

Abstract
Reduction in temperature depolarizes neurons by a partial closure of potassium channels but decreases the vesicle release probability within synapses. Compared with cooling, neuromodulators produce qualitatively similar effects on intrinsic neuronal properties and synapses in the cortex. We used this similarity of neuronal action in ketamine–xylazine-anesthetized mice and non-anesthetized mice to manipulate the thalamocortical activity. We recorded cortical electroencephalogram/local field potential (LFP) activity and intracellular activities from the somatosensory thalamus in control conditions, during cortical cooling and on rewarming. In the deeply anesthetized mice, moderate cortical cooling was characterized by reversible disruption of the thalamocortical slow-wave pattern rhythmicity and the appearance of fast LFP spikes, with frequencies ranging from 6 to 9 Hz. These LFP spikes were correlated with the rhythmic IPSP activities recorded within the thalamic ventral posterior medial neurons and with depolarizing events in the posterior nucleus neurons. Similar cooling of the cortex during light anesthesia rapidly and reversibly eliminated thalamocortical silent states and evoked thalamocortical persistent activity; conversely, mild heating increased thalamocortical slow-wave rhythmicity. In the non-anesthetized head-restrained mice, cooling also prevented the generation of thalamocortical silent states. We conclude that moderate cortical cooling might be used to manipulate slow-wave network activity and induce neuromodulator-independent transition to activated states.
SIGNIFICANCE STATEMENT In this study, we demonstrate that moderate local cortical cooling of lightly anesthetized or naturally sleeping mice disrupts thalamocortical slow oscillation and induces the activated local field potential pattern. Mild heating has the opposite effect; it increases the rhythmicity of thalamocortical slow oscillation. Our results demonstrate that slow oscillation can be influenced by manipulations to the properties of cortical neurons without changes in neuromodulation.
Keywords: cortex, homeostasis, neuromodulation, oscillations, sleep, thalamus
Introduction
Electrical stimulation of ascending brainstem systems disrupts the high-voltage slow-wave electroencephalogram (EEG) pattern and induces cortical activation (Moruzzi and Magoun, 1949). Previous studies have shown that the transitions between sleep and waking are controlled by neuromodulatory systems (Steriade et al., 1993b; Saper, 2006; Lee and Dan, 2012). During slow-wave sleep or anesthesia, when neuromodulatory tone is low, the cortical network generates slow waves that, at an intracellular level, appear as an alternation of hyperpolarized or silent states with minimal levels of synaptic activity and depolarized or active states with high levels of synaptic activities (Steriade et al., 2001; Timofeev et al., 2001a; Chauvette et al., 2011). An elevated level of neuromodulators during wakefulness and rapid eye movement (REM) sleep strongly reduces the activity of the membrane potassium channels, which depolarizes multiple types of cortical neurons (Krnjević et al., 1971; McCormick, 1992; Hasselmo and McGaughy, 2004; Steriade and McCarley, 2005). In these conditions, the cortical network activity switches to persistent active states (Timofeev et al., 2000; Steriade et al., 2001). In modeling studies, the light increase in the resting membrane potential evoked by the partial blocking of potassium leakage channels was sufficient to transform thalamocortical slow-wave activity into wake-like persistent activity (Bazhenov et al., 2002; Compte et al., 2003; Hill and Tononi, 2005). At the level of synapses, acetylcholine reduces the release probability in the cortex and reduces short-term synaptic depression (Gil et al., 1997; Hasselmo and McGaughy, 2004; Eggermann and Feldmeyer, 2009; Chauvette et al., 2012). However, the high efficacy of strong intracortical connections remains preserved in elevated levels of acetylcholine (Hasselmo and McGaughy, 2004; Runfeldt et al., 2014). As a net effect, the mean membrane potential of cortical neurons remains the same during the waking stage, REM sleep, and active phases of slow-wave sleep (Steriade et al., 2001; Timofeev et al., 2001a).
A moderate reduction in temperature below 37°C has been found to reduce activities of potassium channels, resulting in increased neuronal input resistance and the depolarization of resting membrane potential (Volgushev et al., 2000; Trevelyan and Jack, 2002). A similar decrease in temperature has been found to reduce the vesicle release probability within synapses, increase latency, or lead to asynchronous release of neurotransmitters (Katz and Miledi, 1965; Jasper et al., 1970; Hardingham and Larkman, 1998; Volgushev et al., 2004; Yang et al., 2005). Only deep cooling, below 10°C, completely silenced the network with a depolarizing block of sodium channels and abolished synaptic release (Jasper et al., 1970; Volgushev et al., 2000). We hypothesized that mild/moderate cooling (heating) of the cortex can be used as a fast and reversible tool to manipulate network activities via its bidirectional effects on intrinsic neuronal excitability and synaptic release properties. In the present study, we found that moderate cortical cooling indeed eliminated silent states and caused an activated EEG pattern in lightly anesthetized and non-anesthetized sleeping animals. Under the lesser physiological condition of deep anesthesia, cooling disrupted the field potential slow waves and promoted appearance of bursts of fast local field potential (LFP) spikes, which were correlated with thalamic activities.
Materials and Methods
Experiments on anesthetized mice.
Experiments were performed in accordance with the guidelines of the Canadian Council on Animal Care and approved by the Université Laval Committee on Ethics and Animal Research. Adult CD1 (white) and C57BL/6 (black) mice of both sexes were used in experiments as described previously (Sheroziya and Timofeev, 2014). Briefly, the mice were anesthetized with ketamine and xylazine (100 and 10 mg/kg, i.p.), and the head was placed in a stereotaxic frame. Two screws (stainless steel, 1 mm diameter) were inserted into the bone over the right somatosensory cortex and cerebellum (reference electrode) to monitor the EEG slow oscillation. Additional anesthesia was administrated every 30–40 min when the EEG slow waves began to weaken. In some experiments, we used light anesthesia defined by EEG activity (spontaneous alterations of rhythmic slow waves and short periods of activated-like EEG, with no additional injections of ketamine–xylazine during the recording; see below). The temperature of the platform in the stereotaxic frame on which the mice were placed was maintained at 37°C. The gentle opening of the skull was done over the area of interest, and the dura was removed carefully. To reduce the brain pressure and pulsation, we opened the fourth ventricle. The stability of the intracellular and LFP recordings was also reinforced by covering the opened cortical surface with 4% agar in saline. At the end of the recording session, the mice were killed using a lethal dose of sodium pentobarbital and perfused with 4% of paraformaldehyde (PFA) dissolved in 0.1 m phosphate buffer (PB).
Recordings and labeling.
AM 3000 amplifiers (A-M Systems) with custom modifications were used for the EEG and LFP recordings (sampling rate of 10 kHz). The LFPs were recorded using monopolar tungsten electrodes (10–12 MΩ; FHC). The EEG recordings were obtained from a screw inserted into the bone over the right somatosensory cortex. The LFP recordings were made in the left primary motor cortex (M1; lateral, 1.5–2 mm and anteroposterior, 0.7–0.2 mm) and primary somatosensory cortex (S1; lateral, 1–3 mm and anteroposterior, −1 to −2 mm) according to atlas coordinates (Paxinos and Franklin, 2001). Intracellular recordings from the cortices and the thalamus were performed using glass micropipettes pulled from borosilicate glass capillaries (WPI; P-97 from Sutter Instruments) and filled with a solution of 2 m potassium acetate (40–60 MΩ). For the recordings in the thalamus, the pipettes were also filled with 1% biocytin (Sigma-Aldrich). A high-impedance amplifier (Neurodata IR-283 amplifiers; Cygnus Technology) with an active bridge circuitry was used to record the membrane potential and to inject current into the neurons. To stain the thalamic cells, positive current pulses (200 ms on–off duty cycle, 0.5–1.5 nA, 10 min) were applied through the recording pipette. After 1–2 h of survival, the deeply anesthetized mice were perfused through the heart with saline, followed by 4% PFA in PB. The brains were then removed and stored overnight in 4% PFA in PB at 4°C. Then, 50-μm-thick frontal sections were cut.
To stimulate whiskers or fur, we used a small tube (1 mm diameter) connected to a compressed air balloon and equipped with a trigger.
Immunohistochemistry.
To visualize the biocytin-labeled cells, the free-floating sections were incubated in an ABC kit (1:200; Vector Laboratories) in Tris-buffered saline (TBS) for 2 h. After several rinses, the sections were incubated in TBS containing 0.05% 3,3-diaminobenzidine tetrahydrochloride (Sigma-Aldrich), 0.005% CoCl2, and 0.00125% H2O2 for 5–10 min. In the end, all of the sections were mounted onto gelatin-coated glass slides, dehydrated, and coverslipped.
Cooling of the cortex and cortical inactivation with tetrodotoxin.
To cool the cortex in acute experiments, we used the U-shaped duralumin plate (Fig. 1A, left) equipped with the Peltier module connected to the radiator. To monitor the temperature, the temperature sensor was fixed on the duralumin plate between the Peltier module and the tip of the plate (∼2 mm diameter) touching the cortex. The cortex and the plate were covered with agar. The temperature of the plate was controlled by the amperage and the polarity of the Peltier module inputs. Because the temperature sensor was fixed on the plate (not on the cortex), it did not detect the actual cortical temperature. To estimate the cortical temperature corresponding to the readings of the temperature sensor used in all experiments, we performed calibration. We positioned the second temperature sensor (∼0.6 mm diameter) either between the cortex and the duralumin plate or inserted it into the cortex (∼0.5–0.7 mm depth) next to the plate and covered them with agar. Changing the temperature of the plate, we then plotted the dependency between the temperatures from the sensors (3–4 min after the change of amperage of the Peltier module inputs; Fig. 1B). The temperature measured from the cortical surface yielded the maximal possible change in the cortical temperature (Fig. 1C). The temperature from the depth yielded the average change in the cortical temperature corresponding to the readings of the sensor on the duralumin plate (Fig. 1C). We used the obtained curves to estimate the actual cortical temperature in other experiments. To reach fast, but safe, cooling and heating, we used an average cortical temperature ranging from 29–31°C to 39–40°C. In control conditions, the plate temperature was set at 37–39°C (corresponding to 36–37°C in the cortex; Fig. 1C). To cool the cortex, the plate temperature was lowered to 16–21°C (corresponding to 29–31°C in the cortex and 25–26°C on the surface; Fig. 1C). To either rewarm or heat the cortex, the plate temperature was increased to the control conditions or set at 50°C (corresponding to 39–40°C in the cortex and ∼42°C on the surface; Fig. 1C). The duration of cooling/heating usually lasted 3–5 min. Membrane potential depolarization evoked by cooling can be estimated using previously obtained in vitro results (Volgushev et al., 2000), showing that a 1°C drop in temperature produced ∼1.33 mV depolarization of the resting membrane potential.
Figure 1.

Cortical cooling in acute and chronic experiments. A, Drawing shows the shape and position of cooling duralumin plates in acute (left, the plate shown with gray) and chronic (right) experiments. B, Calibration of the cooling plate. The black and red curves show the temperature of the plate and the cortex, respectively. C, The average cortical temperature (red curve) and the surface temperature (blue curve) plotted against the cooling plate temperature. The temperatures used in control, during cooling and heating, are shown with gray (see Materials and Methods).
In some of the acute experiments, we inactivated the cortex with tetrodotoxin (TTX; 0.3–1 mm; Sigma-Aldrich). A paper napkin moistened with TTX in saline was laid on the cortex (the dura was removed) for at least 40 min.
Cooling of the cortex of the non-anesthetized head-restrained mice.
For the chronic experiments, the mice were anesthetized with isoflurane and placed in the stereotaxic frame as described above. Then, the bone was removed to position the 3 × 6 mm concave duralumin plate over the cortical surface (Fig. 1A, right). Two LFP and two electromyogram (EMG) electrodes were inserted into the frontal motor cortex of both hemispheres and in the neck muscles, respectively. The stainless steel screw (1 mm diameter) fastened in the skull above the cerebellum was used as a reference electrode. The duralumin plate and the electrodes were fixed with dental cement. The upper tip of the duralumin plate was left uncovered so it could be connected to the Peltier module. The temperature sensor was connected between the Peltier module and the duralumin plate. After 3 d of recovery within the home cage, the mice were connected to the Peltier module (the implanted duralumin plate was used to fix the head of the mouse and to cool the cortex), and the initial temperature of the cage was maintained at 37–38°C. To cool the cortex, we used the same temperature of the plate (16–21°C) that was used in the acute experiments, and we did not use cortical heating in the chronic experiments. Because the working surface of the implanted plates was higher than the plates in the acute experiments whereas the thickness of the plate and the power of the cooling module were the same, the actual cortical temperature drop in the chronic experiments was obviously smaller than in the acute experiments, but it was sufficient to confirm the results we obtained in the anesthetized animals (see below). To decrease the movement activity of the mice during the recording sessions, the animals were placed in a glass tube (∼4 cm diameter) while their head movements were restrained. It usually took 4–6 d to train the mice to be still and to sleep under these head-restrained conditions.
Electrophysiological data analysis.
To analyze the cortical LFP activities, we calculated the absolute values (γ2)1/2 of the cortical high-frequency gamma oscillation (bandpass filter, 40–300 Hz) LFP activities and plotted their autocorrelations. To obtain the smoothed gamma LFP signal, we then filtered the obtained absolute value (bandpass filter, 0–10 Hz). To plot the distributions of the intracellular membrane potentials and to average the distributions, we used the dimensionless z score: z(x) = (x − mean(x))/SD(x), where x is the filtered membrane potential (bandpass filter, 0–10 Hz), mean(x) is the average value of the membrane potential, and SD(x) is the SD of the membrane potential calculated for each condition (Hahn et al., 2006). To analyze the thalamic intracellular recordings, we extracted postsynaptic potentials (PSPs)/noise from intracellular activities. The negative peaks of the IPSPs within the thalamic ventral posterior medial nucleus (VPM) neurons were detected manually (Sheroziya and Timofeev, 2014). To extract the EPSPs within the posterior nucleus (PO) neurons, the intracellular activity was filtered between 0 and 1000 Hz and then differentiated, and the threshold EPSP detection was set to 0.1 V/s. To reveal the slow-wave modulation of the intracellular thalamic activity, we plotted the autocorrelograms of the extracted postsynaptic events and averaged the intracellular thalamic activities against the extracted events (see Figs. 5, 6, 8, 9). To reveal the changes in thalamocortical synchrony, we averaged the cortical EEG and LFP (correlograms) against the extracted thalamic PSPs. The delta power was calculated from a 5 s sliding window (1 s sliding step) as an integral power between 0.2 and 4 Hz of the full spectrogram.
Figure 5.
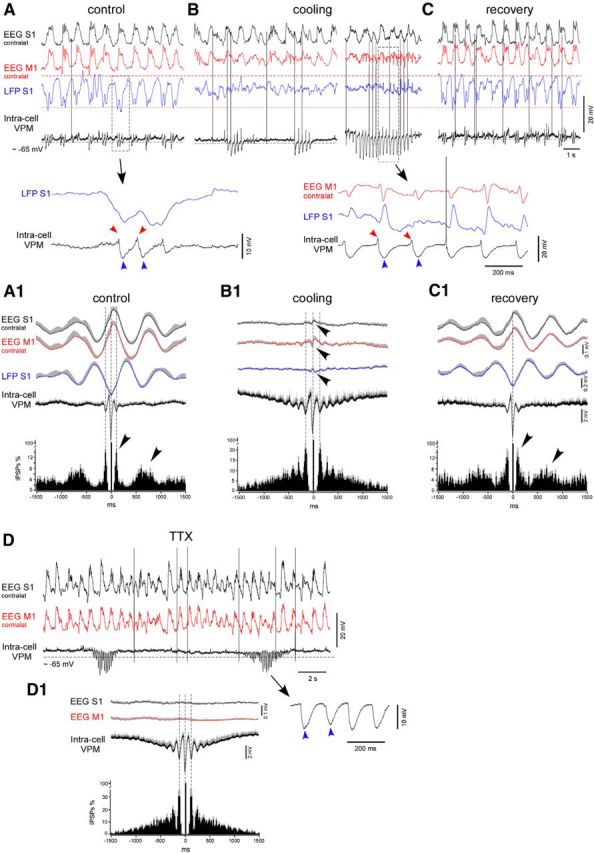
Cortical cooling disrupted the slow-wave pattern and evoked spindle-like activity within VPM neurons in deeply anesthetized mice. A–C, An example of simultaneously recorded EEG (contralateral M1 and S1), LFP (ipsilateral S1), and intracellular activities from the thalamic VPM neuron in control, during cooling, and after recovery, respectively. Bottom traces show one slow-wave cycle in control (left) and cooling-triggered intracellular spindle-like IPSPs correlated to LFP spikes (right). A, Control. B, Cooling of the somatosensory cortex. Electrographic activities of the same electrodes are shown 1 min (left) and 2 min (right) after the beginning of cooling. C, Recovery from the cooling. Electrographic activities of the same electrodes are shown 1 min after the switch from cooling to heating. A1–C1, Averaged EEG, LFP, intracellular activity, and IPSP autocorrelograms (n = 7 neurons from 7 mice). EEG, LFP, and intracellular activities were averaged against (triggered by) extracted IPSPs. Before averaging in a group, each IPSPs autocorrelogram was normalized to 100%. A1, Control. B1, Cooling. C1, Recovery. D, An example of thalamic VPM intracellular activity recorded after (40 min) TTX application over a large ipsilateral cortical area (motor, somatosensory, visual). D1, Average IPSPs autocorrelogram and intracellular thalamic and field potential cortical activities (n = 7 neurons, at least 40 min after TTX application). In all panels, the positive part of the SEM is shown with gray (negative part is not shown).
Figure 6.

Cortical cooling disrupted the slow-wave pattern and evoked rhythmic depolarizing events within PO neurons in deeply anesthetized mice. A–C, An example of simultaneously recorded EEG (contralateral M1 and S1), LFP (ipsilateral S1), and intracellular activities from the thalamic PO neuron in control (A), during cooling (B), and after recovery (C). Bottom traces show one slow-wave cycle in control (left) and cooling-triggered intracellular EPSPs correlated to LFP spikes (right). A1–C1, Averaged EEG, LFP, intracellular activity, and EPSP autocorrelograms (n = 6 recordings from 6 mice). EEG, LFP, and intracellular activities were averaged against (triggered by) extracted EPSPs. Before averaging in a group, each autocorrelogram was normalized to 100%. A1, Control. B1, Cooling. C1, Recovery. In all panels, the positive part of SEM is shown with gray (negative part is not shown).
Figure 8.

VPM intracellular activity of a lightly anesthetized mouse during cortical heating and cooling. A, Simultaneously recorded contralateral EEG, ipsilateral LFP, and intracellular VPM activities in control, during cortical heating, and after cortical cooling. Heating increased the rhythmicity of VPM IPSPs, whereas cooling eliminated silent states, evoked depolarization, and increased synaptic noise. A typical response with a low-threshold spike-burst elicited by a hyperpolarizing current pulse application (see inset) confirmed the quality of recording. B, IPSP autocorrelogram and averaged (triggered by IPSPs) cortical activities. The histograms were normalized to 100%. C, Identification of the VPM neuron. The neuron responded to whisker stimulation (air puff).
Figure 9.

Higher-order thalamic PO intracellular activity of a lightly anesthetized mouse in control, during cortical cooling, and after heating. A, Simultaneously recorded contralateral EEG, ipsilateral LFP, and intracellular PO activities in control, during cortical cooling, and after cortical heating. Cooling eliminated silent states and evoked persistent activity. Note the persistent synaptic noise compared with the silent states in control (bottom traces). B, EPSP autocorrelogram and averaged (triggered by EPSPs) cortical activities. Cortical heating increased the thalamocortical synchronization. C, Light microscopic image of the stained PO neuron.
Wave analysis was performed using Igor Pro version 6.2 software (WaveMetrics). Basic statistical analysis and other data analyses were performed using SigmaPlot (Systat Software). Data are expressed as means ± SD or SEM when indicated. If the data passed the normality and equal variance tests (p > 0.05), then a two-tail t test was used; otherwise, a Mann–Whitney rank-sum test was used (significance level was set at p < 0.05).
Results
Moderate cooling of the cortex eliminates silent states in anesthetized mice
We recorded cortical slow-wave activity with monopolar LFP electrode, in the vicinity of the cooling plate (Fig. 2A) in the control condition, during cooling and during recovery in either deeply or lightly anesthetized mice. The EEG was recorded contralateral to the cooled hemisphere. In the deeply anesthetized animals, as estimated from the rhythmic cortical EEG slow waves, in the control condition and after rewarming without additional injection of ketamine–xylazine (Fig. 2B, left), the cooling reduced the slow-wave amplitude, disrupted the slow-wave rhythmicity, and evoked the fast LFP spikes (20–50 ms duration, ∼1 mV amplitude) associated with an increase in gamma activity (n = 3 mice × 2 times; Fig. 2C,C1). The SD of the LFP signal significantly diminished during cooling, indicating a decrease in slow-wave amplitude (control vs cooling vs recovery, 0.482 ± 0.036 vs 0.213 ± 0.01 vs 0.367 ± 0.028 mV, respectively, mean ± SEM, p < 0.001, paired t test, n = 3 mice × 2 times). The average of the autocorrelation of the LFP recordings revealed the cooling-triggered deceleration of the slow waves (Fig. 2C2). Because gamma LFP activity is a more reliable indicator of active and silent states than the slow-wave LFP component (Mukovski et al., 2007), we analyzed the smoothed LFP gamma signals (see Materials and Methods; Fig. 2C1). The value of the autocorrelation function of the smoothed gamma signal at its minima (the time lag 250–600 ms) decreased significantly during cooling (autocorrelation coefficient: control vs cooling vs recovery, −0.341 ± 0.035 vs −0.16 ± 0.032 vs −0.304 ± 0.04, respectively, mean ± SEM, p < 0.01 and p < 0.05, respectively, paired t test; Fig. 2C3). In the control, the active/silent states correspond to the increase/decrease in gamma activity and the lower/upper parts of each LFP wave, respectively (Fig. 2C1). The gamma LFP revealed stronger neuronal activation during the cortical LFP spikes compared with the active states in the control condition (Fig. 2C1).
Figure 2.

Cortical cooling eliminated silent states in lightly anesthetized mice and disrupted slow waves in deeply anesthetized mice. A, Drawing indicates the positions of the cooling plate and electrodes. B, The examples of EEG and LFP activity recorded during deep (left) and light (right) anesthesia. C, Cortical cooling during deep anesthesia decreased the amplitude of slow-wave activity and evoked LFP spikes (C1, left). C1, LFP and the absolute value of gamma LFP (bottom trace) activities in control and during cooling in deeply anesthetized mouse. The black curve represents the smoothed absolute value of gamma LFP. Cooling disrupted cortical slow waves and evoked fast LFP spikes. C2–C4, Grouped data: three mice, two times. C2, Autocorrelations of LFP signal plotted in control, during cooling, and recovery (deep anesthesia). Autocorrelation (C3) and distribution (C4) of smoothed absolute value of gamma signal in control, during cooling, and recovery (deep anesthesia). D, Cortical cooling during light anesthesia evoked persistent activity (the shown example was obtained from the same mouse as in C). D1, LFP and the absolute value of gamma LFP activities (bottom trace) in control and during cooling (light anesthesia). The black curve represents the smoothed absolute value of gamma LFP. Cooling eliminated cortical silent states and evoked persistent activity. D2–D4, Grouped data: three mice, three times. D2, Autocorrelations of LFP activity plotted in control, during cooling, and recovery (light anesthesia). Autocorrelations (D3) and distributions (D4) of smoothed absolute value of gamma activity (right) plotted in control, during cooling, and recovery (light anesthesia). Dashed red line in D4 shows a distribution of absolute value of gamma activity in lightly anesthetized mice during periods of spontaneous EEG activation. In all the panels, the positive part of SEM is shown with gray (negative part is not shown).
During light anesthesia, characterized by spontaneous alterations of rhythmic slow waves and periods of activated EEG/LFP (persistent activity with duration at least 3–5 s) in the control condition (Fig. 2B, right), cortical mild cooling reversibly eliminated the slow waves (Fig. 2D). In these experiments, we also occasionally observed LFP spikes (data not shown). The SD of the LFP signal diminished significantly during cooling, indicating a decrease in the slow-wave activity (control vs cooling vs recovery, 0.453 ± 0.026 vs 0.195 ± 0.022 vs 0.364 ± 0.029 mV, respectively, mean ± SEM, p < 0.001, paired t test). The average of the autocorrelation of the LFP in the lightly anesthetized mice revealed the cooling-triggered reduction or abolishment of the slow waves (Fig. 2D2). As noted above, we analyzing the smoothed gamma activity, and we confirmed that the LFP pattern evoked by cooling represented persistent network activity in the lightly anesthetized mice. At its minimum, the value of the autocorrelation function of the smoothed gamma activity (the time lag 250–600 ms) decreased significantly during cooling (autocorrelation coefficient: control vs cooling vs recovery, −0.32 ± 0.046 vs −0.046 ± 0.017 vs −0.242 ± 0.057, respectively, mean ± SEM, p < 0.001 and p < 0.01 respectively, paired t test; Fig. 2D3). The level of gamma activity was the highest and the smallest during the rhythmic alternations of the active and silent states in the control condition, respectively, and the histogram of the absolute values of the gamma activity revealed bimodal distribution (Fig. 2D4, control and recovery). During spontaneous periods of persistent activity and during the cooling-triggered persistent activity (Fig. 2D4, red dotted and blue solid lines), the gamma LFP level displayed a monomodal distribution with a maximum located between the peaks corresponding to the bimodal distribution of the active and silent states of the slow oscillation. The locations of the distribution peaks in the control condition and during cooling differed significantly [silent state (control) vs persistent activity (cooling) vs active state (control), 6.8 ± 0.4 vs 14.9 ± 1.5 vs 22.0 ± 1.2 μV, respectively, mean ± SEM, p = 0.004 and p < 0.001, respectively, signed-rank test and paired t test, respectively, n = 9; Fig. 2D4]. These results show that light cortical cooling reduced the cortical slow-wave activity and induced the cortical activated states dominated by the gamma activities. In deeply anesthetized animals, light cortical cooling was also associated with the generation of fast LFP spikes.
Compared with the active states of the slow oscillation, the gamma LFP level during cooling-triggered persistent activity (Fig. 2D4) could decrease as a result of either the artificial conditions of the cooling or differences in the natural persistent neuronal dynamics. Our previous study demonstrated that instantaneous firing rates during active periods of slow oscillation are higher compared with the firing rates in the waking state (Steriade et al., 2001). However, in that study, the levels of synaptic activities were not analyzed. In the present study, we were unable to record intracellular neuronal potentials in the cortex during cooling most likely because of the cooling-induced vasoconstriction, which provoked tissue movement and impaired the stability of the recordings. To compare the natural levels of the neuronal activity during active states of the slow oscillation and persistent activity, we recorded the intracellular membrane potentials of 10 cells in different cortical areas (the frontal, motor, and somatosensory cortices) of lightly anesthetized mice during slow oscillation and during spontaneous brief activated states. To increase the amplitude of the synaptic activities, the majority of neurons were hyperpolarized with an intracellular negative current injection. To average the intracellular activities of different neurons, we used the dimensionless z-score scale (see Materials and Methods). As expected, the membrane potential showed bimodal and monomodal distribution for the slow-wave segments and the persistent activity segments, respectively (Fig. 3). To strengthen the distribution peak detection and before building histograms, we filtered the intracellular signals with a bandpass filter of 0–10 Hz (Fig. 3B, red lines). During the persistent activity, the membrane potential of the direct current hyperpolarized cells was more hyperpolarized compared with the active states of the slow oscillation (Fig. 3A,B,D). Similar to the gamma LFP activities (Fig. 2D4, right), the distribution peak corresponding to the persistent activity was located between the peaks corresponding to the silent and active states of the rhythmic slow oscillations (location of the peaks: silent state vs persistent activity vs active state, −1.01 ± 0.08 vs −0.09 ± 0.11 vs 0.88 ± 0.10 z-score units, mean ± SEM, p < 0.001, paired t test, n = 10 cells; Fig. 3D). With other conditions remaining the same, the summated amplitude of the PSPs depends on the total firing of the presynaptic neurons, which is higher during active states of slow-wave sleep compared with activated brain states (Steriade et al., 2001). These data are in agreement with the higher level of LFP gamma activity during spontaneous active states (Fig. 2). In the shown example of the spiking neuron (Fig. 3A,C), the instantaneous frequency of the spikes achieved higher values during the periodic active states compared with the states of persistent activation (Fig. 3C). Thus, the decreased level of gamma LFP during the persistent activity, compared with active states of the robust slow oscillation, can be associated with neuronal dynamic and not with the specific effects of cooling.
Figure 3.

Membrane potential distribution during the slow oscillation and persistent activity in the cortex. A, An example of the intracellular membrane potential recorded in the somatosensory cortex of a lightly anesthetized mouse (top traces, zero injected current; bottom traces, the same cell was hyperpolarized with injected current). Periods of spontaneous persistent activity are indicated with gray. B, The membrane potential distribution plotted for the spiking trace shown in A (top). The membrane potential distribution of a hyperpolarized cell during slow-wave activities (middle). The red curve represents the distribution of the filtered membrane potential during the same period (see Results). The bottom shows the membrane potential distribution of a hyperpolarized cell during persistent activity. The red curve represents the distribution of the filtered membrane potential during the same period (see Results). C, Interspike intervals (ISIs) of the same neuron during persistent activity (left) and slow oscillation (right). D, Averaged (n = 10 neurons) distributions of the membrane potentials (subthreshold conditions) during slow oscillation and persistent activity.
We conclude that the mild cooling eliminated the cortical silent states in lightly anesthetized mice and evoked persistent network activity, but it did not block neuronal activity as in the case of deep cooling (Carrasco et al., 2013; Nassi et al., 2013). On the contrary, a mild cortical heating (up to ∼39–40°C) in lightly anesthetized mice reversibly strengthened the slow waves, eliminating spontaneous persistent periods (Fig. 4A). Similar to a previous in vitro study (Reig et al., 2010), the value of the autocorrelation function of LFP at its minima (the time lag 200–400 ms) significantly increased during heating (autocorrelation coefficient: control vs heating vs recovery, −0.442 ± 0.052 vs −0.589 ± 0.042 vs −0.427 ± 0.055, respectively, mean ± SEM, p = 0.003 and p = 0.02, respectively, paired t test, n = 3 mice × 2 times; Fig. 4B).
Figure 4.
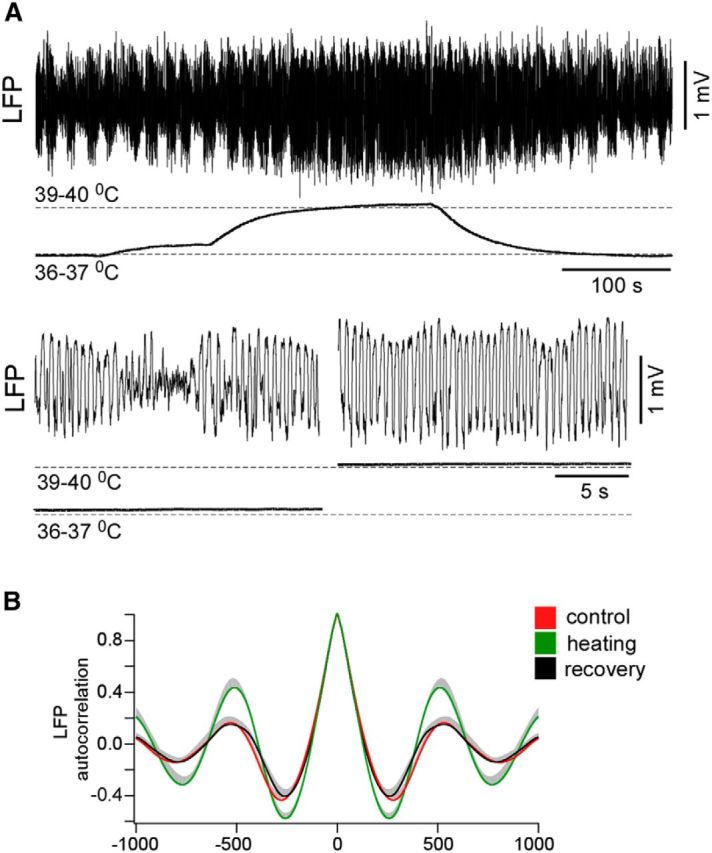
Cortical heating reversibly strengthened slow waves in lightly anesthetized mice. A, An example of effects of cortical heating during light anesthesia. Heating reversibly strengthened slow waves in lightly anesthetized mice and eliminated spontaneous persistent periods (bottom traces). B, Averaged LFP autocorrelations in control, during heating, and after recovery.
Intracellular thalamic activities of deeply anesthetized mice during cortical cooling
To reveal the cortical influence on thalamic neuronal activity in different conditions, we performed intracellular recordings within first-order (VPM) and higher-order (PO) thalamic neurons during deep anesthesia in the control condition, during cooling, and after recovery. The VPM neurons were identified either by their response to the stimulation of the animals' whiskers or after histological identification of biocytin-stained cells (Sheroziya and Timofeev, 2014). Similar to the findings presented in previous studies (Groh et al., 2014; Sheroziya and Timofeev, 2014) in the control conditions of the deeply anesthetized mice, the thalamic VPM neurons (n = 7 neurons from 7 mice) displayed unitary and/or rhythmic IPSPs and low-amplitude (slow or modulator) EPSPs, which followed cortical slow waves (Fig. 5A). The autocorrelograms for the IPSPs showed two peaks corresponding to spindle and slow-wave frequencies in the control and after recovery (Fig. 5A1,C1, arrowheads). The extracted thalamic IPSPs occurred simultaneously with the positive peaks of the averaged EEG and the negative peaks of cortical LFP activity, which represent cortical active states (Fig. 5A1,C1) in the control and after recovery. Cortical cooling disrupted the thalamic slow-wave pattern but evoked groups of spindle-like events with a frequency range of 6–7 Hz within the VPM neurons, which correlated to the bilateral cortical LFP spikes (Fig. 5B). Each postsynaptic event of the cooling-triggered spindle-like series was composed of EPSPs, followed by IPSPs (Fig. 5B, red and blue arrowheads, respectively). The averaged cortical LFP spikes (Fig. 5B1, small-amplitude field potential waves marked with arrowheads) were time-locked with the extracted IPSPs. Between the series of spindle-like events, the intracellular activity of the VPM neurons showed low levels of spontaneous synaptic activity. On average, the rate of all the detectable IPSPs decreased during cooling (control vs cooling vs recovery, 1.60 ± 0.20 vs 0.87 ± 0.09 vs 1.75 ± 0.35 events/s, respectively, mean ± SEM). Excluding the spindle-like IPSPs correlated to the cortical LFP spikes, the mean frequency of the detectable IPSPs decreased to 0.1 ± 0.03 events/s during cooling (p = 0.002 and p = 0.01 compared with the control and recovery, respectively, paired t test).
The thalamus of the decorticated and anesthetized animals generates spontaneous spindles (Morison and Bassett, 1945; Timofeev and Steriade, 1996). To compare the thalamic spindles with the spindle-like activities evoked by cortical cooling, we suppressed cortical activity by applying TTX (200 μm, ∼10 μl) on the ipsilateral hemisphere (surface). At least 40 min after the application of TTX, we recorded the VPM intracellular activities. Under these conditions, the VPM thalamic neurons (n = 7 neurons from 2 mice) displayed spindle-like oscillations composed of rhythmic IPSPs (Fig. 5D, IPSPs shown with blue arrowheads). The spindle-like frequencies of the IPSP calculated from the IPSP autocorrelograms (Fig. 5A1,B1,D1) in the control conditions, during cooling, and after the application of TTX over the cortex differed significantly [control, cooling, TTX, 111 ± 3 (∼9 Hz), 172 ± 10 (∼6 Hz), 132 ± 4 (∼7.5 Hz) ms, respectively, mean ± SEM, p < 0.01, paired t test and t test]. Different from typical spindles recorded in the thalamus of other species (Bal et al., 1995; Timofeev and Steriade, 1996; Timofeev et al., 2001b; Barthó et al., 2014), in all our experiments, we did not observe any rebound low-threshold spikes at the end of the IPSP.
The PO neurons receive driver corticothalamic input, and their activity during the slow oscillation strongly depends on the cortex. Unlike first-order thalamocortical neurons (VPM in this study), the neurons from the PO nucleus do not show phasic IPSPs during active phases of slow oscillation (Groh et al., 2014; Sheroziya and Timofeev, 2014). We did not obtain intracellular recordings in the cortex with the cooling, but we did obtain them in the PO that approximately mimic the activities of cortical neurons (Sheroziya and Timofeev, 2014). Thalamic PO neurons were identified by biocytin staining in the control conditions (n = 6 neurons from 6 mice), and they displayed the depolarized active states that accompanied the cortical slow waves, although occasionally we could also identify IPSPs as shown in previous studies (Groh et al., 2014; Sheroziya and Timofeev, 2014). The autocorrelograms of the extracted EPSPs showed a slow-wave modulation in the control condition and after recovery, and the extracted PSPs were time-locked to the cortical slow waves (Fig. 6A). As noted above, cortical cooling reduces significantly the slow-wave modulation of the thalamic activity (Fig. 6A1,B1). The cooling-triggered LFP spikes were accompanied by groups of EPSPs in the PO neurons (Fig. 6B). The averaged cortical LFP spikes (Fig. 6B1, small-amplitude field potential waves marked with arrowheads) were time-locked with the extracted EPSPs. On average, the incidence of large-amplitude (drivers) EPSPs per 1 s decreased during cortical cooling within the PO neurons compared with the control and the recovery conditions (control vs cooling vs recovery, 10 ± 1.8 vs 7.2 ± 1.1 vs 11.3 ± 1.9 events/s, respectively, mean ± SEM, p = 0.006 and p = 0.02, respectively, paired t test). Thus, the cortical cooling diminished the slow-wave modulation of the thalamic activity and evoked the intracellular potentials correlated to the cortical LFP spikes: rhythmic IPSPs in the first-order (VPM) nucleus and rhythmic EPSPs in the higher-order (PO) nucleus.
Intracellular thalamic activities of lightly anesthetized mice in control conditions and during cortical temperature modulation
One of the objectives of this study was to investigate thalamic activity during cortical transitions between slow-wave activities and persistent activities. We performed intracellular recordings in the thalamus of lightly anesthetized mice and used cortical temperature modulation. The thalamic activity of lightly anesthetized mice followed cortical EEG/LFP activities (Fig. 7). During periods of rhythmic slow-wave EEG activity, the thalamic ventral posterior lateral nucleus (VPL)/VPM neurons displayed a mixture of IPSPs and small-amplitude “modulator” EPSPs (synaptic noise) and noncorrelated to cortical slow waves large-amplitude “driver” EPSPs (Fig. 7A,B). Spontaneous reduction of cortical slow-wave activity or a switch to brief periods of persistent activity was accompanied by the depolarization of the VPM neurons and the disappearance of spontaneous IPSPs (Fig. 7A,B). To quantify the detected slow depolarization, we filtered intracellular signals (bandpass filter of 0–1 Hz; Fig. 7B1,B2, red curve) and plotted distributions of the filtered membrane potential (data not shown). The mean amplitude of the slow depolarization measured between the two peaks of the distributions was 3.3 ± 0.8 mV (n = 8 neurons); five VPM/VPL neurons were identified by location and/or response to sensory stimulation, and three other neurons displayed the same characteristic pattern of membrane potential fluctuations and generated low-threshold spikes in response to injection of hyperpolarizing current pulses (Fig. 7A3, inset). We plotted the average distribution of the membrane potentials using the dimensionless z score (see Materials and Methods; Fig. 7C). Intracellular activities of the thalamic PO neurons identified by location in the lightly anesthetized mice also followed cortical EEG. Disruption of the cortical slow-wave activity and/or a switch to brief periods of persistent activity led to a persistent-like intracellular activity within the PO neurons (Fig. 7D).
Figure 7.

Spontaneous thalamic VPM, VPL, and PO intracellular activity in lightly anesthetized mice. A1, Simultaneously recorded contralateral EEG and intracellular activity of a VPL neuron. Spontaneous disturbance of the cortical slow-wave activity evoked a slow depolarization of the membrane potential. A2, A transition from a more depolarized to a hyperpolarized state. The neuron displayed large-amplitude EPSPs and IPSPs (marked with blue arrows). Note the decrease in the persistent synaptic noise during hyperpolarization. A3, Identification of the VPL neuron. The neuron was stained with biocytin (data not shown), responded to sensory stimulation (air puff, hair on the back), and displayed a low-threshold spike (LTS) in response to a hyperpolarized current pulse (100 ms). B1, An example of the spontaneous intracellular activity of a VPM neuron. Disturbance of the cortical slow-wave activity was associated with a slow depolarization of the membrane potential and a strong reduction in the presence of IPSPs. B2, A transition from a depolarized to a hyperpolarized state. Groups of IPSPs time-locked to cortical slow waves are marked with blue arrowheads. Note the decrease in the persistent synaptic noise during hyperpolarization. The filtered intracellular signal is shown in red (bandpass filter of 0–1 Hz). B3, Identification of the VPM neuron. The neuron was stained with biocytin and generated an LTS in response to hyperpolarized current pulses (data not shown). C, Average membrane potential distribution plotted for filtered activities (red trace in B2; n = 7 VPM and 1 VPL neurons) in lightly anesthetized mice. The two peaks correspond to depolarized and hyperpolarized states. D1, Simultaneously recorded contralateral EEG and PO intracellular activity. Spontaneous disturbance of the cortical slow-wave activity and/or a switch to persistent activity evoked persistent-like activity within a PO neuron. D2, Transition from persistent activity to rhythmic alternations of active and silent states. The PO neuron generated characteristic large-amplitude driver EPSPs. D3, The PO neuron was identified by the electrode trace (data not shown) and generated an LTS in response to hyperpolarized current pulses (100 ms).
We managed to record three identified thalamic cells during cortical cooling/heating in the lightly anesthetized mice (several recorded non-identified thalamic cells displayed similar behavior as shown below; Figs. 8, 9). Two thalamic VPM neurons were identified by either location or the response to sensory stimulation (air puff on the whiskers; only one neuron is shown). In control conditions of light anesthesia, the VPM neurons displayed slow alternations of weak depolarization and lightly hyperpolarized periods with spindle-like IPSP sequences (Fig. 8A). In contrast, cortical heating increased the IPSP rhythmicity and corticothalamic synchronization of the VPM neurons, as shown by the IPSP-triggered EEG/LFP averages (Fig. 8A,B). Cortical cooling eliminated the IPSPs and evoked persistent activity characterized by high levels of synaptic activities (Fig. 8A,B). Within the identified higher-order thalamic PO neurons, cortical cooling eliminated silent states and evoked persistent activity and persistent synaptic noise (Fig. 9A). After heating, corticothalamic synchronization increased compared with the control condition, as shown by the EPSP-triggered averages (Fig. 9B). Because thalamic intracellular activity primarily depends on corticothalamic input, we conclude that the moderate cortical cooling in the lightly anesthetized mice evoked persistent thalamocortical activation characterized by increased gamma cortical activity and a relatively stable and slightly depolarized level of membrane potential in the thalamocortical neurons from the first- and second-order nuclei.
Mild cortical cooling prevented generation of silent states in the non-anesthetized animals
To confirm that the results obtained on the anesthetized animals can be applied to natural physiological states, we performed cortical cooling in non-anesthetized head-restrained mice. We found that moderate cortical cooling reversibly prevented the generation of large-amplitude depth-positive LFP waves during sleep and reversibly decreased the delta power calculated as an integral power between 0.2 and 4 Hz of the full spectrogram (n = 2 mice, tens of cooling–rewarming cycles; Fig. 10). The delta power of LFP was reduced systematically during cortical cooling (Fig. 10A1). The peaks of the depth-positive LFP waves were detected by their maximal amplitude that exceeded a threshold of 7 SDs (Fig. 10A). During cooling, slow waves were detected only occasionally (Fig. 10D). We also calculated the absolute value of the gamma (40–300 Hz) LFP activities and averaged them against the detected peaks of the slow waves (Fig. 10C). The results obtained in two mice showed almost identical dynamics; the gamma activity decreased during the generation of the depth-positive LFP waves, indicating that a majority of the detected waves represented silent states. During cortical cooling, the mice usually remained still and their eyes were either closed or ajar, although the animals jerked every 10–30 s. Sometimes cortical cooling led to an increase in the mice's muscle activity (Fig. 10E, left); at other times, it decreased the frequency of muscle contractions (Fig. 10E, right, the data in the left and right panels were obtained from the same mouse). On all occasions, the LFP in the cooled area was activated for the duration of the cooling period. In this experiment, the activation of LFP could depend on at least two factors. First, the cooling evoked cortical activation, as in the anesthetized animals, and, second, the cooling produced a sensory experience that excited the animals. We always detected the rapid rebound of slow-wave activity during rewarming (Fig. 10A,B), suggesting that the affected cortex was unable to generate silent states during cooling. Because muscle activity could either increase or decrease during local cortical cooling, the cooling likely did not change the entire behavioral state of the animals but reduced local slow-wave activity. In contrast to the anesthetized animals, we did not detect LFP spikes in the non-anesthetized mice during cooling.
Figure 10.

Cortical cooling eliminated silent states in non-anesthetized head-restrained mice. A, LFP activity recorded from the motor cortex of a non-anesthetized mouse in control and during cooling. Cooling reversibly eliminated slow waves. Slow waves were extracted by a threshold (shown with a dotted line, right). A1, The delta power (0.2–4 Hz) calculated for the M1 LFP shown with red in A. B, Averaged delta power in control, during cooling (left, n = 2 mice × 4 times), and rewarming (right, n = 2 mice × 4 times). The positive part of SEM is shown with gray (negative part is not shown). C, Slow wave (top traces) and the absolute value of gamma LFP (bottom traces) averaged against detected slow-wave peaks (2 mice, left and right). D, Averaged slow-wave rate in control, during cooling (left, n = 2 mice × 4 times), and rewarming (right, n = 2 mice × 4 times, the same data as in B). The positive part of SEM is shown with gray (negative part is not shown). D, Simultaneously recorded LFP and EMG (neck muscles) during cooling. The muscle activity could increase (left) or decrease (right) during cooling.
Discussion
In the present study, we showed that a mild cooling of the cortex reversibly eliminated silent states and led to persistent thalamocortical activity in lightly anesthetized mice. Spontaneous IPSPs within the VPM neurons disappeared during cooling-induced cortical activation. Similarly, mild cortical cooling prevented the generation of cortical silent states in non-anesthetized head-restrained mice. Under the lesser physiological condition of deep anesthesia, a moderate cooling of the cortex reduced slow-wave synchrony but did not prevent the generation of slow waves. It also evoked fast cortical LFP spikes with frequencies ranging from 6 to 9 Hz, which correlated to spindle-like IPSPs within the VPM neurons and depolarizing events within the PO neurons. Our data point to a strong cortical control of thalamic activities.
The essential finding of this study is that a moderate cortical cooling in lightly anesthetized or naturally sleeping mice transforms the slow-wave pattern of neuronal activity in activated states. The natural states of vigilance depend on activities of the neuromodulatory systems (Steriade et al., 1993b; Saper, 2006; Lee and Dan, 2012). In contrast to the waking state and REM sleep, during slow-wave sleep, neuromodulatory systems maintain low levels of activity (Hassani et al., 2009; Sakai, 2011; Eschenko et al., 2012; Boucetta et al., 2014). As we described in the Introduction, essentially both moderate cortical cooling and neuromodulator release have a similar action on neurons: they depolarize neurons and reduce the synaptic release. Therefore, the finding that cooling induced a cortical activation in lightly anesthetized mice or during slow-wave sleep was expected.
One of the major unknown questions is whether the local cooling was sufficient to induce activation in widespread zones. Indeed, a certain balance of excitation and inhibition is needed to generate the cortical slow oscillation (Haider et al., 2006; Rudolph et al., 2007; David et al., 2013; Lemieux et al., 2014). In our experiments, the cortical activation in response to cooling was observed not only locally but also in the contralateral hemisphere (Fig. 9). The onset of silent phases has an active inhibitory component (Steriade et al., 1993a; Lemieux et al., 2015). Network-wide, a tonic reduction of inhibition shortens active states (Sanchez-Vives et al., 2010; Chen et al., 2012). However, when a subset of inhibitory neurons is activated simultaneously, the network goes to the silent state (Chen et al., 2012; Lemieux et al., 2015). Compared with pyramidal cells, the interneurons are less sensitive to cooling (Vizi and Sperlágh, 1999; Motamedi et al., 2012). Therefore, a moderate local cortical hypothermia might be sufficient to induce persistent cortical activity attributable to different actions on interneurons and pyramidal cells.
The other question is whether a local cortical activation provides sufficient conditions to induce activation in the corresponding thalamic structures. During REM sleep and the waking state, thalamocortical neurons are depolarized (Hirsch et al., 1983; Woody et al., 2003). There are at least two main causes for such a depolarization. One cause is that neuromodulatory systems projecting to the thalamus can directly depolarize thalamocortical neurons (Curró Dossi et al., 1991; McCormick, 1992; Saper et al., 2005; Steriade and McCarley, 2005; Lee and Dan, 2012). The second cause is that a steady firing of corticothalamic neurons provides a continuous excitatory drive that keeps thalamocortical neurons depolarized. However, it is unclear which of these is the leading cause of depolarization. Indeed, in vivo, in an intact thalamocortical network, stimulation of a cholinergic structure projecting to the thalamus (laterodorsal tegmentum) induced long-lasting depolarization of thalamocortical neurons (Curró Dossi et al., 1991). In contrast, in decorticated cats, pedunculopontine tegmentum (another cholinergic structure) stimulation induced a dramatic reduction in inhibitory responses without a major change in the membrane potential (Timofeev and Steriade, 1998), thereby suggesting that corticothalamic feedback could play a major role in the depolarization of thalamocortical neurons induced by the activation of cholinergic system. Stimulation of glutamatergic pathways terminating in the thalamus (Steriade, 1995) or direct optogenetic stimulation of thalamocortical neurons (Poulet et al., 2012) have been found to induce activated cortical states. Inactivation of the thalamus has been shown to reduce or abolish cortical spontaneous oscillations (Poulet et al., 2012; David et al., 2013; Lemieux et al., 2014). Therefore, the cortex exerts a powerful effect on the thalamus.
Removing the cortex takes the edge off the depolarizing corticothalamic tone; thalamocortical neurons become hyperpolarized and display spindle and delta oscillations (Timofeev and Steriade, 1996). Depolarizing action of the asynchronous cortical drive contributes to the depolarization of thalamocortical neurons and to spindle termination (Timofeev et al., 2001b; Bonjean et al., 2011). The slow depolarization might be evoked by a stronger activation of corticothalamic modulators (Li et al., 2003; Reichova and Sherman, 2004). In this study, we show that cooling induced a cortical activation without activation of the neuromodulatory systems, which was sufficient to induce a persistent active state in the thalamus and a dramatic reduction of detectable IPSPs.
The morphological features and the frequency (6–9 Hz) of the cooling-induced spiky LFP oscillations show their resemblance to spike-wave epileptic activity in rodents. Multiple evidence points to the cortical origin of rodent spike-wave activities (Pinault et al., 1998; Meeren et al., 2002; Polack et al., 2007; Avoli, 2012; Leresche et al., 2012; Zheng et al., 2012), which shares many neuronal features with spike-wave activities in cats (Timofeev and Steriade, 2004). In GAERS (genetic absence epilepsy rats from Strasbourg) rats, a well accepted rodent model of spike-wave epilepsy, TTX inactivation of the somatosensory cortex resulted in a complete disappearance of paroxysmal thalamic oscillations (Polack et al., 2009). In our study, TTX application to the somatosensory cortex did not abolish thalamic oscillations in the VPM nucleus, although the earliest excitatory component of the oscillatory cycles, which likely had a cortical origin, disappeared and the frequency of these oscillations slightly decreased (Fig. 5). Therefore, similar to the findings presented in other animal models, the spike-wave activities in this study were recorded in both the thalamus and the cortex. However, different from other rodent models of spike-waves seizures, in our study, cortical inactivation did not abolish thalamic oscillations, thus pointing to an active thalamic contribution in the generation of the LFP spiky discharges with a frequency ranging from 6 to 9 Hz.
The origin of cortical cooling-induced LFP spikes recorded in this study, which occurs in mice anesthetized deeply with ketamine and xylazine, might be explained as follows. Xylazine is a known agonist of α2 adrenoceptors. Agonists of α2 adrenoceptors suppress hyperpolarization-activated cyclic nucleotide-gated (HCN) channels and evoke light hyperpolarization of the resting membrane potential (Carr et al., 2007; Dembrow et al., 2010). HCN currents, as well as potassium-leaking and inwardly rectifying currents, control neuronal intrinsic properties at resting conditions (Pape, 1996; Day et al., 2005; Nolan et al., 2007). Ketamine is an antagonist of NMDA receptors, with hypnotic action involving inhibition of HCN channels and light hyperpolarization of the resting membrane potential (Chen et al., 2009). The loss of HCN channels increases electric somatodendritic coupling and evokes intrinsic bursting in layer V neurons. This was proposed to be a potential mechanism of generation for absent-like spike-wave discharges (Kole et al., 2007). Cooling down to 25°C also increases somatodendritic coupling and evokes intrinsic bursting in regular firing layer V neurons (Hedrick and Waters, 2011), most likely because of the inactivation of the HCN or potassium channels. Synergistically amplified by cooling and deep anesthesia (both of which target somatodendritic coupling), intrinsic bursting could lead to cortically generated LFP spikes.
Therapeutic hypothermia is often used in patients with refractory epilepsy and in in vivo and in vitro animal models to prevent or control seizures/paroxysmal activity (Motamedi et al., 2013). It is known that, during generalized seizures, the energy consumption reaches its highest values and the therapeutic hypothermia attenuates seizures by a reduction in general metabolism (Duffy et al., 1975; Motamedi et al., 2013). A moderate cooling, lasting hours in vivo, does not produce detectable damage to brain tissue (Erecinska et al., 2003; Motamedi et al., 2013). Moderate cooling maintains a better energy level; that is, energy-consuming reactions are reduced more than ATP synthesis, and this prevents damage (Erecinska et al., 2003). Our study shows that a moderate cortical cooling during deep anesthesia might have a light epileptogenic effect.
We show that the net effect of a moderate cortical cooling during slow-wave sleep or light anesthesia is the field potential activation. Similar effects are produced by activities of neuromodulatory systems during the waking stage or REM sleep. This similarity likely occurs because, with some exceptions, both cooling and neuromodulators produce similar effects on cortical neurons: a reduction of the synaptic release and membrane depolarization. Activation of a cortical network also induced activation of corresponding thalamic regions. Under the lesser physiological condition of deep anesthesia, cortical cooling disrupted thalamocortical slow waves and promoted generation of fast rhythmic LFP spikes.
Footnotes
This study was supported by National Sciences and Engineering Research Council of Canada Grant 298475 and Canadian Institutes of Health Research Grants MOP-325213 and MOP-324941. We are thankful to Serge Ftomov for technical assistance, Josée Seigneur for management assistance, and Sylvain Chauvette for help in the analysis of and comments on this manuscript.
The authors declare no competing financial interests.
References
- Avoli M. A brief history on the oscillating roles of thalamus and cortex in absence seizures. Epilepsia. 2012;53:779–789. doi: 10.1111/j.1528-1167.2012.03421.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bal T, von Krosigk M, McCormick DA. Role of the ferret perigeniculate nucleus in the generation of synchronized oscillations in vitro. J Physiol. 1995;483:665–685. doi: 10.1113/jphysiol.1995.sp020613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barthó P, Slézia A, Mátyás F, Faradzs-Zade L, Ulbert I, Harris KD, Acsády L. Ongoing network state controls the length of sleep spindles via inhibitory activity. Neuron. 2014;82:1367–1379. doi: 10.1016/j.neuron.2014.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazhenov M, Timofeev I, Steriade M, Sejnowski TJ. Model of thalamocortical slow-wave sleep oscillations and transitions to activated states. J Neurosci. 2002;22:8691–8704. doi: 10.1523/JNEUROSCI.22-19-08691.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonjean M, Baker T, Lemieux M, Timofeev I, Sejnowski T, Bazhenov M. Corticothalamic feedback controls sleep spindle duration in vivo. J Neurosci. 2011;31:9124–9134. doi: 10.1523/JNEUROSCI.0077-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucetta S, Cissé Y, Mainville L, Morales M, Jones BE. Discharge profiles across the sleep–waking cycle of identified cholinergic, GABAergic, and glutamatergic neurons in the pontomesencephalic tegmentum of the rat. J Neurosci. 2014;34:4708–4727. doi: 10.1523/JNEUROSCI.2617-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr DB, Andrews GD, Glen WB, Lavin A. α2-Noradrenergic receptors activation enhances excitability and synaptic integration in rat prefrontal cortex pyramidal neurons via inhibition of HCN currents. J Physiol. 2007;584:437–450. doi: 10.1113/jphysiol.2007.141671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasco A, Brown TA, Kok MA, Chabot N, Kral A, Lomber SG. Influence of core auditory cortical areas on acoustically evoked activity in contralateral primary auditory cortex. J Neurosci. 2013;33:776–789. doi: 10.1523/JNEUROSCI.1784-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauvette S, Crochet S, Volgushev M, Timofeev I. Properties of slow oscillation during slow-wave sleep and anesthesia in cats. J Neurosci. 2011;31:14998–15008. doi: 10.1523/JNEUROSCI.2339-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauvette S, Seigneur J, Timofeev I. Sleep oscillations in the thalamocortical system induce long-term neuronal plasticity. Neuron. 2012;75:1105–1113. doi: 10.1016/j.neuron.2012.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JY, Chauvette S, Skorheim S, Timofeev I, Bazhenov M. Interneuron-mediated inhibition synchronizes neuronal activity during slow oscillation. J Physiol. 2012;590:3987–4010. doi: 10.1113/jphysiol.2012.227462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Shu S, Bayliss DA. HCN1 channel subunits are a molecular substrate for hypnotic actions of ketamine. J Neurosci. 2009;29:600–609. doi: 10.1523/JNEUROSCI.3481-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compte A, Sanchez-Vives MV, McCormick DA, Wang XJ. Cellular and network mechanisms of slow oscillatory activity (<1 Hz) and wave propagations in a cortical network model. J Neurophysiol. 2003;89:2707–2725. doi: 10.1152/jn.00845.2002. [DOI] [PubMed] [Google Scholar]
- Curró Dossi R, Paré D, Steriade M. Short-lasting nicotinic and long-lasting muscarinic depolarizing responses of thalamocortical neurons to stimulation of mesopontine cholinergic nuclei. J Neurophysiol. 1991;65:393–406. doi: 10.1152/jn.1991.65.3.393. [DOI] [PubMed] [Google Scholar]
- David F, Schmiedt JT, Taylor HL, Orban G, Di Giovanni G, Uebele VN, Renger JJ, Lambert RC, Leresche N, Crunelli V. Essential thalamic contribution to slow waves of natural sleep. J Neurosci. 2013;33:19599–19610. doi: 10.1523/JNEUROSCI.3169-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day M, Carr DB, Ulrich S, Ilijic E, Tkatch T, Surmeier DJ. Dendritic excitability of mouse frontal cortex pyramidal neurons Is shaped by the Interaction among HCN, Kir2, and Kleak channels. J Neurosci. 2005;25:8776–8787. doi: 10.1523/JNEUROSCI.2650-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dembrow NC, Chitwood RA, Johnston D. Projection-specific neuromodulation of medial prefrontal cortex neurons. J Neurosci. 2010;30:16922–16937. doi: 10.1523/JNEUROSCI.3644-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy TE, Howse DC, Plum F. Cerebral energy metabolism during experimental status epilepticus. J Neurochem. 1975;24:925–934. doi: 10.1111/j.1471-4159.1975.tb03657.x. [DOI] [PubMed] [Google Scholar]
- Eggermann E, Feldmeyer D. Cholinergic filtering in the recurrent excitatory microcircuit of cortical layer 4. Proc Natil Acad Sci USA. 2009;106:11753–11758. doi: 10.1073/pnas.0810062106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erecinska M, Thoresen M, Silver IA. Effects of hypothermia on energy metabolism in mammalian central nervous system. J Cereb Blood Flow Metab. 2003;23:513–530. doi: 10.1097/01.WCB.0000066287.21705.21. [DOI] [PubMed] [Google Scholar]
- Eschenko O, Magri C, Panzeri S, Sara SJ. Noradrenergic neurons of the locus coeruleus are phase locked to cortical Up-Down states during sleep. Cereb Cortex. 2012;22:426–435. doi: 10.1093/cercor/bhr121. [DOI] [PubMed] [Google Scholar]
- Gil Z, Connors BW, Amitai Y. Differential regulation of neocortical synapses by neuromodulators and activity. Neuron. 1997;19:679–686. doi: 10.1016/S0896-6273(00)80380-3. [DOI] [PubMed] [Google Scholar]
- Groh A, Bokor H, Mease RA, Plattner VM, Hangya B, Stroh A, Deschenes M, Acsády L. Convergence of cortical and sensory driver inputs on single thalamocortical cells. Cereb Cortex. 2014;24:3167–3179. doi: 10.1093/cercor/bht173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn TT, Sakmann B, Mehta MR. Phase-locking of hippocampal interneurons' membrane potential to neocortical up-down states. Nat Neurosci. 2006;9:1359–1361. doi: 10.1038/nn1788. [DOI] [PubMed] [Google Scholar]
- Haider B, Duque A, Hasenstaub AR, McCormick DA. Neocortical network activity in vivo is generated through a dynamic balance of excitation and inhibition. J Neurosci. 2006;26:4535–4545. doi: 10.1523/JNEUROSCI.5297-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham NR, Larkman AU. The reliability of excitatory synaptic transmission in slices of rat visual cortex in vitro is temperature dependent. J Physiol. 1998;507:249–256. doi: 10.1111/j.1469-7793.1998.249bu.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassani OK, Lee MG, Henny P, Jones BE. Discharge profiles of identified GABAergic in comparison to cholinergic and putative glutamatergic basal forebrain neurons across the sleep–wake cycle. J Neurosci. 2009;29:11828–11840. doi: 10.1523/JNEUROSCI.1259-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasselmo ME, McGaughy J. High acetylcholine levels set circuit dynamics for attention and encoding and low acetylcholine levels set dynamics for consolidation. Prog Brain Res. 2004;145:207–231. doi: 10.1016/S0079-6123(03)45015-2. [DOI] [PubMed] [Google Scholar]
- Hedrick T, Waters J. Spiking patterns of neocortical L5 pyramidal neurons in vitro change with temperature. Front Cell Neurosci. 2011;5:1. doi: 10.3389/fncel.2011.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill S, Tononi G. Modeling sleep and wakefulness in the thalamocortical system. J Neurophysiol. 2005;93:1671–1698. doi: 10.1152/jn.00915.2004. [DOI] [PubMed] [Google Scholar]
- Hirsch JC, Fourment A, Marc ME. Sleep-related variations of membrane potential in the lateral geniculate body relay neurons of the cat. Brain Res. 1983;259:308–312. doi: 10.1016/0006-8993(83)91264-7. [DOI] [PubMed] [Google Scholar]
- Jasper HH, Shacter DG, Montplaisir J. The effect of local cooling upon spontaneous and evoked electrical activity of cerebral cortex. Can J Physiol Pharmacol. 1970;48:640–652. doi: 10.1139/y70-094. [DOI] [PubMed] [Google Scholar]
- Katz B, Miledi R. The effect of temperature on the synaptic delay at the neuromuscular junction. J Physiol. 1965;181:656–670. doi: 10.1113/jphysiol.1965.sp007790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kole MH, Bräuer AU, Stuart GJ. Inherited cortical HCN1 channel loss amplifies dendritic calcium electrogenesis and burst firing in a rat absence epilepsy model. J Physiol. 2007;578:507–525. doi: 10.1113/jphysiol.2006.122028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krnjević K, Pumain R, Renaud L. The mechanism of excitation by acetylcholine in the cerebral cortex. J Physiol. 1971;215:247–268. doi: 10.1113/jphysiol.1971.sp009467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Dan Y. Neuromodulation of brain states. Neuron. 2012;76:209–222. doi: 10.1016/j.neuron.2012.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemieux M, Chen JY, Lonjers P, Bazhenov M, Timofeev I. The impact of cortical deafferentation on the neocortical slow oscillation. J Neurosci. 2014;34:5689–5703. doi: 10.1523/JNEUROSCI.1156-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemieux M, Chauvette S, Timofeev I. Neocortical inhibitory activities and long-range afferents contribute to the synchronous onset of silent states of the neocortical slow oscillation. J Neurophysiol. 2015;113:768–779. doi: 10.1152/jn.00858.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leresche N, Lambert RC, Errington AC, Crunelli V. From sleep spindles of natural sleep to spike and wave discharges of typical absence seizures: is the hypothesis still valid? Pflugers Arch. 2012;463:201–212. doi: 10.1007/s00424-011-1009-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Bickford ME, Guido W. Distinct firing properties of higher order thalamic relay neurons. J Neurophysiol. 2003;90:291–299. doi: 10.1152/jn.01163.2002. [DOI] [PubMed] [Google Scholar]
- McCormick DA. Neurotransmitter actions in the thalamus and cerebral cortex and their role in neuromodulation of thalamocortical activity. Prog Neurobiol. 1992;39:337–388. doi: 10.1016/0301-0082(92)90012-4. [DOI] [PubMed] [Google Scholar]
- Meeren HK, Pijn JP, Van Luijtelaar EL, Coenen AM, Lopes da Silva FH. Cortical focus drives widespread corticothalamic networks during spontaneous absence seizures in rats. J Neurosci. 2002;22:1480–1495. doi: 10.1523/JNEUROSCI.22-04-01480.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morison RS, Bassett DL. Electrical activity of the thalamus and basal ganglia in decorticate cats. J Neurophysiol. 1945;8:309–314. [Google Scholar]
- Moruzzi G, Magoun HW. Brain stem reticular formation and activation of the EEG. Electroencephalogr Clin Neurophysiol. 1949;1:455–473. doi: 10.1016/0013-4694(49)90219-9. [DOI] [PubMed] [Google Scholar]
- Motamedi GK, Lesser RP, Vicini S. Therapeutic brain hypothermia, its mechanisms of action, and its prospects as a treatment for epilepsy. Epilepsia. 2013;54:959–970. doi: 10.1111/epi.12144. [DOI] [PubMed] [Google Scholar]
- Motamedi GK, Gonzalez-Sulser A, Dzakpasu R, Vicini S. Cellular mechanisms of desynchronizing effects of hypothermia in an in vitro epilepsy model. Neurotherapeutics. 2012;9:199–209. doi: 10.1007/s13311-011-0078-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukovski M, Chauvette S, Timofeev I, Volgushev M. Detection of active and silent states in neocortical neurons from the field potential signal during slow-wave sleep. Cereb Cortex. 2007;17:400–414. doi: 10.1093/cercor/bhj157v. [DOI] [PubMed] [Google Scholar]
- Nassi JJ, Lomber SG, Born RT. Corticocortical feedback contributes to surround suppression in V1 of the alert primate. J Neurosci. 2013;33:8504–8517. doi: 10.1523/JNEUROSCI.5124-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan MF, Dudman JT, Dodson PD, Santoro B. HCN1 channels control resting and active integrative properties of stellate cells from layer II of the entorhinal cortex. J Neurosci. 2007;27:12440–12451. doi: 10.1523/JNEUROSCI.2358-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pape HC. Queer current and pacemaker: the hyperpolarization-activated cation current in neurons. Annu Rev Physiol. 1996;58:299–327. doi: 10.1146/annurev.ph.58.030196.001503. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin K. The mouse brain in stereotaxic coordinates. Ed 4. New York: Academic; 2001. [Google Scholar]
- Pinault D, Leresche N, Charpier S, Deniau JM, Marescaux C, Vergnes M, Crunelli V. Intracellular recordings in thalamic neurones during spontaneous spike and wave discharges in rats with absence epilepsy. J Physiol. 1998;509:449–456. doi: 10.1111/j.1469-7793.1998.449bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polack PO, Guillemain I, Hu E, Deransart C, Depaulis A, Charpier S. Deep layer somatosensory cortical neurons initiate spike-and-wave discharges in a genetic model of absence seizures. J Neurosci. 2007;27:6590–6599. doi: 10.1523/JNEUROSCI.0753-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polack PO, Mahon S, Chavez M, Charpier S. Inactivation of the somatosensory cortex prevents paroxysmal oscillations in cortical and related thalamic neurons in a genetic model of absence epilepsy. Cereb Cortex. 2009;19:2078–2091. doi: 10.1093/cercor/bhn237. [DOI] [PubMed] [Google Scholar]
- Poulet JF, Fernandez LM, Crochet S, Petersen CC. Thalamic control of cortical states. Nat Neurosci. 2012;15:370–372. doi: 10.1038/nn.3035. [DOI] [PubMed] [Google Scholar]
- Reichova I, Sherman SM. Somatosensory corticothalamic projections: distinguishing drivers from modulators. J Neurophysiol. 2004;92:2185–2197. doi: 10.1152/jn.00322.2004. [DOI] [PubMed] [Google Scholar]
- Reig R, Mattia M, Compte A, Belmonte C, Sanchez-Vives MV. Temperature modulation of slow and fast cortical rhythms. J Neurophysiol. 2010;103:1253–1261. doi: 10.1152/jn.00890.2009. [DOI] [PubMed] [Google Scholar]
- Rudolph M, Pospischil M, Timofeev I, Destexhe A. Inhibition determines membrane potential dynamics and controls action potential generation in awake and sleeping cat cortex. J Neurosci. 2007;27:5280–5290. doi: 10.1523/JNEUROSCI.4652-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runfeldt MJ, Sadovsky AJ, MacLean JN. Acetylcholine functionally reorganizes neocortical microcircuits. J Neurophysiol. 2014;112:1205–1216. doi: 10.1152/jn.00071.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai K. Sleep-waking discharge profiles of dorsal raphe nucleus neurons in mice. Neuroscience. 2011;197:200–224. doi: 10.1016/j.neuroscience.2011.09.024. [DOI] [PubMed] [Google Scholar]
- Sanchez-Vives MV, Mattia M, Compte A, Perez-Zabalza M, Winograd M, Descalzo VF, Reig R. Inhibitory modulation of cortical up states. J Neurophysiol. 2010;104:1314–1324. doi: 10.1152/jn.00178.2010. [DOI] [PubMed] [Google Scholar]
- Saper CB. Staying awake for dinner: hypothalamic integration of sleep, feeding, and circadian rhythms. Prog Brain Res. 2006;153:243–252. doi: 10.1016/S0079-6123(06)53014-6. [DOI] [PubMed] [Google Scholar]
- Saper CB, Scammell TE, Lu J. Hypothalamic regulation of sleep and circadian rhythms. Nature. 2005;437:1257–1263. doi: 10.1038/nature04284. [DOI] [PubMed] [Google Scholar]
- Sheroziya M, Timofeev I. Global intracellular slow-wave dynamics of the thalamocortical system. J Neurosci. 2014;34:8875–8893. doi: 10.1523/JNEUROSCI.4460-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steriade M. Two channels in the cerebellothalamocortical system. J Comp Neurol. 1995;354:57–70. doi: 10.1002/cne.903540106. [DOI] [PubMed] [Google Scholar]
- Steriade M, McCarley RW. Brainstem control of wakefulness and sleep. New York: Plenum; 2005. [Google Scholar]
- Steriade M, Nuñez A, Amzica F. A novel slow (<1 Hz) oscillation of neocortical neurons in vivo: depolarizing and hyperpolarizing components. J Neurosci. 1993a;13:3252–3265. doi: 10.1523/JNEUROSCI.13-08-03252.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steriade M, McCormick DA, Sejnowski TJ. Thalamocortical oscillations in the sleeping and aroused brain. Science. 1993b;262:679–685. doi: 10.1126/science.8235588. [DOI] [PubMed] [Google Scholar]
- Steriade M, Timofeev I, Grenier F. Natural waking and sleep states: a view from inside neocortical neurons. J Neurophysiol. 2001;85:1969–1985. doi: 10.1152/jn.2001.85.5.1969. [DOI] [PubMed] [Google Scholar]
- Timofeev I, Steriade M. Low-frequency rhythms in the thalamus of intact-cortex and decorticated cats. J Neurophysiol. 1996;76:4152–4168. doi: 10.1152/jn.1996.76.6.4152. [DOI] [PubMed] [Google Scholar]
- Timofeev I, Steriade M. Cellular mechanisms underlying intrathalamic augmenting responses of reticular and relay neurons. J Neurophysiol. 1998;79:2716–2729. doi: 10.1152/jn.1998.79.5.2716. [DOI] [PubMed] [Google Scholar]
- Timofeev I, Steriade M. Neocortical seizures: initiation, development and cessation. Neuroscience. 2004;123:299–336. doi: 10.1016/j.neuroscience.2003.08.051. [DOI] [PubMed] [Google Scholar]
- Timofeev I, Grenier F, Steriade M. Impact of intrinsic properties and synaptic factors on the activity of neocortical networks in vivo. J Physiol Paris. 2000;94:343–355. doi: 10.1016/S0928-4257(00)01097-4. [DOI] [PubMed] [Google Scholar]
- Timofeev I, Grenier F, Steriade M. Disfacilitation and active inhibition in the neocortex during the natural sleep-wake cycle: an intracellular study. Proc Natl Acad Sci U S A. 2001a;98:1924–1929. doi: 10.1073/pnas.98.4.1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timofeev I, Bazhenov M, Sejnowski T, Steriade M. Contribution of intrinsic and synaptic factors in the desynchronization of thalamic oscillatory activity. Thalamus Relat Syst. 2001b;1:53–69. doi: 10.1017/S1472928801000048. [DOI] [Google Scholar]
- Trevelyan AJ, Jack J. Detailed passive cable models of layer 2/3 pyramidal cells in rat visual cortex at different temperatures. J Physiol. 2002;539:623–636. doi: 10.1113/jphysiol.2001.013291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizi ES, Sperlágh B. Separation of carrier mediated and vesicular release of GABA from rat brain slices. Neurochem Int. 1999;34:407–413. doi: 10.1016/S0197-0186(99)00047-9. [DOI] [PubMed] [Google Scholar]
- Volgushev M, Vidyasagar TR, Chistiakova M, Yousef T, Eysel UT. Membrane properties and spike generation in rat visual cortical cells during reversible cooling. J Physiol. 2000;522:59–76. doi: 10.1111/j.1469-7793.2000.0059m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volgushev M, Kudryashov I, Chistiakova M, Mukovski M, Niesmann J, Eysel UT. Probability of transmitter release at neocortical synapses at different temperatures. J Neurophysiol. 2004;92:212–220. doi: 10.1152/jn.01166.2003. [DOI] [PubMed] [Google Scholar]
- Woody CD, Gruen E, Wang XF. Electrical properties affecting discharge of units of the mid and posterolateral thalamus of conscious cats. Neuroscience. 2003;122:531–539. doi: 10.1016/j.neuroscience.2003.08.011. [DOI] [PubMed] [Google Scholar]
- Yang XF, Ouyang Y, Kennedy BR, Rothman SM. Cooling blocks rat hippocampal neurotransmission by a presynaptic mechanism: observations using 2-photon microscopy. J Physiol. 2005;567:215–224. doi: 10.1113/jphysiol.2005.088948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng TW, O'Brien TJ, Morris MJ, Reid CA, Jovanovska V, O'Brien P, van Raay L, Gandrathi AK, Pinault D. Rhythmic neuronal activity in S2 somatosensory and insular cortices contribute to the initiation of absence-related spike-and-wave discharges. Epilepsia. 2012;53:1948–1958. doi: 10.1111/j.1528-1167.2012.03720.x. [DOI] [PubMed] [Google Scholar]