

Abstract
Pyruvate dehydrogenase kinases (PDK1–4) are mitochondrial metabolic regulators that serve as decision makers via modulation of pyruvate dehydrogenase (PDH) activity to convert pyruvate either aerobically to acetyl-CoA or anaerobically to lactate. Metabolic dysregulation and inflammatory processes are two sides of the same coin in several pathophysiological conditions. The lactic acid surge associated with the metabolic shift has been implicated in diverse painful states. In this study, we investigated the role of PDK-PDH-lactic acid axis in the pathogenesis of chronic inflammatory pain. Deficiency of Pdk2 and/or Pdk4 in mice attenuated complete Freund's adjuvant (CFA)-induced pain hypersensitivities. Likewise, Pdk2/4 deficiency attenuated the localized lactic acid surge along with hallmarks of peripheral and central inflammation following intraplantar administration of CFA. In vitro studies supported the role of PDK2/4 as promoters of classical proinflammatory activation of macrophages. Moreover, the pharmacological inhibition of PDKs or lactic acid production diminished CFA-induced inflammation and pain hypersensitivities. Thus, a PDK-PDH-lactic acid axis seems to mediate inflammation-driven chronic pain, establishing a connection between metabolism and inflammatory pain.
SIGNIFICANCE STATEMENT The mitochondrial pyruvate dehydrogenase (PDH) kinases (PDKs) and their substrate PDH orchestrate the conversion of pyruvate either aerobically to acetyl-CoA or anaerobically to lactate. Lactate, the predominant end product of glycolysis, has recently been identified as a signaling molecule for neuron-glia interactions and neuronal plasticity. Pathological metabolic shift and subsequent lactic acid production are thought to play an important role in diverse painful states; however, their contribution to inflammation-driven pain is still to be comprehended. Here, we report that the PDK-PDH-lactic acid axis constitutes a key component of inflammatory pain pathogenesis. Our findings establish an unanticipated link between metabolism and inflammatory pain. This study unlocks a previously ill-explored research avenue for the metabolic control of inflammatory pain pathogenesis.
Keywords: chronic inflammatory pain, inflammation, macrophages, metabolism, pain, PDK-PDH-lactic acid axis
Introduction
Chronic inflammation has been identified as a central feature of the metabolic syndrome (Hotamisligil, 2006; Lumeng and Saltiel, 2011). Emerging evidence has outlined the crosstalk between inflammatory processes and metabolic dysregulation under pathological conditions (Palomer et al., 2013). Cells participating in inflammation experience diverse metabolic changes, including a shift toward enhanced glucose uptake and glycolysis. Altered metabolism may participate in signal-directed programs that promote or inhibit inflammation (O'Neill and Hardie, 2013). Mitochondrial metabolic shift has been identified as a hallmark of highly prevalent diseases, such as obesity/diabetes, cancer (Ramos-Nino, 2013), and cardiovascular disease (Nisoli et al., 2007). Mitochondrial malfunction, aberrant glucose metabolism, and the subsequent dysregulation of bioenergetics are consistent antecedents to the pathophysiology of the nervous system as well (Jha et al., 2012). Accumulating evidence has implicated the metabolic shift (Joseph and Levine, 2006), reactive oxygen species (Kim et al., 2004), genetic defects (Joseph and Levine, 2010), diabetes (Fernyhough et al., 2010), chemotherapeutics (Bennett, 2010), and HIV infection (Ferrari and Levine, 2010) in the pathogenesis of painful peripheral neuropathies. These studies suggest that modulation of mitochondrial function is a promising therapeutic strategy for the management of diverse pain states (Baloh, 2008).
Pain hypersensitivity that results from infection, inflammation, or peripheral injury to tissue is collectively called inflammatory pain (Kopach et al., 2012). The development of pain hypersensitivity in response to inflammatory insults/stimuli is due to peripheral (Basbaum et al., 2009) and central sensitizations (Kuner, 2010). Recruitment of immune/inflammatory cells in response to inflammatory insult and the subsequent release of proinflammatory/algesic mediators activate peripheral nociceptors resulting in peripheral sensitization, whereas the long-term potentiation in the spinal cord is regarded as a unique form of central sensitization (Ji et al., 2013). Lactic acid, the predominant end product of glycolysis (Rogatzki et al., 2015), has recently been identified as a signaling molecule for neuron–glia interactions (Tang et al., 2014) and neuronal plasticity (Yang et al., 2014). Lactic acid also contributes to pathological pain by facilitating nociceptor activation via lowered pH (Kim et al., 2007). Muscle ischemia or microvascular dysfunction generates lactic acid and inflammatory mediators that play a crucial role in the development of cutaneous tactile allodynia by activating acid sensing ion channels (ASICs) and other receptors on muscle primary afferent fibers (Laferrière et al., 2008). Inflammatory cells are active sources of lactic acid and other proalgesic mediators (Haji-Michael et al., 1999; Adeva-Andany et al., 2014). Lactic acid has also been shown to exert proinflammatory effects (Shime et al., 2008). Nevertheless, the precise role of lactic acid in the development of chronic inflammatory pain is not clearly understood.
The mitochondrial pyruvate dehydrogenase (PDH) complex (PDC) is a gatekeeping enzyme that plays a key regulatory role in cellular metabolism, by linking the citric acid cycle and subsequent oxidative phosphorylation with glycolysis and gluconeogenesis (Patel and Roche, 1990; Takubo et al., 2013). Not surprisingly given its central role in metabolism, PDC is under tight and complex regulation by reversible phosphorylation in response to the physiological state. PDC activity is inhibited by site-specific phosphorylation at three serine residues on the E1α subunit (Ser232, Ser293, and Ser300), which is catalyzed by four different PDH kinases (PDK1-4) having expression in peripheral and central tissues (Jha et al., 2012). Each of the four kinases has a different reactivity for these three sites, and phosphorylation at any site leads to the inhibition of PDH activity. Dysregulation of the PDH/PDK system has been implicated in life-threatening neurometabolic disorders, including Alzheimer's disease, traumatic brain injury, brain aging, and glioblastoma (Jha et al., 2012; Palamiuc et al., 2015). PDK-induced phosphorylation and subsequent inactivation of PDC result in metabolic shift toward glycolysis and lactate production, which led us to hypothesize that lactic acid produced under this condition may have tremendous effects on inflammation and pain behaviors. Thus, a PDK-PDH-lactic acid axis may be a key component that governs chronic inflammatory pain development. This possibility was tested using Pdk2- and Pdk4-deficient mice and chemical inhibitors of PDK or lactic acid production in the complete Freund's adjuvant (CFA)-injected rodent model of inflammatory pain. Macrophages in culture were also used to examine the mechanistic relationship between PDKs and proinflammatory phenotypes.
Materials and Methods
Mouse breeding and maintenance.
All experiments were conducted in accordance with approved animal protocols and guidelines established by the Animal Care Committee of Kyungpook National University (approval no. KNU-2012–73/66). All efforts were made to minimize the number of animals used and animal suffering. Male wild-type (WT, Pdk2+/+ Pdk4+/+), Pdk2 knock-out (Pdk2−/−), Pdk4 knock-out (Pdk4−/−), and Pdk2/4 double knock-out (DKO, Pdk2−/− Pdk4−/−) mice 8–10 weeks of age were used. Pdk2 KO and Pdk4 KO mice were generated as previously described (Jeoung et al., 2012). Pdk2 KO mice were crossed with Pdk4 KO mice to produce Pdk2/4 DKO mice. Age-matched WT mice were produced from the C57BL/6J black mice (The Jackson Laboratory) that were used to stabilize the genetic backgrounds of the Pdk2 KO and Pdk4 KO mice. Heterozygous animals were crossed to generate littermate control animals, which were used to confirm the findings of behavioral studies. Genotypes were confirmed by PCR of the genomic DNA as described previously (Herbst et al., 2014). Animals were housed under a 12 h light/dark cycle (lights on 07:00–19:00) at a constant ambient temperature of 23 ± 2°C with food and water provided ad libitum. Each individual animal was used in only one experiment.
CFA-induced chronic/persistent inflammation.
Chronic/persistent inflammation was induced by CFA injection, as described previously (Pan et al., 2014). Briefly, mice were gently anesthetized with isoflurane (5% for induction and 2% for maintenance), and then administered with a unilateral 30 μl intraplantar injection of CFA (0.5 mg/ml; Sigma-Aldrich) into the left hindpaws (ipsilateral paws). Mice serving as the control group were injected with 30 μl of saline to left hindpaws. Mechanical paw withdrawal thresholds (PWTs) and thermal paw withdrawal latencies (PWLs) were determined before and up to 2 weeks after CFA administration.
Measurement of CFA-induced paw edema.
CFA-induced paw edema (a measure of the degree of inflammation) was quantified using paw thicknesses and/or widths. One experimenter, who was blinded to animal genotypes or treatment conditions, handled and tested all the animals. The dorsoventral thicknesses and laterolateral widths of the middle portions of the hindpaws were measured using a caliper, as described previously (Jha et al., 2014). Paw area was calculated as the product of thickness and width. Paw edema was measured up to 2 weeks after injection.
Behavioral testing.
After arrival in the animal care unit, mice were allowed to acclimate to the testing room, equipment, and experimenter for 1 week and before the actual testing on the same testing day. One experimenter, who was unaware of the animal genotypes or treatment conditions, handled and tested all the animals. Any possible impact of the genetic modifications in mice on acute mechanical and thermal nociception was assessed by using paw withdrawal frequency and tail flick tests, respectively. The sensitivity to respond to noxious mechanical stimuli was assessed using the withdrawal frequency method, by calculating the frequency of paw withdrawal for each monofilament (2.0 or 4.0 g) (Jeon et al., 2013). Thermal pain sensitivity was assessed using the tail immersion test. Mice were gently restrained in a 50 ml conical tube, and the distal third of the tail was immersed into a water bath at 45°C, 50°C, or 55°C. The tail flick latencies to respond as indicated by vigorous tail flexion were calculated by averaging three measurements. A cutoff time of 15 s was used to prevent tissue damage. Open-field test was conducted as described previously (Jang et al., 2013a). Motor coordination was assessed with a RotaRod Treadmill for mice (Ugo Basile) at a constant rotating speed of 32 rpm. All mice had five training sessions before the day of the experiment. The fall-off latency was averaged from three tests. The cutoff time of 90 s was used.
Thermal hyperalgesia and mechanical allodynia associated with inflammation were assessed by measuring the hindpaw withdrawal latencies or thresholds, respectively. Thermal hyperalgesia induced by CFA was defined as a decrease in PWL to a noxious thermal stimulus. PWLs were assessed, as previously described (Jha et al., 2014), using the Hargreaves' Plantar Test Analgesy-Meter (Ugo Basile). Mice were acclimatized for 30 min before testing under a clear inverted plastic chamber on a glass surface maintained at 30°C. The plantar surface of each hindpaw was then exposed to an electrically generated thermal stimulus (set at 5.0 A), as previously described (Hargreaves et al., 1988). The time required for a mouse to withdraw its paw from the stimulus (PWL) was automatically recorded. The intensity of the radiant heat was adjusted so that basal PWL was between 9 and 12 s, with a cutoff of 20 s to avoid any possible tissue injury. To obtain the mean PWL values, tests were conducted three times and averaged using a 5 min interval between tests to avoid thermal stimulus-induced sensitization (Dirig et al., 1997). Mechanical allodynia was evaluated on two consecutive days before the scheduled experiment day, immediately before CFA injection, and at different time points afterward. Mechanical allodynia was tested using calibrated von Frey filaments (Bioseb), as described previously (Chaplan et al., 1994). In brief, mice were acclimated for 20 min in inverted individual acrylic boxes with wire mesh floors to provide access to the ventral side of hindpaws. von Frey filaments were presented perpendicularly to the plantar surface and held in position for ∼5 s with enough force to cause a slight bend. Two trials per paw were conducted with an interval of at least 3 min. A positive response was defined as abrupt paw withdrawal. PWTs were determined by increasing and decreasing the forces, and estimated using the Dixon's up-down method (Dixon, 1980).
Motor nerve conduction velocity measurement.
The measurement of motor nerve conduction velocity was performed as described previously (McGuire et al., 2009). Briefly, 8-week old male WT and DKO mice were anesthetized with ketamine. Animals' body temperature was automatically maintained at a mean rectal temperature of 37 ± 0.5°C with a heated water circulating system (T/Pump, model TP500, Gaymar). The sciatic nerve was stimulated (5–10 mA, 0.05 ms single square-wave pulses with low and high filter settings of 0.3 and 3 kHz) proximally at the level of the sciatic notch and distally at the level of the ankle with bipolar electrodes. Compound muscle action potentials were recorded from the first interosseous muscle (between digits 1 and 2) of the hindpaw, amplified (16-channel Microelectrode amplifier, model 3600, A-M Systems), stored, displayed, and digitized by a personal computer using AxoScope software (Molecular Devices). Latencies were measured from the stimulus to the onset of the negative M-wave deflection of the compound muscle action potentials. Distal and proximal motor latencies from 20 separate recordings were averaged. Motor nerve conduction velocity (in meters/s) was calculated by dividing the distance between stimulating electrodes by the average latency difference.
Immunohistochemistry and histopathology.
Deeply anesthetized mice were perfused through the aorta with 0.1 m PBS followed by 4% PFA fixative. Lumbar spinal cord segments (L4-L6) and hindpaw tissues were dissected out, postfixed in the same PFA fixative overnight, and cryoprotected in 30% sucrose in 0.1 m PBS overnight at 4°C. A cryostat was used to prepare 10 μm sections of hindpaw tissues, which were mounted on gelatin-coated slides, and 30 μm sections of spinal cord, which were placed in 0.1 m PBS. Sections were then blocked with 4% normal serum in 0.3% Triton X-100 for 90 min at room temperature. For immunofluorescence staining, sections were incubated with primary antibodies against PDK2 (rabbit, 1:200; Acris Antibodies), PDK4 (rabbit, 1:200; Atlas Antibodies), phospho-Ser293-PDHE1α (pyruvate dehydrogenase E1α) (rabbit, 1:200; Calbiochem), phospho-Ser300-PDHE1α (pyruvate dehydrogenase E1α) (rabbit, 1:200; Calbiochem), Iba-1 (rabbit, 1:1000; Wako; or goat, 1:200; Novus Biologicals [for costainings]), GFAP (mouse, 1:500; BD Biosciences), Ly6G (rat, 1:200; eBioscience) or iNOS (rabbit, 1:200; BD Biosciences), MYH1/2/3 (mouse, 1:200; Santa Cruz Biotechnology) and β3 tubulin (2G10) (mouse, 1:200; Santa Cruz Biotechnology), overnight at 4°C, and then incubated with FITC- or Cy3-conjugated secondary antibodies (1:200; Jackson ImmunoResearch Laboratories). Slides were washed, coverslipped with Vectashield mounting medium (Vector Laboratories), and visualized under a fluorescence microscope. For H&E staining, hindpaw sections (10 μm) fixed in 4% PFA were processed routinely, stained with H&E, and analyzed under a light microscope (Olympus DP70). For histological analysis of peripheral nerves, sciatic nerves were dissected, postfixed in PFA overnight at 4°C, and processed for paraffin embedding. Sciatic nerves were cross-sectioned at 5 μm thickness and stained with H&E.
Quantitative real-time reverse transcription-PCR.
Deeply anesthetized mice were perfused through the aorta with 0.1 m PBS to remove the blood, and the lumbar spinal cord and hindpaw tissues were rapidly dissected. The spinal cord portion, corresponding to segments L4-L6, was divided into ipsilateral and contralateral sides. Samples were then immediately frozen in liquid nitrogen and instantly homogenized in TRIzol reagent (Invitrogen) for total RNA isolation. Total RNA (2 μg) from each sample was reverse-transcribed into cDNA using a first-strand cDNA synthesis kit (MBI Fermentas). Real-time RT-PCR was performed using the one-step SYBR PrimeScript RT-PCR kit (Perfect Real-Time; Takara Bio) and the ABI Prism 7000 sequence detection system (Applied Biosystems), according to the manufacturer's instructions. The 2−ΔΔCT method was used to calculate relative changes in gene expressions (Livak and Schmittgen, 2001), and Gapdh was used as an internal control. The nucleotide sequences of the primers used in the real-time RT-PCR were as follows: Pdk1, forward, 5′-CAC CAC GCG GAC AAA GG-3′, reverse, 5′-GCC CAG CGT GAC GTG AA-3′; Pdk2, forward, 5′-CCC CGT CCC CGT TGT C-3′, reverse, 5′-TCG CAG GCA TTG CTG GAT-3′; Pdk3, forward, 5′-GGA GCA ATC CCA GCA GTG AA-3′, reverse, 5′-TGA TCT TGT CCT GTT TAG CCT TGT-3′; Pdk4, forward, 5′-CCA TGA GAA GAG CCC AGA AGA-3′, reverse, 5′-GAA CTT TGA CCA GCG TGT CTA CAA-3′; IL-1β, forward, 5′-AAG TTG ACG GAC CCC AAA AGA T-3′, reverse, 5′-TGT TGA TGT GCT GCT GCG A-3′; IL-6, forward, 5′-AGT TGC CTT CTT GGG ACT GA-3′, reverse, 5′-TCC ACG ATT TCC CAG AGA AC-3′; TNF-α, forward, 5′-ATG GCC TCC TCA TCA GTT C-3′, reverse, 5′-TTG GTT TGC TAC GAC GTG-3′; IL-10, forward, 5′-AGT GAA CTG CGC TGT CAA TG-3′, reverse, 5′-TTC AGG GTC AAG GCA AAC TT-3′; Ym-1, forward, 5′-GGG CAT ACC TTT ATC CTG AG-3′, reverse, 5′-CCA CTG AAG TCA TCC ATG TC-3′; Arg-1, forward, 5′-CGC CTT TCT CAA AAG GAC AG-3′, reverse, 5′-CCA GCT CTT CAT TGG CTT TC-3′; Irf4, forward, 5′-CTG CGG CAA TGG GAA ACT CC-3′, reverse, 5′-GCT CTT GTT CAG AGC ACA TCG T-3′; Irf8, forward, 5′-GGA TAT GCC GCC TAT GAC ACA-3′, reverse, 5′-CAT CCG GCC CAT ACA ACT TAG-3′; Gapdh, forward, 5′-TGG GCT ACA CTG AGC ACC AG-3′, reverse, 5′-GGG TGT CGC TGT TGA AGT CA-3′.
Western blot analysis.
Hindpaw tissues (at 3 d after CFA injection) of similar weights were isolated, washed in ice-cold PBS, and placed in 300 μl of lysis buffer (150 mm sodium chloride, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 50 mm Tris-HCl, pH 7.5, 2 mm EDTA) (GenDEPOT) containing Halt protease inhibitor (1×) and phosphatase protease inhibitor cocktails (1×) (Thermo Scientific). Specimens were individually homogenized and then centrifuged at 13,400 × g at 4°C for 15 min. Protein concentration was determined with a Bio-Rad Laboratories Protein Assay Kit using BSA as standard. Proteins (20–30 μg) from each sample were separated on 8% or 15% SDS-PAGE gels and transferred to PVDF membranes (Bio-Rad) by the semidry electroblotting method. The membranes were blocked with 5% skim milk and sequentially incubated with primary antibodies against PDK2 (rabbit monoclonal antibody; Acris Antibodies), PDK4 (rabbit monoclonal antibody; Acris Antibodies), phospho-Ser293-PDHE1α (pyruvate dehydrogenase E1α) (rabbit monoclonal antibody; Calbiochem), phospho-Ser300-PDHE1α (pyruvate dehydrogenase E1α) (rabbit monoclonal antibody; Calbiochem), α-tubulin (mouse monoclonal antibody; Sigma-Aldrich), and HRP-conjugated secondary antibodies (anti-rabbit and anti-mouse IgG antibody; GE Healthcare) followed by enhanced chemiluminescence detection (GE Healthcare). Western blotting was repeated three times (n = 3) for each condition.
Measurement of PDH complex activity.
The PDH complex activity was determined as described previously (Jeoung et al., 2012). Briefly, tissues were pulverized in liquid nitrogen and homogenized with a motor-driven Teflon homogenizer in 5 volumes (w/v) of extraction buffer containing 30 mm HEPES/KOH, pH 7.5, 3% (v/v) Triton X-100, 2 mm EDTA, 2% (v/v) rat serum, 5 mm DTT, 10 μm tosylphenylalanylchloromethane, 10 μg/ml trypsin inhibitor, and 1 μm leupeptin. For the determination of actual PDH complex activity (i.e., the activity that exists in the intact tissue as a result of the phosphorylation state), an aliquot of tissue extract (16 μl) was mixed with an equal volume of resuspension buffer containing 30 mm HEPES/KOH, pH 7.5, 1% Triton X-100, 0.2 mm EDTA, 2% (v/v) bovine serum, 1 μm leupeptin, 5 mm DTT, 20 mm dichloroacetate, and 100 mm KF. For the determination of total PDH complex activity (i.e., the activity of the complex after complete dephosphorylation), an aliquot of tissue extract (8 μl) was mixed with an equal volume of an activation buffer (resuspension buffer containing 40 mm MgCl2, 1.5 mm CaCl2, and 1 μg of recombinant pyruvate dehydrogenase phosphatase 1 protein). Complete activation of the PDH complex by dephosphorylation was achieved by incubating this mixture at 30°C for 20 min. Activity of the PDH complex was measured spectrophotometically in a 96-well plate reader (Spectra Max 190, Molecular Devices) with a coupled assay based on the reaction catalyzed by arylamine acetyltransferase as described previously (Jeoung et al., 2006). One unit of PDH complex activity corresponds to the acetylation of 1 μmol of p-(p-aminophenylazo)-benzenesulfonate per minute at 30°C. PDH activity state was calculated in percentage by using actual and total PDH activities as numerator and denominator, respectively.
Lactate measurements.
Mice were killed by cervical dislocation, and the hindpaw tissues were taken quickly. All of the tissue samples were snap-frozen in liquid nitrogen. On the day of the experiment, tissues were homogenized into 500 μl of lactate assay buffer (Lactate Colorimetric kit, Abcam) and centrifuged at 4°C at 10,000 × g for 4 min. Samples were tested according to the manufacturer's protocol with subtraction of the NADH/NADPH background readings. Lactate levels were normalized to control samples.
Macrophage depletion.
Clodronate liposomes (8 mg/ml; FormuMax Scientific) were administered intraperitoneally to mice 48 h before (150 μl) and after (100 μl) the intraplantar injection of CFA to deplete macrophages (Jha et al., 2014). Macrophage in vivo depletion occurs within 24 h after clodronate liposomal treatment and begins to slowly repopulate ∼5–7 d after the last administration (Summan et al., 2006).
Collection, culture, and treatment of peritoneal macrophages.
Primary mouse peritoneal macrophages were isolated as described previously (Jha et al., 2014). Briefly, mice were intraperitoneally injected with 3 ml of 3% sterile thioglycollate (BD Biosciences), and 4 d later, peritoneal exudate cells were collected by lavage with PBS. Aliquots of 1 × 106 cells were seeded in 6-well polystyrene culture plates at 37°C and 5% CO2 atmosphere and allowed to adhere for 3 h before washing. Afterward, nonadherent cells were removed by vigorous washing three times with PBS and incubated for an additional 24 h under the same conditions. Cells were incubated with LPS (100 ng/ml) plus IFN-γ (50 U/ml) or IL-4 (10 ng/ml) for 8 h and then used for total RNA extraction. In a separate experiment, macrophages were pretreated with dichloroacetate (DCA) (5 mm) for 8 h before LPS stimulation. The entire procedure was performed under sterile conditions.
Nitrite quantification.
Peritoneal macrophages were stimulated in 96-well plate for 24 h, and nitrite (NO2−) levels in culture media were measured using the Griess reaction, as previously described (Jang et al., 2013b), to assess nitric oxide (NO) production levels. Samples (50 μl) were mixed with 50 μl Griess reagent (1% sulfanilamide/0.1% naphthylethylene diamine dihydrochloride/2% phosphoric acid) in 96-well plates and incubated at 25°C for 10 min. Absorbances at 540 nm were measured using a microplate reader (Anthos Labtec Instruments), and NaNO2 standards were used to calculate nitrite levels.
Assessment of extracellular acidification.
XF24 Extracellular Flux Analyzer (Seahorse Biosciences) was used to determine the extracellular acidification rate (ECAR) as described previously (Haschemi et al., 2012). Briefly, RAW264.7 cells were seeded in XF24 plates and allowed to recover for 24 h before the ECAR was assessed in glucose-containing media (Seahorse Biosciences). Result was normalized to the total protein concentrations, extracted immediately after the ECAR reading.
DCA application.
To ascertain the effect of pharmacological inhibition of PDKs on chronic inflammatory pain, CFA injection was coupled with DCA (a PDK inhibitor) treatment. DCA at the dose of 25 mg/kg/d has been found to exert excessive peripheral nerve toxicity (Kaufmann et al., 2006), which is the common side effect during chronic DCA treatment (Kurlemann et al., 1995). Several previous studies have advocated the safe use of DCA at the dosage regimen of 10 mg/kg/d at least for short-term application (Stacpoole et al., 1998a, b), which we have also confirmed to be safe in the current study. In this study, administration of either DCA (10 mg/kg body weight, 10 μl) or vehicle (saline, 10 μl) was done by intraplantar injection daily.
Inhibition of lactic acid production.
To assure the role played by lactic acid in the inflammation-driven chronic pain, CFA-induced lactic acid production was inhibited by using a small-molecule inhibitor (FX11; [2, 3-dihydroxy-6-methyl-7-(phenylmethyl)-4-propyl-1-naphthalenecarboxylic acid]) of lactate dehydrogenase A (LDHA), which converts pyruvate to lactate coupled with the recycling of NAD+ (Laferrière et al., 2008; Le et al., 2010). Mice were injected daily with either FX11 (2 mg/kg body weight) or vehicle (2% [v/v] DMSO) via intraplantar injection from the day of CFA injection for 3 d.
Quantification and statistical analysis.
For the immunohistochemical analysis, microscopic images of the hindpaws were taken (using a 20× objective lens) around the sites of CFA injection in 3 or 4 tissue sections per animal. Similarly, microscopic images of the dorsal horn of the spinal cord were taken from the L4-L6 region in 3 or 4 sections per animal (using a 20× objective lens). Image quantification was performed on images of immunostained tissues captured with an Olympus DP70 camera and DP Controller software (Olympus). For the determination of immunofluorescence intensity, images (at least three microscopic images) were selected randomly, and the mean intensity of each image was measured using the ImageJ software (National Institutes of Health) to obtain the better representation of the tissue as a whole. Likewise, band intensities of images obtained from Western blot analysis were also quantified using the ImageJ software. The background intensity of Western blot image was also determined from the region having no band and deducted from the values obtained. The presented graphs represent average of all data. All results are presented as the mean ± SEM from three or more independent experiments, unless stated otherwise. Statistical comparisons between treatments were performed by a Student's t test, Mann–Whitney test, or one-way ANOVA with Dunnett's/Bonferroni multiple-comparison tests by using the SPSS version 22.0K program (SPSS). The Mann–Whitney test was used to compare PWTs measured by von Frey filaments with the up-down paradigm at a single interval. Differences in the probability values of <0.05 were considered statistically significant.
Results
Enhanced expression and activity of PDK2 and PDK4 in hindpaw tissues following exposure to chronic inflammatory insult
To investigate the role of PDK-PDH-lactic acid axis in the pathogenesis of chronic inflammatory pain, we first examined the expression of PDK isoforms (PDK1–4) in the hindpaw tissues, which encompass skin, subcutaneous fat, muscle, and potentially infiltrated immune cells, following CFA injection by using real-time RT-PCR, Western blot analysis, and immunostaining. Real-time RT-PCR analysis revealed significantly enhanced expression of Pdk2 and Pdk4 mRNAs, but not that of Pdk1 or Pdk3 mRNAs (data not shown), from the first day to day 7, peaking at day 3 after CFA injection (Fig. 1A). Furthermore, the levels of PDK2 and PDK4 protein were significantly enhanced in the CFA injected hindpaw tissues at day 3 after injection (Fig. 1B). We also confirmed the substantial upregulation of PDK2 and PDK4 expression in the hindpaw tissues following CFA administration by immunofluorescence analysis (Fig. 1C,D). PDKs were found to be colocalized with Iba-1-positive macrophages, Ly6G-positive neutrophils, myosin heavy chain (MYH)-positive skeletal muscle or β3 tubulin-positive nerves (data not shown). As PDKs inhibit PDH activity by site-specific phosphorylation on the E1α subunits, we assessed the phosphorylation of PDH-E1α. The degree of phosphorylated PDH-E1α (such as p-S293-PDH and p-S300-PDH) in the hindpaw tissue following the CFA injection was notably increased compared with those of vehicle-injected control animals (Fig. 2A). Immunostaining of CFA-injected hindpaws revealed highly upregulated p-S293-PDH and p-S300-PDH immunoreactivities compared with the vehicle-injected control hindpaws (Fig. 2B). Thus, the increased PDK2 and PDK4 expression may be responsible for the enhanced PDH-E1α phosphorylation and possibly downregulated PDC activity in the CFA-injected hindpaw tissues. Assessment of PDC activity in the hindpaw tissues showed the decreased enzyme activity after CFA injection, and PDC activity in CFA-injected hindpaw of the WT was lower than that of Pdk2/4 DKO mice (Fig. 2C). The phosphorylated PDH-E1α (p-S293-PDH and p-S300-PDH) was found to be partially colocalized with infiltrated neutrophils (Fig. 2D) and macrophages (Fig. 2E) in the hindpaw tissues, suggesting that PDK expression and activity in these inflammatory cells were increased following the CFA injection.
Figure 1.
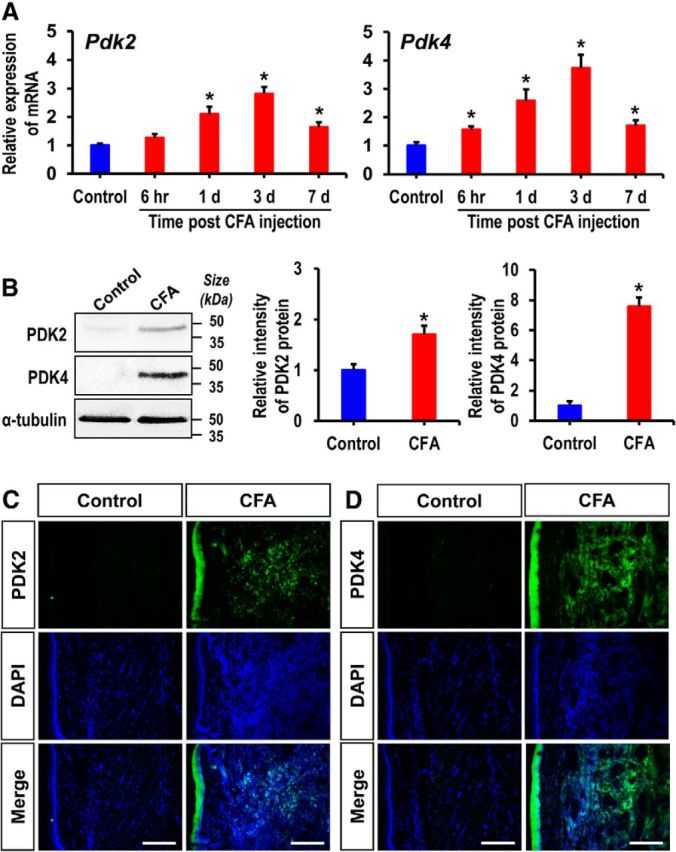
Expression of PDK2 and PDK4 in hindpaw tissues after CFA injection. A, The expression of Pdk2 and Pdk4 mRNAs in hindpaw tissues at different time points after CFA injection was assessed by real-time RT-PCR. Pdk2 and Pdk4 mRNA levels in CFA-injected hindpaw tissues were significantly upregulated after injection, peaking at 3 d and then subsiding. Results for mRNA expression are displayed as the fold increase of gene expression normalized to GAPDH. B, Protein levels of PDK2 and PDK4 in hindpaw tissues 3 d after CFA injection were assessed by Western blot analysis. Quantifications of the band intensities are presented in the adjacent graphs. C, D, Immunofluorescence analysis detected a strong expression of PDK2 and PDK4 in the hindpaw tissues of CFA-injected mice at 3 d after injection, but not in the vehicle-treated control animals. *p < 0.05 versus the vehicle-treated control animals (Student's t test). n = 3. Data are mean ± SEM. Scale bars, 200 μm. Images show the representative results of at least three independent experiments.
Figure 2.
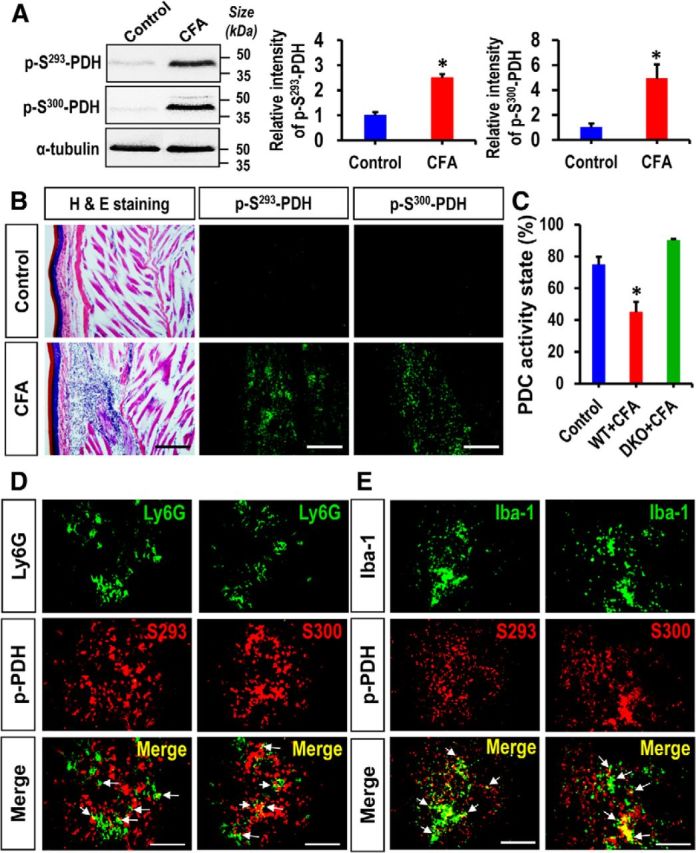
Expression of phosphorylated PDH-E1α in hindpaw tissues after CFA injection. A, Protein levels of phosphorylated PDH-E1α (p-S293-PDH and p-S300-PDH) in hindpaw tissues 3 d after CFA injection were assessed by Western blot analysis. Quantifications of the band intensities are presented in the adjacent graphs. B, Immunofluorescence analysis detected a strong expression of phosphorylated-PDH-E1α (p-S293-PDH and p-S300-PDH) in the hindpaw tissues of CFA-injected mice at 3 d after injection, but not in the vehicle-treated control animals. H&E staining revealed a considerable infiltration of inflammatory cells in the CFA-injected hindpaws. The H&E and immunofluorescence images were obtained from the same area of the hindpaw sections. C, PDH complex activity in CFA-injected hindpaw tissues was assessed at day 3 after CFA injection and is presented as activity state in percentage. D, E, Double immunostaining showed that phosphorylated-PDH-E1α (p-S293-PDH and p-S300-PDH) (red) expression in hindpaw tissue partially colocalized with Ly6G (green, a neutrophil marker) at 1 d (D) and Iba-1 (green, a macrophage marker in the periphery) at 3 d (E) after injection. Arrows indicate double-labeled cells in the merged images. Scale bars: B, 200 μm; D, E, 100 μm. *p < 0.05 versus the vehicle-treated control animals (Student's t test). n = 3. Data are mean ± SEM. Images show the representative results of at least three independent experiments.
Pdk2/4 deficiency diminished inflammatory infiltration and proinflammatory cytokine expression following exposure to chronic inflammatory insult
As the expression and activity of PDKs were enhanced in the CFA-injected hindpaw tissues, we next investigated the ongoing pathophysiological changes at the site of inflammation. The histopathological evaluation of CFA-injected hindpaws revealed infiltration of inflammatory cells and tissue damage, which were notably attenuated in the Pdk2/4 DKO mice. Any mentionable inflammatory infiltration was not detected in the contralateral sides (data not shown) or vehicle-injected control animals. Furthermore, extensive accumulation of Ly6G-positive neutrophils and infiltration of Iba-1-positive macrophages were observed in the hindpaw tissues at 1 d after injection in WT mice, whereas the number of these infiltrated inflammatory cell types was reduced in Pdk2/4-deficient mice. Similarly, substantially increased inducible nitric oxide synthase (iNOS) immunoreactivity, which was observed at 3 d after injection in the hindpaw tissues of the WT animals, was diminished in the Pdk2/4-deficient mice. The iNOS expression was found to be partly colocalized with Iba-1-positive macrophages. Infiltration of neither neutrophils nor macrophages was observed in the hindpaw tissues of vehicle-injected control mice (Fig. 3A; quantifications and statistical analyses are presented in adjacent graphs). The Iba-1 immunoreactivity in CFA-injected WT hindpaws persisted until day 7 after injection, indicating the substantial infiltration of macrophages (Fig. 3B; quantifications and statistical analyses are presented in adjacent graphs). Furthermore, CFA administration prominently increased the expression of TNF-α, IL-1β, and IL-6 mRNAs in the hindpaws at 6 h (Fig. 3C) and 3 d (Fig. 3D) after injection in the WT mice, and CFA-induced proinflammatory cytokine expression was significantly reduced in the Pdk2/4 DKO mice. The crucial role of macrophages and macrophage-derived proinflammatory cytokines in the pathogenesis of chronic inflammatory pain was demonstrated by a macrophage depletion experiment. Clodronate-mediated depletion of macrophages reduced the CFA-induced pain behavior. Intraperitoneal administration of clodronate liposomes 48 h before and after the CFA administration depleted macrophages in the hindpaw tissue, as confirmed by Iba-1 (a macrophage marker) immunohistochemistry (data not shown). Macrophage-depleted mice exhibited significantly attenuated CFA-induced paw edema, thermal hyperalgesia, and mechanical allodynia at least from days 1 to 6 following CFA administration, compared with vehicle-injected control mice (Fig. 4A). Moreover, the clodronate-mediated macrophage depletion significantly reduced the CFA-induced expression of TNF-α, IL-1β, and IL-6 mRNAs in the hindpaws (Fig. 4B). These data suggest that proalgesic mediators, such as proinflammatory cytokines derived from tissue-infiltrating inflammatory neutrophils (at the very early stage of inflammation) and macrophages (proinflammatory macrophages in particular) in the peripheral tissue, may play an important role in the sensitization of peripheral nociceptors and the subsequent initiation and persistence of inflammation-induced chronic pain hypersensitivities.
Figure 3.
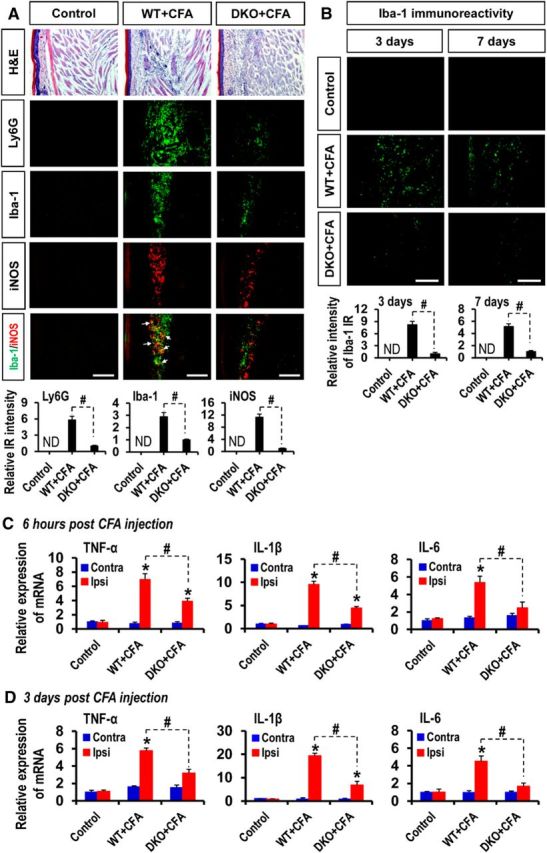
Effect of PDK2/4 deficiency on inflammatory cell infiltration and proinflammatory cytokine expression in CFA-injected hindpaws. A, H&E staining revealed an accumulation of inflammatory cells in the ipsilateral hindpaw tissues at 1 d after injection in the WT mice, but this accumulation was attenuated in the DKO mice. Pdk2/4 deficiency considerably lessened the immunoreactivities of Ly6G (green, a neutrophil marker) and Iba-1 (green, a macrophage marker) in CFA-injected hindpaws at 1 d after injection compared with those of WT animals. Similarly, notably reduced immunoreactivities of iNOS (red, a M1 macrophage phenotype marker) were detected in the CFA-injected hindpaws of Pdk2/4 DKO mice compared with those of WT animals at 3 d after injection. B, Iba-1 immunoreactivity in the ipsilateral hindpaws was also detected at days 3 and 7 after CFA injection. Quantifications and statistical analyses of stained images are presented in adjacent graphs. C, D, The relative mRNA expression of TNF-α, IL-1β, and IL-6 in the hindpaw tissues at 6 h (C) and 3 d (D) after injection was evaluated by real-time RT-PCR. Results for mRNA expression are displayed as the fold increase of gene expression normalized to GAPDH. *p < 0.05 versus the vehicle-treated control animals. #p < 0.05 between indicated groups (Student's t test). n = 3. Data are mean ± SEM. IR, Immunoreactivity; ND, not detected. Scale bars, 200 μm. Images show the representative results of at least three independent experiments.
Figure 4.
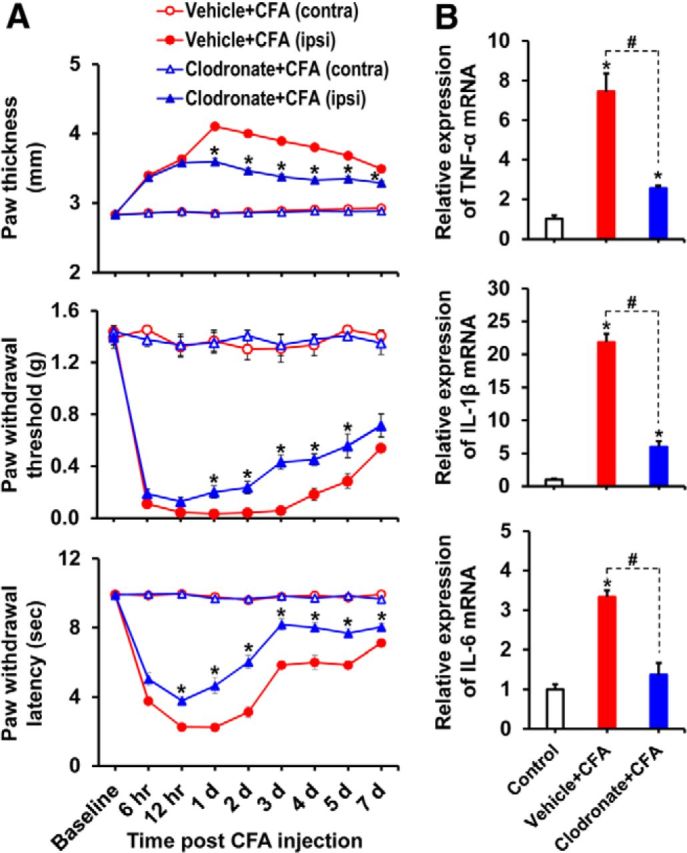
Clodronate-mediated depletion of macrophages attenuated CFA-induced paw edema, pain behaviors, and expression of proinflammatory cytokines in CFA-injected hindpaws. A, Clodronate liposomes were administered intraperitoneally to mice 48 h before (150 μl) and after (100 μl) the intraplantar injection of CFA, and then paw edema and pain responses were measured. In the ipsilateral sides, CFA injection increased paw thickness and reduced PWT to force as well as PWL to heat. The CFA-induced effects were attenuated in the macrophage-depleted mice compared with control animals. No significant change in paw edema or pain-related behavior was observed in the contralateral sides. B, Hindpaw tissues were collected at day 3 after CFA administration from vehicle-injected or clodronate-injected (macrophage-depleted) animals to assess the expression of proinflammatory cytokines. The relative mRNA expression of TNF-α, IL-1β, and IL-6 in the hindpaw tissues was evaluated by real-time RT-PCR. Results for mRNA expression are displayed as the fold increase of gene expression normalized to GAPDH. *p < 0.05 versus the vehicle-treated control animals. #p < 0.05 between indicated groups (Student's t test, Mann–Whitney test for PWT). A, n = 6; B, n = 3. Data are mean ± SEM.
Role of PDKs in skewing macrophages toward proinflammatory phenotypes
The immense infiltration of proinflammatory macrophages and increased expression of proinflammatory cytokines in ipsilateral hindpaws following CFA injection led us to speculate that PDKs may regulate macrophage functional polarization. Macrophage polarization to an M1 phenotype is often accompanied by a cellular metabolic shift from oxidative phosphorylation to aerobic glycolysis as well as nitric oxide (NO) and citrulline production (Orihuela et al., 2015). This metabolic switch increases glucose uptake, which leads to enhanced lactic acid production (Rodríguez-Prados et al., 2010). Conversely, M2 macrophages experience substantially decreased glucose consumption (Rodríguez-Prados et al., 2010), as macrophages of these phenotypes primarily use fatty acid oxidation and oxidative respiration for energy production (Odegaard and Chawla, 2011). M1 macrophages produce proinflammatory cytokines (IL-1α, IL-1β, IL-6, IL-12, IL-23, TNF-α), chemokines, scavenger receptors, costimulatory proteins, and MHC-II (Orihuela et al., 2015). Moreover, immune cell-derived NO is reported to inhibit the enzyme PDH (Klimaszewska-Lata et al., 2015). M2 macrophages exhibit mRNA profiles for Arg-1, Mrc, Ym-1, Fizz1, and Ppar-γ (Orihuela et al., 2015). To investigate the role of PDKs in the proinflammatory phenotypic polarization of macrophages, peritoneal macrophage cultures prepared from Pdk2/4+/+ and Pdk2/4−/− mice were challenged with either LPS plus IFN-γ or IL-4, well-known inducers of the M1 and M2 phenotypes, respectively, for 8 h, and then the expression of M1- or M2-related genes was assessed. The mRNA levels of M1-related genes TNF-α, IL-1β, and IL-6 were higher in macrophages isolated from Pdk2/4+/+ mice compared with those of Pdk2/4−/− animals, whereas the mRNA levels of M2-related genes Ym-1, Arg-1, and IL-10 were lower in Pdk2/4+/+ macrophages compared with Pdk2/4−/− macrophages (Fig. 5A). These results suggest that PDK2/4 play an important role in the induction of the proinflammatory phenotype and inhibition of the anti-inflammatory phenotype of peripheral macrophages. Interferon regulatory factor-8 (IRF8) and IRF4 are the phenotype-determining transcription factors, which determine whether macrophages will adopt classically activated or alternatively activated phenotypes, respectively, during the early differentiation (Lawrence and Natoli, 2011). In the current study, following exposure to LPS plus IFN-γ, the mRNA level of Irf8 was higher in macrophages isolated from Pdk2/4+/+ mice compared with those of Pdk2/4−/− animals, whereas following the exposure to IL-4, the mRNA level of Irf4 was lower in Pdk2/4+/+ macrophages compared with Pdk2/4−/− macrophages (Fig. 5B). Furthermore, an exposure to inflammatory stimuli (LPS plus IFN-γ) increased the expression of Pdk2 and Pdk4 in cultured peritoneal macrophages (Fig. 5C). The inhibition of PDKs by using their inhibitor, DCA, significantly lessened the expression of proinflammatory cytokines TNF-α (by 65.49 ± 7.06%, p < 0.05 vs the LPS-stimulated group; one-way ANOVA followed by Bonferroni corrections; n = 3; mean ± SEM) and IL-1β (by 66.38 ± 3.11%, p < 0.05 vs the LPS-stimulated group; one-way ANOVA followed by Bonferroni corrections; n = 3; mean ± SEM) as well as nitric oxide (nitrite levels in control, 2.08 ± 0.11 μm; LPS only, 16.29 ± 1.90 μm, p < 0.05 vs the control group; and LPS + DCA, 10.20 ± 0.41 μm, p < 0.05 vs the control group and p < 0.05 vs the LPS-stimulated group; one-way ANOVA followed by Bonferroni corrections; n = 3; mean ± SEM). These findings further advocate that PDKs contribute to skewing macrophages toward the proinflammatory phenotypes.
Figure 5.

Role of PDK2/4 in regulating the phenotypes of cultured macrophages. Peritoneal macrophage cultures prepared from WT and Pdk2/4 DKO mice were treated with M1-phenotype inducer mixture [LPS (100 ng/ml) plus IFN-γ (50 U/ml)] for 8 h. A, The mRNA levels of M1-related genes TNF-α, IL-1β, and IL-6 were assessed by real-time RT-PCR (left). Similarly, peritoneal macrophage cultures prepared from WT and Pdk2/4 DKO mice were treated with M2-phenotype inducer [IL-4 (10 ng/ml)] for 8 h, and the mRNA levels of M2-related genes Ym-1, Arg-1, and IL-10 were then assessed by real-time RT-PCR (right). B, The expression of IRF8 or IRF4 mRNAs in the cultured peritoneal macrophages (prepared from WT and Pdk2/4 DKO mice) following stimulation with LPS (100 ng/ml) plus IFN-γ (50 U/ml) or IL-4 (10 ng/ml) for 8 h was assessed by real-time RT-PCR. C, The expression of Pdk2 and Pdk4 mRNAs in the WT peritoneal macrophages following stimulation with LPS (100 ng/ml) plus IFN-γ (50 U/ml) for 8 h was assessed by real-time RT-PCR. Results for mRNA expression are displayed as the fold increase of gene expression normalized to GAPDH. *p < 0.05 versus the control group. #p < 0.05 between indicated groups (Student's t test). n = 3. Data are mean ± SEM.
Pdk2/4 deficiency attenuated the peripheral inflammation-induced glial activation and proinflammatory cytokine expression in spinal cord
We next examined the effect of PDK2/4 on glial activation in the dorsal horn of lumbar segment of spinal cord following the intraplantar injection of CFA. Although CFA-induced peripheral inflammation did not cause any significant changes in the expression as well as activity of Pdk1–4 isoforms in the DRG and the spinal cord (data not shown), a substantial increase in the number of Iba-1-positive microglial cells was found in the ipsilateral dorsal horn of the lumbar segment of the spinal cord in the CFA-injected mice. Those microglia had short and thick processes and exhibited intense Iba-1 immunoreactivity, which was increased from baseline within 1 d, peaked at 3 d, and subsided at 7 d following CFA administration. These morphological features of the microglia and the increased Iba-1 immunoreactivity were attenuated in the Pdk2/4 DKO spinal cord. No significant changes in microglial morphology or Iba-1 immunoreactivity were observed in the control animals or in the contralateral sides of the spinal cords (Fig. 6A; quantifications and statistical analyses are shown in adjacent graph). Similarly, the number of GFAP-positive astrocytes was markedly increased, and they exhibited intense immunoreactivity and hypertrophic morphology with thick processes in the ipsilateral dorsal horn of the spinal cord after 3–7 d from CFA injection in WT mice, but the number and GFAP immunoreactivity of astrocytes were greatly reduced in the Pdk2/4 DKO spinal cord. No significant changes in astrocyte morphology or GFAP immunoreactivity were observed in the control animals or the contralateral sides of the spinal cords (Fig. 6B; quantifications and statistical analyses are shown in adjacent graph). No significant change in the spinal GFAP immunoreactivity was observed at day 1 after CFA injection in any of the genotypes (data not shown). In addition, the peripheral administration of CFA significantly increased the expression of TNF-α, IL-1β, and IL-6 mRNAs in the lumbar segment of the spinal cord at 3 d after injection in the WT mice, whereas the expression of these mRNAs was significantly attenuated in the Pdk2/4-deficient mice (Fig. 6C). These findings indicate that the activation of glial cells and the enhanced expression of proinflammatory cytokines in the spinal cord might be a direct consequence of peripheral inflammation, which is itself modulated by PDK2/4 activity.
Figure 6.
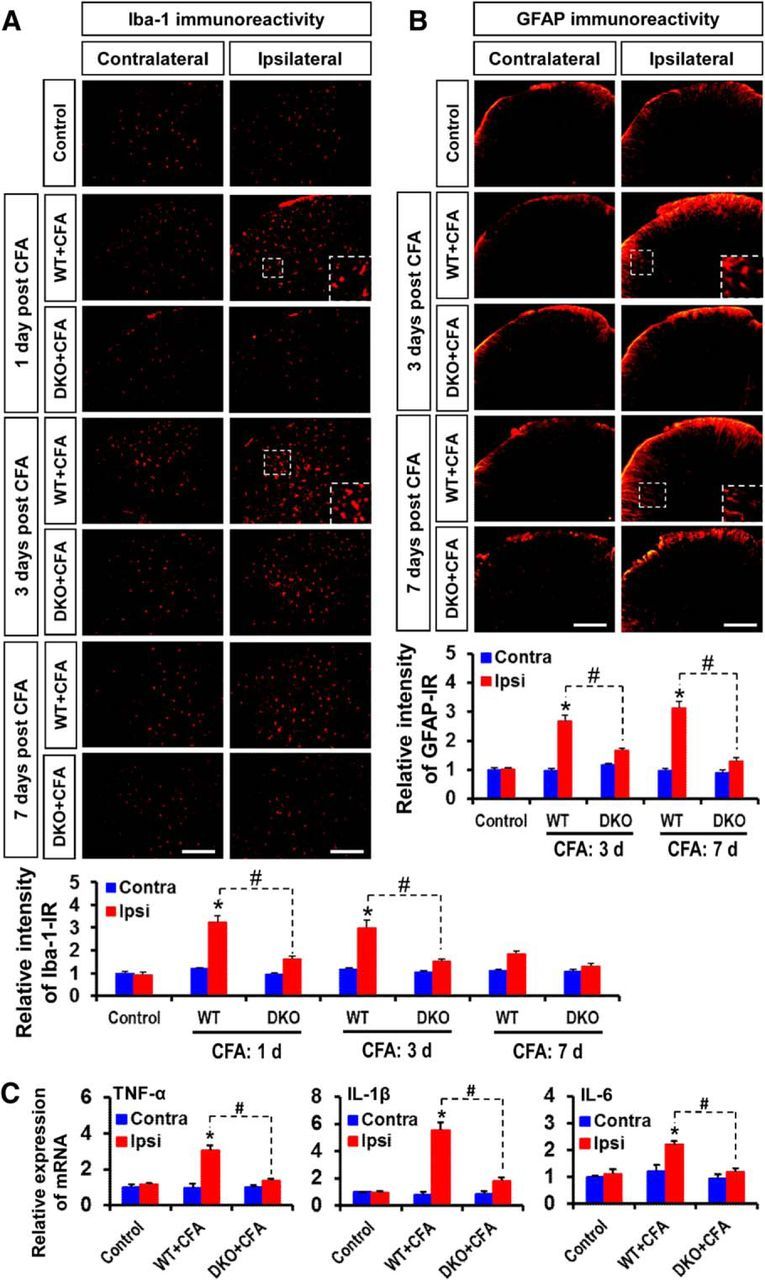
Role of PDK2/4 in glial activation and expression of proinflammatory cytokines in the spinal cord after intraplantar administration of CFA. A, Iba-1 (red, a microglia marker) immunoreactivity was significantly increased in the ipsilateral, but not in the contralateral, dorsal horn of the lumbar segment of the spinal cord at 1–3 d. The increased Iba-1 immunoreactivity persisted at a low level for 7 d after CFA injection. However, CFA-induced Iba-1 immunoreactivities were markedly attenuated in the Pdk2/4 DKO mice at all the time points. B, GFAP (red, an astrocyte marker) immunoreactivity was significantly increased in the ipsilateral, but not in the contralateral, dorsal horn of the lumbar segment of the spinal cord at 3–7 d after CFA injection. The CFA-induced GFAP immunoreactivities were markedly attenuated in the Pdk2/4 DKO mice at these time points. Insets, Magnified images (original magnification × 200). Quantifications and statistical analyses of stained images are presented in adjacent graphs. C, The relative mRNA expression of TNF-α, IL-1β, and IL-6 in the lumbar segment of the spinal cord at 3 d after injection was evaluated by real-time RT-PCR. Results for mRNA expression are displayed as the fold increase of gene expression normalized to GAPDH. *p < 0.05 versus the vehicle-treated control animals. #p < 0.05 between indicated groups (Student's t test). n = 3. Data are mean ± SEM. Scale bar, 200 μm. Images show the representative results of at least three independent experiments.
Attenuation of CFA-induced localized inflammation and pain behaviors in Pdk2/4 DKO mice
Genetically modified mice were used to further evaluate PDK2/4 functions. PDK-mediated inflammatory reactions in CFA-injected hindpaws and the subsequent spinal glial activation led us to compare the development of localized inflammation and nociceptive responses between WT and Pdk2/4-deficient mice. Before the assessment of nociceptive behaviors and edema formation, the open field test and the RotaRod test were performed to determine any possible impact of Pdk2/4 gene deletion on motor coordination, which is an important prerequisite for proper behavioral testing. Pdk2/4-deficient mice revealed unchanged velocity and distanced traveled in the open field test (Fig. 7A). In addition, Pdk2/4-deficient mice showed no difference in their ability to balance on the rotating rod compared with WT mice (Fig. 7B), demonstrating that the genetic modifications did not impair their motor functions. RotaRod experiment was performed to confirm the data obtained from the open field test. A cutoff time of 90 s was used for RotaRod experiments as practiced previously (Holland et al., 2011; Möser et al., 2015). Moreover, Nicol et al. (2015) have recently reported that WT and GPR84 KO mice were unable to remain on the RotaRod apparatus for >100 s at a similar rate of rotation, supporting that 90 s is long enough to determine a motor deficit. Paw withdrawal frequency and tail flick test were used to assess the impact of the genetic modifications on acute mechanical and thermal nociception, respectively. The frequency of paw withdraw in response to von Frey monofilaments of forces 2.0 g as well as 4.0 g (Fig. 7C) and time required to withdraw tail from 45°C, 50°C, and 55°C (Fig. 7E) were found unchanged, indicating that the physiologically important response to acute noxious mechanical and thermal stimulation is unaffected in the Pdk2/4-deficient mice. In addition, the basal level of mechanical and thermal hypersensitivities, which were assessed by measuring PWTs and PWLs, respectively, were found to be unchanged by genetic deletion of Pdk2/4 (Fig. 7D,F). As per some recent reports, mitochondrial metabolism in Schwann cells is necessary for proper function of peripheral nerves (Beirowski et al., 2014; Pooya et al., 2014; Duregotti et al., 2015). This led us to analyze conduction velocity and histological features of nerve to determine any possible impact of Pdk2/4 genetic deletion on peripheral nerve conduction. We measured the basal level motor nerve conduction velocity of WT and DKO mice (Fig. 7G) and found no significant difference between the two genotypes, demonstrating that ablation of Pdk2/4 genes did not affect the nerve function. Furthermore, we also performed the H&E staining of the sciatic nerves from WT and DKO mice and found no remarkable difference in their histological features between the two genotypes (Fig. 7H), which ruled out the possibility that knock-out of Pdk2/4 affects the proper nerve function and that the nerves themselves mediate pain in this model of inflammatory pain.
Figure 7.
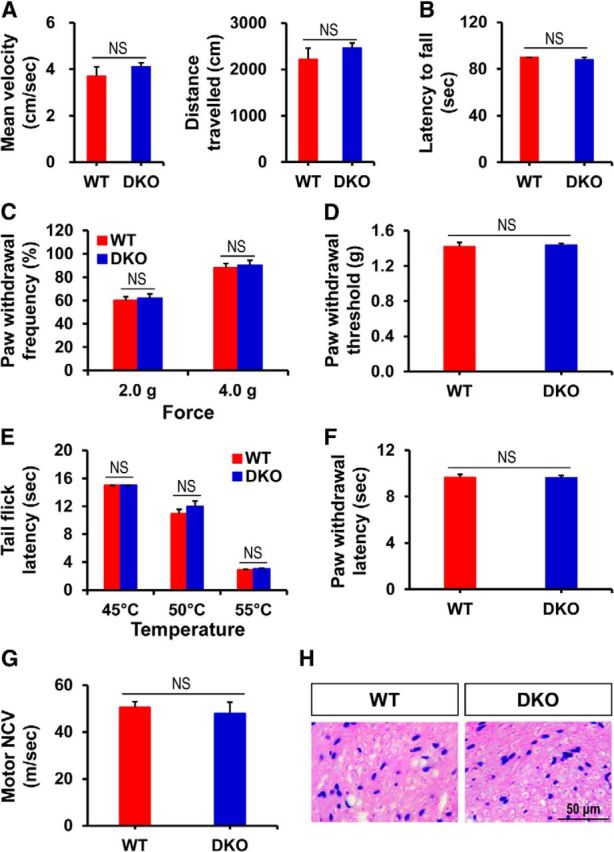
Impact of Pdk2/4 gene knock-out on motor coordination, mechanical as well as thermal nociception, and peripheral nerve function. A, Open field test was performed to measure the mean velocity and distance traveled. B, Ability of mice to balance on the rotating rod was assessed by RotaRod test. C, E, The frequency of paw withdraw in response to von Frey monofilaments of forces 2.0 g as well as 4.0 g and the time required to withdraw tail from 45°C, 50°C, and 55°C were assessed. D, F, PWT to force and PWL to heat were measured in hindpaws. G, Motor nerve conduction velocity was measured and expressed in m/s. H, Cross-sections of sciatic nerve were stained with H&E. All of these behavioral/basal studies were performed in unmanipulated WT and Pdk2/4 DKO mice. *p < 0.05 (Student's t test). NS, Not significant. n = 5. Data are mean ± SEM. Scale bars, 50 μm. Images show the representative results of at least three independent experiments.
Based on the enhanced expression of PDK2 and PDK4 in the hindpaw tissues following CFA injection, we next compared the localized inflammation and pain behaviors of WT, Pdk2 KO, Pdk4 KO, and Pdk2/4 DKO mice at 6 h to 15 d after CFA injection because the unilateral intraplantar injection of CFA induces chronic/persistent pain hypersensitivity as well as edema formation (Dimitrov et al., 2013). An intraplantar administration of CFA led to the induction of paw edema, which became detectable from 6 h after injection and remained for ∼2 weeks, indicating localized inflammation. This induction of paw edema, as measured by dorsoventral paw thickness, was unilateral (i.e., no significant change was observed in the contralateral sides). The paw edema was significantly reduced in the Pdk2/4 DKO (Fig. 8A) and Pdk2 KO and Pdk4 KO (Fig. 9A) mice compared with WT mice. We also confirmed the significantly reduced CFA-induced edema formation in Pdk2/4 DKO compared with WT by assessing the paw area (thickness × width) (data not shown). Similarly, the deficiency of Pdk2 and/or Pdk4 substantially attenuated, but did not completely abolish, unilateral mechanical allodynia (Figs. 8B, 9B) and thermal hyperalgesia (Figs. 8C, 9C) from 6 h to at least 1 week after injection. It is worth mentioning that the CFA administration slightly, but not significantly, upregulated the expression of Pdk2 (1.27-fold of control, p = 0.14 vs the control, Student's t test; n = 3; mean ± SEM) at 6 h after injection. The genetic deletion of Pdk2 ameliorated the CFA-induced pain hypersensitivities even at 6 h after injection. The slight increase of Pdk2 expression at 6 h following CFA administration might be sufficient to inhibit the PDH synergistically with Pdk4 to initiate the inflammatory cascade in the peripheral tissue, thereby driving the pain pathogenesis. These findings suggest the potential involvement of PDK2/4 in the localized inflammation and abnormal pain hypersensitivity following exposure to inflammatory insults.
Figure 8.
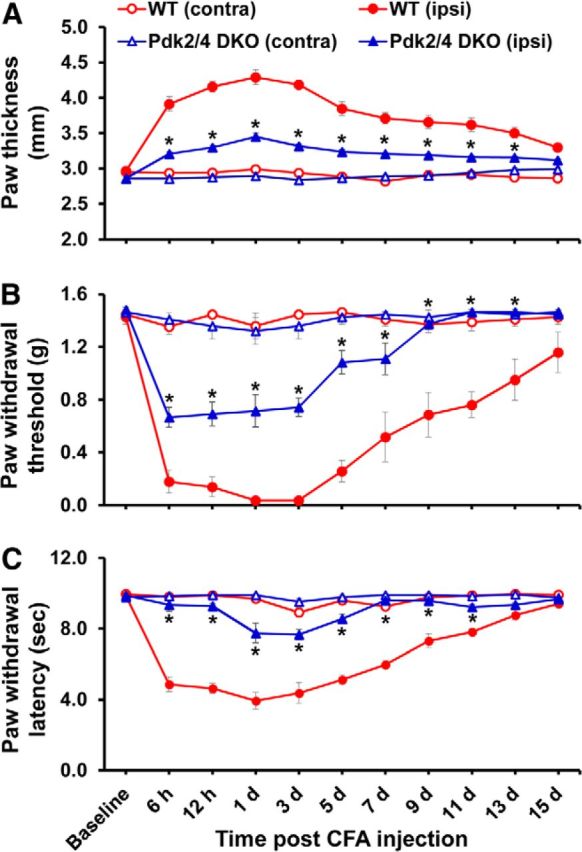
Pdk2/4 DKO mice displayed attenuated paw edema and pain responses to chronic inflammatory insult. To induce local inflammation, CFA was injected into the plantar surface of the left hindpaws (ipsilateral side). A, Paw thicknesses were assessed after injection and were found to be significantly greater in the ipsilateral sides than contralateral sides. The paw edema persisted for >1 week. No change in thickness was observed in the contralateral sides. Pdk2/4 deficiency significantly reduced the CFA-induced increase in paw thickness compared with that of WT animals. PWT to force and PWL to heat were measured in contralateral and ipsilateral sides. In the ipsilateral sides, CFA injection reduced PWT to force (B) and PWL to heat (C). The chronic inflammation-induced pain hypersensitivities were attenuated in Pdk2/4 DKO mice compared with WT animals. No significant change in pain-related behavior was observed in the contralateral sides. *p < 0.05 between ipsilateral sides of WT and DKO mice (one-way ANOVA with Dunnett's procedure for paw thickness and PWL, Mann–Whitney test for PWT). n = 7. Data are mean ± SEM.
Figure 9.
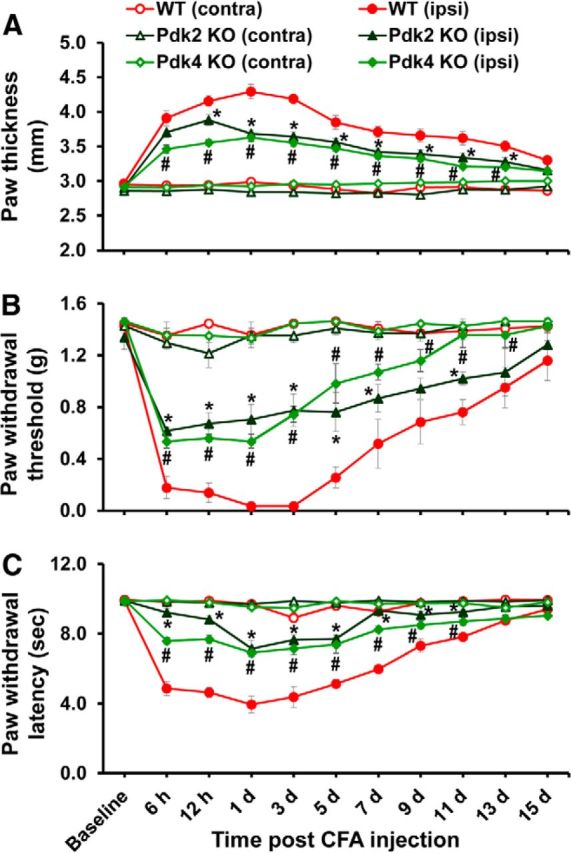
Pdk2 or Pdk4 single knock-out mice showed attenuated paw edema and pain responses to chronic inflammatory insult. A, CFA was injected into the plantar surface of the left hindpaws (ipsilateral side) to induce local inflammation. Paw thicknesses were significantly greater in the ipsilateral sides than contralateral sides. Pdk2 or Pdk4 single-gene deficiency significantly reduced the CFA-induced increase in paw thickness compared with that of WT animals. PWT to force and PWL to heat were measured in contralateral and ipsilateral sides. In the ipsilateral sides, CFA injection reduced PWT to force (B) and PWL to heat (C). The chronic inflammation-induced pain hypersensitivities were attenuated in Pdk2 or Pdk4 single-gene KO mice compared with WT animals. No significant change in pain-related behavior was observed in the contralateral sides. *p < 0.05 between ipsilateral sides of WT and Pdk2 KO mice. #p < 0.05 between ipsilateral sides of WT and Pdk4 KO mice (one-way ANOVA with Dunnett's procedure for paw thickness and PWL, Mann–Whitney test for PWT). n = 7. Data are mean ± SEM.
Pharmacological inhibition of PDKs attenuated CFA-induced inflammation and pain hypersensitivities
To further confirm the pivotal role of PDKs in inflammation-driven chronic pain pathogenesis, we investigated the effect of pharmacological inhibition of PDKs. Injection of DCA into the hindpaws of WT mice daily after CFA administration significantly diminished paw edema formation (Fig. 10A) and the development of mechanical allodynia as well as thermal hyperalgesia (Fig. 10B). Paw edema, withdrawal threshold to mechanical stimuli, and withdrawal latency to thermal stimuli were unchanged in the contralateral hindpaws following CFA/DCA treatment (data not shown). Because DCA has been previously reported to cause a reversible peripheral neuropathy in a dose- and duration-dependent manner (Calcutt et al., 2009), we confirmed that the dosage regimen we practiced was safe. The intraplantar injection of DCA alone did not induce paw edema or pain behavior (Fig. 10A,B). Furthermore, we also performed the H&E staining of the sciatic nerves from control and DCA-injected mice (data not shown) and found no remarkable difference in their histological features between the two groups, which again confirmed that the dosage regimen was not directly affecting the nerve. The intraplantar administration of DCA significantly lessened the expression of proinflammatory cytokines TNF-α, IL-1β, and IL-6 mRNAs in the CFA-injected hindpaw tissues (Fig. 10C). These results obtained from the pharmacological inhibition of PDKs support the crucial role of PDKs in chronic inflammatory pain.
Figure 10.
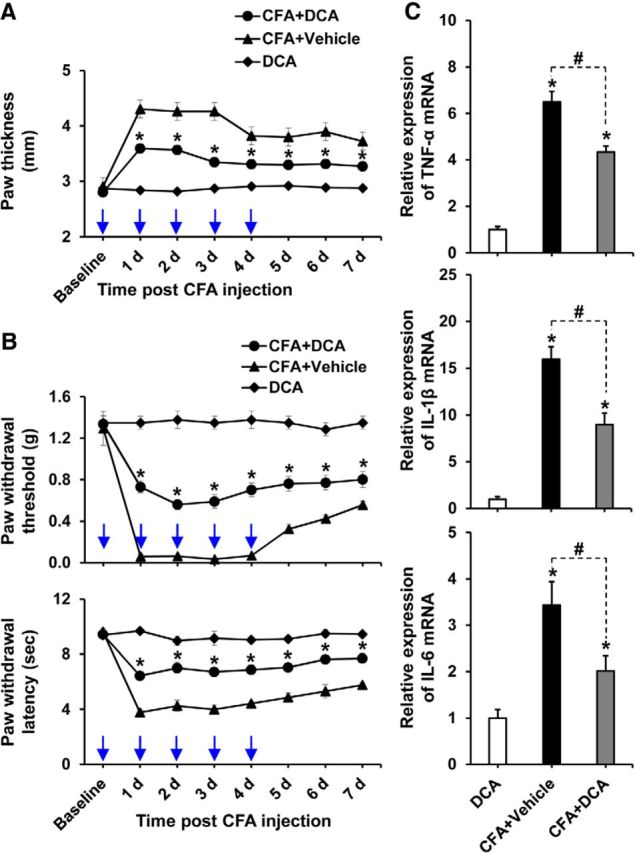
Pharmacological inhibition of PDKs attenuated CFA-induced paw edema, pain behaviors, and expression of proinflammatory cytokines. A, B, To examine the role of PDKs, DCA (10 mg/kg body weight, 10 μl) or vehicle (saline, 10 μl) was administered daily to the CFA-injected hindpaws. A group of animals received only DCA in left hindpaws. CFA-induced increase in paw thickness (A), PWT to force, and PWL to heat (B) was assessed up to 7 d after CFA injection. C, Hindpaw tissues were collected at day 3 after CFA administration from vehicle or DCA-treated animals to assess the expression of proinflammatory cytokines. The relative mRNA expression of TNF-α, IL-1β, and IL-6 in the hindpaw tissues was evaluated by real-time RT-PCR. Results for mRNA expression are displayed as the fold increase of gene expression normalized to GAPDH. *p < 0.05, between CFA+DCA and CFA+Vehicle groups, or versus the control animals. #p < 0.05 between indicated groups (Student's t test, Mann–Whitney test for PWT). A, B, n = 6; C, n = 3. Data are mean ± SEM. A, B, Arrows indicate the time points of DCA or vehicle administration.
A key role of lactic acid in the inflammation-mediated chronic pain
An enhanced expression of PDKs and the subsequent inhibition of PDH favor a cellular metabolic shift from oxidative phosphorylation to aerobic glycolysis, which leads to augmented lactate production (Kim et al., 2006; Rodríguez-Prados et al., 2010; Jha et al., 2012). Thus, an increase in PDK in the inflamed tissue might cause the metabolic shift toward lactate production. When we measured the lactate level in hindpaw tissues following CFA injection, the lactate level in the CFA-injected hindpaws of WT mice was significantly increased by more than threefold compared with the vehicle-injected hindpaws. Pdk2/4 deficiency diminished the inflammation-induced lactic acid surge in the hindpaw tissues (Fig. 11A). CFA-induced inflammation is a localized inflammation. We speculate that this process of localized insult, at least in this inflammation model, may not induce the production of lactic acid in the contracting muscle. However, contracting/overworking muscle cells undergo anaerobic energy production, which results in the accumulation of lactic acid. It creates acidic microenvironment surrounding the muscle cells, disrupts other metabolites, and results in the painful burning sensation. Lactic acid has been previously reported to play a key role in the exercise-related pains, including exercise-induced muscle pain, soreness, and cramps (Miles and Clarkson, 1994). Furthermore, DCA-mediated PDK inhibition and clodronate-mediated macrophage depletion significantly decreased lactate levels in the CFA-injected hindpaws (Fig. 11B). The crucial role of macrophages and their release of lactic acid in inflammatory pain were further supported by LPS-induced increase of ECAR (Fig. 11C). To further demonstrate the role of lactic acid surge in the inflammation and the subsequent pain pathogenesis, a small-molecule inhibitor (FX11) of LDHA was used. LDHA favors the conversion of pyruvate to lactate, and FX11 has been previously shown to block lactate production by inhibiting LDHA (Le et al., 2010). Intraplantar administration of FX11, but not vehicle, daily after CFA injection significantly diminished paw edema formation (Fig. 11D) and mechanical as well as thermal hypersensitivities (Fig. 11E). The CFA/FX11 administration did not induce or affect paw edema, mechanical allodynia, or thermal hyperalgesia in the contralateral side of hindpaws (data not shown). These findings strongly support the critical role of PDK-PDH-lactic acid axis in chronic inflammation and the resulting pain hypersensitivity.
Figure 11.
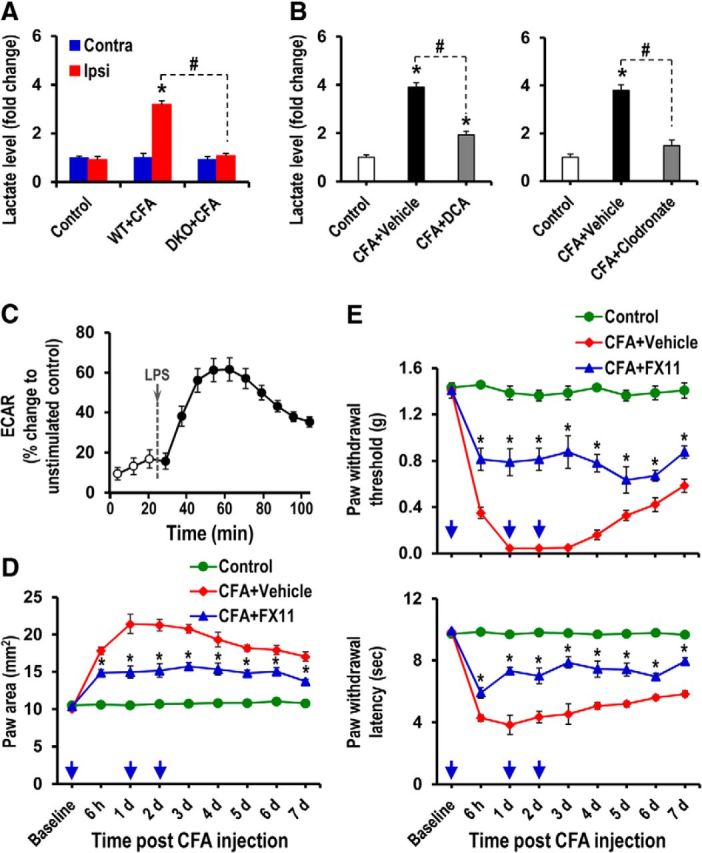
Role of lactic acid in the CFA-induced paw edema and pain hypersensitivities. A, The lactate assay was performed to compare the relative accumulation of lactate in the hindpaw tissues of WT and Pdk2/4 DKO mice at 3 d after CFA injection. B, The lactate assay was performed to measure the relative accumulation of lactate in the hindpaw tissues at 3 d after CFA injection (following either DCA or clodronate administration). The results presented are the fold change relative to control. Control animals were injected with either DCA alone (left) or vehicle (right). C, ECAR was recorded for RAW264.7 cells during LPS (100 ng/ml) stimulation and is presented as percentage change to unstimulated control. D, E, FX11 (a small-molecule inhibitor of lactate dehydrogenase A) (2 mg/kg body weight) or vehicle (2% [v/v] DMSO) was administered intraplantar daily to CFA-injected mice for 2 d. CFA-induced increase in paw area (D), PWT to force, and PWL to heat (E) was assessed up to 7 d after CFA injection. *p < 0.05 versus the vehicle-treated control animals, between CFA+Vehicle and CFA+FX11 groups. #p < 0.05 between indicated groups (Student's t test). A, B, n = 3; C, n = 5; D, E, n = 4. Data are mean ± SEM. Arrows indicate the time points of FX11 or vehicle administration.
Discussion
This study demonstrates that a PDK2/4-PDH-lactic acid axis plays a pivotal role in chronic inflammation-driven pain hypersensitivities, reinforcing the connection between metabolism and inflammation. Induction of local inflammation significantly upregulated the expression of PDK2, PDK4, and phosphorylated-PDH-E1α. Furthermore, CFA-induced infiltration of immune cells, augmented expression of inflammatory mediators, and production of lactic acid in hindpaws were attenuated in Pdk2/4 DKO mice. In addition, the CFA-induced activation of glial cells and the expression of proinflammatory cytokines in the spinal cord were reduced in Pdk2/4 DKO mice. Moreover, Pdk2/4 deficiency skewed the phenotype of macrophages toward M2 (alternative activation), whereas the enhanced expression of PDK2/4 shifted their phenotype toward M1 (classical activation). Our study using Pdk2/4 DKO mice revealed that the absence of PDK2/4 attenuated localized inflammation and pain hypersensitivities following exposure to inflammatory insult. In addition, enhanced expression of PDKs in hindpaw tissues augmented the lactic acid accumulation via inhibition of PDH. Subsequent studies using pharmacological inhibitors of PDKs and lactic acid production supported the crucial role of a PDK-PDH-lactic acid axis in chronic inflammation and pain hypersensitivities. It is worth mentioning that this study is limited to CFA-induced inflammation model. Hence, it is not completely certain that the results obtained in this study are true for all inflammation. These findings suggest that a PDK-PDH-lactic acid axis plays an imperative role in the pathogenesis of chronic/persistent inflammatory pain via multiple proinflammatory events: (1) local enhancement of proinflammatory cytokines and augmentation of lactic acid production; (2) functional polarization of macrophages toward the proinflammatory phenotype; and (3) activation of spinal glial cells (Fig. 12).
Figure 12.

A proposed schematic outlining the implications of a PDK-PDH-lactic acid axis in the pathogenesis of chronic inflammatory pain. Inflammatory stimulus enhances the expression and activity of PDK2 and PDK4 at the site of inflammation, thereby decreasing the oxidation of pyruvate and increasing its conversion into lactate via phosphorylation/inhibition of PDH. This metabolic shift-associated lactic acid production and resulting acidic microenvironment favor the recruitment of inflammatory cells to the site of inflammation and amplify the local inflammation ensuing the nociceptive responses. Enhanced PDK2/4 skew macrophages toward the M1 (proinflammatory) phenotype via activation of proinflammatory phenotype-determining transcription factor IRF8, resulting in the increased secretion of proinflammatory mediators, such as TNF-α, IL-1β, iNOS, and IL-6 as well as nitric oxide. The proalgesic mediators thus released activate nociceptors and spinal glia to cause peripheral and central sensitizations, respectively. These findings suggest that the PDK-PDH-lactic acid axis plays a key role in the pathogenesis of inflammation-induced chronic pain via the modulation of multifaceted proinflammatory events.
Following the administration of CFA, the expression of PDK2 and PDK4 as well as phosphorylated PDH-E1α were found to be significantly upregulated in the hindpaw tissues. An intraplantar injection of CFA results in the edema formation from the interactions of various inflammatory mediators with local tissues, which potently increases vascular permeability and/or blood flow (Sautebin et al., 1995). In this study, H&E staining revealed a significant difference between the WT and Pdk2/4 DKO animals with respect to the infiltration of inflammatory cells, suggesting the pivotal role of local PDK2/4-PDH axis in recruitment of the inflammatory cells. Further investigations showed the accumulation of Ly6G-positive neutrophils and Iba-1-positive macrophages in the CFA-injected hindpaws. These observations are consistent with previous findings suggesting the crucial role of neutrophils and macrophages in the onset and the chronification of inflammation-induced pain, respectively (Jha et al., 2014). Moreover, our own data advocate the crucial role of macrophages in the pathogenesis of chronic inflammatory pain as the clodronate-mediated depletion of macrophages attenuated the CFA-induced nociceptive behaviors, lactic acid accumulation, and expression of proinflammatory cytokines. We have also observed the colocalization of iNOS with infiltrated macrophages. Interestingly, the CFA-induced infiltration of neutrophils and macrophages was substantially reduced in Pdk2/4 DKO mice, supporting the proinflammatory nature of PDK2/4 induction in the inflammatory sites and the connection between metabolism and local inflammation.
In our in vitro studies, the peritoneal macrophages isolated from Pdk2/4 DKO mice showed an M2-biased phenotype, as the Pdk2/4-deficient macrophages expressed significantly higher levels of M2-related genes (Ym-1, Arg-1, and IL-10) and lower levels of M1-related genes (TNF-α, IL-1β, and IL-6) compared with WT macrophages. In addition, DCA inhibited LPS-induced proinflammatory cytokines and nitric oxide in primary macrophages. These findings led us to speculate that PDK2/4 may play an important role in inducing the functional polarization of the macrophages toward the proinflammatory phenotype. Macrophage functional polarity is important in the development of hyperalgesia during chronic inflammation (Godai et al., 2014). Phenotypic switch of muscle macrophages with an increased frequency of the M1 phenotype (proinflammatory) and reduced frequency of the M2 phenotype (anti-inflammatory) has also been reported following carrageenan exposure (da Silva et al., 2015). As macrophages are the major infiltrating cell type to the site of inflammation after CFA injection, PDK2/4-mediated functional polarization of macrophages toward the classical/proinflammatory M1 phenotype at the inflammatory scene provides a mechanistic basis for the interactions between metabolism, inflammation, and chronic pain. Moreover, IRFs play pivotal and functionally diverse roles in the transcriptional regulation of the immune system (Honda and Taniguchi, 2006). The M1 macrophage phenotype is controlled by IRF8 and IRF5, whereas M2 macrophage polarization is regulated by IRF4. The expression of IRF5 in CNS resident macrophages is reported to be regulated by IRF8 (Masuda et al., 2014). The IRF5 is well acknowledged to induce the expression of proinflammatory cytokines (Takaoka et al., 2005) and M1 polarization of macrophages (Krausgruber et al., 2011). Conversely, IRF4 is a key transcriptional factor that controls the M2 macrophage polarization (El Chartouni et al., 2010). In the current study, the Pdk2/4−/− macrophages revealed significantly enhanced expression of Irf4 in response to M2 inducer and substantially reduced expression of Irf8 in response to M1 inducer compared with Pdk2/4+/+ macrophages, suggesting that PDK2/4 potentially contribute to the switching of macrophage phenotype toward M1.
The PDK inhibitor DCA has been well documented to manage lactic acidosis (Morten et al., 1998; Abdelmalak et al., 2013), inhibit proliferation/activation of immune cells, and hinder the progression of inflammation (Ohashi et al., 2013). In the current study, intraplantar administration of DCA significantly attenuated the CFA-induced paw edema and pain hypersensitivities, suggesting that the PDKs are of pivotal importance in the pathogenesis of inflammation-driven persistent pain. Acidic microenvironment is a hallmark of the pain-generating pathological states (Loike et al., 1993; Wemmie et al., 2013). The metabolic shift leading to augmented lactic acid production has been implicated in the pathogenesis of peripheral neuropathy. During phagocytosis, macrophages primarily use the glycolysis pathway to regenerate their energy stores (Loike et al., 1993) and secrete lactate (Young and Zygas, 1987). Similarly, activated neutrophils and macrophages were shown to be the source of lactate in a murine wound model (Sheikh et al., 2000). Ischemia/reperfusion injury-induced allodynia in rat hindpaw has been associated with increased muscle lactic acid production (Laferrière et al., 2008). Under diverse chronic pain conditions, including fibromyalgia syndrome (Bengtsson, 2002) and chronic trapezius myalgia (Larsson et al., 2004), lactate concentration is increased due to insufficient oxygen supply and anaerobic state. In the acidic environment, lactic acid has been shown to induce the release of IL-1β and TNF-α in macrophages and glial cells, suggesting that the local levels of lactic acid may be a regulator of cytokine secretion (Jensen et al., 1990). In line with this, lactic acid and low pH have been reported to boost NF-κB-dependent inflammatory gene expression (Samuvel et al., 2009; Gerry and Leake, 2014). The metabolic shift-associated elevated lactate level has been reported to be a marker of the progression of inflammatory conditions (Finn and Oerther, 2010). The increased level of lactate has been detected in synovial fluids after carrageenan or CFA injection in rodents, suggesting that lactate, as a biomarker of inflammatory processes, plays a central role in cellular, regional, and whole-body metabolism (Gladden, 2004). In the present study, CFA-induced inflammation caused an increased production of lactic acid at the site of inflammation, which was significantly diminished in Pdk2/4-deficient animals as well as DCA-treated or macrophage-depleted mice, demonstrating the role of PDK2/4 in the lactic acid production and subsequent occurrence of the chronic inflammatory pain. These findings are consistent with previous studies reporting that CFA injection causes acidosis in the hindpaw tissues, leading to hyperalgesia (Nagae et al., 2006), and that macrophages cultured in the presence of PDH stimulator or PDK inhibitor show reduced lactate formation (Young and Zygas, 1987). Furthermore, the acidic pH seen in the inflamed tissues (Lee et al., 2005; Eisenhut and Wallace, 2011) has been reported to play a dominant role in the inflammatory excitation and sensitization of nociceptors (Steen et al., 1995). Several studies have demonstrated that an acidic microenvironment, at least in part, contributes to the induction of pain hypersensitivity (Birklein et al., 2000; Julius and Basbaum, 2001; Nagae et al., 2006; Kim et al., 2007; Nagae et al., 2007). ASICs are important transducers for nociception and mechanosensation (Wemmie et al., 2013). The elevated lactic acid level activates ASICs by facilitating their response to low pH (Sluka et al., 2009) and has similar effects on other receptors expressed by muscle primary afferent fibers, such as transient receptor potential vanilloid (Tominaga et al., 1998; Vriens et al., 2009). Meanwhile, the crucial role of lactic acid in inflammatory pain pathogenesis and the subsequent nociceptive behaviors was confirmed by using an inhibitor of lactic acid production. The application of LDHA inhibitor, which kinetically favors the conversion of pyruvate to lactate (Le et al., 2010), ameliorated the CFA-induced inflammation and pain hypersensitivities. Nevertheless, it should be noted that the inhibition of LDHA also inhibits the production of pyruvate by indirectly suppressing glycolysis at the level of GAPDH. Together, these findings advocate that the PDK-PDH-lactic acid axis governs the pathogenesis of inflammation-driven chronic pain.
In conclusion, our results suggest that PDK2/4 are indispensably involved in the pathogenesis of chronic inflammatory pain. At sites of local inflammation, the enhanced expression and activity of PDK2/4 mediate the activation and recruitment of inflammatory cells, biasing the infiltrated macrophages toward the proinflammatory phenotype, and they concurrently increase the production of lactic acid and other proinflammatory mediators that further amplify the localized inflammation. As a direct consequence of PDK-PDH-lactic acid axis-modulated peripheral inflammation, the activation of spinal glia and their production of proalgesic mediators contribute to the central sensitization. This study substantiates the PDK2/4-PDH-lactic acid axis as an essential link between metabolism and chronic inflammation-driven pain hypersensitivities. Therefore, these findings identify the PDK2/4-PDH-lactic acid axis as a promising novel therapeutic target for inflammatory pain.
Footnotes
This work was supported by Korea Healthcare Technology R&D Project, Ministry of Health and Welfare, Republic of Korea Grants A111345 and HI14C3331 and the Basic Science Research Program through the National Research Foundation funded by the Korean Ministry of Education, Science, and Technology 2008-0062282.
The authors declare no competing financial interests.
References
- Abdelmalak M, Lew A, Ramezani R, Shroads AL, Coats BS, Langaee T, Shankar MN, Neiberger RE, Subramony SH, Stacpoole PW. Long-term safety of dichloroacetate in congenital lactic acidosis. Mol Genet Metab. 2013;109:139–143. doi: 10.1016/j.ymgme.2013.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adeva-Andany M, López-Ojén M, Funcasta-Calderón R, Ameneiros-Rodríguez E, Donapetry-García C, Vila-Altesor M, Rodríguez-Seijas J. Comprehensive review on lactate metabolism in human health. Mitochondrion. 2014;17:76–100. doi: 10.1016/j.mito.2014.05.007. [DOI] [PubMed] [Google Scholar]
- Baloh RH. Mitochondrial dynamics and peripheral neuropathy. Neuroscientist. 2008;14:12–18. doi: 10.1177/1073858407307354. [DOI] [PubMed] [Google Scholar]
- Basbaum AI, Bautista DM, Scherrer G, Julius D. Cellular and molecular mechanisms of pain. Cell. 2009;139:267–284. doi: 10.1016/j.cell.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beirowski B, Babetto E, Golden JP, Chen YJ, Yang K, Gross RW, Patti GJ, Milbrandt J. Metabolic regulator LKB1 is crucial for Schwann cell-mediated axon maintenance. Nat Neurosci. 2014;17:1351–1361. doi: 10.1038/nn.3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengtsson A. The muscle in fibromyalgia. Rheumatology (Oxford) 2002;41:721–724. doi: 10.1093/rheumatology/41.7.721. [DOI] [PubMed] [Google Scholar]
- Bennett GJ. Pathophysiology and animal models of cancer-related painful peripheral neuropathy. Oncologist. 2010;15(Suppl 2):9–12. doi: 10.1634/theoncologist.2009-S503. [DOI] [PubMed] [Google Scholar]
- Birklein F, Weber M, Neundörfer B. Increased skin lactate in complex regional pain syndrome: evidence for tissue hypoxia? Neurology. 2000;55:1213–1215. doi: 10.1212/WNL.55.8.1213. [DOI] [PubMed] [Google Scholar]
- Calcutt NA, Lopez VL, Bautista AD, Mizisin LM, Torres BR, Shroads AL, Mizisin AP, Stacpoole PW. Peripheral neuropathy in rats exposed to dichloroacetate. J Neuropathol Exp Neurol. 2009;68:985–993. doi: 10.1097/NEN.0b013e3181b40217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- da Silva MD, Bobinski F, Sato KL, Kolker SJ, Sluka KA, Santos AR. IL-10 cytokine released from M2 macrophages is crucial for analgesic and anti-inflammatory effects of acupuncture in a model of inflammatory muscle pain. Mol Neurobiol. 2015;51:19–31. doi: 10.1007/s12035-014-8790-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrov EL, Kuo J, Kohno K, Usdin TB. Neuropathic and inflammatory pain are modulated by tuberoinfundibular peptide of 39 residues. Proc Natl Acad Sci U S A. 2013;110:13156–13161. doi: 10.1073/pnas.1306342110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirig DM, Salami A, Rathbun ML, Ozaki GT, Yaksh TL. Characterization of variables defining hindpaw withdrawal latency evoked by radiant thermal stimuli. J Neurosci Methods. 1997;76:183–191. doi: 10.1016/S0165-0270(97)00097-6. [DOI] [PubMed] [Google Scholar]
- Dixon WJ. Efficient analysis of experimental observations. Annu Rev Pharmacol Toxicol. 1980;20:441–462. doi: 10.1146/annurev.pa.20.040180.002301. [DOI] [PubMed] [Google Scholar]
- Duregotti E, Negro S, Scorzeto M, Zornetta I, Dickinson BC, Chang CJ, Montecucco C, Rigoni M. Mitochondrial alarmins released by degenerating motor axon terminals activate perisynaptic Schwann cells. Proc Natl Acad Sci U S A. 2015;112:E497–E505. doi: 10.1073/pnas.1417108112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenhut M, Wallace H. Ion channels in inflammation. Pflugers Arch. 2011;461:401–421. doi: 10.1007/s00424-010-0917-y. [DOI] [PubMed] [Google Scholar]
- El Chartouni C, Schwarzfischer L, Rehli M. Interleukin-4 induced interferon regulatory factor (Irf) 4 participates in the regulation of alternative macrophage priming. Immunobiology. 2010;215:821–825. doi: 10.1016/j.imbio.2010.05.031. [DOI] [PubMed] [Google Scholar]
- Fernyhough P, Roy Chowdhury SK, Schmidt RE. Mitochondrial stress and the pathogenesis of diabetic neuropathy. Expert Rev Endocrinol Metab. 2010;5:39–49. doi: 10.1586/eem.09.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari LF, Levine JD. Alcohol consumption enhances antiretroviral painful peripheral neuropathy by mitochondrial mechanisms. Eur J Neurosci. 2010;32:811–818. doi: 10.1111/j.1460-9568.2010.07355.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn A, Oerther SC. Can L(+)-lactate be used as a marker of experimentally induced inflammation in rats? Inflamm Res. 2010;59:315–321. doi: 10.1007/s00011-009-0107-6. [DOI] [PubMed] [Google Scholar]
- Gerry AB, Leake DS. Effect of low extracellular pH on NF-kappaB activation in macrophages. Atherosclerosis. 2014;233:537–544. doi: 10.1016/j.atherosclerosis.2014.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladden LB. Lactate metabolism: a new paradigm for the third millennium. J Physiol. 2004;558:5–30. doi: 10.1113/jphysiol.2003.058701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godai K, Hasegawa-Moriyama M, Kurimoto T, Saito T, Yamada T, Sato T, Kojima M, Kanmura Y. Peripheral administration of morphine attenuates postincisional pain by regulating macrophage polarization through COX-2-dependent pathway. Mol Pain. 2014;10:36. doi: 10.1186/1744-8069-10-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haji-Michael PG, Ladrière L, Sener A, Vincent JL, Malaisse WJ. Leukocyte glycolysis and lactate output in animal sepsis and ex vivo human blood. Metabolism. 1999;48:779–785. doi: 10.1016/S0026-0495(99)90179-8. [DOI] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Haschemi A, Kosma P, Gille L, Evans CR, Burant CF, Starkl P, Knapp B, Haas R, Schmid JA, Jandl C, Amir S, Lubec G, Park J, Esterbauer H, Bilban M, Brizuela L, Pospisilik JA, Otterbein LE, Wagner O. The sedoheptulose kinase CARKL directs macrophage polarization through control of glucose metabolism. Cell Metab. 2012;15:813–826. doi: 10.1016/j.cmet.2012.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst EA, MacPherson RE, LeBlanc PJ, Roy BD, Jeoung NH, Harris RA, Peters SJ. Pyruvate dehydrogenase kinase-4 contributes to the recirculation of gluconeogenic precursors during postexercise glycogen recovery. Am J Physiol Regul Integr Comp Physiol. 2014;306:R102–R107. doi: 10.1152/ajpregu.00150.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland S, Coste O, Zhang DD, Pierre SC, Geisslinger G, Scholich K. The ubiquitin ligase MYCBP2 regulates transient receptor potential vanilloid receptor 1 (TRPV1) internalization through inhibition of p38 MAPK signaling. J Biol Chem. 2011;286:3671–3680. doi: 10.1074/jbc.M110.154765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda K, Taniguchi T. IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat Rev Immunol. 2006;6:644–658. doi: 10.1038/nri1900. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- Jang E, Lee S, Kim JH, Kim JH, Seo JW, Lee WH, Mori K, Nakao K, Suk K. Secreted protein lipocalin-2 promotes microglial M1 polarization. FASEB J. 2013a;27:1176–1190. doi: 10.1096/fj.12-222257. [DOI] [PubMed] [Google Scholar]
- Jang E, Kim JH, Lee S, Kim JH, Seo JW, Jin M, Lee MG, Jang IS, Lee WH, Suk K. Phenotypic polarization of activated astrocytes: the critical role of lipocalin-2 in the classical inflammatory activation of astrocytes. J Immunol. 2013b;191:5204–5219. doi: 10.4049/jimmunol.1301637. [DOI] [PubMed] [Google Scholar]
- Jensen JC, Buresh C, Norton JA. Lactic acidosis increases tumor necrosis factor secretion and transcription in vitro. J Surg Res. 1990;49:350–353. doi: 10.1016/0022-4804(90)90036-2. [DOI] [PubMed] [Google Scholar]
- Jeon S, Jha MK, Ock J, Seo J, Jin M, Cho H, Lee WH, Suk K. Role of lipocalin-2-chemokine axis in the development of neuropathic pain following peripheral nerve injury. J Biol Chem. 2013;288:24116–24127. doi: 10.1074/jbc.M113.454140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeoung NH, Sanghani PC, Zhai L, Harris RA. Assay of the pyruvate dehydrogenase complex by coupling with recombinant chicken liver arylamine N-acetyltransferase. Anal Biochem. 2006;356:44–50. doi: 10.1016/j.ab.2006.06.017. [DOI] [PubMed] [Google Scholar]
- Jeoung NH, Rahimi Y, Wu P, Lee WN, Harris RA. Fasting induces ketoacidosis and hypothermia in PDHK2/PDHK4-double-knockout mice. Biochem J. 2012;443:829–839. doi: 10.1042/BJ20112197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha MK, Jeon S, Suk K. Pyruvate dehydrogenase kinases in the nervous system: their principal functions in neuronal-glial metabolic interaction and neuro-metabolic disorders. Curr Neuropharmacol. 2012;10:393–403. doi: 10.2174/157015912804499528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha MK, Jeon S, Jin M, Ock J, Kim JH, Lee WH, Suk K. The pivotal role played by lipocalin-2 in chronic inflammatory pain. Exp Neurol. 2014;254:41–53. doi: 10.1016/j.expneurol.2014.01.009. [DOI] [PubMed] [Google Scholar]
- Ji RR, Berta T, Nedergaard M. Glia and pain: is chronic pain a gliopathy? Pain. 2013;154(Suppl 1):S10–S28. doi: 10.1016/j.pain.2013.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph EK, Levine JD. Mitochondrial electron transport in models of neuropathic and inflammatory pain. Pain. 2006;121:105–114. doi: 10.1016/j.pain.2005.12.010. [DOI] [PubMed] [Google Scholar]
- Joseph EK, Levine JD. Multiple PKCepsilon-dependent mechanisms mediating mechanical hyperalgesia. Pain. 2010;150:17–21. doi: 10.1016/j.pain.2010.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julius D, Basbaum AI. Molecular mechanisms of nociception. Nature. 2001;413:203–210. doi: 10.1038/35093019. [DOI] [PubMed] [Google Scholar]
- Kaufmann P, Engelstad K, Wei Y, Jhung S, Sano MC, Shungu DC, Millar WS, Hong X, Gooch CL, Mao X, Pascual JM, Hirano M, Stacpoole PW, DiMauro S, De Vivo DC. Dichloroacetate causes toxic neuropathy in MELAS: a randomized, controlled clinical trial. Neurology. 2006;66:324–330. doi: 10.1212/01.wnl.0000196641.05913.27. [DOI] [PubMed] [Google Scholar]
- Kim HK, Park SK, Zhou JL, Taglialatela G, Chung K, Coggeshall RE, Chung JM. Reactive oxygen species (ROS) play an important role in a rat model of neuropathic pain. Pain. 2004;111:116–124. doi: 10.1016/j.pain.2004.06.008. [DOI] [PubMed] [Google Scholar]
- Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177–185. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Kim TJ, Freml L, Park SS, Brennan TJ. Lactate concentrations in incisions indicate ischemic-like conditions may contribute to postoperative pain. J Pain. 2007;8:59–66. doi: 10.1016/j.jpain.2006.06.003. [DOI] [PubMed] [Google Scholar]
- Klimaszewska-Łata J, Gul-Hinc S, Bielarczyk H, Ronowska A, Zyœk M, Gru¿ewska K, Pawełczyk T, Szutowicz A. Differential effects of lipopolysaccharide on energy metabolism in murine microglial N9 and cholinergic SN56 neuronal cells. J Neurochem. 2015;133:284–297. doi: 10.1111/jnc.12979. [DOI] [PubMed] [Google Scholar]
- Kopach O, Viatchenko-Karpinski V, Belan P, Voitenko N. Development of inflammation-induced hyperalgesia and allodynia is associated with the upregulation of extrasynaptic AMPA receptors in tonically firing lamina II dorsal horn neurons. Front Physiol. 2012;3:391. doi: 10.3389/fphys.2012.00391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krausgruber T, Blazek K, Smallie T, Alzabin S, Lockstone H, Sahgal N, Hussell T, Feldmann M, Udalova IA. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat Immunol. 2011;12:231–238. doi: 10.1038/ni.1990. [DOI] [PubMed] [Google Scholar]
- Kuner R. Central mechanisms of pathological pain. Nat Med. 2010;16:1258–1266. doi: 10.1038/nm.2231. [DOI] [PubMed] [Google Scholar]
- Kurlemann G, Paetzke I, Möller H, Masur H, Schuierer G, Weglage J, Koch HG. Therapy of complex I deficiency: peripheral neuropathy during dichloroacetate therapy. Eur J Pediatr. 1995;154:928–932. doi: 10.1007/BF01957508. [DOI] [PubMed] [Google Scholar]
- Laferrière A, Millecamps M, Xanthos DN, Xiao WH, Siau C, de Mos M, Sachot C, Ragavendran JV, Huygen FJ, Bennett GJ, Coderre TJ. Cutaneous tactile allodynia associated with microvascular dysfunction in muscle. Mol Pain. 2008;4:49. doi: 10.1186/1744-8069-4-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson B, Björk J, Kadi F, Lindman R, Gerdle B. Blood supply and oxidative metabolism in muscle biopsies of female cleaners with and without myalgia. Clin J Pain. 2004;20:440–446. doi: 10.1097/00002508-200411000-00009. [DOI] [PubMed] [Google Scholar]
- Lawrence T, Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol. 2011;11:750–761. doi: 10.1038/nri3088. [DOI] [PubMed] [Google Scholar]
- Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM, Royer RE, Vander Jagt DL, Semenza GL, Dang CV. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci U S A. 2010;107:2037–2042. doi: 10.1073/pnas.0914433107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Lee CH, Oh U. Painful channels in sensory neurons. Mol Cells. 2005;20:315–324. [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Loike JD, Kaback E, Silverstein SC, Steinberg TH. Lactate transport in macrophages. J Immunol. 1993;150:1951–1958. [PubMed] [Google Scholar]
- Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. J Clin Invest. 2011;121:2111–2117. doi: 10.1172/JCI57132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda T, Iwamoto S, Yoshinaga R, Tozaki-Saitoh H, Nishiyama A, Mak TW, Tamura T, Tsuda M, Inoue K. Transcription factor IRF5 drives P2X4R+-reactive microglia gating neuropathic pain. Nat Commun. 2014;5:3771. doi: 10.1038/ncomms4771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire JF, Rouen S, Siegfreid E, Wright DE, Dobrowsky RT. Caveolin-1 and altered neuregulin signaling contribute to the pathophysiological progression of diabetic peripheral neuropathy. Diabetes. 2009;58:2677–2686. doi: 10.2337/db09-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles MP, Clarkson PM. Exercise-induced muscle pain, soreness, and cramps. J Sports Med Phys Fitness. 1994;34:203–216. [PubMed] [Google Scholar]
- Morten KJ, Caky M, Matthews PM. Stabilization of the pyruvate dehydrogenase E1alpha subunit by dichloroacetate. Neurology. 1998;51:1331–1335. doi: 10.1212/WNL.51.5.1331. [DOI] [PubMed] [Google Scholar]
- Möser CV, Stephan H, Altenrath K, Kynast KL, Russe OQ, Olbrich K, Geisslinger G, Niederberger E. TANK-binding kinase 1 (TBK1) modulates inflammatory hyperalgesia by regulating MAP kinases and NF-kappaB dependent genes. J Neuroinflammation. 2015;12:100. doi: 10.1186/s12974-015-0319-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagae M, Hiraga T, Wakabayashi H, Wang L, Iwata K, Yoneda T. Osteoclasts play a part in pain due to the inflammation adjacent to bone. Bone. 2006;39:1107–1115. doi: 10.1016/j.bone.2006.04.033. [DOI] [PubMed] [Google Scholar]
- Nagae M, Hiraga T, Yoneda T. Acidic microenvironment created by osteoclasts causes bone pain associated with tumor colonization. J Bone Miner Metab. 2007;25:99–104. doi: 10.1007/s00774-006-0734-8. [DOI] [PubMed] [Google Scholar]
- Nicol LS, Dawes JM, La Russa F, Didangelos A, Clark AK, Gentry C, Grist J, Davies JB, Malcangio M, McMahon SB. The role of G-protein receptor 84 in experimental neuropathic pain. J Neurosci. 2015;35:8959–8969. doi: 10.1523/JNEUROSCI.3558-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nisoli E, Clementi E, Carruba MO, Moncada S. Defective mitochondrial biogenesis: a hallmark of the high cardiovascular risk in the metabolic syndrome? Circ Res. 2007;100:795–806. doi: 10.1161/01.RES.0000259591.97107.6c. [DOI] [PubMed] [Google Scholar]
- Odegaard JI, Chawla A. Alternative macrophage activation and metabolism. Annu Rev Pathol. 2011;6:275–297. doi: 10.1146/annurev-pathol-011110-130138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi T, Akazawa T, Aoki M, Kuze B, Mizuta K, Ito Y, Inoue N. Dichloroacetate improves immune dysfunction caused by tumor-secreted lactic acid and increases antitumor immunoreactivity. Int J Cancer. 2013;133:1107–1118. doi: 10.1002/ijc.28114. [DOI] [PubMed] [Google Scholar]
- O'Neill LA, Hardie DG. Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature. 2013;493:346–355. doi: 10.1038/nature11862. [DOI] [PubMed] [Google Scholar]
- Orihuela R, McPherson CA, Harry GJ. Microglial M1/M2 polarization and metabolic states. Br J Pharmacol. 2015 doi: 10.1111/bph.13139. doi: 10.1111/bph.13139. Advance online publication. Retrieved Mar. 20, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palamiuc L, Schlagowski A, Ngo ST, Vernay A, Dirrig-Grosch S, Henriques A, Boutillier AL, Zoll J, Echaniz-Laguna A, Loeffler JP, René F. A metabolic switch toward lipid use in glycolytic muscle is an early pathologic event in a mouse model of amyotrophic lateral sclerosis. EMBO Mol Med. 2015;7:526–546. doi: 10.15252/emmm.201404433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palomer X, Salvadó L, Barroso E, Vázquez-Carrera M. An overview of the crosstalk between inflammatory processes and metabolic dysregulation during diabetic cardiomyopathy. Int J Cardiol. 2013;168:3160–3172. doi: 10.1016/j.ijcard.2013.07.150. [DOI] [PubMed] [Google Scholar]
- Pan Z, Zhu LJ, Li YQ, Hao LY, Yin C, Yang JX, Guo Y, Zhang S, Hua L, Xue ZY, Zhang H, Cao JL. Epigenetic modification of spinal miR-219 expression regulates chronic inflammation pain by targeting CaMKIIgamma. J Neurosci. 2014;34:9476–9483. doi: 10.1523/JNEUROSCI.5346-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel MS, Roche TE. Molecular biology and biochemistry of pyruvate dehydrogenase complexes. FASEB J. 1990;4:3224–3233. doi: 10.1096/fasebj.4.14.2227213. [DOI] [PubMed] [Google Scholar]
- Pooya S, Liu X, Kumar VB, Anderson J, Imai F, Zhang W, Ciraolo G, Ratner N, Setchell KD, Yoshida Y, Yutaka Y, Jankowski MP, Dasgupta B. The tumour suppressor LKB1 regulates myelination through mitochondrial metabolism. Nat Commun. 2014;5:4993. doi: 10.1038/ncomms5993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos-Nino ME. The role of chronic inflammation in obesity-associated cancers. ISRN Oncol. 2013;2013:697521. doi: 10.1155/2013/697521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Prados JC, Través PG, Cuenca J, Rico D, Aragonés J, Martín-Sanz P, Cascante M, Boscá L. Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. J Immunol. 2010;185:605–614. doi: 10.4049/jimmunol.0901698. [DOI] [PubMed] [Google Scholar]
- Rogatzki MJ, Ferguson BS, Goodwin ML, Gladden LB. Lactate is always the end product of glycolysis. Front Neurosci. 2015;9:22. doi: 10.3389/fnins.2015.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuvel DJ, Sundararaj KP, Nareika A, Lopes-Virella MF, Huang Y. Lactate boosts TLR4 signaling and NF-kappaB pathway-mediated gene transcription in macrophages via monocarboxylate transporters and MD-2 up-regulation. J Immunol. 2009;182:2476–2484. doi: 10.4049/jimmunol.0802059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sautebin L, Ialenti A, Ianaro A, Di Rosa M. Endogenous nitric oxide increases prostaglandin biosynthesis in carrageenin rat paw oedema. Eur J Pharmacol. 1995;286:219–222. doi: 10.1016/0014-2999(95)00581-5. [DOI] [PubMed] [Google Scholar]
- Sheikh AY, Gibson JJ, Rollins MD, Hopf HW, Hussain Z, Hunt TK. Effect of hyperoxia on vascular endothelial growth factor levels in a wound model. Arch Surg. 2000;135:1293–1297. doi: 10.1001/archsurg.135.11.1293. [DOI] [PubMed] [Google Scholar]
- Shime H, Yabu M, Akazawa T, Kodama K, Matsumoto M, Seya T, Inoue N. Tumor-secreted lactic acid promotes IL-23/IL-17 proinflammatory pathway. J Immunol. 2008;180:7175–7183. doi: 10.4049/jimmunol.180.11.7175. [DOI] [PubMed] [Google Scholar]
- Sluka KA, Winter OC, Wemmie JA. Acid-sensing ion channels: a new target for pain and CNS diseases. Curr Opin Drug Discov Devel. 2009;12:693–704. [PMC free article] [PubMed] [Google Scholar]
- Stacpoole PW, Henderson GN, Yan Z, James MO. Clinical pharmacology and toxicology of dichloroacetate. Environ Health Perspect. 1998a;106(Suppl 4):989–994. doi: 10.1289/ehp.98106s4989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stacpoole PW, Henderson GN, Yan Z, Cornett R, James MO. Pharmacokinetics, metabolism and toxicology of dichloroacetate. Drug Metab Rev. 1998b;30:499–539. doi: 10.3109/03602539808996323. [DOI] [PubMed] [Google Scholar]
- Steen KH, Steen AE, Reeh PW. A dominant role of acid pH in inflammatory excitation and sensitization of nociceptors in rat skin, in vitro. J Neurosci. 1995;15:3982–3989. doi: 10.1523/JNEUROSCI.15-05-03982.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summan M, Warren GL, Mercer RR, Chapman R, Hulderman T, Van Rooijen N, Simeonova PP. Macrophages and skeletal muscle regeneration: a clodronate-containing liposome depletion study. Am J Physiol Regul Integr Comp Physiol. 2006;290:R1488–R1495. doi: 10.1152/ajpregu.00465.2005. [DOI] [PubMed] [Google Scholar]
- Takaoka A, Yanai H, Kondo S, Duncan G, Negishi H, Mizutani T, Kano S, Honda K, Ohba Y, Mak TW, Taniguchi T. Integral role of IRF-5 in the gene induction programme activated by Toll-like receptors. Nature. 2005;434:243–249. doi: 10.1038/nature03308. [DOI] [PubMed] [Google Scholar]
- Takubo K, Nagamatsu G, Kobayashi CI, Nakamura-Ishizu A, Kobayashi H, Ikeda E, Goda N, Rahimi Y, Johnson RS, Soga T, Hirao A, Suematsu M, Suda T. Regulation of glycolysis by Pdk functions as a metabolic checkpoint for cell cycle quiescence in hematopoietic stem cells. Cell Stem Cell. 2013;12:49–61. doi: 10.1016/j.stem.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang F, Lane S, Korsak A, Paton JF, Gourine AV, Kasparov S, Teschemacher AG. Lactate-mediated glia-neuronal signalling in the mammalian brain. Nat Commun. 2014;5:3284. doi: 10.1038/ncomms4284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tominaga M, Caterina MJ, Malmberg AB, Rosen TA, Gilbert H, Skinner K, Raumann BE, Basbaum AI, Julius D. The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron. 1998;21:531–543. doi: 10.1016/S0896-6273(00)80564-4. [DOI] [PubMed] [Google Scholar]
- Vriens J, Appendino G, Nilius B. Pharmacology of vanilloid transient receptor potential cation channels. Mol Pharmacol. 2009;75:1262–1279. doi: 10.1124/mol.109.055624. [DOI] [PubMed] [Google Scholar]
- Wemmie JA, Taugher RJ, Kreple CJ. Acid-sensing ion channels in pain and disease. Nat Rev Neurosci. 2013;14:461–471. doi: 10.1038/nrn3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Ruchti E, Petit JM, Jourdain P, Grenningloh G, Allaman I, Magistretti PJ. Lactate promotes plasticity gene expression by potentiating NMDA signaling in neurons. Proc Natl Acad Sci U S A. 2014;111:12228–12233. doi: 10.1073/pnas.1322912111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young PR, Zygas AP. Secretion of lactic acid by peritoneal macrophages during extracellular phagocytosis: the possible role of local hyperacidity in inflammatory demyelination. J Neuroimmunol. 1987;15:295–308. doi: 10.1016/0165-5728(87)90123-8. [DOI] [PubMed] [Google Scholar]