

Abstract
Synaptotagmin-1 (Syt1) is the principal Ca2+ sensor for vesicle fusion and is also essential for vesicle docking in chromaffin cells. Docking depends on interactions of the Syt1-C2B domain with the t-SNARE SNAP25/Syntaxin1 complex and/or plasma membrane phospholipids. Here, we investigated the role of the positively charged “bottom” region of the C2B domain, proposed to help crosslink membranes, in vesicle docking and secretion in mouse chromaffin cells and in cell-free assays. We expressed a double mutation shown previously to interfere with lipid mixing between proteoliposomes and with synaptic transmission, Syt1-R398/399Q (RQ), in syt1 null mutant cells. Ultrastructural morphometry revealed that Syt1-RQ fully restored the docking defect observed previously in syt1 null mutant cells, similar to wild type Syt1 (Syt1-wt). Small unilamellar lipid vesicles (SUVs) that contained the v-SNARE Synaptobrevin2 and Syt1-R398/399Q also docked to t-SNARE-containing giant vesicles (GUVs), similar to Syt1-wt. However, unlike Syt1-wt, Syt1-RQ-induced docking was strictly PI(4,5)P2-dependent. Unlike docking, neither synchronized secretion in chromaffin cells nor Ca2+-triggered SUV–GUV fusion was restored by the Syt1 mutants. Finally, overexpressing the RQ-mutant in wild type cells produced no effect on either docking or secretion. We conclude that the positively charged bottom region in the C2B domain—and, by inference, Syt1-mediated membrane crosslinking—is required for triggering fusion, but not for docking. Secretory vesicles dock by multiple, PI(4,5)P2-dependent and PI(4,5)P2-independent mechanisms. The R398/399 mutations selectively disrupt the latter and hereby help to discriminate protein regions involved in different aspects of Syt1 function in docking and fusion.
SIGNIFICANCE STATEMENT This study provides new insights in how the two opposite sides of the C2B domain of Synaptotagmin-1 participate in secretory vesicle fusion, and in more upstream steps, especially vesicle docking. We show that the “bottom” surface of the C2B domain is required for triggering fusion, but not for docking. Synaptotagmin-1 promotes docking by multiple, PI(4,5)P2-dependent and PI(4,5)P2-independent mechanisms. Mutations in the C2B bottom surface (R398/399) selectively disrupt the latter. These mutations help to discriminate protein regions involved in different aspects of Synaptotagmin-1 function in docking and fusion.
Keywords: mouse chromaffin cells, patch-clamp technique, synaptotagmin-1, ultrastructural analysis
Introduction
Fusion of secretory vesicles with the plasma membrane depends on the assembly of v-SNARE (Synaptobrevin2) and t-SNAREs (SNAP25/Syntaxin1) into a SNARE-complex (for review, see Jahn and Scheller, 2006; Südhof and Rothman, 2009). Vesicles go through several upstream steps, referred to as docking and priming, to gain fusion competence (Verhage and Sørensen, 2008). The vesicular protein Synaptotagmin-1 (Syt1) is the principal Ca2+ sensor for fusion in many CNS neurons and in neuroendocrine cells (Geppert et al., 1994; Fernández-Chacón et al., 2001; Voets et al., 2001) and is also part of the docking complex for secretory granules (de Wit et al., 2009). Interactions between Syt1 and the t-SNAREs mediate docking in neuroendocrine cells (de Wit et al., 2009; Mohrmann et al., 2013), consistent with previous findings in invertebrate neurons (Jorgensen et al., 1995; Reist et al., 1998; Loewen et al., 2006). Previously, Syt1 deficiency was shown to also impair synaptic vesicle accumulation near the active zone in mammalian CNS neurons (Imig et al., 2014). Syt1 contains a transmembrane domain and a tandem of C2 domains, which bind Ca2+ ions and contain many positively and negatively charged amino acids for potential electrostatic interactions with t-SNAREs, PI(4,5)P2, and other interactors (Fernandez et al., 2001; Pang et al., 2006; Chapman, 2008; Pang and Südhof, 2010; Jahn and Fasshauer, 2012; Park et al., 2012).
The electrostatic interactions supporting the role of Syt1 in docking are probably located in a polybasic patch, centered around K326/327 on the Syt1-C2B domain, that interacts with t-SNAREs (Rickman et al., 2004). The same polybasic stretch also binds PI(4,5)P2 (Fernandez et al., 2001; Rizo et al., 2006; Chapman, 2008; Jahn and Fasshauer, 2012; Park et al., 2012), and this interaction occurs at sites where Syntaxin1 and PI(4,5)P2 form clusters (Honigmann et al., 2013). Syt1 might also mediate docking by simultaneous binding of two opposing membranes. This property of Syt1 was shown in vitro and depends on the calcium-binding region on the “top” and on two arginines at the “bottom” of the C2B domain (Araç et al., 2006; Xue et al., 2008). Indeed, mutations in this bottom region (R398Q, R399Q, called the RQ mutant) abolished neurotransmitter release in neurons (Xue et al., 2008), and the single mutation (R398A) impaired Ca2+-dependent Syt1 function in a lipid mixing assay (Gaffaney et al., 2008). However, it is unknown whether these mutations also affect vesicle docking and whether potential docking defects contribute to the observed effects on release. A homologous mutation in Syt2 (R399Q, R400Q) was proposed to affect positional priming (Young and Neher, 2009). Again, it is unknown whether this overexpression of Syt2-RQ affects vesicle distribution.
In this study we aimed to resolve the role of the Syt1-C2B bottom domain in docking and secretion by expressing wild type Syt1 and the RQ mutant in syt1 null chromaffin cells and by comparing both Syt1 variants in an in vitro liposome docking and fusion assay (Malsam et al., 2012). We found that the bottom residues are critical for fast Syt1-dependent fusion, but dispensable for docking. However, docking becomes critically dependent on PI(4,5)P2. Hence, the RQ mutant helps to segregate different aspects of Syt1 function in docking and fusion.
Materials and Methods
Cell culture and transfection of chromaffin cells
Mouse chromaffin cells were obtain by isolating adrenal glands of wild type or synaptotagmin-1 null littermates [embryonic day 18 (E18) pups of either sex] and cultured as described previously (Sørensen et al., 2003; de Wit et al., 2009). Cells were infected on day in vitro 3 for 8–12 h with Semliki Forest virus (SFV; Ehrengruber and Lundstrom, 2007) containing full-length synaptotagmin-1-IRES-eGFP or syt1 (R398,399Q)-IRES-eGFP (a gift from Dr. C. Rosenmund, Charite Medical University, Berlin, Germany; Xue et al., 2008). In ultramorphology analysis experiments, SFV expressing IRES-eGFP was used as a control.
Confocal microscopy
Cells were fixed using 4% paraformaldehyde (Merck) in PBS, pH 7.4, for 1 h, permeabilized with 1% Triton X-100 in PBS for 5 min, and blocked for 30 min with solution containing PBS, 2% NGS, and 1% Triton X-100. Cells were then incubated with primary antibody [polyclonal rabbit anti-synaptotagmin 1, 1:2000 (W855; a gift from T. C. Südhof, Stanford, CA) or polyclonal rabbit anti-chromoganinB, 1:500 (Synaptic System)] for 1 h, washed, and then incubated with secondary antibody (goat-anti-rabbit Alexa Fluor 546, 1:1000; Sigma) for 1 h. Cell fixation and staining procedures were performed at room temperature. Coverslips were mounted on glass slides using Mowiol 4–88 (Aldrich). A confocal laser scanning microscope (LSM 500 meta; Carl Zeiss Microimaging) was used to image stained cells. Infected cells were identified based on eGFP fluorescence. Images were taken using a 63×/1.4 numerical aperture oil-immersion objective lens. Fluorescence intensity was calculated using Fiji (Schindelin et al., 2012).
Electrophysiological recordings
Transfected cells were identified by green fluorescent response to 475 nm illumination by a monochromator (Polychrome IV; TILL Photonics). Amperometry measurements were performed with 5-μm-diameter polyethylene insulated carbon fibers (Thornel P-650/42; Cytec; Bruns, 2004). The tip of the fiber was gently pressed against the cell while a constant voltage of 700 mV was applied. Currents were amplified by an EPC-7 amplifier (HEKA Elektronik), filtered at 2.9 kHz, and sampled at 11.5 kHz. Cell membrane capacitance was measured via whole-cell patch-clamp technique as described by Lindau and Neher (1988). Pulse software (version 8.53) and an EPC-9 amplifier (HEKA Elektronik) were used in “sine+dc” mode to record and inject currents. Currents were filtered at 3 kHz and sampled at 11.5 kHz. Secretion was stimulated by UV photolysis of the Ca2+ cage nitrophenyl-EGTA by using UV light from a UV flash lamp (JML-C2; Rapp Optoelectronics) or by applying depolarizing pulses both controlled by the Pulse software and triggered by the EPC-9 amplifier.
Chromaffin cells were visualized using a Zeiss Axiovert 10 with a 40× Fluar objective (Carl Zeiss). Ca2+ measurements were conducted by exciting Fura dyes at 350 and 380 nm. Emitted light was detected by a photo diode (Till Photonics) in an area around the cell defined by a View Finder (Till Photonics). The output of the photo diode was connected to an auxilliary input channel on the EPC-9 amplifier. The signal was filtered at 3 kHz and sampled at 11.5 kHz. The Ca2+ concentration was measured with a mixture of high- and low-affinity Fura dyes as described previously (Voets, 2000). The free Ca2+ concentrations were calculated using a custom-written macro for IGOR Pro (Wavemetrics), assuming a KD of 0.222 μm for BAPTA and of 80 μm for DPTA while taking into account the additional buffering of Ca2+ by the dyes, nitrophenyl-EGTA, and ATP. The patch pipette solution contained the following (in mm): 100 Cs-glutamate, 8 NaCl, 4 CaCl2, 32 HEPES, 2 Mg-ATP, 0.3 NaGTP, 5 nitrophenyl-EGTA, 1 ascorbic acid, 0.4 fura-4F (Invitrogen), and 0.4 furaptra (Invitrogen), adjusted to pH 7.2 with CsOH.
Electron microscopy of cultured chromaffin cells
Cells were fixed using conventional aldehyde fixation protocol with 2.5% glutaraldehyde in 0.1 m cacodylate buffer, pH 7.4, for 1 h and subsequently washed with 0.1 m cacodylate buffer, pH 7.4. Postfixation was done for 1.5 h using 1% osmium tetroxide and 1% ruthenium tetroxide mixture in 0.1 m cacodylate buffer. Due to cell detachment in high-pressure freezing/freeze substitution (HPF/FS) protocols, HPF/FS could not be applied. Since a side-by-side comparison between aldehyde fixation and HPF/FS on large dense core vesicles docking revealed no differences in conclusions (de Wit et al., 2009), we relied on chemical fixation in this study. Dehydration using a series of ethanol concentrations (30–100%) was performed followed by embedding in Epon and polymerization for at least 15 h at 65°C. The coverslip was removed from Epon resin by alternatively dipping it into liquid nitrogen and hot water. Flat Epon surface region (2 × 3 mm) with a high cell density monolayer was selected, cut, and mounted on prepolymerized Epon blocks for thin sectioning. Ultrathin sections (∼90 nm) were collected on single-slot, formvar-coated copper grids and stained with uranyl acetate and lead citrate. A Jeol 1010 transmission electron microscope was used for high-resolution imaging. Vesicle membrane attached to the plasma membrane was considered docked vesicles. An infected cell was identified based on presence of SFV (droplet shape with dense core) on the plasma membrane. Distance measurements were performed using 10,000× or 30,000× magnification images on iTEM software (Olympus).
Reconstituted SUV–GUV docking and fusion assays
Proteins were expressed, purified, and reconstituted into liposomes as described previously (Weber et al., 1998; Malsam et al., 2012). The following modifications were made: The C-terminal His6-tag of full length mouse Synaptobrevin2 was replaced by an N-terminal GST-tag, which was removed by thrombin cleavage before reconstitution of Synaptobrevin2 into v-SNARE/Syt1 liposomes. Therefore, a cDNA encoding mouse Synaptobrevin2 (pTW2) was amplified by PCR and subcloned into pGEX4T3 using BamHI and XhoI sites resulting in pSK28. To introduce the R398/399Q (RQ) and R398/399A (Syt1-RA) double mutations into Syt1, the template plasmid pLM6, encoding Syt1 (Mahal et al., 2002), was modified using the QuickChange DNA mutagenesis method (Qiagen). The resulting constructs pJM80 and pJM45 encode His6-rat Syt1 R398/399Q and His6-rat Syt1 R398/399A, respectively.
For the preparation of giant unilamellar t-SNARE vesicles (GUVs), indium tin oxid (ITO)-coated glass slides were replaced by Pt-coated glass slides (GeSIM). In addition, in the GUV preparation buffer 1 mm HEPES-KOH, pH 7.4, was replaced by 0.5 mm EPPS-KOH [3-[4-(2-hydroxyethyl)-1-piperazinyl]propanesulfonic acid hydrate], pH 8.0. In the lipid mix used for the reconstitution of the v-SNARE/Syt1 small unilamellar lipid vesicles (SUVs), the fluorophores rhodamine-DPPE [N-(lissamine rhodamine B sulfonyl)1,2-dipalmitoyl phosphatidylethanolamine] and NBD-DPPE [N-(7-nitro-2,1,3-benzoxadiaziole-4-yl)-1,2-dipalmitoyl phosphatidylethanolamine] were replaced by 0.5 mol% each of Atto488-DPPE and Atto550-DPPE [Atto-dye labeled phospholipids 1,2-dipalmitol-sn-glycero-3-phosphoethanolamine] from Atto-Tec.
SUV–GUV docking and fusion reactions and data analysis were performed as described previously (Malsam et al., 2012). Briefly, incubations occurred in an iso-osmolar buffer containing 25 mm HEPES-KOH, pH 7.4, 135 mm KCl, 0.5 mm MgCl2, 1 mm dithiothreithol, and 100 μm EGTA. Calcium was added to a free concentration of 100 μm using the Maxchelator software (http://maxchelator.stanford.edu).
Statistics
Physiology data.
When three groups were compared (typically WT overexpression, RQ mutant overexpression, and a control group), a nonparametric Kruskal–Wallis test for independent samples was used to compare the distributions. If the test was significant, a post-test with significance levels corrected for multiple comparisons was used for pairwise testing (IBM SPSS Statistics, version 22). A significance level of 0.05 was assumed throughout.
Ultrastructural morphometry.
The data obtained from individual cells was nested within individual embryos (more than one observation drawn from one independent sample, an embryo). To accommodate potential dependency, we tested the effects by multilevel analysis using SPSS (IBM) software (Aarts et al., 2014). The number obtained per embryo for each data set is noted in each figure legend; overall cell numbers and embryo numbers are annotated in the figures.
Results
Syt1 null chromaffin cells (E18) show impaired docking and a Ca2+-dependent fast burst release component (Voets et al., 2001; Nagy et al., 2006; Schonn et al., 2008; de Wit et al., 2009). We used this model to analyze the role of the Syt1-C2B bottom domain in docking and secretion by introducing exogenous Syt1-wt-IRES-eGFP or Syt1-RQ-IRES-eGFP. We assessed the level and localization of the overexpressed Syt1 by confocal microscopy. Endogenous Syt1 labeling shows a punctate pattern that is concentrated in the vicinity of the plasma membrane (Fig. 1A). This labeling is absent in syt1 null cells, indicating that this labeling is specific for Syt1 (Fig. 1B,C). Syt1 null cells expressing the Syt1-wt and Syt1-RQ constructs showed a distribution of Syt1 close to plasma membrane (Fig. 1D,E), comparable to the endogenous Syt1 pattern. This indicates that the RQ mutation had no effect on the subcellular distribution of Syt1. Both constructs yielded a significant increase in Syt1 levels of a factor of four over wild type levels (multilevel test against wild type; syt1 null + eGFP, p = 0.389; syt1 null + Syt1-wt and syt1 null + Syt1-RQ, p ≤ 0.001; Fig. 1F).
Figure 1.

SFV transfection leads to overexpression of Syt1-wt and the Syt1-RQ mutant. A, B, Syt1 is located near the plasma membrane in wild type chromaffin cells (A) and is absent in syt1 null cells (B). C–E, eGFP overexpression does not change Syt1 expression in the syt1 null cell (C), whereas Syt1-wt (D) and Syt1-RQ mutant (E) transfection result in 4–5× overexpression (OE). Scale bars: 2 μm. F, Quantification of Syt1 levels by multilevel analysis. Between 3 and 14 cells were collected from two to four embryos per condition, and overall cell numbers and embryo numbers are annotated. ***p ≤ 0.001. Error bars indicate SEM. Flu. Int., Fluorescence intensity.
Syt1-C2B bottom region is dispensable for secretory vesicle docking
We first assessed the secretory vesicle distribution in syt1 null cells expressing Syt1-wt or the Syt1-RQ mutant by electron microscopy. In line with previously reported observations in syt1 null cells (de Wit et al., 2009), we found a >50% reduction in docked vesicles in syt1 null cells compared to wild type (p ≤ 0.001; Fig. 2A–D). Moreover, Syt1-wt restored docking in syt1 null cells to the wild type level, without affecting the total number of vesicles: wild type, 29 ± 2.5 (p ≤ 0.001 against syt1 null + eGFP); syt1 null + Syt1-wt, 26.3 ± 2.9 (p ≤ 0.001 against syt1 null + eGFP); syt1 null, 12.4 ± 1.8 (p = 0.656 against syt1 null + eGFP); syt1 null + eGFP, 14.8 ± 1.4 docked vesicles/section. Total number of vesicles ranged from 104 ± 7 to 114 ± 5 vesicles/section in all conditions (mean ± SEM; all not significantly different; Figs. 2C,D). Syt1 null cells expressing the Syt1-RQ mutant showed a similar number of docked vesicles as compared to wild type cells (syt1 null + Syt1-RQ, 33.9 ± 2.9 docked vesicles/section, mean ± SEM, p = 0.154, multilevel analysis; Fig. 2A,C). Importantly, expressing Syt1-RQ or Syt1-wt on the syt1 null background does not change the total number of vesicles, but shifts the distribution pattern of vesicles to the wild type pattern (Fig. 2B). This indicates that the positive-charged arginines of the Syt1-C2B domain are dispensable for Syt1 function in secretory vesicle docking.
Figure 2.
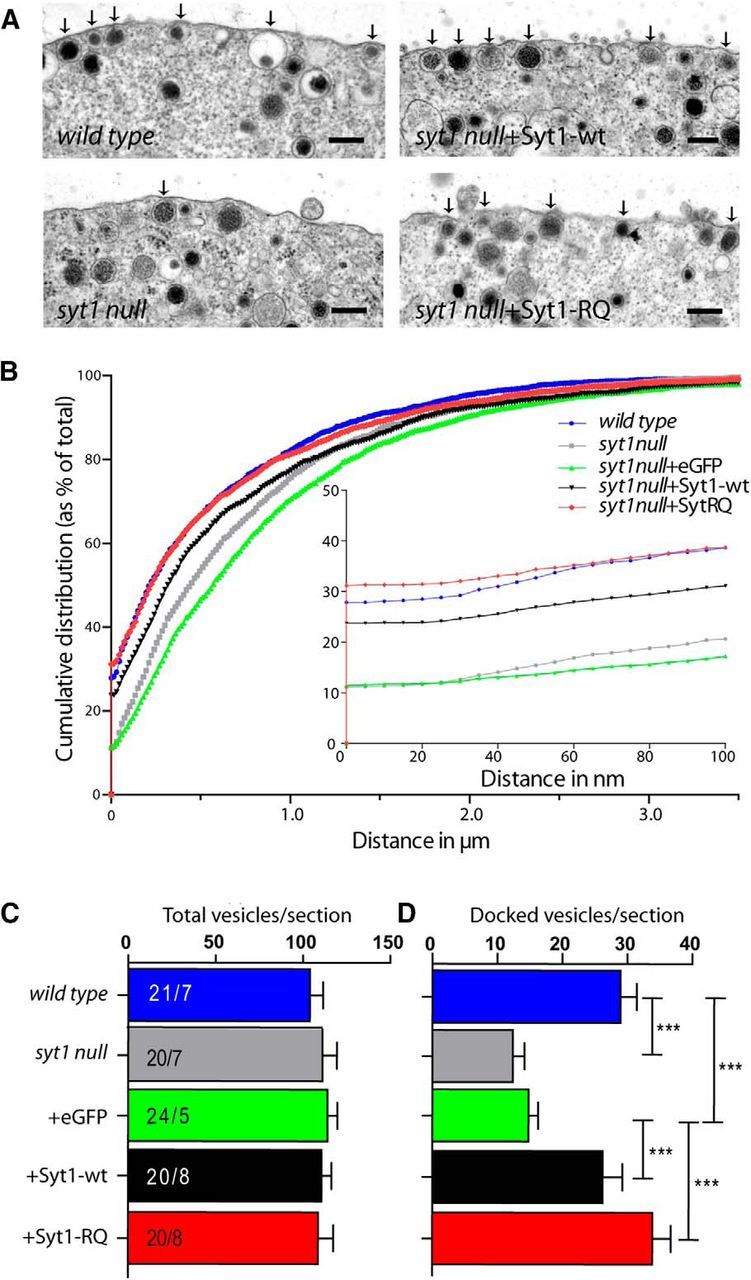
Syt1-RQ mutant rescues docking in syt1 null cells. A, Ultrastructural images of different noninfected and infected conditions on the syt1 null cell background. Arrows mark docked vesicles. Scale bars: 200 nm. B, Normalized cumulative distribution of vesicles as a function of the distance from the plasma membrane. Inset, Distance in the range of 0–100 nm from membrane. C, D, Number of total (C) and docked (D) vesicles per section showing no difference in docking for Syt1-RQ mutant. ***p ≤ 0.001 by multilevel analysis (between 1–8 cells were collected from 5–8 embryos per condition; overall cell numbers and embryo numbers are annotated in C). Error bars indicate SEM.
The normal docking functions of Syt1-RQ and RA critically depend on PI(4,5)P2 in vitro
Docking in cells might be affected by multiple proteins and protein–protein as well as protein–lipid interactions. To investigate the role of the Syt1-C2B bottom region in a reduced system, we recapitulated Syt1's role in docking in vitro in a cell-free assay by analyzing docking of SUVs that contain the v-SNARE Synaptobrevin2 and membrane-anchored Syt1 to reconstituted GUVs containing the t-SNARE Syntaxin1/SNAP-25 (Fig. 3A; Malsam et al., 2012). Syt1-R398/399Q was reconstituted into SUVs with similar efficiency as Syt1-wt (Fig. 3B) and SUVs containing Syt1-RQ docked equally well to GUVs as wild type Syt1 (Fig. 3C) in the presence of 2% PI(4,5)P2. For docking efficiency, the presence or absence of Complexin II was irrelevant. In the absence of PI(4,5)P2, a large functional difference between Syt1-wt and Syt1-RQ was observed. Whereas Syt1-wt showed a minor impairment in docking efficiency as observed before for synthetic vesicle-vesicle tethering (Vennekate et al., 2012, Parisotto et al., 2012), Syt1-RQ was completely defective in docking SUVs to GUVs lacking PI(4,5)P2 (Fig. 3C). In addition to the Syt1-R398/399Q mutation, a previous study used a similar mutation, but converted the arginines to alanines (Syt1-R398/399A; Gaffaney et al., 2008). We tested this mutation and found indistinguishable effects on SUV-docking to GUVs with the A or Q mutant (Fig. 3B,C). Hence, in this reduced system an intact Syt1-C2B bottom region is critical to mediate Syt1-dependent liposome docking in the absence of PI(4,5)P2.
Figure 3.

Syt1–R398/399Q- and Syt1–R398/399A-dependent vesicle docking requires the presence of PI(4,5)P2 in a reconstituted SUV–GUV docking assay. A, Scheme of vesicle docking assay. t-SNARE-GUVs (28 nmol lipid, 28 pmol Syntaxin1/SNAP-25) lacking or containing 2% PI(4,5)P2 were mixed with 3H-labeled v-SNARE/Syt1-SUVs (5 nmol lipid, 16.6 pmol Synaptobrevin2, 6.2 pmol Syt1), containing either Syt1-wt or Syt1–R398/399Q or Syt1-RA in the presence and absence of 6 μm Complexin II (CpxII). Samples were incubated in a final volume of 100 μl for 5 min on ice to allow docking, followed by centrifugation at 5000 × g for 5 min to reisolate GUVs. B, Reconstituted SUVs, containing Synaptobrevin2 (Syb2) and either Syt1-wt or Syt1–R398/399Q or Syt1-RA, were analyzed by SDS-PAGE and Coomassie blue staining. C, 3H-labeled SUVs bound to GUVs in the pellet were quantified and normalized to maximum binding. Error bars indicate SEM (n = 3).
Syt1-C2B bottom region is crucial for secretion
Syt1 null cells lack the Ca2+-dependent fast burst phase of release in chromaffin cells (Voets et al., 2001), which can be reestablished by overexpression of Syt1-wt (Nagy et al., 2006). We tested whether the Syt1-RQ mutant has the same capacity. We analyzed vesicle release using membrane capacitance measurements upon flash photolysis of caged calcium. As anticipated, in Syt1-overexpressing (i.e., rescued) syt1 null cells, the burst phase (i.e., release within the first second) was reestablished compared to syt1 null cells (Fig. 4A–J). In contrast, the Syt1-RQ mutant showed a strong reduction of burst release as compared to Syt1-wt (syt1 null, mean ΔCm = 87 ± 17 fF, n = 28; syt1 null + Syt1-wt, mean ΔCm = 147 ± 19 fF, n = 23; syt1 null + Syt1-RQ, mean ΔCm = 58 ± 10 fF; n = 23; mean ± SEM; Kruskal–Wallis test, p < 0.001; Fig. 4A,C). We observed a similar pattern for the total release (syt1 null, mean ΔCm = 165 ± 28 fF; syt1 null + Syt1-wt, mean ΔCm = 249 ± 26 fF; syt1 null + Syt1-RQ, mean ΔCm = 119 ± 13 fF; mean ± SEM; Kruskal–Wallis test, p < 0.01; Fig. 4A,D). For the sustained release, a strong tendency to a difference between groups was noted (not significantly different; syt1 null, mean ΔCm = 78 ± 12 fF; syt1 null + Syt1-wt, mean ΔCm = 99 ± 13 fF; syt1 null + Syt1-RQ, mean ΔCm = 58 ± 7 fF; mean ± SEM; Kruskal–Wallis test, p = 0.051; Fig. 4A,E). Indeed, in a pairwise test, the sustained release in Syt1-RQ-expressing cells was significantly smaller than in WT-rescued cells (Mann–Whitney U test, p = 0.007), confirming lack of rescue also for this parameter.
Figure 4.

Syt1-RQ mutant impaired secretion in syt1 null cells. A, Mean membrane capacitance and Ca2+ measurements of syt1 null (gray) with Syt1-wt overexpression (black) and Syt1-RQ mutant (red). B, Quantification of preflash [Ca2+]. C, Burst size was higher in Syt1-wt overexpression than Syt1-RQ overexpression and syt1 null (ΔCm at 1 s; Kruskal–Wallis test, p < 0.001; result of pairwise post-tests, syt1 null vs Syt1-wt overexpression, p = 0.016; Syt1-wt overexpression vs Syt1-RQ, p < 0.001). D, The same was true of total release size (ΔCm at 5.5 s, Kruskal–Wallis test, p = 0.004; result of pairwise post-tests, syt1 null vs Syt1-wt overexpression, p = 0.035; Syt1-wt overexpression vs Syt1-RQ, p = 0.005). E, For sustained release, the difference was almost, but not quite, significant (ΔCm at 5.5–1 s; Kruskal–Wallis test, p = 0.051). F, G, I, J, Fitting individual capacitance traces with a sum of exponentials allowed separating the amplitude and time constant of fast burst secretion from the amplitude and time constant of slow burst secretion. Only the amplitude of the fast burst (Afast) was significantly different between groups (Kruskal–Wallis test, p < 0.001; result of pairwise post-tests, syt1 null vs Syt1-wt overexpression, p < 0.001; Syt1-wt overexpression vs Syt1-RQ, p < 0.001). H, Scaled version of the mean capacitance traces in A clearly show the fast component in the Syt1-wt overexpression, which is missing from syt1 null and Syt1-RQ overexpression. Error bars indicate SEM.
The kinetic properties of release were analyzed by fitting capacitance changes with a sum of exponentials. We found that fast burst phase secretion was significantly increased in Syt1-wt-overexpressing syt1 null cells compared to control syt1 null or Syt1-RQ-overexpressing syt1 null cells (syt1 null, mean Afast = 16 ± 4 fF; syt1 null + Syt1-wt, mean Afast = 67 ± 12 fF; syt1 null + Syt1-RQ, Afast = 14 ± 4 fF; mean ± SEM; Kruskal–Wallis test, p < 0.001; Fig. 4I). Furthermore, the Syt1-RQ mutant showed a reduced slow burst phase compared to the other groups, but this was not significant (syt1 null, mean Aslow = 85 ± 21 fF; syt1 null + Syt1-wt, mean Aslow = 48 ± 11 fF; syt1 null + Syt1-RQ, mean Aslow = 34 ± 7 fF; mean ± SEM; Kruskal–Wallis test, p = 0.116; Fig. 4J). The time constants of the fast and slow release components were unaffected in all the tested conditions (syt1 null, τfast = 22.3 ± 3.7 ms, τslow = 0.375 ± 0.080s; syt1 null + Syt1-wt, τfast = 18.8 ± 2.7 ms, τslow = 0.203 ± 0.048 s; syt1 null + Syt1-RQ, τfast = 24.3 ± 4.8 ms, τslow = 0.282 ± 0.056 s; mean ± SEM; Fig. 4F,G). The fast burst of release is assumed to originate from the readily releasable pool (RRP) of vesicles (Voets, 2000), whereas the slow burst is due to an upstream pool, which is probably not available for immediate release (Walter et al., 2013). Our data indicate that the Syt1-C2B bottom arginines, although being dispensable for docking, are crucial for the role of Syt1 in enabling fast burst secretion. These data suggest that the generation or release of the RRP is impaired by the Syt1-RQ mutation in a post-docking step.
Syt1-RQ and Syt1-RA support Ca2+-independent, but abolish Ca2+-dependent, fusion in a cell-free assay
To analyze whether the essential role of the Syt1 bottom arginine residues in synchronous release can be reproduced in the cell-free system, we tested the Syt1-RQ and Syt1-RA mutants in Ca2+-synchronized lipid mixing in an in vitro liposome fusion assay. We found that SUVs containing Synaptobrevin2 and Syt1-RQ or Syt1-RA fused readily with GUVs in a Ca2+-independent manner, albeit with ∼30% lower efficiency (Fig. 5), in line with previous observations for the single mutations (R398A or R399A; Gaffaney et al., 2008). Since it was shown previously that fusion in this v/t-SNARE SUV/GUV assay depends on the presence of Syt1, a minus Syt1 control was omitted (Malsam et al., 2012; Parisotto et al., 2012). Addition of 6 μm Complexin II effectively suppressed (clamped) fusion for SUVs containing wild-type Syt1, but less so in SUVs containing Syt1-RQ and Syt1-RA. Moreover, consistent with our observations in intact cells (Fig. 4) and again in line with previous observations for the single mutations (R398A or R399A; Gaffaney et al., 2008), SUVs containing Syt1-RQ and Syt1-RA, in contrast to SUVs containing Syt1-wt, showed little increase in lipid mixing upon addition of 100 μm Ca2+ (Fig. 5). Hence, although the Syt1-RQ and Syt1-RA mutations support to a significant degree Ca2+-independent fusion, the Syt1 bottom arginine residues are critical for the Ca2+-dependent reaction.
Figure 5.

Syt1-RQ and Syt1-RA mutants fail to promote Ca2+-triggered lipid mixing in a reconstituted SUV–GUV fusion assay. t-SNARE-GUVs (14 nmol lipid, 14 pmol Syntaxin1/SNAP25) were mixed with 3H-labeled v-SNARE-SUVs (2.5 nmol lipid, 8.3 pmol Synaptobrevin2, 3.1 pmol Syt1) containing either Syt1-wt or Syt1–R398/399Q or Syt1-RA in the presence and absence of 6 μm CpxII in a final volume of 100 μl. The increase of Atto488 fluorescence was monitored. After 2 min at 37°C, Ca2+ was added to a final concentration of 100 μm and the measurement continued for another 2 min. Values were normalized to the maximum fluorescent signal after detergent lysis. Error bars indicate SEM. n = 3.
No dominant effects of Syt1-RQ when overexpressed in wild type chromaffin cells
It was reported previously that the overexpression of the Syt2-RQ (R399, 400Q) mutation in the calyx of Held produces dominant-negative effects on secretion when expressed on a wild type background (Young and Neher, 2009). We tested whether this was also the case in our system by overexpressing the Syt1-RQ mutant or Syt1-wt on wild type chromaffin cells. Before analyzing its effect on secretion, we investigated the potential effects on the docked pool of vesicles under these conditions. The morphology of chromaffin cells was similar in all conditions, and the distribution of secretory vesicles was also similar in all conditions except wild type + Syt1-wt (Fig. 6A,B). Surprisingly, wild type cells overexpressing wild type Syt1, but not cells overexpressing Syt1-RQ, showed a significant decrease in docked vesicles [wild type, 26 ± 2.0 docked vesicles/section; wild type + eGFP, 31 ± 3.5 docked vesicles/section; wild type + Syt1-wt = 16 ± 2.7 docked vesicles/section (Student's t test against wild type + eGFP, p ≤ 0.001, against wild type and wild type +Syt1-RQ, p ≤ 0.01); wild type + Syt1-RQ, 26 ± 2.6 docked vesicles/section; mean ± SEM; one-way ANOVA, p = 0.002; Fig. 6A–D]. This unexpected reduction of docking in cells overexpressing wild type Syt1 but not Syt1-RQ may relate to the observation in our cell-free system (Fig. 3) that the docking mechanisms differ between wild type Syt1 and Syt1-RA: Whereas Syt1-RA critically depends on PI(4,5)P2, Syt1 wild type can also dock vesicles in the absence of PI(4,5)P2, probably via interaction with t-SNAREs. Hence, overexpression of Syt1-wt may compete with these PI(4,5)P2-independent interactions in wild-type cells, whereas overexpression of Syt1-RQ cannot. In any case, no evidence was observed for a dominant-negative effect of the Syt1-RQ mutant on the total vesicle pool or vesicle docking.
Figure 6.
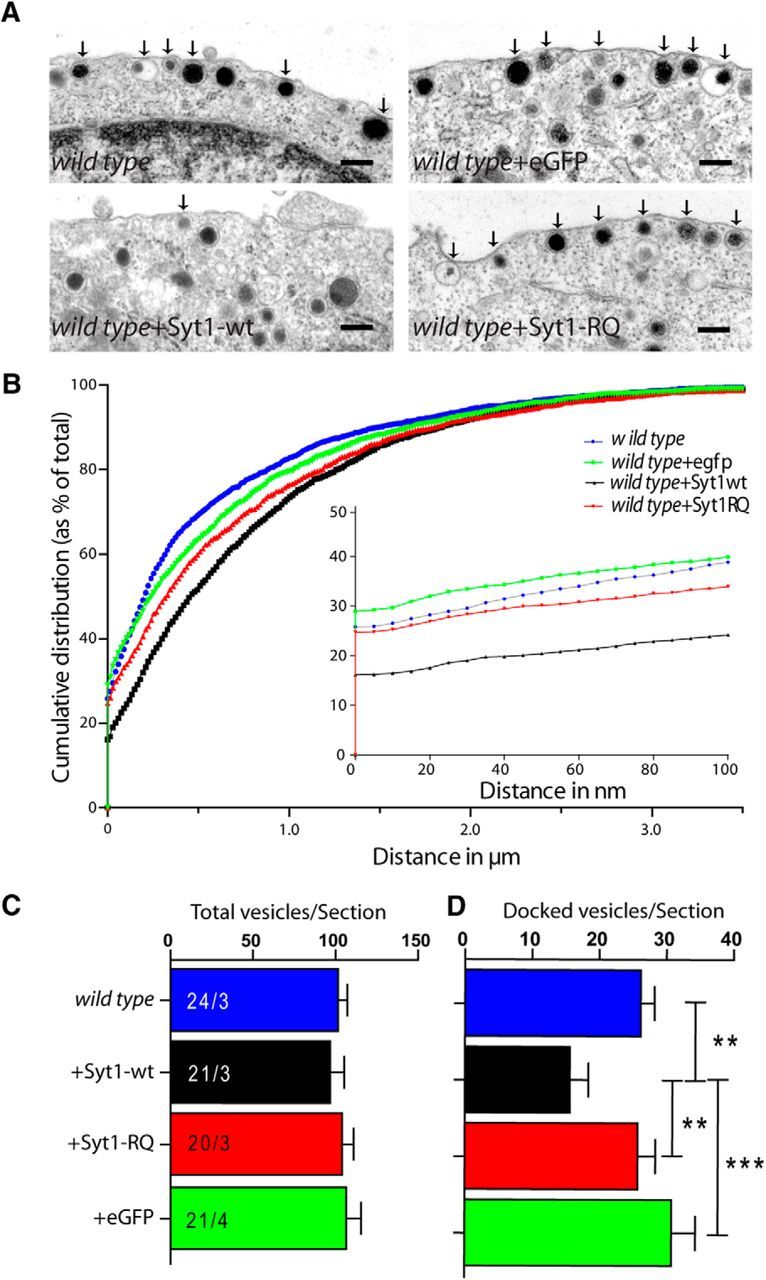
Syt1-wt overexpression in wild type chromaffin cells reduces docking. A, Ultrastructural images of different noninfected and infected conditions on the wild-type cell background. Scale bars: 200 nm. Arrows mark docked vesicles. B, Normalized cumulative distribution of vesicles as a function of the distance from the plasma membrane. Inset, The distance range of 0–100 nm from the membrane. C, D, Numbers of total (C) and docked (D) vesicles per section showing a significant reduction in docking in Syt1-wt-overexpressing wild-type cells. Error bars indicate SEM.
Vesicle secretion evoked by calcium uncaging was unaffected by overexpression of Syt1-wt or Syt1-RQ in wild type chromaffin cells (total release, wild type, mean ΔCm = 319 ± 31 fF, n = 20; wild type + Syt1-wt, mean ΔCm = 447 ± 43 fF, n = 17; wild type + Syt1-RQ, mean ΔCm = 374 ± 68 fF, n = 20; mean ± SEM; Kruskal–Wallis, n.s.; Fig. 7A–J), although a nonsignificant increase in burst secretion upon Syt1-wt overexpression (within 1 s of the flash) was observed (burst release, wild type, mean ΔCm = 125 ± 17 fF, n = 20; wild type + Syt1-wt, mean ΔCm = 176 ± 20 fF, n = 17; wild type + Syt1-RQ, mean ΔCm = 144 ± 29 fF, n = 20; mean ± SEM; Kruskal–Wallis, p = 0.149; Fig. 7C). Consistently, both fast and slow bursts were unchanged between the three groups (fast burst, wild type, Afast = 62 ± 10 fF; wild type + Syt1-wt, Afast = 85 ± 16 fF; wild type + Syt1-RQ, Afast = 53 ± 9 fF; mean ± SEM, Kruskal–Wallis, n.s., Fig. 7I; slow burst, wild type, mean Aslow = 55 ± 11 fF; wild type + Syt1-wt, mean Aslow = 81 ± 12 fF; wild type + Syt1-RQ, mean Aslow = 56 ± 9 fF; Kruskal–Wallis, n.s.; Fig. 7J). No differences in time constants for any conditions were observed (wild type, mean τfast = 18 ± 3 ms, mean τslow = 0.46 ± 0.20 s; wild type + Syt1-wt, mean τfast = 15 ± 4 ms, mean τslow = 0.23 ± 0.06 s; wild type + Syt1-RQ, mean τfast = 18 ± 4 ms, mean τslow = 0.24 ± 0.04 ms; Kruskal–Wallis, n.s. for both parameters; Fig. 7F,G).
Figure 7.

Overexpression of the Syt1-RQ mutant does not alter secretion in wild type chromaffin cells. A, Mean membrane capacitance, Ca2+, and amperometry measurements of wild-type (blue), with Syt1-wt overexpression (black) and Syt1-RQ mutant (red). B, Quantification of preflash [Ca2+]. C, The burst size was slightly higher in Syt1-wt overexpression than in Syt1-RQ overexpression and syt1 null, but this was not significant (Kruskal–Wallis, p = 0.149). D, The total release (ΔCm at 5.5 s) was unchanged between conditions. E, No difference was detected for sustained release (ΔCm at 5.5–1 s). F, G, I, J, Fitting individual capacitance traces with a sum of exponentials allowed separating the amplitude and time constant of fast burst secretion from the amplitude and time constant of slow burst secretion. None of the parameters were significantly different between groups. H, Scaled version of the mean capacitance traces in A clearly show similar kinetics in the three groups. Overexpression levels of Syt1 constructs were similar (see Fig. 1). Error bars indicate SEM.
A subpopulation of the RRP, called the immediately releasable pool (IRP), comprises vesicles in close vicinity of voltage-sensitive calcium channels (Alvarez and Marengo, 2011) that fuse first upon depolarization-induced Ca2+ influx. A similar pool was shown to be affected by overexpression of Syt2-RQ in the calyx of Held synapse (Young and Neher, 2009); hence, we investigated the effects of Syt1-RQ overexpression on the IRP in chromaffin cells. The IRP can be distinguished from the RRP by trains of depolarizations of different duration, either 10 ms (for the IRP) or 100 ms (RRP) (Voets et al., 1999). We found no significant differences in vesicle secretion upon depolarization in wild-type cells expressing Syt1-WT or Syt1-RQ (IRP, i.e., capacitance change elicited by five consecutive 10 ms depolarizations; wild type, ΔCm = 29 ± 5 fF, n = 14; wild type + Syt1-wt, ΔCm = 31 ± 4 fF, n = 28; wild type + Syt1-RQ, ΔCm = 25 ± 4 fF, n = 26; Kruskal–Wallis n.s.; Fig. 8A,B). Moreover, the RRP (detected as the capacitance increase until the end of the fourth 100 ms depolarization) and total membrane capacitance after a UV flash had photoreleased Ca2+ to release any remaining vesicles (end of trace) were not significantly different in any condition (Fig. 8C,D). These data indicate that Syt1-RQ overexpression does not result in a dominant-negative phenotype on vesicle fusion, and that the IRP remains unaffected under these conditions. Thus, overall, the Syt1-RQ mutant does not participate in Ca2+-triggered secretion in chromaffin cells and also does not interfere with it.
Figure 8.

Syt1-RQ mutant overexpression in wild type chromaffin cells does not affect the IRP size. A, Mean membrane capacitance, Ca2+, and amperometry measurements of wild-type cells (blue), Syt1-wt overexpression (black), and Syt1-RQ mutant (red) overexpression in wild-type cells. B, There was no difference in IRP size (capacitance increase elicited by the first 6 brief 10 ms depolarizations) between conditions. C, D, RRP (capacitance increase elicited by the 6 10 ms depolarizations, and the 4 100 ms depolarizations) and total membrane capacitance after a Ca2+ uncaging flash were not significantly different in any condition. Error bars indicate SEM.
Discussion
In the current study, we investigated the role of the Syt1-C2B domain bottom residues in secretory granule docking and secretion, using the Syt1 R398Q/399Q mutant. This mutant severely disrupted synaptic transmission in hippocampal neurons (Xue et al., 2008), and, using a similar mutation in Syt2 (R399Q/400Q, homologous positions to R398, R399 in Syt1), it was concluded that C2B domain bottom residues affect positional priming in the calyx of Held (Young and Neher, 2009). Applying ultrastructural analysis, patch-clamp, and cell-free assays, we found that this mutation fully restored docking, but made docking in vitro strictly PI(4,5)P2 dependent and failed to restore secretion of fast burst release in syt1 null chromaffin cells and in a cell-free fusion assay. These impairments of secretion were not the result of a dominant-negative effect of the Syt1-RQ mutant, as docking and secretion in wild-type cells overexpressing this mutant remained unaffected.
The role of Syt1 in vesicle docking
The Syt1-RQ mutant restored docking in syt1 null cells (Fig. 2) and supported docking in a cell-free assay (Fig. 3), but did not restore secretion in syt1 null cells (Fig. 4), and the Syt1-RQ and Syt1-RA mutants did not support Ca2+-dependent lipid mixing in a cell-free assay. This indicates that the molecular properties of Syt1 relevant for vesicle docking and for fusion are different. Mutation of the C2B bottom surface of Syt1 does not affect the extent of docking, but does eliminate Ca2+-dependent membrane fusion. An important indication for the underlying differences between docking and fusion came from the cell-free docking assay. This assay revealed that mutations in the C2B bottom surface make docking critically dependent on PI(4,5)P2. This observation suggests that wild-type Syt1 can use PI(4,5)P2-dependent and PI(4,5)P2-independent principles to dock vesicles, and the mutations disrupt the PI(4,5)P2-independent principle. Hence, the C2B bottom surface appears to be involved in a PI(4,5)P2-independent docking principle. Furthermore, since Syt1-RQ rescues docking in syt1 null cells, docking in living chromaffin cells is probably PI(4,5)P2-dependent.
Given the fact that this PI(4,5)P2-independent docking principle was observed in a highly reduced cell-free system, the number of possible mechanisms is limited: interactions with the t-SNAREs and/or with other phospholipids. Under resting conditions (low Ca2+), Syt1 is reported to bind to (monomeric) SNAP-25 and Syntaxin1A and to the binary and the ternary SNARE complex (Rizo et al., 2006; Chapman, 2008; Jahn and Fasshauer, 2012; Kim et al., 2012; Südhof, 2012). The polybasic region on the side of Syt1-C2B domain, around K325, K326, K327, and K331, is a prominent interaction surface (Rickman et al., 2004; Martens et al., 2007; Lai et al., 2011; Wang et al., 2011; Zhou et al., 2013). Additional contact sites have been proposed, including the C2A domain and the bottom surface of the C2B domain, where R398 and R399 are located (Lai et al., 2011; Zhou et al., 2013). A previous study concluded that this bottom surface is close to the SNARE interface, but still sufficiently exposed to allow other interactions (Choi et al., 2010). Mutations of negative charges in the middle region of SNAP-25, which interact with the Syt1 polybasic region, are sufficient to produce a docking defect in chromaffin cells (Mohrmann et al., 2013), while Syt1-RQ-expressing cells show no docking defect (Fig. 2). Hence, docking tolerates mutations in the C2B domain bottom region, but not the disruption of Syt1–tSNARE interaction. This suggests that the Syt1–tSNARE interaction is probably preserved in Syt1-RQ and Syt1-RA, and the interaction with other phospholipids (Martens et al., 2007; Gaffaney et al., 2008; Kuo et al., 2009; Wang et al., 2011; Park et al., 2012; Vennekate et al., 2012) might be disrupted and exposed upon removal of PI(4,5)P2 in the SUV/GUV docking assay. Indeed, R398A impaired Syt1 binding to SNARE liposomes without PI(4,5)P2 (Gaffaney et al., 2008), and two studies that measured Ca2+-dependent liposome aggregation observed that R398/399Q abolished liposome aggregation (Araç et al., 2006; Xue et al., 2008). In our GUV/SUV assay, docking was preserved with the R398/399Q and RA mutations as long as PI(4,5)P2 was present (Fig. 3). Finally, separate interactions with the tSNAREs might also account for the role of the C2B bottom surface in PI(4,5)P2-dependent docking. Probably the most likely scenario is that docking is effectuated via multiple interactions, which are not necessarily of high affinity, but due to their avidity and a potential involvement of multiple Syt1 molecules on the vesicle (cooperativity), can effectively dock vesicles to the target membrane. It remains to be determined to what extent the strict PI(4,5)P2 dependence of docking by Syt1-RQ- and Syt1-RA-containing vesicles recapitulates the situation in living cells (something that cannot be easily tested), but in any case, our data show that in intact chromaffin cells, docking is probably driven by redundant electrostatic interactions in any of a number of different alternative configurations.
The role of Syt1 in vesicle priming and fusion
In line with previous results, wild-type cells and syt1 null cells expressing Syt1 showed similar secretion levels (Nagy et al., 2006). Syt1-RQ failed to restore the RRP and even tended to reduce remaining release further (although not significantly; Fig. 4), and Syt1-RQ and Syt1-RA mutants did not show Ca2+-induced SUV–GUV lipid mixing (Fig. 5). Hence, our data confirm the importance of the C2B domain bottom residues of Syt1 for calcium-triggered synchronous release, as observed before in neurons (Xue et al., 2008). Similar to this study, none of our experiments in chromaffin cells expressing the Syt1-RQ mutant produced evidence that this mutant retains any fusion activity. However, the SUV–GUV lipid mixing assay revealed that Syt1-RQ and Syt1-RA show substantial Ca2+-independent lipid mixing (but not Ca2+ dependent), consistent with their unaltered vesicle docking properties in the presence of PI(4,5)P2. Although Ca2+-independent lipid mixing is a well known property of Syt1 in cell-free systems (Gaffaney et al., 2008; Malsam et al., 2012), is not known to what extent this activity relates to aspects of Syt1 function in living cells, for instance, to spontaneous synaptic vesicle fusion in synapses [not reported by Xue et al. (2008)]. Chromaffin cells hardly show spontaneous events. In fact, we detected not a single amperometric spike during 180 s recordings of six chromaffin cells expressing either Syt1-wt and Syt1-RQ (data not shown). Together with the overwhelming evidence for the involvement of the Ca2+-binding loops at the “top” of the C2B domain in fusion (Rizo et al., 2006; Chapman, 2008; Jahn and Fasshauer, 2012; Kim et al., 2012; Südhof, 2012), the clear role for the arginines in the bottom domain suggests that these two opposite sides of the C2B domain participate in the final Ca2+-dependent triggering of fusion, as proposed previously (Araç et al., 2006; Xue et al., 2008).
Expression of Syt2 R399Q/400Q in wild type calyx of Held resulted in dominant-negative effects on neurotransmitter release when stimulated by depolarization, but not upon Ca2+ uncaging (Young and Neher, 2009). It was concluded that Syt2 RQ is defective in positional priming, such that vesicles are mislocalized with respect to Ca2+ channels. Isolated chromaffin cells have no active zones as neuronal synapses do, and positional priming is not a major phenomenon. However, a small vesicle pool (IRP) has been identified that can be triggered to fuse with short (10 ms) depolarizations, expected to produce discrete, local increases in intracellular Ca2+ (Voets et al., 1999). These vesicles are colocalized with Ca2+ channels, but it is not clear whether they are molecularly different from other RRP vesicles. IRP vesicles might be drawn from a homogeneous RRP during short depolarizations by chance. In any case, the IRP is not reduced upon expression of the Syt1-RQ mutant in wild-type cells, and Ca2+ uncaging did not result in enhanced secretion of potentially mislocalized vesicles when expressing the Syt1-RQ mutation in syt1 null or wild-type cells. Hence, we conclude that the Syt1-RQ mutation does not affect positional priming in chromaffin cells. Instead, our data support the idea that the Syt1 bottom domain physically and locally couples SNARE or lipid interactions to the Ca2+-dependent reactions mediated by the top domain (Araç et al., 2006; Xue et al., 2008). We conclude that Syt1-C2B bottom domain is dispensable for docking due to the existence of multiple alternative docking mechanisms, but plays a crucial role in vesicle secretion, indicating that the C2B domain bottom residues are required specifically for a post-docking step to achieve calcium-dependent vesicle secretion.
Footnotes
This work was supported by grants from the Erasmus Mundus Joint Doctorate Program (SGA 2011-1632-001-EMJD; G.H.K.), the Fonds Economische Structuurversterking NeuroBasic PharmaPhenomics grant (Agentschap NL FES0908; M.V.), the European Research Council (ERC-ADG-322966-DCVfusion; M.V.), the Netherlands Organization for Scientific Research (MEERVOUD-836.10.002; H.d.W.), the European Neuroscience Campus Network (Cycle 2-project 7; H.d.W., M.V.), the Lundbeck Center for Biomembranes in Nanomedicine (J.B.S.), and the Novo Nordisk Foundation (J.B.S.). We thank Joke Wortel, Christiaan van der Meer, Joost Hoetjes, Frank den Oudsten, and Erik Ceelen for breeding and genotyping mice, Robbert Zalm for cloning and producing viral vectors, and Anne Marie Nordvig Petersen for excellent technical assistance. We thank Prof. Dr. Christian Rosenmund (Charite Medical University, Berlin) for providing the Syt1-RQ mutant construct and Dr. Sophie van der Sluis for advice on the statistical analysis of nested data.
The authors declare no conflict of interest.
References
- Aarts E, Verhage M, Veenvliet JV, Dolan CV, van der Sluis S. A solution to dependency: using multilevel analysis to accommodate nested data. Nat Neurosci. 2014;17:491–496. doi: 10.1038/nn.3648. [DOI] [PubMed] [Google Scholar]
- Alvarez YD, Marengo FD. The immediately releasable vesicle pool: highly coupled secretion in chromaffin and other neuroendocrine cells. J Neurochem. 2011;116:155–163. doi: 10.1111/j.1471-4159.2010.07108.x. [DOI] [PubMed] [Google Scholar]
- Araç D, Chen X, Khant HA, Ubach J, Ludtke SJ, Kikkawa M, Johnson AE, Chiu W, Südhof TC, Rizo J. Close membrane-membrane proximity induced by Ca(2+)-dependent multivalent binding of synaptotagmin-1 to phospholipids. Nat Struct Mol Biol. 2006;13:209–217. doi: 10.1038/nsmb1056. [DOI] [PubMed] [Google Scholar]
- Bruns D. Detection of transmitter release with carbon fiber electrodes. Methods. 2004;33:312–321. doi: 10.1016/j.ymeth.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Chapman ER. How does synaptotagmin trigger neurotransmitter release? Annu Rev Biochem. 2008;77:615–641. doi: 10.1146/annurev.biochem.77.062005.101135. [DOI] [PubMed] [Google Scholar]
- Choi UB, Strop P, Vrljic M, Chu S, Brunger AT, Weninger KR. Single-molecule FRET-derived model of the synaptotagmin 1-SNARE fusion complex. Nat Struct Mol Biol. 2010;17:318–384. doi: 10.1038/nsmb.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit H, Walter AM, Milosevic I, Gulyás-Kovács A, Riedel D, Sørensen JB, Verhage M. Synaptotagmin-1 docks secretory vesicles to syntaxin-1/SNAP-25 acceptor complexes. Cell. 2009;138:935–946. doi: 10.1016/j.cell.2009.07.027. [DOI] [PubMed] [Google Scholar]
- Ehrengruber MU, Lundstrom K. Alphaviruses: Semliki Forest virus and Sindbis virus vectors for gene transfer into neurons. Curr Protoc Neurosci. 2007;4:4.22. doi: 10.1002/0471142301.ns0422s41. [DOI] [PubMed] [Google Scholar]
- Fernández-Chacón R, Königstorfer A, Gerber SH, García J, Matos MF, Stevens CF, Brose N, Rizo J, Rosenmund C, Südhof TC. Synaptotagmin I functions as a calcium regulator of release probability. Nature. 2001;410:41–49. doi: 10.1038/35065004. [DOI] [PubMed] [Google Scholar]
- Fernandez I, Araç D, Ubach J, Gerber SH, Shin O, Gao Y, Anderson RG, Südhof TC, Rizo J. Three-dimensional structure of the synaptotagmin 1 C2B-domain: synaptotagmin 1 as a phospholipid binding machine. Neuron. 2001;32:1057–1069. doi: 10.1016/S0896-6273(01)00548-7. [DOI] [PubMed] [Google Scholar]
- Gaffaney JD, Dunning FM, Wang Z, Hui E, Chapman ER. Synaptotagmin C2B domain regulates Ca2+-triggered fusion in vitro: critical residues revealed by scanning alanine mutagenesis. J Biol Chem. 2008;283:31763–31775. doi: 10.1074/jbc.M803355200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geppert M, Goda Y, Hammer RE, Li C, Rosahl TW, Stevens CF, Südhof TC. Synaptotagmin I: a major Ca2+ sensor for transmitter release at a central synapse. Cell. 1994;79:717–727. doi: 10.1016/0092-8674(94)90556-8. [DOI] [PubMed] [Google Scholar]
- Honigmann A, van den Bogaart G, Iraheta E, Risselada HJ, Milovanovic D, Mueller V, Müllar S, Diederichsen U, Fasshauer D, Grubmüller H, Hell SW, Eggeling C, Kühnel K, Jahn R. Phosphatidylinositol 4,5-bisphosphate clusters act as molecular beacons for vesicle recruitment. Nat Struct Mol Biol. 2013;20:679–686. doi: 10.1038/nsmb.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imig C, Min SW, Krinner S, Arancillo M, Rosenmund C, Südhof TC, Rhee J, Brose N, Cooper BH. The morphological and molecular nature of synaptic vesicle priming at presynaptic active zones. Neuron. 2014;84:416–431. doi: 10.1016/j.neuron.2014.10.009. [DOI] [PubMed] [Google Scholar]
- Jahn R, Scheller RH. SNAREs–engines for membrane fusion. Nat Rev Mol Cell Biol. 2006;7:631–643. doi: 10.1038/nrm2002. [DOI] [PubMed] [Google Scholar]
- Jahn R, Fasshauer D. Molecular machines governing exocytosis of synaptic vesicles. Nature. 2012;490:201–207. doi: 10.1038/nature11320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen EM, Hartwieg E, Schuske K, Nonet ML, Jin Y, Horvitz HR. Defective recycling of synaptic vesicles in synaptotagmin mutants of Caenorhabditis elegans. Nature. 1995;378:196–199. doi: 10.1038/378196a0. [DOI] [PubMed] [Google Scholar]
- Kim JY, Choi BK, Choi MG, Kim SA, Lai Y, Shin YK, Lee NK. Solution single-vesicle assay reveals PIP2-mediated sequential actions of synaptotagmin-1 on SNAREs. EMBO J. 2012;31:2144–2155. doi: 10.1038/emboj.2012.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo W, Herrick DZ, Ellena JF, Cafiso DS. The calcium-dependent and calcium-independent membrane binding of synaptotagmin 1: two modes of C2B binding. J Mol Biol. 2009;387:284–294. doi: 10.1016/j.jmb.2009.01.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai AL, Huang H, Herrick DZ, Epp N, Cafiso DS. Synaptotagmin 1 and SNAREs form a complex that is structurally heterogeneous. J Mol Biol. 2011;405:696–706. doi: 10.1016/j.jmb.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindau M, Neher E. Patch-clamp techniques for time-resolved capacitance measurements in single cells. Pflugers Arch. 1988;411:137–146. doi: 10.1007/BF00582306. [DOI] [PubMed] [Google Scholar]
- Loewen CA, Lee SM, Shin YK, Reist NE. C2B polylysine motif of synaptotagmin facilitates a Ca2+-independent stage of synaptic vesicle priming in vivo. Mol Biol Cell. 2006;17:5211–5226. doi: 10.1091/mbc.E06-07-0622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahal LK, Sequeira SM, Gureasko JM, Söllner TH. Calcium-independent stimulation of membrane fusion and SNAREpin formation by synaptotagmin I. J Cell Biol. 2002;158:273–282. doi: 10.1083/jcb.200203135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malsam J, Parisotto D, Bharat TA, Scheutzow A, Krause JM, Briggs JA, Söllner TH. Complexin arrests a pool of docked vesicles for fast Ca2+-dependent release. EMBO J. 2012;31:3270–3281. doi: 10.1038/emboj.2012.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens S, Kozlov MM, McMahon HT. How synaptotagmin promotes membrane fusion. Science. 2007;316:1205–1208. doi: 10.1126/science.1142614. [DOI] [PubMed] [Google Scholar]
- Mohrmann R, de Wit H, Connell E, Pinheiro PS, Leese C, Bruns D, Davletov B, Verhage M, Sørensen JB. Synaptotagmin interaction with SNAP-25 governs vesicle docking, priming, and fusion triggering. J Neurosci. 2013;33:14417–14430. doi: 10.1523/JNEUROSCI.1236-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy G, Kim JH, Pang ZP, Matti U, Rettig J, Südhof TC, Sørensen JB. Different effects on fast exocytosis induced by synaptotagmin 1 and 2 isoforms and abundance but not by phosphorylation. J Neurosci. 2006;26:632–643. doi: 10.1523/JNEUROSCI.2589-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang Z, Südhof T. Cell biology of Ca2+-triggered exocytosis. Curr Opin Cell Biol. 2010;22:496–505. doi: 10.1016/j.ceb.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang ZP, Sun J, Shin OH, Meyer AC, Rosenmund C, Südhof TC. A gain-of-function mutation in synaptotagmin-1 reveals a critical role of Ca2+-dependent soluble N-ethylmaleimide- sensitive factor attachment protein receptor complex binding in synaptic exocytosis. J Neurosci. 2006;26:12556–12565. doi: 10.1523/JNEUROSCI.3804-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisotto D, Malsam J, Scheutzow A, Krause JM, Söllner TH. SNAREpin assembly by Munc18-1 requires previous vesicle docking by synaptotagmin 1. J Biol Chem. 2012;287:31041–31049. doi: 10.1074/jbc.M112.386805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park Y, Hernandez JM, van den Bogaart G, Ahmed S, Holt M, Riedel D, Jahn R. Controlling synaptotagmin activity by electrostatic screening. Nat Struct Mol Biol. 2012;19:991–997. doi: 10.1038/nsmb.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reist NE, Buchanan J, Li J, DiAntonio A, Buxton EM, Schwarz TL. Morphologically docked synaptic vesicles are reduced in synaptotagmin mutants of Drosophila. J Neurosci. 1998;18:7662–7673. doi: 10.1523/JNEUROSCI.18-19-07662.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickman C, Archer DA, Meunier FA, Craxton M, Fukuda M, Burgoyne RD, Davletov B. Synaptotagmin interaction with the syntaxin/SNAP-25 dimer is mediated by an evolutionarily conserved motif and is sensitive to inositol hexakisphosphate. J Biol Chem. 2004;279:12574–12579. doi: 10.1074/jbc.M310710200. [DOI] [PubMed] [Google Scholar]
- Rizo J, Chen X, Araç D. Unraveling the mechanisms of synaptotagmin and SNARE function in neurotransmitter release. Trends Cell Biol. 2006;16:339–350. doi: 10.1016/j.tcb.2006.04.006. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonn JS, Maximov A, Lao Y, Südhof TC, Sørensen JB. Synaptotagmin-1 and -7 are functionally overlapping Ca2+ sensors for exocytosis in adrenal chromaffin cells. Proc Natl Acad Sci U S A. 2008;105:3998–4003. doi: 10.1073/pnas.0712373105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sørensen JB, Fernández-Chacón R, Südhof TC, Neher E. Examining synaptotagmin 1 function in dense core vesicle exocytosis under direct control of Ca2+ J Gen Physiol. 2003;122:265–276. doi: 10.1085/jgp.200308855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC. Calcium control of neurotransmitter release. Cold Spring Harbor Perspect Biol. 2012;4:a011353. doi: 10.1101/cshperspect.a011353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC, Rothman JE. Membrane fusion: grappling with SNARE and SM proteins. Science. 2009;323:474–477. doi: 10.1126/science.1161748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vennekate W, Schröder S, Lin CC, van den Bogaart G, Grunwald M, Jahn R, Walla PJ. Cis- and trans-membrane interactions of synaptotagmin-1. Proc Natl Acad Sci U S A. 2012;109:11037–11042. doi: 10.1073/pnas.1116326109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhage M, Sørensen JB. Vesicle docking in regulated exocytosis. Traffic. 2008;9:1414–1424. doi: 10.1111/j.1600-0854.2008.00759.x. [DOI] [PubMed] [Google Scholar]
- Voets T. Dissection of three Ca2+-dependent steps leading to secretion in chromaffin cells from mouse adrenal slices. Neuron. 2000;28:537–545. doi: 10.1016/S0896-6273(00)00131-8. [DOI] [PubMed] [Google Scholar]
- Voets T, Neher E, Moser T. Mechanisms underlying phasic and sustained secretion in chromaffin cells from mouse adrenal slices. Neuron. 1999;23:607–615. doi: 10.1016/S0896-6273(00)80812-0. [DOI] [PubMed] [Google Scholar]
- Voets T, Moser T, Lund PE, Chow RH, Geppert M, Südhof TC, Neher E. Intracellular calcium dependence of large dense-core vesicle exocytosis in the absence of synaptotagmin I. Proc Natl Acad Sci U S A. 2001;98:11680–11685. doi: 10.1073/pnas.201398798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter A, Pinheiro P, Verhage M, Sørensen J. A sequential vesicle pool model with a single release sensor and a Ca2+-dependent priming catalyst effectively explains Ca2+-dependent properties of neurosecretion. PLoS Comput Biol. 2013;9:e1003362. doi: 10.1371/journal.pcbi.1003362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Liu H, Gu Y, Chapman ER. Reconstituted synaptotagmin I mediates vesicle docking, priming, and fusion. J Cell Biol. 2011;195:1159–1170. doi: 10.1083/jcb.201104079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber T, Zemelman BV, McNew JA, Westermann B, Gmachl M, Parlati F, Söllner TH, Rothman JE. SNAREpins: minimal machinery for membrane fusion. Cell. 1998;92:759–772. doi: 10.1016/S0092-8674(00)81404-X. [DOI] [PubMed] [Google Scholar]
- Xue M, Ma C, Craig TK, Rosenmund C, Rizo J. The Janus-faced nature of the C(2)B domain is fundamental for synaptotagmin-1 function. Nat Struct Mol Biol. 2008;15:1160–1168. doi: 10.1038/nsmb.1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young SM, Jr, Neher E. Synaptotagmin has an essential function in synaptic vesicle positioning for synchronous release in addition to its role as a calcium sensor. Neuron. 2009;63:482–496. doi: 10.1016/j.neuron.2009.07.028. [DOI] [PubMed] [Google Scholar]
- Zhou A, Brewer KD, Rizo J. Analysis of SNARE complex/synaptotagmin-1 interactions by one-dimensional NMR spectroscopy. Biochemistry. 2013;52:3446–3456. doi: 10.1021/bi400230u. [DOI] [PMC free article] [PubMed] [Google Scholar]