

Abstract
α-synuclein, a protein enriched in Lewy bodies and highly implicated in neurotoxicity in Parkinson's disease, is distributed both at nerve terminals and in the cell nucleus. Here we show that a nuclear derivative of α-synuclein induces more pronounced changes at the gene expression level in mouse primary dopamine (DA) neurons compared to a derivative that is excluded from the nucleus. Moreover, by RNA sequencing we analyzed the extent of genome-wide effects on gene expression resulting from expression of human α-synuclein in primary mouse DA neurons. The results implicated the transcription factor Nurr1 as a key dysregulated target of α-synuclein toxicity. Forced Nurr1 expression restored the expression of hundreds of dysregulated genes in primary DA neurons expressing α-synuclein, and therefore prompted us to test the possibility that Nurr1 can be pharmacologically targeted by bexarotene, a ligand for the retinoid X receptor that forms heterodimers with Nurr1. Although our data demonstrated that bexarotene was ineffective in neuroprotection in rats in vivo, the results revealed that bexarotene has the capacity to coregulate subsets of Nurr1 target genes including the receptor tyrosine kinase subunit Ret. Moreover, bexarotene was able to restore dysfunctional Ret-dependent neurotrophic signaling in α-synuclein-overexpressing mouse DA neurons. These data highlight the role of the Nurr1–Ret signaling pathway as a target of α-synuclein toxicity and suggest that retinoid X receptor ligands with appropriate pharmacological properties could have therapeutic potential in Parkinson's disease.
SIGNIFICANCE STATEMENT How α-synuclein, a protein enriched in Lewy bodies in Parkinson's disease, is causing neuropathology in dopamine neurons remains unclear. This study elucidated how α-synuclein is influencing gene expression and how Nurr1, a transcription factor known to protect dopamine neurons against α-synuclein toxicity, can counteract these effects. Moreover, given the protective role of Nurr1, this study also investigated how Nurr1 could be pharmacologically targeted via bexarotene, a ligand of Nurr1's heterodimerization partner retinoid X receptor (RXR). The results showed that RXR ligands could increase neurotrophic signaling, but provided a mixed picture of its potential in a Parkinson's disease rat model in vivo. However, this study clearly emphasized Nurr1's neuroprotective role and indicated that other RXR ligands could have therapeutic potential in Parkinson's disease.
Introduction
Parkinson's disease (PD) is the most common neurodegenerative motor disorder characterized by the progressive degeneration of dopamine (DA) neurons in the substantia nigra (SN) pars compacta and the formation of intraneuronal Lewy bodies containing misfolded α-synuclein (Lees et al., 2009). Current treatments for PD alleviate motor symptoms by DA replacement using levodopa and/or DA receptor agonists. However, these treatments do not block or slow neurodegeneration, and they become progressively ineffective as disease develops. There is thus a major need for new neuroprotective strategies that can be used alone or in combination with existing treatments (Smith et al., 2012).
Since the initial identification of α-synuclein as a protein localized to the presynapse and in the nucleus, nuclear localization of α-synuclein has been verified in a number of studies (Maroteaux et al., 1988; Goers et al., 2003; Specht et al., 2005; Yu et al., 2007). However, α-synuclein-induced toxicity has mostly been characterized as a consequence of α-synuclein expression in the cytoplasm and nerve terminals, and the impact of nuclear α-synuclein on disease remains largely unexplored. Interestingly, studies in a Drosophila model of DA neuron dysfunction indicated that the nuclear form of α-synuclein is the main mediator of toxicity, and other studies indicate that α-synuclein influences acetylation of histones and may have other detrimental effects in the nucleus linked to dysregulated gene expression (Kontopoulos et al., 2006; Jin et al., 2011; Decressac et al., 2012, 2013b).
Previous studies have pointed out that one of the nuclear targets that may be relevant in PD pathology is the transcription factor Nurr1. Nurr1 is not only essential for the differentiation of midbrain DA neurons during development, but is also required for the maintenance of DA neurons in the adult brain (Zetterstrom et al., 1997; Castillo et al., 1998; Saucedo-Cardenas et al., 1998; Smidt and Burbach, 2007; Kadkhodaei et al., 2009, 2013). Genetic variants of Nurr1 have been associated with PD, and Nurr1 has been shown to be downregulated in DA neurons containing α-synuclein-rich inclusions in postmortem PD brain tissue (Chu et al., 2002; Moran et al., 2007; for review, see Decressac et al., 2013a). Notably, Nurr1 and several of its transcriptional targets including Ret, the signaling receptor for glial cell line-derived neurotrophic factor (GDNF), are downregulated in midbrain DA neurons in rodent PD models expressing high levels of α-synuclein (for review, see Decressac et al., 2013a). Ret downregulation was shown to contribute to the inability of GDNF to exert its normal neurotrophic signaling activity in α-synuclein overexpressing DA neurons. Moreover, Nurr1 gene ablation in adult DA neurons results in increased vulnerability to α-synuclein, and, reciprocally, forced Nurr1 expression by viral vectors provides protection against α-synuclein toxicity in vivo (Decressac et al., 2012).
Based on these previous findings, Nurr1 is potentially interesting as a new target for therapeutic approaches in PD, and compounds that would increase its activity might be used as PD-modifying drugs. Nurr1 belongs to the family of ligand-activated nuclear receptors, but it lacks identified ligands and the structure of its ligand-binding domain indicates that small molecule ligands that can activate Nurr1 may be difficult to identify (Wang et al., 2003). However, Nurr1 can form a heterodimer with the nuclear retinoid X receptor (RXR), suggesting the possibility that RXR ligands could be used to modulate Nurr1 activity (Forman et al., 1995; Perlmann and Jansson, 1995). Interestingly, RXR ligands have provided promising results in neuroprotection in PD models (Yin et al., 2012; McFarland et al., 2013; Reiner et al., 2014). Here we further addressed the significance of Nurr1 as a drug target via activation of its heterodimerization partner RXR in experiments performed both in primary cultures in vitro and PD models in vivo. Moreover, to further understand the context underlying Nurr1 downregulation by α-synuclein, we investigated the impact of α-synuclein on global gene expression in DA neurons, and we characterized the importance of the Nurr1–Ret–GDNF pathway in the cellular defense against α-synuclein pathology.
Materials and Methods
Immunohistochemistry.
Cells were fixed in 2% paraformaldehyde for 30 min at room temperature, washed with PBS, blocked in PBS containing 0.3% Triton X-100 (Sigma) and 1% FCS (PAA Laboratories, GE Healthcare) for 1 h, and then incubated overnight at 4°C with the following primary antibodies diluted in blocking solution: rabbit anti-phospho-S6 Ser 235/236 (Cell Signaling Technology; 1:500), mouse α-synuclein 211 (Santa Cruz Biotechnology; 1:100), and sheep anti-TH (Pel-Freeze; 1:500). Cells were then rinsed with PBS and incubated for 1 h at room temperature with fluorochrome-conjugated secondary antibodies (Alexa, Invitrogen) diluted 1:400 in PBS. Finally, cells were rinsed with PBS and examined using a Zeiss LSM5 Exciter confocal microscope. Cell counting was done manually from eight randomly picked fields with a 10× objective. Statistical significance was calculated by one-way ANOVA, and the data are presented as mean values + SEM of triplicate samples.
Lentiviral infections.
Lentiviral particles were produced as described previously (Zufferey et al., 1997). Ret lentiviral particles were purchased from GenTarget and concentrated by ultracentrifugation at 28000 rpm for 2.5 h. Adherent primary cultures were transduced 48 h after seeding with the indicated lentiviruses at a multiplicity of infection of 30. The virus was removed after 48 h, and the cells were analyzed 5 d post infection, unless specified otherwise.
Constructs.
The lentiviral expression vectors used were constructed by subcloning the cDNAs of WT Nurr1, Nurr1-DIM (dimerization mutant), and Nurr1-DBD (DNA binding mutant) (Castro et al., 2001), of WT-, NES (Nuclar Export Signal)-, and NLS (Nuclear Localization Signal)-α-synuclein (kind gifts from Dr. Mel Feany, Harvard Medical School, Boston) into the lentiviral vector pRRLSIN.cPPT.PGK-GFP.WPRE (Addgene) after removal of the enhanced green fluorescent protein (eGFP) cDNA. The lentiviral red fluorescent protein (RFP) vector (pLenti-III-RenLuc-RFP) was purchased from Abmgood. The mouse Ret cDNA (kind gift from Dr. Carlos Ibanez, Karolinska Institutet, Stockholm, Sweden) was introduced in an AAV2 transfer plasmid (Decressac et al. 2012). For the downregulation of Nurr1, primary cultures were infected with lentiviruses generated from scrambled (pLKO.1) or shNurr1 Mission clones (Sigma) and fluorescence-activated cell sorting (FACS) sorted 48 h after infection. For the coimmunoprecipitation experiment, the ligand-binding domain of RXR together with an HA tag were cloned in a pCMX vector containing the Gal4 DNA-binding domain (Wang et al., 2003).
Transfection and coimmunoprecipitation assay.
HEK293T cells were transfected with the lentiviral constructs for Nurr1 and the lentiviral construct for α-synuclein or pCMX-Gal4-HA-RXR in 10 cm dishes with Lipofectamine/Plus (Invitrogen) according to the manufacturer's protocol. Transfected cells were harvested after 48 h in RIPA buffer (Sigma) supplemented with protease inhibitor cocktail (Roche) and PMSF protease inhibitor (Invitrogen). Protein A/G beads (Sigma) were coated with the mouse anti-α-synuclein 211 (Santa Cruz Biotechnology), rabbit anti-Nurr1 E-20 (Santa Cruz Biotechnology), or mouse anti-HA antibodies (Roche) and then incubated overnight with 50 μg of lysate. Beads were washed three times with RIPA buffer, centrifuged at 5000 × g for 5 min, boiled for 5 min in SDS sample buffer (Bio-Rad) supplemented with 10% β-mercaptoethanol, and subjected to Western blot analysis.
Chemicals.
GDNF was purchased from R & D Systems and resuspended in PBS containing 0.1% BSA. Bexarotene was purchased from Sigma and resuspended in DMSO. The absolute volumes of DMSO added to the cells were minimal (<0.5%) and did not affect cell survival. RPI-1 was purchased from Sigma, resuspended in DMSO, and used at a final concentration of 40 μm.
Real-time PCR analysis.
Total RNA was prepared using the RNeasy micro kit (Qiagen), treated with DNase (Qiagen), and reverse-transcribed using SuperScriptIII (Invitrogen) and oligo (dT)12–18 (Invitrogen) primer. Real-time PCR was performed on the vIIA7 system (Applied Biosystems) using Taqman Gene Expression Master Mix (Applied Biosystems) and Taqman Gene Expression Assays probes (Applied Biosystems). Gene expression values were normalized against Rpl19, and fold change was calculated using the ΔΔCT method (Livak and Schmittgen, 2001). Statistical significance was calculated by one-way ANOVA, and the data are presented as means + SEM (n = 3).
FACS sorting.
Pitx3eGFP/+ primary ventral midbrain cells were harvested using the MACS Neural Tissue Dissociation Kit (P) (Miltenyi Biotec) and resuspended in phenol-free HBSS (Invitrogen). Samples were filtered through cell strainer caps (35 μm; BD Biosciences) and sorted for eGFP expression. Primary ventral midbrain (vMB) cultures from embryonic day 13.5 (E13.5) wild-type C57/BL6 mice were used as a control. FACS was performed using the FACSAria cell sorter and the FACSDiva software (BD Biosciences). The population of interest, excluding debris and duplicates, was identified by forward and side scatter gating. A 100 μm nozzle, sheath pressure of 20–25 psi, and an acquisition rate of 2000–3000 events per second were used. Sorted cells were collected in RNeasy lysis buffer (Qiagen), and samples were vortexed and frozen on dry ice directly after acquisition.
Primary cultures.
Primary ventral midbrain cultures were obtained from E13.5 Pitx3eGFP/+ mouse embryos. Briefly, the anlage of the substantia nigra was microdissected, dissociated with the MACS Neural Tissue Dissociation Kit (P) (Miltenyi Biotec), and plated at 50,000 cells/mm2 on poly-ornithine- and laminin-coated (Sigma) plates in attachment medium consisting of stock medium DMEM/F-12/Neurobasal differentiation medium (Invitrogen) 1:1, 1× B27 (Invitrogen), 1× N2 (Invitrogen), 1 mm sodium pyruvate (Invitrogen), 0.25% bovine serum albumin (Sigma), and 2 mm glutamine (Invitrogen), after addition of 3% FCS (PAA Laboratories, GE Healthcare) and 20 ng/ml bFGF (R & D Systems). After 24 h, the medium was replaced with differentiation medium consisting of stock medium supplemented with 100 μm ascorbic acid (Sigma) and 1% FCS. The first 48 h after adhesion, the stock medium was also supplemented with 10 ng/ml recombinant human GDNF (R & D Systems) and 20 ng/ml recombinant human BDNF (R & D Systems). For the downregulation of Nurr1 expression by shNurr1, E11.5 primary cultures were prepared using the same protocol, with the following modifications: the Hedgehog agonist Hh-Ag1.3 (Curis) and FGF8 (R & D Systems) were added to the attachment medium and the cells received proliferation medium (stock medium supplemented with 20 ng/ml bFGF, 100 ng/ml FGF8, and 100 nm Hedgehog agonist Hh-Ag1.3) for 48 h before the media was replaced with differentiation medium.
RNA sequencing.
RNA concentration and integrity were assessed on an Agilent RNA 6000 Pico chip using the Agilent 2100 BioAnalyzer (Agilent Technologies). For the libraries presented in Figure 3, RNA sequencing was performed using the SMARTer Ultra Low Input RNA Kit for Illumina sequencing (Clontech), and the generated cDNA was tagmentated, amplified, and purified using the Nextera XT DNA sample preparation kit (Illumina), whereas for libraries presented in Figure 4, the Smart-Seq2 protocol (Picelli et al., 2013) was used. The cDNA libraries were run on high-sensitivity DNA chips on the Agilent 2100 BioAnalyzer and sequenced on an Illumina HiSeq 2000 system. The clusters were exported into fastq files and analyzed by DESeq for Figure 4 (Anders and Huber, 2010) and DESeq2 (Love et al., 2014) for Figure 3, after removing the genes with mean RPKM (reads per kilobase of transcript per million mapped reads) values <1 in the DMSO/RFP samples. Pathway enrichment analysis was performed using the DAVID tools (Huang da et al., 2009). The cluster dendrogram in Figure 4 was generated using the hclust script in R. Raw data have been submitted to the NCBI (Gene Expression Omnibus accession number GSE70368).
Figure 3.
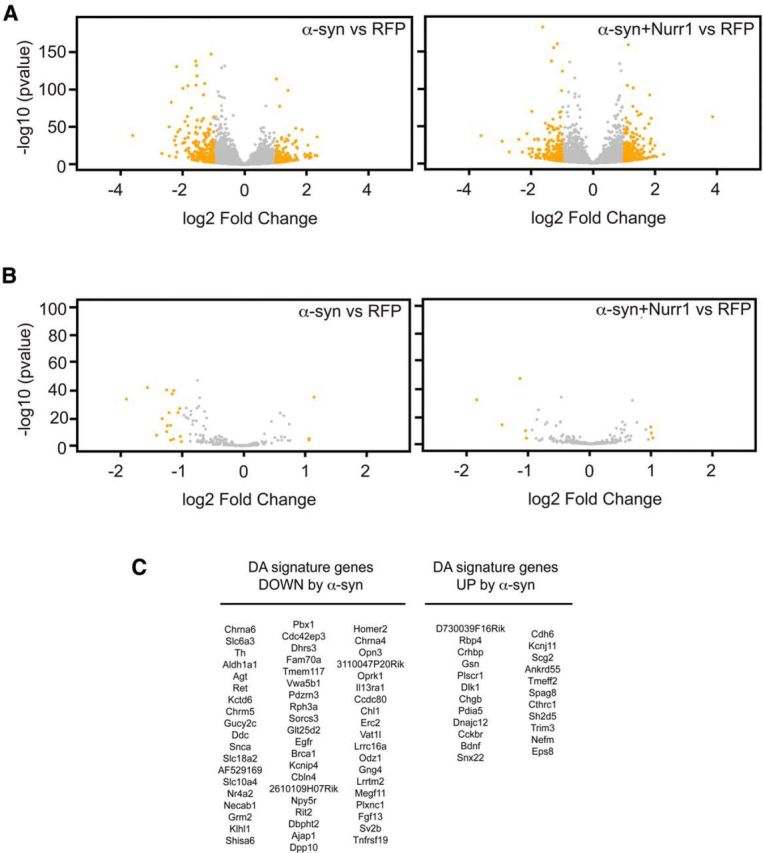
RNA sequencing of Pitx3-eGFP primary midbrain DA cultures infected with α-synuclein or α-synuclein and Nurr1 lentiviruses. A, B, Volcano plots showing all genes (A) or DA signature genes (B) with RPKM values >1 after infection of E13.5 primary ventral midbrain cultures with α-synuclein or α-synuclein and Nurr1 lentiviruses. The negative log10 transformed p values (y-axis) test the null hypothesis of no difference in expression levels and are plotted against the average log2 fold changes in expression (x-axis). Genes in yellow are differentially expressed with an absolute log2 fold change >1. C, Tables showing the DA signature genes significantly upregulated/downregulated after α-synuclein overexpression.
Figure 4.

RNA sequencing of FACS-sorted Pitx3-eGFP DA neurons treated with bexarotene or vehicle. A, Cluster dendrogram showing the similarity between the different RNASeq samples based on their Spearman correlation. B, Table showing the number of genes with an RPKM value >1 that are upregulated or downregulated by bexarotene (padj < 0.05). C, Scatter diagram showing the fold change of the RPKM values for the DA signature genes after infection with shNurr1 lentivirus or treatment with bexarotene. Bar shows the median value. D, Table showing the gene count and p values (after multiple hypothesis testing with the Benjamini–Hochberg method) for the significantly upregulated and downregulated KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways in bexarotene-treated DA neurons. E, Scatter diagram showing the fold change of the RPKM values for oxidative phosphorylation genes after infection with shNurr1 lentivirus or treatment with bexarotene. Bars show median values. F, Left, Venn diagram showing the overlap between the genes significantly (padj < 0.05) upregulated by bexarotene treatment and the genes significantly (padj < 0.05) downregulated in DA neurons of Nurr1-DATCreER animals. Right, Table with the 35 overlapping genes with genes encoding proteins with a function in mitochondria being highlighted. G, Scatter diagram showing the fold change of the RPKM values by bexarotene for the genes significantly downregulated in Nurr1-DATCreER animals. Bar shows the median value.
Statistical analysis.
For comparisons between experimental groups, the Student's t test or one-way ANOVA was used, and the results are presented as mean values + SEM with significance at *p < 0.05 and **p < 0.01. In Figure 4, C, E, and G, the binomial test with two tails was used to calculate the statistical significance. In Figure 4D, the p values are corrected after multiple hypothesis testing with the Benjamini–Hochberg method (Benjamini and Hochberg, 1995). In Figure 4F, the following hypergeometric R phyper script was used was used to calculate the probability of overlap between the two gene lists: phyper (q, m, n, k, lower.tail = FALSE, log.p = FALSE), where q is the size of overlap minus 1, m is the number of genes downregulated in Nurr1-DATCreER animals, n is the total number of genes with an RPKM value >1 in Nurr1-DATCreER animals minus the number of genes downregulated in Nurr1-DATCreER animals, and k is the number of genes upregulated by bexarotene.
Animal strains.
Adult female Sprague Dawley rats (Charles River Laboratories) weighing 225–250 g at the time of surgery were housed two or three per cage with ad libitum access to food and water during a 12 h light/dark cycle. Pitx3eGFP/+ heterozygous mutant mice have been described by Zhao et al. (2004). All procedures were conducted in accordance with the European Union Directive 2010/63/EU and approved by the ethical committee for the use of laboratory animals in the Lund–Malmö region.
Surgical procedures.
All surgical procedures in rats were performed under general anesthesia by intraperitoneal injection of a 20:1 fentanyl citrate and medetomidin hypochloride mixture. Three injections of 7 μg of 6-hydroxydopamine (6-OHDA) dissolved in 0.9% w/v NaCl containing 0.2 mg/ml ascorbic acid (Sigma) were injected in three different sites of the striatum [(1) AP, +1.3; ML, −3.0; DV, −5.0 and −4.5; (2) AP, +0.4; ML, −3.2; DV, −5.0 and −4.0; (3) AP, −0.4; ML, −3.8; DV, −5.0 and −4.0; flat scull position] at a rate of 0.2 μl/min. Human recombinant GDNF (1 μg in 3 μl) was injected in the striatum at the following coordinates according to Bregma, with flat scull position: AP, +0.5 mm; ML, 3 mm; DV, 4 mm below the dural surface. Vector injections were performed unilaterally on the right side above the substantia nigra at the following coordinates (flat skull position): AP, 5.3 mm behind bregma; ML, 1.7 mm; DV, 7.2 mm below dural surface. Rats received 3 μl of AAV-α-synuclein and AAV-GFP (see Fig. 5) or 3 μl of a 1:1 mixture of AAV-α-synuclein and AAV-GFP or AAV-Ret (see Fig. 8) with a Hamilton syringe fitted with a glass capillary at a rate of 0.2 μl/min, and the needle was left in place for an additional 3 min before it was slowly retracted.
Figure 5.
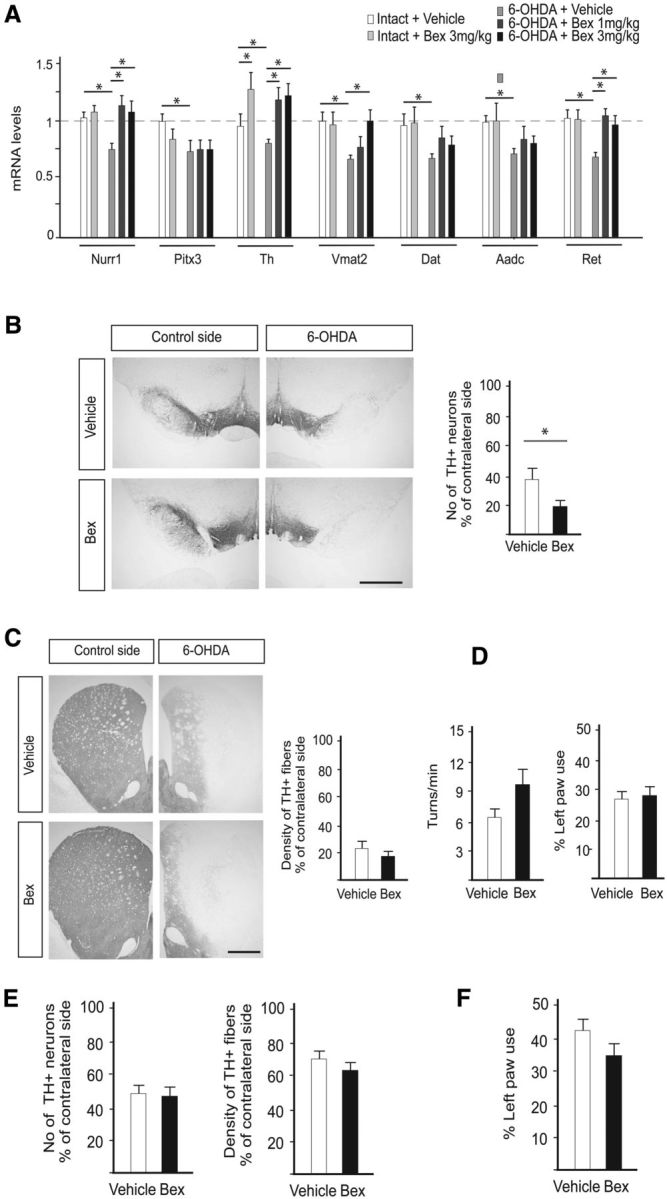
Effect of bexarotene on Nurr1-dependent gene expression, DA neuron cell loss, and motor performance in the 6-OHDA and AAV-α-synuclein rat PD models. A, qPCR analysis of Nurr1, Pitx3, and Nurr1 target genes in the ventral midbrain of rats after 6-OHDA injection and/or daily treatment for 1 week with vehicle or two doses of bexarotene (1 and 3 mg/kg) by oral gavage. Levels of mRNAs in intact rats are set at 1. n = 4. *p < 0.05. B, TH immunostaining in the substantia nigra of rats receiving 6-OHDA plus vehicle or daily 3 mg/kg bexarotene by oral gavage and stereological counts of TH+ neurons in the substantia nigra. C, TH immunostaining in the striatum of rats receiving 6-OHDA plus vehicle or daily 3 mg/kg bexarotene by oral gavage and optical densitometry of TH+ striatal fibers. n = 8 per group. D, Motor performance in the amphetamine rotation (left) and the cylinder tests (right) in rats injected unilaterally with 6-OHDA and treated with vehicle or bexarotene (3 mg/kg) by oral gavage. n = 8 per group. E, Optical densitometry of TH+ striatal fibers and stereological counts of TH+ neurons in the substantia nigra of rats injected unilaterally with AAV-α-synuclein and treated with vehicle or bexarotene (10 mg/kg) by oral gavage. n = 7–8 per group. F, Motor performance in the cylinder test in rats injected unilaterally with AAV-α-synuclein and treated with vehicle or bexarotene (10 mg/kg) by oral gavage. n = 7–8 per group. Mean values + SEM are shown for all panels. *p < 0.05. Scale bars: 1 cm.
Figure 8.

Ret protects nigral DA neurons against α-synuclein toxicity in vivo. A, Stereological counts of substantia nigra TH+ neurons (left) performed on TH-immunostained midbrain sections (right). B, Optical densitometry of the striatal TH+ innervation (left) performed on TH-immunostained striatal sections (right). C, Optical densitometry of the striatal rat α-synuclein+ innervation. Scale bars: 1 cm. *p < 0.05.
Bexarotene treatment.
Bexarotene was administered daily by oral gavage at one of three doses, 1, 3, or 10 mg/kg. In the 6-OHDA lesioned rats, the treatment was started on the day of 6-OHDA injection and continued until the day before the animal was killed. In the AAV-α-synuclein experiments, bexarotene treatment was started 1 week after vector injection and continued until the day before the animal was killed.
AAV vector production.
Production of AAV6-synapsin-α-synuclein, AAV6-synapsin-eGFP, and AAV6-CBA-Ret was performed as described previously (Decressac et al., 2012). Genome copy titers were determined using real-time qPCR and were 1.2 × 1013, 2.0 × 1013, and 1.8 × 1013 genome copies per milliliter for AAV6-synapsin-α-synuclein, AAV6-synapsin-eGFP, and AAV6-synapsin-Ret, respectively. Dilution of the batches was performed so that an equivalent number of genome copies were injected.
Tissue processing and histological procedures.
Animals were deeply anesthetized with intraperitoneal sodium pentobarbital (Apoteksbolaget) and perfused with room-temperature saline (0.9% w/v) through the ascending aorta, followed by ice-cold 4% paraformaldehyde. The brains were subsequently removed, post-fixed in the same fixative, and cryoprotected for 24–48 h in 25% sucrose at 4°C before being cut on a Leica microtome at 35 μm thickness. For immunohistochemistry, endogenous peroxidases were blocked for 30 min in 10% H2O2/10% methanol in PBS, and sections were preincubated for 1 h in blocking solution containing 10% goat or donkey serum and 0.1% Triton X-100 in PBS. The following antibodies diluted in blocking solution were applied overnight at 4°C: rabbit TH (1:1500; Millipore Bioscience Research Reagents), rabbit phospho-S6 Ser235/236 (1:300; Cell Signaling Technology), mouse α-synuclein 211 (1:3000; Santa Cruz Biotechnology), and goat anti-GDNF (1:1000; R & D Systems). After rinses with PBS, biotinylated-conjugated secondary antibodies (1:200; Jackson ImmunoResearch Laboratories) diluted in 10% goat or donkey serum in PBS were applied for 1 h at room temperature, followed by incubation with streptavidin-horseradish peroxidase complex (ABC kit, Vector Laboratories) for 1 h and subsequent exposure to diaminobenzidine (DAB kit, Vector Laboratories).
Analysis of protein expression by Western Blot.
Cells were lysed in RIPA buffer (Sigma) supplemented with phosphatase inhibitor and protease inhibitor cocktail (1:100; Sigma). For the experiment described in Figure 1D, nuclear and cytoplasmic extracts were performed using the protocol described by Dignam et al. (1983). Protein concentration was determined using the BCA Protein Assay Kit (Thermo Fisher Scientific), and equal amounts of protein (20–30 μg) were resolved on SDS-polyacrylamide gels and blotted onto nitrocellulose membranes (Invitrogen). After transfer, the membranes were washed with PBS and saturated with 10% skim milk in TBS containing 0.1% Tween 20 (TBS-T) for 1 h at room temperature and then incubated overnight at 4°C with the following primary antibodies in PBS-T containing 5% milk: rabbit anti-actin (1:5000; Sigma), rabbit anti-H3 (1:1000; Diagenode), mouse anti-tuj1 (1:1000; Covance), and rabbit anti-α-synuclein (1:1000; Millipore). For the coimmunoprecipitation experiment, the following antibodies were used: mouse anti-α-synuclein 211 (Santa Cruz Biotechnology; 1:100), rabbit anti-Nurr1 E-20 (Santa Cruz Biotechnology; 1:1000), and mouse anti-HA (Roche; 1:2000). After washing with TBS-T, membranes were incubated for 1 h at room temperature with horseradish peroxidase-coupled antibodies (1:10,000; Thermo Fisher Scientific). Labeling was performed as described in the GE Healthcare ECL Plus detection kit, and chemiluminescence was determined with an ImageQuant LAS 4000 Mini image analyzer (GE Healthcare; Fujifilm Photo Film).
Figure 1.
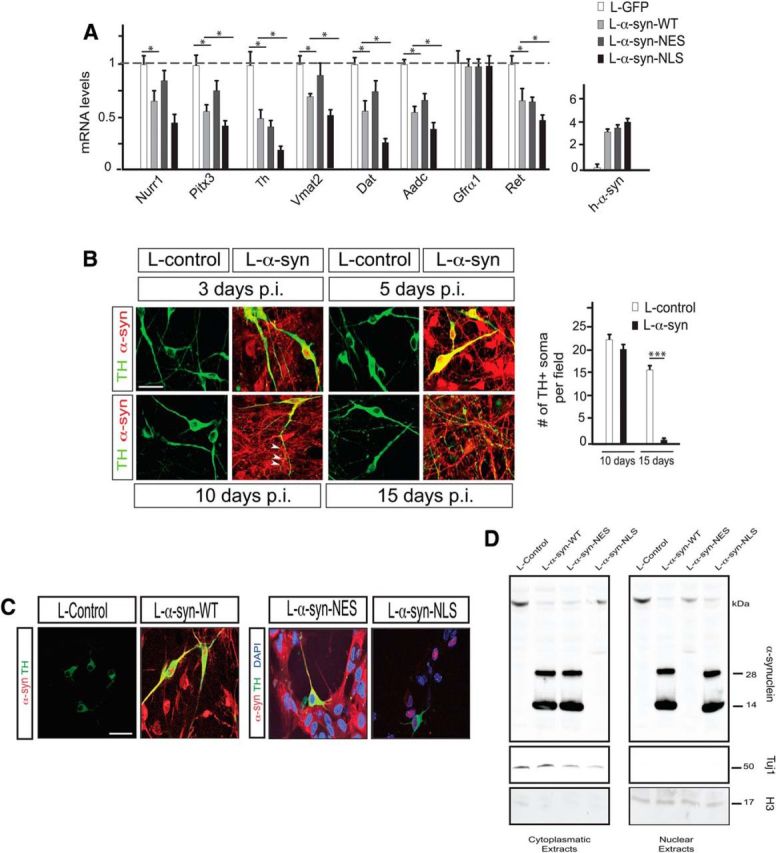
Effects of WT, cytoplasmic (NES), and nuclear (NLS) α-synuclein on DA neuron marker expression. A–D, E13.5 mouse primary ventral midbrain cultures. A, qPCR analysis of α-synuclein, Nurr1, Pitx3, and Nurr1 target genes after infection with α-synuclein/GFP lentiviruses. All data are represented as mean + SEM of the fold change normalized against Rpl19 levels; n = 3. *p < 0.05. B, Immunostaining showing α-synuclein and TH protein expression in primary cultures from dissected E13.5 ventral midbrains at the indicated time points postinfection (p.i.) with α-synuclein or control lentiviruses. The arrowheads point to fragmented dendrites. The diagram on the right shows the number of TH+ neuronal soma per counted field (mean values + SEM; n = 3). ***p < 0.001. C, Expression of TH and α-synuclein analyzed by immunohistochemistry. Cultures were infected with control or α-synuclein lentiviruses. D, Western blot analysis of α-synuclein in cytoplasmic (left) and nuclear (right) cell extracts from E13.5 mouse primary ventral midbrain cultures infected for 5 d with the indicated lentiviruses. Tuj1 is used as a cytoplasmic marker and histone H3 as a nuclear marker. Scale bars: 50 μm.
Behavioral testing.
Assessment of drug-induced rotational behavior was performed as described previously (Ungerstedt and Arbuthnott, 1970). Rats received an intraperitoneal injection of 2.5 mg/kg d-amphetamine sulfate (Apoteksbolaget), and the right and left full body turns were recorded over a period of 90 min in automated bowls coupled to the Rotameter software (AccuScan Instruments). Data are expressed as net full turns per minute, with turns ipsilateral to the injection site assigned a positive value. For the cylinder test, rats were put in a glass cylinder, a total of 20 forepaw touches were counted, and the percentage of left paw touches was determined.
Stereological cell counting and optical densitometry analysis.
The total number of TH-positive neurons in the substantia nigra was determined by unbiased stereology according to the optical fractionator principle using the MicroBrightField StereoInvestigator 10.31 software. Every sixth section covering the entire extent of the substantia nigra was included in the counting procedure. A coefficient error of <0.1 was accepted. The data are expressed as a percentage of the corresponding area from the intact side. The mean density per cell of TH/α-synuclein-positive fibers was measured by optical densitometry in three striatal sections sampled at 0.3, 0.7, and 1.6 mm rostral to bregma using the ImageJ software (NIH). The mean density of phospho-S6 staining per cell was measured in the nigral TH+ neurons. Pixel brightness was measured in a randomly selected brain hemisphere, and the measured values were corrected for nonspecific background staining by subtracting values obtained from the cortex. The data are expressed as a percentage of the corresponding area from the intact side.
Results
Nuclear localization of α-synuclein disrupts gene expression
To gain a deeper insight into the effects of α-synuclein on gene expression, we expressed human α-synuclein in primary vMB cultures obtained from E13.5 embryos derived from a heterozygous mouse strain expressing eGFP from the Pitx3 gene locus (Pitx3eGFP/+) with highly selective eGFP expression in midbrain postmitotic DA neurons (Zhao et al., 2004). Lentivirus-mediated overexpression of full-length human α-synuclein (L-α-syn-WT) recapitulated the effects on gene expression seen after AAV-mediated overexpression of α-synuclein in the substantia nigra in rats (Decressac et al., 2012). Accordingly, the same DA neuron markers were downregulated and one of the analyzed markers (Gfrα1) was unaffected 5 d after infection (Fig. 1A). At this time point, DA neurons appeared morphologically normal (Fig. 1B). At later stages, a progressive toxicity was apparent with fragmented DA neuron dendrites at day 10 and DA neuron degeneration at day 15 (Fig. 1B).
To address whether nuclear or cytoplasmic expression of α-synuclein causes disrupted gene expression, primary ventral midbrain cultures were infected with either L-α-syn-WT or with lentiviruses overexpressing genetically altered variants of α-synuclein. These variants contain either a C-terminal nuclear localization signal (in L-α-syn-NLS lentivirus) or a nuclear export signal (in L-α-syn-NES lentivirus). Immunocytochemical and Western blot analysis of primary neurons showed that the NES- and NLS-containing α-synuclein variants were expressed at similar levels and predominantly localized to the cytoplasm and the nucleus respectively, whereas wild-type α-synuclein localized in both compartments of DA neurons (Fig. 1C,D). Importantly, the nuclear form of α-synuclein had a significantly more profound effect on gene expression compared to wild-type α-synuclein, as seen by the downregulation of a panel of DA neuron markers. In contrast, the NES-coupled α-synuclein variant exhibited a much milder disruption of gene expression (Fig. 1A). These data indicate that nuclear, and to a much lesser extent, cytoplasmic, overexpression of α-synuclein results in rapid disruption of gene expression in DA neurons.
Nurr1 protects against α-synuclein-induced disruption of gene expression in primary DA neurons
We showed previously that forced expression of Nurr1 protects against α-synuclein toxicity in vivo (Decressac et al., 2012). At the level of gene expression, this effect was closely recapitulated in vitro in primary cultures as seen after cotransduction of α-synuclein and Nurr1 using lentivirus vectors (L-α-syn-WT and L-Nurr1, respectively; Fig. 2). The same panel of DA neuron markers was analyzed after transduction with L-α-syn-WT together with vectors for either L-Nurr1 or a mutated derivative of Nurr1 (L-Nurr1-DIM). Nurr1-DIM harbors a specific mutation that renders it unable to form heterodimers with RXR while retaining its ability to regulate transcription as a monomer or homodimer (Castro et al., 2001). Interestingly, L-Nurr1 and L-Nurr1-DIM vectors were equally efficient in protecting cells against α-synuclein-induced overexpression. In contrast, and as expected, a vector for a DNA binding-deficient derivative of Nurr1 (L-Nurr1-DBD) did not protect against effects of α-synuclein on gene expression (Fig. 2A). In additional experiments we noted that L-Nurr1 did not interact with α-synuclein in a coimmunoprecipitation assay, nor did it affect lentivirus-mediated or endogenous α-synuclein expression levels, subcellular localization, or oligomerization (Fig. 2B–D). In conclusion, Nurr1 does not require heterodimerization with RXR for its ability to block the effects of forced α-synuclein expression on gene expression.
Figure 2.
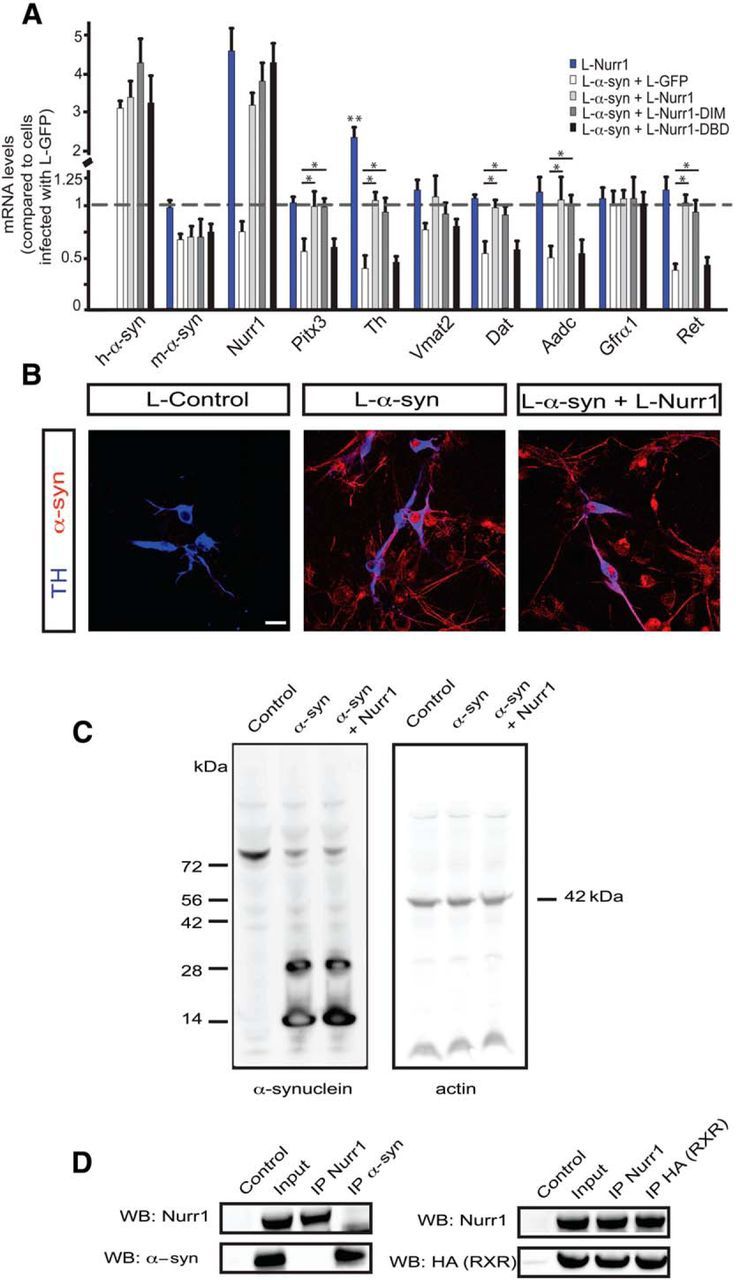
Nurr1-mediated restoration of disrupted DA neuron gene expression after α-synuclein overexpression. A, qPCR analysis of α-synuclein, Nurr1, Pitx3, and selected DA neuron markers after infection with eGFP or the indicated Nurr1 lentiviruses. All data are represented as mean + SEM of the fold change normalized against Rpl19 levels; n = 3. B, Immunostaining showing α-synuclein and TH protein expression in primary cultures from dissected E13.5 ventral midbrains 5 d after infection with control, α-synuclein, or α-synuclein and Nurr1 lentiviruses. Scale bar, 50 μm. C, Western blot analysis of α-synuclein and actin in whole cell extracts from E13.5 mouse primary ventral midbrain cultures infected for 5 d with control, α-synuclein, and α-synuclein, and Nurr1 lentiviruses. D, Coimmunoprecipitation of overexpressed Nurr1 and α-synuclein (left) or Gal4-HA-RXR (positive control; right) in HEK293T cells. *p < 0.05; **p < 0.01.
To analyze Nurr1 protection in more depth, we first analyzed the extent of α-synuclein-disrupted gene expression by global gene expression analysis using massively parallel mRNA sequencing (RNAseq). Primary vMB cultures obtained from E13.5 Pitx3eGFP/+ embryos were infected with lentiviruses for RFP as a control or L-α-syn-WT alone or together with L-Nurr1 for 5 d. To identify differentially expressed transcripts, we used DESeq2 statistical analysis based on a model using negative binomial distribution (Love et al., 2014). This test identified hundreds of genes whose expression changed significantly in cultures infected with L-α-syn-WT (Fig. 3A; p value adjusted for multiple testing (padj) < 0.05). Twenty-five genes were upregulated more than threefold, whereas 57 genes where downregulated more than threefold. Importantly, Nurr1 overexpression restored the expression of approximately half of the genes significantly upregulated or downregulated by α-synuclein, thus underscoring the potency of Nurr1 in protection against the effects of forced α-synuclein expression.
In the infected vMB cultures, DA neurons constitute only a minority of all cells. Thus, the observed dysregulation may to a large extent originate from α-synuclein-induced effects in nondopaminergic cells. To determine the effects of α-synuclein and Nurr1 on gene expression specifically in DA neurons, we therefore focused on DA neuron-specific genes identified by comparing the expression profile of eGFP-positive DA neurons with eGFP-negative cells isolated by FACS from the midbrain of E13.5 Pitx3eGFP/+ embryos. This generated a list of 284 genes that are highly specific for DA neurons and therefore referred to as the “DA signature.” All expected markers including Th, Nurr1 (Nr4a2), Pitx3, α-synuclein, Ret, Vmat2 (Slc18a2), and Dat (Slc6a3) are represented in the DA signature. α-Synuclein overexpression resulted in dysregulation of 28% (81 of 284) of these genes (padj < 0.05,) of which 58 were downregulated and 23 were upregulated (Fig. 3B,C). Interestingly, Nurr1 overexpression rescued the abnormal expression of 63.7% (37 of 58) of the downregulated and 47.8% (11 of 23) of upregulated DA signature genes (Fig. 3B). Together, at this early stage of α-synuclein toxicity (i.e., before any gross morphological abnormalities are seen; Fig. 1B), a distinct battery of DA signature genes is significantly dysregulated. Most importantly, Nurr1 is highly efficient in blocking this toxic effect on transcription and could therefore be considered a key player in protecting against α-synuclein toxicity.
Effects of RXR ligands on gene expression in primary DA neurons
As seen from the experiments above (Fig. 2), heterodimerization with RXR is not essential for the ability of Nurr1 to block α-synuclein-mediated repression of a few selected DA neuron markers in primary culture experiments. However, since we tested the requirement of RXR on only a limited set of selected markers, the experiment did not exclude the possibility that other Nurr1 target genes are normally regulated by Nurr1-RXR heterodimers. Importantly, it also remains possible that RXR, although not being an obligatory requirement for Nurr1's transcriptional activity, may still form functional heterodimers with Nurr1 and that RXR ligands may potentiate the protective effects of Nurr1. To address this possibility, we used bexarotene, a synthetic RXR ligand currently in clinical use to treat cutaneous T-cell lymphoma (Duvic et al., 2001). Bexarotene can cross the blood–brain barrier (Cramer et al., 2012) and was shown previously to efficiently and selectively activate Nurr1-RXR heterodimers and protect DA neurons from neurotoxins in vitro and in the in vivo 6-OHDA rat model of PD (McFarland et al., 2013). Primary vMB cultures obtained from E13.5 Pitx3eGFP/+ embryos were treated with 100 nm bexarotene for 24 h and were then sorted by FACS to isolate the eGFP-positive DA neurons. Isolated mRNA was subjected to RNAseq to analyze the effects on gene expression. After RNAseq, the Spearman correlation coefficient between biological replicates averaged 0.98, indicating high correlation between replicates, which clustered according to treatment in a cluster dendrogram (Fig. 4A). DESeq statistical analysis detected 776 genes significantly upregulated in DA neurons treated with bexarotene, whereas 993 genes were significantly downregulated (Fig. 4B; padj < 0.05).
If RXR is important as a Nurr1 heterodimer partner in DA neurons, Nurr1-regulated genes should be affected by bexarotene. To address this question, vMB cultures were infected with a lentivirus vector harboring a short hairpin (sh) RNA against Nurr1 (L-shNurr1). After RNAseq of eGFP cells isolated by FACS, 74.1% of the DA neuron signature genes were expressed at a lower level compared to control cultures. This is a highly significant trend as determined by negative binomial test (p = 5.08e-13) and is consistent with the significant role for Nurr1 in the regulation of the DA phenotype (Fig. 4C). In contrast, bexarotene treatment did not show a significant trend in upregulation of the DA neuron signature genes (47%; p = 0.393; Fig. 4C). Thus, these data indicate that Nurr1 does not function as a heterodimer with RXR in the regulation of the DA neuron phenotype, as defined by the expression of the DA signature.
However, additional findings indicated that RXR has potential for coregulating other categories of Nurr1 target genes. Thus, the most significant gene ontology term that was found to be upregulated by bexarotene corresponds to genes involved in oxidative phosphorylation (Fig. 4D), a category of genes also regulated by Nurr1 (Kadkhodaei et al., 2013). After infection with L-shNurr1, 90.4% of genes involved in oxidative phosphorylation were lower than in controls, which confirmed our previous results in vivo and is a highly significant trend as determined by negative binomial test (p = 2.38e-11). A similar and highly significant opposite trend (p = 2.2e-16) was seen after bexarotene treatment showing that 93.7% of genes involved in oxidative phosphorylation are expressed at a higher level compared to controls (Fig. 4E). Thus, these observations indicate that Nurr1 and RXR may function together in regulation of oxidative phosphorylation in DA neurons.
Finally, we wished to analyze genes that were significantly (padj < 0.05) upregulated by bexarotene and see to what extent they overlap with genes downregulated in adult Nurr1-ablated DA neurons (Kadkhodaei et al., 2013). Thirty-five of the 168 genes downregulated in Nurr1-ablated DA neurons were significantly upregulated by bexarotene (Fig. 4F). Of note, the majority of Nurr1-regulated genes (76.9%) were expressed at a higher value compared to control cultures. By negative binomial test, this is again highly significant (p = 4.17e-09) and indicates a potential for RXR coregulation also of this category of adult Nurr1-regulated genes (Fig. 4G). Together, the data from global RNAseq analysis of bexarotene-regulated genes in vMB DA neurons in vitro indicate a dynamic regulatory relationship between Nurr1 and RXR. Accordingly, although we did not find evidence for coregulation of the DA neuron signature genes, a strongly significant trend indicated that RXR ligands could exert a positive regulation on a category of Nurr1-regulated genes involved in oxidative phosphorylation and on the battery of genes that are downregulated as a result of Nurr1 ablation in adult DA neurons.
Effect of bexarotene on DA neuron cell loss and motor performance in the 6-OHDA and AAV-α-synuclein rat PD models
To test whether bexarotene can induce DA neuron-specific gene expression in vivo, rats were treated with bexarotene (3 mg/kg) by daily oral gavage for 1 week. Animals were killed 6 h after the last dose and the levels of Nurr1, Th, Vmat2, Dat, Aadc, and Ret were analyzed by qPCR on dissected vMB tissue. As seen in Figure 5A, bexarotene treatment did not affect the expression of Nurr1 mRNA per se, and only showed a weak (25%) but significant upregulation of Th expression, whereas other DA neuron markers remained unchanged. Next, we analyzed the effects of bexarotene (at 1 or 3 mg/kg) on Nurr1 and Nurr1 target genes in the 6-OHDA-lesioned rat model of PD. 6-OHDA was injected in the striatum, which resulted in an expected downregulation of the mRNA levels of Nurr1 and its target genes, as well as of the key DA neuron transcription factor Pitx3 (Fig. 5A). These effects were observed 1 week after 6-OHDA injection, before any apparent DA neuron loss. Bexarotene restored the expression of Nurr1 and three DA neuron markers (Th, Vmat2, Ret) as reported previously (McFarland et al., 2013), but neither Dat, Aadc, nor Pitx3 was significantly changed.
The potential of bexarotene to protect DA neurons in the 6-OHDA model of PD was further tested by administering vehicle or bexarotene daily with oral gavage at a dose of 3 mg/kg, starting on the day of 6-OHDA treatment, to rats that had also received unilateral injections of 6-OHDA (7 μg at three different sites, 21 μg total). During the fourth postoperative week, motor performance of the animals was assayed, and at the end of the fourth week, the animals were killed and the brains were processed for immunohistochemical staining. Stereological counting and optical densitometry respectively revealed that the 6-OHDA lesion induced a cell loss of ∼60% in the SN (Fig. 5B), accompanied by an average 75% loss of TH-positive innervation in the striatum (Fig. 5C). Bexarotene treatment had no protective effect on the density of TH-positive fibers in the striatum (Fig. 5C) and a negative impact on the number of TH-positive cells in the SN (Fig. 5B). Consistent with the lack of protection by bexarotene in terms of DA cell loss and striatum innervation, the motor impairment caused by 6-OHDA, as assayed by the amphetamine rotation test and the paw use in the cylinder test, was not significantly affected by bexarotene (Fig. 5D).
We also assessed the potential protective effect of bexarotene in the AAV-α-synuclein rat PD model that more clearly mimics the early stages and slow development of human PD. In a long-term experiment to analyze neuroprotection, rats were unilaterally injected with AAV-α-synuclein into the SN and treated with vehicle or 10 mg/kg bexarotene daily for 8 weeks, starting 1 week after vector injection. Paw use in the cylinder test was performed during the eighth week. The animals were killed 1 d after the last dose, and the brains were processed for TH immunohistochemistry. Stereology revealed that α-synuclein overexpression resulted in ∼50% loss of TH-positive cells in the SN on the AAV-α-synuclein-injected side, which was similar in magnitude in both treatment groups (Fig. 5E). In line with this, optical densitometry of striatal TH-positive fiber density did not show any difference between vehicle- and bexarotene-treated animals (Fig. 5E). The magnitude of cell loss in these animals was not sufficient to produce any significant impairment in the cylinder test (Fig. 5F). Thus, despite an early trend for a protective effect on gene expression in the 6-OHDA-lesioned rat model, no clear evidence for long-term neuroprotection could be seen in the 6-OHDA or the AAV-α-synuclein-lesioned rat PD models.
Potentiation of the neuroprotective effect of GDNF by bexarotene
Although bexarotene did not exert a neuroprotective effect in vivo, expression of Ret was robustly restored in the 6-OHDA-lesioned PD model (Fig. 5A). Since GDNF signaling is disrupted (via downregulation of the Nurr1-Ret axis) and unable to confer neuroprotection from α-synuclein overexpression (Decressac et al., 2012), we hypothesized that bexarotene could counteract disrupted GDNF signaling. We first assessed the combined effects of bexarotene and GDNF on gene expression in our in vitro α-synuclein toxicity model. Primary vMB cultures were transduced with L-eGFP or L-α-syn-WT lentivirus vectors as previously (Fig. 1), 48 h after seeding. Virus was removed 48 h after infection, and at that point cells were treated with bexarotene or vehicle for 3 additional days before analysis. Bexarotene did not affect DA neuron markers, including Nurr1, Pitx3, and Th, but robustly restored the expression level of Ret (Fig. 6A). Interestingly, consistent with its effect on Ret, when GDNF was combined with bexarotene, a much more efficient restoration of gene expression was observed compared with cultures exposed to GDNF alone (Fig. 6A). Moreover, GDNF-induced phosphorylation of the S6 kinase was blocked by α-synuclein in vitro, as seen previously in vivo (Decressac et al., 2012), but restored by the combined treatment of both GDNF and bexarotene (Fig. 6B). Thus, in in vitro vMB cultures, bexarotene appears to have a robust ability to counteract α-synuclein-induced blockage of GDNF signaling.
Figure 6.

A, B, Potentiation of the neuroprotective effect of GDNF by bexarotene. Primary ventral midbrain cultures of E13.5 mouse embryos. A, qPCR analysis of Nurr1, Pitx3, and Nurr1 target genes after infection with α-synuclein lentivirus and treatment with DMSO, bexarotene (100 nm), GDNF (30 ng/ml), or GDNF together with bexarotene. Levels of mRNAs in cultures infected with eGFP lentivirus are set at 1. Mean values + SEM are shown; n = 3. B, Expression of TH and phospho-S6 analyzed by immunohistochemistry. Cultures were infected with control or α-synuclein lentivirus and treated with DMSO, bexarotene (100 nm), GDNF (30 ng/ml), or GDNF together with bexarotene. The diagram on the right shows the percentage of TH+ neurons expressing phospho-S6. Mean values + SEM are shown; n = 3. C, Immunohistochemistry showing the spread of GDNF after striatal injection of the recombinant peptide and the expression of human α-synuclein in the striatum resulting from nigral delivery of the AAV-α-synuclein vector. D, Immunohistochemistry showing the expression of phospho-S6 in rats receiving AAV-α-synuclein and GDNF plus daily bexarotene or vehicle. The diagram shows the optical density of phospho-S6 staining (mean density per cell) in the nigral TH+ neurons on the treated side, expressed as a percentage of the intensity measured on the noninjected side (n = 5 per group). Scale bars: B, 50 μm; C, D, 1 cm. *p < 0.05; **p < 0.01.
The results in vitro prompted us to test whether bexarotene can restore GDNF signaling in vivo in the rat AAV-α-synuclein model. To address this question, 10 animals were injected with AAV-α-synuclein unilaterally in the SN (Fig. 6C) and divided in two groups of five animals that received either vehicle or 10 mg/kg bexarotene daily by oral gavage from day 2 to day 12 after vector injection. On day 10, all rats received an injection of 1 μg GDNF protein into the striatum (Fig. 6C), and the animals were killed on day 12, i.e., 2 d after GDNF injection. Consistent with our previous observations in vivo (Decressac et al., 2012) showing that the response to GDNF is blocked in DA neurons expressing increased levels of α-synuclein, no detectable response to GDNF measured by the levels of phospho-S6 was detected in the nigral TH-positive neurons in the vehicle-treated group (Fig. 6D). The results indicate a trend to increased expression phospho-S6 in response to GDNF in the bexarotene-treated group (Fig. 6D), but this did not reach statistical significance (p = 0.13).
Ret protects from α-synuclein-induced toxicity in vitro and in vivo
As explained above, previous data suggest that Nurr1 regulates Ret in DA neurons and that α-synuclein toxicity may interfere with GDNF signaling via Nurr1 and Ret downregulation (Decressac et al., 2012). If Ret is a key downstream target gene of Nurr1 in α-synuclein-induced toxicity, forced overexpression of Ret in vMB cultures should be sufficient to rescue α-synuclein-induced changes on Nurr1-regulated genes. We wished to further investigate this hypothesis with additional in vitro and in vivo experiments. Indeed, lentivirus-mediated Ret expression efficiently rescued the gene expression of tested DA neuron markers in in vitro cultured vMB neurons infected with α-synuclein (Fig. 7A). Moreover, when cultures were treated with an inhibitor (RPI-1) of Ret kinase activity, the protective effect of Nurr1 against α-synuclein was attenuated (Fig. 7B), suggesting that Ret is critically important in Nurr1-mediated defense against α-synuclein toxicity. Reciprocally, forced expression of Ret could not fully restore DA markers gene expression in α-synuclein-expressing cultures that had been infected with L-shNurr1 (Fig. 7C), indicating that Nurr1 may also be required for the ability of Ret to confer strong neuroprotection.
Figure 7.

Ret restores DA marker expression after α-synuclein overexpression in vitro. A–C, qPCR analysis of α-synuclein, Nurr1, Pitx3, and Nurr1 target genes after infection of primary ventral midbrain cultures of E13.5 mouse embryos with the indicated lentiviruses, in the presence or absence of the Ret kinase inhibitor RPI-1 (B). Levels of mRNAs in cultures infected with eGFP lentivirus are set at 1. All data is represented as mean + SEM of the fold change normalized against Rpl19 levels; n = 3. *p < 0.05.
Next, we asked whether forced AAV vector-mediated Ret expression could prevent DA cell loss caused by α-synuclein toxicity in vivo. As above, rats received intranigral injection of AAV-α-synuclein together with AAV-Ret or AAV-GFP and were examined 8 weeks later. Stereological counting showed that AAV-mediated Ret expression provided robust protection of TH-positive nigral neurons compared to the GFP control group (Fig. 8A), while densitometry analysis revealed a significant protection of the striatal TH-positive (B) and α-synuclein-positive (C) innervation on the injected side. In conclusion, therefore, forced expression experiments both in vitro and in vivo underscore the central role of Ret in a disrupted pathway resulting from α-synuclein expression in DA neurons.
The results from these comprehensive experiments are summarized in Figure 9. Our data suggest that α-synuclein exerts toxicity, at least in part, by interfering with gene expression in the nucleus. One of the affected genes is Nurr1, which in turn affects genes controlling DA neurotransmission, survival via regulation of Ret, and mitochondrial gene expression [oxidative phosphorylation (OXPHOS)]. Bexarotene can upregulate Ret and Ret-mediated signaling and OXPHOS gene expression. Our data are consistent with the possibility that this upregulation is mediated by Nurr1-RXR heterodimers.
Figure 9.

Protection from nuclear α-synuclein-induced toxicity by Nurr1/Ret overexpression and/or bexarotene treatment. Nuclear α-synuclein interferes with gene expression by targeting Nurr1 and other genes (A,B,C). Nurr1 affects the expression of genes involved in DA neurotransmission, in survival via the GDNF-Ret pathway and in OXPHOS. Nurr1 and Ret overexpression protect from the toxic effects of α-synuclein, whereas bexarotene restores Ret expression in α-synuclein overexpressing neurons and upregulates OXPHOS.
Discussion
The presynaptic role of α-synuclein has been investigated in previous studies, and intense attention has been focused on how high levels of α-synuclein result in PD pathology. Although it is clear that the protein is present both in nerve terminals and the cytoplasm, it is also localized in the cell nucleus where it accumulates under pathological conditions (Goers et al., 2003; Monti et al., 2010). Thus, it is of interest to better understand possible detrimental effects resulting from nuclear α-synuclein accumulation in affected nerve cells. In the present study, we studied the influence of α-synuclein on gene expression in DA neurons and how the nuclear receptor Nurr1 may be exploited as a drug target. We provide evidence that the nuclear localization of α-synuclein results in stronger toxicity in terms of disrupted gene expression compared with a cytoplasmic α-synuclein derivative. These results are in line with findings showing increased degeneration by nuclear targeting of α-synuclein in DA neurons of Drosophila (Kontopoulos et al., 2006).
We used RNAseq to interrogate the changes in gene expression that occur as a result of forced α-synuclein expression in primary vMB DA neurons. It should be noted that gene expression was analyzed at an early time point after lentiviral transduction, before any obvious morphological signs of toxicity were apparent. Importantly, we did not observe a general inhibition of transcription, but rather defined changes with subsets of genes being affected. Gene ontology analysis revealed that α-synuclein could potently downregulate the expression of genes involved in key processes including synaptic transmission and cholesterol biosynthesis, and upregulate genes involved in the cellular response to stress. Axonal and synaptic defects have been seen upon α-synuclein overexpression, and RNAseq data indicate that at least some of these effects could be attributed to changes in gene expression (Lee et al., 2006; Chung et al., 2009; Siebert et al., 2010; Chu et al., 2012; Anichtchik et al., 2013; Games et al., 2013; Prots et al., 2013; O'Donnell et al., 2014). Downregulation of cholesterol biosynthesis genes by α-synuclein is in line with findings that deletion of α-synuclein in mice significantly increases cholesterol in the brain, while abnormal levels of cholesterol metabolites were found in degenerating DA neurons with increased α-synuclein levels (Halliday et al., 2005; Bosco et al., 2006; Barceló-Coblijn et al., 2007; Bar-On et al., 2008). Thus, several previously recognized pathological effects in PD might at least in part be explained by disruption of gene expression programs.
An interesting question that has not been addressed here concerns the mechanisms whereby α-synuclein influences gene expression in neurons. Our finding that a nuclear derivative of α-synuclein was more potent argues in favor of mechanisms influencing gene expression directly in the nucleus and is also consistent with previous observations using α-synuclein derivatives localizing preferentially to the nucleus in Drosophila (Kontopoulos et al., 2006). Previous studies have indicated several possible ways by which α-synuclein may affect gene expression. For example, α-synuclein has been shown to interact with transcription factors, ribonucleoproteins, splicing factors, histones, and DNA (Goers et al., 2003; Zhou et al., 2004; Duce et al., 2006; Kontopoulos et al., 2006; Hegde and Rao, 2007; Jin et al., 2007). In addition, histone acetylation seems to be inhibited by α-synuclein (Kontopoulos et al., 2006; Jin et al., 2011; Decressac et al., 2012), observations that are consistent with data showing that histone deacetylase inhibitors can protect DA neurons from α-synuclein-induced toxicity in mice (Ono et al., 2009; Zhou et al., 2011). Several of these findings are intriguing but require independent verification, and it is evident that further focus on how α-synuclein influences gene expression is essential.
Importantly, Nurr1 expression in cells subjected to α-synuclein toxicity efficiently rescued expression of hundreds (over 50%) of dysregulated genes and also influenced many genes that are not considered classical dopaminergic markers. Thus, the analysis presented here underscores that Nurr1-regulated genes are severely influenced by α-synuclein toxicity (Decressac et al., 2012; Lin et al., 2012). It seems likely that this effect, at least in part, is due to downregulation of Nurr1 by α-synuclein. An important goal in our studies was then to investigate whether, and to what extent, RXR functions as a heterodimer partner in Nurr1-dependent DA neuron gene regulation. It is important to note that Nurr1 has the capacity to bind DNA without RXR, and it has remained uncertain whether Nurr1 even requires RXR in DA neurons. Expression of the mutated Nurr1-DIM derivative indicated that the capacity of Nurr1 to restore gene expression resulting from α-synuclein overexpression does not require heterodimerization with RXR. Bexarotene-induced changes in gene expression, as revealed by RNAseq, uncovered a somewhat more dynamic regulation, indicating that some but not all Nurr1 target genes are influenced by RXR ligands. The total number of genes regulated by bexarotene was very large, but since RXR forms heterodimers with several other nuclear receptors, this in itself is not surprising (Gilardi and Desvergne, 2014). We found that forced expression of Nurr1-shRNA in primary vMB cultures significantly downregulated DA signature genes, but this category of genes were not affected by RXR ligands, arguing against coregulation of typical DA neuron markers by Nurr1-RXR heterodimers. On the other hand, bexarotene upregulated nuclear-encoded mitochondrial genes involved in oxidative phosphorylation. This cohort of genes were also significantly downregulated by Nurr1-shRNA in primary vMB neurons and support previous findings obtained from conditional knock-out mice showing that these genes are regulated by Nurr1 (Kadkhodaei et al., 2013). Although other RXR heterodimer partners including peroxisome proliferator activated receptors may also be involved in regulating mitochondrial gene expression, the results here are consistent with a potential for coregulation of genes important for oxidative phosphorylation (Ravnskjaer et al., 2010; Corona et al., 2014). Finally, the cohort of genes found previously to be downregulated in mature DA neurons of conditional Nurr1 knock-out mice were significantly upregulated by bexarotene (Kadkhodaei et al., 2013). Together, these observations indicate that RXR is not essential for all Nurr1 in vivo functions; however, the upregulation of genes involved in oxidative phosphorylation and some of the other Nurr1-regulated target genes seen following bexarotene treatment suggest a potential for coregulation and pharmacological significance in the context of PD. Notably, mitochondrial dysfunction is highly implicated in disease, and mitochondrial gene expression seems to be affected in PD patients (for review, see Exner et al., 2012).
Despite the results showing a potential for coregulation, our results using bexarotene in vivo provided mixed and somewhat disappointing results. Thus, in contrast to the observations reported previously (McFarland et al., 2013), bexarotene treatment had no effect neither on DA neuron cell loss nor motor impairment in rats receiving unilateral injections of 6-OHDA in the striatum. However, bexarotene treatment resulted in upregulation of mRNA expression of Nurr1, TH, and Ret. We found that the upregulation of Ret was intriguing considering that it is a Nurr1 target gene that is critical for GDNF signaling and disrupted by α-synuclein toxicity. Moreover, we showed here that expression of Ret using an AAV-Ret vector in the rat α-synuclein overexpression model is able to induce efficient neuroprotection, a result that further emphasizes its critical role as an essential downstream target of Nurr1 in protection against α-synuclein-induced toxicity. Along the same line, the constitutive active Ret mutant expressed in MEN2B (Multiple Endocrine Neoplasia type 2B) mice protects DA cell bodies from 6-OHDA-induced toxicity (Mijatovic et al., 2011), while the same mutated Ret was previously shown to rescue disintegration of mitochondria and ATP content and improve electron transport chain complex I function in Pink1 Drosophila mutants (Klein et al., 2014). Upregulation of Ret by bexarotene prompted us to further examine whether bexarotene could rescue the disrupted GDNF signaling following α-synuclein overexpression. Interestingly, bexarotene showed a robust effect on GDNF signaling in in vitro cultured vMB neurons, significantly increasing phosphorylation of S6 kinase and augmenting the ability of GDNF to rescue α-synuclein-disrupted gene expression. Again, these promising effects were not recapitulated in vivo, as bexarotene alone or in combination with GDNF did not result in neuroprotection in the rat α-synuclein overexpression model.
Together, the data presented here indicate a somewhat mixed picture concerning bexarotene as a strategy for pharmacological targeting of Nurr1. Our results support the hypothesis that α-synuclein influences the process of gene expression in the nucleus and extend the conclusion that Nurr1 via its downstream target Ret can interfere with α-synuclein toxicity. Moreover, in vitro analysis demonstrated that bexarotene could affect some but not all Nurr1 target genes. Notably, bexarotene seems to have a capacity for rescuing disrupted Ret expression and GDNF signaling as seen in vMB cultures. However, the negative results in vivo indicate that the neuroprotective effect of bexarotene is not robust or easily reproducible. Despite these negative data, the results from our in vitro experiments indicate that RXR ligands are potentially highly interesting in neuroprotection against PD-induced pathology. We thus envision that future research should be focused on the identification of more selective compounds with appropriate pharmacological properties for being effective in neuroprotection in vivo.
Footnotes
This work was supported by grants from the Swedish Research Council (A.B., T.P.), the Swedish Strategic Research Foundation (A.B., T.P.), the Michael J. Fox Foundation (A.B., T.P.) and the European Union, Seventh Framework Programme under grant agreement mdDANeurodev. We thank Jenny Johansson and Ulla Jarl for technical assistance and members of the Perlmann lab for discussions. We thank Dr. Meng Li and for providing the Pitx3eGFP/+ mouse line.
The authors declare no competing financial interests.
References
- Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anichtchik O, Calo L, Spillantini MG. Synaptic dysfunction in synucleinopathies. CNS Neurol Disord Drug Targets. 2013;12:1094–1100. [PubMed] [Google Scholar]
- Barceló-Coblijn G, Golovko MY, Weinhofer I, Berger J, Murphy EJ. Brain neutral lipids mass is increased in alpha-synuclein gene-ablated mice. J Neurochem. 2007;101:132–141. doi: 10.1111/j.1471-4159.2006.04348.x. [DOI] [PubMed] [Google Scholar]
- Bar-On P, Crews L, Koob AO, Mizuno H, Adame A, Spencer B, Masliah E. Statins reduce neuronal alpha-synuclein aggregation in in vitro models of Parkinson's disease. J Neurochem. 2008;105:1656–1667. doi: 10.1111/j.1471-4159.2008.05254.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Statist Soc B. 1995;57:289–300. [Google Scholar]
- Bosco DA, Fowler DM, Zhang Q, Nieva J, Powers ET, Wentworth P, Jr, Lerner RA, Kelly JW. Elevated levels of oxidized cholesterol metabolites in Lewy body disease brains accelerate alpha-synuclein fibrilization. Nat Chem Biol. 2006;2:249–253. doi: 10.1038/nchembio782. [DOI] [PubMed] [Google Scholar]
- Castillo SO, Baffi JS, Palkovits M, Goldstein DS, Kopin IJ, Witta J, Magnuson MA, Nikodem VM. Dopamine biosynthesis is selectively abolished in substantia nigra/ventral tegmental area but not in hypothalamic neurons in mice with targeted disruption of the Nurr1 gene. Mol Cell Neurosci. 1998;11:36–46. doi: 10.1006/mcne.1998.0673. [DOI] [PubMed] [Google Scholar]
- Castro DS, Hermanson E, Joseph B, Wallén A, Aarnisalo P, Heller A, Perlmann T. Induction of cell cycle arrest and morphological differentiation by Nurr1 and retinoids in dopamine MN9D cells. J Biol Chem. 2001;276:43277–43284. doi: 10.1074/jbc.M107013200. [DOI] [PubMed] [Google Scholar]
- Chu Y, Kompoliti K, Cochran EJ, Mufson EJ, Kordower JH. Age-related decreases in Nurr1 immunoreactivity in the human substantia nigra. J Comp Neurol. 2002;450:203–214. doi: 10.1002/cne.10261. [DOI] [PubMed] [Google Scholar]
- Chu Y, Morfini GA, Langhamer LB, He Y, Brady ST, Kordower JH. Alterations in axonal transport motor proteins in sporadic and experimental Parkinson's disease. Brain. 2012;135:2058–2073. doi: 10.1093/brain/aws133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung CY, Koprich JB, Hallett PJ, Isacson O. Functional enhancement and protection of dopaminergic terminals by RAB3B overexpression. Proc Natl Acad Sci U S A. 2009;106:22474–22479. doi: 10.1073/pnas.0912193106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corona JC, de Souza SC, Duchen MR. PPARgamma activation rescues mitochondrial function from inhibition of complex I and loss of PINK1. Exp Neurol. 2014;253:16–27. doi: 10.1016/j.expneurol.2013.12.012. [DOI] [PubMed] [Google Scholar]
- Cramer PE, Cirrito JR, Wesson DW, Lee CY, Karlo JC, Zinn AE, Casali BT, Restivo JL, Goebel WD, James MJ, Brunden KR, Wilson DA, Landreth GE. ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models. Science. 2012;335:1503–1506. doi: 10.1126/science.1217697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decressac M, Kadkhodaei B, Mattsson B, Laguna A, Perlmann T, Björklund A. alpha-Synuclein-induced down-regulation of Nurr1 disrupts GDNF signaling in nigral dopamine neurons. Sci Transl Med. 2012;4:163ra156. doi: 10.1126/scitranslmed.3004676. [DOI] [PubMed] [Google Scholar]
- Decressac M, Volakakis N, Björklund A, Perlmann T. NURR1 in Parkinson disease–from pathogenesis to therapeutic potential. Nat Rev Neurol. 2013a;9:629–636. doi: 10.1038/nrneurol.2013.209. [DOI] [PubMed] [Google Scholar]
- Decressac M, Mattsson B, Weikop P, Lundblad M, Jakobsson J, Björklund A. TFEB-mediated autophagy rescues midbrain dopamine neurons from alpha-synuclein toxicity. Proc Natl Acad Sci U S A. 2013b;110:E1817–E1826. doi: 10.1073/pnas.1305623110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dignam JD, Lebovitz RM, Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duce JA, Smith DP, Blake RE, Crouch PJ, Li QX, Masters CL, Trounce IA. Linker histone H1 binds to disease associated amyloid-like fibrils. J Mol Biol. 2006;361:493–505. doi: 10.1016/j.jmb.2006.06.038. [DOI] [PubMed] [Google Scholar]
- Duvic M, Hymes K, Heald P, Breneman D, Martin AG, Myskowski P, Crowley C, Yocum RC. Bexarotene is effective and safe for treatment of refractory advanced-stage cutaneous T-cell lymphoma: multinational phase II-III trial results. J Clin Oncol. 2001;19:2456–2471. doi: 10.1200/JCO.2001.19.9.2456. [DOI] [PubMed] [Google Scholar]
- Exner N, Lutz AK, Haass C, Winklhofer KF. Mitochondrial dysfunction in Parkinson's disease: molecular mechanisms and pathophysiological consequences. EMBO J. 2012;31:3038–3062. doi: 10.1038/emboj.2012.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman BM, Umesono K, Chen J, Evans RM. Unique response pathways are established by allosteric interactions among nuclear hormone receptors. Cell. 1995;81:541–550. doi: 10.1016/0092-8674(95)90075-6. [DOI] [PubMed] [Google Scholar]
- Games D, Seubert P, Rockenstein E, Patrick C, Trejo M, Ubhi K, Ettle B, Ghassemiam M, Barbour R, Schenk D, Nuber S, Masliah E. Axonopathy in an alpha-synuclein transgenic model of Lewy body disease is associated with extensive accumulation of C-terminal-truncated alpha-synuclein. Am J Pathol. 2013;182:940–953. doi: 10.1016/j.ajpath.2012.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilardi F, Desvergne B. RXRs: collegial partners. Subcell Biochem. 2014;70:75–102. doi: 10.1007/978-94-017-9050-5_5. [DOI] [PubMed] [Google Scholar]
- Goers J, Manning-Bog AB, McCormack AL, Millett IS, Doniach S, Di Monte DA, Uversky VN, Fink AL. Nuclear localization of alpha-synuclein and its interaction with histones. Biochemistry. 2003;42:8465–8471. doi: 10.1021/bi0341152. [DOI] [PubMed] [Google Scholar]
- Halliday GM, Ophof A, Broe M, Jensen PH, Kettle E, Fedorow H, Cartwright MI, Griffiths FM, Shepherd CE, Double KL. Alpha-synuclein redistributes to neuromelanin lipid in the substantia nigra early in Parkinson's disease. Brain. 2005;128:2654–2664. doi: 10.1093/brain/awh584. [DOI] [PubMed] [Google Scholar]
- Hegde ML, Rao KS. DNA induces folding in alpha-synuclein: understanding the mechanism using chaperone property of osmolytes. Arch Biochem Biophys. 2007;464:57–69. doi: 10.1016/j.abb.2007.03.042. [DOI] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Jin H, Kanthasamy A, Ghosh A, Yang Y, Anantharam V, Kanthasamy AG. alpha-Synuclein negatively regulates protein kinase Cdelta expression to suppress apoptosis in dopaminergic neurons by reducing p300 histone acetyltransferase activity. J Neurosci. 2011;31:2035–2051. doi: 10.1523/JNEUROSCI.5634-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Li GJ, Davis J, Zhu D, Wang Y, Pan C, Zhang J. Identification of novel proteins associated with both alpha-synuclein and DJ-1. Mol Cell Proteomics. 2007;6:845–859. doi: 10.1074/mcp.M600182-MCP200. [DOI] [PubMed] [Google Scholar]
- Kadkhodaei B, Ito T, Joodmardi E, Mattsson B, Rouillard C, Carta M, Muramatsu S, Sumi-Ichinose C, Nomura T, Metzger D, Chambon P, Lindqvist E, Larsson NG, Olson L, Björklund A, Ichinose H, Perlmann T. Nurr1 is required for maintenance of maturing and adult midbrain dopamine neurons. J Neurosci. 2009;29:15923–15932. doi: 10.1523/JNEUROSCI.3910-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadkhodaei B, Alvarsson A, Schintu N, Ramsköld D, Volakakis N, Joodmardi E, Yoshitake T, Kehr J, Decressac M, Björklund A, Sandberg R, Svenningsson P, Perlmann T. Transcription factor Nurr1 maintains fiber integrity and nuclear encoded mitochondrial gene expression in dopamine neurons. Proc Natl Acad Sci U S A. 2013;110:2360–2365. doi: 10.1073/pnas.1221077110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein P, Müller-Rischart AK, Motori E, Schönbauer C, Schnorrer F, Winklhofer KF, Klein R. Ret rescues mitochondrial morphology and muscle degeneration of Drosophila Pink1 mutants. EMBO J. 2014;33:341–355. doi: 10.1002/embj.201284290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontopoulos E, Parvin JD, Feany MB. Alpha-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum Mol Genet. 2006;15:3012–3023. doi: 10.1093/hmg/ddl243. [DOI] [PubMed] [Google Scholar]
- Lee HJ, Khoshaghideh F, Lee S, Lee SJ. Impairment of microtubule-dependent trafficking by overexpression of alpha-synuclein. Eur J Neurosci. 2006;24:3153–3162. doi: 10.1111/j.1460-9568.2006.05210.x. [DOI] [PubMed] [Google Scholar]
- Lees AJ, Hardy J, Revesz T. Parkinson's disease. Lancet. 2009;373:2055–2066. doi: 10.1016/S0140-6736(09)60492-X. [DOI] [PubMed] [Google Scholar]
- Lin X, Parisiadou L, Sgobio C, Liu G, Yu J, Sun L, Shim H, Gu XL, Luo J, Long CX, Ding J, Mateo Y, Sullivan PH, Wu LG, Goldstein DS, Lovinger D, Cai H. Conditional expression of Parkinson's disease-related mutant alpha-synuclein in the midbrain dopaminergic neurons causes progressive neurodegeneration and degradation of transcription factor nuclear receptor related 1. J Neurosci. 2012;32:9248–9264. doi: 10.1523/JNEUROSCI.1731-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroteaux L, Campanelli JT, Scheller RH. Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J Neurosci. 1988;8:2804–2815. doi: 10.1523/JNEUROSCI.08-08-02804.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland K, Spalding TA, Hubbard D, Ma JN, Olsson R, Burstein ES. Low dose bexarotene treatment rescues dopamine neurons and restores behavioral function in models of Parkinson's disease. ACS Chem Neurosci. 2013;4:1430–1438. doi: 10.1021/cn400100f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mijatovic J, Piltonen M, Alberton P, Männistö PT, Saarma M, Piepponen TP. Constitutive Ret signaling is protective for dopaminergic cell bodies but not for axonal terminals. Neurobiol Aging. 2011;32:1486–1494. doi: 10.1016/j.neurobiolaging.2009.08.009. [DOI] [PubMed] [Google Scholar]
- Monti B, Gatta V, Piretti F, Raffaelli SS, Virgili M, Contestabile A. Valproic acid is neuroprotective in the rotenone rat model of Parkinson's disease: involvement of alpha-synuclein. Neurotox Res. 2010;17:130–141. doi: 10.1007/s12640-009-9090-5. [DOI] [PubMed] [Google Scholar]
- Moran LB, Croisier E, Duke DC, Kalaitzakis ME, Roncaroli F, Deprez M, Dexter DT, Pearce RK, Graeber MB. Analysis of alpha-synuclein, dopamine and parkin pathways in neuropathologically confirmed parkinsonian nigra. Acta Neuropathol. 2007;113:253–263. doi: 10.1007/s00401-006-0181-6. [DOI] [PubMed] [Google Scholar]
- O'Donnell KC, Lulla A, Stahl MC, Wheat ND, Bronstein JM, Sagasti A. Axon degeneration and PGC-1alpha-mediated protection in a zebrafish model of alpha synuclein toxicity. Dis Model Mech. 2014;7:571–582. doi: 10.1242/dmm.013185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono K, Ikemoto M, Kawarabayashi T, Ikeda M, Nishinakagawa T, Hosokawa M, Shoji M, Takahashi M, Nakashima M. A chemical chaperone, sodium 4 phenylbutyric acid, attenuates the pathogenic potency in human alpha-synuclein A30P + A53T transgenic mice. Parkinsonism Relat Disord. 2009;15:649–654. doi: 10.1016/j.parkreldis.2009.03.002. [DOI] [PubMed] [Google Scholar]
- Perlmann T, Jansson L. A novel pathway for vitamin A signaling mediated by RXR heterodimerization with NGFI-B and NURR1. Genes Dev. 1995;9:769–782. doi: 10.1101/gad.9.7.769. [DOI] [PubMed] [Google Scholar]
- Picelli S, Björklund ÅK, Faridani OR, Sagasser S, Winberg G, Sandberg R. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat Methods. 2013;10:1096–1098. doi: 10.1038/nmeth.2639. [DOI] [PubMed] [Google Scholar]
- Prots I, Veber V, Brey S, Campioni S, Buder K, Riek R, Böhm KJ, Winner B. alpha-Synuclein oligomers impair neuronal microtubule-kinesin interplay. J Biol Chem. 2013;288:21742–21754. doi: 10.1074/jbc.M113.451815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravnskjaer K, Frigerio F, Boergesen M, Nielsen T, Maechler P, Mandrup S. PPARdelta is a fatty acid sensor that enhances mitochondrial oxidation in insulin secreting cells and protects against fatty acid-induced dysfunction. J Lipid Res. 2010;51:1370–1379. doi: 10.1194/jlr.M001123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner DJ, Yu SJ, Shen H, He Y, Bae E, Wang Y. 9-Cis retinoic acid protects against methamphetamine-induced neurotoxicity in nigrostriatal dopamine neurons. Neurotox Res. 2014;25:248–261. doi: 10.1007/s12640-013-9413-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saucedo-Cardenas O, Quintana-Hau JD, Le WD, Smidt MP, Cox JJ, De Mayo F, Burbach JP, Conneely OM. Nurr1 is essential for the induction of the dopaminergic phenotype and the survival of ventral mesencephalic late dopaminergic precursor neurons. Proc Natl Acad Sci U S A. 1998;95:4013–4018. doi: 10.1073/pnas.95.7.4013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siebert H, Kahle PJ, Kramer ML, Isik T, Schlüter OM, Schulz-Schaeffer WJ, Brück W. Overexpression of alpha-synuclein in the nervous system enhances axonal degeneration after peripheral nerve lesion in a transgenic mouse strain. J Neurochem. 2010;114:1007–1018. doi: 10.1111/j.1471-4159.2010.06832.x. [DOI] [PubMed] [Google Scholar]
- Smidt MP, Burbach JP. How to make a mesodiencephalic dopaminergic neuron. Nat Rev Neurosci. 2007;8:21–32. doi: 10.1038/nrn2039. [DOI] [PubMed] [Google Scholar]
- Smith Y, Wichmann T, Factor SA, DeLong MR. Parkinson's disease therapeutics: new developments and challenges since the introduction of levodopa. Neuropsychopharmacology. 2012;37:213–246. doi: 10.1038/npp.2011.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Specht CG, Tigaret CM, Rast GF, Thalhammer A, Rudhard Y, Schoepfer R. Subcellular localisation of recombinant alpha- and gamma-synuclein. Mol Cell Neurosci. 2005;28:326–334. doi: 10.1016/j.mcn.2004.09.017. [DOI] [PubMed] [Google Scholar]
- Ungerstedt U, Arbuthnott GW. Quantitative recording of rotational behavior in rats after 6-hydroxy-dopamine lesions of the nigrostriatal dopamine system. Brain Res. 1970;24:485–493. doi: 10.1016/0006-8993(70)90187-3. [DOI] [PubMed] [Google Scholar]
- Wang Z, Benoit G, Liu J, Prasad S, Aarnisalo P, Liu X, Xu H, Walker NP, Perlmann T. Structure and function of Nurr1 identifies a class of ligand-independent nuclear receptors. Nature. 2003;423:555–560. doi: 10.1038/nature01645. [DOI] [PubMed] [Google Scholar]
- Yin LH, Shen H, Diaz-Ruiz O, Bäckman CM, Bae E, Yu SJ, Wang Y. Early post-treatment with 9-cis retinoic acid reduces neurodegeneration of dopaminergic neurons in a rat model of Parkinson's disease. BMC Neurosci. 2012;13:120. doi: 10.1186/1471-2202-13-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S, Li X, Liu G, Han J, Zhang C, Li Y, Xu S, Liu C, Gao Y, Yang H, Uéda K, Chan P. Extensive nuclear localization of alpha-synuclein in normal rat brain neurons revealed by a novel monoclonal antibody. Neuroscience. 2007;145:539–555. doi: 10.1016/j.neuroscience.2006.12.028. [DOI] [PubMed] [Google Scholar]
- Zetterström RH, Solomin L, Jansson L, Hoffer BJ, Olson L, Perlmann T. Dopamine neuron agenesis in Nurr1-deficient mice. Science. 1997;276:248–250. doi: 10.1126/science.276.5310.248. [DOI] [PubMed] [Google Scholar]
- Zhao S, Maxwell S, Jimenez-Beristain A, Vives J, Kuehner E, Zhao J, O'Brien C, de Felipe C, Semina E, Li M. Generation of embryonic stem cells and transgenic mice expressing green fluorescence protein in midbrain dopaminergic neurons. Eur J Neurosci. 2004;19:1133–1140. doi: 10.1111/j.1460-9568.2004.03206.x. [DOI] [PubMed] [Google Scholar]
- Zhou W, Bercury K, Cummiskey J, Luong N, Lebin J, Freed CR. Phenylbutyrate up-regulates the DJ-1 protein and protects neurons in cell culture and in animal models of Parkinson disease. J Biol Chem. 2011;286:14941–14951. doi: 10.1074/jbc.M110.211029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Gu G, Goodlett DR, Zhang T, Pan C, Montine TJ, Montine KS, Aebersold RH, Zhang J. Analysis of alpha-synuclein-associated proteins by quantitative proteomics. J Biol Chem. 2004;279:39155–39164. doi: 10.1074/jbc.M405456200. [DOI] [PubMed] [Google Scholar]
- Zufferey R, Nagy D, Mandel RJ, Naldini L, Trono D. Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat Biotechnol. 1997;15:871–875. doi: 10.1038/nbt0997-871. [DOI] [PubMed] [Google Scholar]