

Abstract
The complement system is part of the innate immune response responsible for removing pathogens and cellular debris, in addition to helping to refine CNS neuronal connections via microglia-mediated pruning of inappropriate synapses during brain development. However, less is known about the role of complement during normal aging. Here, we studied the role of the central complement component, C3, in synaptic health and aging. We examined behavior as well as electrophysiological, synaptic, and neuronal changes in the brains of C3-deficient male mice (C3 KO) compared with age-, strain-, and gender-matched C57BL/6J (wild-type, WT) control mice at postnatal day 30, 4 months, and 16 months of age. We found the following: (1) region-specific and age-dependent synapse loss in aged WT mice that was not observed in C3 KO mice; (2) age-dependent neuron loss in hippocampal CA3 (but not in CA1) that followed synapse loss in aged WT mice, neither of which were observed in aged C3 KO mice; and (3) significantly enhanced LTP and cognition and less anxiety in aged C3 KO mice compared with aged WT mice. Importantly, CA3 synaptic puncta were similar between WT and C3 KO mice at P30. Together, our results suggest a novel and prominent role for complement protein C3 in mediating aged-related and region-specific changes in synaptic function and plasticity in the aging brain.
SIGNIFICANCE STATEMENT The complement cascade, part of the innate immune response to remove pathogens, also plays a role in synaptic refinement during brain development by the removal of weak synapses. We investigated whether complement C3, a central component, affects synapse loss during aging. Wild-type (WT) and C3 knock-out (C3 KO) mice were examined at different ages. The mice were similar at 1 month of age. However, with aging, WT mice lost synapses in specific brain regions, especially in hippocampus, an area important for memory, whereas C3 KO mice were protected. Aged C3 KO mice also performed better on learning and memory tests than aged WT mice. Our results suggest that complement C3, or its downstream signaling, is detrimental to synapses during aging.
Keywords: aging, CA3, cognition, complement C3, innate immunity, synapse
Introduction
The complement cascade, of which C3 is a common central component, is part of the innate immune system and mediates the phagocytic removal of opsonized pathogens and cellular debris, as well as the lysis of cell membranes (Fujita, 2002). Complement has also been shown to play a key role in synaptic pruning in the healthy developing brain (Stevens et al., 2007; Schafer et al., 2012). In the postnatal brain, C1q and C3 localize to subsets of immature synapses and promote their elimination via microglial-mediated phagocytosis during developmental refinement of the visual system (Stevens et al., 2007; Schafer et al., 2012).
Complement proteins also play a role in the adult and aging brain. C1q protein, an initiating factor of the classical complement cascade, is significantly increased in human and mouse brains with age (Stephan et al., 2013) and colocalizes with synapses in brain areas vulnerable to degeneration. Moreover, adult C1q knock-out (KO) mice showed enhanced activity-dependent synaptic potentiation (Stephan et al., 2013). C3 gene expression is also upregulated with aging in humans (Cribbs et al., 2012).
Synaptic degeneration is an early hallmark of aging and neurodegenerative disease that often precedes neuron loss (Morrison and Baxter, 2012). It is unclear whether the mechanisms of complement-mediated synaptic pruning during brain development are recapitulated in the aging brain (Zabel and Kirsch, 2013). C1q is dramatically upregulated with aging, but does not appear to mediate synapse elimination (Stephan et al., 2013). However, it remains to be determined whether C3, a downstream central component of the classical complement cascade and an initiating factor of the alternative complement pathway, promotes synapse loss during aging and to what extent this process is brain region specific. Here, we hypothesized that C3 mediates aberrant synapse loss and dysfunction in the aging brain, thereby contributing to age-associated cognitive decline. We found a region-specific increase in C3 deposition at hippocampal (HC) CA3 synapses in young adult (4 months) C57BL/6J (wild-type, WT) mice, accompanied by a subsequent reduction in synapses and neurons in the same region in aged (16 months) WT mice. Importantly, aged (16 months) C3-deficient mice (C3 KO) were protected against this age-dependent synapse and neuron loss and displayed enhanced learning and spatial memory, less anxiety, and were rescued from age-related LTP decline. Together, these results suggest an important role for C3 in mediating synaptic functional integrity and plasticity in the aging brain.
Materials and Methods
Mice
C57BL/6J mice and homozygous C3-deficient mice (C3−/−; line B6.129S4-C3tm1Crr/J; Wessels et al., 1995) were obtained from The Jackson Laboratory. C3 KO mice were backcrossed onto the C57BL/6J background for >10 generations. Homozygous KOs were crossbred for 10–12 generations. However, a separate cohort of C3 KO mice was bred with C57BL/6J mice to generate C3 KO heterozygotes and WT littermates and then the heterozygotes were crossbred with C3 KO mice to generate homozygotes. Mice were bred and maintained in a virus antibody-free animal facility at Harvard Medical School. Mice were genotyped by PCR using the following primers: 5′-ATCTTGAGTGCACCAAGCC-3′ and 5′-GGTTGCAGC AGTCTATGAAGG-3′ (C3 WT); 5′-CTTGGGTGGAG AGGCTATTC-3′ and 5′-AGGTGAGATGACAGGAGATC-3′(C3 mutant) and 5′-CCTCTTTGT GACTATGTGGACTGATGTCGG-3′ and 5′-GTGGATAACC CCTCCCCCAGCCTAGACC-3′. Mice of all genotypes were aged to postnatal day 30 (P30), 4 months, or 16 months of age. Only males were used for this study to reduce gender-specific variability. At the end of the study, each mouse was anesthetized, blood was collected, and the brain was perfused with saline before harvest. The brain was then bisected and one half was snap frozen for biochemical studies; the other half was immersion fixed in 4% paraformaldehyde (PFA; Electron Microscopy Sciences) overnight at 4°C, cryoprotected in 30% sucrose, and then embedded in optimal cutting temperature (OCT) compound for immunohistochemical study.
Immunohistochemistry and quantification of synaptic markers
Mice from each group were killed and brain tissue collected on the same day. Hemibrains were fixed in 4% PFA for 24 h and embedded in OCT compound (Sakura Finetek). Serial 10 μm sagittal cryosections were mounted on glass slides on the same day for mouse brains from each genotype at each age.
Immunofluorescent labeling and confocal microscopy.
Sections were dried, washed in Tris-buffered saline (TBS), and blocked with 20% goat serum + 0.3% Triton X-100 in TBS for 2 h. Primary antibodies were diluted in antibody buffer with 0.03% Triton X-100 and 10% goat serum as follows: Vglut2 (Millipore goat anti-guinea pig, 1:1000), Homer1 (R&D Systems goat anti-mouse, 1:200), GluR1 (Abcam goat anti-rabbit, 1:200), and SYN-1 (Millipore goat anti-rabbit, 1:200) and incubated overnight at 4°C. Secondary Alexa Fluor-conjugated antibodies (Life Technologies) were added at 1:200 in 0.03% Triton X-100 with 10% goat serum for 2 h at room temperature (RT). For C3 staining, immunohistochemistry was performed using a goat anti-rat C3 antibody (1:200; MPBio Cappel) and buffers containing 1–5% BSA instead of goat serum (Schafer et al., 2012). Sections were mounted (Hard Set with DAPI; Vector Laboratories) and coverslipped.
For each confocal experiment (e.g., the comparison of synaptic markers at 3 ages in WT mice as shown in Fig. 1 or the comparison of synaptic markers for each genotype at each of 3 ages as shown in Fig. 2), all brain sections from a single plane were cut on the same day, immunostained together within 1 d of sectioning, and imaged within 48 h of staining. This process was repeated for each of two additional brain sections at equidistant planes (200 μm apart) per mouse. Confocal imaging was performed using a Zeiss LSM710 confocal microscope and a 63× oil objective. The operator was blinded to the genotype and age of each mouse. Images were acquired using a 1.5 airy unit (AU) pinhole while holding constant the gain and offset parameters for all sections and mice per experiment. In general, a detector gain of ∼660, a digital gain of 1, and an offset of ∼−60 were used when capturing the images. The optical plane with the maximum synaptic staining was identified within a z-stack of 7–11 serial optical images (0.8 μm step intervals) collected over a depth of 7–8 μm. The average synaptic puncta density was calculated using the images from the optical plane with maximum staining, as well as the optical planes immediately above and below (i.e., average density from three consecutive optical planes per brain section), using ImageJ version 1.45 as described previously (Ippolito and Eroglu, 2010). Threshold parameters for ImageJ image analysis were identical for all mouse genotypes and ages for each experiment. The average of the puncta density across three equidistant brain tissue sections (200 μm apart) per mouse was used to compare synaptic puncta density between different genotypes and ages. Note that this method varies somewhat from our earlier work in younger mice (Stevens et al., 2007), in which a 1 AU pinhole was used for acquiring images and, by comparison, may have selected somewhat for larger, brighter synapses. However, the relative differences in synaptic puncta density between age and genotype were significant and consistent with our biochemical, immunohistochemical, electrophysiological, and behavioral results.
Figure 1.
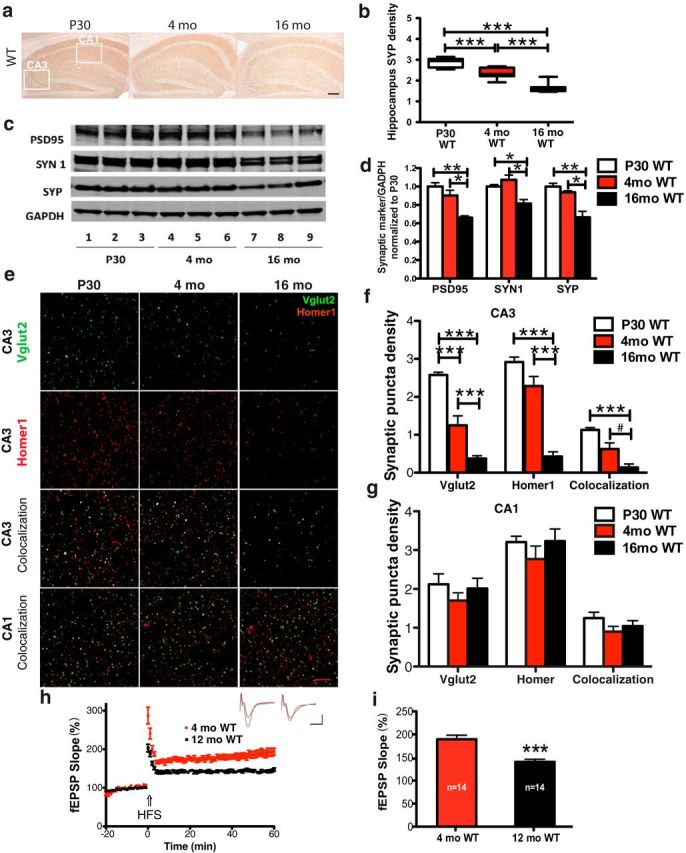
Age-dependent HC synapse loss in male C57BL/6J WT mice. a, Densitometric analysis reveals age-dependent decrease of SYP immunoreactivity in P30, 4-month-old, and 16-month-old WT mice. Scale bar, 100 μm. b, SYP density was lower at 4 months and 16 months of age relative to P30 (***p < 0.001, n = 12, one-way ANOVA). c, d, Western blotting of HC homogenates revealed age-dependent decreases of synaptic proteins (pre: SYN1, SYP; post: PSD95) in 16-month-old versus P30 WT mice (*p < 0.05, **p < 0.01, n = 6; one-way ANOVA). e, Representative confocal microscopy images display an age-dependent reduction in Vglut2 (green, presynaptic marker) and Homer1 (red, postsynaptic marker) IR in CA3 but not CA1 from P30 to 16 months of age. Scale bar, 15 μm. f, g, Quantification of synaptic puncta density revealed age-dependent decreases in Vglut2 and Homer1 IR and colocalization in CA3 but not CA1 (#p = 0.0523, ***p < 0.001, n = 6; 3 equidistant planes 200 μm apart per mouse; one-way ANOVA). h, LTP field recording of HC CA3–CA1 revealed a loss of synaptic plasticity from 4 to 12 months of age. Inset traces are typical fEPSPs recorded before (black) and after (red) high-frequency stimulation recorded in 4-month-old (left) and 12-month-old (right) mice. Horizontal bars: 10 ms; vertical bars: 0.5 mV. i, Statistical analysis of the mean value of the last 10 min of LTP recording confirmed an age-dependent reduction in synaptic potentiation from 4 months to 12 months of age (***p < 0.001, n = 14; unpaired Student's t test).
Figure 2.
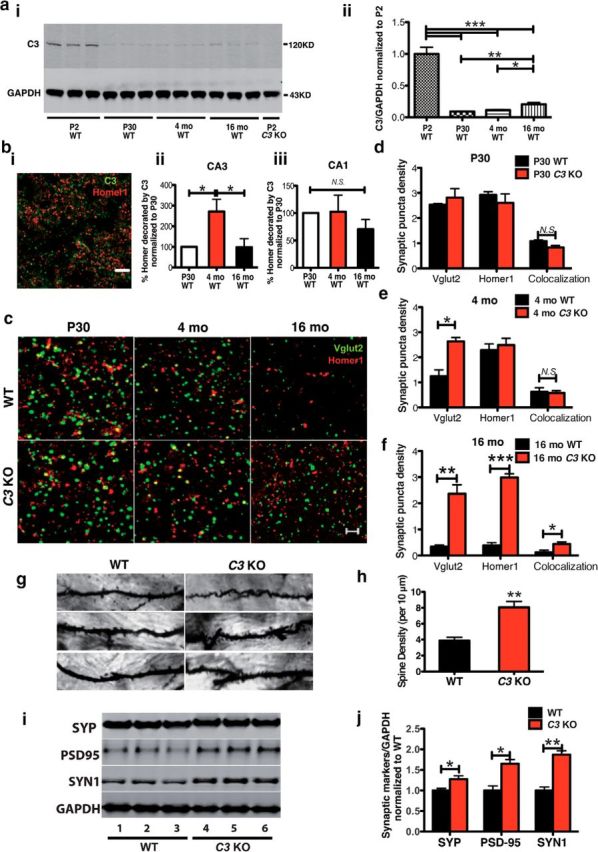
Complement C3 deficiency rescues age-dependent synapse loss in C57BL/6 WT mice. a, Western blotting of C3 in HC homogenates of WT mice at P2, P30, 4-month-old, and 16-month-old WT mice and P2 C3 KO mice. C3 protein levels in WT mice were high at P2, but dropped dramatically by P30. The C3 protein was absent in C3 KO mice. C3 protein levels were modestly but significantly elevated in P30 compared with 16 months of age. (*p < 0.05, **p < 0.01, ***p < 0.0001, one-way ANOVA; n = 3 mice per age). b, Confocal image of C3 (green) and Homer1 (red) synaptic puncta in CA3 of a 4-month-old WT mouse. Scale bar, 10 μm (i). b, The density of C3-tagged Homer1 puncta was significantly increased in CA3 (ii) but not CA1 (iii), of WT mice at 4 months of age compared with P30 and 16 months of age. (*p < 0.05, n = 4 or 5 mice per age; one-way ANOVA). c, High-resolution confocal microscopy of presynaptic (Vglut2, green) and postsynaptic (Homer1, red) IR puncta in CA3 of HC in WT and C3 KO mice at P30, 4 months, and 16 months of age. Scale bar, 5 μm. d, At P30, WT and C3 KO mice had similar synaptic puncta densities. e, f, At 4 months, Vglut2-synaptic density was higher in C3 KO versus WT mice, whereas at 16 months, Homer1, Vglut2, and colocalized puncta were significantly increased in C3 KO mice compared with WT mice (*p < 0.05, **p < 0.01, ***p < 0.001, n = 6 mice per age and genotype; 3 equidistant planes 200 μm apart per mouse; unpaired Student's t test). g, Golgi staining of dendritic spines in CA3 of WT and C3 KO mice at 16 months of age. Scale bar, 10 μm. h, Quantification of Golgi-labeled dendritic spines revealed a significant increase in spine density in C3 KO mice compared with WT at 16 months of age (**p < 0.01, n = 2 mice per genotype; unpaired Student's t test). i, j, Western blotting of HC homogenates revealed elevated SYP, PSD95, and SYN1 synaptic proteins in 16-month-old C3 KO mice compared with age-matched WT mice (*p < 0.05, **p < 0.01, n = 6 mice per age and genotype; unpaired Student's t test). N.S., Not significant.
Synaptophysin immunohistochemistry and optical density analysis.
Immunohistochemistry was performed as described previously (Lemere et al., 2002). Fixed, frozen sections were incubated with anti-synaptophysin (anti-SYP, 1:200; Sigma-Aldrich) mouse monoclonal antibodies overnight at 4°C. After washing with TBS, sections were incubated with a goat anti-mouse biotinylated secondary antibody and developed using Vector ELITE ABC kits (Vector Laboratories) and 3,3-diaminobenzidine (Sigma-Aldrich). Quantification of the optical density of SYP immunoreactivity (IR) was performed using the Bioquant image analysis system with the operator blinded to mouse genotype and age. Images were captured in a single session under a Nikon Eclipse E400 microscope using the 10× objective. The optical density of SYP IR was calculated for the HC and cerebellum (CB) from three equidistant sections 500 μm apart for each animal. The imaging threshold was held constant for the analysis of an entire series of sections.
Electrophysiology on ex vivo mouse HC brain slices
HC slice preparation.
Mice were euthanized with isoflurane at 4 or 12 months of age. The brain was quickly removed and submerged in ice-cold oxygenated sucrose-replaced artificial CSF (ACSF) cutting solution containing the following (in mm): 206 sucrose, 2 KCl, 2 MgSO4, 1.25 NaH2PO4, 1 CaCl2, 1 MgCl2, 26 NaHCO3, and 10 d-glucose, pH 7.4, 315 mOsm. Transverse slices (350 μm thickness) from the middle portion of each HC were cut on a vibroslicer. After dissection, brain slices were incubated in ACSF that contained the following (in mm): 124 NaCl, 2 KCl, 2 MgSO4, 1.25 NaH2PO4, 2.5 CaCl2, 26 NaHCO3, 10 d-glucose, pH 7.4, 310 mOsm, in which they were allowed to recover for at least 90 min before recording. A single slice was then transferred to the recording chamber and continuously perfused with ACSF that had been saturated with 95% O2 and 5% CO2. Slices were incubated in the recording chamber for 20 min at RT (26°C) before stimulation.
Electrophysiological recordings.
Standard procedures were used to record field EPSPs (fEPSPs) of CA3–CA1 synapses in the HC (Li et al., 2011). A bipolar stimulating electrode (FHC) was placed in the Schaffer collaterals to deliver test and conditioning stimuli. A borosilicate glass recording electrode filled with ACSF was positioned in stratum radiatum of CA1 200–300 μm from the stimulating electrode. fEPSPs in the CA1 region were induced by test stimuli at 0.05 Hz with an intensity that elicited an fEPSP amplitude 40–50% of maximum. Test responses were recorded for 30–60 min before the start of the experiment to ensure stability of the response. To induce LTP, 2 consecutive trains (1 s) of stimuli at 100 Hz separated by 20 s were applied to the slices, a protocol that induced LTP lasting ∼1.5 h. The field potentials were amplified 100× using a Molecular Devices 200B amplifier and digitized with Digidata 1322A. Data were sampled at 10 kHz and filtered at 2 kHz. Traces were obtained by pClamp version 9.2 and analyzed using the Clampfit 9.2 program. LTP values reported throughout were measured at 60 min after the conditioning stimulus unless stated otherwise. Paired-pulse responses were monitored at 20, 40, 60, 100, 200, and 500 ms interstimulus intervals. The facilitation ratio was calculated as fEPSP2 slope/fEPSP1 slope.
NeuN immunoreactivity and stereology
NeuN immunohistochemistry was performed as described previously (Maier et al., 2008). Stereological neuron counts were performed on 10 μm sagittal, NeuN-stained sections at each of three planes 500 μm apart per mouse using the optical dissector method (Irizarry et al., 1997). The number of neurons per section in each brain region was estimated using ∼15 optical dissectors and the Bioquant image analysis system according to the principles of Cavalieri (West and Gundersen, 1990). Each optical dissector included a 50 × 50 μm sampling box. Using a 40× objective, neurons with a visible NeuN cell body were counted if they were not visible in the initial plane of focus but came into focus as the optical plane moved through the tissue. The average number of neurons in the HC [CA3, CA1, dentate gyrus (DG)], visual cortex 1 (V1), and CB was determined by calculating the mean of the neuron counts per brain region from each of three sections per animal.
Western blotting
Frozen brain tissues were lysed in 0.2 ml of homogenization buffer containing 5 mm PB, 10% sucrose, and 1% protease inhibitor mixture set I (Calbiochem). Protein concentration was determined by bicinchoninic acid (BCA) assay. Twenty micrograms of total protein was loaded per lane, separated on a 4–12% SDS-PAGE gel, and blotted onto a nitrocellulose membrane. The blot was blocked for 1 h at RT, followed by incubation overnight at 4°C with mouse monoclonal antibodies SYP (1:200; Millipore), PSD95 (1:200; Millipore), GAPDH (1:200; Millipore), rabbit polyclonal Synapsin-1 (SYN-1, 1:200; Millipore), or complement C3 (1:50; Abcam ab11887). Membranes were rinsed and incubated for 1 h with fluorescent-conjugated goat anti-rabbit or mouse IgG (1:5000; Life Technologies). Blots were scanned using a LiCor Odyssey Infrared Imaging System. Intensity of bands was measured by LiCor Odyssey software.
Preparation of synaptosome fractions and quantitative Western blotting
Preparation of synaptosome fractions was performed as described previously (Li et al., 2011). Hippocampi were dissected from C3 KO and WT mice and then the tissues were homogenized in a glass mortar using a rotating Teflon pestle (2000 rpm) with at least 20 passes to create a Dounce homogenate. The homogenate was centrifuged at 1000 × g for 10 min to remove nuclei and incompletely homogenized material (first pellet; P1). The resulting supernatant (S1) was spun at 10,000 × g for 15 min to obtain a second pellet, P2. The supernatant (S2) was defined as the cytosolic fraction. The P2 was subsequently resuspended in 120 μl of sucrose buffer using a motorized pestle mixing/grinding rod (Kontes) directly in the microfuge tube with 30 pulses. The P2 was then subjected to detergent extraction by adding 8 volumes of Triton X-100 buffer (final = 0.5% v/v) containing the following reagents (in mm): 10 Tris, pH 7.4, 1 Na3VO4, 5 NaF, 1 EDTA, and 1 EGTA. This suspension was incubated at 4°C for 20 min with gentle rotation. The resulting suspension was operationally defined as the synaptosome fraction. Protein concentrations were determined using the BCA assay. Twenty micrograms of total protein were loaded per lane, separated by 4%–12% SDS-PAGE, and then transferred onto a nitrocellulose membrane. The blot was blocked for 1 h at RT, followed by incubation overnight at 4°C with rabbit polyclonal antibodies: GluN1, GluR1, SYN-1, or mouse monoclonal antibodies: PSD95, SYP, and GAPDH (all from Millipore). Membranes were rinsed and incubated for 1 h with fluorescent-conjugated goat anti-rabbit or anti-mouse IgG (1:5000; Life Technologies). Blots were scanned using a Li-Cor Odyssey system.
Golgi staining
The FD Rapid Golgi Stain Kit (FD Neurotechnologies), a simplified and reliable kit for Golgi impregnation, was used to label neurons in the CA3 region of HC, as described previously (Li et al., 2013). Upon killing, brains from each of two 16-month-old C3 KO and WT mice were immediately removed and rinsed in MilliQ water (Millipore). After rinsing, the brains were immersed in a Golgi–Cox solution comprising potassium dichromate, mercuric chloride, and potassium chromate. The solution was replaced once after 6 h of initial immersion at RT in darkness and then maintained for an additional 2 weeks. After the immersion period in the Golgi–Cox solution, the brains were transferred to a cryoprotectant solution (FD Rapid Golgi Stain Kit) and stored at 4°C for at least 1 week in the dark before cutting. Then, 250-μm-thick brain sections were cut in the coronal plane using a vibratome. Sections were transferred onto gelatin-coated slides and air dried at RT in the dark for 2 weeks before further processing. After drying, sections were rinsed with distilled water, stained in a developing solution (FD Rapid Golgi Stain Kit) and dehydrated with 50%, 75%, 95%, and 100% ethanol. Finally, the sections were defatted in xylene substitute and coverslipped with Permount (Fisher Scientific). Images were acquired from the stained sections using a Zeiss LSM510 microscope. Each neuron was scanned under high (63×, oil-immersion) magnification. At least five neurons were selected from CA3 per mouse. The density of dendritic spines was measured with ImageJ software.
Behavioral tests
Water T-maze.
The water T-maze (WTM) behavioral paradigm assesses spatial learning and memory by training mice to use the spatial cues in a room to navigate to a hidden platform to escape water. The test also measures cognitive flexibility through a reversal learning procedure in which mice must learn a new location of the hidden platform. The testing apparatus is a plus maze (each arm 14 cm length, 4.6 cm width) made of clear Plexiglas with each arm designated as N, S, E, or W. A divider was placed on the maze to block off the appropriate arm so that the mouse could choose only the E or W arm for escape. The maze was filled with water (25–26°C) and an escape platform was placed on the E side of the maze submerged ∼1 cm below the surface of the water. To ensure that the mice could not see the submerged platform, the water was made opaque by adding white, nontoxic paint. At the start of the trial, the divider was put in place to block off the appropriate arm and mice were carried to the appropriate start point. The experimenter scored a correct or incorrect response for each trial and mice were allowed to stay on the platform for 10 s before being removed. Mice were given 10 trials each day with semirandomized starting points from the N and S positions. The criterion for acquisition was 80% or more correct responses averaged across the 10 trials for 2 consecutive days. The day after all of the mice had reached the acquisition criterion, reversal training began. The platform was moved to the opposite side (W) and the same procedure was repeated until the mice had learned the new position of the platform.
Contextual fear conditioning.
Contextual fear conditioning (CFC) is used to assess memory and is based on the tendency of mice to show a fear response (freezing) when reexposed to the context where they received an aversive stimulus (in this case, foot shock). Mice were exposed to a conditioning chamber (31 cm length × 25 cm width × 25 cm height; Med Associates) with Plexiglas sidewalls, stainless steel end walls, and a floor consisting of steel bars. Mice were allowed to explore the chamber for 2 min and were given 2 presentations of a 2 s foot shock (0.5 mA) separated by 2 min. Mice were removed from the chamber 1 min after the last foot shock. Twenty-four hours after the training session, mice were placed back into the conditioning chamber for 3 min (no electric shock was delivered during this session) and freezing was videotaped and scored with Topscan software (CleverSys). The freezing response was used as a surrogate marker of memory performance because mice remembering receiving the shock during the training session are expected to spend a significant amount of time freezing during the retention session.
Open field test.
The open field (OF) test measures both general locomotor activity and anxiety-like behavior in the mouse. The mouse OF chambers used (Med Associates) are made of clear Plexiglas and consist of a square base (27 cm × 27 cm) with walls 20 cm high. For each testing session, the mouse was allowed free exploration in the environment for 1 h. A computer-assisted infrared tracking system and software (Activity Monitor; Med Associates) was used to record the number of beam breaks and time and entries into center and peripheral zones. An increase in the percentage of ambulatory time (in seconds) spent in the center of the OF was used as a surrogate measure of anxiolytic-like behavior. The total distance traveled (in centimeters) was used as a measure of general locomotor activity.
Elevated plus maze.
The elevated plus maze (EPM) test, which measures anxiety-like behavior in the mouse, consists of two open and two closed arms extended out from a central platform. Each arm of the maze is 30 cm long and 5 cm wide. The maze surface is 85 cm above the floor and the test is performed in dim ambient lighting. Mice were placed on the center platform of the maze facing an open arm and were allowed to explore the apparatus for 5 min. A computer-assisted video-tracking system (TopScan) was used to record the number of open and closed arm entries (all four paws in an arm), as well as the total time spent in open, closed, and center compartments. An increase in the percentage of time spent in the open arms [(open arm time/open + closed arm time) * 100] or an increase in the number of entries into the open arms was used as a surrogate measure of anxiolytic-like behavior. The number of closed arm entries was used as a measure of general locomotor activity.
Object habituation test.
The object habituation (OH) test apparatus is a black, square OF with a white floor (custom made; Plastic Craft). The walls are 50 cm long, 50 cm wide, and 40 cm high. Four OH boxes are used in parallel. The CleverSys computer-assisted video-tracking system (TopScan) was used to record the distance traveled and the time spent exploring the object. Mice were acclimated to the testing room for at least 20 min before testing. Each mouse was given a 30 min habituation to the chamber, followed by a 3 min test with 1 object (a 20 ml glass scintillation vial) placed in the center of the OF. The number of contacts with the object was recorded in 1 min time bins.
Statistical analysis
Data are expressed as mean ± SEM. Significance was assessed with Student's t test or one-way ANOVA followed by Bonferroni's post hoc test using Prism version 6.0 software GraphPad. p < 0.05 was considered significant.
Animal use approval
All animal protocols were approved by the Harvard Medical Area Standing Committee on Animals and studies were performed in accordance with all state and federal regulations. The Harvard Medical School animal management program is accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care, International (AAALAC), and meets all National Institutes of Health standards as demonstrated by an approved Assurance of Compliance (A3431-01) filed at the Office of Laboratory Animal Welfare (OLAW).
Results
Age-dependent synapse loss in HC of WT mice
Previous studies have shown an age-dependent loss of synapses in the HC CA3 region of C57BL/6J WT mice from ∼25 to ∼43 weeks of age (Yu et al., 2011), as well as reduced HC spine density and spine length from 6–7 months to 21–22 months of age (von Bohlen und Halbach et al., 2006). Similarly, age-related loss of HC CA3 synapses has been reported in rats (De Groot et al., 1987; Adams et al., 2008; Adams et al., 2010). To confirm and expand these findings, we examined changes in HC synapses of male C57BL/6J mice (referred to as WT hereafter) at P30, 4 months, and 16 months of age using several different approaches, including morphological imaging for synaptic density, biochemical assays for synaptic protein expression, and electrophysiology for synaptic plasticity. We examined, not only structural, but also functional aspects of HC synapses associated with aging to better understand how aging affects synaptic health.
To determine whether male WT mice display age-dependent synaptic marker loss, we initially performed densitometric analysis of SYP IR in HC and found a steady decline of SYP IR from P30 to 4 months to 16 months of age (percentage of decrease: 15.2 ± 2.6%, 4 months vs P30, p = 0.0005; 20.5 ± 1.2%, 16 months vs 4 months, p < 0.0001; 38.9 ± 1.4%, 16 months vs P30, p < 0.0001; Fig. 1a,b). To confirm the IR findings, we performed Western blotting of HC homogenates and found significant, age-dependent decreases in presynaptic (both SYP and SYN1) and postsynaptic (PSD95) markers from P30 to 16 months of age (PSD95: p = 0.0033; SYN1: p = 0.0181; SYP: p = 0.0035, P30 vs 16 months) and from 4 months to 16 months of age (PSD95: p = 0.0156; SYN1: p = 0.0193; SYP: p = 0.0152, 4 months vs 16 months; Fig. 1c,d). To further confirm the decrease in synaptic markers and to assess their colocalization, we used confocal microscopy to perform synaptic puncta analysis on immunofluorescently labeled brain sections (Ippolito and Eroglu, 2010). We corroborated the reduction in presynaptic (Vglut2) and postsynaptic (Homer1) markers and observed a decrease in colocalized Vglut2- and Homer1 puncta in CA3 stratum radiatum (CA3–SR) of WT mice from P30 to 16 months of age (Vglut2: p < 0.0001; Homer1: p < 0.0001; colocalization: p < 0.0001) and from 4 months to 16 months of age (Vglut2: p = 0.0006; Homer1: p < 0.0001; colocalization: p = 0.0523; Fig. 1e,f). Vglut2 puncta in CA3 were also significantly reduced from P30 to 4 months, as revealed by puncta analysis (Vglut2: p < 0.0001). However, synapse loss was most evident from P30 to 16 months of age (Fig. 1e,f). Age-dependent synapse loss was also confirmed using other synaptic markers, such as the presynaptic marker SYN1 and a postsynaptic marker, GluR1 (data not shown). Together, these three different methods demonstrate age-dependent synapse loss in the CA3 region of HC of WT mice.
To determine whether age-dependent synapse loss in WT mice was region specific, we examined synaptic immunoreactivity in other regions of HC, including CA1-SR and DG stratum moleculare (DG-SM), as well as V1 and CB, using IR densitometry and confocal synaptic puncta analysis (Table 1). We found age-dependent and region-specific changes in synaptic density. Densitometric analysis of SYP IR revealed significant decreases in presynaptic termini in HC and V1 cortex, but not CB, from P30 to 16 months of age in WT mice (Table 1). To determine whether this decrease in the HC presynaptic marker was region specific, we performed confocal puncta analysis on individual densities of Vglut2-IR and Homer1-IR synaptic puncta and their colocalization. We found that both markers and their colocalization were significantly reduced in DG-SM and V1 from P30 to 16 months of age (data not shown) but, interestingly, not in CA1-SR (Fig. 1e,g). Our findings suggest that age-dependent synapse loss occurs in both the HC and cortex in WT mice, some of which is region specific.
Table 1.
Region-specific and age-dependent synapse and neuron loss in WT and C3 KO mice
| Test | Brain region | WT |
C3 KO |
|---|---|---|---|
| P30 → 4 mo → 16 mo | P30 → 4 mo → 16 mo | ||
| Synaptic puncta density (IF), Vglut2/Homer1, GluR1/SYN1 | CA3 | Age-dependent ↓ | Rescued |
| CA1 | N.S. | N.S. | |
| DG | Age-dependent ↓ | Rescued | |
| V1 | Age-dependent ↓ | Rescued | |
| Synaptophysin density (IR) | Hippocampus | Age-dependent ↓ | Rescued |
| CB | N.S. | N.S. | |
| Synaptic proteins (WB) | Hippocampus | Age-dependent ↓ | Rescued |
| PSD95, Synaptophysin, SYN-1 | |||
| Synaptic plasticity, LTP | CA3-CA1 | Age-dependent ↓ | Rescued |
| Neuron number | CA3 | Age-dependent ↓ | Rescued |
| CA1 | N.S. | N.S. | |
| DG | N.S. | N.S. | |
| V1 | N.S. | N.S. | |
| CB | N.S. | N.S. | |
We found age-dependent and region-specific changes in synaptic density in the hippocampus of WT mice. Synaptic puncta analysis revealed significant decreases in synaptic density in CA3-SR, DG-SM, and V1 cortex, but not in CA1-SR, in male WT mice from P30 to 16 months of age. Densitometric analysis of SYP IR showed significant decreases in presynaptic termini in hippocampus and V1 cortex, but not CB. In addition, we observed significantly reduced synaptic protein levels in WT hippocampal homogenates, an age-dependent decline of LTP in hippocampal CA3 to CA1 Schaffer collaterals, as well as a significant loss of neurons in hippocampal CA3 of aged WT mice. However, these phenotypes were rescued in aged male C3 KO mice, which showed significantly more synaptic puncta, higher SYP IR density in hippocampal CA3, higher synaptic protein levels in hippocampal homogenates, higher LTP, and more neurons in hippocampal CA3 compared with age-matched male WT mice. These results suggest that C3 deficiency protects against age-dependent synapse and neuron loss, particularly in the hippocampal CA3 brain region. N.S., Not significant.
To determine whether our observed decrease in synaptic markers represented a decrease in functional synapses, we performed electrophysiological field recordings of HC LTP in the Schaffer collaterals within the HC CA3–CA1 SR region of ex vivo brain slices from 4- and 12-month-old WT mice. We found a significant reduction of LTP from 4 to 12 months of age (189.4 ± 9.0% in 4-month-old mice vs 142.9 ± 3.3% in 12-month-old mice, p < 0.0001; Fig. 1h,i), which is consistent with an age-dependent loss of HC synaptic plasticity in WT mice and suggests that the age-dependent decrease in synaptic puncta colocalization in CA3 is functionally significant.
Complement-deficient mice do not display age-related synapse loss
To determine whether C3 contributes to age-dependent synapse loss of WT mice, we first examined C3 mRNA and protein levels, as well as C3 localization to synapses at different ages in WT mice. It has been reported that C3 mRNA, albeit low throughout life, is elevated in the forebrain of C57BL/6 mice with aging (Reichwald et al., 2009). We failed to find a consistently significant age-related increase in C3 mRNA levels in the whole HC of WT mice (data not shown). However, we did find high C3 protein levels in whole HC homogenates at P2 that dropped dramatically by P30 and were modestly but significantly elevated by 16 months of age (P2 vs P30, 4 months and 16 months: p < 0.0001; P30 vs 16 months: p = 0.0061; 4 months vs 16 months: p = 0.0176; Fig. 2a). In addition, we found a significantly higher level of C3 IR colocalized with postsynaptic Homer1-IR puncta in the HC of 4-month-old WT mice compared with P30 and 16-month-old WT mice by confocal microscopy (4 months vs P30: p = 0.0418; 4 months vs 16 months: p = 0.0399), suggesting the possibility that C3 deposition at 4 months of age in CA3 may be associated with age-dependent synapse elimination in aged WT mice (Fig. 2b). Interestingly, this significant increase in synaptic colocalization with C3 was observed in CA3, but not in CA1 (P > 0.9999; Fig. 2b), suggesting that the localization of complement to synaptic markers may be region specific.
To determine whether C3 mediates age-dependent synapse loss in WT mice, we assessed synaptic puncta density in male C3 KO mice at P30, 4 months, and 16 months of age compared with age-matched male WT mice. Vglut2-IR presynaptic puncta, Homer1-IR postsynaptic puncta, and colocalization of both markers were imaged by confocal microscopy within the HC CA3–SR area (Fig. 2c). Importantly, at P30, we found no differences between WT and C3 KO mice in the densities of Homer1-IR puncta, Vglut2-IR puncta, and their colocalization in the HC CA3 region (Fig. 2c,d). In contrast, the density of Vglut2-IR presynaptic termini in CA3 was significantly higher in C3 KO mice compared with WT mice at 4 months of age (p = 0.0347); however, no differences were seen in postsynaptic Homer1-IR puncta or colocalization of the markers at this age (Fig. 2c,e). By 16 months of age, the densities of Vglut2-IR puncta, Homer1-IR puncta, and their colocalization were significantly increased in HC CA3 of C3 KO mice compared with age-matched WT mice (Vglut2: p = 0.002, Homer1: p < 0.0001 and colocalization: p = 0.0109 in C3 KO vs WT mice; Fig. 2c,f). These data suggest that C3 KO mice are protected against the age-dependent decrease of synaptic puncta and their colocalization in the CA3–SR region of HC observed in WT mice. Similar protective effects against synaptic loss were found in the HC DG-SM and V1 cortex areas in C3 KO mice compared with WT mice at 16 months of age, whereas no differences were observed in CA1 synapses at any age between the two genotypes (Table 1). This region-specific change in HC synaptic density corresponds to a region-specific increase in C3 deposition at synapses in CA3, but not in the CA1 region (Fig. 2b).
To further examine the effect of C3 deficiency on age-dependent synapse loss, we examined 16-month-old WT and C3 KO mice using additional assays. Golgi impregnation was used to compare semiquantitatively dendritic spine density in CA3–SR within a small number of 16-month-old C3 KO and WT mice. Visual inspection and quantification indicated that C3 KO mice had increased spine density compared with WT mice (p = 0.0012; Fig. 2g,h). Consistent with this finding, Western blotting of HC homogenates revealed significant increases in SYP, PSD95, and SYN1 protein levels in aged C3 KO mice compared with WT mice (SYP: p = 0.0271; PSD95: p = 0.0118; SYN1: p = 0.0024 C3 KO vs WT; Fig. 2i,j). In addition, SYP IR density in HC was higher in HC CA3 of 16-month-old C3 KO mice compared with age-matched WT mice (data not shown). Therefore, all three assays confirmed the increase in synapses in 16-month-old C3 KO mice compared with WT mice that was observed by puncta analysis. These results suggest that complement C3 deficiency protects against age-dependent, brain-region-specific synapse loss that occurs with aging in the CA3 and DG of HC and the specific cortical region (V1) that we examined in the WT mice.
Complement-deficient mice do not display age-related HC neuron loss
Age-related neuron loss in the HC has been reported in aged rats (Kerr et al., 1991), particularly in CA3 (Landfield et al., 1981b; Sapolsky et al., 1985), as well as in ICR mice (Institute for Cancer Research, a cancer-prone breed; Himeda et al., 2005). To our knowledge, unlike age-dependent synaptic loss in HC, there is no published evidence of HC neuron loss in aged C57BL/6J WT mice, including a report that specifically focused on CA1 and DG, but did not examine the HC CA3 region (Calhoun et al., 1998). To determine whether neuron loss accompanies the synapse loss that we observed in WT mice during aging, we performed NeuN immunohistochemistry and quantitative, unbiased stereological analyses on brains of P30, 4-month-old, and 16-month-old WT mice. A significant reduction in the number of HC CA3 neurons was observed in WT mice from P30 to 16 months of age (p = 0.0351, 16 months vs P30; p = 0.2272, 16 months vs 4 months; p = 0.7603, 4 months vs P30; Fig. 3a,b). However, no reduction in neuron number was observed in CA1, DG, V1 cortex, or CB in WT mice with aging (Table 1), suggesting that CA3 neurons are more vulnerable to aging than neurons in other brain regions. To determine whether complement deficiency protected against age-dependent HC CA3 neuron loss, we performed stereological analysis of C3 KO mice at P30, 4 months, and 16 months of age. Interestingly, no age-dependent CA3 neuron loss was found in C3 KO mice from P30 to 16 months of age (p = 0.7576, 16 months vs P30; p = 0.8169, 16 months vs 4 months; p = 0.9186, 4 months vs P30; Fig. 3c,d), suggesting that C3 deficiency protected against age-dependent neuron loss in the HC CA3 brain region. Neuron numbers in CA1, V1 cortex, and CB were not significantly different between 16-month-old WT and C3 KO mice (CA1: p = 0.9937; cortex V1, p = 0.7377; CB, p = 0.9670; Fig. 3e–g, Table 1), demonstrating the region specificity of neuron loss.
Figure 3.

C3 deficiency rescues neuron loss in the HC CA3 region in aged WT mice. a, c, NeuN-immunostained neurons in HC of WT (a), C3 KO (c) mice at P30, 4 months and 16 months of age (HC shown in top and CA3 in bottom) Scale bar, 50 μm. b, Unbiased stereological neuron counts revealed an age-dependent decrease in neurons in HC CA3 of WT mice (*p < 0.05, P30 vs 16 months; n = 5 mice per age and genotype; 3 equidistant planes 500 μm apart per mouse; unpaired Student's t test). d, Neuron numbers remained stable from P30 to 16 months of age in C3 KO mice, indicating the absence of age-dependent neuron loss (n = 4–7 mice per age and genotype; 3 equidistant planes 500 μm apart per mouse; unpaired Student's t test). e–h, No significant differences were observed between genotypes in neuron number in HC CA1, DG, cortex V1, and CB of 16-month-old WT and C3 KO mice (n = 8 mice per age and genotype; 3 equidistant planes per mouse; unpaired Student's t test). N.S., Not significant.
C3-deficient mice have enhanced synaptic plasticity at 12 months, but not at 4 months of age, compared with WT mice
One consequence of age-dependent synapse loss is the alteration of synaptic function and plasticity that may underlie age-related cognitive decline. We next investigated whether age-dependent loss of HC synaptic plasticity and LTP in WT mice (Fig. 1h,i) is reduced in C3 KO mice. Field recording of the CA3–CA1 Shaffer collateral (CA3–CA1 SR layer) HC LTP elicited by high-frequency stimulation was performed on brain slices from 4- and 12-month-old WT and C3 KO mice. Although basal synaptic transmission was not significantly different between genotypes, we found a small trend for increased basal synaptic transmission in CA3–CA1 SR in 12-month-old C3 KO mice compared with WT mice (P > 0.05; Fig. 4a). Moreover, we observed an increase in paired pulse facilitation at interstimulation intervals of 40 and 60 ms in 12-month-old C3 KO mice compared with WT mice (p = 0.0408 at 40 ms; p = 0.0916 at 60 ms; Fig. 4b). No significant differences in HC CA3–CA1 LTP were detected between 4-month-old C3 KO and WT mice (Fig. 4c,e). Interestingly, we found a significant enhancement of LTP in 12-month-old C3 KO mice compared with age-matched WT mice (173.5 ± 8.7% vs 141.5 ± 4.5%, p = 0.0078; Fig. 4d,e). HC LTP was significantly reduced from 4 months to 12 months in WT mice (196.8 ± 12.4% vs 141.5 ± 4.5%, p = 0.0007), but not in C3 KO mice (187.0 ± 12.8% vs 173.5 ± 8.7%, p = 0.4066; Fig. 4e). These electrophysiological data indicate that, in contrast to WT mice, C3-deficient mice maintained long-term synaptic plasticity and showed rescue of age-dependent decline in LTP and enhanced presynaptic facilitation in aged mice (Fig. 4a–e, Table 1), consistent with our structural studies.
Figure 4.
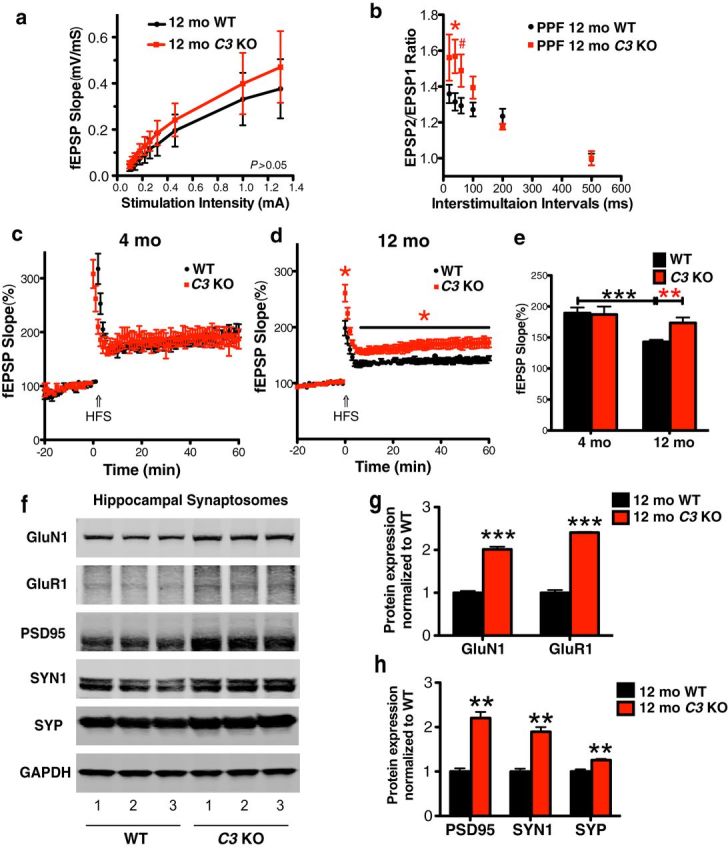
C3 deficiency rescues age-dependent LTP attenuation in 12-month-old mice. a, The I/O curve revealed that the relationship between the range of stimulus intensities and the fEPSP rising slope was similar between 12-month-old WT and C3 KO mice. b, At 12 months of age, C3 KO mice had increased PPF compared with WT mice (*p < 0.05 at 40 ms intervals and #p = 0.0916 at 60 ms intervals; n = 8–10 per genotype; unpaired Student's t test). c, At 4 months, LTP induced by high-frequency stimulation (HFS, arrow) was similar between WT and C3 KO mice (n = 14 slices in total from 7–8 mice per genotype). d, At 12 months, HFS (arrow)-induced LTP was higher in C3 KO mice versus WT mice (*p < 0.05; n = 12 slices in total from 6 mice per genotype; unpaired Student's t test). e, Based on c and d, WT mice showed an age-dependent reduction in LTP that was rescued in C3 KO mice (**p < 0.01, 4-month-old WT vs 12-month-old WT; **p < 0.01, 12-month-old C3 KO vs 12-month-old WT; unpaired Student's t test). The bar graph represents the mean value of the last 10 min of LTP recording. f–h, Western blotting of HC synaptosomes revealed increases in NMDAR GluN1, AMPAR GluR1 (g), and synaptic markers PSD95, SYN1, and SYP (h) in 12-month-old C3 KO versus WT mice (**p < 0.01, ***p < 0.001, n = 6 mice per genotype; unpaired Student's t test). g and h do not have the black line comparing the two bars that are present in all other bar graphs.
Activation of the AMPA-type glutamate receptor (AMPAR) drives the fast EPSP in glutamatergic synapses, whereas the delayed opening of the NMDA-type glutamate receptor (NMDAR), upon concomitant postsynaptic membrane depolarization, leads to LTP induction. Therefore, changes in the number of AMPARs and NMDARs result in modification of postsynaptic strength (Sheng and Kim, 2002). To further elucidate the mechanism of LTP enhancement in C3 KO mice, we examined the protein levels of the NMDAR obligatory subunit GluN1, the AMPAR subunit GluR1, two presynaptic markers (SYN1 and SYP), and a postsynaptic marker (PSD95) in HC synaptosomes isolated from 12-month-old C3 KO and WT mouse brains. Although the level of the control protein GAPDH was similar between the two genotypes for each of the three fractions, synaptic proteins were highly enriched in the synaptosome fraction relative to GAPDH compared with the total protein homogenate and cytosome fractions (data not shown). All five proteins were significantly elevated in synaptosomes from C3 KO vs WT mice. GluN1 and GluR1 were significantly increased in C3 KO mice by ∼2- to 2.5-fold compared with WT mice (GluN1: p = 0.002; GluR1: p < 0.0001; C3 KO vs WT; Fig. 4f,g). PSD95, SYN1, and SYP were significantly increased by ∼1.2- to 2.2-fold in C3 KO mice compared with WT mice (PSD95: p = 0.0015; SYN1: p = 0.0018; SYP: p = 0.0096; Fig. 4f,h). These data suggest that the enhanced LTP found in 12-month-old C3 KO mice may be mediated by increases in both presynaptic and postsynaptic proteins.
Aged C3 KO mice display enhanced learning and spatial memory and an anxiolytic phenotype compared with age-matched WT mice
Loss of HC synapses and neurons is associated with age-related learning and memory deficits. To determine whether complement deficiency affects age-related behavioral changes in mice, we assessed learning and memory using the WTM and CFC tests (Fig. 5, Table 2) in 16-month-old WT and C3 KO male mice. In the WTM, which can measure spatial learning and memory during acquisition and cognitive flexibility during reversal learning (Kleinknecht et al., 2012), there were no significant differences in the percentage of correct choices in locating the hidden platform between 16-month-old C3 KO and WT mice during the 6 d acquisition period (Fig. 5a). However, upon switching the location of the platform in the reversal trial, C3 KO mice showed a significantly higher rate of correct choices by the second day compared with WT mice (day 2: p = 0.0008; day 3: p = 0.0034; day 4: p = 0.093, C3 KO vs WT), indicating the enhanced ability of C3 KO mice to relearn and retain the new location of the platform (Fig. 5a). This finding suggests that aged C3-deficient mice had enhanced cognitive flexibility compared with aged WT mice (Kleinknecht et al., 2012).
Figure 5.
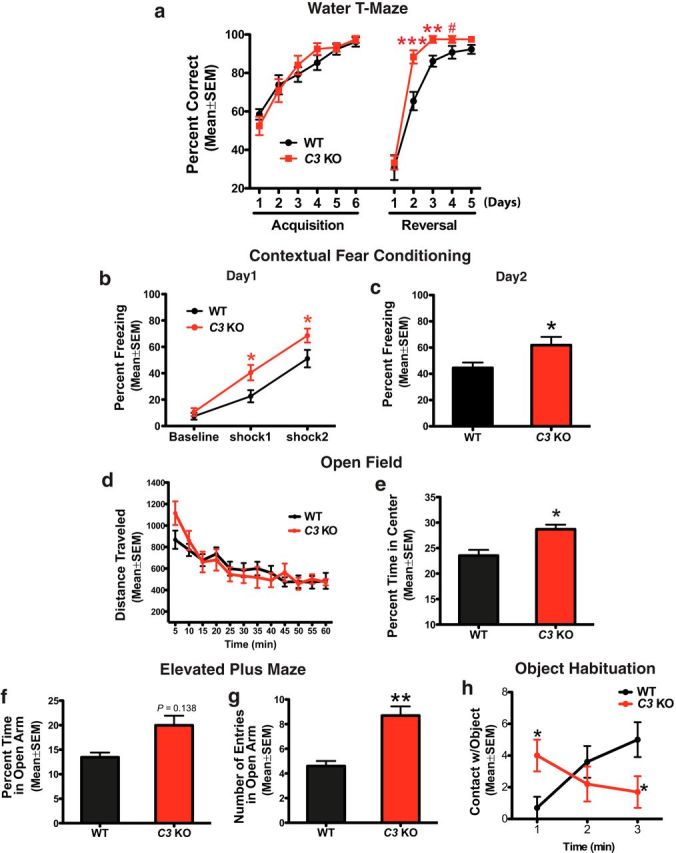
C3 deficiency improved spatial and contextual memory in the WTM and CFC tests at 16 months of age. a, In the WTM test, 16-month-old C3 KO and WT mice were similar in their ability to find the platform location during acquisition. However, the C3 KO mice learned and remembered the new location of the platform in the reversal test significantly sooner (day 2, day 3) than the WT mice (***p < 0.001 at reversal day 2, **p < 0.01 at reversal day 3, #p = 0.093 at reversal day 4, n = 12 male mice per genotype; unpaired Student's t test). b, c, In the CFC test, 16-month-old C3 KO mice demonstrated significantly increased freezing times compared with age-matched WT mice after both the first and second shocks during training on day 1 (b) and appeared to show better retention of the shock-paired context, as indicated by increased freezing times in the absence of a shock, during the conditioning test on day 2 (*p < 0.05, n = 14 male C3 KO mice, n = 12 male WT mice; unpaired Student's t test; c). d, e, C3 KO and WT mice displayed normal locomotor activity in the OF test (d), but C3 KO mice spent significantly more time in the center of the field than WT mice, suggesting an anxiolytic phenotype (*p < 0.05, n = 12 male WT mice, n = 10 male C3 KO mice; unpaired Student's t test; e). f, g, In the EPM, C3 KO mice made significantly more open arm entries (**p < 0.01; g), suggesting that they were less anxious than WT mice (n = 12 male mice per genotype; unpaired Student's t test). h, In the OH test, C3 KO mice showed significantly more contacts with the novel object than WT mice in the first minutes, supporting an anti-anxiety like phenotype and fewer contacts at 3 min, suggesting that these mice may have lost interest in the novel object (*p < 0.05, unpaired Student's t test).
Table 2.
Behavioral test results in C3 KO and WT mice at 4 mo and 16 mo of age
| Behavior tests | 4 mo |
16 mo |
|---|---|---|
| C3 KO vs WT | C3 KO vs WT | |
| No change | ||
| Baseline behavior | ||
| SHIRPA | Normal | Normal |
| Rotarod | Normal | Normal |
| Grip strength | Normal | Normal |
| Social interaction | Normal | Normal |
| Age-dependent | ||
| Cognition/memory | ||
| Water maze | Morris water maze | WTM |
| Normal | Better memory | |
| CFC | Froze more (d 1); better learning | Froze more (d 1 and 2) |
| Better learning and memory | ||
| Anxiety | ||
| OF | Normal | Increased time in center |
| Less anxiety | ||
| EPM | Normal | Increased open arm entries |
| Less anxiety | ||
| OH | Normal | More contacts in min 1 |
| Fewer contacts in min 3 |
For motor and social behavior, we performed SHIRPA, Rotarod, grip strength and social interaction tests, none of which revealed any differences between genotypes at either 4 or 16 months of age. In terms of cognition/memory, 4-month-old C3 KO mice froze longer during CFC training compared with WT mice, suggesting better learning in the C3 KO mice compared with 4-month-old WT mice. No differences were found in the CFC memory test and WTM between 4-month-old C3 KO and WT mice. However, 16-month-old C3 KO mice performed better than 16-month-old WT mice in the reversal phase of the WTM test (i.e. enhanced cognitive flexibility) and spent more time freezing in both the training and testing sessions of the CFC test, suggesting that C3 deficiency rescued age-dependent learning and memory deficits. In addition, 16-month-old C3 KO mice demonstrated anxiolytic-like behavior compared with age-matched WT mice in three different tests. The aged C3 KO mice spent significantly more time in the center of the arena in the OF test, showed more total open arm entries in the EPM test, and made significantly more contacts with the novel object during the first minute and fewer contacts during the third minute in the OH test compared with aged WT mice.
The CFC is an important behavioral paradigm for studying the neurobiology of learning and memory and the mnemonic function of the HC (Rudy et al., 2004). We performed CFC testing in 16-month-old WT and C3 KO mice. During training on day 1, C3 KO mice demonstrated increased freezing time after the first and second shocks compared with age-matched WT mice (shock 1: p = 0.0251; shock 2: p = 0.0472; Fig. 5b). Twenty-four hours later, in the absence of a shock, the C3 KO mice spent significantly more time freezing than WT mice, suggesting that the C3 KO mice showed better retention of the shock-paired context from the day before than did WT mice (p = 0.0319; Fig. 5c). These data provide further support that aged C3-deficient mice showed better performance in an HC-dependent task compared with aged WT mice.
Other behavioral tests were also performed on 16-month-old WT and C3 KO mice and no differences were observed between genotypes in the SHIRPA test, indicating normal basic sensory and motor function, as well as rotarod, grip strength, or social interactions (Table 2). Locomotor activity was similar between genotypes in the OF test. However, 16-month-old C3 KO mice spent significantly more ambulatory time in the center of the field compared with age-matched WT mice, indicative of an anxiolytic-like (anti-anxiety) phenotype (p = 0.0222; Fig. 5d,e). The anxiolytic-like phenotype in C3 KO mice was confirmed in the EPM, in which 16-month-old C3 KO mice made significantly more total open arm entries compared with WT mice (p = 0.0083; Fig. 5g). No significant difference was found in close arm entries (data not shown). In the OH test, 16-month-old C3 KO mice showed significantly more contacts with the novel object compared with age-matched WT mice during the first minute and fewer contacts during the third minute of the test, whereas WT mice showed less contact at 1 min and more contact at 3 min (Fig. 5h). These data further confirmed that 16-month-old C3-deficient mice had less anxiety because the mice appeared more interested in the novel object initially, but lost interest more quickly compared with WT mice. We therefore observed anxiolytic behavior in aged C3 KO mice in three different tests.
Additional behavioral tests were performed on 4-month-old WT and C3 KO mice and some tests were repeated comparing 4-month-old C3 KO mice and WT littermate control mice, but no significant differences were found in the OF (data not shown), general health (SHIRPA), rotarod, grip strength, and social interactions tests (Table 2). No differences were observed between genotypes in the Morris water maze, EPM, and OH tests in 4-month-old mice (Table 2). However, 4-month-old C3 KO mice froze more during training, but not retention, than WT mice in the CFC test; this difference was more pronounced and accompanied by enhanced retention 24 h later in 16-month-old mice (Table 2), suggesting a greater protective effect of C3 deficiency on memory in aged versus young adult mice. Therefore, complement C3 deficiency resulted in age-dependent behavioral enhancement compared with complement C3-sufficient WT mice.
Discussion
Synapse loss and neuronal dysfunction likely underlie age-related cognitive decline, although the molecular pathways remain unclear. Complement C3, an innate immune system molecule, mediates synapse elimination in the developing mouse visual system (Stevens et al., 2007), but it is unknown whether C3 also plays a role in synaptic health during aging. Here, we used molecular, cellular, electrophysiological, and behavioral indices to demonstrate that C3-deficient mice are spared from the synapse and neuron loss and cognitive decline typically observed in older mice, suggesting an important role for C3 in the aging brain.
Region-specific and age-dependent synapse loss in C57BL/6 WT mouse brain
We observed an age-dependent decline in CA3 and DG HC synapses in WT mice that was region specific because the CA1 was largely spared, consistent with other reports showing that age-related CA1 synapse loss was absent (Calhoun et al., 1998; Geinisman et al., 2004) even in the presence of synapse loss in CA3 and DG (Morrison and Baxter, 2012). It remains to be determined which synaptic inputs onto CA3 pyramidal neurons are lost, but, given the age-dependent loss of CA3 neurons described herein, the loss of CA3 recurrent connections may be a contributing factor. Inputs from the medial septum and diagonal brand of Broca may also be involved because both areas display moderate degenerative changes during aging (Baskerville et al., 2006; Schliebs and Arendt, 2006; Li et al., 2008). Consistent with our results, synapse loss in the DG-SM, and associated memory impairments, have been reported previously (Geinisman et al., 1986; Geinisman et al., 1992; Rutten et al., 2005). Our finding of synaptic loss in visual cortex is consistent with age-dependent visual deficits in aged C57BL/6 mice (Stover and Brown, 2012). Therefore, synapses in different brain regions, including specific subregions of HC and cortex, may have different vulnerabilities to aging (Morrison and Baxter, 2012).
Complement C3 may contribute to synapse loss during aging
Although they are dynamically regulated during brain development to allow appropriate synaptic pruning by microglia (Stevens et al., 2007; Schafer et al., 2012), complement proteins have been shown to increase in the CNS during aging (Stoltzner et al., 2000; Wyss-Coray et al., 2002; Reichwald et al., 2009; Stephan et al., 2013). Unlike C1q, which dramatically increases with age (Stephan et al., 2013), C3 is found in much lower abundance in adult and aged brains. Whereas a 9-fold increase in forebrain C3 mRNA from 3 to 24 months of age in C57BL/6 mice has been reported (Reichwald et al., 2009), we did not detect a consistent change in HC C3 mRNA levels from P30 to 16 months in WT mice. At the protein level, we found high C3 levels in brain at P2 that dropped by P30, followed by a small but significant increase by 16 months. Deposition of C3 was selectively enhanced at HC CA3 synapses at 4 months, suggesting that, as in development, C3 activity may contribute to synapse loss in adulthood. Recently, an NF-κB-mediated elevation in C3 secretion by astrocytes was shown to alter neuronal morphology and function (Lian et al., 2015), further underscoring the importance of C3 during brain aging.
We found that C3 KO mice were largely spared from the age-dependent synapse loss seen in CA3 of WT mice, demonstrating a role for C3 in synapse loss in the aging brain. This effect was region specific because CA1 was spared from synapse loss, consistent with a recent study analyzing dendritic spine morphology of young (P17–P30) WT and C3 KO mice (Perez-Alcazar et al., 2014). Importantly, we observed no difference in HC CA3 synaptic puncta between WT and C3 KO mice at P30, strongly arguing against a developmental effect and supporting an age-dependent effect of C3 deficiency. Furthermore, C1q, the classical complement cascade initiator that plays a prominent role in synapse elimination during development, does not appear to play a role in synapse loss during aging, but instead is associated with maintaining brain functionality in the adult mouse (Stephan et al., 2013).
Synaptic integrity is associated with LTP induction and maintenance in the HC (Geinisman et al., 1991; Geinisman et al., 1996). We found that C3 KO mice were spared from the age-dependent LTP reduction observed in WT mice from 4 to 12 months of age, consistent with synapse preservation in C3 KO mice. This effect appears to be specific to older mice because a recent study found no difference in evoked fEPSPs in very young C3 KO compared with WT mice (Perez-Alcazar et al., 2014). Interestingly, C1q deficiency enhanced LTP of the DG perforant pathway of young adult mice, but not aged mice (Stephan et al., 2013), suggesting that adult C1q protein acts in a nonclassical complement fashion.
Region-specific, age-dependent neuron loss may be a consequence of complement-mediated synapse elimination
Neuron loss during aging has been reported in some brain regions of humans, nonhuman primates, and rodents, including the HC (Landfield et al., 1981a; Coleman and Flood, 1987; Meaney et al., 1988; Kerr et al., 1991; Himeda et al., 2005); however, technical differences have yielded inconsistent results (Morrison and Hof, 2002; von Bohlen und Halbach and Unsicker, 2002). Here, we report that the selective reduction in neuron number in HC CA3 from P30 to 16 months in WT mice is spared in C3 KO mice, consistent with a recent report showing that C3 deficiency protected against neuron loss in a ganglioside double-KO mouse model of spinal cord degeneration (Ohmi et al., 2014).
CA3 neuronal viability is selectively affected by stress and immune signaling challenges (Sapolsky et al., 1985; Brunson et al., 2001; Yamada et al., 2001) and our data implicate C3 as potentially mediating this death. Our finding that early CA3 synapse loss precedes late neuron loss in WT mice suggests that the age-dependent loss of CA3 neurons is due to postdevelopment complement-mediated synapse elimination in the adult brain. Further studies are needed to fully understand the mechanisms underlying the selective vulnerability of CA3 neurons during stress, aging, and neurodegeneration.
Complement C3-deficient mice do not display age-dependent behavioral deficits
Changes in HC synapses, dendritic spine densities, and neuron numbers represent morphological correlates of altered HC-dependent learning and memory (Gerstein et al., 2012). Many studies report age-related deficits in HC-dependent learning and spatial memory tasks (Magnusson et al., 2003; von Bohlen und Halbach et al., 2006; Wong and Brown, 2007; Filali and Lalonde, 2009; Yu et al., 2011; Pistell et al., 2012). Here, we found that C3 deficiency significantly protected mice against age-related cognitive decline. Recently, young (2–3 months) C3 KO mice were shown to have enhanced place and reversal learning (Perez-Alcazar et al., 2014), consistent with our observation that 4-month-old C3 KO mice displayed increased learning during day 1 training in the CFC test. Our finding that aged C3 KO mice display an anxiolytic-like (i.e., anti-anxiety) phenotype compared with aged WT mice may be attributed to the preservation of synapses and enhanced presynaptic facilitation (Aroniadou-Anderjaska et al., 2012) in HC and cortex of aged C3 KO mice. Therefore, it appears that complement C3, and/or its downstream signaling, contributes to age-dependent cognitive decline, possibly by mediating synaptic degeneration and subsequent neuron loss in the CA3 region of HC.
Our findings of protection against age-dependent cognitive decline in C3 KO mice are consistent with those reported by Stephan el al. (2013), in which aged C1q KO mice showed less HC-dependent cognitive decline than WT mice. However, in their study, cognitive protection in aged C1q KO mice appeared to act through a nonclassical pathway. Although Stephan el al. (2013) suggest that C1q affects neuronal circuitry in DG in a novel, complement-independent way during aging, our data suggest that C3 activation, perhaps via the alternative pathway, may induce the phagocytic removal of complement-tagged HC CA3 synapses weakened by the aging process. Further studies are under way.
C3 deficiency in neurodegeneration
Aging is a significant risk factor for neurodegenerative disorders typically associated with early synapse loss and subsequent neuron loss and our data implicate C3 as potentially playing a role in these processes. In the context of Alzheimer's disease, we and others have shown that C3 deficiency reduced Aβ peptide uptake and clearance by microglia (Fu et al., 2012), whereas C3 deficiency or C3 inhibition in hAPP-transgenic mice increased Aβ deposition and promoted neurodegeneration (Wyss-Coray et al., 2002; Maier et al., 2008), suggesting that, in some AD-like transgenic mouse models, C3 may be protective. This contrasts strongly with our current data regarding C3 deficiency in aged WT mice and a recent report (Ohmi et al., 2014) showing that C3 deficiency spared spinal cord neuron loss in a ganglioside double-KO mouse model of neurodegeneration, both of which suggest that C3 inhibition may be protective. Further studies are under way to better understand the mechanistic effects of C3 deficiency in WT mice and various disease models.
In summary, we describe age-dependent and region-specific synapse and neuron loss in WT mice that appear to contribute to cognitive decline. These age-related effects were spared in C3 KO mice, suggesting that complement C3 affects synaptic health in adult brain. Our findings shed light on mechanisms of HC vulnerability and neurodegeneration, identifying C3 as a potential therapeutic target for brain aging.
Footnotes
This work was supported by an anonymous foundation (C.A.L. and B.S.). We thank Saba Chowdhury and Kriti Gupta for technical assistance.
The authors declare no competing financial interests.
References
- Adams MM, Shi L, Linville MC, Forbes ME, Long AB, Bennett C, Newton IG, Carter CS, Sonntag WE, Riddle DR, Brunso-Bechtold JK. Caloric restriction and age affect synaptic proteins in hippocampal CA3 and spatial learning ability. Exp Neurol. 2008;211:141–149. doi: 10.1016/j.expneurol.2008.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams MM, Donohue HS, Linville MC, Iversen EA, Newton IG, Brunso-Bechtold JK. Age-related synapse loss in hippocampal CA3 is not reversed by caloric restriction. Neuroscience. 2010;171:373–382. doi: 10.1016/j.neuroscience.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aroniadou-Anderjaska V, Pidoplichko VI, Figueiredo TH, Almeida-Suhett CP, Prager EM, Braga MF. Presynaptic facilitation of glutamate release in the basolateral amygdala: a mechanism for the anxiogenic and seizurogenic function of GluK1 receptors. Neuroscience. 2012;221:157–169. doi: 10.1016/j.neuroscience.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baskerville KA, Kent C, Nicolle MM, Gallagher M, McKinney M. Aging causes partial loss of basal forebrain but no loss of pontine reticular cholinergic neurons. Neuroreport. 2006;17:1819–1823. doi: 10.1097/WNR.0b013e32800fef5a. [DOI] [PubMed] [Google Scholar]
- Brunson KL, Eghbal-Ahmadi M, Bender R, Chen Y, Baram TZ. Long-term, progressive hippocampal cell loss and dysfunction induced by early-life administration of corticotropin-releasing hormone reproduce the effects of early-life stress. Proc Natl Acad Sci U S A. 2001;98:8856–8861. doi: 10.1073/pnas.151224898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calhoun ME, Kurth D, Phinney AL, Long JM, Hengemihle J, Mouton PR, Ingram DK, Jucker M. Hippocampal neuron and synaptophysin-positive bouton number in aging C57BL/6 mice. Neurobiol Aging. 1998;19:599–606. doi: 10.1016/S0197-4580(98)00098-0. [DOI] [PubMed] [Google Scholar]
- Coleman PD, Flood DG. Neuron numbers and dendritic extent in normal aging and Alzheimer's disease. Neurobiol Aging. 1987;8:521–545. doi: 10.1016/0197-4580(87)90127-8. [DOI] [PubMed] [Google Scholar]
- Cribbs DH, Berchtold NC, Perreau V, Coleman PD, Rogers J, Tenner AJ, Cotman CW. Extensive innate immune gene activation accompanies brain aging, increasing vulnerability to cognitive decline and neurodegeneration: a microarray study. J Neuroinflammation. 2012;9:179. doi: 10.1186/1742-2094-9-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Groot DMG BE. Numerical changes in rat hippocampal synapses: an effect of ‘aging’? Acta Stereol. 1987;6:53–58. [Google Scholar]
- Filali M, Lalonde R. Age-related cognitive decline and nesting behavior in an APPswe/PS1 bigenic model of Alzheimer's disease. Brain Res. 2009;1292:93–99. doi: 10.1016/j.brainres.2009.07.066. [DOI] [PubMed] [Google Scholar]
- Fu H, Liu B, Frost JL, Hong S, Jin M, Ostaszewski B, Shankar GM, Costantino IM, Carroll MC, Mayadas TN, Lemere CA. Complement component C3 and complement receptor type 3 contribute to the phagocytosis and clearance of fibrillar Abeta by microglia. Glia. 2012;60:993–1003. doi: 10.1002/glia.22331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita T. Evolution of the lectin-complement pathway and its role in innate immunity. Nat Rev Immunol. 2002;2:346–353. doi: 10.1038/nri800. [DOI] [PubMed] [Google Scholar]
- Geinisman Y, de Toledo-Morrell L, Morrell F. Loss of perforated synapses in the dentate gyrus: morphological substrate of memory deficit in aged rats. Proc Natl Acad Sci U S A. 1986;83:3027–3031. doi: 10.1073/pnas.83.9.3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geinisman Y, deToledo-Morrell L, Morrell F. Induction of long-term potentiation is associated with an increase in the number of axospinous synapses with segmented postsynaptic densities. Brain Res. 1991;566:77–88. doi: 10.1016/0006-8993(91)91683-R. [DOI] [PubMed] [Google Scholar]
- Geinisman Y, deToledo-Morrell L, Morrell F, Persina IS, Rossi M. Age-related loss of axospinous synapses formed by two afferent systems in the rat dentate gyrus as revealed by the unbiased stereological dissector technique. Hippocampus. 1992;2:437–444. doi: 10.1002/hipo.450020411. [DOI] [PubMed] [Google Scholar]
- Geinisman Y, Detoledo-Morrell L, Morrell F, Persina IS, Beatty MA. Synapse restructuring associated with the maintenance phase of hippocampal long-term potentiation. J Comp Neurol. 1996;368:413–423. doi: 10.1002/(SICI)1096-9861(19960506)368:3<413::AID-CNE7>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Geinisman Y, Ganeshina O, Yoshida R, Berry RW, Disterhoft JF, Gallagher M. Aging, spatial learning, and total synapse number in the rat CA1 stratum radiatum. Neurobiol Aging. 2004;25:407–416. doi: 10.1016/j.neurobiolaging.2003.12.001. [DOI] [PubMed] [Google Scholar]
- Gerstein H, O'Riordan K, Osting S, Schwarz M, Burger C. Rescue of synaptic plasticity and spatial learning deficits in the hippocampus of Homer1 knockout mice by recombinant Adeno-associated viral gene delivery of Homer1c. Neurobiol Learn Mem. 2012;97:17–29. doi: 10.1016/j.nlm.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himeda T, Mizuno K, Kato H, Araki T. Effects of age on immunohistochemical changes in the mouse hippocampus. Mech Ageing Dev. 2005;126:673–677. doi: 10.1016/j.mad.2004.12.004. [DOI] [PubMed] [Google Scholar]
- Ippolito DM, Eroglu C. Quantifying synapses: an immunocytochemistry-based assay to quantify synapse number. J Vis Exp. 2010;pii:2270. doi: 10.3791/2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry MC, Soriano F, McNamara M, Page KJ, Schenk D, Games D, Hyman BT. Abeta deposition is associated with neuropil changes, but not with overt neuronal loss in the human amyloid precursor protein V717F (PDAPP) transgenic mouse. J Neurosci. 1997;17:7053–7059. doi: 10.1523/JNEUROSCI.17-18-07053.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr DS, Campbell LW, Applegate MD, Brodish A, Landfield PW. Chronic stress-induced acceleration of electrophysiologic and morphometric biomarkers of hippocampal aging. J Neurosci. 1991;11:1316–1324. doi: 10.1523/JNEUROSCI.11-05-01316.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinknecht KR, Bedenk BT, Kaltwasser SF, Grünecker B, Yen YC, Czisch M, Wotjak CT. Hippocampus-dependent place learning enables spatial flexibility in C57BL/6N mice. Front Behav Neurosci. 2012;6:87. doi: 10.3389/fnbeh.2012.00087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landfield PW, Baskin RK, Pitler TA. Brain aging correlates: retardation by hormonal-pharmacological treatments. Science. 1981a;214:581–584. doi: 10.1126/science.6270791. [DOI] [PubMed] [Google Scholar]
- Landfield PW, Braun LD, Pitler TA, Lindsey JD, Lynch G. Hippocampal aging in rats: a morphometric study of multiple variables in semithin sections. Neurobiol Aging. 1981b;2:265–275. doi: 10.1016/0197-4580(81)90034-8. [DOI] [PubMed] [Google Scholar]
- Lemere CA, Spooner ET, Leverone JF, Mori C, Clements JD. Intranasal immunotherapy for the treatment of Alzheimer's disease: Escherichia coli LT and LT(R192G) as mucosal adjuvants. Neurobiol Aging. 2002;23:991–1000. doi: 10.1016/S0197-4580(02)00127-6. [DOI] [PubMed] [Google Scholar]
- Li D, Wang J, Yew DT, Lucy Forster E, Yao Z. Age-related alterations of Nestin-immunoreactive neurons in rat basal forebrain with aged memory deficit. Neurochem Int. 2008;53:270–277. doi: 10.1016/j.neuint.2008.08.006. [DOI] [PubMed] [Google Scholar]
- Lian H, Yang L, Cole A, Sun L, Chiang AC, Fowler SW, Shim DJ, Rodriguez-Rivera J, Taglialatela G, Jankowsky JL, Lu HC, Zheng H. NFkappaB-activated astroglial release of complement C3 compromises neuronal morphology and function associated with Alzheimer's disease. Neuron. 2015;85:101–115. doi: 10.1016/j.neuron.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Jin M, Koeglsperger T, Shepardson NE, Shankar GM, Selkoe DJ. Soluble Aβ oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J Neurosci. 2011;31:6627–6638. doi: 10.1523/JNEUROSCI.0203-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Jin M, Zhang D, Yang T, Koeglsperger T, Fu H, Selkoe DJ. Environmental novelty activates beta2-adrenergic signaling to prevent the impairment of hippocampal LTP by Abeta oligomers. Neuron. 2013;77:929–941. doi: 10.1016/j.neuron.2012.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnusson KR, Scruggs B, Aniya J, Wright KC, Ontl T, Xing Y, Bai L. Age-related deficits in mice performing working memory tasks in a water maze. Behav Neurosci. 2003;117:485–495. doi: 10.1037/0735-7044.117.3.485. [DOI] [PubMed] [Google Scholar]
- Maier M, Peng Y, Jiang L, Seabrook TJ, Carroll MC, Lemere CA. Complement C3 deficiency leads to accelerated amyloid beta plaque deposition and neurodegeneration and modulation of the microglia/macrophage phenotype in amyloid precursor protein transgenic mice. J Neurosci. 2008;28:6333–6341. doi: 10.1523/JNEUROSCI.0829-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meaney MJ, Aitken DH, van Berkel C, Bhatnagar S, Sapolsky RM. Effect of neonatal handling on age-related impairments associated with the hippocampus. Science. 1988;239:766–768. doi: 10.1126/science.3340858. [DOI] [PubMed] [Google Scholar]
- Morrison JH, Baxter MG. The ageing cortical synapse: hallmarks and implications for cognitive decline. Nat Rev Neurosci. 2012;13:240–250. doi: 10.1038/nrn3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison JH, Hof PR. Selective vulnerability of corticocortical and hippocampal circuits in aging and Alzheimer's disease. Prog Brain Res. 2002;136:467–486. doi: 10.1016/S0079-6123(02)36039-4. [DOI] [PubMed] [Google Scholar]
- Ohmi Y, Ohkawa Y, Tajima O, Sugiura Y, Furukawa K, Furukawa K. Ganglioside deficiency causes inflammation and neurodegeneration via the activation of complement system in the spinal cord. J Neuroinflammation. 2014;11:61. doi: 10.1186/1742-2094-11-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Alcazar M, Daborg J, Stokowska A, Wasling P, Björefeldt A, Kalm M, Zetterberg H, Carlström KE, Blomgren K, Ekdahl CT, Hanse E, Pekna M. Altered cognitive performance and synaptic function in the hippocampus of mice lacking C3. Exp Neurol. 2014;253:154–164. doi: 10.1016/j.expneurol.2013.12.013. [DOI] [PubMed] [Google Scholar]
- Pistell PJ, Spangler EL, Kelly-Bell B, Miller MG, de Cabo R, Ingram DK. Age-associated learning and memory deficits in two mouse versions of the Stone T-maze. Neurobiol Aging. 2012;33:2431–2439. doi: 10.1016/j.neurobiolaging.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichwald J, Danner S, Wiederhold KH, Staufenbiel M. Expression of complement system components during aging and amyloid deposition in APP transgenic mice. J Neuroinflammation. 2009;6:35. doi: 10.1186/1742-2094-6-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudy JW, Huff NC, Matus-Amat P. Understanding contextual fear conditioning: insights from a two-process model. Neurosci Biobehav Rev. 2004;28:675–685. doi: 10.1016/j.neubiorev.2004.09.004. [DOI] [PubMed] [Google Scholar]
- Rutten BP, Van der Kolk NM, Schafer S, van Zandvoort MA, Bayer TA, Steinbusch HW, Schmitz C. Age-related loss of synaptophysin immunoreactive presynaptic boutons within the hippocampus of APP751SL, PS1M146L, and APP751SL/PS1M146L transgenic mice. Am J Pathol. 2005;167:161–173. doi: 10.1016/S0002-9440(10)62963-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapolsky RM, Krey LC, McEwen BS. Prolonged glucocorticoid exposure reduces hippocampal neuron number: implications for aging. J Neurosci. 1985;5:1222–1227. doi: 10.1523/JNEUROSCI.05-05-01222.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, Ransohoff RM, Greenberg ME, Barres BA, Stevens B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74:691–705. doi: 10.1016/j.neuron.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schliebs R, Arendt T. The significance of the cholinergic system in the brain during aging and in Alzheimer's disease. J Neural Transm. 2006;113:1625–1644. doi: 10.1007/s00702-006-0579-2. [DOI] [PubMed] [Google Scholar]
- Sheng M, Kim MJ. Postsynaptic signaling and plasticity mechanisms. Science. 2002;298:776–780. doi: 10.1126/science.1075333. [DOI] [PubMed] [Google Scholar]
- Stephan AH, Madison DV, Mateos JM, Fraser DA, Lovelett EA, Coutellier L, Kim L, Tsai HH, Huang EJ, Rowitch DH, Berns DS, Tenner AJ, Shamloo M, Barres BA. A dramatic increase of C1q protein in the CNS during normal aging. J Neurosci. 2013;33:13460–13474. doi: 10.1523/JNEUROSCI.1333-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SW, Barres BA. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131:1164–1178. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- Stoltzner SE, Grenfell TJ, Mori C, Wisniewski KE, Wisniewski TM, Selkoe DJ, Lemere CA. Temporal accrual of complement proteins in amyloid plaques in Down's syndrome with Alzheimer's disease. Am J Pathol. 2000;156:489–499. doi: 10.1016/S0002-9440(10)64753-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stover KR, Brown RE. Age-related changes in visual acuity, learning and memory in the APPswe/PS1dE9 mouse model of Alzheimer's disease. Behav Brain Res. 2012;231:75–85. doi: 10.1016/j.bbr.2012.02.044. [DOI] [PubMed] [Google Scholar]
- von Bohlen und Halbach O, Unsicker K. Morphological alterations in the amygdala and hippocampus of mice during ageing. Eur J Neurosci. 2002;16:2434–2440. doi: 10.1046/j.1460-9568.2002.02405.x. [DOI] [PubMed] [Google Scholar]
- von Bohlen und Halbach O, Zacher C, Gass P, Unsicker K. Age-related alterations in hippocampal spines and deficiencies in spatial memory in mice. J Neurosci Res. 2006;83:525–531. doi: 10.1002/jnr.20759. [DOI] [PubMed] [Google Scholar]
- Wessels MR, Butko P, Ma M, Warren HB, Lage AL, Carroll MC. Studies of group B streptococcal infection in mice deficient in complement component C3 or C4 demonstrate an essential role for complement in both innate and acquired immunity. Proc Natl Acad Sci U S A. 1995;92:11490–11494. doi: 10.1073/pnas.92.25.11490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West MJ, Gundersen HJ. Unbiased stereological estimation of the number of neurons in the human hippocampus. J Comp Neurol. 1990;296:1–22. doi: 10.1002/cne.902960102. [DOI] [PubMed] [Google Scholar]
- Wong AA, Brown RE. Age-related changes in visual acuity, learning and memory in C57BL/6J and DBA/2J mice. Neurobiol Aging. 2007;28:1577–1593. doi: 10.1016/j.neurobiolaging.2006.07.023. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Yan F, Lin AH, Lambris JD, Alexander JJ, Quigg RJ, Masliah E. Prominent neurodegeneration and increased plaque formation in complement-inhibited Alzheimer's mice. Proc Natl Acad Sci U S A. 2002;99:10837–10842. doi: 10.1073/pnas.162350199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada M, Takeshita T, Miura S, Murata K, Kimura Y, Ishii N, Nose M, Sakagami H, Kondo H, Tashiro F, Miyazaki JI, Sasaki H, Sugamura K. Loss of hippocampal CA3 pyramidal neurons in mice lacking STAM1. Mol Cell Biol. 2001;21:3807–3819. doi: 10.1128/MCB.21.11.3807-3819.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu YF, Zhai F, Dai CF, Hu JJ. The relationship between age-related hearing loss and synaptic changes in the hippocampus of C57BL/6J mice. Exp Gerontol. 2011;46:716–722. doi: 10.1016/j.exger.2011.04.007. [DOI] [PubMed] [Google Scholar]
- Zabel MK, Kirsch WM. From development to dysfunction: microglia and the complement cascade in CNS homeostasis. Ageing Res Rev. 2013;12:749–756. doi: 10.1016/j.arr.2013.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]