000; Horne and Baulk, 2004; Anderson and Horne, 2006; Afaghi et al., 2007).
Our results provide evidence that high-energy stores are important to maintain an excitation of neurons required to trigger and maintain SWS. Conversely, we would expect that a depletion of energy supplies in the VLPO could decrease the firing rate of these cells and would thus facilitate awakening. The novel mechanisms regulating the activity of VLPO neurons described herein reinforce the perspectives of a crucial link between sleep and metabolism.
Footnotes
This work was supported by the Fondation Pierre-Gilles de Gennes pour la Recherché (Grant ANR-12-JSV4-0001-01), the Centre National de la Recherche Scientifique, and Ecole Supérieure de Physique et de Chimie Industrielles ParisTech. We thank Brandon Loveall of Improvence for English proofreading and Karim Benchenane for invaluable comments on the manuscript.
The authors declare no competing financial interests.
References
- Adamantidis AR, Zhang F, Aravanis AM, Deisseroth K, de Lecea L. Neural substrates of awakening probed with optogenetic control of hypocretin neurons. Nature. 2007;450:420–424. doi: 10.1038/nature06310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afaghi A, O'Connor H, Chow CM. High-glycemic-index carbohydrate meals shorten sleep onset. Am J Clin Nutr. 2007;85:426–430. doi: 10.1093/ajcn/85.2.426. [DOI] [PubMed] [Google Scholar]
- Anderson C, Horne JA. A high sugar content, low caffeine drink does not alleviate sleepiness but may worsen it. Hum Psychopharmacol. 2006;21:299–303. doi: 10.1002/hup.769. [DOI] [PubMed] [Google Scholar]
- Ashcroft FM, Gribble FM. New windows on the mechanism of action of KATP channel openers. Trends Pharmacol Sci. 2000;21:439–445. doi: 10.1016/S0165-6147(00)01563-7. [DOI] [PubMed] [Google Scholar]
- Benington JH, Heller HC. Restoration of brain energy metabolism as the function of sleep. Prog Neurobiol. 1995;45:347–360. doi: 10.1016/0301-0082(94)00057-O. [DOI] [PubMed] [Google Scholar]
- Bouzier-Sore AK, Voisin P, Canioni P, Magistretti PJ, Pellerin L. Lactate is a preferential oxidative energy substrate over glucose for neurons in culture. J Cereb Blood Flow Metab. 2003;23:1298–1306. doi: 10.1097/01.WCB.0000091761.61714.25. [DOI] [PubMed] [Google Scholar]
- Burdakov D, Alexopoulos H. Metabolic state signalling through central hypocretin/orexin neurons. J Cell Mol Med. 2005;9:795–803. doi: 10.1111/j.1582-4934.2005.tb00380.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdakov D, Luckman SM, Verkhratsky A. Glucose-sensing neurons of the hypothalamus. Philos Trans R Soc Lond B Biol Sci. 2005;360:2227–2235. doi: 10.1098/rstb.2005.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdakov D, Jensen LT, Alexopoulos H, Williams RH, Fearon IM, O'Kelly I, Gerasimenko O, Fugger L, Verkhratsky A. Tandem-pore K+ channels mediate inhibition of orexin neurons by glucose. Neuron. 2006;50:711–722. doi: 10.1016/j.neuron.2006.04.032. [DOI] [PubMed] [Google Scholar]
- Danguir J, Nicolaidis S. Dependence of sleep on nutrients' availability. Physiol Behav. 1979;22:735–740. doi: 10.1016/0031-9384(79)90240-3. [DOI] [PubMed] [Google Scholar]
- Dentico D, Amici R, Baracchi F, Cerri M, Del Sindaco E, Luppi M, Martelli D, Perez E, Zamboni G. c-Fos expression in preoptic nuclei as a marker of sleep rebound in the rat. Eur J Neurosci. 2009;30:651–661. doi: 10.1111/j.1460-9568.2009.06848.x. [DOI] [PubMed] [Google Scholar]
- de Vries MG, Arseneau LM, Lawson ME, Beverly JL. Extracellular glucose in rat ventromedial hypothalamus during acute and recurrent hypoglycemia. Diabetes. 2003;52:2767–2773. doi: 10.2337/diabetes.52.11.2767. [DOI] [PubMed] [Google Scholar]
- Dewasmes G, Duchamp C, Minaire Y. Sleep changes in fasting rats. Physiol Behav. 1989;46:179–184. doi: 10.1016/0031-9384(89)90252-7. [DOI] [PubMed] [Google Scholar]
- Dunn-Meynell AA, Routh VH, Kang L, Gaspers L, Levin BE. Glucokinase is the likely mediator of glucosensing in both glucose-excited and glucose-inhibited central neurons. Diabetes. 2002;51:2056–2065. doi: 10.2337/diabetes.51.7.2056. [DOI] [PubMed] [Google Scholar]
- Dunn-Meynell AA, Sanders NM, Compton D, Becker TC, Eiki J, Zhang BB, Levin BE. Relationship among brain and blood glucose levels and spontaneous and glucoprivic feeding. J Neurosci. 2009;29:7015–7022. doi: 10.1523/JNEUROSCI.0334-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dworak M, McCarley RW, Kim T, Kalinchuk AV, Basheer R. Sleep and brain energy levels: ATP changes during sleep. J Neurosci. 2010;30:9007–9016. doi: 10.1523/JNEUROSCI.1423-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates. Ed 3. Amsterdam: Academic; 2007. [Google Scholar]
- Gallopin T, Fort P, Eggermann E, Cauli B, Luppi PH, Rossier J, Audinat E, Mühlethaler M, Serafin M. Identification of sleep-promoting neurons in vitro. Nature. 2000;404:992–995. doi: 10.1038/35010109. [DOI] [PubMed] [Google Scholar]
- Gallopin T, Luppi PH, Rambert FA, Frydman A, Fort P. Effect of the wake-promoting agent modafinil on sleep-promoting neurons from the ventrolateral preoptic nucleus: an in vitro pharmacologic study. Sleep. 2004;27:19–25. [PubMed] [Google Scholar]
- Gallopin T, Luppi PH, Cauli B, Urade Y, Rossier J, Hayaishi O, Lambolez B, Fort P. The endogenous somnogen adenosine excites a subset of sleep-promoting neurons via A2A receptors in the ventrolateral preoptic nucleus. Neuroscience. 2005;134:1377–1390. doi: 10.1016/j.neuroscience.2005.05.045. [DOI] [PubMed] [Google Scholar]
- Gallopin T, Geoffroy H, Rossier J, Lambolez B. Cortical sources of CRF, NKB, and CCK and their effects on pyramidal cells in the neocortex. Cereb Cortex. 2006;16:1440–1452. doi: 10.1093/cercor/bhj081. [DOI] [PubMed] [Google Scholar]
- Gonzàlez JA, Reimann F, Reimann F, Burdakov D. Dissociation between sensing and metabolism of glucose in sugar sensing neurones. J Physiol. 2009;587:41–48. doi: 10.1113/jphysiol.2008.163410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gvilia I, Xu F, McGinty D, Szymusiak R. Homeostatic regulation of sleep: a role for preoptic area neurons. J Neurosci. 2006;26:9426–9433. doi: 10.1523/JNEUROSCI.2012-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horne JA, Baulk SD. Awareness of sleepiness when driving. Psychophysiology. 2004;41:161–165. doi: 10.1046/j.1469-8986.2003.00130.x. [DOI] [PubMed] [Google Scholar]
- Jacobs BL, McGinty DJ. Effects of food deprivation on sleep and wakefulness in the rat. Exp Neurol. 1971;30:212–222. doi: 10.1016/S0014-4886(71)80002-X. [DOI] [PubMed] [Google Scholar]
- Jenkins JB, Omori T, Guan Z, Vgontzas AN, Bixler EO, Fang J. Sleep is increased in mice with obesity induced by high-fat food. Physiol Behav. 2006;87:255–262. doi: 10.1016/j.physbeh.2005.10.010. [DOI] [PubMed] [Google Scholar]
- Kang L, Routh VH, Kuzhikandathil EV, Gaspers LD, Levin BE. Physiological and molecular characteristics of rat hypothalamic ventromedial nucleus glucosensing neurons. Diabetes. 2004;53:549–559. doi: 10.2337/diabetes.53.3.549. [DOI] [PubMed] [Google Scholar]
- Kang L, Dunn-Meynell AA, Routh VH, Gaspers LD, Nagata Y, Nishimura T, Eiki J, Zhang BB, Levin BE. Glucokinase is a critical regulator of ventromedial hypothalamic neuronal glucosensing. Diabetes. 2006;55:412–420. doi: 10.2337/diabetes.55.02.06.db05-1229. [DOI] [PubMed] [Google Scholar]
- Kong D, Vong L, Parton LE, Ye C, Tong Q, Hu X, Choi B, Brüning JC, Lowell BB. Glucose stimulation of hypothalamic MCH neurons involves K(ATP) channels, is modulated by UCP2, and regulates peripheral glucose homeostasis. Cell Metab. 2010;12:545–552. doi: 10.1016/j.cmet.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambolez B, Audinat E, Bochet P, Crépel F, Rossier J. AMPA receptor subunits expressed by single Purkinje cells. Neuron. 1992;9:247–258. doi: 10.1016/0896-6273(92)90164-9. [DOI] [PubMed] [Google Scholar]
- Landström U, Knutsson A, Lennernäs M. Field studies on the effects of food content on wakefulness. Nutr Health. 2000;14:195–204. doi: 10.1177/026010600001400401. [DOI] [PubMed] [Google Scholar]
- Levin BE, Routh VH, Kang L, Sanders NM, Dunn-Meynell AA. Neuronal glucosensing: what do we know after 50 years? Diabetes. 2004;53:2521–2528. doi: 10.2337/diabetes.53.10.2521. [DOI] [PubMed] [Google Scholar]
- Liu YW, Li J, Ye JH. Histamine regulates activities of neurons in the ventrolateral preoptic nucleus. J Physiol. 2010;588:4103–4116. doi: 10.1113/jphysiol.2010.193904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Greco MA, Shiromani P, Saper CB. Effect of lesions of the ventrolateral preoptic nucleus on NREM and REM sleep. J Neurosci. 2000;20:3830–3842. doi: 10.1523/JNEUROSCI.20-10-03830.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch RM, Tompkins LS, Brooks HL, Dunn-Meynell AA, Levin BE. Localization of glucokinase gene expression in the rat brain. Diabetes. 2000;49:693–700. doi: 10.2337/diabetes.49.5.693. [DOI] [PubMed] [Google Scholar]
- Matschinsky FM. Glucokinase as glucose sensor and metabolic signal generator in pancreatic beta-cells and hepatocytes. Diabetes. 1990;39:647–652. doi: 10.2337/diab.39.6.647. [DOI] [PubMed] [Google Scholar]
- Matsuo S, Jang IS, Nabekura J, Akaike N. alpha 2-Adrenoceptor-mediated presynaptic modulation of GABAergic transmission in mechanically dissociated rat ventrolateral preoptic neurons. J Neurophysiol. 2003;89:1640–1648. doi: 10.1152/jn.00491.2002. [DOI] [PubMed] [Google Scholar]
- McCarren HS, Chalifoux MR, Han B, Moore JT, Meng QC, Baron-Hionis N, Sedigh-Sarvestani M, Contreras D, Beck SG, Kelz MB. α2-adrenergic stimulation of the ventrolateral preoptic nucleus destabilizes the anesthetic state. J Neurosci. 2014;34:16385–16396. doi: 10.1523/JNEUROSCI.1135-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki T, Liss B, Minami K, Shiuchi T, Saraya A, Kashima Y, Horiuchi M, Ashcroft F, Minokoshi Y, Roeper J, Seino S. ATP-sensitive K+ channels in the hypothalamus are essential for the maintenance of glucose homeostasis. Nat Neurosci. 2001;4:507–512. doi: 10.1038/87455. [DOI] [PubMed] [Google Scholar]
- Minet-Ringuet J, Le Ruyet PM, Tomé D, Even PC. A tryptophan-rich protein diet efficiently restores sleep after food deprivation in the rat. Behav Brain Res. 2004;152:335–340. doi: 10.1016/j.bbr.2003.10.018. [DOI] [PubMed] [Google Scholar]
- Moore JT, Chen J, Han B, Meng QC, Veasey SC, Beck SG, Kelz MB. Direct activation of sleep-promoting VLPO neurons by volatile anesthetics contributes to anesthetic hypnosis. Curr Biol. 2012;22:2008–2016. doi: 10.1016/j.cub.2012.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau C, Prost AL, Dérand R, Vivaudou M. SUR, ABC proteins targeted by KATP channel openers. J Mol Cell Cardiol. 2005;38:951–963. doi: 10.1016/j.yjmcc.2004.11.030. [DOI] [PubMed] [Google Scholar]
- Netchiporouk L, Shram N, Salvert D, Cespuglio R. Brain extracellular glucose assessed by voltammetry throughout the rat sleep-wake cycle. Eur J Neurosci. 2001;13:1429–1434. doi: 10.1046/j.0953-816x.2001.01503.x. [DOI] [PubMed] [Google Scholar]
- Nicolaidis S. Metabolic mechanism of wakefulness (and hunger) and sleep (and satiety): Role of adenosine triphosphate and hypocretin and other peptides. Metab Clin Exp. 2006;55:S24–S29. doi: 10.1016/j.metabol.2006.07.009. [DOI] [PubMed] [Google Scholar]
- Pellerin L, Pellegri G, Bittar PG, Charnay Y, Bouras C, Martin JL, Stella N, Magistretti PJ. Evidence supporting the existence of an activity-dependent astrocyte-neuron lactate shuttle. Dev Neurosci. 1998;20:291–299. doi: 10.1159/000017324. [DOI] [PubMed] [Google Scholar]
- Roky R, Kapás L, Taishi TP, Fang J, Krueger JM. Food restriction alters the diurnal distribution of sleep in rats. Physiol Behav. 1999;67:697–703. doi: 10.1016/S0031-9384(99)00137-7. [DOI] [PubMed] [Google Scholar]
- Routh VH. Glucose-sensing neurons: are they physiologically relevant? Physiol Behav. 2002;76:403–413. doi: 10.1016/S0031-9384(02)00761-8. [DOI] [PubMed] [Google Scholar]
- Saint-Mleux B, Eggermann E, Bisetti A, Bayer L, Machard D, Jones BE, Mühlethaler M, Serafin M. Nicotinic enhancement of the noradrenergic inhibition of sleep-promoting neurons in the ventrolateral preoptic area. J Neurosci. 2004;24:63–67. doi: 10.1523/JNEUROSCI.0232-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saint-Mleux B, Bayer L, Eggermann E, Jones BE, Mühlethaler M, Serafin M. Suprachiasmatic modulation of noradrenaline release in the ventrolateral preoptic nucleus. J Neurosci. 2007;27:6412–6416. doi: 10.1523/JNEUROSCI.1432-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saper CB, Chou TC, Scammell TE. The sleep switch: hypothalamic control of sleep and wakefulness. Trends Neurosci. 2001;24:726–731. doi: 10.1016/S0166-2236(00)02002-6. [DOI] [PubMed] [Google Scholar]
- Saper CB, Cano G, Scammell TE. Homeostatic, circadian, and emotional regulation of sleep. J Comp Neurol. 2005;493:92–98. doi: 10.1002/cne.20770. [DOI] [PubMed] [Google Scholar]
- Saper CB, Fuller PM, Pedersen NP, Lu J, Scammell TE. Sleep state switching. Neuron. 2010;68:1023–1042. doi: 10.1016/j.neuron.2010.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharf MT, Naidoo N, Zimmerman JE, Pack AI. The energy hypothesis of sleep revisited. Prog Neurobiol. 2008;86:264–280. doi: 10.1016/j.pneurobio.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seino S. ATP-sensitive potassium channels: a model of heteromultimeric potassium channel/receptor assemblies. Annu Rev Physiol. 1999;61:337–362. doi: 10.1146/annurev.physiol.61.1.337. [DOI] [PubMed] [Google Scholar]
- Sherin JE, Shiromani PJ, McCarley RW, Saper CB. Activation of ventrolateral preoptic neurons during sleep. Science. 1996;271:216–219. doi: 10.1126/science.271.5246.216. [DOI] [PubMed] [Google Scholar]
- Sherin JE, Elmquist JK, Torrealba F, Saper CB. Innervation of histaminergic tuberomammillary neurons by GABAergic and galaninergic neurons in the ventrolateral preoptic nucleus of the rat. J Neurosci. 1998;18:4705–4721. doi: 10.1523/JNEUROSCI.18-12-04705.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shindo T, Yamada M, Isomoto S, Horio Y, Kurachi Y. SUR2 subtype (A and B)-dependent differential activation of the cloned ATP-sensitive K+ channels by pinacidil and nicorandil. Br J Pharmacol. 1998;124:985–991. doi: 10.1038/sj.bjp.0701927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shram N, Netchiporouk L, Cespuglio R. Lactate in the brain of the freely moving rat: voltammetric monitoring of the changes related to the sleep-wake states. Eur J Neurosci. 2002;16:461–466. doi: 10.1046/j.1460-9568.2002.02081.x. [DOI] [PubMed] [Google Scholar]
- Siegel JM. Sleep viewed as a state of adaptive inactivity. Nat Rev Neurosci. 2009;10:747–753. doi: 10.1038/nrn2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver IA, Erecińska M. Extracellular glucose concentration in mammalian brain: continuous monitoring of changes during increased neuronal activity and upon limitation in oxygen supply in normo-, hypo-, and hyperglycemic animals. J Neurosci. 1994;14:5068–5076. doi: 10.1523/JNEUROSCI.14-08-05068.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steininger TL, Gong H, McGinty D, Szymusiak R. Subregional organization of preoptic area/anterior hypothalamic projections to arousal-related monoaminergic cell groups. J Comp Neurol. 2001;429:638–653. [PubMed] [Google Scholar]
- Szymusiak R, McGinty D. Hypothalamic regulation of sleep and arousal. Ann N Y Acad Sci. 2008;1129:275–286. doi: 10.1196/annals.1417.027. [DOI] [PubMed] [Google Scholar]
- Szymusiak R, Alam N, Steininger TL, McGinty D. Sleep-waking discharge patterns of ventrolateral preoptic/anterior hypothalamic neurons in rats. Brain Res. 1998;803:178–188. doi: 10.1016/S0006-8993(98)00631-3. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Lin JS, Sakai K. Characterization and mapping of sleep-waking specific neurons in the basal forebrain and preoptic hypothalamus in mice. Neuroscience. 2009;161:269–292. doi: 10.1016/j.neuroscience.2009.02.075. [DOI] [PubMed] [Google Scholar]
- Thomzig A, Laube G, Prüss H, Veh RW. Pore-forming subunits of K-ATP channels, Kir6.1 and Kir6.2, display prominent differences in regional and cellular distribution in the rat brain. J Comp Neurol. 2005;484:313–330. doi: 10.1002/cne.20469. [DOI] [PubMed] [Google Scholar]
- Tsujino N, Sakurai T. Orexin/hypocretin: a neuropeptide at the interface of sleep, energy homeostasis, and reward system. Pharmacol Rev. 2009;61:162–176. doi: 10.1124/pr.109.001321. [DOI] [PubMed] [Google Scholar]
- Tsuzuki K, Lambolez B, Rossier J, Ozawa S. Absolute quantification of AMPA receptor subunit mRNAs in single hippocampal neurons. J Neurochem. 2001;77:1650–1659. doi: 10.1046/j.1471-4159.2001.00388.x. [DOI] [PubMed] [Google Scholar]
- Wang Q, Yue XF, Qu WM, Tan R, Zheng P, Urade Y, Huang ZL. Morphine inhibits sleep-promoting neurons in the ventrolateral preoptic area via mu receptors and induces wakefulness in rats. Neuropsychopharmacology. 2013;38:791–801. doi: 10.1038/npp.2012.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Liu X, Hentges ST, Dunn-Meynell AA, Levin BE, Wang W, Routh VH. The regulation of glucose-excited neurons in the hypothalamic arcuate nucleus by glucose and feeding-relevant peptides. Diabetes. 2004;53:1959–1965. doi: 10.2337/diabetes.53.8.1959. [DOI] [PubMed] [Google Scholar]
- Wheeler A, Wang C, Yang K, Fang K, Davis K, Styer AM, Mirshahi U, Moreau C, Revilloud J, Vivaudou M, Liu S, Mirshahi T, Chan KW. Coassembly of different sulfonylurea receptor subtypes extends the phenotypic diversity of ATP-sensitive potassium (KATP) channels. Mol Pharmacol. 2008;74:1333–1344. doi: 10.1124/mol.108.048355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka A, Beuckmann CT, Willie JT, Hara J, Tsujino N, Mieda M, Tominaga M, Yagami Ki, Sugiyama F, Goto K, Yanagisawa M, Sakurai T. Hypothalamic orexin neurons regulate arousal according to energy balance in mice. Neuron. 2003;38:701–713. doi: 10.1016/S0896-6273(03)00331-3. [DOI] [PubMed] [Google Scholar]